Патент на изобретение №2228332
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ 4-МЕТИЛ-5-ФОРМИЛТИАЗОЛА
(57) Реферат: Изобретение относится к способу получения 4-метил-5-формил-тиазола путем окисления 4-метил-5-(2-гидроксиэтил)-тиазола при помощи водного раствора окиси хрома или неорганических бихроматов в присутствии серной кислоты и окисление проводят в двухфазной системе вода-органический растворитель, при температуре 20-50°С, при этом в качестве органического растворителя используют диэтиловый эфир, бензол, хлороформ и хлористый метилен. Технический результат – новый способ получения 4-метил-5-формил-тиазола. 2 з.п. ф-лы, 1 табл. Область техники, к которой относится изобретение. Изобретение относится к области синтеза органических соединений, в частности, предмет настоящего исследования – разработка нового способа получения 4-метил-5-формилтиазола. Предпосылки создания изобретения. 4-Метил-5-формилтиазол является одним из важнейших синтонов, использующихся в синтезе современных лекарственных препаратов цефалоспоринов [К.Atsumi, К.Sakagami, Y.Yamamoto, T.Yoshida, К.Nichihata, S.Kondo, S.Fukatsu. Preparation of (methylthiazolyl) vinylcephem carboxylic acid derivatives as antibiotics. (Meyi Seika Kaisha, Ltd.) Eur. Pat. Appl. EP 236231 (C1 C 07 D 501/24) 09 Sep. 1987. С.А.: V 108: 94289 z] и нейропротекторов [B.R.Boar, A.J.Cross, D.A.Gray, A.R.Green. Novel 1-(heteroazolyl) alkanes and their use as neuroprotective agents. (Astra AB) PCT Int. Appl. WO 01,979 (C1 C 07 D 413/06) 19 Jan. 1995. С.А.: V. 123: 169608 y]. Однако до настоящего времени синтез его представляет значительные трудности. В связи с тем, что способ получения 4-метил-5-формилтиазола не освоен промышленностью, в качестве базового объекта принимается известный из литературы способ получения этого продукта, который рассматривается нами как прототип. Существующие методы получения 4-метил-5-формилтиазола основаны на использовании двух исходных соединений: 1) 4-метил-5-метоксикарбонил-тиазола (МТ-СООМе) и 2) 4-метил-5-(2-гидроксиэтил)тиазола (МТ-СН2СН2OН) 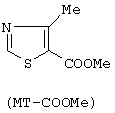 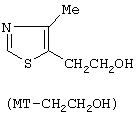
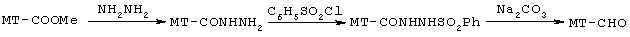 Суммарный выход продукта в этих условиях составляет лишь 23%. Прямое восстановление эфира МТ-СООМе до альдегида МТ-СНО молекулярным водородом в газовой фазе в присутствии специального катализатора запатентовано в 1989 г. [Т.Yokoyama, Т.Setoyama, N.Setoyama, N.Matsuyama, T.Maki. Process for producing heterocyclic aldehyde’s. (Mitsubishi Kasei Corp). Eur. Pat. Appl. EP 343640 (C1 C 07 D 213/48). 29 Nov. 1989. С.А.: V. 112: 198133 t]: Суммарный выход продукта в этих условиях составляет лишь 23%. Прямое восстановление эфира МТ-СООМе до альдегида МТ-СНО молекулярным водородом в газовой фазе в присутствии специального катализатора запатентовано в 1989 г. [Т.Yokoyama, Т.Setoyama, N.Setoyama, N.Matsuyama, T.Maki. Process for producing heterocyclic aldehyde’s. (Mitsubishi Kasei Corp). Eur. Pat. Appl. EP 343640 (C1 C 07 D 213/48). 29 Nov. 1989. С.А.: V. 112: 198133 t]:
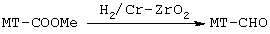 Процесс газофазного гидрирования протекает в жестких температурных условиях (315
Процесс газофазного гидрирования протекает в жестких температурных условиях (315 С). При этом конверсия субстрата составляет 74%; кроме того, селективность восстановления не превышает 80%.
Наиболее близким по своим характеристикам к предлагаемому изобретению является способ, основанный на использовании в качестве исходного соединения 4-метил-5-(2-гидроксиэтил)тиазола (МТ-СН2СН2OН), широко применяемого в органическом синтезе, в частности в синтезе витамина B1.
Попытка получения 4-метил-5-формил-тиазола из 4-метил-5-(2-гидроксиэтил)тиазола (МТ-СН2СН2 С). При этом конверсия субстрата составляет 74%; кроме того, селективность восстановления не превышает 80%.
Наиболее близким по своим характеристикам к предлагаемому изобретению является способ, основанный на использовании в качестве исходного соединения 4-метил-5-(2-гидроксиэтил)тиазола (МТ-СН2СН2OН), широко применяемого в органическом синтезе, в частности в синтезе витамина B1.
Попытка получения 4-метил-5-формил-тиазола из 4-метил-5-(2-гидроксиэтил)тиазола (МТ-СН2СН2
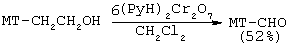 1) применение дорогостоящего окислителя, синтез которого требует дополнительных затрат и увеличивает число стадий процесса,
2) применение избытка окислителя (6 моль окислителя на 1 моль субстрата), что приводит к образованию большого количества твердых отходов и загрязнению окружающей среды,
3) трудности при выделении продукта, в частности экстракция целевого соединения из смолообразной смеси, образующейся по окончании реакции.
