Патент на изобретение №2228180
|
||||||||||||||||||||||||||
(54) СПОСОБ ПРЕДОТВРАЩЕНИЯ ЗЛОУПОТРЕБЛЕНИЯ СОДЕРЖАЩИМИ ОПИОИДЫ ЛЕКАРСТВЕННЫМИ ФОРМАМИ
(57) Реферат: Изобретение относится к способу уменьшения возможности злоупотребления оральной лекарственной формой опиоидного анальгетика, в котором анальгетически эффективное количество орально активного опиоидного агониста соединяют с опиоидным антагонистом в лекарственную форму для орального применения, причем потребуется, по крайней мере, двухступенчатая экстракция для выделения опиоидного агониста, а количество введенного в лекарственную форму опиоидного антагониста достаточно, чтобы подавить действие опиоида при совместной экстракции с опиоидным агонистом и парентеральном введении. Изобретение обеспечивает вызывающий отвращение опыт, когда большое количество продукта применяется лицом со сформировавшейся физической зависимостью. 57 з.п. ф-лы, 7 табл. Опиоиды, известные также как опиоидные агонисты, составляют группу лекарств, которые проявляют опиум- или морфиноподобные свойства. Опиоиды применяются главным образом в качестве умеренно- и сильнодействующих анальгетиков, но также фармакологически эффективны, например, в отношении гиперсомнии (сонливость), угнетения дыхания, депрессивных состояний и помрачения ума, не ведущего к потере сознания. Опиоиды действуют как агонисты, взаимодействуя со стереоспецифичными и способными к насыщению связывающими центрами в головном мозге и в других тканях. Эндогенные опиоидподобные пептиды присутствуют, в основном, в тех участках центральной нервной системы, которые, как полагают, связаны с восприятием болевых ощущений, способностью двигаться, с настроением и поведением, а также регуляцией нейроэндокринологических функций. Опиум содержит более 20 отдельных алкалоидов. Морфины, кодеины и папаверин также входят в эту группу. К середине девятнадцатого века в медицине стало распространяться использование чистых алкалоидов, таких как морфин, взамен субстанций, выделенных из неочищенного опиума. Возможность парентерального применения морфина стала причиной использования более сильной разновидности компульсивных лекарственных средств. Проблема привыкания к опиоидам стимулировала поиск потенциальных анальготических средств, которые не вызывали бы наркотических пристрастий к ним. К 1967 году было сделано заключение о том, что сложные взаимодействия между морфиноподобными лекарствами, антагонистами, и то, что было впоследствии названо “смешанный агонист – антагонист” лучше всего можно объяснить, если принять без доказательств, что существует несколько типов рецепторов для опиоидов и родственных лекарственных средств. С появлением новых полностью синтетических форм морфиноподобного действия термин “опиоид”, в основном, сохранился как родовое понятие для всех экзогенных веществ, которые стереоспецифически связываются с любым из нескольких подвидов опиоидных рецепторов и действуют как агонисты. Возможность развития устойчивости и физической зависимости при неоднократном использовании опиоидов является характерной чертой всех опиоидных лекарств, и возможность возникновения физиологической зависимости (т.е. привыкания) – одна из главных проблем при использовании опиоидов для облегчения страданий и боли, даже несмотря на то, что ятрогенное привыкание является редким. Другой важной проблемой, связанной с применением опиоидов, является распространение или передача этих лекарственных средств от больного (пациента) к другому лицу (непациенту) с развлекательными целями, например наркоману. Возможность неправильного использования (или злоупотребления) опиоидом не обуславливается всецело только каким-то одним-единственным фактором. Напротив, существует комплекс факторов, включающий способность лекарства приводить к выработке рода физической зависимости, при которой отмена лекарства вызывает сильный дистресс, побуждающий к попыткам розыска этого лекарства; способность (умение) подавить симптомы отмены, связанные с этой отменой, с помощью других средств; степень индуцирования эйфории подобна эффекту от морфина и других опиоидов; характер токсичности, которая проявляется при передозировке свыше нормального терапевтического уровня; и физические характеристики лекарственных средств, например растворимость в воде. По таким физическим характеристикам можно судить о вероятности неправильного употребления лекарства парентеральным путем. В Соединенных Штатах попытка контролировать лиц, использующих компульсивные лекарства, включает и усилия регулировать доступность лекарств путем установления ограничений на использование опиоидов при обезболивании лицами, использующими компульсивные лекарства. На практике врач часто сталкивается с необходимостью делать выбор, назначать ли сильнодействующие опиоидные анальгетики даже тем лицам, кто, по всей видимости, предрасположен к возникновению и развитию физиологической зависимости, т.е. к привыканию к лекарствам подобного рода. В связи с этой проблемой было рекомендовано, что этим пациентам не следует назначать опиоиды, если есть другое лекарство, вполне подходящее и которое не может быть неправильно употреблено; и, кроме того, не следует допускать самостоятельного употребления этими пациентами таких лекарств парентеральным путем, для них следует обеспечить однократную поставку дозы, соответствующей дозе за несколько дней. Различают, по крайней мере, три основных типа использования опиоидов и опиоидной зависимости. К первому типу относятся лица, для которых употребление (наркотических) лекарственных средств начинается в связи с лечением, и кто получает свои самые первые дозы через врачей, например. Другой тип начинает с экспериментирования или “развлечения” (наркотическим) лекарственным средством и затем расширяет его потребление. К третьему типу относятся лица, начинающие использовать лекарственные средства по первому или второму из вышеуказанных способов, а затем переключаются на опиоиды, применяемые оральным путем, например метадон, получаемые по каналам организованного наркобизнеса. Толерантность характеризуется необходимостью увеличения время от времени дозы опиоида для того, чтобы достигнуть того же уровня обезболивания или эйфории, или наблюдением того факта, что неоднократное введение одной и той же дозы приводит к уменьшению обезболивающего, вызывающего эйфорию или др. опиоидных эффектов. Было установлено, что высокий уровень толерантности развивается при респираторно-депрессантном анальгетическом, седативном, рвотном и вызывающем эйфорию эффектах опиоидов. Однако скорость, с которой может развиваться эта толерантность как у наркомана, так и у пациента, которому необходимо обезболивание, зависит от схемы приема опиоида. При частом применении опиоида может требоваться увеличение дозы. Толерантность не развивается в одинаковой степени или с одинаковой скоростью в отношении всех аспектов действия опиоидов, и даже у лиц, высокотолерантных в отношении респираторно-депрессантного действия, продолжает проявляться миоз (сужение зрачков) и констипация. Толерантность к опиоидам в значительной степени исчезает по завершении периода абстиненции (синдрома отмены). Физическая зависимость может развиться в результате неоднократно вводимых доз или передозировки опиоидов. Физическая зависимость проявляется постепенно после прекращения приема опиоидов или внезапно (например, в течение 20 минут) после введения антагониста наркотика (что называется “ускоренной абстиненцией”). В зависимости от того, по отношению к какому лекарственному средству установлена зависимость, продолжительности употребления и величины дозы, симптомы синдрома отмены различаются количеством, силой, типом и длительностью своего проявления. Подавляющее количество общих симптомов, характерных для синдрома отмены, включают анорексию, потерю веса, расширение зрачков, озноб, перемежающийся с потоотделением, спазмы в животе, тошноту, рвоту, мышечные спазмы, гиперчувствительность, слезотечение, обильные выделения из носа, “гусиную кожу” и увеличенную частоту сердечных сокращений. Абстинентный синдром обычно возникает спустя 24-48 ч после последней дозы и этот синдром достигает максимума интенсивности примерно на третий день и может не снижаться вплоть до третьей недели. Опиоидная физиологическая зависимость (т.е. наркомания) характеризуется попытками разыскать наркотик с целью достижения состояния эйфории и избежать, например, мер психосоциоэкономического осуждения. Наркоман будет продолжать употреблять наркотики (опиоиды) с нелечебными целями и несмотря на вред, причиняемый самому себе. С точки зрения фармакологии антагонисты опиоидов обычно блокируют или делают обратимым характер всех эффектов опиоидных агонистов. Один вариант использования опиоидных антагонистов состоит в однократном в течение дня приеме налтрексона, чтобы блокировать эйфорические эффекты, которые могут в противном случае развиться при введении опиоидов наркоманам. Небольшие дозы опиоидных антагонистов используются для определения, имеется ли у человека физическая зависимость от опиоидов. Наиболее часто опиоидные антагонисты применяются, чтобы сделать обратимым действие опиоидов у лиц, получивших передозировку лекарственных средств, включающих опиоидные агонисты. До настоящего момента делались попытки контролировать возможность неправильного употребления лекарственных средств на основе опиоидных анальгетиков. Обычно отдельная доза опиоидного анальгетика является более действенной при парантеральном введении по сравнению с той же дозой, введенной орально. Поэтому популярный способ злоупотребления медикаментами, предназначенными для применения оральным путем, включает экстракцию опиоида из лекарственной формы с последующей инъекцией опиоида (с использованием любого “подходящего” для инъекции средства) с целью достижения “подъема”. Попытки ограничить эти злоупотребления поэтому обычно сконцентрированы на включении в лекарственную форду для орального применения опиоидного антагониста, который орально не активен, но который будет заметно блокировать анальгетическое действие опиоида, если кто-то попытается растворить опиоид и ввести его парентерально. Например, комбинация пентазоцина и налоксона была использована в таблетках, продаваемых в Соединенных Штатах, под коммерческим названием Talwin  Nx от Sanofi-Winthrop. Talwin Nx содержит гидрохлорид пентазоцина в количестве, эквивалентном 50 мг основания, и гидрохлорид налоксона в количестве, эквивалентном 0,5 мг основания. Talwin Nx показан для ослабления боли от умеренной до сильной степени. Количество налоксона, содержащегося в этой композиции, не проявляет никакого действия при приеме оральным путем и не будет мешать проявлению фармакологического эффекта пентазоцина. Однако это же количество налоксона, введенного в виде инъекции, проявило антагонистический эффект по отношении к наркотическим анальгетикам. Так что включение налоксона имеет целью ограничить злоупотребление орально применяемым пентазоцином, что имеет место, когда дозированную форму растворяют и вводят в виде инъекции. Поэтому это лекарство обусловливает меньше возможностей для злоупотребления парентеральным путем по сравнению с известными ранее составами, включающими пентазоцин и применяемыми орально. Тем не менее оно еще остается уязвимым с точки зрения злоупотреблений и неправильного употребления оральным путем со стороны пациентов, например пациента, имеющего многократную дозу одномоментно.
