Патент на изобретение №2233842
|
||||||||||||||||||||||||||
(54) ПРОИЗВОДНЫЕ ПУРИНА, ОБЛАДАЮЩИЕ ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ
(57) Реферат: Изобретение относится к производным пурина, обладающим противовирусной активностью в отношении цитомегаловируса человека и вируса иммунодефицита человека типа 1, общей формулы: 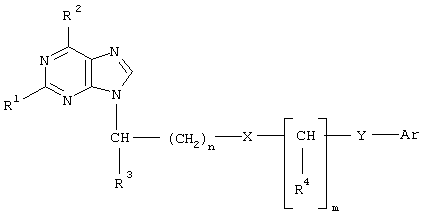 где n = 0 – 4; m = 0 – 3; R1 = Н, ОН или NH2; R2 = ОН, NH2, ацетиламино или бензоиламино; R3 = Н или низший алкил C1–С4; R4 = Н, низший алкил C1-С4 или фенил; Х = СН2, О, S, NH или С(O)O; Y = СН2, СН=СН, С(O), О или ординарная связь; Ar = фенил, пиридил, нафтил или замещенный фенил формулы
где n = 0 – 4; m = 0 – 3; R1 = Н, ОН или NH2; R2 = ОН, NH2, ацетиламино или бензоиламино; R3 = Н или низший алкил C1–С4; R4 = Н, низший алкил C1-С4 или фенил; Х = СН2, О, S, NH или С(O)O; Y = СН2, СН=СН, С(O), О или ординарная связь; Ar = фенил, пиридил, нафтил или замещенный фенил формулы
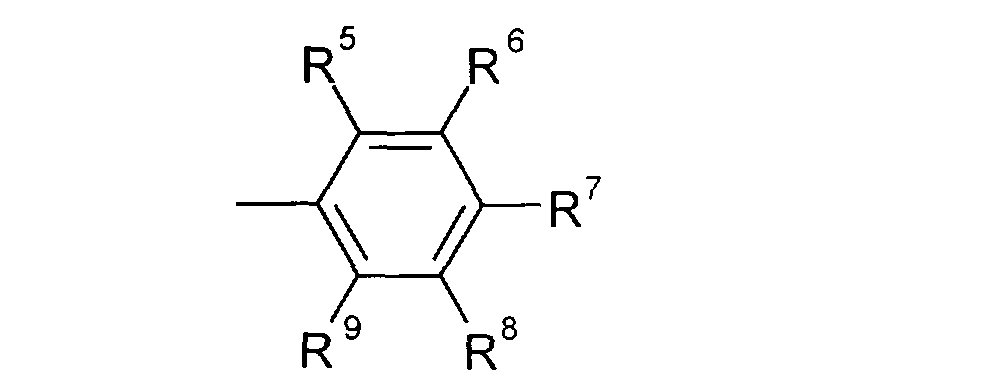 где независимо R5–R9 = алкил С1–C8, циклоалкил C5–С6, 1-адамантил, аллил, фенил, бензил, F, Cl, Br, J, трифторметил, алкокси C1–С5, фенокси, бензилокси, бензоилокси, циано, карбокси, ацетил или нитро, противовирусное действие наиболее активных веществ в отношении цитомегаловируса человека in vitro проявляется в концентрациях 0,01-0,0005
где независимо R5–R9 = алкил С1–C8, циклоалкил C5–С6, 1-адамантил, аллил, фенил, бензил, F, Cl, Br, J, трифторметил, алкокси C1–С5, фенокси, бензилокси, бензоилокси, циано, карбокси, ацетил или нитро, противовирусное действие наиболее активных веществ в отношении цитомегаловируса человека in vitro проявляется в концентрациях 0,01-0,0005  М и характеризуется селективностью 1-400 тысяч. 5 табл.
Изобретение относится к области медицины и химии азотсодержащих гетероциклических соединений, в частности к новым производным пурина общей формулы М и характеризуется селективностью 1-400 тысяч. 5 табл.
Изобретение относится к области медицины и химии азотсодержащих гетероциклических соединений, в частности к новым производным пурина общей формулы
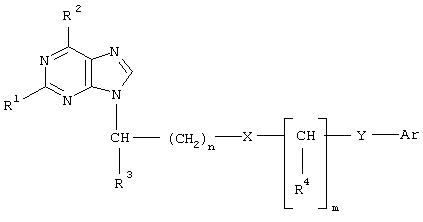 где n=0-4;
m=0-3;
R1=Н, ОН или NH2;
R2=ОН, NH2, ацетиламино или бензоиламино;
R3=Н или низший алкил C1-С4;
R4=Н, низший алкил С1-С4 или фенил;
Х=СН2, О, S, NH или С(O)O;
Y=СН2, СН=СН, С(O), О или ординарная связь;
Аr = фенил, пиридил, нафтил или замещенный фенил формулы
где n=0-4;
m=0-3;
R1=Н, ОН или NH2;
R2=ОН, NH2, ацетиламино или бензоиламино;
R3=Н или низший алкил C1-С4;
R4=Н, низший алкил С1-С4 или фенил;
Х=СН2, О, S, NH или С(O)O;
Y=СН2, СН=СН, С(O), О или ординарная связь;
Аr = фенил, пиридил, нафтил или замещенный фенил формулы
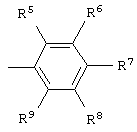 где независимо R5-R9 = алкил C1-C8, циклоалкил С5-С6, 1-адамантил, аллил, фенил, бензил, F, Cl, Br, J, трифторметил, алкокси C1-С5, фенокси, бензилокси, бензоилокси, циано, карбокси, ацетил или нитро, которые обладают противовирусной активностью, в том числе в отношении цитомегаловируса человека и вируса иммунодефицита человека типа 1, и могут найти применение в медицине в качестве лекарственных веществ в лекарственных средствах, применяющихся в комплексной терапии СПИД и оппортунистических вирусных инфекций.
Описаны изомерные аналоги ацикловира и ганцикловира, обладающие противовирусной активностью и имеющие принципиально иное строение ациклической цепи, соединенной с положением N9 пуринового цикла непосредственно через атом кислорода [Пат. США №4965270 (1990), МКИ С 07 D 473/18, А 61 К 31/52 / Hamden M.R., Wyatt P.G., Bailey S. Purine derivatives].
Однако все описанные выше производные пурина, за исключением ганцикловира, не обладают уровнем анти-ЦМВ действия, достаточным для создания на их основе лекарственных средств для лечения ЦМВ инфекции.
Среди активных анти-ЦМВ агентов, имеющих иную, не цуриновую химическую природу, следует выделить производные 1,6-нафтиридина, проявляющие ингибиторные свойства в отношении ЦМВ in vitro в минимальной концентрации 0,0004
Целью предлагаемого изобретения является создание новых, высокоэффективных, селективных и малотоксичных антивирусных агентов для лечения ЦМВ и других вирусных инфекций.
Сущность изобретения заключается в синтезе новых производных пурина, в том числе аденина, гуанина, гипоксантина и 2,6-диаминопурина, содержащих в качестве заместителя незамещенное или замещенное ароматическое ядро (фенил, нафтил или пиридил), соединенное с атомом азота N9 пуриновой гетероциклической системы ациклической цепью, состоящей из 1-5 атомов углерода, которая может дополнительно включать атомы кислорода, серы или азота, и отвечающих указанной выше общей формуле.
Синтез новых соединений был осуществлен путем алкилирования соответствующего пуринового основания (аденина, гуанина, гипоксантина или 2,6-диаминопурина) тозилатами
где независимо R5-R9 = алкил C1-C8, циклоалкил С5-С6, 1-адамантил, аллил, фенил, бензил, F, Cl, Br, J, трифторметил, алкокси C1-С5, фенокси, бензилокси, бензоилокси, циано, карбокси, ацетил или нитро, которые обладают противовирусной активностью, в том числе в отношении цитомегаловируса человека и вируса иммунодефицита человека типа 1, и могут найти применение в медицине в качестве лекарственных веществ в лекарственных средствах, применяющихся в комплексной терапии СПИД и оппортунистических вирусных инфекций.
Описаны изомерные аналоги ацикловира и ганцикловира, обладающие противовирусной активностью и имеющие принципиально иное строение ациклической цепи, соединенной с положением N9 пуринового цикла непосредственно через атом кислорода [Пат. США №4965270 (1990), МКИ С 07 D 473/18, А 61 К 31/52 / Hamden M.R., Wyatt P.G., Bailey S. Purine derivatives].
Однако все описанные выше производные пурина, за исключением ганцикловира, не обладают уровнем анти-ЦМВ действия, достаточным для создания на их основе лекарственных средств для лечения ЦМВ инфекции.
Среди активных анти-ЦМВ агентов, имеющих иную, не цуриновую химическую природу, следует выделить производные 1,6-нафтиридина, проявляющие ингибиторные свойства в отношении ЦМВ in vitro в минимальной концентрации 0,0004
Целью предлагаемого изобретения является создание новых, высокоэффективных, селективных и малотоксичных антивирусных агентов для лечения ЦМВ и других вирусных инфекций.
Сущность изобретения заключается в синтезе новых производных пурина, в том числе аденина, гуанина, гипоксантина и 2,6-диаминопурина, содержащих в качестве заместителя незамещенное или замещенное ароматическое ядро (фенил, нафтил или пиридил), соединенное с атомом азота N9 пуриновой гетероциклической системы ациклической цепью, состоящей из 1-5 атомов углерода, которая может дополнительно включать атомы кислорода, серы или азота, и отвечающих указанной выше общей формуле.