Сущность изобретения.
Заявитель предлагаемого изобретения обнаружил, что указанные недостатки могут быть устранены при получении МТ-СНО окислением МТ-СН2СН2ОН в двухфазной системе с использованием дешевых и доступных окислителей, каковыми являются хромовая кислота и ее неорганические соли в водном растворе кислоты.
Интенсификация процесса окисления МТ-СН2СН2ОН, оптимизация теплового режима, удобство и простота выделения целевого продукта, а также увеличение его выхода достигаются за счет применения водорастворимых неорганических окислителей, оптимизации соотношения МТ-СН2СН2ОН:окислитель и благодаря использованию двухфазной системы.
Процесс окисления МТ-СН2СН2ОН окисью хрома или неорганическими бихроматами описывается уравнениями, которые представлены на схемах (1) и (2) соответственно. Окисление МТ-СН2СН2ОН сопровождается расщеплением С-С-связи в боковой цепи молекулы исходного спирта (схема 1, уравнения 1, 3, 5, 7 и схема 2, уравнения 1, 3, 5, 7). При этом оксиметильный фрагмент, содержащий концевой атом углерода, в условиях реакции может быть превращен в соль муравьиной кислоты, а в более жестких условиях может быть окислен в углекислоту.
Главная часть молекулы преобразуется в альдегид МТ-СНО, который является устойчивым в предложенных условиях реакции благодаря аллильному резонансу С=O-группы и С=С-связи гетероциклического ядра. Наличие избытка окислителя и/или H2SO4, а также температура выше 50
Заявитель предлагаемого изобретения обнаружил, что распределение исходного спирта 4-метил-5-(2-гидроксиэтил)тиазола и продукта реакции – 4-метил-5-формил-тиазола в двухфазной системе благоприятно для проведения окисления водорастворимыми окислителями. Данные по распределению МТ-СН2СН2ОН и МТ-СНО в двухфазной системе органический растворитель-водный раствор 2М H2SO4 приведены в таблице. В изученной системе исходный спирт МТ-СН2СH2ОН, так же как и водорастворимый окислитель, находится в кислой водной фазе, в то время как альдегид МТ-СНО преимущественно содержится в органическом растворе, что препятствует его дальнейшему окислению. Заявитель применял в реакции ряд органических растворителей – диэтиловый эфир, бензол, хлороформ и хлористый метилен. Все они удобны для выполнения реакции и могут использоваться для извлечения образующегося МТ-СНО из водного раствора окислителя, но хлороформ и хлористый метилен наиболее пригодны для полного извлечения продукта реакции из кислого водного раствора.
Выбор соотношения реагентов МТ-СН2СН2ОН:окислитель:H2SO4 для осуществления реакции проведен с учетом уравнений 1, 3 схемы 1 (для СrО3) и в соответствии с уравнениями 1, 3 схемы 2 (для К2Сr2O7 и Na2Cr3O7). Оптимальное соотношение реагентов в случае СrО3 составляет 1:2:3, а в случае К2Сr2O7 и Na2Cr3O7 составляет 1:1:4. Дальнейшее увеличение количеств окислителя и/или H2SO4 приводит лишь к снижению выхода за счет превращения целевого альдегида в соответствующую 4-метил-тиазолил-5-карбоновую кислоту.
Недостаточное количество окислителя и/или Н2SO4 также приводит к снижению выхода целевого продукта за счет неполного окисления исходного спирта, при этом непрореагировавший исходный спирт не загрязняет продукт, т.к. остается в кислом водном слое и может быть выделен из него после соответствующей обработки.
Строение и индивидуальность синтезированных соединений подтверждены результатами тонкослойной хроматографии (ТСХ), высокоэффективной жидкостной хроматографии (ВЭЖХ) и данными спектроскопии ЯМР 1Н и 13С.
Спектры ЯМР 1H и 13С зарегистрированы на ЯМР-спектрометре DPX-200 (BRUKER) в CDCl3 и ДМСО-D6, внутренний стандарт – TMS. Температура плавления измерена на приборе BOETIUS РНМК-05. Результаты ВЭЖХ получены на хроматографе HP-1100 (Hewlett Packard) с детектором УФ-излучения 254 нм, элюент 30% МеОН+70% Н2О и 70% МеОН+30% Н2О. Контроль за ходом реакции осуществлялся также методом ТСХ на алюминиевых пластинах 5
1) применение дорогостоящего окислителя, синтез которого требует дополнительных затрат и увеличивает число стадий процесса,
2) применение избытка окислителя (6 моль окислителя на 1 моль субстрата), что приводит к образованию большого количества твердых отходов и загрязнению окружающей среды,
3) трудности при выделении продукта, в частности экстракция целевого соединения из смолообразной смеси, образующейся по окончании реакции.
Сущность изобретения.
Заявитель предлагаемого изобретения обнаружил, что указанные недостатки могут быть устранены при получении МТ-СНО окислением МТ-СН2СН2ОН в двухфазной системе с использованием дешевых и доступных окислителей, каковыми являются хромовая кислота и ее неорганические соли в водном растворе кислоты.