Sunshine, et al. “Analgesic Efficacy of Pentazocine Versus a Pentazocine-Naloxone Combination Following Oral Administration”, Clin. J. Pain, 1988:4:35-40, сообщили о влиянии добавления 0,5 мг налоксона на анальгетическое действие пентазоцина в количестве 50 мг. Было установлено, что эта комбинация значительно менее эффективна, чем пентазоцин, в отношении суммарной разности интенсивности боли (SPID) и в отношении остаточной боли и разности интенсивности боли (PID) на четвертый час.
Применительно к пациентам с умеренным порогом болевой чувствительности эта комбинация обеспечивала значительно меньшее ослабление боли, чем пентазоцин в отношении SPID и в отношении остаточной боли и PID на 3-й и 4-й час. Для пациентов с высоким порогом чувствительности (испытывающим более жестокие боли) особой разницы между пентазоцином и комбинацией пентазоцина с налоксаном не наблюдалось.
Wang, et al. “Grossover and Parallel Study of Oral Analgesics”, J.Clin Pharmacol 1981; 21:162-8, изучали комбинацию налоксона 0,25 мг и Percodan (состоящего из 4,5 мг оксикодон-HCl, 0,28 мг терефталата оксикодона, 224 мг аспирина, 160 мг фенацетина и 32 мг кофеина) в сравнении с Percodan самим по себе и плацебо в перекрестном опыте с пациентами, испытывающими хронические боли. Эта комбинация имела более низкие показатели по сравнению с одним Percodan для большей части ежечасных измерений анальгетической активности в более поздние часы испытания. В то же время, что касается итоговых величин, комбинация не показала значительной разницы по сравнению с плацебо и с Percodan.
Связанная композиция бупренорфина и налоксона для обезболивания была представлена в 1991 г. в Новой Зеландии Temgesig Nx, Reckitt & Colman.
Лечение связанной композицией, содержащей тилидин (50 мг) и налоксон (4 мг), было испытано в Германии для контроля за сильными болями, начиная с 1978 г. (ValoronN, Goedecke). Рациональное значение композиции из этих лекарств заключается в эффективном обезболивании и предотвращении наркотической зависимости от тилидина благодаря индуцируемому налоксаном антагонизму со стороны рецептора морфина.
Патент US 3773955 (Pachter, et al.) описывает оральные композиции анальгического действия, которые при парентеральном применении не вызывают обезболивания, эйфории или физической зависимости и предотвращают тем самым парентеральное использование (злоупотребление) этими анальготическими средствами. Такие композиции содержали от около 0,1 мг до около 10 мг налоксона в одной обезболивающей оральной дозе. В этой ссылке не сообщается о злоупотреблениях в результате орального приема опиоидов.
В патенте US 3493657 (Lewenstein, et al.) описаны композиции, содержащие налоксон и морфин или оксиморфон, о которых сообщалось, что они обеспечивают сильный обезболивающий эффект, не сопровождающийся побочными эффектами, такими как галлюцинации.
В патенте US 4457933 (Gordon, et al) описан способ уменьшения возможности неправильного употребления оральным или парентеральным путем сильнодействующих анальгетических средств, таких как оксикодон, пропоксифен и пентазоцин, сочетанием анальгетически действенной дозы опиоида с налоксоном в определенном, относительно узком интервале соотношений. Предпочтительными были композиции оксикодона и налоксона с соотношением (по весу) 2,5-5:1 и пентазоцина и налоксона с соотношением (по весу) 16-50:1. Дозу налоксона, которую нужно было сочетать с опиоидом, устанавливали такой, которая практически исключала возможность как орального, так и парентерального злоупотребления, не оказывая существенного влияния на проявляющееся орально анальгетическое действие.
В US патенте 4582835 (Lewis) описан способ обезболивания путем введения подъязычно эффективной дозы бупренорфина с налоксоном. Lewis приводит соотношение налоксона к бупренорфину в дозировке как от 1:3 до 1:1 для парентерального введения и от 1:2 до 2:1 для подъязычного введения.
Все больше и больше признают тот факт, что применяемые орально опиоидные составы могут быть неправильно употреблены не только парентеральным путем, но и также с помощью и орального способа, когда пациент или наркоман самостоятельно принимают орально дозу, превышающую предназначенную дозу в рамках какого-то дозировочного интервала.
Объекты и краткая сущность изобретения
Цель изобретения – обеспечить создание лекарственной формы опиоидного анальгетика для орального применения, которая предназначена, чтобы уменьшить возможности злоупотребления, имеющие место при парентеральном способе введения, по сравнению с уже имеющимися в продаже лекарственными формами.
Другая цель изобретения – обеспечить способ обезболивания пациентов с помощью применяемой орально лекарственной формы, содержащей опиоидный анальгетик, уменьшая при этом возможности парентерального злоупотребления лекарственной формой.
Еще одна цель изобретения – разработать способ изготовления лекарственной формы опиоидного анальгетика для орального применения с тем, чтобы она была менее уязвима с точки зрения парентерального и/или орального неправильного употребления.
Эти, а также другие цели изобретения достигаются использованием настоящего изобретения, которое направлено отчасти на способ уменьшения возможности неправильного употребления орально применяемой лекарственной формы опиоидного анальгетика, предусматривающий соединение анальгетически эффективного количества опиоидного агониста с опиоидным антагонистом в лекарственной форме для орального применения, которая будет нуждаться, по крайней мере, в двухступенчатом процессе экстракции для выделения опиоидного агониста; причем введенное количество опиоидного антагониста является практически таковым, которое обеспечивает подавление опиоидных эффектов при его совместном с опиоидным агонистом экстрагировании и парентеральном введении. Предпочтительно сочетание опиоидного агониста и опиоидного антагониста таково, что из лекарственной формы их можно экстрагировать только совместно, и для отделения их друг от друга требуется отдельная стадия экстракции. Например, и опиоидный агонист, и опиоидный антагонист могут быть растворимы в кислоте, и для их разделения необходимо использовать раствор с высоким значением рН. В одном предпочтительном варианте опиоидный агонист – битартрат гидрокодона, а опиоидный антагонист – гидрохлорид налтрексона, причем оба они, и гидрокодон, и налтрексон, растворяются при рН менее 8, и около 80% указанного гидрокодона и около 10% указанного налтрексона могут быть экстрагированы при высоком рН, например, значительно выше, чем рН 10, и предпочтительно выше рН 11.
В других вариантах опиоидный агонист – гидрохлорид гидроморфона, а опиоидный антагонист – гидрохлорид налтрексона, или опиоидный антагонист – гидрохлорид оксикодона, а опиоидный антагонист – гидрохлорид налтрексона; или опиоидный агонист – сульфат морфина, а опиоидный антагонист – гидрохлорид налтрексона.
В следующих вариантах способ далее предусматривает включение в лекарственную форму еще одного ингредиента, который делает отделение опиоидного агониста от опиоидного антагониста более трудным. К таким ингредиентам относятся желирующие агенты, воски или другие фармацевтически пригодные наполнители.
В других вариантах изобретения способ дополнительно предусматривает включение в процесс приготовления лекарственного средства дополнительной обработки, состоящей из одной или нескольких стадий, которая впоследствии должна затруднить отделение опиоидного агониста от опиоидного антагониста.
В некоторых предпочтительных вариантах способа опиоидгидрокодон, гидроморфон, оксикодон, морфин или их фармацевтически приемлемые соли.
В некоторых предпочтительных вариантах способа опиоидный агонист и опиоидный антагонист соединены в соотношении опиоидного антагониста к опиоидному агонисту (анальгетику), которое анальгетически эффективно при введении композиции орально, но зато обладает свойством вызывать отвращение у лиц с физической зависимостью. В этом смысле композиционный продукт (антагонист/агонист) может быть по существу лекарством для одной группы лиц (страдающих от боли пациентов), будучи при этом неприемлемым (до отвращения) для другой группы лиц (например, физически зависимых) при оральном способе введения одной и той же дозы или при превышении обычно предписываемой дозы, например, примерно в 2-3 раза, чем обычно предписываемая доза опиоида. Таким образом, эта лекарственная форма, предназначенная для орального введения, будет потенциально менее привлекательной для злоупотребления как при оральном приеме, так и при парентеральном введении. В тех вариантах, где опиоид – гидрокодон, а антагонист – налтрексон, отношение налтрексона к гидрокодону предпочтительно составляет от приблизительно 0,03-0,27:1 по весу и более предпочтительно от около 0,05-0,20:1 по весу. В тех вариантах, где опиоидный антагонист – налтрексон, а опиоидный агонист – гидроморфон, отношение налтрексона к гидроморфону предпочтительно составляет от около 0,148:1 до около 1,185:1, и более предпочтительно от около 0,222:1 до около 0,889:1. В тех вариантах, где опиоидный антагонист – налтрексон, а опиоидный агонист – морфин, отношение налтрексона к морфину предпочтительно составляет от около 0,018:1 до около 0,148:1, и более предпочтительно от около 0,028:1 до около 0,111:1. В тех вариантах, где опиоидный антагонист – налтрексон, а опиоидный агонист – оксикодон, отношение налтрексона к оксикодону предпочтительно составляет от около 0,037:1 до около 0,296:1, и более предпочтительно от около 0,056:1 до около 0,222:1.
