Патент на изобретение №2237479
|
||||||||||||||||||||||||||
(54) НУКЛЕОЗИДЫ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ ПРОТИВ ВИРУСА ГЕПАТИТА В
(57) Реферат:
Изобретение относится к медицине, в частности гепатологии и вирусологии, и касается лечения инфекции, вызванной вирусом гепатита В. Для этого разработаны соединения и фармацевтические композиции, обладающие специфической противовирусной активностью, а также способ лечения вирусной инфекции гепатита В. Эти разработки касаются введения эффективного количества производных
Данное изобретение относится к области способов борьбы с вирусом гепатита В (также обозначаемом как HBV), включающих введение эффективного количества одного или нескольких Предпосылки изобретения HBV является второй после табака причиной рака у человека. Механизм индуцирования рака HBV неизвестен, несмотря на предположение, что он может непосредственно или опосредованно вызывать развитие опухоли в результате хронического воспаления, цирроза и регенерации клеток, связанных с инфекцией. Вирус гепатита В достиг эпидемического уровня во всем мире. После двух-шестимесячного инкубационного периода, на протяжении которого носитель не знает об инфекции, HBV-инфекция может привести к острому гепатиту и повреждению печени, вызывающему боль в брюшной полости, желтухе и повышенному уровню некоторых ферментов в крови. HBV может вызвать молниеносный (фульминантный) гепатит, быстро прогрессирующую, зачастую фатальную форму заболевания, при которой поражаются большие участки печени. Обычно пациенты излечиваются от острого вирусного гепатита. Однако у некоторых пациентов высокий уровень вирусного антигена сохраняется в крови в течение продолжительного или неопределенного периода времени, вызывая хроническую инфекцию. Хроническая инфекция может привести к хроническому персистирующему гепатиту. Пациенты, инфицированные хроническим персистирующим HBV, наиболее часто встречаются в развивающихся странах. К середине 1991 года насчитывалось приблизительно 225 млн. хронических носителей HBV в одной только Азии, а по всему миру – почти 300 млн. носителей. Хронический персистирующий гепатит может вызывать усталость, цирроз печени и печеночно-клеточный рак, первичный рак печени. В западных индустриально развитых странах в группы высокого риска заражения HBV входят люди, находящиеся в контакте с носителями HBV или образцами их крови. Фактически эпидемиология HBV весьма напоминает эпидемиологию синдрома приобретенного иммунодефицита, что объясняет тот факт, почему HBV-инфекция распространена среди больных СПИДом или ВИЧ-ассоциируемыми инфекциями. Однако HBV более контагиозен, чем ВИЧ. Ежедневное лечение а-интерфероном, генетически сконструированным белком, оказалось перспективным. Для иммунизации пациентов против HBV также была разработана вакцина, получаемая из сыворотки человека. Вакцины были получены в результате генной инженерии. Несмотря на ее эффективность, получение вакцины является трудоемким, поскольку получение сыворотки человека от хронических носителей ограничено, а процедура очистки является длительной и дорогостоящей. Кроме того, каждая партия вакцины, получаемая из различных сывороток, должна быть проверена на шимпанзе с целью безопасности. Помимо этого такая вакцина не помогает пациентам, уже инфицированным вирусом. Был разработан ряд синтетических нуклеозидов, проявляющих активность против HBV. (-)-Энантиомер ВСН-189 (2′,3′-дидеокси-3′-тиацитидин), известный как ЗТС, заявленный в патенте США №5 539 116 на имя Liotta et al., в настоящее время проходит клинические испытания на лечение гепатита В. См. также ЕРА 0 494 119 A1, поданную BioChem Pharma, Inc.
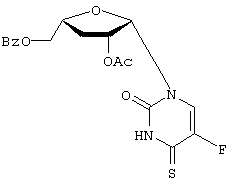
Патенты США №№5 565 438, 5 567 688 и 5 587 362 (Chu et al.) описывают применение 2′-фтор-5-метил- Пенцикловир (2-амино-1,9-дигидро-9-[4-окси-3-оксиметил)-бутил]6Н-пурин-6-он; PCV) обладает установленной активностью против гепатита В. См. патенты США 5 075 445 и 5 684 153. Адефовир (9-[2-(фосфонометокси)этил]аденин, также обозначаемый как РМЕА или [2-(6-амино-9Н-пурин-9-ил)этокси] метилфосфоновая кислота), также обладает установленной активностью против гепатита В. См., например, патенты США №№5 641 763 и 5 142 051. Yale University и The University of Georgia Research Foundation, Inc. раскрывают применение L-FDDC (5-фтор-3′-тиа-2’3′-дидеоксицитидин) для лечения вируса гепатита В в WO 92/18517. Другие лекарственные препараты, применяемые для лечения HBV, включают аденозин арабинозид, тимозин, ацикловир, фосфоноформиат, зидовудин, (+)-цианиданол, хинакрин и 2′-фторарабинозил-5-иодурацил. Патенты США №№ 5 444 063 и 5 684 010 на имя Emory University раскрывают применение энантиомерно чистых нуклеозидов WO 96/40164, поданная Emory University, UAB Research Foundation и the Centre National de la Recherche Scientifique, описывает ряд WO 95/07287, также поданная Emory University, UAB Research Foundation и the Centre National de la Recherche Scientifique, раскрывает нуклеозиды 2′ или 3′ деокси и 2′,3′-дидеокси- WO 96/13512, поданная Genencor International, Inc., и Lipitek, Inc., описывает получение нуклеозидов L-рибофуранозила в качестве противоопухолевых агентов и вируцидов. WO 95/32984 описывает сложные липидные эфиры монофосфатов нуклеозидов в качестве иммуносупрессорных лекарственных препаратов. DE 4224737 описывает нуклеозиды цитозина и их фармацевтическое применение. Tsai et al. в Biochem. Pharmacol. 48(7), pages 1477-81, 1994, описывают действие аналогов 2′- Galvez, J. Chem. Inf. Comput. Sci. (1994), 35(5), 1198-203 описывает молекулярный подсчет Mahmoudian, Pharm. Research 8(1), 43-6 (1991) описывает количественный структурно-активный анализ связи ВИЧ-агентов, таких как Патент США №5 703 058 описывает нуклеозиды (5-карбоксимидо или 5-фтор)-(2′,3′-ненасыщенного или 3′-модифицированного) пиримидина для лечения ВИЧ или HBV. Lin et al. в J. Med. Chem. 31(2), 336-340 (1988) описывают синтез и противовирусную активность 3′-азидо-аналогов Существенным шагом в способе действия пуриновых и пиримидиновых нуклеозидов против вирусных заболеваний, в частности HBV и ВИЧ, является их метаболическое активирование клеточными и вирусными киназами для получения моно-, ди- и трифосфатных производных. Биологически активным видом многих нуклеозидов является трифосфатная форма, ингибирующая ДНК-полимеразу или обратную транскриптазу, либо обрывающая цепь. Нуклеозидные производные, разработанные до настоящего времени для лечения HBV и ВИЧ, представлены для введения пациенту в нефосфорилированном виде, несмотря на то, что нуклеозид должен быть фосфорилирован в клетке до проявления своего антивирусного действия, поскольку трифосфатную форму обычно дефосфорилируют прежде, чем она достигнет клетки, в противном случае она плохо абсорбируется клеткой. Нуклеотиды в целом пересекают клеточные мембраны с большим трудом и обычно не очень эффективны in vitro. Попытки модифицирования нуклеотидов для повышения их абсорбции и эффективности описаны R. Jones and N. Bischofberger, Antiviral Research, 27 (1995) 1-17. Ввиду того, что вирус гепатита В достиг во всем мире эпидемического уровня и оказывает сильное, а зачастую и трагическое действие на инфицированного пациента, сохраняется высокая потребность в разработке новых эффективных низкотоксичных фармацевтических агентов для лечения людей, инфицированных вирусом. Следовательно, целью настоящего изобретения является разработка соединений, композиций и способов для лечения людей или других пациентов, инфицированных HBV. Краткое описание изобретения Описан способ лечения HBV-инфекции у людей и других животных, включающий введение эффективного количества В соответствии с одним из вариантов осуществления данного изобретения активное соединение представляет собой где R представляет Н, ацил, монофосфат, дифосфат или трифосфат либо стабилизированное фосфатное производное (для образования стабилизированного нуклеотидного пролекарства), а R’ представляет Н, ацил или алкил. В соответствии с другим вариантом осуществления данного изобретения активное соединение представляет собой где R представляет Н, ацил, монофосфат, дифосфат или трифосфат либо стабилизированное фосфатное производное (для получения стабилизированного нуклеотидного пролекарства), а R’ представляет Н, ацил или алкил. Описываемые нуклеозиды либо их фармацевтически приемлемые пролекарства, сложные эфиры или соли, либо фармацевтически приемлемые составы, содержащие такие соединения, могут быть использованы для профилактики и лечения HBV-инфекций и подобных им состояний, таких как анти-HBV антитело-положительные и HBV-положительные состояния, хроническое воспаление печени, вызываемое HBV, цирроз, острый гепатит, фульминантный гепатит, хронический персистирующий гепатит и усталость. Такие соединения или составы также могут быть использованы профилактически для предотвращения или