Интенсификация процесса окисления МТ-СН2СН2ОН, оптимизация теплового режима, удобство и простота выделения целевого продукта, а также увеличение его выхода достигаются за счет применения водорастворимых неорганических окислителей, оптимизации соотношения МТ-СН2СН2ОН:окислитель и благодаря использованию двухфазной системы.
Процесс окисления МТ-СН2СН2ОН окисью хрома или неорганическими бихроматами описывается уравнениями, которые представлены на схемах (1) и (2) соответственно. Окисление МТ-СН2СН2ОН сопровождается расщеплением С-С-связи в боковой цепи молекулы исходного спирта (схема 1, уравнения 1, 3, 5, 7 и схема 2, уравнения 1, 3, 5, 7). При этом оксиметильный фрагмент, содержащий концевой атом углерода, в условиях реакции может быть превращен в соль муравьиной кислоты, а в более жестких условиях может быть окислен в углекислоту.
Главная часть молекулы преобразуется в альдегид МТ-СНО, который является устойчивым в предложенных условиях реакции благодаря аллильному резонансу С=O-группы и С=С-связи гетероциклического ядра. Наличие избытка окислителя и/или H2SO4, а также температура выше 50
Заявитель предлагаемого изобретения обнаружил, что распределение исходного спирта 4-метил-5-(2-гидроксиэтил)тиазола и продукта реакции – 4-метил-5-формил-тиазола в двухфазной системе благоприятно для проведения окисления водорастворимыми окислителями. Данные по распределению МТ-СН2СН2ОН и МТ-СНО в двухфазной системе органический растворитель-водный раствор 2М H2SO4 приведены в таблице. В изученной системе исходный спирт МТ-СН2СH2ОН, так же как и водорастворимый окислитель, находится в кислой водной фазе, в то время как альдегид МТ-СНО преимущественно содержится в органическом растворе, что препятствует его дальнейшему окислению. Заявитель применял в реакции ряд органических растворителей – диэтиловый эфир, бензол, хлороформ и хлористый метилен. Все они удобны для выполнения реакции и могут использоваться для извлечения образующегося МТ-СНО из водного раствора окислителя, но хлороформ и хлористый метилен наиболее пригодны для полного извлечения продукта реакции из кислого водного раствора.
Выбор соотношения реагентов МТ-СН2СН2ОН:окислитель:H2SO4 для осуществления реакции проведен с учетом уравнений 1, 3 схемы 1 (для СrО3) и в соответствии с уравнениями 1, 3 схемы 2 (для К2Сr2O7 и Na2Cr3O7). Оптимальное соотношение реагентов в случае СrО3 составляет 1:2:3, а в случае К2Сr2O7 и Na2Cr3O7 составляет 1:1:4. Дальнейшее увеличение количеств окислителя и/или H2SO4 приводит лишь к снижению выхода за счет превращения целевого альдегида в соответствующую 4-метил-тиазолил-5-карбоновую кислоту.
Недостаточное количество окислителя и/или Н2SO4 также приводит к снижению выхода целевого продукта за счет неполного окисления исходного спирта, при этом непрореагировавший исходный спирт не загрязняет продукт, т.к. остается в кислом водном слое и может быть выделен из него после соответствующей обработки.
Строение и индивидуальность синтезированных соединений подтверждены результатами тонкослойной хроматографии (ТСХ), высокоэффективной жидкостной хроматографии (ВЭЖХ) и данными спектроскопии ЯМР 1Н и 13С.
Спектры ЯМР 1H и 13С зарегистрированы на ЯМР-спектрометре DPX-200 (BRUKER) в CDCl3 и ДМСО-D6, внутренний стандарт – TMS. Температура плавления измерена на приборе BOETIUS РНМК-05. Результаты ВЭЖХ получены на хроматографе HP-1100 (Hewlett Packard) с детектором УФ-излучения 254 нм, элюент 30% МеОН+70% Н2О и 70% МеОН+30% Н2О. Контроль за ходом реакции осуществлялся также методом ТСХ на алюминиевых пластинах 5 10 см. Силикагель 60 F254 (Merck), элюент – СНСl3-ЕtOН (10:1). Колоночная хроматография выполнена на колонках Wakogel С-200, 75-150 мкм, элюенты – СНСl3 и СНСl3-ЕtOН (10:1). В качестве окислителей использовались реактивы марки чда. Все использованные растворители были промыты концентрированной Н2SO4 и водой, высушены CaCl2 и перегнаны.
Приведенные ниже примеры иллюстрируют, но не ограничивают применение данного изобретения.
Пример 1.
Раствор 1,60 г (16 ммоль) СrО3 и 1,77 г (18 ммоль) Н2SO4 в 10 мл Н2О добавили при перемешивании к раствору 1,15 г (8 ммоль) MT-CH2CH2OH в 30 мл бензола. После добавления всего окислителя реакционную смесь перемешивали 4 ч при температуре 35С. Через каждый час органический слой отделяли и заменяли новой порцией бензола. Все органические слои объединили, высушили Na2SO4 и растворитель удалили в вакууме. Получили 0,69 г МТ-СНО, выход 68%. Продукт очистили методом колоночной хроматографии. Получили 0,55 г МТ-СНО, выход 54%. Т. пл. 73-74С. Спектр ЯМР 1Н ( 10 см. Силикагель 60 F254 (Merck), элюент – СНСl3-ЕtOН (10:1). Колоночная хроматография выполнена на колонках Wakogel С-200, 75-150 мкм, элюенты – СНСl3 и СНСl3-ЕtOН (10:1). В качестве окислителей использовались реактивы марки чда. Все использованные растворители были промыты концентрированной Н2SO4 и водой, высушены CaCl2 и перегнаны.