Синтез новых соединений был осуществлен путем алкилирования соответствующего пуринового основания (аденина, гуанина, гипоксантина или 2,6-диаминопурина) тозилатами  -арилалканолов или -арилалкилбромидами в среде безводного диметилформамида в присутствии безводного карбоната калия или гидрида натрия по схеме: -арилалканолов или -арилалкилбромидами в среде безводного диметилформамида в присутствии безводного карбоната калия или гидрида натрия по схеме:
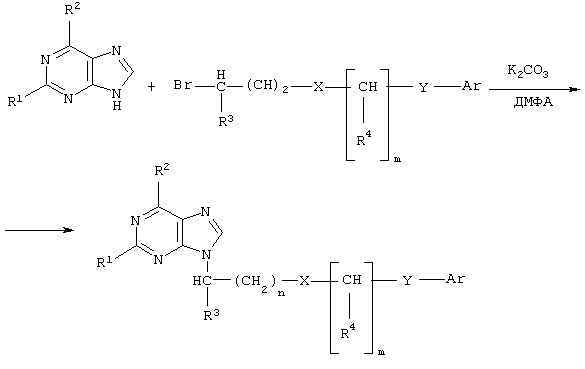 где n=0-4;
m=0-3;
R1=Н, ОН или NH2;
R2=ОН, NH2, ацетиламино или бензоиламино;
R3=Н или низший алкил C1-С4;
R4=Н, низший алкил C1-С4 или фенил;
Х=СН2, О, S, NH или С(O)O;
Y=СН2, СН=СН, С(O), О или ординарная связь;
Аr = фенил, пиридил, нафтил или замещенный фенил формулы
где n=0-4;
m=0-3;
R1=Н, ОН или NH2;
R2=ОН, NH2, ацетиламино или бензоиламино;
R3=Н или низший алкил C1-С4;
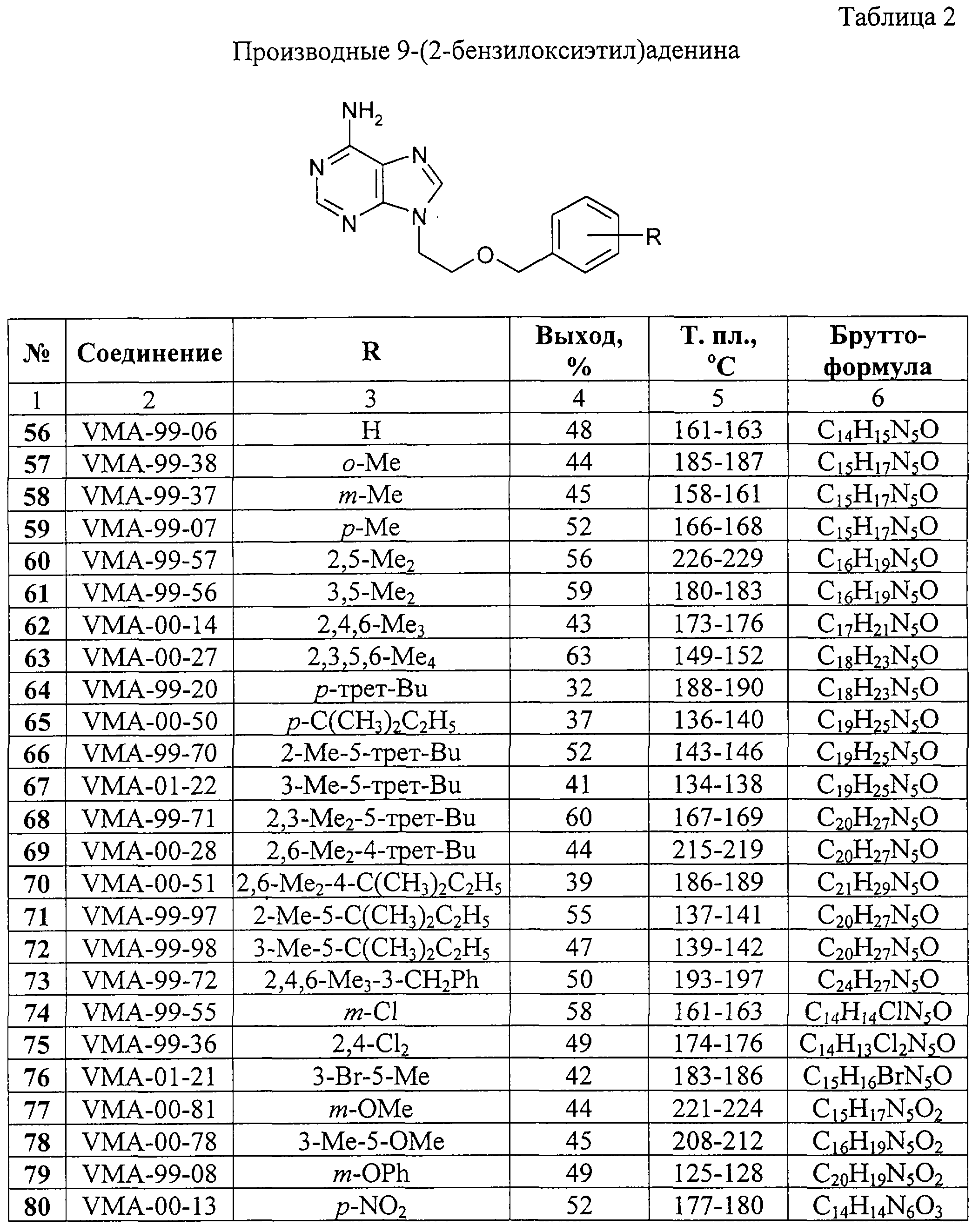
R4=Н, низший алкил C1-С4 или фенил;
Х=СН2, О, S, NH или С(O)O;
Y=СН2, СН=СН, С(O), О или ординарная связь;
Аr = фенил, пиридил, нафтил или замещенный фенил формулы
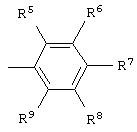 где независимо R5-R9 = алкил C1-C8, циклоалкил С5-С6, 1-адамантил, аллил, фенил, бензил, F, Cl, Br, J, трифторметил, алкокси C1-С5, фенокси, бензилокси, бензоилокси, циано, карбокси, ацетил или нитро.
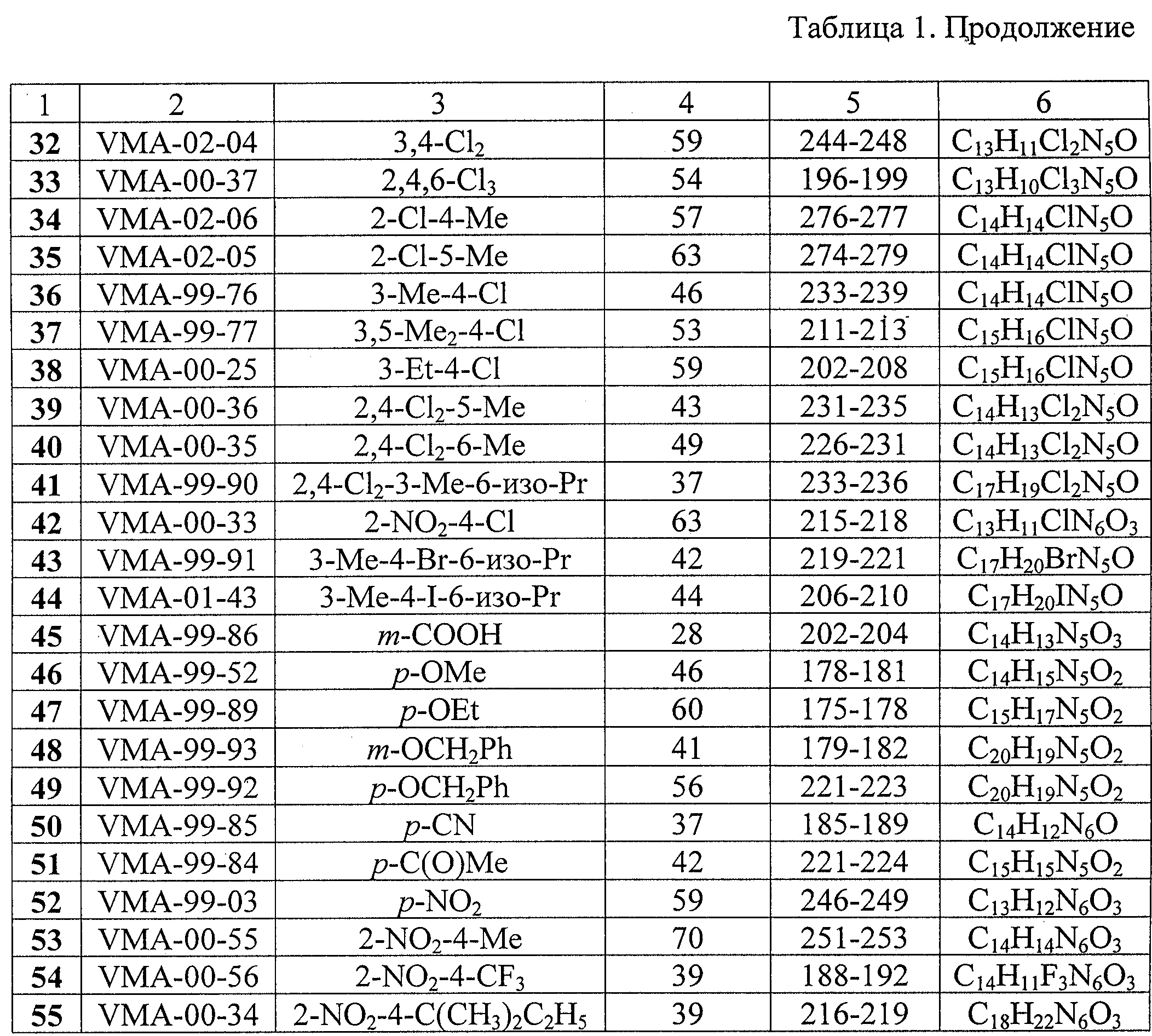
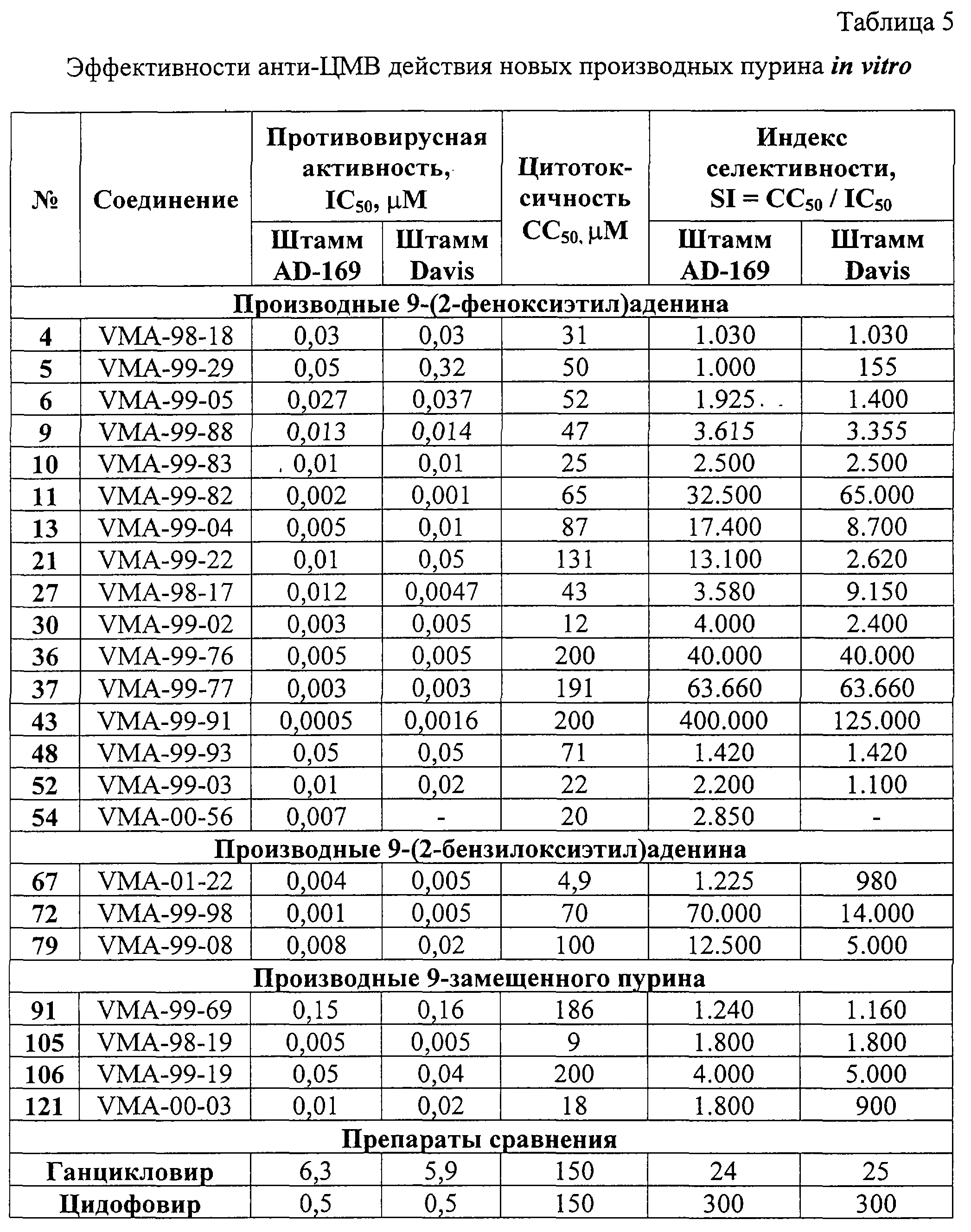
Химическое строение, выход и физико-химические свойства синтезированных соединений 1-130 представлены в таблицах 1-3. Структура и элементный состав соединений доказаны методами ПМР-спектроскопии и масс-спектрометрии, чистота и индивидуальность – методом тонкослойной хроматографии.
Пример 1. 9-[2-(4-Изопропилфенокси)этил]аденин (11, VMA-99-82).
2-(4-Изопропилфенокси)этанол.
