Патент на изобретение №2235118
|
||||||||||||||||||||||||||
(54) ЦЕТАНПОВЫШАЮЩАЯ ПРИСАДКА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ
(57) Реферат: Изобретение относится к нефтеперерабатывающей и нефтехимической промышленности, а конкретно к способу получения нитроэфиров спиртов, которые находят применение в качестве эффективных цетанповышающих присадок (ЦПП) к дизельным топливам. В качестве сырья для получения цетанповышающей присадки используют фракцию, кипящую в пределах 166-190  С, выделенную из кубового остатка производства бутиловых спиртов (КОБС), получаемых методом оксосингеза и содержащую 60-90% 2-этилгексанола, 1,3-5,0% 2-этил-4-метилпентанола, 3-16% ацеталей, 5,7-19% моногликолевых эфиров. Фракцию, содержащую в качестве основного субстрата для нитрования 2-этилгексанол, нитруют нитрующей смесью азотной и серной кислот при температуре 8-18С в течение 2,5-1,5 часов. Полученные продукты нитрования отделяют от отработанных в процессе нитрации кислот. Выделенный органический слой (целевой продукт) промывают дважды 10% (мас.) раствором солей, полученных нейтрализацией отработанных кислот и единожды этим же раствором с добавлением 10% (мас.) раствора гидроксида натрия. 3 мас. части раствора солей добавляют на 10 мас. частей целевого продукта. Изобретение позволяет упростить процесс, а также снизить затраты на процесс получения целевого продукта. 2 н. и 3 з.п. ф-лы, 1 табл.
Изобретение относится к нефтеперерабатывающей и нефтехимической промышленности, конкретно к способу получения нитроэфиров спиртов, которые находят применение в качестве цетанповышающей присадки (ЦПП) к дизельным топливам.
Известен способ получения ЦПП – циклогексилнитрата путем нитрования циклогексанола в безводной среде смесями серной и азотной кислот, при этом нитрование возможно только при температуре не выше – 20С (А.с. №687791 СССР, МКИ С 07 С 68/04).
Недостатками данного способа являются необходимость проведения процесса при низких температурах из-за низкой стабильности целевого продукта циклогексилнитрата в условиях синтеза, кроме того, процесс проводят с использованием хлористого метилена или гексана.
Известны способы получения циклогексилнитрата с использованием обводненных нитрующих смесей кислот при температуре не выше 0-5С. Процесс нитрования проводят в среде растворителей – хлористого метилена, гексана, присутствие которых облегчает теплоотвод из реакционной смеси (Питеркин Р.Н., Чесалов А.М. Нитрование циклогексанола в среде растворителей. / Химическая промышленность, №9, 2000 г., с. 483-486).
Описанные выше способы имеют существенные недостатки:
– необходимость проведения процесса при минусовых температурах,
– использование растворителей,
– использование дефицитного дорогостоящего сырья циклогексанола. Названные недостатки усложняют технологический процесс, уменьшают его стабильность и надежность, снижают эффективность производства, повышают стоимость присадки.
Наиболее близким по технической сущности и достигаемому эффекту является способ получения ЦПП – 2-этилгексилнитрата путем нитрования 2-этилгексанола с содержанием основного вещества не менее 99,0 мас.% нитрующей смесью азотной и серной кислот при добавлении карбамида, с отделением отработанных кислот от целевого продукта – 2-этилгексилнитрата и последующей его промывкой последовательно дважды 10% (мас.) водным раствором сульфата натрия и затем однократно 10% (мас.) раствором смеси сульфата и карбоната натрия, взятых в соотношении 1:1, используя каждый раз на 2 части 2-этилгексилнитрата 1 часть раствора солей (пат. США №5162568).
Описанная присадка и способ ее получения имеют следующие недостатки: для ее получения используют дорогостоящее и дефицитное сырье – 2-этилгексанол, большой расход реагентов для промывки, низкая устойчивость и стабильность процесса нитрования, что обусловлено подачей нитруемого вещества – 2-этилгексанола в нитрующую смесь.
Задачей настоящего изобретения является расширение сырьевой базы для получения цетанповышающей присадки и упрощение процесса, повышение его надежности и стабильности, снижение расхода реагентов для производства присадки и повышение экономичности от ее применения, квалифицированное использование кубового остатка производства бутиловых спиртов и отработанных в процессе нитрования кислот.