Приведенные ниже примеры иллюстрируют, но не ограничивают применение данного изобретения.
Пример 1.
Раствор 1,60 г (16 ммоль) СrО3 и 1,77 г (18 ммоль) Н2SO4 в 10 мл Н2О добавили при перемешивании к раствору 1,15 г (8 ммоль) MT-CH2CH2OH в 30 мл бензола. После добавления всего окислителя реакционную смесь перемешивали 4 ч при температуре 35С. Через каждый час органический слой отделяли и заменяли новой порцией бензола. Все органические слои объединили, высушили Na2SO4 и растворитель удалили в вакууме. Получили 0,69 г МТ-СНО, выход 68%. Продукт очистили методом колоночной хроматографии. Получили 0,55 г МТ-СНО, выход 54%. Т. пл. 73-74С. Спектр ЯМР 1Н ( , м.д.): 2,82 с (Me, 3Н), 9,02 с (СНО, 1Н), 10,15 с (CHN, 1H). Спектр ЯМР 13С (, м.д.): 16,15 (СН3), 132,74 (CS), 158,89 (NCS), 161,80 (CN), 182,44 (СНО). Содержание МТ-СНО по данным ВЭЖХ 99,7%.
Пример 2.
Раствор 0,55 г (5,5 ммоль) СrО3 и 0,65 г (6,6 ммоль) H2SO4 в 5 мл Н2O добавили по каплям при перемешивании к раствору 0,36 г (2,5 ммоль) МТ-СН2СН2ОН в 15 мл бензола. После добавления всего окислителя реакционную смесь перемешивали 20 мин при температуре 45-50С. Водный слой отделили и экстрагировали хлороформом (210 мл). Все органические слои объединили, промыли водой, высушили Na2SO4 и растворители удалили в вакууме. Получили 0,19 г МТ-СНО, выход 58%.
Пример 3.
Раствор 1,60 г (16 ммоль) СrО3 и 2,35 г (24 ммоль) H2SO4 в 20 мл Н2O добавили по каплям при перемешивании к раствору 0,86 г (6 ммоль) МТ-СН2СН2ОН в 30 мл бензола при температуре 40-45С. После добавления всего окислителя реакционную смесь перемешивали 2 ч при той же температуре. Водный слой отделили и экстрагировали хлороформом (215 мл). Все органические слои объединили, промыли водой, высушили Na2SO4 и растворители удалили в вакууме. Продукт очистили методом колоночной хроматографии на силикагеле. Получили 0,50 г МТ-СНО, выход 66% и 0,05 г МТ-СООН, выход 6%.
Пример 4.
Раствор 2,00 г (20 ммоль) СrО3 и 1,96 г (20 ммоль) H2SO4 в 10 мл Н2O прибавили порциями по 4 мл при перемешивании к раствору 1,43 г (10 ммоль) R-СН2СН2OН в бензоле. После добавления каждой порции окислителя реакционную массу перемешивали 60 мин при 20С, органический слой отделяли и заменяли новой порцией бензола. После завершения реакции органические слои объединили, высушили Na2SO4 и растворитель удалили в вакууме. Получили 0,80 г МТ-СНО, выход 63%. Водный слой нейтрализовали 1М КОН и экстрагировали хлороформом (210 мл) 0,30 г (21%) исходного спирта МТ-СН2СН2ОН.
Пример 5.
Раствор 1,00 г (10 ммоль) СrО3 и 1,47 г (15 ммоль) Н2SO4 в 10 мл Н2О прибавили при перемешивании к раствору 0,72 г (5 ммоль) МТ-СН2СН2ОН в 25 мл хлористого метилена. После добавления окислителя реакционную массу перемешивали 3 ч при 20С, органический слой отделили, промыли водой, высушили Na2SO4 и растворитель удалили в вакууме. Получили 0,37 г МТ-СНО, выход 58%. Водный слой нейтрализовали 1М КОН и экстрагировали хлороформом (210 мл) 0,07 г (10%) исходного спирта МТ-СН2СН2ОН.
Пример 6.
Раствор 1,50 г (15 ммоль) СrО3 в 3 мл Н2O добавили к раствору 1,08 г (7,5 ммоль) R-CH2CH2OH в 20 мл диэтилового эфира и перемешивали 3 ч при 20С. В течение этого периода в реакционную смесь добавили тремя равными порциями раствор 2,21 г (22,5 ммоль) Н2SO4 в 10 мл Н2О. Органический слой отделяли и заменяли новой порцией эфира перед каждым прибавлением H2SO4. После завершения реакции все органические слои объединили, высушили Na2SO4, растворитель удалили в вакууме. Получили 0,59 г МТ-СНО, выход 63%. Водный слой нейтрализовали 1М КОН и экстрагировали хлороформом (210 мл) 0,29 г (27%) МТ-СН2СН2ОН.
Пример 7.