Лекарственные формы настоящего изобретения могут быть жидкими, таблетками, составами, состоящими из мультичастиц, с использованием любых желаемых фармацевтически приемлемых наполнителей, известных специалисту в данной области. Однако предпочтительно, чтобы опиоидный агонист и опиоидный антагонист были включены в принимаемую орально лекарственную форму таким образом, который не позволял бы легко разделить оба лекарства.
В некоторых вариантах эти лекарственные средства настоящего изобретения представляют собой непрерывно выделяющиеся составы. Это может быть достигнуто, например, включением непрерывно выделяющего носителя, содержащего опиоидный агонист и опиоидный антагонист, внутрь матрицы; или же нанесение покрытия из вещества, непрерывно выделяющего опиоидный агонист и опиоидный антагонист, где непрерывно выделяющее покрытие состоит, по крайней мере, из части непрерывно выделяющего носителя, входящего в состав лекарственного средства. В любом случае предпочтительно непрерывно выделяющийся препарат был приготовлен таким образом, при котором опиоидный агонист и опиоидный антагонист соединены в матрице или диспергированы так, чтобы наркоману нужно было применять методы экстракции при разделении этих лекарств.
Настоящее изобретение относится также к способу обезболивания, применяемому для людей, который до некоторой степени ограничивает вероятность орального злоупотребления опиоидными анальгетиками и который предусматривает введение пациенту – человеку оральной лекарственной формы на основе заявленных композиций опиоидного агониста и опиоидного антагониста, для экстракции которых необходимы, по крайней мере, две отдельные стадии экстракции.
В некоторых вариантах опиоидный антагонист включают в количестве (i), которое не должно вызывать уменьшения уровня обезболивания, обусловленного данной лекарственной формой при ее оральном применении до нетерапевтического уровня, и (ii), которое обеспечивает, по меньшей мере, легкое чувство отвращения у лиц с физической зависимостью (например, рвотный абстинентный синдром), если кто-то попытается принять, по крайней мере, удвоенную дозу за один раз (и часто дозу, превышающую обычную в 2-3 раза или больше) по сравнению с сопоставимой дозой опиоида в отсутствие опиоидного антагониста. В некоторых предпочтительных вариантах количество налтрексона, включаемого в лекарственную форму для орального применения, является менее позитивно укрепляющим (например, менее “любимым”) для наркомана с нефизической опиоидной зависимостью, чем аналогичная оральная лекарственная форма без включения антагониста. Предпочтительно технология приготовления лекарственного средства обеспечивает эффективное обезболивание при оральном применении.
В некоторых предпочтительных вариантах способ также предусматривает включение опиоидного агониста и опиоидного антагониста в лекарственную форму, которая содержит непрерывно выделяющий носитель, либо включенный в матрицу, либо в качестве непрерывно выделяющего покрытия, так что оральная лекарственная форма может вводиться один или два раза в день.
Фармацевтические композиции для орального использования, используемые в способах настоящего изобретения, могут быть в форме таблеток, лепешек, леденцов, водных или масляных суспензий, дисперсных порошков или гранул, эмульсий, твердых или мягких капсул или сиропов, или эликсиров, микрочастиц (например, микрокапсул, микросфер и т.п.), защечных таблеток и т.д.
Термин “парентерально” в данном здесь смысле включает подкожные инъекции, методы внутривенного, внутримышечного, внутригрудинного инъецирования или инфузирования.
Термин “эффективное обезболивание” для целей настоящего изобретения определяется как удовлетворительное уменьшение или устранение боли наряду с приемлемым уровнем побочных эффектов при определении этого пациентом-человеком. Признают, что соотношения опиоидного антагониста и опиоидного агониста, включенные в некоторые варианты изобретения (например, где опиоидный агонист включается в количестве (i), которое не вызывает уменьшение уровня обезболивания, обуславливаемого лекарственной формой при ее оральном применении до нетерапевтического уровня, и (ii), которое обеспечивает, по крайней мере, легкое чувство отвращения у лиц с физической зависимостью, если принято большое количество опиоида, например в 2-3 раза превышающее обычную дозу, и введено орально лицом с физической зависимостью), может уменьшать анальгезию до некоторой степени, если лекарственная форма введена орально, что определяется при непосредственной оценке пациентов или использованием одного или нескольких критериев замещения опиоидной анальгетической действенности в отношении человеческих существ, таких как Visual Analogue Scale (VAS) на “лекарственное действие”. Пациент, страдающий от боли, может заметить или может не заметить ощутимой разницы между составом, вводимым согласно такому варианту изобретения, и аналогичным составом, который включает ту же самую дозу опиоидного агониста без опиоидного антагониста, но будет получать облегчение боли от такой комбинации. Показатели замещения опиоидной действенности (обезболивание) включают седативный эффект, частоту дыхания и/или размер зрачков (посредством пупиллометрии) и аналоговую шкалу (“VAS”) для “действия лекарства”. В таких вариантах такие показатели замещения изменены из-за сниженного опиоидного эффекта по сравнению с такой же дозой опиоида без сопутствующей дозы опиоидного антагониста. Фармакодинамический эффект (анальгезия) составов, введенных в соответствии с изобретением, может быть описан с помощью заметок, взятых из ответов из анкет пациентов, отражающих эффект обезболивания, сделанных по мере течения времени после введения лекарственной формы. Суммарные оценки обезболивания включают сумму разности интенсивности боли (SPID) и полное исчезновение боли (ТОТ PAR).
Термин “непрерывное выделение” означает для целей настоящего изобретения выделение лекарства (опиоидного анальгетика) из трансдермального состава при такой скорости, когда его концентрация (уровень) в крови (например, в плазме) сохраняется на терапевтическом уровне (выше минимума эффективной обезболивающей концентрации или “МЕАС”), но ниже токсических уровней в течение периода времени, характерного для состава, применяемого дважды или один раз в день.
Для целей настоящего изобретения термин “опиоидный агонист” равнозначен термину “опиоид” или “опиоидный анальгетик” и сюда же следует включать опиоид как основание, его фармацевтически приемлемые соли, его стереоизомеры, его простые и сложные эфиры, смешанные агонист-антагонисты и частичные агонисты.
Для целей настоящего изобретения под термином “опиоидный антагонист” следует подразумевать основание, его фармацевтически приемлемые соли, его стереоизомеры, эфиры и их смеси.
Подробное описание изобретения
Принято считать, что существует, по крайней мере, три подвида опиоидных рецепторов, обозначенных “мю”, “каппа” и “дельта”. В рамках этой системы считают, что мю-рецептор участвует в процессе суперспинального обезболивания, угнетения дыхания, эйфории и физической зависимости. Каппа-рецептор, как полагают, участвует в процессе спинального обезболивания, миоза и седативного эффекта. Активация гамма-рецепторов вызывает дисфорию и галлюцинации, а также эффекты респираторного и вазомоторного стимулирования. Рецептор, отличающийся от “мю-рецептора” и обозначенный “гамма”, – в vas deferens мыши был описан в (Lord, et al. Nature, 1977, 267, 495-99). Полагают, что опиоидные агонисты проявляют свои агонистные действия сначала в отношении мю-рецептора и в меньшей степени в отношении каппа-рецептора. Появились несколько лекарств, действующие как частичные агонисты в отношении одного типа рецептора или другого. Такие лекарства проявляют максимальный эффект. Такие лекарства включают налорфин, пропирам и бупренорфин. Однако другие лекарства действуют как конкурирующие антагонисты на мю-рецептор и тормозят действие морфиноподобных лекарств, действуя как агонисты на каппа- и омега-рецепторы. Термин “агонист-антагонист” возник с целью описания таких механизмов действия. Считается, что концепция антагонизма применительно к действиям, производимым опиоидами, носит сложный характер.
Было установлено, что при введении опиоидных агонистов-антагонистов и частичных агонистов развивается толерантность к действию агонистов, но не к действию антагонистов в лекарствах. Даже после продолжительного введения высоких доз прерывание введения налоксона не характеризуется каким-либо заметным синдромом отмены, а отмена налтрексона, другого относительно чистого опиоидного антагониста приводит к этим самым признакам и симптомам. Тем не менее, после продолжительного приема высоких доз резкая отмена опиоидных агонистов-антагонистов налорфина или циклазоцина вызывает характерный синдром отмены, как в случае обоих этих лекарств.
Налоксон – опиоидный антагонист, который почти не обладает агонистическим действием. Подкожные дозы вплоть до 12 мг налоксона вызывают незаметные субъективные эффекты, а 24 мг налоксона вызывают только небольшую сонливость. Маленькие дозы (0,4-0,8 мг) налоксона, введенные внутримышечно или внутривенно человеку, предотвращают или быстро делают обратимым действие морфиноподобного опиоидного агониста. Сообщается, что 1 мг налоксона, введенного внутривенно, абсолютно блокирует эффект от 25 мг героина. Эффект от налоксона виден сразу же после внутривенного введения. Лекарство абсорбируется после орального введения, но нет сообщений о его быстром переходе в неактивную форму при первом его прохождении через печень, так что сообщалось, что только одна пятидесятая его часть является такой же сильнодействующей, как и при парентеральном введении. Сообщается, что оральная доза менее чем 1 г почти полностью метаболизируется менее чем через 24 часа.
Другие опиоидные антагонисты, например циклазоцин и налтрексон, оба имеют циклопропилметильные заместители у атома азота и сохраняют свою эффективность при оральном приеме и продолжительность их действия гораздо больше, достигая 24 часов после орального применения доз. Самый предпочтительный опиоидный антагонист – налтрексон. Однако равноантагонистичные оральные дозы других опиоидных антагонистов, включая (но без ограничения ими) налоксон, налмефен, циклазоцин и леваллорфан, могут использоваться согласно настоящему изобретению. Соотношение таких других антагонистов к частичному опиоидному агонисту может быть легко определено без какого-либо экспериментирования любым специалистом в данной области, кто желает использовать не налтрексон, а другой какой-нибудь опиоидный антагонист, соотношение которого к опиоидным антагонистам показано здесь в виде примеров и обсуждается детально. Специалисты в данной области могут определить такие соотношения других антагонистов к опиоидным агонистам, например, проведением таких же самых или подобных клинических исследований, отраженных в прилагаемых примерах. Таким образом, считается, что сочетание опиоидных антагонистов/опиоидных агонистов, которые вводят орально в соотношениях, эквивалентных соотношению, например, налтрексона к гидрокодону, изложенные здесь, соответствуют объему настоящего изобретения и объему формулы изобретения. Например, в некоторых вариантах изобретения налоксон используется в качестве опиоидного антагониста, количество налоксона, включаемое в лекарственную форму, должно быть достаточно велико для обеспечения равноантагонистичного эффекта, как если бы налтрексон был включен в состав.