Поставленную задачу решают следующим образом. В качестве сырья для получения цетанповышающей присадки предлагается применять фракцию, кипящую в пределах 166-190С, выделенную из кубового остатка производства бутиловых спиртов (КОБС), которые получают методом оксосинтеза. Фракция содержит, мас.%:
2-этилгексанол 60,0-90,0
2-этил-4-метилпентанол 1,3-5,0
ацетали 3,0-16,0
моногликолевые эфиры 5,7-19,0
Подачу сырья и реагентов в реактор предложено проводить в следующем порядке: вначале в охлажденный реактор загружают карбамид, затем при постоянном охлаждении и перемешивании одновременно отдельно друг от друга дозируют (0,30-0,60 г/мин) предварительно приготовленную нитрующую смесь серной и азотной кислот и вышеназванную фракцию, тщательно контролируя при этом температуру, которая поддерживается в интервале 8-18С. По окончании загрузки в реактор нитрующей смеси и сырья (вышеназванной фракции) реакционную массу выдерживают при температуре 8 С, выделенную из кубового остатка производства бутиловых спиртов (КОБС), получаемых методом оксосингеза и содержащую 60-90% 2-этилгексанола, 1,3-5,0% 2-этил-4-метилпентанола, 3-16% ацеталей, 5,7-19% моногликолевых эфиров. Фракцию, содержащую в качестве основного субстрата для нитрования 2-этилгексанол, нитруют нитрующей смесью азотной и серной кислот при температуре 8-18С в течение 2,5-1,5 часов. Полученные продукты нитрования отделяют от отработанных в процессе нитрации кислот. Выделенный органический слой (целевой продукт) промывают дважды 10% (мас.) раствором солей, полученных нейтрализацией отработанных кислот и единожды этим же раствором с добавлением 10% (мас.) раствора гидроксида натрия. 3 мас. части раствора солей добавляют на 10 мас. частей целевого продукта. Изобретение позволяет упростить процесс, а также снизить затраты на процесс получения целевого продукта. 2 н. и 3 з.п. ф-лы, 1 табл.
Изобретение относится к нефтеперерабатывающей и нефтехимической промышленности, конкретно к способу получения нитроэфиров спиртов, которые находят применение в качестве цетанповышающей присадки (ЦПП) к дизельным топливам.
Известен способ получения ЦПП – циклогексилнитрата путем нитрования циклогексанола в безводной среде смесями серной и азотной кислот, при этом нитрование возможно только при температуре не выше – 20С (А.с. №687791 СССР, МКИ С 07 С 68/04).
Недостатками данного способа являются необходимость проведения процесса при низких температурах из-за низкой стабильности целевого продукта циклогексилнитрата в условиях синтеза, кроме того, процесс проводят с использованием хлористого метилена или гексана.
Известны способы получения циклогексилнитрата с использованием обводненных нитрующих смесей кислот при температуре не выше 0-5С. Процесс нитрования проводят в среде растворителей – хлористого метилена, гексана, присутствие которых облегчает теплоотвод из реакционной смеси (Питеркин Р.Н., Чесалов А.М. Нитрование циклогексанола в среде растворителей. / Химическая промышленность, №9, 2000 г., с. 483-486).
Описанные выше способы имеют существенные недостатки:
– необходимость проведения процесса при минусовых температурах,
– использование растворителей,
– использование дефицитного дорогостоящего сырья циклогексанола. Названные недостатки усложняют технологический процесс, уменьшают его стабильность и надежность, снижают эффективность производства, повышают стоимость присадки.
Наиболее близким по технической сущности и достигаемому эффекту является способ получения ЦПП – 2-этилгексилнитрата путем нитрования 2-этилгексанола с содержанием основного вещества не менее 99,0 мас.% нитрующей смесью азотной и серной кислот при добавлении карбамида, с отделением отработанных кислот от целевого продукта – 2-этилгексилнитрата и последующей его промывкой последовательно дважды 10% (мас.) водным раствором сульфата натрия и затем однократно 10% (мас.) раствором смеси сульфата и карбоната натрия, взятых в соотношении 1:1, используя каждый раз на 2 части 2-этилгексилнитрата 1 часть раствора солей (пат. США №5162568).
Описанная присадка и способ ее получения имеют следующие недостатки: для ее получения используют дорогостоящее и дефицитное сырье – 2-этилгексанол, большой расход реагентов для промывки, низкая устойчивость и стабильность процесса нитрования, что обусловлено подачей нитруемого вещества – 2-этилгексанола в нитрующую смесь.