Раствор 1,77 г (6 ммоль) К2Сr2O7 и 2,36 г (24 ммоль) H2SO4 в 18 мл Н2О прибавили по каплям при перемешивании к раствору 0,86 г (6 ммоль) R-СН2СН2ОН в 30 мл CH2Cl2 при 20С и перемешивали при той же температуре 20 ч. Органический слой отделили, промыли водой, высушили Na2SO4 и растворитель удалили в вакууме. Получили 0,45 г МТ-СНО, выход 59%.
Пример 8.
Раствор 1,77 г (6 ммоль) К2Сr2O7 и 2,36 г (24 ммоль) Н2SO4 в 18 мл Н2О прибавили по каплям при перемешивании к раствору 0,86 г (6 ммоль) МТ-СН2СН2ОН в 30 мл бензола при 50С и перемешивали при той же температуре 1 ч. Водный слой экстрагировали хлороформом (210 мл). Объединенные органические слои промыли водой, высушили Na2SO4 и растворители удалили в вакууме. Получили 0,40 г R-CHO, выход 53%.
Пример 9.
Раствор 1,77 г (6 ммоль) К2Сr2О7 и 2,36 г (24 ммоль) H2SO4 в 18 мл H2O прибавили по каплям при перемешивании к раствору 0,86 г (6 ммоль) МТ-СН2СН2ОН в 30 мл диэтилового эфира при слабом кипении растворителя и перемешивали при той же температуре 2 ч. Водный слой экстрагировали хлороформом (210 мл). Объединенные органические слои промыли водой, высушили Na2SO4 и растворители удалили в вакууме. Получили 0,44 г МТ-СНО, выход 58%.
Пример 10.
Раствор 1,77 г (6 ммоль) К2Сr2O7 и 1,18 г (12 ммоль) Н2SO4 в 8 мл H2О прибавили порциями по 3 мл при перемешивании к раствору 0,86 г (6 ммоль) R-СН2СН2ОН в 30 мл СН2Cl2 при 38С. После добавления каждой порции окислителя реакционную смесь перемешивали 30 мин, органический слой отделяли и заменяли новой порцией растворителя. После завершения реакции органические слои объединили, высушили Na2SO4 и растворитель удалили в вакууме. Получили 0,42 г МТ-СНО, выход 54%. Водный слой нейтрализовали 1М КОН и экстрагировали хлористым метиленом (210 мл) 0,31 г (36%) непрореагировавшего МТ-СН2СН2ОН.
Пример 11.
Раствор 1,60 г (16 ммоль) СrO3 и 3,02 г (48 ммоль) HNO3 в 10 мл Н2О прибавили при перемешивании к раствору 1,15 г (8 ммоль) MT-CH2CH2OH в 30 мл хлороформа. После добавления всего окислителя реакционную смесь перемешивали 4 ч при температуре 35С. Через каждый час органический слой отделяли и заменяли новой порцией хлороформа. Все органические слои объединили, высушили Na2SO4 и растворитель удалили в вакууме. Получили 0,65 г МТ-СНО, выход 64%.
Пример 12.
Раствор 1,60 г (16 ммоль) СrО3 и 1,70 г (48 ммоль) НСl в 10 мл Н2О добавили при перемешивании к раствору 1,15 г (8 ммоль) МТ-СН2СН2ОН в 30 мл хлороформа. После добавления всего окислителя реакционную смесь перемешивали 4 ч при температуре 35С. Через каждый час органический слой отделяли и заменяли новой порцией хлороформа. Все органические слои объединили, высушили Na2SO4 и растворитель удалили в вакууме. Получили 0,63 г МТ-СНО, выход 62%.
Таким образом, приведенные данные позволяют предложить способ получения 4-метил-5-формил-тиазола окислением 4-метил-5-(2-гидроксиэтил)тиазола в двухфазной системе с использованием окиси хрома или неорганических бихроматов в качестве окислителей. , м.д.): 2,82 с (Me, 3Н), 9,02 с (СНО, 1Н), 10,15 с (CHN, 1H). Спектр ЯМР 13С (, м.д.): 16,15 (СН3), 132,74 (CS), 158,89 (NCS), 161,80 (CN), 182,44 (СНО). Содержание МТ-СНО по данным ВЭЖХ 99,7%.
Пример 2.
Раствор 0,55 г (5,5 ммоль) СrО3 и 0,65 г (6,6 ммоль) H2SO4 в 5 мл Н2O добавили по каплям при перемешивании к раствору 0,36 г (2,5 ммоль) МТ-СН2СН2ОН в 15 мл бензола. После добавления всего окислителя реакционную смесь перемешивали 20 мин при температуре 45-50С. Водный слой отделили и экстрагировали хлороформом (210 мл). Все органические слои объединили, промыли водой, высушили Na2SO4 и растворители удалили в вакууме. Получили 0,19 г МТ-СНО, выход 58%.
Пример 3.
Раствор 1,60 г (16 ммоль) СrО3 и 2,35 г (24 ммоль) H2SO4 в 20 мл Н2O добавили по каплям при перемешивании к раствору 0,86 г (6 ммоль) МТ-СН2СН2ОН в 30 мл бензола при температуре 40-45С. После добавления всего окислителя реакционную смесь перемешивали 2 ч при той же температуре. Водный слой отделили и экстрагировали хлороформом (215 мл). Все органические слои объединили, промыли водой, высушили Na2SO4 и растворители удалили в вакууме. Продукт очистили методом колоночной хроматографии на силикагеле. Получили 0,50 г МТ-СНО, выход 66% и 0,05 г МТ-СООН, выход 6%.