К раствору 40,0 г (0,294 моль) 4-изопропилфенола в 200 мл воды, содержащей 30,0 г (0,535 моль) КОН, при температуре 75-80
где независимо R5-R9 = алкил C1-C8, циклоалкил С5-С6, 1-адамантил, аллил, фенил, бензил, F, Cl, Br, J, трифторметил, алкокси C1-С5, фенокси, бензилокси, бензоилокси, циано, карбокси, ацетил или нитро.
Химическое строение, выход и физико-химические свойства синтезированных соединений 1-130 представлены в таблицах 1-3. Структура и элементный состав соединений доказаны методами ПМР-спектроскопии и масс-спектрометрии, чистота и индивидуальность – методом тонкослойной хроматографии.
Пример 1. 9-[2-(4-Изопропилфенокси)этил]аденин (11, VMA-99-82).
2-(4-Изопропилфенокси)этанол.
К раствору 40,0 г (0,294 моль) 4-изопропилфенола в 200 мл воды, содержащей 30,0 г (0,535 моль) КОН, при температуре 75-80 С и интенсивном перемешивании в течение 30 мин добавляют 20,6 мл (0,307 моль) 2-хлорэтанола. Реакционную массу перемешивают при той же температуре еще 2 ч и оставляют на ночь при комнатной температуре. Органическую фазу отделяют, водную фазу экстрагируют хлороформом (4 С и интенсивном перемешивании в течение 30 мин добавляют 20,6 мл (0,307 моль) 2-хлорэтанола. Реакционную массу перемешивают при той же температуре еще 2 ч и оставляют на ночь при комнатной температуре. Органическую фазу отделяют, водную фазу экстрагируют хлороформом (4 25 мл), органическую фазу и экстракты объединяют, сушат Na2SO4, фильтруют и упаривают в вакууме водоструйного насоса на кипящей водяной бане. Остаток перегоняют в вакууме, собирая фракцию, кипящую при 157-162С (3 мм рт.ст.). Получают 39,0 г бесцветной вязкой жидкости, n20D 1,5246, выход 74%.
1-(4-Изопропилфенокси)-2-(n-толуолсульфонилокси)этан.
К раствору 30,0 г (0,166 моль) 2-(4-изопропилфенокси)этанола в 150 мл безводного дихлорметана и 40 мл пиридина при интенсивном перемешивании и охлаждении до 0С добавляют порциями в течение 20 мин 38,0 г (0,199 моль) n-толуолсульфохлорида с такой скоростью, чтобы температура реакционной смеси не поднималась выше 5С. После этого реакционную массу перемешивают при той же температуре еще 2 ч и оставляют на ночь при комнатной температуре. Реакционную массу последовательно промывают 2%-ным водным раствором соляной кислоты (2100 мл), водой (280 мл), насыщенным раствором соды (2100 мл) и вновь водой до нейтральной реакции, сушат Na2SO4, фильтруют и упаривают в вакууме водоструйного насоса. Остаток дополнительно вакуумируют при 40-50С (5 мм рт.ст.) в течение 1 ч. Получают 47,4 г вязкого сиропообразного продукта светло-желтого цвета, выход 85%.
1-Бром-2-(4-изопропилфенокси)этан. Метод А.
К раствору 14,0 г (0,078 моль) 2-(4-изопропилфенокси)этанола в 100 мл безводного хлороформа и 10 мл пиридина при интенсивном перемешивании и охлаждении до -10С прибавляют по каплям в течение 10 мин 2,7 мл (0,029 моль) трехбромистого фосфора. После этого реакционную массу перемешивают при 0С еще 2 ч, затем 2 ч при комнатной температуре и оставляют на ночь. Реакционную массу выливают в 250 мл холодной воды, органический слой промывают насыщенным раствором соды (2100 мл) и водой до нейтральной реакции, сушат Na2SO4, фильтруют и упаривают в вакууме водоструйного насоса. Остаток перегоняют в вакууме, собирая фракцию, кипящую при 158-162С (3 мм рт.ст.). Получают 10,2 г бесцветной прозрачной жидкости, n20D 1,5112, выход 54%.
1-Бром-2-(4-изопропилфенокси)этан. Метод Б.
Смесь 15,0 г (0,110 моль) 4-изопропилфенола, 20,0 мл (0,232 моль) 1,2-дибромэтана и раствора 6,2 г (0,110 моль) КОН в 10 мл воды кипятят в течение 8 ч до нейтральной реакции. Реакционную массу разбавляют 100 мл хлороформа, органический слой промывают 3%-ным раствором NaOH (100 мл) и водой (2100 мл) до нейтральной реакции, сушат Na2SO4, фильтруют, растворитель и избыток 1,2-дибромэтана отгоняют в вакууме водоструйного насоса на кипящей водяной бане. Остаток перегоняют в вакууме, собирая фракцию, кипящую при 158-163С (3 мм рт.ст.). Получают 13,5 г бесцветной прозрачной жидкости, n20D 1,5118, выход 51%.
9-[2-(4-Изопропилфенокси)этил]аденин (11, VMA-99-82). Метод А.
Суспензию 15,0 г (0,111 моль) аденина и 17,3 г (0,125 моль) свежепрокаленного карбоната калия в 100 мл ДМФА перемешивают при температуре 80-85С в течение 1,5 ч, добавляют раствор 37,0 г (0,111 моль) 1-(4-изопропилфенокси)-2-(n-толуолсульфонилокси)этана в 50 мл ДМФА и полученную смесь перемешивают при той же температуре еще 8 ч. Реакционную массу оставляют на ночь при комнатной температуре, фильтруют, фильтрат упаривают в вакууме, остаток обрабатывают 200 мл холодной воды и охлаждают при 0-5С в течение суток. Закристаллизовавшийся остаток отфильтровывают и после сушки на воздухе перекристаллизовывают из смеси 100 мл ДМФА и 10 мл воды. Получают 22,1 г VMA-99-82 в виде белого мелкокристаллического продукта с выходом 67%. Rf (метиленхлорид – метанол, 10 : 1) 0,64. Т. пл. 231-233С.
Масс-спектр: M+ 297 (6%), 282 (2%), 254 (2%), 178 (9%), 163 (81%), 149 (84%), 134 (100%), 119 (30%), 107 (50%), 105 (47%), 92 (99%), 91 (80%).
9-[2-(4-Изопропилфенокси)этил]аденин (11, VMA-99-82). Метод Б.
Суспензию 5,0 г (0,037 моль) аденина и 6,3 г (0,045 моль) свежепрокаленного карбоната калия в 60 мл ДМФА перемешивают при температуре 105-110С в течение 1,5 ч, добавляют раствор 9,0 г (0,037 моль) 1-бром-2-(4-изопропилфенокси)этана в 20 мл ДМФА и полученную смесь перемешивают при той же температуре еще 2 ч. Выделение и очистка аналогична описанному выше. Получают 6,1 г VMA-99-82 в виде белого мелкокристаллического продукта, выход 55%.
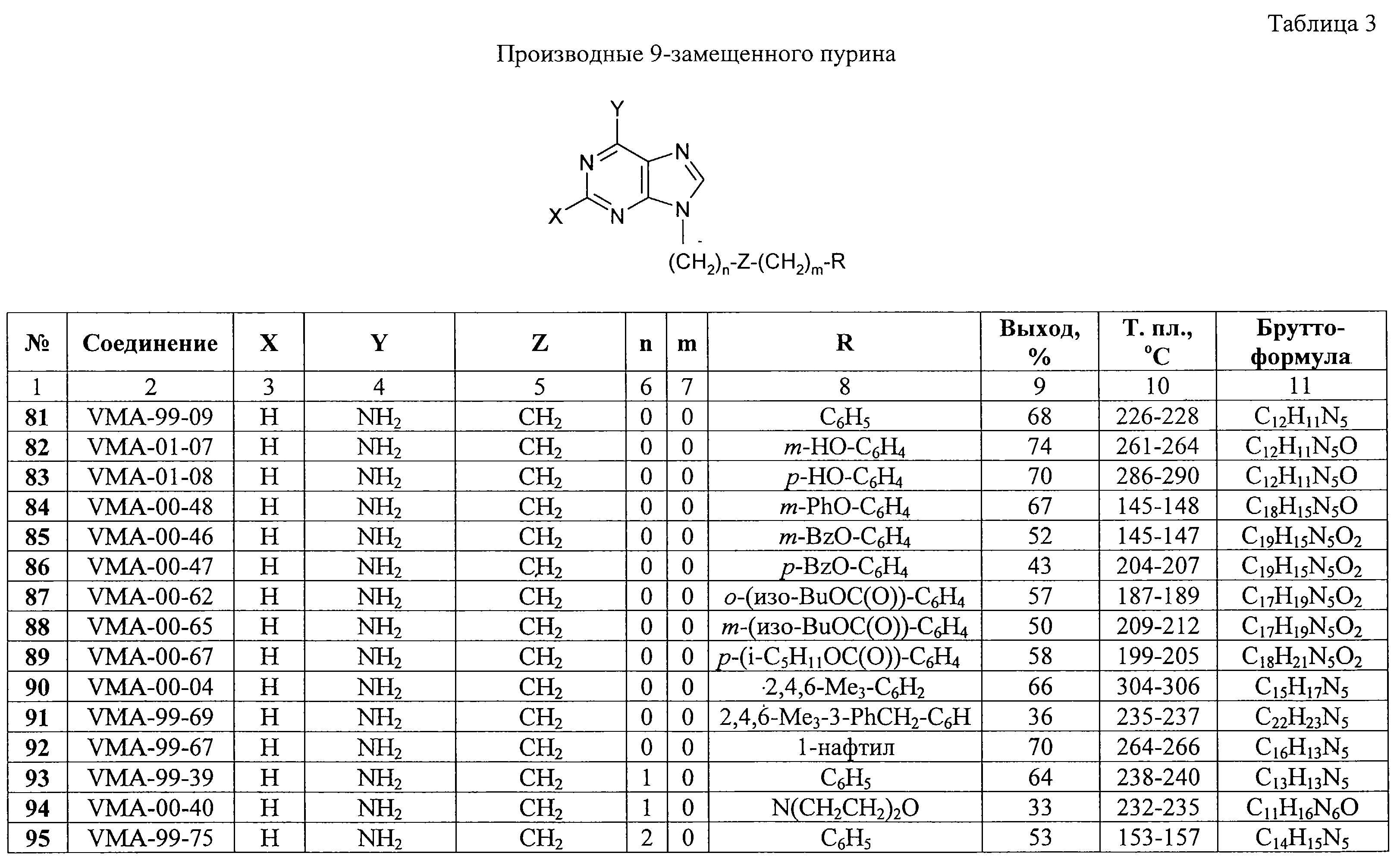
Пример 2. 9-[2-(3,5-Диметилбензилокси)этил]аденин (61, VMA-99-56).
2-(3,5-Диметилбензилокси)этанол.
К раствору 210 мл (1,509 моль) мезитилена и 100 мл ССl4 при кипячении и облучении светом добавляют в течение 30 мин раствор 14,0 мл (0,272 моль) брома в 25 мл ССl4. Реакционную массу упаривают в вакууме водоструйного насоса на водяной бане, к остатку прибавляют раствор 16,0 г (0,285 моль) КОН в 100 мл (1,793 моль) этиленгликоля и полученную смесь перемешивают на водяной бане при температуре 80-90С в течение 10 ч. Реакционную массу разбавляют 250 мл холодной воды, органическую фазу отделяют, водную фазу экстрагируют мезитиленом (330 мл), органическую фазу и экстракты объединяют, промывают солевым раствором (100 мл), сушат Na2SO4, фильтруют и упаривают в вакууме водоструйного насоса. Остаток перегоняют в вакууме, собирая фракцию, кипящую при 168-172С (3 мм рт.ст.). Получают 27,3 г бесцветной жидкости, n20D 1,5227, выход 56%.
1-(3,5-Диметилбензилокси)-2-(n-толуолсульфонилокси)этан.