замедления прогрессирования клинического заболевания у анти-НВV-антитело или HBV-антиген-положительных пациентов, или тех, кто находится в контакте с HBV. В соответствии с одним из вариантов его осуществления настоящее изобретение включает способ лечения людей, инфицированных HBV, включающий введение необходимого для лечения HBV количества пролекарства конкретных L-(2′ или 3′)-А-5-Fddc-нуклеозидов. Используемый в данном описании термин “пролекарство” относится к фармацевтически приемлемому производному конкретно описываемого нуклеозида, превращаемого в нуклеозид при введении in vivo либо обладающего собственной активностью. Неограничивающими примерами являются 5′ и N4-цитозин-ацилированные или алкилированные производные активного соединения, а также производные 5′-монофосфата, дифосфата или трифосфата, другие фосфаты либо стабилизированные нуклеотидные пролекарства, более подробно описываемые ниже. Например, нуклеозид присутствует в виде монофосфата, дифосфата или трифосфата в составе, защищающем соединение от дефосфорилирования. Составы включают липосомы, липосферы, микросферы или наносферы (из которых последние три могут быть нацелены на инфицированные клетки). В соответствии с одним из вариантов осуществления настоящего изобретения одно или несколько активных соединений вводят попеременно или в сочетании с одним или несколькими другими анти-НВV-агентами, осуществляя эффективное анти-НВV-лечение. Примеры анти-НВV-агентов, которые могут быть использованы в чередующейся или сочетанной терапии, включают, но не ограничиваются ими, цис-2-оксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолан, предпочтительно по существу в виде (-)-оптического изомера (“FTC”, см. WO 92/14743); (-)-энантиомер цис-2-оксиметил-5- (цитозин-1-ил) -1, 3-оксатиолан (ЗТС); нуклеозиды Может быть применен любой способ чередования, обеспечивающий лечение пациента. Неограничивающие примеры схем чередования включают 1-6 недель введения эффективного количества агента, а затем 1-6 недель введения эффективного количества второго анти-НВV-агента. Сочетанная терапия обычно включает одновременное введение эффективного соотношения доз двух и более анти-НВV-агентов. Ввиду того, что HBV часто обнаруживается у пациентов, также имеющих положительную реакцию на анти-ВИЧ-антитело или ВИЧ-антиген либо контактирующих с ВИЧ, описываемые здесь активные анти-НВV-соединения либо их производные или пролекарства могут быть введены в соответствующих обстоятельствах в сочетании или попеременно с анти-ВИЧ-веществами. В соответствии с одним из вариантов осуществления настоящего изобретения вторым антивирусным агентом для лечения ВИЧ является ингибитор обратной транскриптазы (“RTI”), который может представлять собой либо синтетический нуклеозид (“NRTI”), либо ненуклеозидное соединение (“NNRTI”), в соответствии с альтернативным вариантом в случае с ВИЧ второй (или третий) антивирусный агент может представлять собой ингибитор протеазы. В соответствии с другими вариантами второе (или третье) соединение может представлять собой аналог пирофосфата или ингибитор связывания слияния. Данные по резистентности, собранные in vitro и in vivo для многих антивирусных соединений, приведены Schinazi et al., Mutations in retroviral genes associated with drug resistance, International Antiviral News, Volume 1(4), International Medical Press, 1996. Предпочтительные примеры антивирусных агентов, которые могут быть использованы в сочетании либо попеременно с описываемыми здесь соединениями для HBV-терапии, включают 2-оксиметил-5-(5-фторцитозин-1-ил)-1,3-оксатиолан (FTC); (-)-энантиомер 2-оксиметил-5-(цитозин-1-ил)-1,3-оксатиолана (3ТС); карбовир, ацикловир, интерферон, L-FMAU и нуклеозиды Активные анти-НВV-агенты также могут быть введены в сочетании с антибиотиками, другими антивирусными соединениями, противогрибковыми агентами либо другими фармацевтическими агентами, вводимыми для лечения вторичных инфекций. Краткое описание фигур Фигура 1 – иллюстрация общей реакционной схемы для стереоспецифического синтеза 3′-замещенных Фигура 2 – иллюстрация общей реакционной схемы для стереоспецифического синтеза 2′-замещенных Фигура 3 – иллюстрация способа получения Фигура 4 – иллюстрация способа получения Подробное описание изобретения Используемые в данном описании термины “по существу в виде”, “по существу при отсутствии” или “по существу свободный от” относятся к нуклеозидной композиции, включающей по меньшей мере приблизительно 95%, предпочтительно приблизительно 97, 98, 99 или 100% одного энантиомера определенного нуклеозида. Термин “алкил” в данном описании, если не указано иначе, относится к насыщенному прямолинейному, разветвленному или циклическому, первичному, вторичному или третичному углеводороду с C1-С10 и конкретно включает метил, этил, пропил, изопропил, циклопропил, бутил, изобутил, т-бутил, циклобутил, пентил, циклопентил, изопентил, неопентил, гексил, изогексил, циклогексил, циклогексилметил, 3-метилпентил, 2,2-диметилбутил и 2,3-диметилбутил. Алкильная группа может быть необязательно замещена одним или несколькими остатками, выбираемыми из группы, включающей гидроксил, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфоновую кислоту, сульфат, фосфоновую кислоту, фосфат или фосфонат, незащищенные или защищенные надлежащим образом, как известно специалистам в данной области, например, как указывает Greene et al., “Protective Groups in Organic Synthesis”, John Wiley and Sons, Second. Edition., 1991. Термин “низший алкил” в данном описании, если не указано иначе, относится к C1-C4 этил, пропил, бутил, пентил, гексил, изопропил, изобутил, втор-бутил или т-бутилгруппе. В соответствии с данным описанием термин “ацил” конкретно включает, но не ограничивается ими, С(О)алкил, С(O)арил, ацетил, пропионил, бутирил, пентаноил, 3-метилбутирил, водородсукцинат, 3-хлорбензоат, бензоил, ацетил, пивалоил, мезилат, пропионил, валерил, капроновый, каприловый, каприновый, лауриновый, миристиновый, пальмитиновый, стеариновый и олеиновый либо остаток аминокислотной части. Термин “арил” в данном описании, если не указано иначе, относится к фенилу, бифенилу или нафтилу, предпочтительно фенилу. Арильная группа может быть необязательно замещена одним или несколькими остатками, выбираемыми из группы, включающей гидроксил, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфоновую кислоту, сульфат, фосфоновую кислоту, фосфат или фосфонат, незащищенные или защищенные надлежащим образом, как известно специалистам в данной области, например, в соответствии с указаниями Greene et al., “Protective Groups in Organic Synthesis”, John Wiley and Sons, Second Edition, 1991. В данном описании термин “аминокислота” относится к природным и неприродным аминокислотам и включает, но не ограничивается ими, аланил, валинил, лейцинил, изолейцинил, пролинил, фенилаланинил, триптофанил, метионинил, глицинил, серинил, треонинил, цистеинил, тирозинил, аспарагинил, глутаминил, аспартоил, глутаоил, лизинил, артининил и гистидинил. Термин “пролекарство” в данном описании относится к фармацевтически приемлемому производному конкретно описываемого нуклеозида, превращаемого в нуклеозид при введении in vivo или обладающего собственной активностью. Неограничивающими примерами являются 5′- и N4-цитозинацилированные или алкилированные производные активного соединения, а также производные 5′-монофосфата, дифосфата или трифосфата, другие фосфаты, либо стабилизированные нуклеотидные пролекарства, либо 5′-эфирные липиды, как подробно описано ниже. Например, нуклеозид присутствует в виде монофосфата, дифосфата или трифосфата в составе, защищающем соединение от дефосфорилирования. Составы включают липосомы, липосферы, микросферы или наносферы (из которых три последних могут быть нацелены на инфицированные клетки). Описываемое изобретение включает способ и композицию для лечения HBV-инфекции и других вирусов, воспроизводимых подобным образом, у людей или других животных, включающий введение эффективного для лечения HBV количества одного или нескольких вышеуказанных соединений, либо их физиологически приемлемых солей, необязательно с фармацевтически приемлемым носителем. Соединения в соответствии с настоящим изобретением либо обладают анти-НВV-активностыо, либо подвергаются метаболизму до соединения или соединений, проявляющих анти-HBV-активность. Структура и получение активных нуклеозидов Стереохимия Поскольку 1’ и 4′ атомы углерода сахара (в дальнейшем называемого в целом “сахарным остатком”) нуклеозидов являются хиральными, то их неводородные заместители (CH2OR и пиримидиновое или пуриновое основание соответственно) могут иметь либо цис- (на одной и той же стороне) либо трансконфигурацию (на противоположных сторонах) по отношению к кольцевой системе сахара. Следовательно, четыре оптических изомера представлены следующими конфигурациями (при ориентации сахарного