При лечении пациентов, до того злоупотреблявших опиоидами, налтрексон назначают орально в больших дозах (свыше 100 мг), чтобы предотвратить вызывающее эйфорию действие опиоидных агонистов. Сообщается, что налтрексон проявляет сильный блокирующий эффект преимущественно в отношении мю-сайтов по сравнению с дельта-сайтами. Налтрексон известен как синтетический представитель того же ряда, что и оксиморфон, не обладающий свойствами опиоидного агониста, но отличающийся по структуре от оксиморфона заменой метильной группы у атома азота оксиморфона на циклопропилметильную группу. Солянокислая соль налтрексона растворима в воде почти до 100 мг/см3. Фармакологические и фармакокинетические свойства налтрексона изучались на различных животных и в клинике. См., например, Gonzalez J.P., et al. Naltrexone: A review of its Pharmacodynamic and Pharmacokinetic Properties and Therapeutic Efficacy in the Management of Opioid Dependence. Drugs 1988; 35:192-213, включенную здесь в качестве ссылки. После орального введения налтрексон быстро абсорбируется (в течение 1 часа) и является орально биодоступным, варьируя от 5-40%. Процент связывания налтрексона белками составляет приблизительно 21%, а объем распределения после введения одной дозы составляет 16,1 л/кг.
Налтрексон продается в форме таблеток (Revia®, DuPont) для лечения алкогольной зависимости и для блокады экзогенно введенных опиоидов. См., например, Revia (таблетки соляно-кислого налтрексона). Physician’s Desk Reference 51st ed., Montvale, NJ. Medical Economics 1997; 51:957-959. Доза 50 мг Revia® блокирует фармакологические эффекты 25 мг в/в введенного героина в течение времени, не превышающего 24 часов.
Известно, что при совместном введении морфина, героина или других опиоидов на основе хронического состояния налтрексон блокирует развитие физической зависимости от опиоидов. Считают, что способ блокирования налтрексоном действия героина заключается в конкурентном связывании опиоидных рецепторов. Налтрексон используют для лечения наркотической зависимости путем полной блокады опиоидных рецепторов. Установлено, что наиболее успешное применение налтрексона в отношении наркомании – это благоприятный прогноз для наркоманов как часть всесторонней профессиональной или реабилитационной программы, включающей контроль за поведением и другие методы, повышающие степень соблюдения режима потребления лекарственных средств. Для лечения наркотической зависимости налтрексоном желательно, чтобы пациент не принимал опиоиды в течение, по крайней мере, 7-10 дней. Начальная доза налтрексона для таких целей обычно составляет около 25 мг, и если нет признаков синдрома отмены, доза может быть увеличена до 50 мг/день. Считают, что дневная доза в 50 мг обеспечивает адекватную клиническую блокаду действия парентерально введенных опиоидов. Налтрексон применяется также для лечения алкоголизма как вспомогательное средство вместе с методами социальной и психотерапии.
В лекарственных формах и способах изобретения количество включенного налтрексона значительно меньше, чем в коммерчески доступных на сегодня лекарственных формах. Отчасти это потому, что использование налтрексона в настоящем изобретении иное: задача не в том, чтобы блокировать действие опиоидов, а скорее в том, чтобы обеспечить негативный, вызывающий отвращение опыт, когда большое количество композиционного продукта, например, почти в 2-3 раза превышающее обычную дозу, принимается лицом со сформировавшейся физической зависимостью или вводится ему.
Таким образом, например, в составах настоящего изобретения, в которых опиоид представлен 15 мг битартрата гидрокодона, количество солянокислого налтрексона, включаемое в состав, составляет от около 0,5 мг до около 4 мг, предпочтительно от около 0,75 мг до около 3 мг налтрексона на 15 мг гидрокодона.
Опиоидные анальгетики, которые используются в настоящем изобретении, включают все опиоидные агонисты или смешанные агонист-антагонисты, частичные агонисты, а именно (но без ограничения): альфентанил, аллилпродин, альфапродин, анилеридин, бензилморфин, безитрамид, бупренорфин, буторфанол, клонитазен, кодеин, дезоморфин, декстроморамид, дезоцин, диампромид, диаморфон, дигидрокодеин, дигидроморфин, дименоксадол, димефептанол, диметилтиамбутен, бутират диоксафетила, дипипанон, эптазоцин, этогептазин, этилметилтиамбутен, этилморфин, этонитазен, фентанил, героин, гидрокодон, гидроморфон, гидроксипетидин, изометадон, кетобемидон, леворфанол, левофенапилморфан, лофентанил, меперидин, мептазинол, метазоцин, метадон, метопон, морфин, мирофин, нарцеин, никоморфин, норлеворфанол, норметадон, налорфин, налбуфен, норморфин, норпипанон, опиум, оксикодон, оксиморфон, папаверетум, пентазоцин, фенадоксон, феноморфан, феназоцин, феноперидин, пиминодин, пиритрамид, профептазин, промедол, проперидин, пропоксифен, суфентанил, тилидин, трамадон, смеси любых из выше перечисленных веществ, их соли и т.п.
В некоторых предпочтительных вариантах опиоидный агонист или анальгетик выбирается из группы, включающей гидрокодон, морфин, гидроморфон, оксикодон, кодеин, леворфанол, меперидин, метадон или их соли, или их смеси. В определенных предпочтительных вариантах опиоидный агонист – гидрокодон. Эквианальгетические дозы этих опиоидов в сравнении с 15 мг дозой гидрокодона приведены в табл.1. Nx от Sanofi-Winthrop. Talwin Nx содержит гидрохлорид пентазоцина в количестве, эквивалентном 50 мг основания, и гидрохлорид налоксона в количестве, эквивалентном 0,5 мг основания. Talwin Nx показан для ослабления боли от умеренной до сильной степени. Количество налоксона, содержащегося в этой композиции, не проявляет никакого действия при приеме оральным путем и не будет мешать проявлению фармакологического эффекта пентазоцина. Однако это же количество налоксона, введенного в виде инъекции, проявило антагонистический эффект по отношении к наркотическим анальгетикам. Так что включение налоксона имеет целью ограничить злоупотребление орально применяемым пентазоцином, что имеет место, когда дозированную форму растворяют и вводят в виде инъекции. Поэтому это лекарство обусловливает меньше возможностей для злоупотребления парентеральным путем по сравнению с известными ранее составами, включающими пентазоцин и применяемыми орально. Тем не менее оно еще остается уязвимым с точки зрения злоупотреблений и неправильного употребления оральным путем со стороны пациентов, например пациента, имеющего многократную дозу одномоментно.
Sunshine, et al. “Analgesic Efficacy of Pentazocine Versus a Pentazocine-Naloxone Combination Following Oral Administration”, Clin. J. Pain, 1988:4:35-40, сообщили о влиянии добавления 0,5 мг налоксона на анальгетическое действие пентазоцина в количестве 50 мг. Было установлено, что эта комбинация значительно менее эффективна, чем пентазоцин, в отношении суммарной разности интенсивности боли (SPID) и в отношении остаточной боли и разности интенсивности боли (PID) на четвертый час.
Применительно к пациентам с умеренным порогом болевой чувствительности эта комбинация обеспечивала значительно меньшее ослабление боли, чем пентазоцин в отношении SPID и в отношении остаточной боли и PID на 3-й и 4-й час. Для пациентов с высоким порогом чувствительности (испытывающим более жестокие боли) особой разницы между пентазоцином и комбинацией пентазоцина с налоксаном не наблюдалось.
Wang, et al. “Grossover and Parallel Study of Oral Analgesics”, J.Clin Pharmacol 1981; 21:162-8, изучали комбинацию налоксона 0,25 мг и Percodan (состоящего из 4,5 мг оксикодон-HCl, 0,28 мг терефталата оксикодона, 224 мг аспирина, 160 мг фенацетина и 32 мг кофеина) в сравнении с Percodan самим по себе и плацебо в перекрестном опыте с пациентами, испытывающими хронические боли. Эта комбинация имела более низкие показатели по сравнению с одним Percodan для большей части ежечасных измерений анальгетической активности в более поздние часы испытания. В то же время, что касается итоговых величин, комбинация не показала значительной разницы по сравнению с плацебо и с Percodan.
Связанная композиция бупренорфина и налоксона для обезболивания была представлена в 1991 г. в Новой Зеландии Temgesig Nx, Reckitt & Colman.
Лечение связанной композицией, содержащей тилидин (50 мг) и налоксон (4 мг), было испытано в Германии для контроля за сильными болями, начиная с 1978 г. (ValoronN, Goedecke). Рациональное значение композиции из этих лекарств заключается в эффективном обезболивании и предотвращении наркотической зависимости от тилидина благодаря индуцируемому налоксаном антагонизму со стороны рецептора морфина.
Патент US 3773955 (Pachter, et al.) описывает оральные композиции анальгического действия, которые при парентеральном применении не вызывают обезболивания, эйфории или физической зависимости и предотвращают тем самым парентеральное использование (злоупотребление) этими анальготическими средствами. Такие композиции содержали от около 0,1 мг до около 10 мг налоксона в одной обезболивающей оральной дозе. В этой ссылке не сообщается о злоупотреблениях в результате орального приема опиоидов.