К раствору 51,0 г (0,283 моль) 2-(3,5-диметилбензилокси)этанола в 250 мл безводного хлороформа и 80 мл пиридина при интенсивном перемешивании и охлаждении до 0С добавляют порциями в течение 30 мин 64,7 г (0,339 моль) n-толуолсульфохлорида с такой скоростью, чтобы температура реакционной смеси не поднималась выше 5С. После этого реакционную массу перемешивают при той же температуре еще 2 ч и оставляют на ночь при комнатной температуре. Затем реакционную массу последовательно промывают 2%-ным водным раствором соляной кислоты (2150 мл), водой (2100 мл), насыщенным раствором соды (2100 мл) и вновь водой до нейтральной реакции, сушат Na2SO4, фильтруют и упаривают в вакууме водоструйного насоса. Остаток растворителя удаляют в вакууме при 40-50С (5 мм рт.ст.) в течение 1 ч. Получают 77,6 г вязкого сиропообразного продукта светло-желтого цвета, выход 82%.
1-Бром-2-(3,5-диметилбензилокси)этан.
К раствору 18,75 г (0,104 моль) 2-(3,5-диметилбензилокси)этанола в 100 мл безводного хлороформа и 10 мл пиридина при интенсивном перемешивании и охлаждении до 10С добавляют в течение 10 мин 3,6 мл (0,0379 моль) трехбромистого фосфора. После этого реакционную массу перемешивают при 0С еще 1 ч, 2 ч при комнатной температуре и оставляют на ночь. Затем реакционную массу выливают в 300 мл холодной воды, органический слой промывают насыщенным раствором соды (2100 мл) и водой до нейтральной реакции, сушат Na2SO4, фильтруют и упаривают в вакууме водоструйного насоса. Остаток перегоняют в вакууме, собирая фракцию, кипящую при 167-173С (3 мм рт.ст.). Получают 12,9 г бесцветной прозрачной жидкости, n20D 1,5391, выход 51%.
9-[2-(3,5-Диметилбензилокси)этил]аденин (61, VMA-99-56). Метод А.
Суспензию 23,0 г (0,170 моль) аденина и 27,0 г (0,195 моль) свежепрокаленного карбоната калия в 250 мл ДМФА перемешивают при температуре 80-85С в течение 1,5 ч, добавляют раствор 60,0 г (0,176 моль) 1-(3,5-диметилбензилокси)-2-(n-толуолсульфонилокси)этана в 50 мл ДМФА и полученную смесь перемешивают при той же температуре еще 8 ч. Реакционную массу оставляют на ночь при комнатной температуре, фильтруют, твердый осадок обрабатывают 0,5 л холодной воды, фильтруют и после сушки на воздухе получают 17,8 г белого кристаллического продукта-сырца. Фильтрат первичной реакционной массы упаривают в вакууме, к остатку добавляют 0,5 л холодной воды, перемешивают и охлаждают при 0-5С в течение суток. Закристаллизовавшийся остаток отфильтровывают и после сушки на воздухе дополнительно получают 22,2 г целевого продукта-сырца. Обе партии продукта объединяют и перекристаллизовывают из смеси 100 мл ДМФА и 15 мл воды. Получают 29,6 г белого мелкокристаллического продукта с выходом 59%. Rf (метиленхлорид – метанол, 10:1) 0,60. Т.пл. 180-183С.