остатка в горизонтальной плоскости таким образом, чтобы “первичный” кислород (между атомами С1′ и С4′) находился сзади): “ Активные нуклеозиды в соответствии с настоящим изобретением имеют Составы пролекарства Описываемые здесь нуклеозиды могут быть введены в виде любого производного, которое после введения реципиенту способно прямо или косвенно образовывать активное родительское соединение либо проявляет собственную активность. В соответствии с одним из вариантов осуществления настоящего изобретения водород 5′-ОН-группы замещают C1-C20 алкилом; ацилом, включающим такой ацил, в котором некарбонильный остаток сложной эфирной группы выбирают из прямолинейного, разветвленного или циклического C1-C20 алкила, фенила или бензила; природной или неприродной аминокислотой; 5′-сложным эфирным липидом или 5′-сложным фосфоэфирным липидом; алкоксиалкилом, включающим метоксиметил; аралкилом, включающим бензил; арилоксиалкилом, таким как феноксиметил; арилом, включающим фенил, необязательно замещенный галогеном, C1-C4 алкилом или C1-C4 алкокси; дикарбоновой кислотой, такой как янтарная кислота; сложными эфирами сульфоната, такими как алкил или аралкилсульфонил, включая метансульфонил; либо моно-, ди- или трифосфатным эфиром. Один или оба водорода аминогрупп на пуриновом или пиримидиновом основании могут быть замещены C1-C20 алкилом; ацилом, в котором некарбонильный остаток сложноэфирной группы выбирают из прямолинейного, разветвленного или циклического C1-C20 алкила, фенила или бензила; алкоксиалкилом, включая метоксиметил; аралкилом, включая бензил; арилоксиалкилом, таким как феноксиметил; арилом, включая фенил, необязательно замещенный галогеном, C1-C4 алкилом или C1-C4 алкокси. Активный нуклеозид также может быть в виде 5′ -сложноэфирного липида, как описано в следующих ссылках: Kucera, L.S., N. lyer, E. Leake, A. Raben, Modest E.J., D. L.W., and C. Piantadosi, 1990. Novel membrane-interactive ether lipid analogs that inhibit infectious HIV-1 production and induce defective virus formation. AIDS Res Hum Retroviruses. 6:491-501; Piantadosi, C., J. Marasco C.J., S.L. Morris-Natschke, K.L. Meyer, F. Gumus, J.R. Surles, K.S. Ishaq, L.S. Kucera, N. lyer, C.A. Wallen, S. Piantadosi, and E.J. Modest, 1991. Synthesis and evaluation of novel ether lipid nucleoside conjugates for anti-HIV activity. J. Med. Chem. 34:1408.1414; Hostetler, K.Y., D.D. Richman, D.A. Carson, L.M. Stuhmiller, G.M. T. van Wijk, and H. van den Bosch. 1992. Greatly enhanced inhibition of human immunodeficiency virus type 1 replication in СЕМ and HT4-6C cells by 3′-deoxythymidine diphosphate dimyristoylglycerol, a lipid prodrug of 3′-deoxythymidine. Antimicrob Agents Chemother. 36:2025.2029; Hostetler, K.Y., L.M. Stuhmiller, H.B. Lenting, H. van den Bosch, and D.D. Richman, 1990. Synthesis and antiretroviral activity of phospholipid analogs of azidothymidine and other antiviral nucleosides. J. Biol. Chem. 265:6112.7. Стабилизированные нуклеотиды Любые описываемые здесь нуклеозиды могут быть введены в виде нуклеотидного пролекарства или фосфолипидного пролекарства для повышения активности, биодоступности, стойкости или какого-либо другого изменения свойств нуклеозида. Количество лигандов нуклеотидного пролекарства известно. В целом алкилирование, ацилирование либо другая липофильная модификация моно-, ди- или трифосфата нуклеозида усиливает стойкость нуклеотида. Примерами групп-заместителей, которые могут замещать один или несколько атомов водорода в фосфатном остатке, являются алкил, арил, стероиды, углеводы, включая сахара, 1,2-диацилглицерин и спирты. Многие из них описаны В. Jones and H. Bischofberger, Antiviral Research, 27 (1995) 1-17. Любые из них могут быть использованы в сочетании с описываемыми нуклеозидами для получения желаемого эффекта. Неограничивающие примеры нуклеотидных пролекарств описаны в следующих ссылках: Но, D.H.W. (1973) Distribution of Kinase and deaminase of Ib-D-arabinofuranosylcytosine in tissues of man and muse. Cancer Res. 33,2816-2820; Holy, A. (1993) Isopolar phosphorous-modified nucleotide analogues. In: De Clercq (Ed.), Advances in Antiviral Drug Design, Vol. I, JAI Press, pp. 179-231; Hong, C.I., Nechaev, A., and West, C.R. (1979a) Synthesis and antitumor activity of Ib-D-arabinofuranosylcytosine conjugates of cortisol and cortisone. Biochem. Biophys. Rs. Commun. 88,1223-1229; Hong, C.I., Nechaev, A., Kirisits, A.J. Buchheit, D.J. and West, C.R. (1980) Nucleoside conjugates as potential antitumor agents. 3. Synthesis and antitumor activity of l-(b-D-arabinofuranosyl)cytosine conjugates of corticosteriods and selected lipophilic alcohols. J. Med. Chem. 28,171-177; Hostetler, K.Y. Stuhmiller, L.M., Lenting, H.B.M. van den Bosch, H. and Richman, D.D. (1990) Synthesis and antiretrioviral activity of phospholipid analogs of azidothymidine and other antiviral nucleosides. J. Biol. Chem. 265, 6112-6117; Hostetler, K..Y., Carson, D.A. and Richman, D.D. (1991); Phosphatidylazidothymidine: mechanism of antiretroviral action in СЕМ cells. J. Biol. Chem. 266, 11714-11717; Hostetler, K.Y., Korba, B. Sridhar, C., Gardener, M. (1994a) Antiviral activity of phosphatidyl-dideoxycytidine in hepatitis B-infected cells and enhanced hepatic uptake in mice. Antiviral Res. 24, 59-67; Hostetler, K.Y., Richman, D.D., Sridhar, C.N. Felgner, P.L, Felgner, J., Ricci, J., Gardener, M.F. Selleseth, D.W. and Ellis, M.N. (1994b) Phosphatidylazidothymidine and phosphatidyl-ddC: Assessment of uptake in mouse lymphoid tissues and antiviral activities in human immunodeficiency virus-infected cells and in rauscher leukemia virus-infected mice. Antimicrobial Agents Chemolher. 38, 2792-2797; Hunston, R.N., Jones, A.A. McGuigan, C., Walker, R.T., Balzarini, J., and De Clercq, E. (1984) Synthesis and biological properties of some cyclic phosphotrieslers derived from 2′-deoxy-5-fluorouridine. J. Med. Chem. 27, 440-444; Ji, Y.H., Moog, C., Schmitt, G., Bischoff, P. and Luu, B. (1990); Monophosphoric acid diesters of 7b-hydroxycholesterol and of pyrimidine nucleosides as potential antitumor agents: synthesis and preliminary evaluation of antitumor activity. J. Med. Chem. 33, 2264-2270; Jones, A.S„ McGuigan, C., Walker, R.T. Balzarini, J. and DeClercq, E. (1984) Synthesis, properties, and biological activity of some nucleoside cyclic phosphoramidates. J. Chem. Soc. Perkin Trans. I, 1471-1474; Juodka, B.A. and Smrt, J. (1974) Synthesis of ditribonucleoside phosph(P®N) amino acid derivatives. Coll. Czech. Chem. Comm. 39, 363-968; Kataoka, S., Imai, J., Yamaji, N., Kato, M., Saito, M., Kawada, T. and Imai, S. (1989) Alkylacted cAMP derivatives; selective synthesis and biological activities. Nucleic Acids Res. Sym. Ser., 21, 1-2; Kataoka, S., Uchida, R. and Yamaji, N. (1991) A convenient synthesis of adenosine 3′,5’cyclic phosphate (cAMP) benzyl and methyl triesters. Heterocycles 32, 1351-1356; Kinchington, D., Harvey, J.J., O’Connor, T.J., Jones, B.C.N.M., Devine, K.G., Taylor-Robinson, D., Jeffries, D.J. and McGuigan, C. (1992) Comparison of antiviral effects of zidovudine phosphoramidate and phosphorodiamidate derivatives against HIV and ULV in vitro. Antiviral Chem. Chemother. 3,107-112; Kodama, К., Morozumi, M., Saitoh, K.I., Kuninaka, H., Yoshino, H. and Saneyoshi, M. (1989) Antitumor activity and pharmacology of 1 -b-D-arabinofuranosylcytosine -5′-stearylphosphate; an orally active derivative of 1-b-D-arabinofuranosylcytosine. Jpn. J. Cancer Res. 80, 679-685; Korty, M. and Engels, J. (1979) The effects ofadenosine- and guanosine 3′, 5’phosphoric and acid benzyl esters on guinea-pig ventricular myocardium. Naunyn-Schmiedeberg’s Arch. Pharmacol. 310, 103-111; Kumar, A., Goe, P.L., Jones, A.S. Walker, R.T. Balzarini, J. and De Clercq, E. (1990) Synthesis and biological evaluation of some cyclic phosphoramidate nucleoside derivatives. J. Med. Chem. 33,2368-2375; LeBec, С., and Huynh-Dinh, T. (1991) Synthesis of lipophilic phosphate triester derivatives of 5-fluorouridine and arabinocytidine as anticancer prodrugs. Tetrahedron Lett. 32,6553-6556; Lichtenstein, J., Bamer, H.D. and Cohen, S.S. (I960) The metabolism of exogenously supplied nucleotides by Escherichia co!i„ J. Biol. Chem. 235, 457-465; Lucthy, J., Von Daeniken, A., Friederich, J. Manthey, В., Zweifel, J., Schlatter, C. and Benn, M.H. (1981) Synthesis and toxicological properties of three naturally occurring cyanoepithioalkanes. Mitt. Geg. Lebensmittelunters. Hyg. 72,131-133 (Chem. Abstr. 95,127093); McGuigan, C. Tollerfield, S.M. and Riley, P.A. (1989) Synthesis and biological evaluation of some phosphate triester derivatives of the antiviral drug Ara. Nucleic Acids Res. 17,6065-6075; McGuigan, C., Devine, K.G., O’Connor, T.J., Galpin, S.A„ Jeffries, D.J. and Kinchington, D. (1990a) Synthesis and evaluation of some novel phosphoramidate derivatives of3′-azido-3′-deoxythymidine (AZT) as ani-HIV compounds. Antiviral Chem. Chemother. 