В патенте US 3493657 (Lewenstein, et al.) описаны композиции, содержащие налоксон и морфин или оксиморфон, о которых сообщалось, что они обеспечивают сильный обезболивающий эффект, не сопровождающийся побочными эффектами, такими как галлюцинации.
В патенте US 4457933 (Gordon, et al) описан способ уменьшения возможности неправильного употребления оральным или парентеральным путем сильнодействующих анальгетических средств, таких как оксикодон, пропоксифен и пентазоцин, сочетанием анальгетически действенной дозы опиоида с налоксоном в определенном, относительно узком интервале соотношений. Предпочтительными были композиции оксикодона и налоксона с соотношением (по весу) 2,5-5:1 и пентазоцина и налоксона с соотношением (по весу) 16-50:1. Дозу налоксона, которую нужно было сочетать с опиоидом, устанавливали такой, которая практически исключала возможность как орального, так и парентерального злоупотребления, не оказывая существенного влияния на проявляющееся орально анальгетическое действие.
В US патенте 4582835 (Lewis) описан способ обезболивания путем введения подъязычно эффективной дозы бупренорфина с налоксоном. Lewis приводит соотношение налоксона к бупренорфину в дозировке как от 1:3 до 1:1 для парентерального введения и от 1:2 до 2:1 для подъязычного введения.
Все больше и больше признают тот факт, что применяемые орально опиоидные составы могут быть неправильно употреблены не только парентеральным путем, но и также с помощью и орального способа, когда пациент или наркоман самостоятельно принимают орально дозу, превышающую предназначенную дозу в рамках какого-то дозировочного интервала.
Объекты и краткая сущность изобретения
Цель изобретения – обеспечить создание лекарственной формы опиоидного анальгетика для орального применения, которая предназначена, чтобы уменьшить возможности злоупотребления, имеющие место при парентеральном способе введения, по сравнению с уже имеющимися в продаже лекарственными формами.
Другая цель изобретения – обеспечить способ обезболивания пациентов с помощью применяемой орально лекарственной формы, содержащей опиоидный анальгетик, уменьшая при этом возможности парентерального злоупотребления лекарственной формой.
Еще одна цель изобретения – разработать способ изготовления лекарственной формы опиоидного анальгетика для орального применения с тем, чтобы она была менее уязвима с точки зрения парентерального и/или орального неправильного употребления.
Эти, а также другие цели изобретения достигаются использованием настоящего изобретения, которое направлено отчасти на способ уменьшения возможности неправильного употребления орально применяемой лекарственной формы опиоидного анальгетика, предусматривающий соединение анальгетически эффективного количества опиоидного агониста с опиоидным антагонистом в лекарственной форме для орального применения, которая будет нуждаться, по крайней мере, в двухступенчатом процессе экстракции для выделения опиоидного агониста; причем введенное количество опиоидного антагониста является практически таковым, которое обеспечивает подавление опиоидных эффектов при его совместном с опиоидным агонистом экстрагировании и парентеральном введении. Предпочтительно сочетание опиоидного агониста и опиоидного антагониста таково, что из лекарственной формы их можно экстрагировать только совместно, и для отделения их друг от друга требуется отдельная стадия экстракции. Например, и опиоидный агонист, и опиоидный антагонист могут быть растворимы в кислоте, и для их разделения необходимо использовать раствор с высоким значением рН. В одном предпочтительном варианте опиоидный агонист – битартрат гидрокодона, а опиоидный антагонист – гидрохлорид налтрексона, причем оба они, и гидрокодон, и налтрексон, растворяются при рН менее 8, и около 80% указанного гидрокодона и около 10% указанного налтрексона могут быть экстрагированы при высоком рН, например, значительно выше, чем рН 10, и предпочтительно выше рН 11.
В других вариантах опиоидный агонист – гидрохлорид гидроморфона, а опиоидный антагонист – гидрохлорид налтрексона, или опиоидный антагонист – гидрохлорид оксикодона, а опиоидный антагонист – гидрохлорид налтрексона; или опиоидный агонист – сульфат морфина, а опиоидный антагонист – гидрохлорид налтрексона.
В следующих вариантах способ далее предусматривает включение в лекарственную форму еще одного ингредиента, который делает отделение опиоидного агониста от опиоидного антагониста более трудным. К таким ингредиентам относятся желирующие агенты, воски или другие фармацевтически пригодные наполнители.
В других вариантах изобретения способ дополнительно предусматривает включение в процесс приготовления лекарственного средства дополнительной обработки, состоящей из одной или нескольких стадий, которая впоследствии должна затруднить отделение опиоидного агониста от опиоидного антагониста.
В некоторых предпочтительных вариантах способа опиоидгидрокодон, гидроморфон, оксикодон, морфин или их фармацевтически приемлемые соли.
В некоторых предпочтительных вариантах способа опиоидный агонист и опиоидный антагонист соединены в соотношении опиоидного антагониста к опиоидному агонисту (анальгетику), которое анальгетически эффективно при введении композиции орально, но зато обладает свойством вызывать отвращение у лиц с физической зависимостью. В этом смысле композиционный продукт (антагонист/агонист) может быть по существу лекарством для одной группы лиц (страдающих от боли пациентов), будучи при этом неприемлемым (до отвращения) для другой группы лиц (например, физически зависимых) при оральном способе введения одной и той же дозы или при превышении обычно предписываемой дозы, например, примерно в 2-3 раза, чем обычно предписываемая доза опиоида. Таким образом, эта лекарственная форма, предназначенная для орального введения, будет потенциально менее привлекательной для злоупотребления как при оральном приеме, так и при парентеральном введении. В тех вариантах, где опиоид – гидрокодон, а антагонист – налтрексон, отношение налтрексона к гидрокодону предпочтительно составляет от приблизительно 0,03-0,27:1 по весу и более предпочтительно от около 0,05-0,20:1 по весу. В тех вариантах, где опиоидный антагонист – налтрексон, а опиоидный агонист – гидроморфон, отношение налтрексона к гидроморфону предпочтительно составляет от около 0,148:1 до около 1,185:1, и более предпочтительно от около 0,222:1 до около 0,889:1. В тех вариантах, где опиоидный антагонист – налтрексон, а опиоидный агонист – морфин, отношение налтрексона к морфину предпочтительно составляет от около 0,018:1 до около 0,148:1, и более предпочтительно от около 0,028:1 до около 0,111:1. В тех вариантах, где опиоидный антагонист – налтрексон, а опиоидный агонист – оксикодон, отношение налтрексона к оксикодону предпочтительно составляет от около 0,037:1 до около 0,296:1, и более предпочтительно от около 0,056:1 до около 0,222:1.
Лекарственные формы настоящего изобретения могут быть жидкими, таблетками, составами, состоящими из мультичастиц, с использованием любых желаемых фармацевтически приемлемых наполнителей, известных специалисту в данной области. Однако предпочтительно, чтобы опиоидный агонист и опиоидный антагонист были включены в принимаемую орально лекарственную форму таким образом, который не позволял бы легко разделить оба лекарства.
В некоторых вариантах эти лекарственные средства настоящего изобретения представляют собой непрерывно выделяющиеся составы. Это может быть достигнуто, например, включением непрерывно выделяющего носителя, содержащего опиоидный агонист и опиоидный антагонист, внутрь матрицы; или же нанесение покрытия из вещества, непрерывно выделяющего опиоидный агонист и опиоидный антагонист, где непрерывно выделяющее покрытие состоит, по крайней мере, из части непрерывно выделяющего носителя, входящего в состав лекарственного средства. В любом случае предпочтительно непрерывно выделяющийся препарат был приготовлен таким образом, при котором опиоидный агонист и опиоидный антагонист соединены в матрице или диспергированы так, чтобы наркоману нужно было применять методы экстракции при разделении этих лекарств.
Настоящее изобретение относится также к способу обезболивания, применяемому для людей, который до некоторой степени ограничивает вероятность орального злоупотребления опиоидными анальгетиками и который предусматривает введение пациенту – человеку оральной лекарственной формы на основе заявленных композиций опиоидного агониста и опиоидного антагониста, для экстракции которых необходимы, по крайней мере, две отдельные стадии экстракции.
В некоторых вариантах опиоидный антагонист включают в количестве (i), которое не должно вызывать уменьшения уровня обезболивания, обусловленного данной лекарственной формой при ее оральном применении до нетерапевтического уровня, и (ii), которое обеспечивает, по меньшей мере, легкое чувство отвращения у лиц с физической зависимостью (например, рвотный абстинентный синдром), если кто-то попытается принять, по крайней мере, удвоенную дозу за один раз (и часто дозу, превышающую обычную в 2-3 раза или больше) по сравнению с сопоставимой дозой опиоида в отсутствие опиоидного антагониста. В некоторых предпочтительных вариантах количество налтрексона, включаемого в лекарственную форму для орального применения, является менее позитивно укрепляющим (например, менее “любимым”) для наркомана с нефизической опиоидной зависимостью, чем аналогичная оральная лекарственная форма без включения антагониста. Предпочтительно технология приготовления лекарственного средства обеспечивает эффективное обезболивание при оральном применении.
В некоторых предпочтительных вариантах способ также предусматривает включение опиоидного агониста и опиоидного антагониста в лекарственную форму, которая содержит непрерывно выделяющий носитель, либо включенный в матрицу, либо в качестве непрерывно выделяющего покрытия, так что оральная лекарственная форма может вводиться один или два раза в день.
Фармацевтические композиции для орального использования, используемые в способах настоящего изобретения, могут быть в форме таблеток, лепешек, леденцов, водных или масляных суспензий, дисперсных порошков или гранул, эмульсий, твердых или мягких капсул или сиропов, или эликсиров, микрочастиц (например, микрокапсул, микросфер и т.п.), защечных таблеток и т.д.
Термин “парентерально” в данном здесь смысле включает подкожные инъекции, методы внутривенного, внутримышечного, внутригрудинного инъецирования или инфузирования.