Масс-спектр: M+ 297 (3%), 178 (14%), 163 (31%), 149 (22%), 134 (100%), 119 (67%), 105 (47%), 92 (64%), 91 (46%).
9-[2-(3,5-Диметилбензилокси)этил]аденин (61, VMA-99-56). Метод Б.
Суспензию 5,0 г (0,037 моль) аденина и 6,3 г (0,045 моль) свежепрокаленного карбоната калия в 60 мл ДМФА перемешивают при температуре 105-110С в течение 1,5 ч, добавляют раствор 9,0 г (0,037 моль) 1-бром-2-(3,5-диметилбензилокси)этана в 20 мл ДМФА и полученную смесь перемешивают при той же температуре еще 2 ч. Выделение и очистка аналогична описанному выше. Получают 5,6 г VMA-99-56 в виде белого мелкокристаллического продукта, выход 51%.
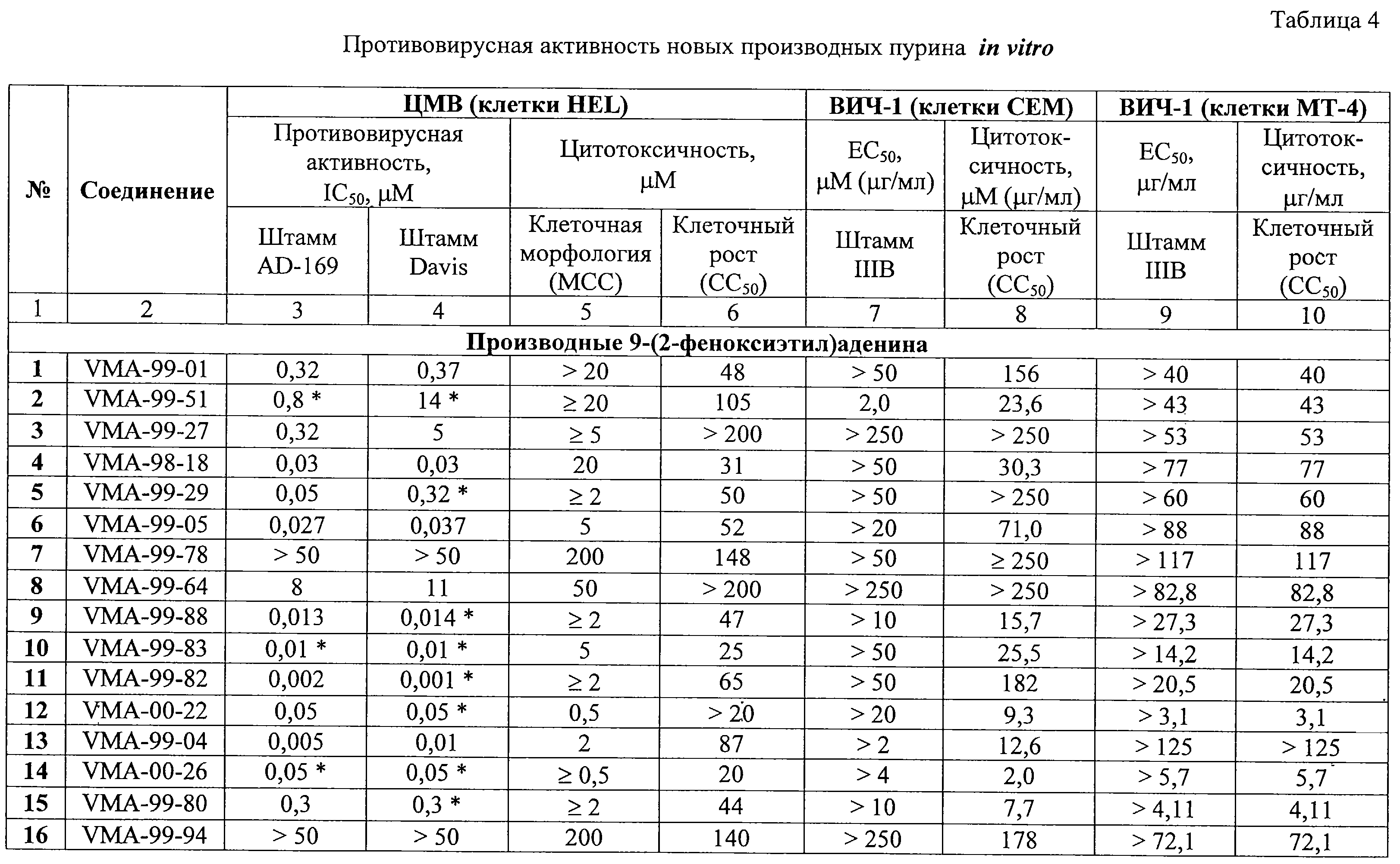
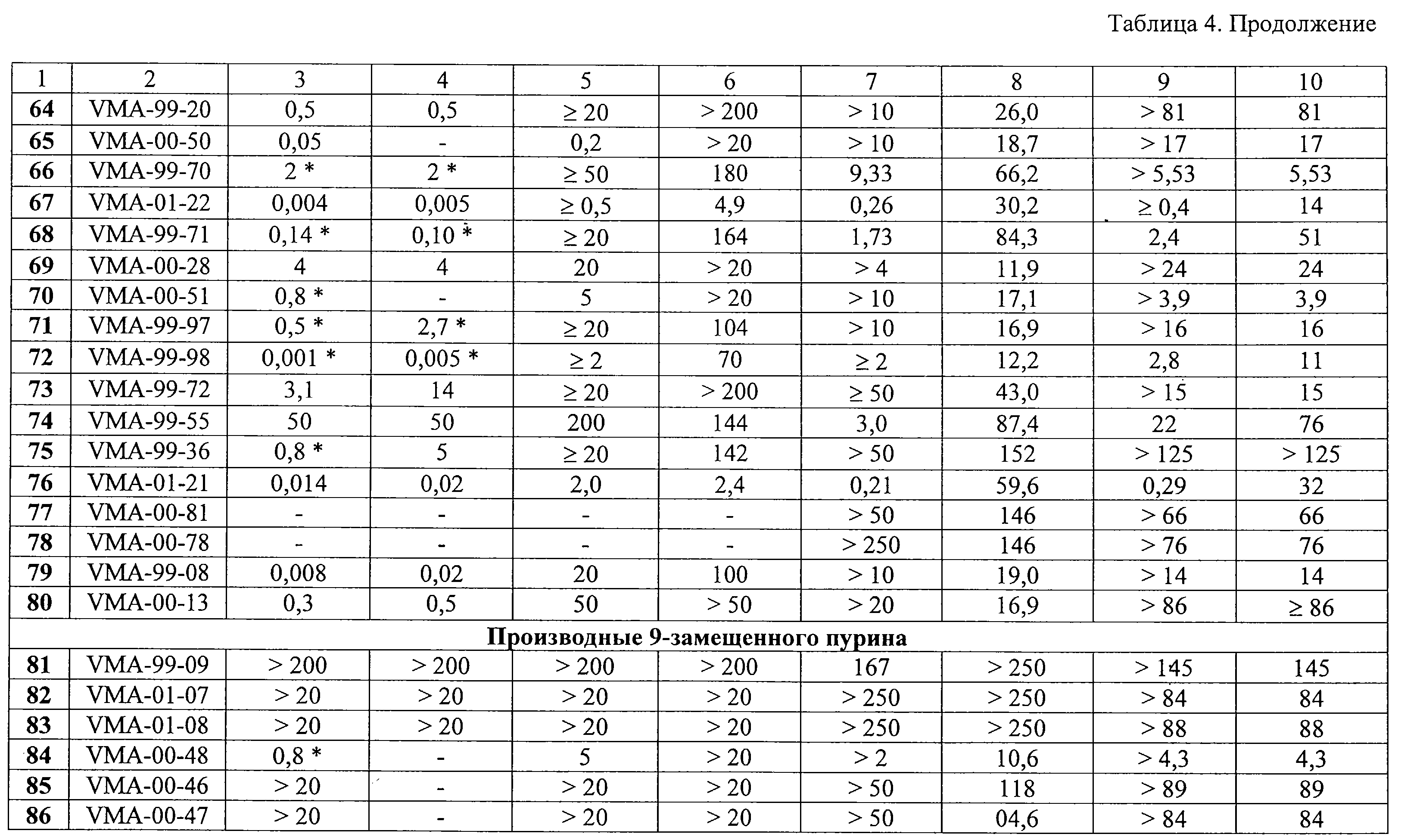
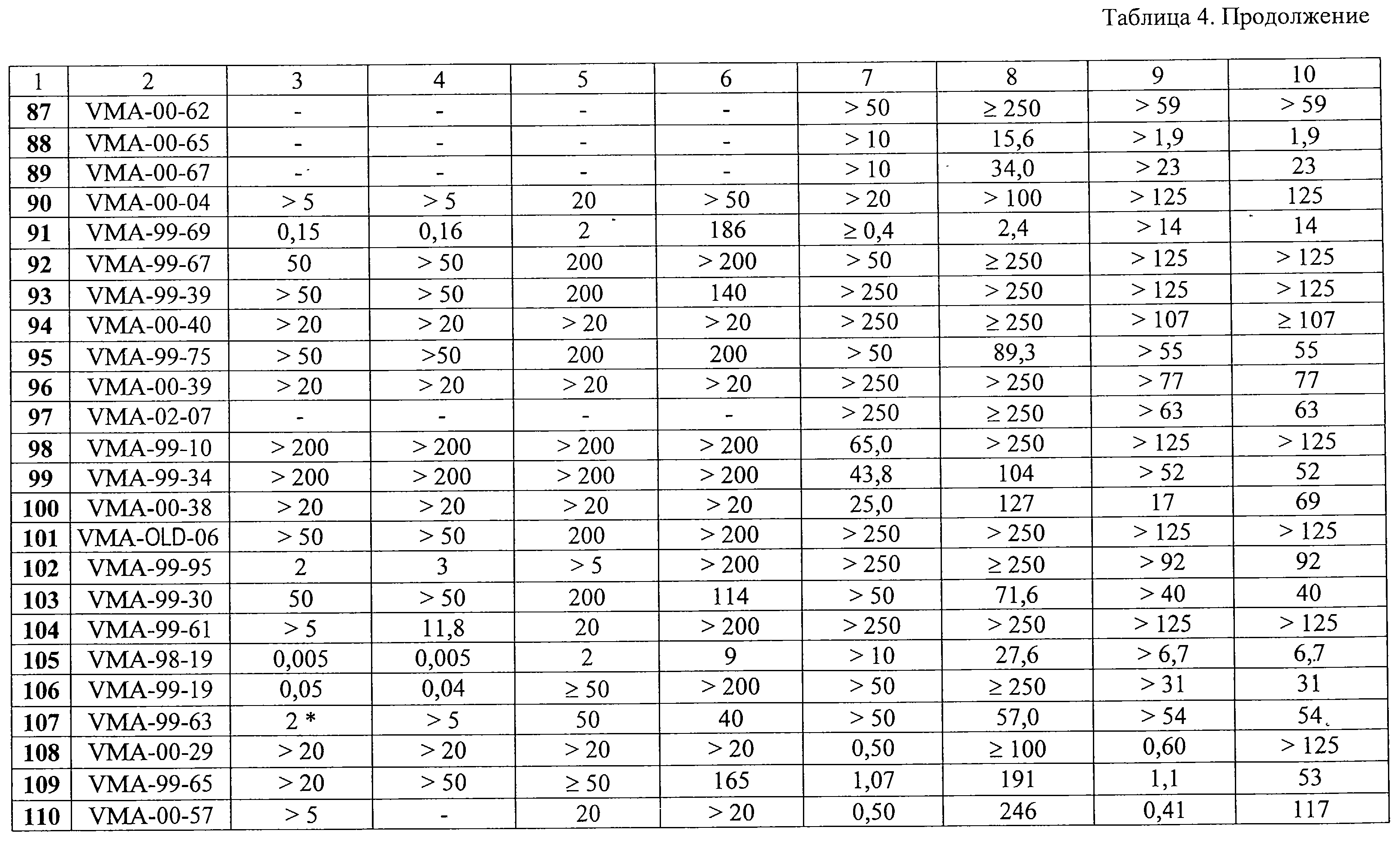
Противовирусная активность in vitro синтезированных соединений 1-130 в отношении ЦМВ, ВИЧ-1 и ряда других ДНК- и РНК-содержащих вирусов была исследована в Rega Institute for Medical Research (Католический университет, Лювен, Бельгия). Результаты скрининга веществ в отношении ЦМВ и ВИЧ-1 представлены в таблице 4.
Анти-ЦМВ активность.
Производные 9-(2-феноксиэтил)аденина оказались исключительно активными ингибиторами репликации ЦМВ в культуре клеток HEL. В частности, соединения 11, 13, 27, 30, 36, 37, 43 и 54 ингибировали ЦМВ в концентрациях, равных или значительно ниже 0,01 М, хотя в случае некоторых веществ, например 11, 37, 43, не было достигнуто полного подавления вирусной репликации. Для ряда производных 9-(2-феноксиэтил)аденина была достигнута селективность противовирусного действия (отношение СС50/IС50), равная или значительно превышающая 1000 (таблица 5).
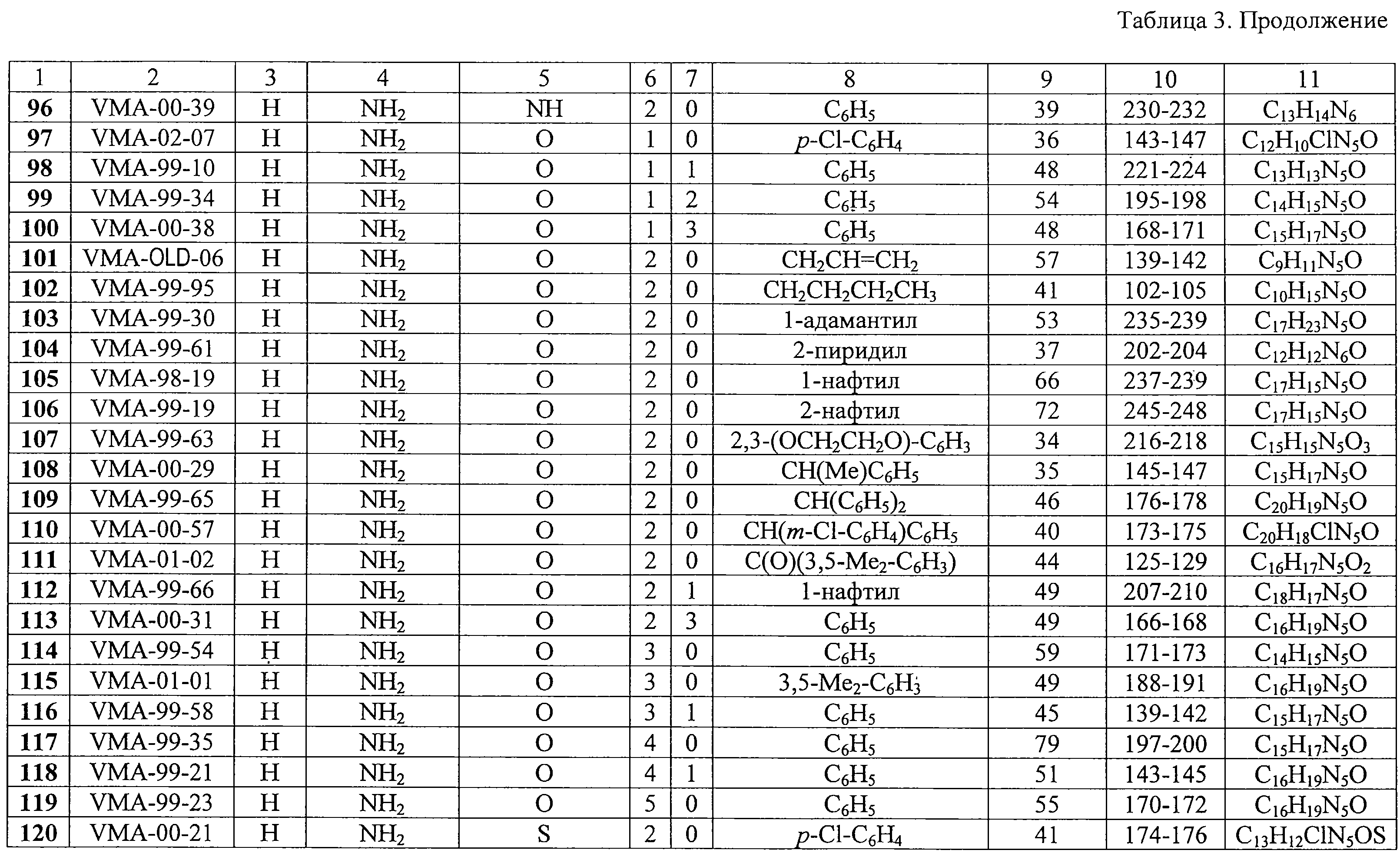
Среди производных 9-(2-бензилоксиэтил)аденина некоторые соединения также были высокоактивны против ЦМВ. В частности, соединения 67 и 72 проявляли ингибиторные свойства в концентрациях ниже наномолярного уровня.
Некоторые 9-замещенные производные пурина были высокоактивными в отношении репликации ЦМВ, в том числе соединения 91 (IС50 0,15 M), 105 (IC50 0,005 M), 106 (IC50 0,05 M), 121 (IC50 0,01 M) и 126 (IC50 0,05 М). В частности, соединения 105 и 106 показали резко выраженную селективность (свыше 1000).
Анти-ВИЧ-1 активность.
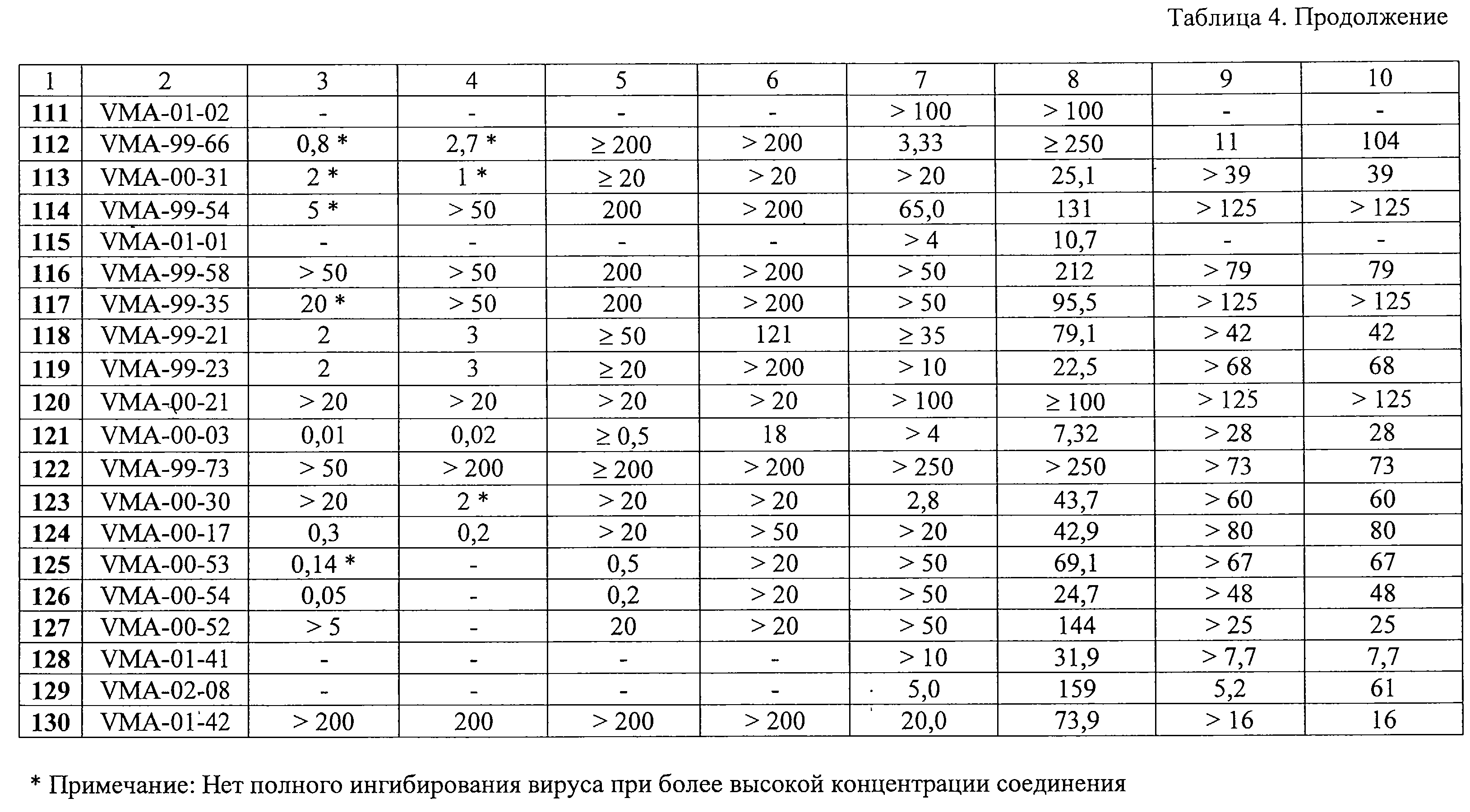
За исключением соединения 2, которое имело индекс селективности около 10 в отношении ВИЧ-1 в культуре СЕМ-клеток, ни одно из производных 9-(2-феноксиэтил)аденина, имеющих заместитель в фенильном ядре и оказывающих высокую активность в отношении ЦМВ, не ингибировало репликацию ВИЧ-1 в культурах клеток СЕМ и МТ-4. Однако некоторые соединения (например, 14, 29, 30, 39, 40, 54 и 55) обладали высокой цитотоксичностью в использованных клеточных культурах (50%-ная цитотоксическая концентрация СС50 находилась в диапазоне 1-10 М).