1,107-113; McGuigan, C., O’Connor, T.J., Nicholls, S.R. Nickson, C. and Kinchington, D. (1990b) Synthesis and anti-HIV activity of some novel substituted dialky phosphate derivatives of AZT and ddCyd. Antiviral Chem. Chemother. 1, 355-360; McGuigan, С., Nicholls, S.R., O’Connor, T.J., and Kinchingion, D. (1990с) Synthesis of some novel dialkyi phosphate derivative of 3′-modified nucleosides as potential anti-AIDS drugs. Antiviral Chem. Chemother. 1, 25-33; McGuigan, C., Devine, K..G., O’Connor, T.J., and Kinchington, D.(1991) Synthesis and anti-HIV activity of some haloalky phosphoramidate derivatives of 3′-azido-3’deoxythylmidine (AZT); potent activity of the trichloroethyl methoxyalaninyl compound. Antiviral Res. 15,255-263; McGuigan, С., Pathirana, R.N., Mahmood, N., Devine, K.G. and Hay, A.J. (1992) Aryl phosphate derivatives of AZT retain activity against HIV1 in cell lines which are resistant to the action of AZT. Antiviral Res. 17, 311-321; McGuigan, С., Pathirana, R.N., Choi, S.M., Kinchington, D. and O’Connor, T.J. (1993a) Phosphoramidate derivatives of AZT as inhibitors ofHIV; studies on the carboxyl terminus. Antiviral Chem. Chemother. 4, 97-101; McGuigan, С., Pathirana, R.N., Balzarini, J. and De Clercq, E. (1993b) Intracellular delivery ofbioactive AZT nucleotides by aryl phosphate derivatives of AZT. J. Med. Chem. 36, 1048-1052. Производные алкилгидрофосфоната анти-ВИЧ-агента AZT могут быть менее токсичны, чем родительский нуклеозидный аналог. Antiviral Chem. Chemother. 5,271-277; Meyer, R. В., Jr., Shuman, D.A. and Robins, R.K. (1973) Synthesis of purine nucleoside 3′,5′-cyclic phosphoramidates. Tetrahedron Lett. 269-272; Nagyvary, J. Gohil, R.N., Kirchner, C.R. and Stevens, J.D. (1973) Studies on neutral esters of cyclic AMP, Biochem. Biophys. Res. Commun. 55, 1072-1077; Namane, A. Gouyette, C., Fillion, M.P., Fillion, G. and Huynh-Dinh, T. (1992) Improved brain delivery of AZT using a glycosyi phosphotriester prodrug. J. Med. Chem. 35, 3039-3044; Nargeot, J. Nerbonne, J.M. Engels, J. and Leser, H.A. (1983) Natl. Acad. Sci. U.S.A. 80, 2395-2399; Nelson, K.A., Bentrude, W.G., Stser, W.N. and Hutchinson, J.P. (1987) The question of chair-twist equilibria for the phosphate rings of nucleoside cyclic 3′,5’monophosphates. ‘HNMR and x-ray crystallographic study of the diasteromers of thymidine phenyl cyclic 3′,5′-monophosphate. J. Am. Chem. Soc. 109, 4058-4064; Nerbonne, J.M., Richard, S., Nargeot, J. and Lester, H.A. (1984) New photoactivatable cyclic nucleotides produce intracellular jumps in cyclic AMP and cyclic GMP concentrations. Nature 301, 74-76; Neumann, J.M., Herve, M., Debouzy, J.C., Guerra, F.I., Gouyette, C., Dupraz. B. and Huynh-Dinh, T. (1989) Synthesis and transmembrane transport studies by NMR of aglucosyi phospholipid of thymidine. J. Am. Chem. Soc. Ill, 4270-4277; Ohno, R., Tatsumi, N.. Hirano, M., Imai, K. Mizoguchi, H., Nakamura, Т., Kosaka, M, Takatuski, К., Yamaya, Т., Toyama, К., Yoshida, Т., Masaoka, Т., Hashimoto, S., Ohshima, Т., Kimura, I., Yamada, K. and Kimura, J. (1991) Treatment of myelodysplastic syndromes with orally administered 1-b-D-rabinofuranosylcytosine -5’stearylphosphate. Oncology 48,451-455. Palomino, E., Kessle, D. and Horwitz, J.P. (1989) A dihydropyridine carrier system for sustained delivery of 2′, 3’dideoxynucleosides to the brain. J. Med. Chem. 32,622-625; Perkins, R.M., Barney, S., Wittrock, R., Clark, P.H., Levin, R. Lambert, D.M., Petteway, S.R., Serafinowska, H.T., Bailey, S.M„ Jackson, S., Harnden, M.R. Ashton, R., Sutton. D., Harvey, J.J. and Brown, A.G. (1993) Activity of BRL47923 and its oral prodrug, SB203657A against a rauscher murine leukemia virus infection in mice. Antiviral Res. 20 (Suppl. I). 84; Piantadosi, C., Marasco, C.J., Jr., Morris-Natschke, S.L., Meyer, K.L., Gumus, F., Surles, J.R., Ishaq, K.S., Kucera, L.S. lyer, N., Wallen, C.A., Piantadosi, S. and Modest, E.J. (1991) Synthesis and evaluation of novel ether lipid nucleoside conjugates for anti-HIV-1 activity. J. Med. Chem. 34,1408-1414; Pompon, A., Lefebvre, I., Imbach, J.L., Kahn, S. and Farquhar, D. (1994) Decomposition pathways of the mono- and bis(pivaloyloxymethyl) esters of azidothymidine-5′-monophosphate in cell extract and in tissue culture medium; an application of the on-line ISRP-cleaning’ HPLC technique. Antiviral Chem. Chemother. 5,91-98; Postemark, Т. (1974) Cyclic AMP and cyclic GMP. Annu. Rev. Pharmacol. 14,23-33; Prisbe, E.J., Martin, J.C.M., McGee, D.P.C., Barker, M.F., Smee, D.F. Duke, A.E., Matthews, T.R. and Verheyden, J.P.J. (1986) Synthesis and antiherpes virus activity of phosphate an phosphonate derivatives of 9-[(l,3-dihydroxy-2-propoxy)memyl] guanine. J. Med. Chem. 29, 671-675; Pucch, F., Gosselin, G., Lefebvre, I., Pompon, A., Aubertin, A.M. Dirn, A. and Imbach, J.L. (1993) Intracellular delivery of nucleoside monophosphate through a reductase-mediated activation process. Antiviral Res. 22,155-174; Pugaeva, V.P., Klochkeva, S.I., Mashbits, F.D. and Eizengart, R.S. (1969). Toxicological assessment and health standard ratings for ethylene sulfide in the industrial atmosphere. Gig. Trf. Prof. Zabol. 13,47-48 (Chem. Abstr. 72,212); Robins, R.K. (1984) The potential of nucleotide analogs as inhibitors of retroviruses and tumors. Pharm. Res. 11-18; Rosowsky, A., Kirn. S.H., Ross and J. Wick, M.M. (1982) Lipophilic 5′-(alkylphosphate) esters of l-b-D-arabinofuranosylcytosine and its N4-acyl and 2.2′-anhydro-3’0-acyl derivatives as potential prodrugs. J. Med. Chem. 25,171-178; Ross, W. (1961) Increased sensitivity of the walker turnout towards aromatic nitrogen mustards carrying basic side chains following glucose pretreatment. Biochem. Pharm. 8,235-240; Ryu, e.K., Ross, R.J. Matsushita, Т., MacCoss, M., Hong, C.I. and West, C.R. (1982). Phospholipidnucleoside conjugates. 3. Synthesis and preliminary biological evaluation of 1-b-D-arabinofuranosylcytosine 5’diphosphate[-], 2-diacylglycerols. J. Med. Chem. 25, 1322-1329; Saffhill, R. and Hume, W.J. (1986) The degradation of 5-iododeoxyurindine and 5-bromoeoxyuridine by serin from different sources and its consequences for the use of these compounds for incorporation into DNA. Chem. Biol. Interact. 57, 347-355; Saneyoshi, M., Morozumi, M., Kodama, K., Machida, J., Kuninaka, A. and Yoshino, H. (1980) Synthetic nucleosides and nucleotides. XVI. Synthesis and biological evaluations of a series of 1-b-D-arabinofuranosylcytosine 5′-alky or arylphosphates. Chem. Pharm. Bull. 28, 2915-2923; Sastry, J.K., Nehete, P.N., Khan, S., Nowak, B.J., Plunkett, W., Arlinghaus, R.B. and Farquhar, D. (1992) Membrane-permeable dideoxyuridine 5′-monophosphate analogue inhibits human immunodeficiency virus infection. Mol. Pharmacol. 41,441-445; Shaw, J.P., Jones, R.J. Arimilli, M.N., Louie, M.S., Lee, W.A. and Cundy, K.C. (1994) Oral bioavailability of PMEA from PMEA prodrugs in male Sprague-Dawley rats. 9th Annual AAPS Meeting. San Diego, CA (Abstract). Shuto, S., Ueda, S., Imamura, S., Fukukawa, K. Matsuda, A. and Ueda, T. (1987) A facile one-step synthesis of 5’phosphatidylnucleosides by an enzymatic two-phase reaction. Tetrahedron Lett. 28,199-202; Shuto, S., Itoh, H., Ueda, S., Imamura, S., Kukukawa, K., Tsujino, M., Matsuda, A. and Ueda, T. (1988) A facile enzymatic synthesis of 5′-(3-sn-phosphatidyl)nucleosides and their antileukemic activities. Chem. Pharm. Bull. 36,209-217. Предпочтительной фосфатной группой пролекарства является 3-ацил-2-тиоэтилгруппа, также обозначаемая как “SATE”. Получение активных соединений Нуклеозиды, применяемые в описанном способе для лечения HBV-инфекций в организме хозяина, могут быть получены в соответствии с известными способами. Общий процесс стереоспецифического синтеза 3′ -замещенных Пример 1 Получение Температуру плавления определяют в открытых капиллярных трубках на приборе Gallenkamp MFB-595-010 М и не корректируют. УФ-спектр поглощения записывают на спектрофотометре Uvikon 931 (KONTRON) в этаноле. 1H-ЯМР спектр определяют при комнатной температуре в ДМСО-d6 на спектрометре Bruker AC 250 или 400. Химические сдвиги приведены в м.д., при этом ДМСО-d5 настроен на 2,49 м.