Термин “эффективное обезболивание” для целей настоящего изобретения определяется как удовлетворительное уменьшение или устранение боли наряду с приемлемым уровнем побочных эффектов при определении этого пациентом-человеком. Признают, что соотношения опиоидного антагониста и опиоидного агониста, включенные в некоторые варианты изобретения (например, где опиоидный агонист включается в количестве (i), которое не вызывает уменьшение уровня обезболивания, обуславливаемого лекарственной формой при ее оральном применении до нетерапевтического уровня, и (ii), которое обеспечивает, по крайней мере, легкое чувство отвращения у лиц с физической зависимостью, если принято большое количество опиоида, например в 2-3 раза превышающее обычную дозу, и введено орально лицом с физической зависимостью), может уменьшать анальгезию до некоторой степени, если лекарственная форма введена орально, что определяется при непосредственной оценке пациентов или использованием одного или нескольких критериев замещения опиоидной анальгетической действенности в отношении человеческих существ, таких как Visual Analogue Scale (VAS) на “лекарственное действие”. Пациент, страдающий от боли, может заметить или может не заметить ощутимой разницы между составом, вводимым согласно такому варианту изобретения, и аналогичным составом, который включает ту же самую дозу опиоидного агониста без опиоидного антагониста, но будет получать облегчение боли от такой комбинации. Показатели замещения опиоидной действенности (обезболивание) включают седативный эффект, частоту дыхания и/или размер зрачков (посредством пупиллометрии) и аналоговую шкалу (“VAS”) для “действия лекарства”. В таких вариантах такие показатели замещения изменены из-за сниженного опиоидного эффекта по сравнению с такой же дозой опиоида без сопутствующей дозы опиоидного антагониста. Фармакодинамический эффект (анальгезия) составов, введенных в соответствии с изобретением, может быть описан с помощью заметок, взятых из ответов из анкет пациентов, отражающих эффект обезболивания, сделанных по мере течения времени после введения лекарственной формы. Суммарные оценки обезболивания включают сумму разности интенсивности боли (SPID) и полное исчезновение боли (ТОТ PAR).
Термин “непрерывное выделение” означает для целей настоящего изобретения выделение лекарства (опиоидного анальгетика) из трансдермального состава при такой скорости, когда его концентрация (уровень) в крови (например, в плазме) сохраняется на терапевтическом уровне (выше минимума эффективной обезболивающей концентрации или “МЕАС”), но ниже токсических уровней в течение периода времени, характерного для состава, применяемого дважды или один раз в день.
Для целей настоящего изобретения термин “опиоидный агонист” равнозначен термину “опиоид” или “опиоидный анальгетик” и сюда же следует включать опиоид как основание, его фармацевтически приемлемые соли, его стереоизомеры, его простые и сложные эфиры, смешанные агонист-антагонисты и частичные агонисты.
Для целей настоящего изобретения под термином “опиоидный антагонист” следует подразумевать основание, его фармацевтически приемлемые соли, его стереоизомеры, эфиры и их смеси.
Подробное описание изобретения
Принято считать, что существует, по крайней мере, три подвида опиоидных рецепторов, обозначенных “мю”, “каппа” и “дельта”. В рамках этой системы считают, что мю-рецептор участвует в процессе суперспинального обезболивания, угнетения дыхания, эйфории и физической зависимости. Каппа-рецептор, как полагают, участвует в процессе спинального обезболивания, миоза и седативного эффекта. Активация гамма-рецепторов вызывает дисфорию и галлюцинации, а также эффекты респираторного и вазомоторного стимулирования. Рецептор, отличающийся от “мю-рецептора” и обозначенный “гамма”, – в vas deferens мыши был описан в (Lord, et al. Nature, 1977, 267, 495-99). Полагают, что опиоидные агонисты проявляют свои агонистные действия сначала в отношении мю-рецептора и в меньшей степени в отношении каппа-рецептора. Появились несколько лекарств, действующие как частичные агонисты в отношении одного типа рецептора или другого. Такие лекарства проявляют максимальный эффект. Такие лекарства включают налорфин, пропирам и бупренорфин. Однако другие лекарства действуют как конкурирующие антагонисты на мю-рецептор и тормозят действие морфиноподобных лекарств, действуя как агонисты на каппа- и омега-рецепторы. Термин “агонист-антагонист” возник с целью описания таких механизмов действия. Считается, что концепция антагонизма применительно к действиям, производимым опиоидами, носит сложный характер.
Было установлено, что при введении опиоидных агонистов-антагонистов и частичных агонистов развивается толерантность к действию агонистов, но не к действию антагонистов в лекарствах. Даже после продолжительного введения высоких доз прерывание введения налоксона не характеризуется каким-либо заметным синдромом отмены, а отмена налтрексона, другого относительно чистого опиоидного антагониста приводит к этим самым признакам и симптомам. Тем не менее, после продолжительного приема высоких доз резкая отмена опиоидных агонистов-антагонистов налорфина или циклазоцина вызывает характерный синдром отмены, как в случае обоих этих лекарств.
Налоксон – опиоидный антагонист, который почти не обладает агонистическим действием. Подкожные дозы вплоть до 12 мг налоксона вызывают незаметные субъективные эффекты, а 24 мг налоксона вызывают только небольшую сонливость. Маленькие дозы (0,4-0,8 мг) налоксона, введенные внутримышечно или внутривенно человеку, предотвращают или быстро делают обратимым действие морфиноподобного опиоидного агониста. Сообщается, что 1 мг налоксона, введенного внутривенно, абсолютно блокирует эффект от 25 мг героина. Эффект от налоксона виден сразу же после внутривенного введения. Лекарство абсорбируется после орального введения, но нет сообщений о его быстром переходе в неактивную форму при первом его прохождении через печень, так что сообщалось, что только одна пятидесятая его часть является такой же сильнодействующей, как и при парентеральном введении. Сообщается, что оральная доза менее чем 1 г почти полностью метаболизируется менее чем через 24 часа.
Другие опиоидные антагонисты, например циклазоцин и налтрексон, оба имеют циклопропилметильные заместители у атома азота и сохраняют свою эффективность при оральном приеме и продолжительность их действия гораздо больше, достигая 24 часов после орального применения доз. Самый предпочтительный опиоидный антагонист – налтрексон. Однако равноантагонистичные оральные дозы других опиоидных антагонистов, включая (но без ограничения ими) налоксон, налмефен, циклазоцин и леваллорфан, могут использоваться согласно настоящему изобретению. Соотношение таких других антагонистов к частичному опиоидному агонисту может быть легко определено без какого-либо экспериментирования любым специалистом в данной области, кто желает использовать не налтрексон, а другой какой-нибудь опиоидный антагонист, соотношение которого к опиоидным антагонистам показано здесь в виде примеров и обсуждается детально. Специалисты в данной области могут определить такие соотношения других антагонистов к опиоидным агонистам, например, проведением таких же самых или подобных клинических исследований, отраженных в прилагаемых примерах. Таким образом, считается, что сочетание опиоидных антагонистов/опиоидных агонистов, которые вводят орально в соотношениях, эквивалентных соотношению, например, налтрексона к гидрокодону, изложенные здесь, соответствуют объему настоящего изобретения и объему формулы изобретения. Например, в некоторых вариантах изобретения налоксон используется в качестве опиоидного антагониста, количество налоксона, включаемое в лекарственную форму, должно быть достаточно велико для обеспечения равноантагонистичного эффекта, как если бы налтрексон был включен в состав.
При лечении пациентов, до того злоупотреблявших опиоидами, налтрексон назначают орально в больших дозах (свыше 100 мг), чтобы предотвратить вызывающее эйфорию действие опиоидных агонистов. Сообщается, что налтрексон проявляет сильный блокирующий эффект преимущественно в отношении мю-сайтов по сравнению с дельта-сайтами. Налтрексон известен как синтетический представитель того же ряда, что и оксиморфон, не обладающий свойствами опиоидного агониста, но отличающийся по структуре от оксиморфона заменой метильной группы у атома азота оксиморфона на циклопропилметильную группу. Солянокислая соль налтрексона растворима в воде почти до 100 мг/см3. Фармакологические и фармакокинетические свойства налтрексона изучались на различных животных и в клинике. См., например, Gonzalez J.P., et al. Naltrexone: A review of its Pharmacodynamic and Pharmacokinetic Properties and Therapeutic Efficacy in the Management of Opioid Dependence. Drugs 1988; 35:192-213, включенную здесь в качестве ссылки. После орального введения налтрексон быстро абсорбируется (в течение 1 часа) и является орально биодоступным, варьируя от 5-40%. Процент связывания налтрексона белками составляет приблизительно 21%, а объем распределения после введения одной дозы составляет 16,1 л/кг.
Налтрексон продается в форме таблеток (Revia®, DuPont) для лечения алкогольной зависимости и для блокады экзогенно введенных опиоидов. См., например, Revia (таблетки соляно-кислого налтрексона). Physician’s Desk Reference 51st ed., Montvale, NJ. Medical Economics 1997; 51:957-959. Доза 50 мг Revia® блокирует фармакологические эффекты 25 мг в/в введенного героина в течение времени, не превышающего 24 часов.
Известно, что при совместном введении морфина, героина или других опиоидов на основе хронического состояния налтрексон блокирует развитие физической зависимости от опиоидов. Считают, что способ блокирования налтрексоном действия героина заключается в конкурентном связывании опиоидных рецепторов. Налтрексон используют для лечения наркотической зависимости путем полной блокады опиоидных рецепторов. Установлено, что наиболее успешное применение налтрексона в отношении наркомании – это благоприятный прогноз для наркоманов как часть всесторонней профессиональной или реабилитационной программы, включающей контроль за поведением и другие методы, повышающие степень соблюдения режима потребления лекарственных средств. Для лечения наркотической зависимости налтрексоном желательно, чтобы пациент не принимал опиоиды в течение, по крайней мере, 7-10 дней. Начальная доза налтрексона для таких целей обычно составляет около 25 мг, и если нет признаков синдрома отмены, доза может быть увеличена до 50 мг/день. Считают, что дневная доза в 50 мг обеспечивает адекватную клиническую блокаду действия парентерально введенных опиоидов. Налтрексон применяется также для лечения алкоголизма как вспомогательное средство вместе с методами социальной и психотерапии.