Задачей настоящего изобретения является расширение сырьевой базы для получения цетанповышающей присадки и упрощение процесса, повышение его надежности и стабильности, снижение расхода реагентов для производства присадки и повышение экономичности от ее применения, квалифицированное использование кубового остатка производства бутиловых спиртов и отработанных в процессе нитрования кислот.
Поставленную задачу решают следующим образом. В качестве сырья для получения цетанповышающей присадки предлагается применять фракцию, кипящую в пределах 166-190С, выделенную из кубового остатка производства бутиловых спиртов (КОБС), которые получают методом оксосинтеза. Фракция содержит, мас.%:
2-этилгексанол 60,0-90,0
2-этил-4-метилпентанол 1,3-5,0
ацетали 3,0-16,0
моногликолевые эфиры 5,7-19,0
Подачу сырья и реагентов в реактор предложено проводить в следующем порядке: вначале в охлажденный реактор загружают карбамид, затем при постоянном охлаждении и перемешивании одновременно отдельно друг от друга дозируют (0,30-0,60 г/мин) предварительно приготовленную нитрующую смесь серной и азотной кислот и вышеназванную фракцию, тщательно контролируя при этом температуру, которая поддерживается в интервале 8-18С. По окончании загрузки в реактор нитрующей смеси и сырья (вышеназванной фракции) реакционную массу выдерживают при температуре 8 18С в течение 2,5-1,5 часов. После отделения отработанного раствора кислот от органического слоя – продукта нитрования, последний дважды обрабатывают (промывают) 10% (мас.) раствором солей (сульфата и нитрата натрия), полученных при нейтрализации отработанных в процессе нитрования кислот (кислотность среды нейтральная), и единожды этим же раствором с добавлением 10% (мас.) водного раствора гидроксида натрия (кислотность среды щелочная). При промывке на 10 мас. частей продукта нитрования используют 3 мас. части 10% (мас.) раствора солей.
Общим с предлагаемым изобретением является использование 2-этилгексанолсодержащего сырья, проведение процесса нитрования с использованием серной и азотной кислот и карбамида, наличие стадий отделения органического слоя – продукта нитрования от отработанных кислот и его промывка растворами солей.
Отличительной особенностью изобретения является: применение другого 2-этилгексанолсодержащего сырья (фракция, кипящая в пределах 166-190С, выделенная из кубового остатка производства бутиловых спиртов (КОБС), полученных методом оксосинтеза, и содержащая от 60 до 90 мас.% 2-этилгексанола, 2-этил-4-метилпентанола, а также ацетали, моногликолевые эфиры, использование для промывки полученного продукта нитрования других реагентов – отработанных в процессе нитрования и нейтрализованных гидроксидом натрия кислот, другие порядок загрузки (дозирования) реагентов в реактор, температура и продолжительность процесса нитрования.
Решение поставленной задачи с использованием полученной предложенным способом цетанповышающей присадки позволяет сократить затраты на сырье и реагенты (квалифицированно используются отход производства бутиловых спиртов и отработанные растворы кислот), повысить надежность и стабильность процесса, увеличить эффективность применения присадки и расширить сырьевую базу для ее производства.
Предлагаемый способ получения ЦПП и ее эффективность при добавлении в дизельные топлива иллюстрируются следующими примерами.
Пример 1. В реактор, снабженный мешалкой и охлаждающей баней, при температуре 8-12С загружают 21,15 г азотной кислоты концентрации 69,2 мас.%, 36,6 г серной кислоты концентрации 93,6 мас.%. В полученную нитрующую смесь добавляют 1,5 г карбамида. В качестве сырья для нитрования используют фракцию 166-190С следующего состава, мас.%:
2-этил-гексанола 89,2
2-этил-4-метилпентанола 1,3
ацеталей 3,0
моногликолевых эфиров 6,5
В реактор с нитрующей смесью со скоростью 0,30-0,35 г/мин, при температуре 8-12С вводят (дозируют) названную фракцию в количестве 25,0 г. После введения всего ее количества реакционную смесь при непрерывном перемешивании выдерживают при температуре 8-18С в течение 2,5-1,5 часов. По окончании процесса нитрования и расслоения реакционной смеси отработанные кислоты отделяют от органического слоя продукта нитрования.