д. в качестве стандарта. Дейтерообмен, эксперименты по расщеплению или 2D-COSY проводят с целью подтверждения отделения протонов. Мультиплетные сигналы представлены s (синглет), d (дублет), dd (дублет дублетов), t (триплет), q (квадруплет), br (широкий), m (мультиплет). Все величины J обозначены в Hz. Масс-спектр FAB записан в режиме положительных (FAB>0) или отрицательных (FAB<0) ионов на масс-спектрометре JEOL DX 300. Матрица представляет собой 3-нитробензиловый спирт (NBA) или смесь (50:50 об./об.) глицерина и тиоглицерина (GT). Удельное вращение измеряют на спектрополяриметре Perkin-Elmer 241 (длина шага – 1 см) и обозначают в единицах 10-1 град. см2 г-1. Элементный анализ проводился “Service de Microanalyses du CNRS, Division de Vemaison” (France). Анализы, обозначенные символами элементов или функций, составляют ±0,4% от теоретических величин. Для тонкослойной хроматографии используют алюминиевые листы с предварительно нанесенным покрытием Silica Gel 60 F254 (Merck, Art. 5554), визуализацию продуктов осуществляют, применяя УФ-поглощение с последующим обугливанием 10% этанольной серной кислотой и нагреванием. Колоночную хроматографию осуществляют на Silica Gel 60 (Merck, Art. 9385) при атмосферном давлении. 1-(2-О-Ацетил-3,5-ди-0-бензоил- Суспензию 5-фторурацила (5,0 г, 38,4 ммоль) обрабатывают гексаметилдисилазаном (HMDS, 260 мл) и каталитическим количеством сульфата аммония в течение 18 ч при кипячении с обратным холодильником. Охладив до комнатной температуры, смесь выпаривают при пониженном давлении, а остаток, получаемый в виде бесцветного масла, разбавляют безводным 1,2-дихлорэтаном (260 мл). К полученному раствору добавляют 1,2-ди-О-ацетил-3,5-ди-О-бензоил-L-ксилофуранозу 1 (11,3 г, 25,6 ммоль.) [см.: Gosselin, G.; Bergogne, M. – C; Imbach, J.-.L., “Synthesis and Antiviral Evaluation of 1-(3,5-Ди-О-бензоил- Гидразингидрат (2,80 мл, 57,4 ммоль) добавляют к раствору 1- (2-O-ацетил-3,5-ди-O-бензоил- УФ (этанол): 1-(2-Деокси-3,5-ди-O-бензоил- К раствору 1-(3,5-ди-О-бензоил- (C6H5CO)+; FAB<0 (матрица GT) m/z 452 (М-Н)–, 129 (В)–; [ 1-(2-Деокси-3,5-ди-о-бензоил- Реактив Лавессона (Lawesson’s) (3,1 г, 7, 70 ммоль) добавляют в атмосфере аргона к раствору соединения 5 (5,0 г, 11,0 ммоль.) в безводном 1,2-дихлорэтане (200 мл) и реакционную смесь перемешивают в течение ночи при кипячении с обратным холодильником. Затем растворитель выпаривают при пониженном давлении и остаток очищают колоночной хроматографией на силикагеле [растворитель для элюирования: ступенчатый градиент метанола (0-2%) в хлороформе], получая 4-тио-промежуточное соединение 6 (выход 80%) в виде желтой пены; т.пл.=178-179°С; УФ (этанол): 1-(2-Деокси- Раствор 4-тио-промежуточного соединения 6 (1,0 г, 2,13 ммоль) в метанольном аммиаке (предварительно насыщенном при -10 1-(2-Деокси-5-O-т-бутилдиметил силил- К раствору соединения 7_ (1,69 г, 6,89 ммоль) в сухом пиридине (35 мл) по каплям в атмосфере аргона добавляют хлористый т-бутилдиметилсилил (1,35 г, 8,96 ммоль) и смесь перемешивают в течение 5 ч при комнатной температуре. Затем смесь выливают в насыщенный водный раствор бикарбоната натрия (100 мл) и экстрагируют хлороформом (3 1-(2-Деокси-3-O-мезил-5-O-т-бутилдиметилсилил- Суспензию соединения 8 (0,70 г, 1,96 ммоль) в сухом пиридине (30 мл) перемешивают в атмосфере аргона и охлаждают до 0 1-(2,3-Дидеокси-3-азидо-5-O-т-бутилдиметилсилил- К раствору соединения 9 (520 мг, 1,19 ммоль.) в безводном диметилформамиде (12 мл) добавляют азид лития, увлажненный 10% метанолом (300 мг, 5,31 ммоль). Реакционную смесь перемешивают при 100 1-(2,3-Дидеокси-3-азидо- 1 M раствор трифторида тетрабутиламмония в тетрагидрофуране (TBAF/ТГФ, 1,53 мл, 1,53 ммоль) добавляют к раствору соединения 10 (295 мг, 0,67 ммоль) в безводном ТГФ (4 мл). Полученную смесь перемешивают при комнатной температуре в течение 1,5 ч и упаривают при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле [растворитель для элюирования: ступенчатый градиент метанола (4-8%) в хлороформе]. Наконец, соответствующие фракции выпаривают при пониженном давлении, разбавляют метанолом и фильтруют через установку Millex HV-4 (0,45 мкм, Millipore), получая чистое соединение 11 (199 мг, 96%), кристаллизуемое из этанола: т.пл. 188-189 Аналитические данные представлены в таблице А. Пример 2 Получение Применяемые общие процедуры и приборы описаны в примере 1 в экспериментально-протокольной части, описывающей синтез 3′-изомера (3′-N3– 1-(2-O-Ацетил-3-деокси-5-O-бензоил- Суспензию 5-фторурацила (5,15 г, 39,6 ммоль) обрабатывают гексаметилдисилазаном (HMDS, 257 мл) и каталитическим количеством сульфата аммония на протяжении 18 ч при кипячении с обратным холодильником. После охлаждения до комнатной температуры смесь упаривают при пониженном давлении и остаток, получаемый в виде бесцветного масла, разбавляют безводным 1,2-дихлорэтаном (290 мл). К полученному раствору добавляют 1,2-ди-O-ацетил-3-деокси-5-O-бензоил-L-эритро-пентофуранозу 12 (8,5 г, 26,4 ммоль.) [См. Mathe, C., Ph.D. Dissertation, Universite de Montpellier II -Sciences et Techniques du Languedoc, Montpellier (France), September 13, 1994; Gosselin, G.; Mathe, C.; Bergogne, M.-C.; Aubertin, A.M.; Kirn, A.; Sommadossi, J.P.; Schinazi, R.F.; Imbach, J.L., “2′- and/or 3′-deoxy- 1-(3-Деокси-5-O-бензоил- К раствору соединения 13 (5,90 г, 15,0 ммоль) в тетрагидрофуране (ТГФ, 175 мл) добавляют метилат натрия (2,84 г, 52,6 ммоль.). Полученную суспензию перемешивают при комнатной температуре в течение 5 ч, а затем нейтрализуют, добавляя Dowex 50 W X 2 (Н+-форма). Смолу фильтруют и промывают теплым метанолом, а соединенные фильтраты упаривают досуха. В результате колоночной хроматографии остатка на силикагеле [растворитель для элюирования: ступенчатый градиент метанола (0-8%) в хлористом метилене] получают соединение 14 (4,11 г, 78%), кристаллизуемое из смеси хлористого метилена/метанола: т.пл. 154-156 1-(3-Деокси-5-O-бензоил- Дициклогексикарбодиимид (DCC, 3,53 г, 17,1 ммоль) и дихлоруксусную кислоту (0,235 мл, 2,56 ммоль) добавляют к раствору соединения 14 (2,00 г, 5,71 ммоль.) в безводном бензоле (50 мл), ДМСО (35 мл) и пиридине (0,46 мл). Полученный раствор перемешивают при комнатной температуре в атмосфере аргона в течение 4 ч и разбавляют этилацетатом (300 мл). Добавляют щавелевую кислоту (1,54 г, 17,1 ммоль.), растворенную в метаноле (4,6 мл), и реакционную смесь перемешивают при комнатной температуре в течение 1 ч, а затем фильтруют для удаления осажденной дициклогексилмочевины (DCU). Перед высушиванием над сульфатом натрия и упариванием при пониженном давлении фильтрат промывают насыщенным раствором соли (3 1-(2-O-Ацетил-3-деокси-5-O-бензоил- Уксусный ангидрид (0,88 мл, 9,28 ммоль) добавляют в атмосфере аргона к раствору соединения 15 (2,50 г, 7,14 ммоль) в сухом пиридине (50 мл) и полученную смесь перемешивают при комнатной температуре в течение 22 ч. Затем добавляют этанол и растворители выпаривают при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле [растворитель для элюирования: ступенчатый градиент метанола (0-2%) в хлористом метилене), получая чистое соединение 16 (2,69 г, 96%) в виде белой пены; т.пл.=68-70 1-(2-O-Ацетил-3-деокси-5-O-бензоил- Реактив Лавессона (Lawesson’s) (1,9 г, 4,69 ммоль) добавляют в атмосфере аргона к раствору соединения 16 (2,63 г, 6,70 ммоль) в безводном 1,2-дихлорэтане (165 мл) и реакционную смесь перемешивают в течение ночи при кипячении с обратным холодильником. Затем растворитель выпаривают при пониженном давлении, а остаток очищают колоночной хроматографией на силикагеле [растворитель для элюирования: ступенчатый градиент метанола (0-3%) в хлористом метилене], получая 4-тиопроизводное 17 (2,65 г, выход 96%) в виде желтой пены; т.пл.=78-79 1-(3-Деокси- Раствор 4-тиопроизводного 17 (0,86 г, 2,19 ммоль.) в метанольном аммиаке (предварительно насыщенном при -10 1-(3-Деокси-5-O-т-бутилдиметилсилил- К раствору соединения 18 (1,38 г, 5,63 ммоль) в сухом пиридине (30 мл) по каплям в атмосфере аргона добавляют хлористый т-бутилдиметилсилил (1,10 г, 7,32 ммоль) и смесь перемешивают в течение 10 ч при комнатной температуре. Затем смесь выливают в насыщенный водный раствор бикарбоната натрия (100 мл) и экстрагируют хлороформом (3 1-(3-Деокси-2-O-мезил-5-O-т-бутил-диметилсилил- Суспензию соединения 19 (1,70 г, 4,73 ммоль.) в сухом пиридине (80 мл) перемешивают в атмосфере аргона и охлаждают до 0 1-(2,3-Дидеокси-2-азидо-5-O-т-бутилдиметилсилил- К раствору соединения 20 (442 мг, 1,01 ммоль) в безводном диметилформамиде (12 мл) добавляют азид лития, увлажненный 10% метанолом (265 мг, 4,87 ммоль). Реакционную смесь перемешивают при 100 1-(2,3-Дидеокси-2-азидо- 1M раствор трифтористого тетрабутиламмония в тетрагидрофуране (TBAF/ТГФ, 1,90 мл, 1,90 ммоль) добавляют к раствору соединения 21 (480 мг, 1,25 ммоль) в безводном ТГФ (8 мл). Полученную смесь перемешивают при комнатной температуре в течение 1,5 ч и упаривают при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле [растворитель для элюирования: ступенчатый градиент метанола (4-8%) в хлороформе]. Наконец, соответствующие фракции упаривают при пониженном давлении, разбавляют метанолом и фильтруют через установку Millex HV-4 (0,45 мкм, Millipore), получая чистое соединение 22 (304 мг, выход 90%), кристаллизуемое из этанола: т.