Пример 4.
Раствор 2,00 г (20 ммоль) СrО3 и 1,96 г (20 ммоль) H2SO4 в 10 мл Н2O прибавили порциями по 4 мл при перемешивании к раствору 1,43 г (10 ммоль) R-СН2СН2OН в бензоле. После добавления каждой порции окислителя реакционную массу перемешивали 60 мин при 20С, органический слой отделяли и заменяли новой порцией бензола. После завершения реакции органические слои объединили, высушили Na2SO4 и растворитель удалили в вакууме. Получили 0,80 г МТ-СНО, выход 63%. Водный слой нейтрализовали 1М КОН и экстрагировали хлороформом (210 мл) 0,30 г (21%) исходного спирта МТ-СН2СН2ОН.
Пример 5.
Раствор 1,00 г (10 ммоль) СrО3 и 1,47 г (15 ммоль) Н2SO4 в 10 мл Н2О прибавили при перемешивании к раствору 0,72 г (5 ммоль) МТ-СН2СН2ОН в 25 мл хлористого метилена. После добавления окислителя реакционную массу перемешивали 3 ч при 20С, органический слой отделили, промыли водой, высушили Na2SO4 и растворитель удалили в вакууме. Получили 0,37 г МТ-СНО, выход 58%. Водный слой нейтрализовали 1М КОН и экстрагировали хлороформом (210 мл) 0,07 г (10%) исходного спирта МТ-СН2СН2ОН.
Пример 6.
Раствор 1,50 г (15 ммоль) СrО3 в 3 мл Н2O добавили к раствору 1,08 г (7,5 ммоль) R-CH2CH2OH в 20 мл диэтилового эфира и перемешивали 3 ч при 20С. В течение этого периода в реакционную смесь добавили тремя равными порциями раствор 2,21 г (22,5 ммоль) Н2SO4 в 10 мл Н2О. Органический слой отделяли и заменяли новой порцией эфира перед каждым прибавлением H2SO4. После завершения реакции все органические слои объединили, высушили Na2SO4, растворитель удалили в вакууме. Получили 0,59 г МТ-СНО, выход 63%. Водный слой нейтрализовали 1М КОН и экстрагировали хлороформом (210 мл) 0,29 г (27%) МТ-СН2СН2ОН.
Пример 7.
Раствор 1,77 г (6 ммоль) К2Сr2O7 и 2,36 г (24 ммоль) H2SO4 в 18 мл Н2О прибавили по каплям при перемешивании к раствору 0,86 г (6 ммоль) R-СН2СН2ОН в 30 мл CH2Cl2 при 20С и перемешивали при той же температуре 20 ч. Органический слой отделили, промыли водой, высушили Na2SO4 и растворитель удалили в вакууме. Получили 0,45 г МТ-СНО, выход 59%.
Пример 8.
Раствор 1,77 г (6 ммоль) К2Сr2O7 и 2,36 г (24 ммоль) Н2SO4 в 18 мл Н2О прибавили по каплям при перемешивании к раствору 0,86 г (6 ммоль) МТ-СН2СН2ОН в 30 мл бензола при 50С и перемешивали при той же температуре 1 ч. Водный слой экстрагировали хлороформом (210 мл). Объединенные органические слои промыли водой, высушили Na2SO4 и растворители удалили в вакууме. Получили 0,40 г R-CHO, выход 53%.
Пример 9.
Раствор 1,77 г (6 ммоль) К2Сr2О7 и 2,36 г (24 ммоль) H2SO4 в 18 мл H2O прибавили по каплям при перемешивании к раствору 0,86 г (6 ммоль) МТ-СН2СН2ОН в 30 мл диэтилового эфира при слабом кипении растворителя и перемешивали при той же температуре 2 ч. Водный слой экстрагировали хлороформом (210 мл). Объединенные органические слои промыли водой, высушили Na2SO4 и растворители удалили в вакууме. Получили 0,44 г МТ-СНО, выход 58%.
Пример 10.
Раствор 1,77 г (6 ммоль) К2Сr2O7 и 1,18 г (12 ммоль) Н2SO4 в 8 мл H2О прибавили порциями по 3 мл при перемешивании к раствору 0,86 г (6 ммоль) R-СН2СН2ОН в 30 мл СН2Cl2 при 38С. После добавления каждой порции окислителя реакционную смесь перемешивали 30 мин, органический слой отделяли и заменяли новой порцией растворителя. После завершения реакции органические слои объединили, высушили Na2SO4 и растворитель удалили в вакууме. Получили 0,42 г МТ-СНО, выход 54%. Водный слой нейтрализовали 1М КОН и экстрагировали хлористым метиленом (210 мл) 0,31 г (36%) непрореагировавшего МТ-СН2СН2ОН.
Пример 11.
Раствор 1,60 г (16 ммоль) СrO3 и 3,02 г (48 ммоль) HNO3 в 10 мл Н2О прибавили при перемешивании к раствору 1,15 г (8 ммоль) MT-CH2CH2OH в 30 мл хлороформа. После добавления всего окислителя реакционную смесь перемешивали 4 ч при температуре 35С. Через каждый час органический слой отделяли и заменяли новой порцией хлороформа. Все органические слои объединили, высушили Na2SO4 и растворитель удалили в вакууме. Получили 0,65 г МТ-СНО, выход 64%.