Для обработки органического слоя используют 10% (мас.) раствор смеси солей сульфата натрия и нитрата натрия, полученный нейтрализацией отработанных в процессе нитрования кислот гидроксидом натрия (кислотность среды нейтральная), в количестве по 10 см3 для двукратной обработки и третий раз в количестве 10 см3 раствора солей с добавлением 4 см3 10% (мас.) раствора гидроксида натрия (кислотность среды щелочная). Каждую обработку проводят при перемешивании в течение 15 минут.
Конверсия 2-этилгексанола, содержащегося в сырье, составляет 99,6 %, выход 2-этилгексилнитрата 99,1 мас.%.
Пример 2. (Прототип). По примеру 1 с тем отличием, что для нитрования вместо фракции 166-190С, выделенной из КОБС, используют 2-этилгексанол чистотой 99,5 мас.%.
Конверсия 2-этилгексанола составляет 99,7%, выход 2-этилгексилнитрата 97,8 мас.%.
Пример 3. В реактор для приготовления нитрующей смеси, снабженный мешалкой и охлаждающей баней, загружают 42,3 г азотной кислоты концентрации 69 мас.% и в нее дозируют 73,2 г серной кислоты концентрации 93 мас.%. Полученную нитрующую смесь переливают в мерник нитрующей смеси. В другой мерник заливают фракцию 166-190С, выделенную из КОБС бутиловых спиртов и содержащую, мас.%:
2-этил-гексанола 71,0
2-этил-4-метилпентанола 1,8
ацеталей 12,0
моногликолевых эфиров 15,2
В реактор нитрования, снабженный мешалкой и охлаждающей баней, загружают 3 г карбамида. Затем при постоянном перемешивании и охлаждении в реактор одновременно дозируют нитрующую смесь в количестве 115,5 г и вышеназванную фракцию 166-190С в количестве 50 г. Дозировку компонентов ведут со скоростью 0,3-0,6 г/мин, поддерживая температуру в реакторе в пределах 816С. После введения всего количества реагентов реакционную смесь выдерживают при постоянном перемешивании при температуре 8-16С в течение 2,5-1,5 часов. Обработку (промывку) органического слоя проводят по примеру 1.
Конверсия 2-этилгексанола, содержащегося в сырье составляет 99,6%, выход 2-этилгексилнитрата – 99,0 мас.%.
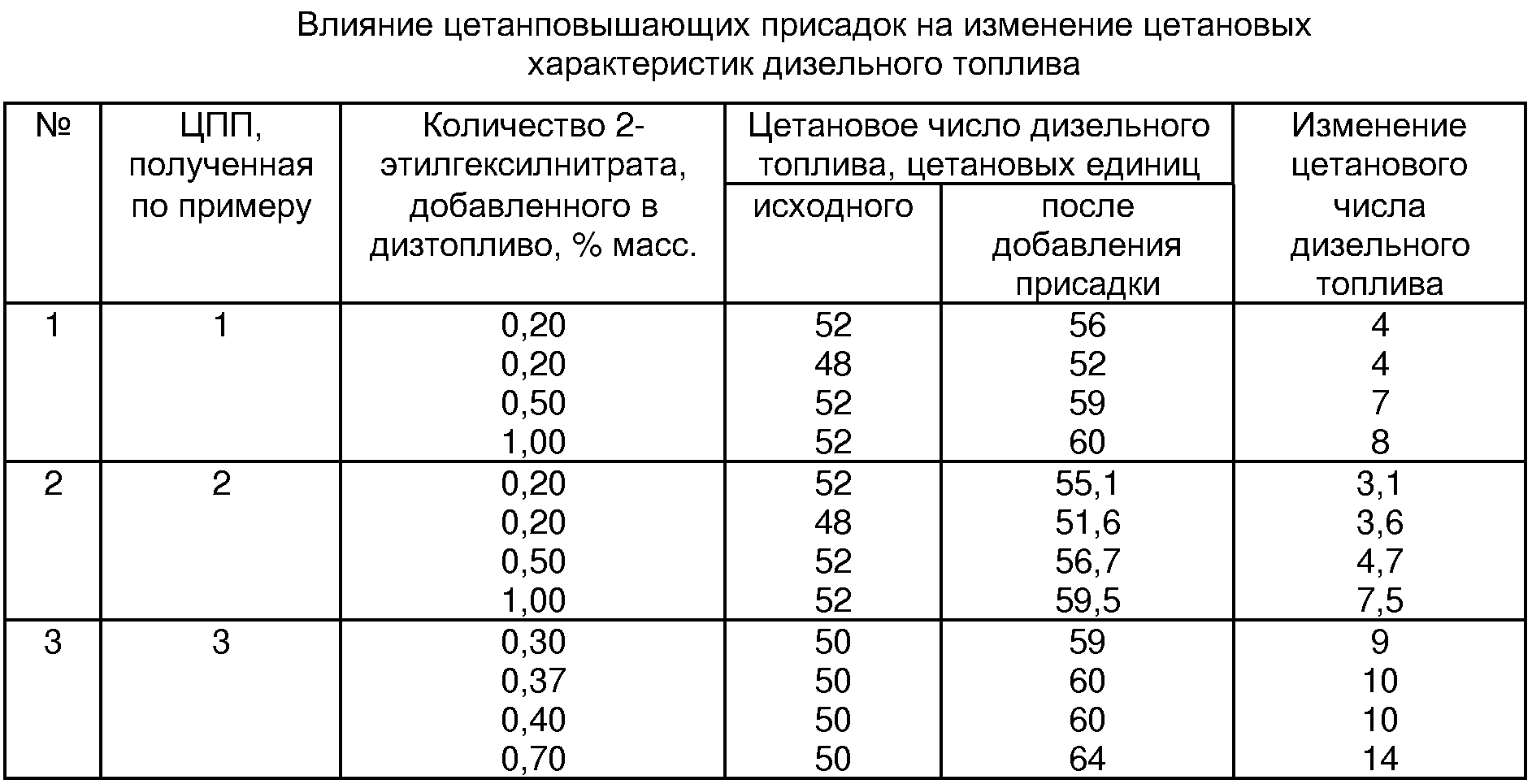
Пример 4. Полученные ЦПП добавлены в дизельное топливо и определены цетановые характеристики дизтоплива. Результаты испытаний приведены в таблице. 18С в течение 2,5-1,5 часов. После отделения отработанного раствора кислот от органического слоя – продукта нитрования, последний дважды обрабатывают (промывают) 10% (мас.) раствором солей (сульфата и нитрата натрия), полученных при нейтрализации отработанных в процессе нитрования кислот (кислотность среды нейтральная), и единожды этим же раствором с добавлением 10% (мас.) водного раствора гидроксида натрия (кислотность среды щелочная). При промывке на 10 мас. частей продукта нитрования используют 3 мас. части 10% (мас.) раствора солей.