пл. 219-221 Аналитические данные представлены в таблице Б. Анти-НВV-активность Способность соединений Антивирусную оценку осуществляют на двух отдельных пассажах клеток, по две культуры на пассаж (в целом 4 культуры). Все ячейки на всех планшетах засевают с одинаковой плотностью и в одно и то же время. Из-за присущих колебаний уровней как внутриклеточной, так и внеклеточной HBV-ДНК, только подавление более чем в 3,0 раза (для ДНК HBV-вириона) или 2,5 раза (для интермедиатов репликации ДНК HBV), отличающееся от среднего уровня для таких форм ДНК HBV в необработанных клетках, обычно считают статистически значимым [Р<0,05] (Korba and Gerin, Antiviral Res. 19:55-70, 1992). Уровень интегрированной ДНК HBV в каждом клеточном ДНК-препарате (остающемся постоянным в таких экспериментах в расчете на клетку) используют для определения уровня внутриклеточных форм ДНК HBV, таким образом устраняя технические колебания, присущие анализам на блот-гибридизацию. Обычный уровень внеклеточной ДНК вириона HBV в необработанных клетках варьируется от 50 до 150 пкг/мл культуральной среды (средний уровень составляет приблизительно 76 пкг/мл). Уровень интермедиатов репликации внутриклеточных ДНК HBV в необработанных клетках варьируется от 50 до 100 пкг/мкг клеток ДНК (в среднем уровень составляет приблизительно 74 пкг/мкг клеток ДНК). В целом снижение уровня внутриклеточной ДНК HBV благодаря обработке антивирусными соединениями менее выражено и происходит медленнее, чем подавление уровня ДНК вириона HBV. Для сравнения результаты анализов по гибридизации, полученные в данных экспериментах, соответствуют приблизительно 1,0 пкг внутриклеточной ДНК HBV/мкг клеточной ДНК к 2-3 геномным копиям на клетку, и 1,0 пкг внеклеточной ДНК HBV/мл культуральной среды к 3 Анализы на токсичность проводят для того, чтобы определить, происходит ли любое наблюдаемое антивирусное действие благодаря общему влиянию на жизнеспособность клеток. Это может быть установлено путем поглощения нейтрального красного красителя, т.е. стандартного и широко применяемого теста на жизнеспособность клеток в различных системах вирус-хозяин, включая HSV (вирус герпеса) и ВИЧ. Тест-соединения используют в виде маточных растворов ДМСО (замороженного на сухом льду). Подготавливают суточные аликвоты тест-образцов и замораживают их при -20 Пример 3 Токсичность соединений Определяют способность активных соединений ингибировать рост вируса в культурах клеток 2.2.15 (клетки Hep G2, трансформированные вирионом гепатита). Как следует из таблицы 1, при концентрации 100 мкм не наблюдается какой-либо существенной токсичности (превышающей 50% подавление уровня поглощения красителя, наблюдаемого у необработанных клеток) всех тестируемых соединений. Анализы на токсичность осуществляют в 96-луночных планшетах с плоским дном для тканевых культур. Клетки для анализа на токсичность культивируют и обрабатывают тест-соединениями таким же образом, как и при определении антивирусной активности. Каждое соединение исследуют в четырех концентрациях и на трех культурах. Для определения относительного уровня токсичности применяют поглощение нейтрального красного красителя. Количественный анализ проводят по поглощению при 510 нм (А510) интернализованного красителя. Результаты представлены в виде процентных величин средних значений А510 (± стандартное отклонение) 9 различных культур необработанных клеток, находящихся в этом же 96-луночном планшете в качестве тест-соединений. Процент поглощения красителя в 9 контрольных культурах на планшете 40 составляет 100±3. При 150-190 мкм Пример 4 Влияние аналогов анти-HBV Влияние аналогов анти-HBV Получение фармацевтических композиций Описываемые здесь соединения и их фармацевтически приемлемые соли, пролекарства и производные могут быть использованы для профилактики и лечения HBV-инфекций и подобных им состояний, таких как анти-НВV-антитело положительные и HBV-положительные состояния, вызываемое HBV хроническое воспаление печени, цирроз, острый гепатит, фульминантный гепатит, хронический персистирующий гепатит и усталость. Такие соединения или составы также могут быть использованы профилактически для предотвращения или замедления клинического заболевания у анти-НВV-антитело или HBV-антиген-положительных пациентов либо находящихся в контакте с HBV. Пациенты, страдающие от какого-либо из вышеуказанных состояний, могут быть подвергнуты лечению путем введения эффективного для лечения HBV количества одного из описываемых здесь активных соединений или их смеси, либо их фармацевтически приемлемого производного или соли, необязательно вместе с фармацевтически приемлемым носителем или разбавителем. Активные материалы могут быть введены любым подходящим способом, например, пероральным, парентеральным, внутривенным, интрадермальным, подкожным или локальным, в жидком или твердом виде. Активное соединение включают в фармацевтически приемлемый носитель или разбавитель в количестве, достаточном для обеспечения пациента терапевтически активным количеством, не вызывая у него серьезного токсического действия. Предпочтительная доза активного соединения для всех вышеуказанных состояний составляет приблизительно от 1 до 60 мг/кг, предпочтительно от 1 до 20 мг/кг массы тела в сутки, более широко, от 0,1 до приблизительно 100 мг на килограмм массы тела реципиента в сутки. Эффективный интервал дозы фармацевтически приемлемых производных может быть рассчитан на основе массы вводимого родительского нуклеозида. Если производное обладает самостоятельной активностью, то эффективная дозировка может быть рассчитана, как указано выше, на основании массы производного либо другими способами, известными специалистам в данной области. В соответствии с одним из вариантов осуществления настоящего изобретения активное соединение вводят, как указано в прилагаемой к продукту инструкции или в Physician’s Desk Reference для 3′-азидо-3′-деокситимидина (AZT), 2′,3′-дидеоксиинозина (DDI), 2′,3′-дидеоксицитидина (DDC) или 2′,3′-дидеокси-2′,3′-дидегидротимидина (D4T) для назначения при ВИЧ. Соединение целесообразно вводить в виде любой дозированной лекарственной формы, включая, но не ограничиваясь ею, форму, содержащую от 7 до 3000 мг, предпочтительно от 70 до 1400 мг активного ингредиента на стандартную дозу. Обычно подходящей является пероральная доза, содержащая 50-1000 мг. В идеале вводимый активный ингредиент должен обеспечивать наивысшую концентрацию активного соединения в плазме, составляющую приблизительно от 0,2 до 70 мМ, предпочтительно приблизительно от 1,0 до 10 мМ. Это может быть достигнуто, например, в результате внутривенной инъекции 0,1-5% раствора активного ингредиента, необязательно в солевом растворе, либо введено в виде болюса активного ингредиента. Активное соединение может быть получено в виде фармацевтически приемлемых солей. В данном описании термин “фармацевтически приемлемые соли или комплексы” относится к солям или комплексам нуклеозидов, сохраняющим необходимую биологическую активность родительского соединения и не оказывающим либо оказывающим минимальное нежелательное токсикологическое действие. Неограничивающие примеры таких солей включают (а) соли присоединения кислот, образуемые с неорганическими кислотами (например, соляная, бромисто-водородная, серная, фосфорная, азотная кислота и т.п.), а также соли, образуемые с органическими кислотами, такими как уксусная, щавелевая, винная, янтарная, яблочная, аскорбиновая, бензойная, дубильная, памовая, альгиновая, полиглутаминовая, нафталинсульфоновые, нафталиндисульфоновые кислоты и полигалактуроновая кислота; (b) соли присоединения основания, образуемые с катионами, такими как натрий, калий, цинк, кальций, висмут, барий, магний, алюминий, медь, кобальт, никель, кадмий и т.п., либо с органическим катионом, получаемым из N,N-дибензилэтилендиамина, аммония или этилендиамина; либо (с) сочетания (а) и (b); например соль танната цинка или т.п. Модификации активного соединения конкретно в позициях N6 или N4 и 5′-О могут обеспечить биодоступность и необходимый уровень метаболизма активных веществ, таким образом обеспечивая контроль за их доставкой. Концентрация активного соединения в составе лекарственного препарата зависит от уровня его поглощения, инактивации и выделения, а также от других факторов, известных специалистам в данной области. Следует отметить, что величина дозы также варьируется в зависимости от тяжести подвергаемого лечению состояния. Также следует отметить, что для каждого отдельного пациента со временем необходимо подбирать конкретные схемы приема в соответствии с индивидуальной потребностью и профессиональным мнением лица, вводящего или контролирующего введение составов, и что указанные здесь интервалы концентрации являются всего лишь иллюстративными и не предназначены для ограничения объема или применения заявленной композиции. Активный ингредиент может быть введен за один раз либо может быть разделен на несколько меньших доз, вводимых через различные промежутки времени. Предпочтительным способом введения активного соединения является пероральный. Композиции для перорального применения обычно включают инертный разбавитель или пищевой носитель. Они могут быть заключены в желатиновые капсулы либо спрессованы в таблетки. Для перорального терапевтического введения активное соединение может быть смешано с формообразующими веществами и использовано в виде таблеток, пастилок или капсул. Фармацевтически совместимые связующие и/или адъюванты могут также составлять часть композиции. Таблетки, пилюли, капсулы, пастилки и т.