В противоположность этому, некоторые производные 9-(2-бензилоксиэтил)аденина показали выраженную и селективную анти-ВИЧ-1 активность. В частности, соединения 61, 67 и 76, проявили себя как наиболее мощные и селективные ингибиторы репликации ВИЧ-1 в культурах клеток СЕМ и МТ-4. Величина их ингибиторной концентрации ЕС50 составляла 0,1-0,3 г/мл, тогда как клеточная токсичность СС50 была в пределах 13-75 г/мл. В целом, цитостатическая активность была более выражена в культуре лимфоцитов МТ-4, чем в СЕМ клетках. Во всех трех случаях заместители (Me, трет-Bu или Вr) были в положениях 3 и 5 фенильного кольца. Перемещение указанных заместителей из положения 3 в положение 2 приводило к заметному понижению антивирусной активности соединений.
Соединения 108, 109 и 110, содержащие соответственно 2-(1-фенилэтокси)этильный, 2-(дифенилметокси)этильный и 2-(фенил-м-Сl-фенилметокси)этильный заместители, также проявили заметную активность в отношении ВИЧ-1 в культурах клеток СЕМ и МТ-4 (ЕС50 0,4-1,1 г/мл, СС50 53-191 г/мл). Таким образом, некоторые из синтезированных соединений продемонстрировали выраженную способность ингибировать цитопатический эффект ВИЧ-1 в клеточных культурах в наномолярных концентрациях и имели индекс селективности (СС50/ЕС50) в пределах от 200 до 750, в зависимости от природы клеточной линии, включенной в исследование.
Из изучения закономерности структура – противовирусная активность следует, что у полученных пуриновых производных однозначной корреляции между активностью против ВИЧ-1 и против ЦМВ нет. Некоторые соединения, которые показали мощную активность в отношении ВИЧ-1, были неактивными против ВИЧ-2 (данные не приводятся). У отдельных высокоактивных ингибиторов ЦМВ отсутствует какая-либо анти-ВИЧ активность (например, у соединений 30, 36, 37, 105). Другие соединения этого же химического ряда были заметно активны против обоих вирусов (например, соединения 61, 67, 68, 72, 76). Это в целом отличает синтезированный новый класс пуриновых производных от известных ненуклеозидных ингибиторов обратной транскриптазы ВИЧ. Вероятно, некоторые из полученных производных 9-замещенного аденина имеют новый механизм противовирусного действия.
Таким образом, открыт новый класс противовирусных агентов ненуклеозидной природы, которые проявляют выраженную активность в отношении ВИЧ-1 и ЦМВ in vitro и по эффективности анти-ЦМВ действия в десятки и сотни раз превосходят использующиеся в клинике современные лекарственные средства. Наличие у одних и тех же соединений одновременно высокой анти-ВИЧ-1 и анти-ЦМВ активности позволяет считать их перспективными для комплексного лечения СПИД и оппортунистических цитомегаловирусных инфекций. 25 мл), органическую фазу и экстракты объединяют, сушат Na2SO4, фильтруют и упаривают в вакууме водоструйного насоса на кипящей водяной бане. Остаток перегоняют в вакууме, собирая фракцию, кипящую при 157-162С (3 мм рт.ст.). Получают 39,0 г бесцветной вязкой жидкости, n20D 1,5246, выход 74%.
1-(4-Изопропилфенокси)-2-(n-толуолсульфонилокси)этан.
К раствору 30,0 г (0,166 моль) 2-(4-изопропилфенокси)этанола в 150 мл безводного дихлорметана и 40 мл пиридина при интенсивном перемешивании и охлаждении до 0С добавляют порциями в течение 20 мин 38,0 г (0,199 моль) n-толуолсульфохлорида с такой скоростью, чтобы температура реакционной смеси не поднималась выше 5С. После этого реакционную массу перемешивают при той же температуре еще 2 ч и оставляют на ночь при комнатной температуре. Реакционную массу последовательно промывают 2%-ным водным раствором соляной кислоты (2100 мл), водой (280 мл), насыщенным раствором соды (2100 мл) и вновь водой до нейтральной реакции, сушат Na2SO4, фильтруют и упаривают в вакууме водоструйного насоса. Остаток дополнительно вакуумируют при 40-50С (5 мм рт.ст.) в течение 1 ч. Получают 47,4 г вязкого сиропообразного продукта светло-желтого цвета, выход 85%.
1-Бром-2-(4-изопропилфенокси)этан. Метод А.
К раствору 14,0 г (0,078 моль) 2-(4-изопропилфенокси)этанола в 100 мл безводного хлороформа и 10 мл пиридина при интенсивном перемешивании и охлаждении до -10С прибавляют по каплям в течение 10 мин 2,7 мл (0,029 моль) трехбромистого фосфора. После этого реакционную массу перемешивают при 0С еще 2 ч, затем 2 ч при комнатной температуре и оставляют на ночь. Реакционную массу выливают в 250 мл холодной воды, органический слой промывают насыщенным раствором соды (2100 мл) и водой до нейтральной реакции, сушат Na2SO4, фильтруют и упаривают в вакууме водоструйного насоса. Остаток перегоняют в вакууме, собирая фракцию, кипящую при 158-162С (3 мм рт.ст.). Получают 10,2 г бесцветной прозрачной жидкости, n20D 1,5112, выход 54%.
1-Бром-2-(4-изопропилфенокси)этан. Метод Б.
Смесь 15,0 г (0,110 моль) 4-изопропилфенола, 20,0 мл (0,232 моль) 1,2-дибромэтана и раствора 6,2 г (0,110 моль) КОН в 10 мл воды кипятят в течение 8 ч до нейтральной реакции. Реакционную массу разбавляют 100 мл хлороформа, органический слой промывают 3%-ным раствором NaOH (100 мл) и водой (2100 мл) до нейтральной реакции, сушат Na2SO4, фильтруют, растворитель и избыток 1,2-дибромэтана отгоняют в вакууме водоструйного насоса на кипящей водяной бане. Остаток перегоняют в вакууме, собирая фракцию, кипящую при 158-163С (3 мм рт.ст.). Получают 13,5 г бесцветной прозрачной жидкости, n20D 1,5118, выход 51%.
9-[2-(4-Изопропилфенокси)этил]аденин (11, VMA-99-82). Метод А.
Суспензию 15,0 г (0,111 моль) аденина и 17,3 г (0,125 моль) свежепрокаленного карбоната калия в 100 мл ДМФА перемешивают при температуре 80-85С в течение 1,5 ч, добавляют раствор 37,0 г (0,111 моль) 1-(4-изопропилфенокси)-2-(n-толуолсульфонилокси)этана в 50 мл ДМФА и полученную смесь перемешивают при той же температуре еще 8 ч. Реакционную массу оставляют на ночь при комнатной температуре, фильтруют, фильтрат упаривают в вакууме, остаток обрабатывают 200 мл холодной воды и охлаждают при 0-5С в течение суток. Закристаллизовавшийся остаток отфильтровывают и после сушки на воздухе перекристаллизовывают из смеси 100 мл ДМФА и 10 мл воды. Получают 22,1 г VMA-99-82 в виде белого мелкокристаллического продукта с выходом 67%. Rf (метиленхлорид – метанол, 10 : 1) 0,64. Т. пл. 231-233С.
Масс-спектр: M+ 297 (6%), 282 (2%), 254 (2%), 178 (9%), 163 (81%), 149 (84%), 134 (100%), 119 (30%), 107 (50%), 105 (47%), 92 (99%), 91 (80%).
9-[2-(4-Изопропилфенокси)этил]аденин (11, VMA-99-82). Метод Б.
Суспензию 5,0 г (0,037 моль) аденина и 6,3 г (0,045 моль) свежепрокаленного карбоната калия в 60 мл ДМФА перемешивают при температуре 105-110С в течение 1,5 ч, добавляют раствор 9,0 г (0,037 моль) 1-бром-2-(4-изопропилфенокси)этана в 20 мл ДМФА и полученную смесь перемешивают при той же температуре еще 2 ч. Выделение и очистка аналогична описанному выше. Получают 6,1 г VMA-99-82 в виде белого мелкокристаллического продукта, выход 55%.
Пример 2. 9-[2-(3,5-Диметилбензилокси)этил]аденин (61, VMA-99-56).
2-(3,5-Диметилбензилокси)этанол.
К раствору 210 мл (1,509 моль) мезитилена и 100 мл ССl4 при кипячении и облучении светом добавляют в течение 30 мин раствор 14,0 мл (0,272 моль) брома в 25 мл ССl4. Реакционную массу упаривают в вакууме водоструйного насоса на водяной бане, к остатку прибавляют раствор 16,0 г (0,285 моль) КОН в 100 мл (1,793 моль) этиленгликоля и полученную смесь перемешивают на водяной бане при температуре 80-90С в течение 10 ч. Реакционную массу разбавляют 250 мл холодной воды, органическую фазу отделяют, водную фазу экстрагируют мезитиленом (330 мл), органическую фазу и экстракты объединяют, промывают солевым раствором (100 мл), сушат Na2SO4, фильтруют и упаривают в вакууме водоструйного насоса. Остаток перегоняют в вакууме, собирая фракцию, кипящую при 168-172С (3 мм рт.ст.). Получают 27,3 г бесцветной жидкости, n20D 1,5227, выход 56%.
1-(3,5-Диметилбензилокси)-2-(n-толуолсульфонилокси)этан.
К раствору 51,0 г (0,283 моль) 2-(3,5-диметилбензилокси)этанола в 250 мл безводного хлороформа и 80 мл пиридина при интенсивном перемешивании и охлаждении до 0С добавляют порциями в течение 30 мин 64,7 г (0,339 моль) n-толуолсульфохлорида с такой скоростью, чтобы температура реакционной смеси не поднималась выше 5С. После этого реакционную массу перемешивают при той же температуре еще 2 ч и оставляют на ночь при комнатной температуре. Затем реакционную массу последовательно промывают 2%-ным водным раствором соляной кислоты (2150 мл), водой (2100 мл), насыщенным раствором соды (2100 мл) и вновь водой до нейтральной реакции, сушат Na2SO4, фильтруют и упаривают в вакууме водоструйного насоса. Остаток растворителя удаляют в вакууме при 40-50С (5 мм рт.ст.) в течение 1 ч. Получают 77,6 г вязкого сиропообразного продукта светло-желтого цвета, выход 82%.
1-Бром-2-(3,5-диметилбензилокси)этан.