Общим с предлагаемым изобретением является использование 2-этилгексанолсодержащего сырья, проведение процесса нитрования с использованием серной и азотной кислот и карбамида, наличие стадий отделения органического слоя – продукта нитрования от отработанных кислот и его промывка растворами солей.
Отличительной особенностью изобретения является: применение другого 2-этилгексанолсодержащего сырья (фракция, кипящая в пределах 166-190С, выделенная из кубового остатка производства бутиловых спиртов (КОБС), полученных методом оксосинтеза, и содержащая от 60 до 90 мас.% 2-этилгексанола, 2-этил-4-метилпентанола, а также ацетали, моногликолевые эфиры, использование для промывки полученного продукта нитрования других реагентов – отработанных в процессе нитрования и нейтрализованных гидроксидом натрия кислот, другие порядок загрузки (дозирования) реагентов в реактор, температура и продолжительность процесса нитрования.
Решение поставленной задачи с использованием полученной предложенным способом цетанповышающей присадки позволяет сократить затраты на сырье и реагенты (квалифицированно используются отход производства бутиловых спиртов и отработанные растворы кислот), повысить надежность и стабильность процесса, увеличить эффективность применения присадки и расширить сырьевую базу для ее производства.
Предлагаемый способ получения ЦПП и ее эффективность при добавлении в дизельные топлива иллюстрируются следующими примерами.
Пример 1. В реактор, снабженный мешалкой и охлаждающей баней, при температуре 8-12С загружают 21,15 г азотной кислоты концентрации 69,2 мас.%, 36,6 г серной кислоты концентрации 93,6 мас.%. В полученную нитрующую смесь добавляют 1,5 г карбамида. В качестве сырья для нитрования используют фракцию 166-190С следующего состава, мас.%:
2-этил-гексанола 89,2
2-этил-4-метилпентанола 1,3
ацеталей 3,0
моногликолевых эфиров 6,5
В реактор с нитрующей смесью со скоростью 0,30-0,35 г/мин, при температуре 8-12С вводят (дозируют) названную фракцию в количестве 25,0 г. После введения всего ее количества реакционную смесь при непрерывном перемешивании выдерживают при температуре 8-18С в течение 2,5-1,5 часов. По окончании процесса нитрования и расслоения реакционной смеси отработанные кислоты отделяют от органического слоя продукта нитрования.