п. могут содержать любой из следующих ингредиентов или соединений, имеющих подобную природу: связующее, такое как микрокристаллическая целлюлоза, трагакант или желатин; формообразующее вещество, такое как крахмал или лактоза, разрыхлитель, такой как альгиновая кислота, Primogel или кукурузный крахмал; смазывающий агент, такой как стеарат магния или Sterotes; глидант (вещество, обеспечивающее скольжение), такой как коллоидная двуокись кремния; подсластитель, такой как сахароза или сахарин; или отдушка, такая как мята, метилсалицилат или апельсин. Если дозированной лекарственной формой является капсула, она может содержать, помимо материалов вышеуказанного типа, жидкий носитель, такой как жирное масло. Кроме того, дозированные лекарственные формы могут содержать и другие различные материалы, модифицирующие его физическую форму, например, покрытие из сахара, шеллака или других энтеросолюбильных агентов. Активное соединение либо его фармацевтически приемлемая соль или производное могут быть введены в виде компонента эликсира, суспензии, сиропа, воды, жевательной резинки или т.п. Помимо активных соединений сироп может содержать сахарозу в качестве подсластителя и некоторые консерванты, красители, пигменты и отдушки. Активное соединение либо его фармацевтически приемлемое производное или соль также могут быть смешаны с другими активными материалами, не ухудшающими желаемое действие, либо с материалами, дополняющими желаемое действие, такими как антибиотики, противогрибковые, противовоспалительные или другие антивирусные препараты, включая анти-HBV, анти-цитомегаловирусные или анти-ВИЧ-агенты. Растворы или суспензии, применяемые для парентерального, интрадермального, подкожного или местного введения, могут включать следующие компоненты: стерильный разбавитель, такой как вода для инъекций, физиологический раствор, жирные масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные агенты, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты или фосфаты, и агенты для регулирования тонуса, такие как хлористый натрий или декстроза. Парентеральный препарат может быть заключен в ампулы, разовые шприцы или пузырьки для нескольких доз, изготовленные из стекла или пластика. При внутривенном введении предпочтительными носителями являются физиологический солевой раствор или фосфатно-солевой буферный раствор (PBS). В соответствии с предпочтительным вариантом осуществления настоящего изобретения активные соединения получают вместе с носителями, защищающими соединение от быстрого выведения из организма, например, в виде состава с контролируемым высвобождением, включая имплантаты и системы для микрокапсулированной доставки. Могут быть использованы биорасщепляемые, биосовместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, сложные полиортоэфиры и полимолочная кислота. Способы получения таких составов очевидны специалистам в данной области. Материалы также могут быть приобретены у Alza Corporation и Nova Pharmaceuticals, Inc. Липосомальные суспензии (включая липосомы, нацеленные на инфицированные клетки с моноклональными антителами к вирусным антигенам) также являются предпочтительными в качестве фармацевтически приемлемых носителей. Они могут быть получены в соответствии со способами, известными специалистам в данной области, например, описанными в патенте США №4 522 811. К примеру, составы из липосом могут быть получены в результате растворения соответствующего липида (липидов) (таких как стеароилфосфатидилэтаноламин, стеароилфосфатидилхолин, араха-доилфосфатидилхолин и холестерин) в неорганическом растворителе, который затем выпаривают, получая тонкую пленку сухого липида на поверхности контейнера. Затем в контейнер помещают водный раствор активного соединения либо его монофосфатного, дифосфатного и/или трифосфатного производного. Затем контейнер встряхивают вручную, чтобы отделить липидный материал от стенок контейнера и диспергировать липидные агрегаты, получая таким образом липосомальную суспензию. Настоящее изобретение описано со ссылкой на предпочтительные варианты его осуществления. Вариации и модификации изобретения очевидны специалистам в данной области из вышеприведенного подробного описания изобретения. Подразумевается, что все эти вариации и модификации входят в объем прилагаемой формулы изобретения. Формула изобретения
1. Способ лечения инфекции, вызванной вирусом гепатита В в организме, включающий введение эффективного количества соединения где R представляет Н, ацил, монофосфат, дифосфат или трифосфат либо стабилизированное фосфатное производное; R’ представляет Н, ацил или алкил. 2. Способ по п.1, в котором R представляет Н. 3. Способ по п.1, в котором R представляет ацил. 4. Способ по п.1, в котором R представляет монофосфат. 5. Способ по п.1, в котором R представляет дифосфат. 6. Способ по п.1, в котором R представляет трифосфат. 7. Способ по п.1, в котором R представляет стабилизированное фосфатное производное. 8. Способ лечения инфекции, вызванной вирусом гепатита В в организме, включающий введение эффективного количества соединения где R представляет Н, ацил, монофосфат, дифосфат или трифосфат либо стабилизированное фосфатное производное; R’ представляет Н, ацил или алкил. 9. Способ по п.8, в котором R представляет Н. 10. Способ по п.8, в котором R представляет ацил. 11. Способ по п.8, в котором R представляет монофосфат. 12. Способ по п.8, в котором R представляет дифосфат. 13. Способ по п.8, в котором R представляет трифосфат. 14. Способ по п.8, в котором R представляет стабилизированное фосфатное производное. 15. где R представляет Н, ацил, монофосфат, дифосфат или трифосфат либо стабилизированное фосфатное производное; R’ представляет Н, ацил или алкил. 16. Соединение по п.15, в котором R представляет Н. 17. Соединение по п.15, в котором R представляет ацил. 18. Соединение по п.15, в котором R представляет монофосфат. 19. Соединение по п.15, в котором R представляет дифосфат. 20. Соединение по п.15, в котором R представляет трифосфат. 21. Соединение по п.15, в котором R представляет стабилизированное фосфатное производное. 22. где R представляет Н, ацил, монофосфат, дифосфат или трифосфат либо стабилизированное фосфатное производное; R’ представляет Н, ацил или алкил. 23. Соединение по п.22, в котором R представляет Н. 24. Соединение по п.22, в котором R представляет ацил. 25. Соединение по п.22, в котором R представляет монофосфат. 26. Соединение по п.22, в котором R представляет дифосфат. 27. Соединение по п.22, в котором R представляет трифосфат. 28. Соединение по п.22, в котором R представляет стабилизированное фосфатное производное. 29. Фармацевтическая композиция для лечения инфекции, вызванной вирусом гепатита В в организме, включающая эффективное количество соединения где R представляет Н, ацил, монофосфат, дифосфат или трифосфат либо стабилизированное фосфатное производное; R’ представляет Н, ацил или алкил, в сочетании с фармацевтически приемлемым носителем. 30. Композиция по п.29, в которой R представляет Н. 31. Композиция по п.29, в которой R представляет ацил. 32. Композиция по п.29, в которой R представляет монофосфат. 33. Композиция по п.29, в которой R представляет дифосфат. 34. Композиция по п.29, в которой R представляет трифосфат. 35. Композиция по п.29, в которой R представляет стабилизированное фосфатное производное. 36. Фармацевтическая композиция для лечения инфекции, вызванной вирусом гепатита В в организме, включающая эффективное количество соединения где R представляет Н, ацил, монофосфат, дифосфат или трифосфат либо стабилизированное фосфатное производное; R’ представляет Н, ацил или алкил, в сочетании с фармацевтически приемлемым носителем. 37. Композиция по п.36, в которой R представляет Н. 38. Композиция по п.36, в которой R представляет ацил. 39. Композиция по п.36, в которой R представляет монофосфат. 40. Композиция по п.36, в которой R представляет дифосфат. 41. Композиция по п.36, в которой R представляет трифосфат. 42. Композиция по п.36, в которой R представляет стабилизированное фосфатное производное. 43. где R представляет Н, ацил, монофосфат, дифосфат или трифосфат либо стабилизированное фосфатное производное; R’ представляет Н, ацил или алкил, в качестве действующего начала для лечения инфекции, вызванной вирусом гепатита В в организме человека или другого животного. 