Пример 12.
Раствор 1,60 г (16 ммоль) СrО3 и 1,70 г (48 ммоль) НСl в 10 мл Н2О добавили при перемешивании к раствору 1,15 г (8 ммоль) МТ-СН2СН2ОН в 30 мл хлороформа. После добавления всего окислителя реакционную смесь перемешивали 4 ч при температуре 35С. Через каждый час органический слой отделяли и заменяли новой порцией хлороформа. Все органические слои объединили, высушили Na2SO4 и растворитель удалили в вакууме. Получили 0,63 г МТ-СНО, выход 62%.
Таким образом, приведенные данные позволяют предложить способ получения 4-метил-5-формил-тиазола окислением 4-метил-5-(2-гидроксиэтил)тиазола в двухфазной системе с использованием окиси хрома или неорганических бихроматов в качестве окислителей.
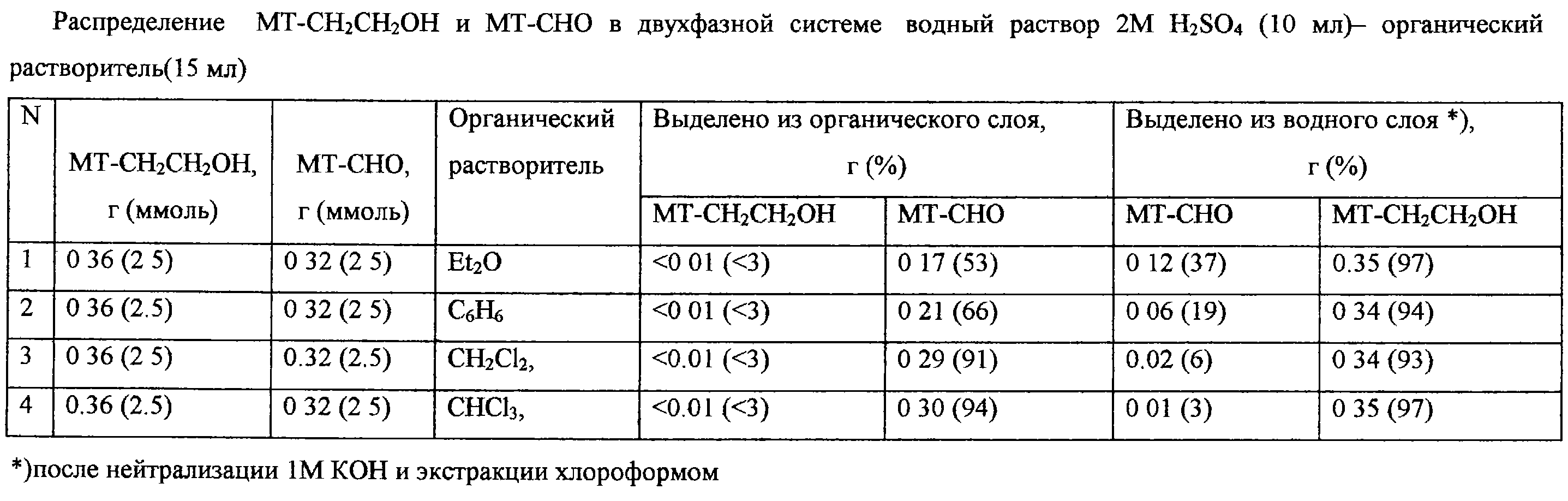 Схема 1. Уравнения, которыми описывается реакция окисления МТ-СН2СН2ОН действием Сr2O3 (или Н2Сr2O7) в растворе Н2SO4
6RCH2CH2OH+12CrO3+15H2SO4
Схема 1. Уравнения, которыми описывается реакция окисления МТ-СН2СН2ОН действием Сr2O3 (или Н2Сr2O7) в растворе Н2SO4
6RCH2CH2OH+12CrO3+15H2SO4  6RCHO+2Сr(СООН)3+5Cr2(SO4)3+24Н2O (1)
2Сr(СООН)3+3H2SO4 6НСООН+Сr2(SO4)3 (2)
6RCH2CH2OH+12СrO3+18H2SO4 6RCHO+6НСООН+6Сr2(SO4)3+24Н2O (3)
6НСООН+4СrO3+6H2SO4 6СO2+6Сr2(SO4)3+14Н2O (4)
6RCH2CH2OH+16СrO3+24H2SO4 6RCHO+6СO2+8Cr2(SO4)3+36Н2O (5)
6RCHO+4СrO3+6H2SO4 6RCOOH+2Сr2(SO4)3+6Н2O (6)
6RCH2CH2OH+20СrO3+30H2SO4 6RCOOH+6СO2+10Cr2(SO4)3+42Н2O (7)
Схема 2. Уравнения, которыми описывается реакция окисления МТ-СН2СН2ОН действием К2Cr2О7 (или Na2Cr2O7·2Н2О) в растворе H2SO4
6RСН2СН2ОН+6Nа2Cr2О7·2Н2O+21H2SO4 6RСНО+2Сr(СООН)3+6Nа2SO4+5Сr2(SO4)3 +42Н2O (1)
2Сr(СООН)3+3H2SO4 6НСООН+Сr2(SO4)3 (2)
6RCH2CH2OH+6Na2Cr2O7·2H2O+24H2SO4 6RCHO+6НСООН+8Nа2SO4+6Сr2(SO4)3
+21Н2O (3)
6НСООН+2Na2Cr2O7·2H2O+8H2SO4 6СO2+2Сr2(SO4)3+2Na2SO4+18Н2O (4)
6RCH2CH2OH+8Na2Cr2O7·2Н2О+32H2SO4 6RCHO+6СO2+8Na2SO4+8Сr2(SO4)3+60Н2O (5)
6RCHO+2Na2Cr2O7·2H2O+8H2SO4 6RCOOH+2Na2SO4+2Сr2(SO4)3+8Н2O (6)
6RCH2CH2OH+10Nа2Сr2O7·2Н2O+40Н2SO4 6RCOOH+6СO2+10Сr2(SO4)3+10Na2SO4 +72Н2O (7) 6RCHO+2Сr(СООН)3+5Cr2(SO4)3+24Н2O (1)
2Сr(СООН)3+3H2SO4 6НСООН+Сr2(SO4)3 (2)
6RCH2CH2OH+12СrO3+18H2SO4 6RCHO+6НСООН+6Сr2(SO4)3+24Н2O (3)
6НСООН+4СrO3+6H2SO4 6СO2+6Сr2(SO4)3+14Н2O (4)
6RCH2CH2OH+16СrO3+24H2SO4 6RCHO+6СO2+8Cr2(SO4)3+36Н2O (5)
6RCHO+4СrO3+6H2SO4 6RCOOH+2Сr2(SO4)3+6Н2O (6)
6RCH2CH2OH+20СrO3+30H2SO4 6RCOOH+6СO2+10Cr2(SO4)3+42Н2O (7)
Схема 2. Уравнения, которыми описывается реакция окисления МТ-СН2СН2ОН действием К2Cr2О7 (или Na2Cr2O7·2Н2О) в растворе H2SO4