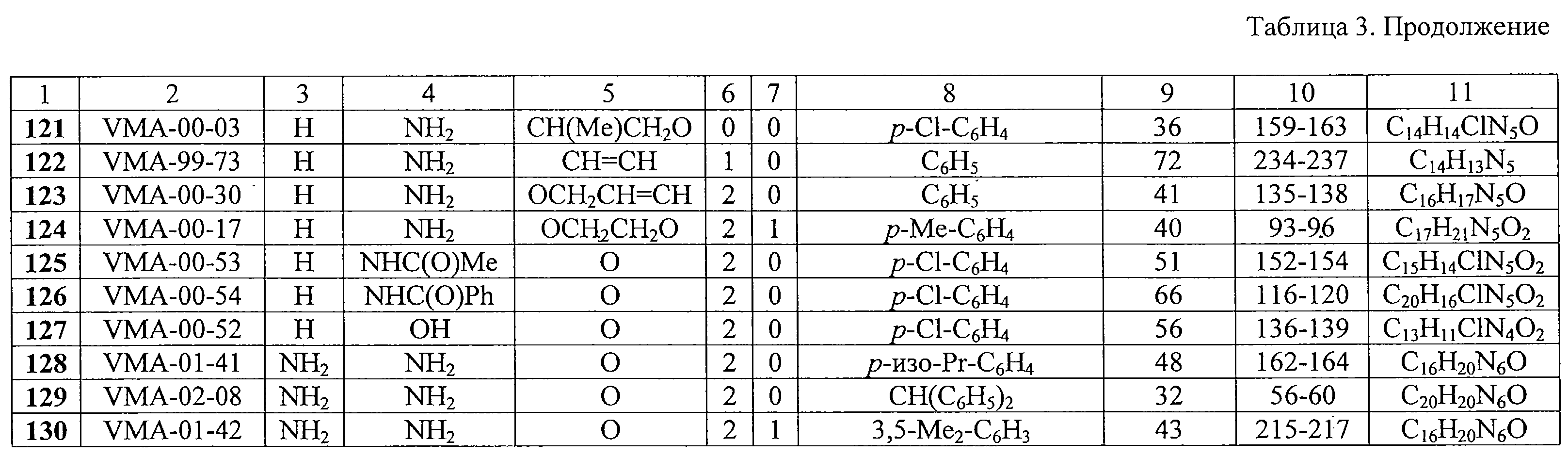
К раствору 18,75 г (0,104 моль) 2-(3,5-диметилбензилокси)этанола в 100 мл безводного хлороформа и 10 мл пиридина при интенсивном перемешивании и охлаждении до 10С добавляют в течение 10 мин 3,6 мл (0,0379 моль) трехбромистого фосфора. После этого реакционную массу перемешивают при 0С еще 1 ч, 2 ч при комнатной температуре и оставляют на ночь. Затем реакционную массу выливают в 300 мл холодной воды, органический слой промывают насыщенным раствором соды (2100 мл) и водой до нейтральной реакции, сушат Na2SO4, фильтруют и упаривают в вакууме водоструйного насоса. Остаток перегоняют в вакууме, собирая фракцию, кипящую при 167-173С (3 мм рт.ст.). Получают 12,9 г бесцветной прозрачной жидкости, n20D 1,5391, выход 51%.
9-[2-(3,5-Диметилбензилокси)этил]аденин (61, VMA-99-56). Метод А.
Суспензию 23,0 г (0,170 моль) аденина и 27,0 г (0,195 моль) свежепрокаленного карбоната калия в 250 мл ДМФА перемешивают при температуре 80-85С в течение 1,5 ч, добавляют раствор 60,0 г (0,176 моль) 1-(3,5-диметилбензилокси)-2-(n-толуолсульфонилокси)этана в 50 мл ДМФА и полученную смесь перемешивают при той же температуре еще 8 ч. Реакционную массу оставляют на ночь при комнатной температуре, фильтруют, твердый осадок обрабатывают 0,5 л холодной воды, фильтруют и после сушки на воздухе получают 17,8 г белого кристаллического продукта-сырца. Фильтрат первичной реакционной массы упаривают в вакууме, к остатку добавляют 0,5 л холодной воды, перемешивают и охлаждают при 0-5С в течение суток. Закристаллизовавшийся остаток отфильтровывают и после сушки на воздухе дополнительно получают 22,2 г целевого продукта-сырца. Обе партии продукта объединяют и перекристаллизовывают из смеси 100 мл ДМФА и 15 мл воды. Получают 29,6 г белого мелкокристаллического продукта с выходом 59%. Rf (метиленхлорид – метанол, 10:1) 0,60. Т.пл. 180-183С.
Масс-спектр: M+ 297 (3%), 178 (14%), 163 (31%), 149 (22%), 134 (100%), 119 (67%), 105 (47%), 92 (64%), 91 (46%).
9-[2-(3,5-Диметилбензилокси)этил]аденин (61, VMA-99-56). Метод Б.
Суспензию 5,0 г (0,037 моль) аденина и 6,3 г (0,045 моль) свежепрокаленного карбоната калия в 60 мл ДМФА перемешивают при температуре 105-110С в течение 1,5 ч, добавляют раствор 9,0 г (0,037 моль) 1-бром-2-(3,5-диметилбензилокси)этана в 20 мл ДМФА и полученную смесь перемешивают при той же температуре еще 2 ч. Выделение и очистка аналогична описанному выше. Получают 5,6 г VMA-99-56 в виде белого мелкокристаллического продукта, выход 51%.
Противовирусная активность in vitro синтезированных соединений 1-130 в отношении ЦМВ, ВИЧ-1 и ряда других ДНК- и РНК-содержащих вирусов была исследована в Rega Institute for Medical Research (Католический университет, Лювен, Бельгия). Результаты скрининга веществ в отношении ЦМВ и ВИЧ-1 представлены в таблице 4.
Анти-ЦМВ активность.
Производные 9-(2-феноксиэтил)аденина оказались исключительно активными ингибиторами репликации ЦМВ в культуре клеток HEL. В частности, соединения 11, 13, 27, 30, 36, 37, 43 и 54 ингибировали ЦМВ в концентрациях, равных или значительно ниже 0,01 М, хотя в случае некоторых веществ, например 11, 37, 43, не было достигнуто полного подавления вирусной репликации. Для ряда производных 9-(2-феноксиэтил)аденина была достигнута селективность противовирусного действия (отношение СС50/IС50), равная или значительно превышающая 1000 (таблица 5).
Среди производных 9-(2-бензилоксиэтил)аденина некоторые соединения также были высокоактивны против ЦМВ. В частности, соединения 67 и 72 проявляли ингибиторные свойства в концентрациях ниже наномолярного уровня.
Некоторые 9-замещенные производные пурина были высокоактивными в отношении репликации ЦМВ, в том числе соединения 91 (IС50 0,15 M), 105 (IC50 0,005 M), 106 (IC50 0,05 M), 121 (IC50 0,01 M) и 126 (IC50 0,05 М). В частности, соединения 105 и 106 показали резко выраженную селективность (свыше 1000).
Анти-ВИЧ-1 активность.
За исключением соединения 2, которое имело индекс селективности около 10 в отношении ВИЧ-1 в культуре СЕМ-клеток, ни одно из производных 9-(2-феноксиэтил)аденина, имеющих заместитель в фенильном ядре и оказывающих высокую активность в отношении ЦМВ, не ингибировало репликацию ВИЧ-1 в культурах клеток СЕМ и МТ-4. Однако некоторые соединения (например, 14, 29, 30, 39, 40, 54 и 55) обладали высокой цитотоксичностью в использованных клеточных культурах (50%-ная цитотоксическая концентрация СС50 находилась в диапазоне 1-10 М).
В противоположность этому, некоторые производные 9-(2-бензилоксиэтил)аденина показали выраженную и селективную анти-ВИЧ-1 активность. В частности, соединения 61, 67 и 76, проявили себя как наиболее мощные и селективные ингибиторы репликации ВИЧ-1 в культурах клеток СЕМ и МТ-4. Величина их ингибиторной концентрации ЕС50 составляла 0,1-0,3 г/мл, тогда как клеточная токсичность СС50 была в пределах 13-75 г/мл. В целом, цитостатическая активность была более выражена в культуре лимфоцитов МТ-4, чем в СЕМ клетках. Во всех трех случаях заместители (Me, трет-Bu или Вr) были в положениях 3 и 5 фенильного кольца. Перемещение указанных заместителей из положения 3 в положение 2 приводило к заметному понижению антивирусной активности соединений.
Соединения 108, 109 и 110, содержащие соответственно 2-(1-фенилэтокси)этильный, 2-(дифенилметокси)этильный и 2-(фенил-м-Сl-фенилметокси)этильный заместители, также проявили заметную активность в отношении ВИЧ-1 в культурах клеток СЕМ и МТ-4 (ЕС50 0,4-1,1 г/мл, СС50 53-191 г/мл). Таким образом, некоторые из синтезированных соединений продемонстрировали выраженную способность ингибировать цитопатический эффект ВИЧ-1 в клеточных культурах в наномолярных концентрациях и имели индекс селективности (СС50/ЕС50) в пределах от 200 до 750, в зависимости от природы клеточной линии, включенной в исследование.
Из изучения закономерности структура – противовирусная активность следует, что у полученных пуриновых производных однозначной корреляции между активностью против ВИЧ-1 и против ЦМВ нет. Некоторые соединения, которые показали мощную активность в отношении ВИЧ-1, были неактивными против ВИЧ-2 (данные не приводятся). У отдельных высокоактивных ингибиторов ЦМВ отсутствует какая-либо анти-ВИЧ активность (например, у соединений 30, 36, 37, 105). Другие соединения этого же химического ряда были заметно активны против обоих вирусов (например, соединения 61, 67, 68, 72, 76). Это в целом отличает синтезированный новый класс пуриновых производных от известных ненуклеозидных ингибиторов обратной транскриптазы ВИЧ. Вероятно, некоторые из полученных производных 9-замещенного аденина имеют новый механизм противовирусного действия.
Таким образом, открыт новый класс противовирусных агентов ненуклеозидной природы, которые проявляют выраженную активность в отношении ВИЧ-1 и ЦМВ in vitro и по эффективности анти-ЦМВ действия в десятки и сотни раз превосходят использующиеся в клинике современные лекарственные средства. Наличие у одних и тех же соединений одновременно высокой анти-ВИЧ-1 и анти-ЦМВ активности позволяет считать их перспективными для комплексного лечения СПИД и оппортунистических цитомегаловирусных инфекций.