Для обработки органического слоя используют 10% (мас.) раствор смеси солей сульфата натрия и нитрата натрия, полученный нейтрализацией отработанных в процессе нитрования кислот гидроксидом натрия (кислотность среды нейтральная), в количестве по 10 см3 для двукратной обработки и третий раз в количестве 10 см3 раствора солей с добавлением 4 см3 10% (мас.) раствора гидроксида натрия (кислотность среды щелочная). Каждую обработку проводят при перемешивании в течение 15 минут.
Конверсия 2-этилгексанола, содержащегося в сырье, составляет 99,6 %, выход 2-этилгексилнитрата 99,1 мас.%.
Пример 2. (Прототип). По примеру 1 с тем отличием, что для нитрования вместо фракции 166-190С, выделенной из КОБС, используют 2-этилгексанол чистотой 99,5 мас.%.
Конверсия 2-этилгексанола составляет 99,7%, выход 2-этилгексилнитрата 97,8 мас.%.
Пример 3. В реактор для приготовления нитрующей смеси, снабженный мешалкой и охлаждающей баней, загружают 42,3 г азотной кислоты концентрации 69 мас.% и в нее дозируют 73,2 г серной кислоты концентрации 93 мас.%. Полученную нитрующую смесь переливают в мерник нитрующей смеси. В другой мерник заливают фракцию 166-190С, выделенную из КОБС бутиловых спиртов и содержащую, мас.%:
2-этил-гексанола 71,0
2-этил-4-метилпентанола 1,8
ацеталей 12,0
моногликолевых эфиров 15,2
В реактор нитрования, снабженный мешалкой и охлаждающей баней, загружают 3 г карбамида. Затем при постоянном перемешивании и охлаждении в реактор одновременно дозируют нитрующую смесь в количестве 115,5 г и вышеназванную фракцию 166-190С в количестве 50 г. Дозировку компонентов ведут со скоростью 0,3-0,6 г/мин, поддерживая температуру в реакторе в пределах 816С. После введения всего количества реагентов реакционную смесь выдерживают при постоянном перемешивании при температуре 8-16С в течение 2,5-1,5 часов. Обработку (промывку) органического слоя проводят по примеру 1.
Конверсия 2-этилгексанола, содержащегося в сырье составляет 99,6%, выход 2-этилгексилнитрата – 99,0 мас.%.
Пример 4. Полученные ЦПП добавлены в дизельное топливо и определены цетановые характеристики дизтоплива. Результаты испытаний приведены в таблице.
Формула изобретения 1. Цетанповышающая присадка к дизельным топливам на основе 2-этилгексилнитрата, отличающаяся тем, что цетанповышающая присадка представляет собой 2-этилгексилнитратсодержащий продукт нитрования фракции 166-190 С, выделенной из кубового остатка производства бутиловых спиртов, полученных методом оксосинтеза, следующего компонентного состава, мас.%:
2-Этилгексанол 60,0 – 90,0
2-Этил-4-метилпентанол 1,3 – 5,0
Ацетали 3,0 – 16,0
Моногликолевые эфиры 5,7 – 19,0
2. Способ получения цетанповышающей присадки путем нитрования 2-этилгексанола, отличающийся тем, что в качестве 2-этилгексанолсодержащего сырья используют фракцию 166-190С состава по п.1.
3. Способ по п.2, отличающийся тем, что процесс нитрования осуществляют при температуре 8-18С в течение 2,5-1,5 ч.
4. Способ по п.2, oтличaющийся тем, что нитрующую смесь и вышеназванную фракцию дозируют в содержащий карбамид реактор нитрования одновременно со скоростью 0,3-0,6 г/мин.
5. Способ по п.2, отличающийся тем, что обработку продукта нитрования (органического слоя) проводят путем двукратной его промывки 10-ным мас.% раствором смеси солей сульфата и нитрата натрия, полученных путем нейтрализации отработанных кислот процесса нитрования гидроксидом натрия (реакция среды нейтральная), однократной – указанной смесью солей с добавлением 10-ного мас.% раствора гидроксида натрия (реакция среды щелочная), указанную смесь солей подают в количестве 3 мас.ч. на 10 мас.ч. органического слоя.
MM4A Досрочное прекращение действия патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 15.11.2005
Извещение опубликовано: 10.12.2006 БИ: 34/2006
|
||||||||||||||||||||||||||