6RСН2СН2ОН+6Nа2Cr2О7·2Н2O+21H2SO4 6RСНО+2Сr(СООН)3+6Nа2SO4+5Сr2(SO4)3 +42Н2O (1)
2Сr(СООН)3+3H2SO4 6НСООН+Сr2(SO4)3 (2)
6RCH2CH2OH+6Na2Cr2O7·2H2O+24H2SO4 6RCHO+6НСООН+8Nа2SO4+6Сr2(SO4)3
+21Н2O (3)
6НСООН+2Na2Cr2O7·2H2O+8H2SO4 6СO2+2Сr2(SO4)3+2Na2SO4+18Н2O (4)
6RCH2CH2OH+8Na2Cr2O7·2Н2О+32H2SO4 6RCHO+6СO2+8Na2SO4+8Сr2(SO4)3+60Н2O (5)
6RCHO+2Na2Cr2O7·2H2O+8H2SO4 6RCOOH+2Na2SO4+2Сr2(SO4)3+8Н2O (6)
6RCH2CH2OH+10Nа2Сr2O7·2Н2O+40Н2SO4 6RCOOH+6СO2+10Сr2(SO4)3+10Na2SO4 +72Н2O (7)
Формула изобретения 1. Способ получения 4-метил-5-формилтиазола, заключающийся в окислении 4-метил-5-(2-гидроксиэтил)тиазола, отличающийся тем, что в качестве окислителя используют раствор окиси хрома или неорганических бихроматов в водном растворе серной кислоты и реакцию проводят в двухфазной системе вода – органический растворитель при температуре 20-50 С.
2. Способ по п.1, отличающийся тем, что в качестве органического растворителя используют диэтиловый эфир, бензол и хлористый метилен.
3. Способ по пп.1 и 2, отличающийся тем, что в качестве неорганического бихромата используют бихромат калия.
TK4A – Поправки к публикациям сведений об изобретениях в бюллетенях “Изобретения (заявки и патенты)” и “Изобретения. Полезные модели”
Страница: 462
Напечатано: (57) 1. …в водном растворе серной кислоты…
Следует читать: (57) 1. …в водном растворе кислоты…
Номер и год публикации бюллетеня: 19-2004 Номер и год публикации бюллетеня: 13-2004
Извещение опубликовано: 10.07.2004
TK4A – Поправки к публикациям сведений об изобретениях в бюллетенях “Изобретения (заявки и патенты)” и “Изобретения. Полезные модели”
Страница: 462
Напечатано: 2. …используют диэтиловый эфир, бензол и хлористый метилен.
Следует читать: 2. …используют диэтиловый эфир, бензол, хлороформ и хлористый метилен.
Номер и год публикации бюллетеня: 19-2004 Номер и год публикации бюллетеня: 13-2004
Извещение опубликовано: 10.07.2004
TZ4A – Поправки к описаниям изобретений
Часть описания, где обнаружена ошибка: Текст реферата, Кол. 2, строка 7
Напечатано: …в присутствии серной кислоты…
Следует читать: …в присутствии водного раствора кислоты…
Номер и год публикации бюллетеня: 19-2004 Номер и год публикации бюллетеня: 13-2004
Извещение опубликовано: 10.07.2004
PC4A – Регистрация договора об уступке патента Российской Федерации на изобретение
(73) Патентообладатель(и):
(73) Патентообладатель:
Дата и номер государственной регистрации перехода исключительного права: 18.10.2007 № РД0027902
Извещение опубликовано: 27.11.2007 БИ: 33/2007
|
||||||||||||||||||||||||||