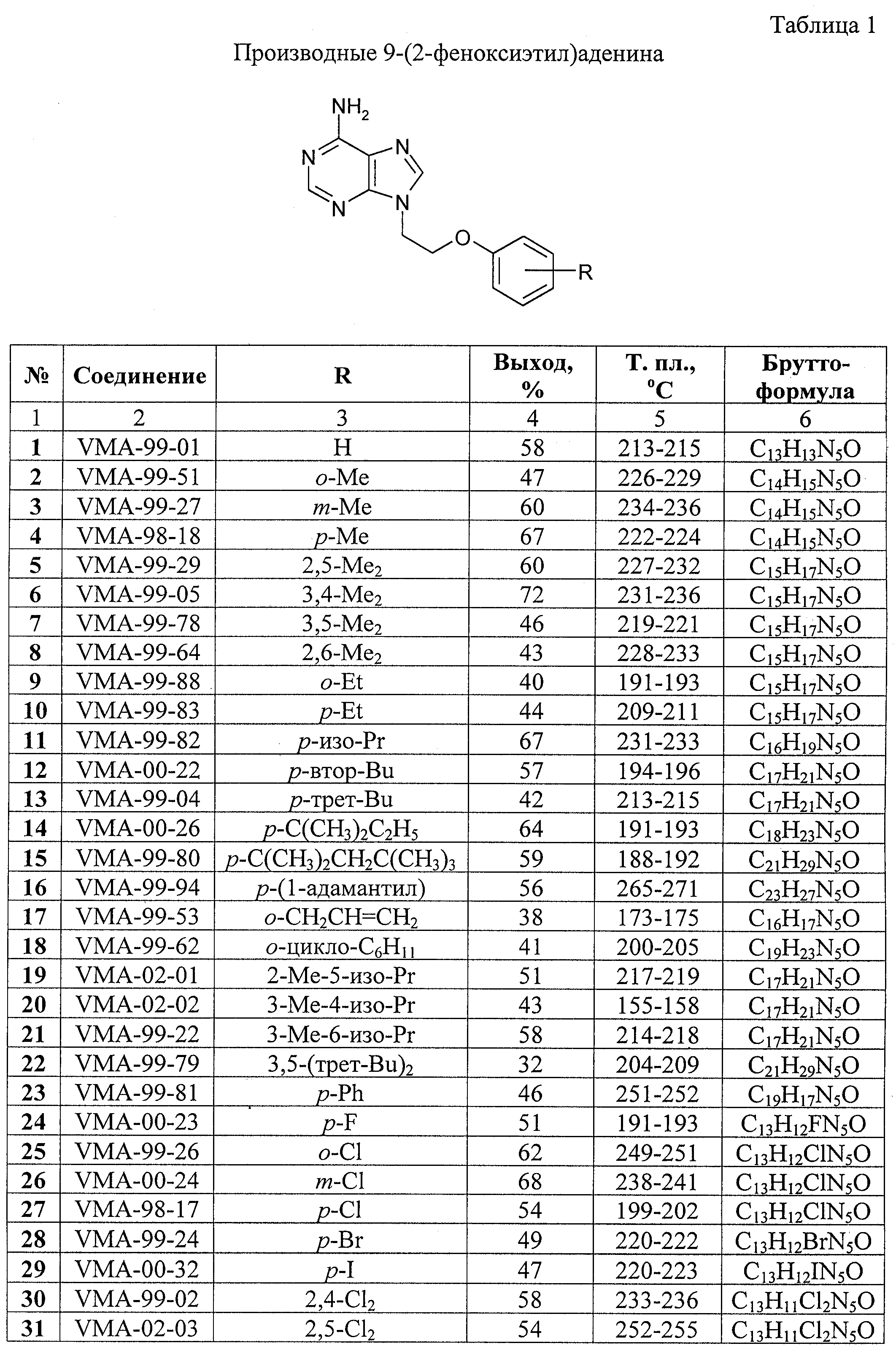       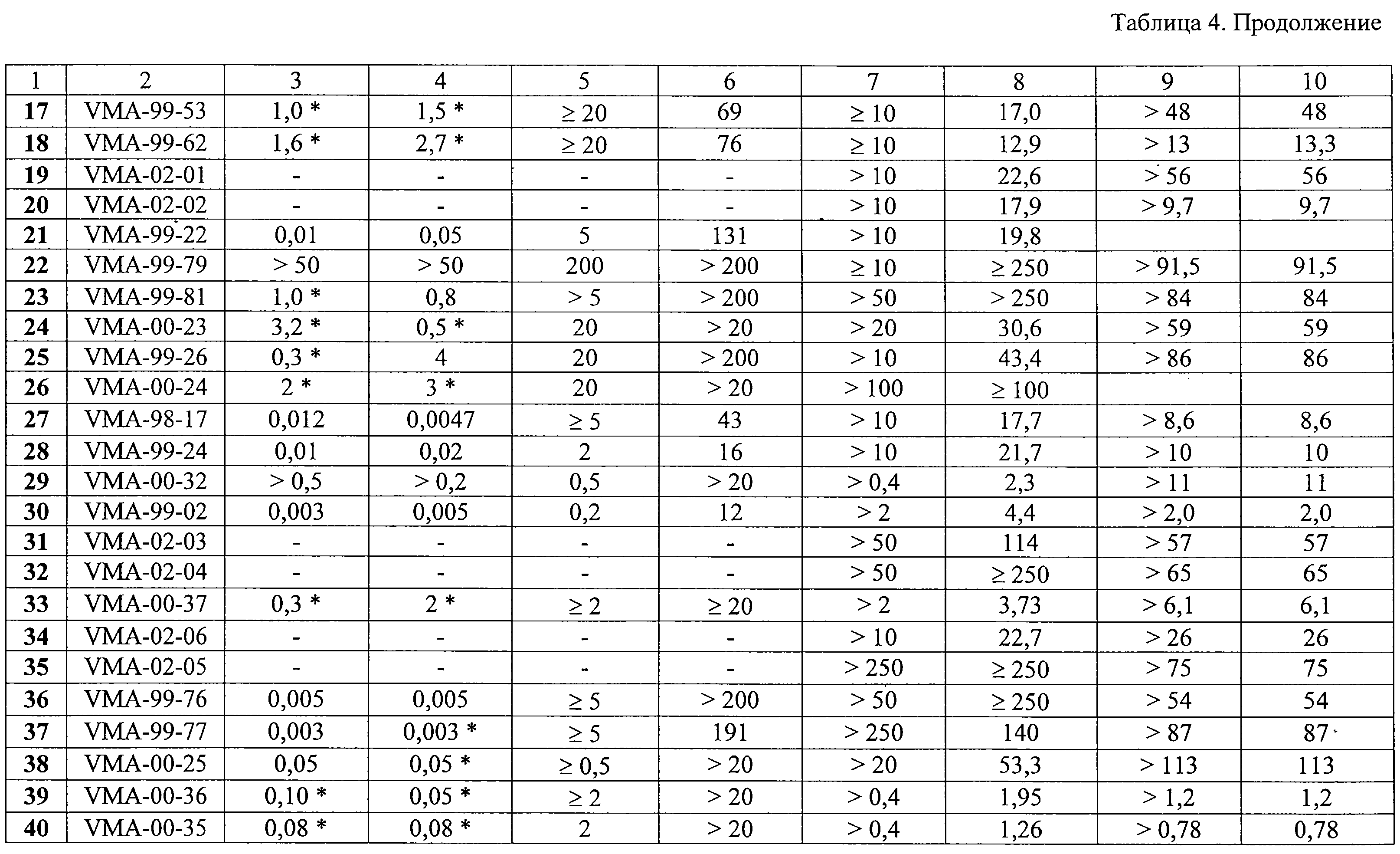 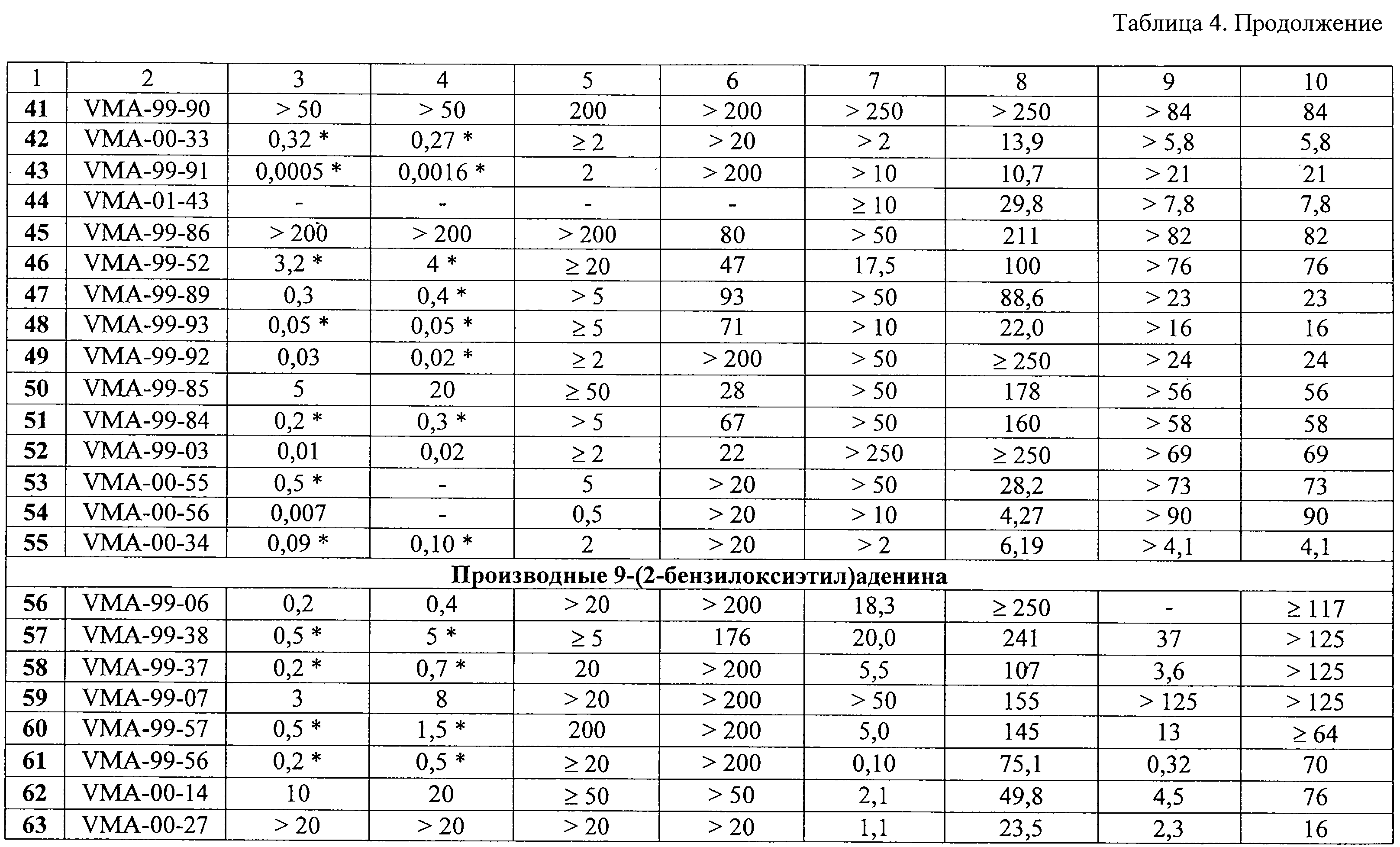    
Формула изобретения Производные пурина общей формулы 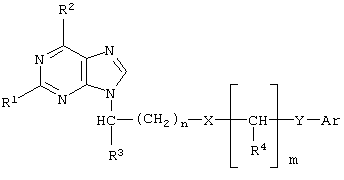 где n = 0 – 4;
m = 0 – 3;
R1 = Н, ОН или NH2;
R2 = ОН, NH2, ацетиламино или бензоиламино;
R3 = Н или низший алкил C1–С4;
R4 = Н, низший алкил C1-С4 или фенил;
Х = СН2, О, S, NH или С(O)O;
Y = СН2, СН=СН, С(O), О или ординарная связь;
Ar = фенил, пиридил, нафтил или замещенный фенил формулы
где n = 0 – 4;
m = 0 – 3;
R1 = Н, ОН или NH2;
R2 = ОН, NH2, ацетиламино или бензоиламино;
R3 = Н или низший алкил C1–С4;
R4 = Н, низший алкил C1-С4 или фенил;
Х = СН2, О, S, NH или С(O)O;
Y = СН2, СН=СН, С(O), О или ординарная связь;
Ar = фенил, пиридил, нафтил или замещенный фенил формулы
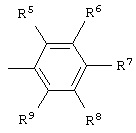 где независимо R5–R9 = алкил С1–C8, циклоалкил C5–С6, 1-адамантил, аллил, фенил, бензил, F, CI, Br, J, трифторметил, алкокси C1–С5, фенокси, бензилокси, бензоилокси, циано, карбокси, ацетил или нитро, обладающие противовирусной активностью.
где независимо R5–R9 = алкил С1–C8, циклоалкил C5–С6, 1-адамантил, аллил, фенил, бензил, F, CI, Br, J, трифторметил, алкокси C1–С5, фенокси, бензилокси, бензоилокси, циано, карбокси, ацетил или нитро, обладающие противовирусной активностью.
|
||||||||||||||||||||||||||