В лекарственных формах и способах изобретения количество включенного налтрексона значительно меньше, чем в коммерчески доступных на сегодня лекарственных формах. Отчасти это потому, что использование налтрексона в настоящем изобретении иное: задача не в том, чтобы блокировать действие опиоидов, а скорее в том, чтобы обеспечить негативный, вызывающий отвращение опыт, когда большое количество композиционного продукта, например, почти в 2-3 раза превышающее обычную дозу, принимается лицом со сформировавшейся физической зависимостью или вводится ему.
Таким образом, например, в составах настоящего изобретения, в которых опиоид представлен 15 мг битартрата гидрокодона, количество солянокислого налтрексона, включаемое в состав, составляет от около 0,5 мг до около 4 мг, предпочтительно от около 0,75 мг до около 3 мг налтрексона на 15 мг гидрокодона.
Опиоидные анальгетики, которые используются в настоящем изобретении, включают все опиоидные агонисты или смешанные агонист-антагонисты, частичные агонисты, а именно (но без ограничения): альфентанил, аллилпродин, альфапродин, анилеридин, бензилморфин, безитрамид, бупренорфин, буторфанол, клонитазен, кодеин, дезоморфин, декстроморамид, дезоцин, диампромид, диаморфон, дигидрокодеин, дигидроморфин, дименоксадол, димефептанол, диметилтиамбутен, бутират диоксафетила, дипипанон, эптазоцин, этогептазин, этилметилтиамбутен, этилморфин, этонитазен, фентанил, героин, гидрокодон, гидроморфон, гидроксипетидин, изометадон, кетобемидон, леворфанол, левофенапилморфан, лофентанил, меперидин, мептазинол, метазоцин, метадон, метопон, морфин, мирофин, нарцеин, никоморфин, норлеворфанол, норметадон, налорфин, налбуфен, норморфин, норпипанон, опиум, оксикодон, оксиморфон, папаверетум, пентазоцин, фенадоксон, феноморфан, феназоцин, феноперидин, пиминодин, пиритрамид, профептазин, промедол, проперидин, пропоксифен, суфентанил, тилидин, трамадон, смеси любых из выше перечисленных веществ, их соли и т.п.
В некоторых предпочтительных вариантах опиоидный агонист или анальгетик выбирается из группы, включающей гидрокодон, морфин, гидроморфон, оксикодон, кодеин, леворфанол, меперидин, метадон или их соли, или их смеси. В определенных предпочтительных вариантах опиоидный агонист – гидрокодон. Эквианальгетические дозы этих опиоидов в сравнении с 15 мг дозой гидрокодона приведены в табл.1.
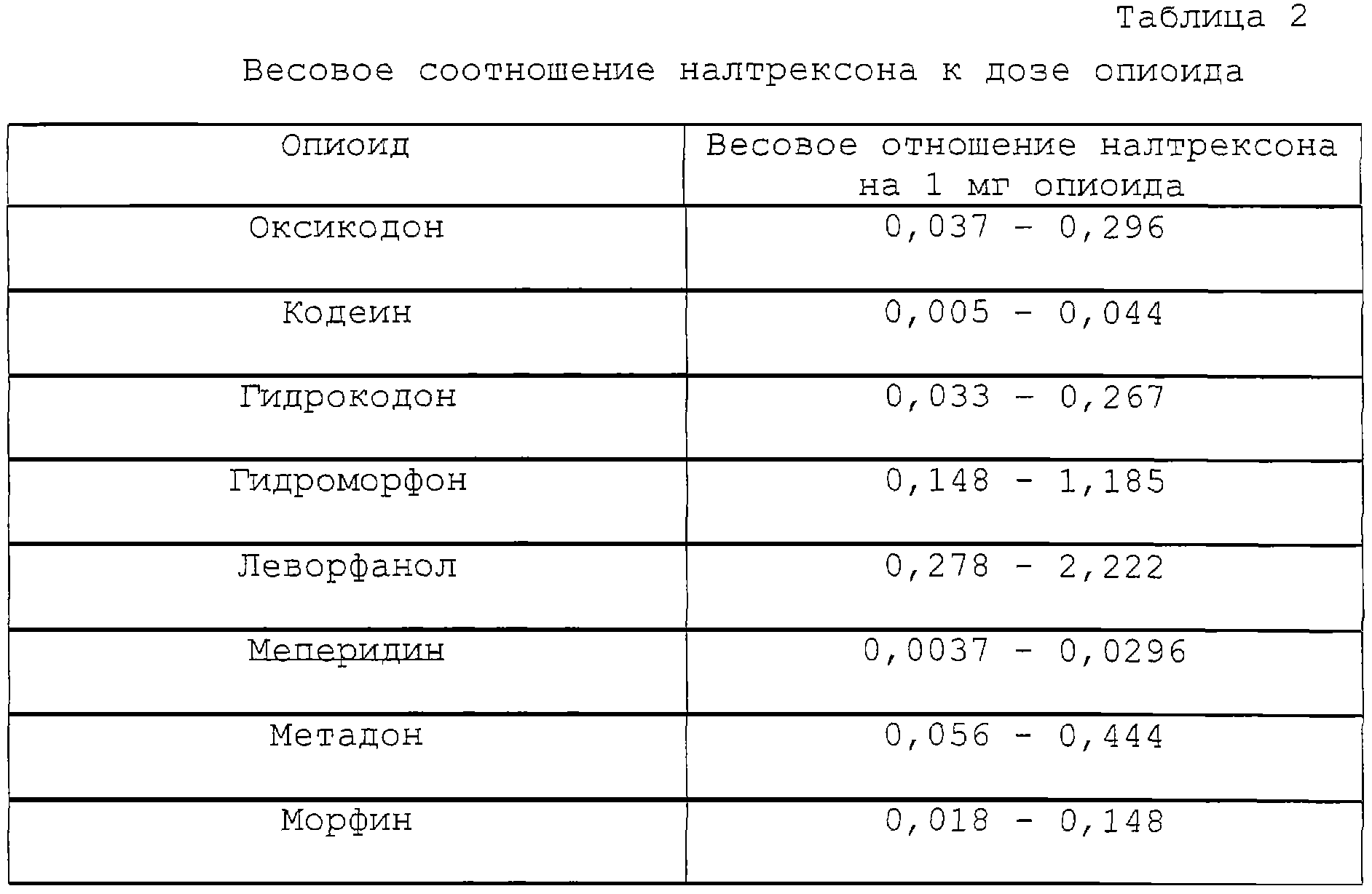
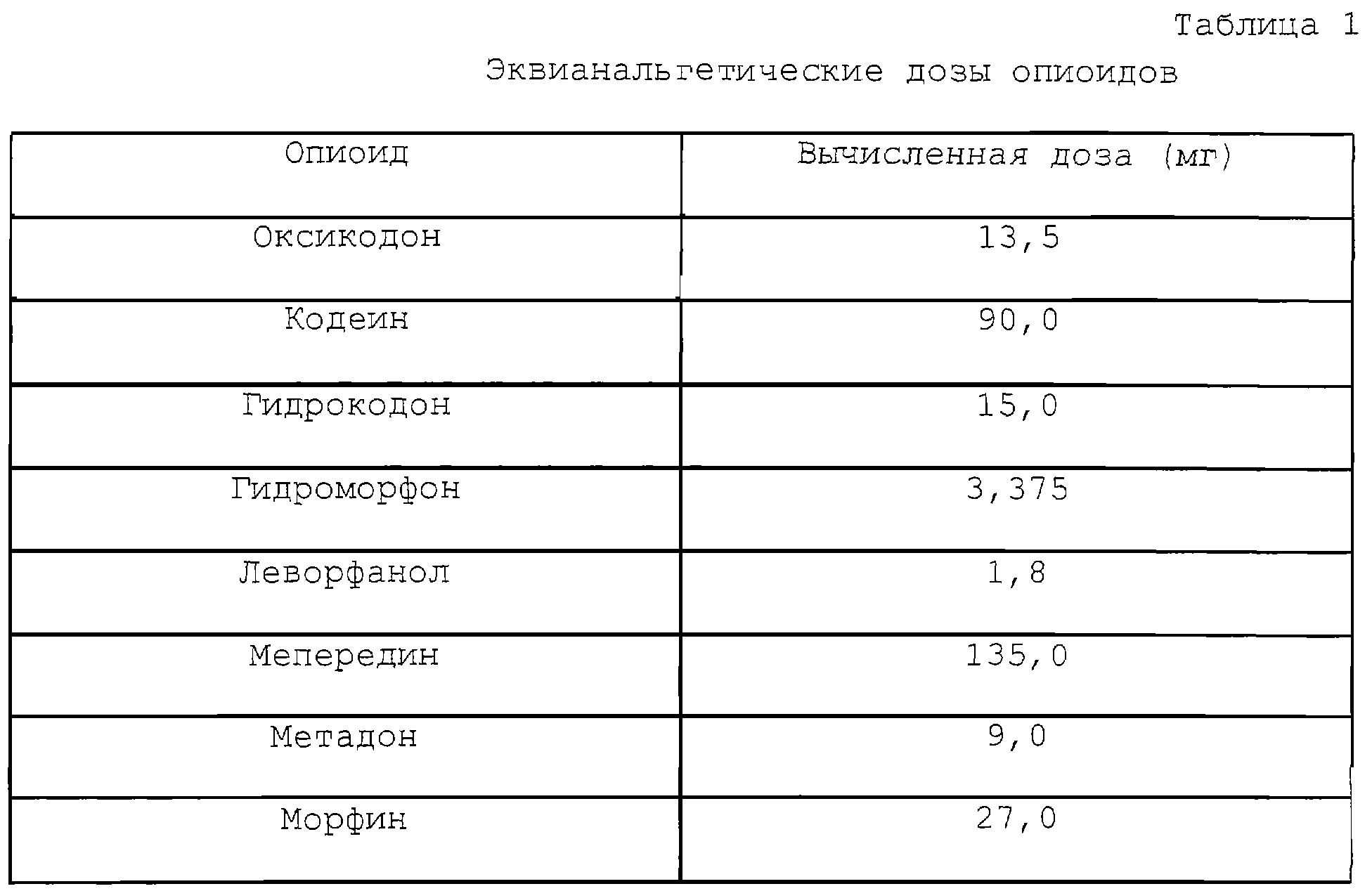 Исходя из предпочтительного соотношения налтрексона в количестве от около 0,5 до около 4 мг на 15 мг гидрокодона, соответствующее соотношение налтрексона на 1 мг каждого опиоида приведено в табл.2.