44. где R представляет Н, ацил, монофосфат, дифосфат или трифосфат либо стабилизированное фосфатное производное; R’ представляет Н, ацил или алкил, в качестве действующего начала для лечения инфекции, вызванной вирусом гепатита В в организме человека или другого животного. 45. где R представляет Н, ацил, монофосфат, дифосфат или трифосфат либо стабилизированное фосфатное производное; R’ представляет Н, ацил или алкил, в качестве активного ингредиента препарата для лечения инфекции, вызванной вирусом гепатита В в организме человека или другого животного. 46. где R представляет Н, ацил, монофосфат, дифосфат или трифосфат либо стабилизированное фосфатное производное; R’ представляет Н, ацил или алкил, в качестве активного ингредиента препарата для лечения инфекции, вызванной вирусом гепатита В в организме человека или другого животного. 47. Способ по п.1 или 8, в котором R и R’ представляют Н. 48. Способ по п.1 или 8, в котором R представляет ацил, а R’ представляет Н. 49. Способ по п.1 или 8, в котором R представляет монофосфат, а R’ представляет Н. 50. Способ по п.1 или 8, в котором R представляет дифосфат, а R’ представляет Н. 51. Способ по п.1 или 8, в котором R представляет трифосфат, а R’ представляет Н. 52. Способ по п.1 или 8, в котором R представляет стабилизированное фосфатное производное, а R’ представляет Н. 53. Способ по п.1 или 8, в котором R представляет Н, а R’ представляет алкил. 54. Способ по п.1 или 8, в котором R представляет ацил, а R’ представляет алкил. 55. Способ по п.1 или 8, в котором R представляет монофосфат, а R’ представляет алкил. 56. Способ по п.1 или 8, в котором R представляет дифосфат, а R’ представляет алкил. 57. Способ по п.1 или 8, в котором R представляет трифосфат, а R’ представляет алкил. 58. Способ по п.1 или 8, в котором R представляет стабилизированное фосфатное производное, а R’ представляет алкил. 59. Способ по п.1 или 8, в котором R представляет Н, а R’ представляет ацил. 60. Способ по п.1 или 8, в котором R и R’ представляют ацил. 61. Способ по п.1 или 8, в котором R представляет монофосфат, а R’ представляет ацил. 62. Способ по п.1 или 8, в котором R представляет дифосфат, а R’ представляет ацил. 63. Способ по п.1 или 8, в котором R представляет трифосфат, а R’ представляет ацил. 64. Способ по п.1 или 8, в котором R представляет стабилизированное фосфатное производное, а R’ представляет ацил. 65. Соединение по п.15 или 22, в котором R и R’ представляют Н. 66. Соединение по п.15 или 22, в котором R представляет ацил, a R’ представляет Н. 67. Соединение по п.15 или 22, в котором R представляет монофосфат, а R’ представляет Н. 68. Соединение по п.15 или 22, в котором R представляет дифосфат, а R’ представляет Н. 69. Соединение по п.15 или 22, в котором R представляет трифосфат, а R’ представляет Н. 70. Соединение по п.15 или 22, в котором R представляет стабилизированное фосфатное производное, а R’ представляет Н. 71. Соединение по п.15 или 22, в котором R представляет Н, а R’ представляет алкил. 72. Соединение по п.15 или 22, в котором R представляет ацил, а R’ представляет алкил. 73. Соединение по п.15 или 22, в котором R представляет монофосфат, а R’ представляет алкил. 74. Соединение по п.15 или 22, в котором R представляет дифосфат, а R’ представляет алкил. 75. Соединение по п.15 или 22, в котором R представляет трифосфат, а R’ представляет алкил. 76. Соединение по п.15 или 22, в котором R представляет стабилизированное фосфатное производное, а R’ представляет алкил. 77. Соединение по п.15 или 22, в котором R представляет Н, а R’ представляет ацил. 78. Соединение по п.15 или 22, в котором R и R’ представляют ацил. 79. Соединение по п.15 или 22, в котором R представляет монофосфат, а R’ представляет ацил. 80. Соединение по п.15 или 22, в котором R представляет дифосфат, а R’ представляет ацил. 81. Соединение по п.15 или 22, в котором R представляет трифосфат, а R’ представляет ацил. 82. Соединение по п.15 или 22, в котором R представляет стабилизированное фосфатное производное, а R’ представляет ацил. 83. Композиция по п.29 или 36, в которой R и R’ представляют Н. 84. Композиция по п.29 или 36, в которой R представляет ацил, а R’ представляет Н. 85. Композиция по п.29 или 36, в которой R представляет монофосфат, а R’ представляет Н. 86. Композиция по п.29 или 36, в которой R представляет дифосфат, а R’ представляет Н. 87. Композиция по п.29 или 36, в которой R представляет трифосфат, а R’ представляет Н. 88. Композиция по п.29 или 36, в которой R представляет стабилизированное фосфатное производное, а R’ представляет Н. 89. Композиция по п.29 или 36, в которой R представляет Н, а R’ представляет алкил. 90. Композиция по п.29 или 36, в которой R представляет ацил, а R’ представляет алкил. 91. Композиция по п.29 или 36, в которой R представляет монофосфат, а R’ представляет алкил. 92. Композиция по п.29 или 36, в которой R представляет дифосфат, а R’ представляет алкил. 93. Композиция по п.29 или 36, в которой R представляет трифосфат, а R’ представляет алкил. 94. Композиция по п.29 или 36, в которой R представляет стабилизированное фосфатное производное, а R’ представляет алкил. 95. Композиция по п.29 или 36, в которой R представляет H, a R’ представляет ацил. 96. Композиция по п.29 или 36, в которой R и R’ представляют ацил. 97. Композиция по п.29 или 36, в которой R представляет монофосфат, а R’ представляет ацил. 98. Композиция по п.29 или 36, в которой R представляет дифосфат, а R’ представляет ацил. 99. Композиция по п.29 или 36, в которой R представляет трифосфат, а R’ представляет ацил. 100. Композиция по п.29 или 36, в которой R представляет стабилизированное фосфатное производное, а R’ представляет ацил. РИСУНКИ
|
||||||||||||||||||||||||||
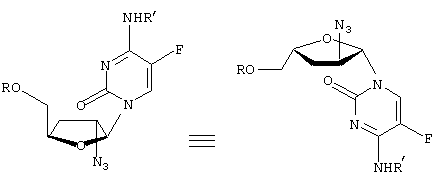
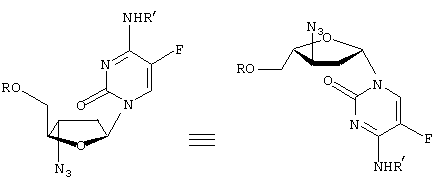
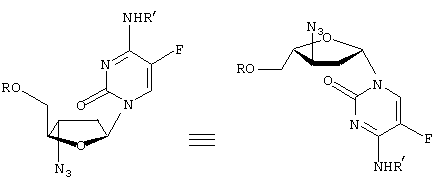
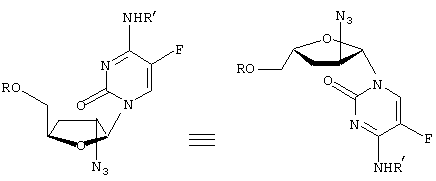
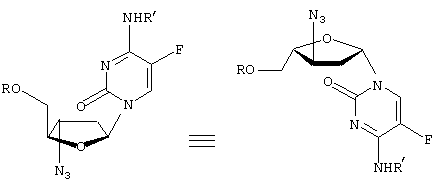
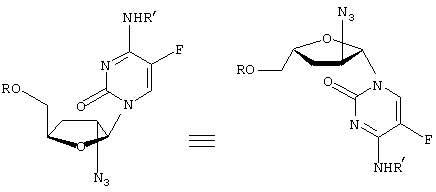
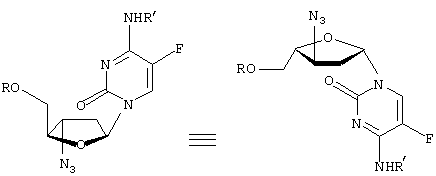
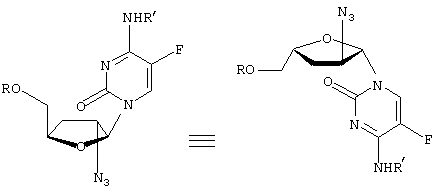
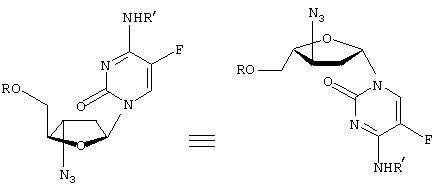
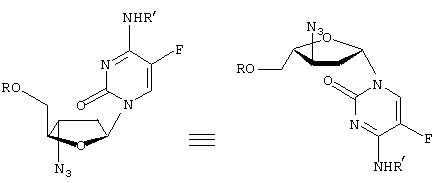
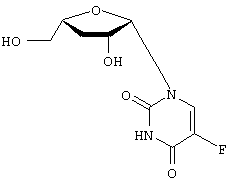
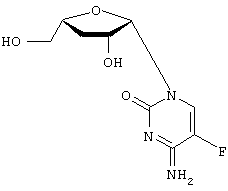
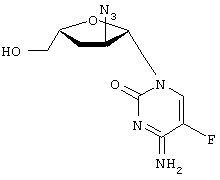
 -L-(2′ или 3(-азидо)-2
-L-(2′ или 3(-азидо)-2 ,3
,3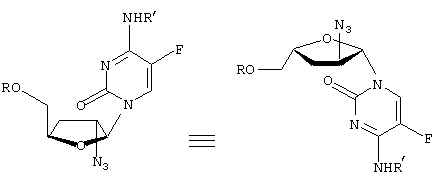
 ” или “транс” (когда заместитель С2 расположен “вверху”, а заместитель С5 – “внизу”), а также “
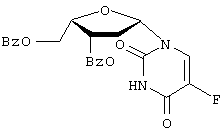
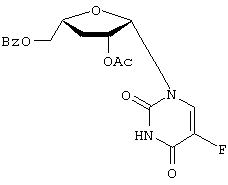
” или “транс” (когда заместитель С2 расположен “вверху”, а заместитель С5 – “внизу”), а также “ 800 мл). Органическую фазу сушат над сульфатом натрия, а затем выпаривают при пониженном давлении. Полученный сырой материал очищают колоночной хроматографией на силикагеле [растворитель для элюирования: ступенчатый градиент метанола (0-4%) в метиленхлориде], получая соединение 2 (11,0 г, выход 84%) в виде белой пены; т.пл.=96-98
800 мл). Органическую фазу сушат над сульфатом натрия, а затем выпаривают при пониженном давлении. Полученный сырой материал очищают колоночной хроматографией на силикагеле [растворитель для элюирования: ступенчатый градиент метанола (0-4%) в метиленхлориде], получая соединение 2 (11,0 г, выход 84%) в виде белой пены; т.пл.=96-98 С; УФ (этанол):
С; УФ (этанол):  mах=228 hm (
mах=228 hm ( =25900) 266 nm (
=25900) 266 nm ( 11,1 (br s, 1H, NH), 8,05 (1H, H-6, J6-FS=6,8 Hz), 7,9-7,4 (m, 10H, 2 С6Н5СО), 5,99 (d, 1H, Н-1’, J1’-2’=3,1 Hz), 5,74 (dd, 1H, H-3′, J3’-2’=4,2 Hz и J3’-4’=2,3 Hz), 5,54 (t, 1H, H-2′, J2’-1’=J2’-3’=2,9 Hz), 4,8-4,6 (m, 3H, H-4′, H-5′ and H-5″); MS: FAB>0 (матрица GT) m/z 513 (M+H)+, 383 (S)+ 105 (C6H5CO)+; FAB<0 (матрица GT) m/z 511 (M-H)–, 469 (М-СН3СО)–, 129 (В)–, 121 (С6Н5СO2)–; [
11,1 (br s, 1H, NH), 8,05 (1H, H-6, J6-FS=6,8 Hz), 7,9-7,4 (m, 10H, 2 С6Н5СО), 5,99 (d, 1H, Н-1’, J1’-2’=3,1 Hz), 5,74 (dd, 1H, H-3′, J3’-2’=4,2 Hz и J3’-4’=2,3 Hz), 5,54 (t, 1H, H-2′, J2’-1’=J2’-3’=2,9 Hz), 4,8-4,6 (m, 3H, H-4′, H-5′ and H-5″); MS: FAB>0 (матрица GT) m/z 513 (M+H)+, 383 (S)+ 105 (C6H5CO)+; FAB<0 (матрица GT) m/z 511 (M-H)–, 469 (М-СН3СО)–, 129 (В)–, 121 (С6Н5СO2)–; [

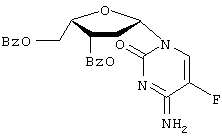
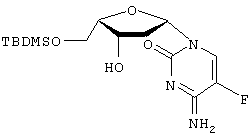
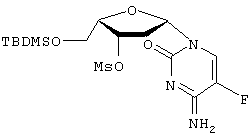
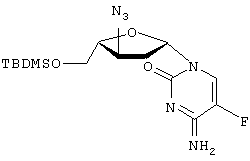
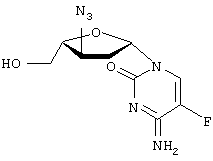
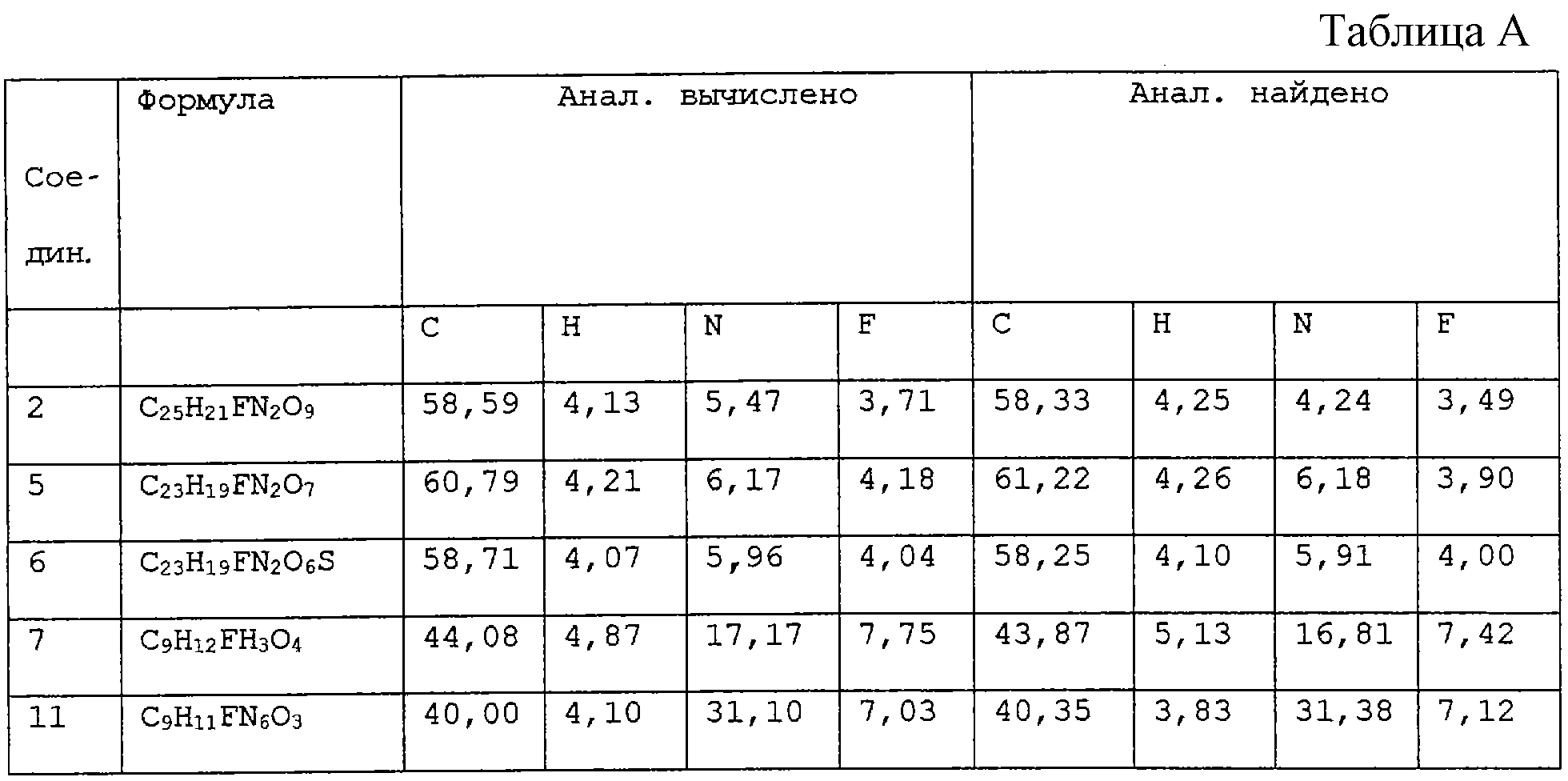
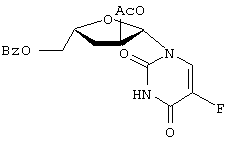
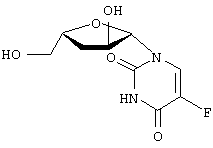
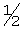 H2O; Вычислено: 42,52; Н=5,15; N=16,53; F=7,47; Найдено С=43,16; H=5,32; N=16,97; F=6,92
H2O; Вычислено: 42,52; Н=5,15; N=16,53; F=7,47; Найдено С=43,16; H=5,32; N=16,97; F=6,92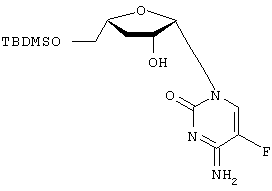
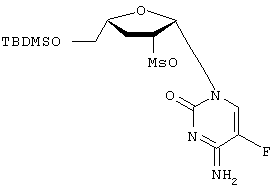
 105 вирусных частиц/мл.
105 вирусных частиц/мл.