Исходя из предпочтительного соотношения налтрексона в количестве от около 0,5 до около 4 мг на 15 мг гидрокодона, соответствующее соотношение налтрексона на 1 мг каждого опиоида приведено в табл.2.
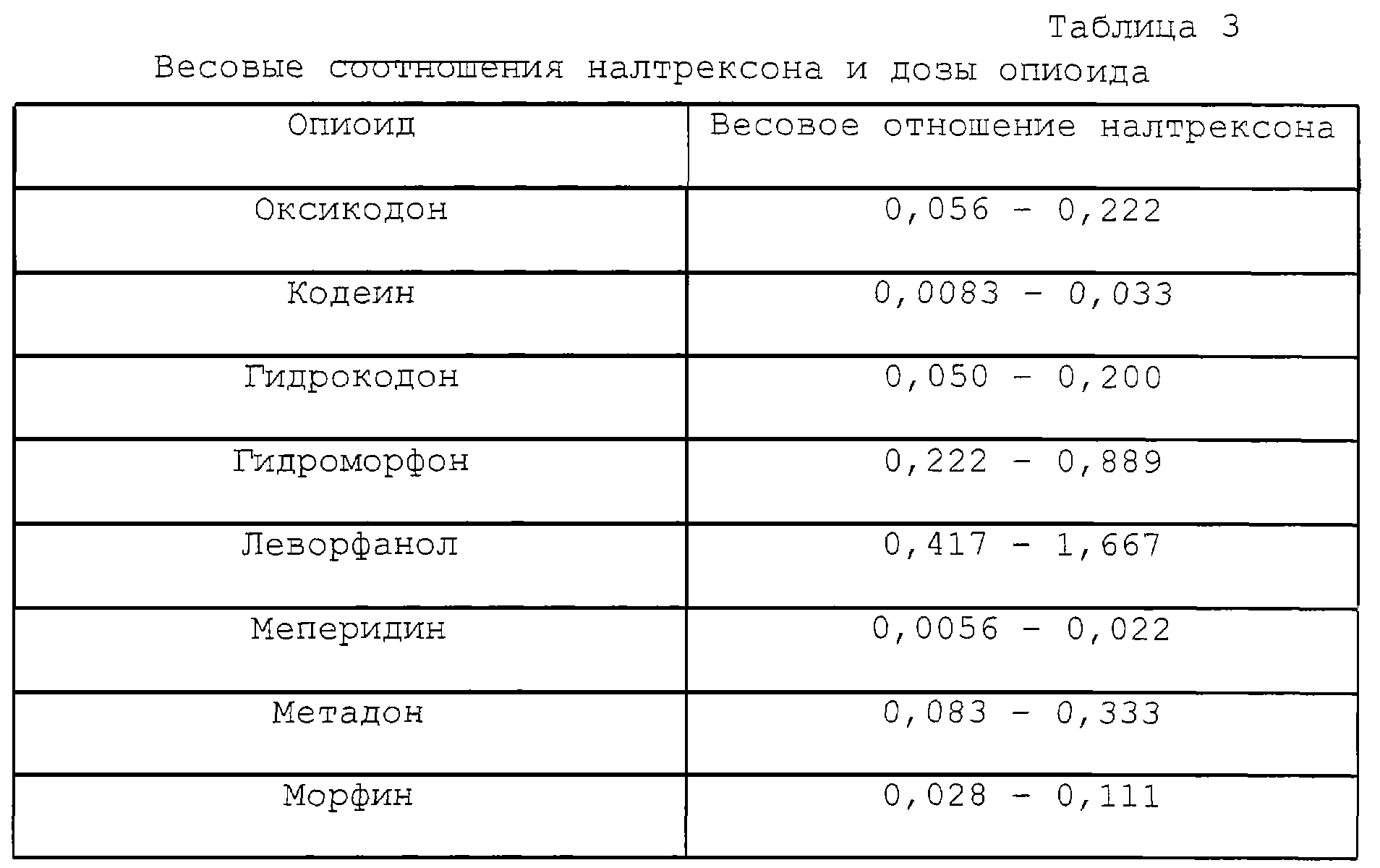
 Исходя из наиболее предпочтительного соотношения около 0,75 мг до около 3 мг налтрексона на 15 мг гидрокодона, соответствующее соотношение налтрексона к 1 мг каждого опиоида приводится в табл.3.
Исходя из наиболее предпочтительного соотношения около 0,75 мг до около 3 мг налтрексона на 15 мг гидрокодона, соответствующее соотношение налтрексона к 1 мг каждого опиоида приводится в табл.3.
 Хотя гидрокодон эффективно побеждает боль, имеет место повышение злоупотребления им со стороны лиц с физической зависимостью от опиоидов или тех, кто злоупотребляет опиоидами в нетерапевтических целях. Предыдущая практика с другими опиоидами показала, что есть возможность уменьшения их использования не по назначению, если вводить опиоиды в сочетании с антагонистом наркотика, особенно пациентам, которые являются бывшими наркоманами.
Weinhold L.L., et al. Buprenorphine Alone and in Combination with Naltrexone in Non-Dependent Humans, Drug and Alcohol Dependence 1992; 30:263-274; Mendelson J., et. al. Buprenorphine and Naloxone Interactions in Opiate-Dependent Volunteers, Clin Pharm Ther 1996; 60:105-114; приведенные здесь в качестве ссылки.
Гидрокодон представляет собой полусинтетический наркотик с обезболивающим и противокашлевым эффектом, оказывающий множественные эффекты на центральную нервную систему и желудочно-кишечный тракт. Химически гидрокодон представляет собой 4,5-эпокси-3-метокси-17-метилморфинан-6-он и известен, кроме того, как дигидрокодеинон. Подобно другим опиоидам, гидрокодон может иметь свойство формировать и может вызывать лекарственную зависимость морфинового типа. При избыточных дозах гидрокодон, как и другие опиаты, будет угнетать дыхание.
Гидрокодон для орального применения доступен также в Европе (Бельгии, Германии, Греции, Италии, Люксембурге, Норвегии и Швейцарии) как противокашлевое средство. Состав для парентерального применения есть также в Германии в качестве противокашлевого средства. Для использования в качестве анальгетика битартрат гидрокодона продается в Соединенных Штатах только в виде связанной композиции с неопиатными лекарствами (т.е. ибупрофен, ацетаминофен, аспирин и т.д.) для облегчения средней или умеренно-сильной боли.
Обычная лекарственная форма гидрокодона представляет собой комбинацию с ацетаминофеном и она продается, например, как Lortab в US от UCB Pharma, Ins. как таблетки 2,5/500 мг, 5/500 мг, 7,5/500 мг и 10/500 мг гидрокодона и ацетаминофена. Доступны также таблетки при соотношении 7,5 мг битартрата гидрокодона и 650 мг ацетаминофена и 7,5 мг битартрата гидрокодона и 750 мг ацетаминофена. Гидрокодон в сочетании с аспирином назначают орально взрослым, в основном, в дозе 1-2 таблетки каждые 4-6 часов, если требуется смягчить боль. Таблетка содержит 5 мг битартрата гидрокодона и 224 мг аспирина вместе с 32 мг кофеина или – 5 мг битартрата гидрокодона и 500 мг аспирина. Относительно новый состав включает битартрат гидрокодона и ибупрофен. Викопрофен, коммерчески доступный в US от Knoll Laboratories, представляет собой таблетку, содержащую 7,5 мг битартрата гидрокодона и 200 мг ибупрофена. Настоящее изобретение следует расценивать как охватывающее все такие составы, в которые включен орально активный опиоидный антагонист в пределах заявленных количеств, указанных ниже.
Возможность неправильного употребления опиоидных анальгетиков, например, гидрокодона, удивительно сокращается за счет предлагаемых композиций согласно настоящему изобретению. В частности, установлено, что можно соединить в отдельную оральную лекарственную форму опиоидный анальгетик вместе с небольшим количеством опиоидного антагониста, чтобы получить продукт, который не только обеспечит обезболивание, но и который значительно снизит возможность того, что человек с физической зависимостью будет продолжать злоупотребление лекарством, принимая больше одной таблетки за 1 раз, например в 2-3 раза больше обычно предназначаемой дозы.
Оральные лекарственные формы изобретения включают орально терапевтически эффективное количество опиоидного агониста с опиоидным антагинистом, таким как налтрексон, в количестве (i), которое не вызывает уменьшения уровня обезболивания, проявляемого лекарственной формой при оральном введении не терапевтического уровня, и (ii), которое обеспечивает по крайней мере неприятное чувство отвращения у лиц с физической зависимостью, например физически зависимых наркоманов (например, с отчетливым синдромом абстиненции) при употреблении за один раз дозы, превышающей предназначенную дозу. Предпочтительно количество антагониста, включенное в оральную лекарственную форму, является (iii) менее решительно используемым, т.е. менее “привлекательным” со стороны лиц с физической зависимостью, например, наркоманом, употребляющим опиаты, чем аналогичная оральная лекарственная форма, в которую антагонист не включен.
Количество антагониста, которое используют для выполнения условий (i)-(iii), приведенных в предшествующем абзаце, можно определить, по крайней мере, частично, с помощью “замещающих” тестов, таких как VAS шкала (когда субъект соотносит по шкале свое ощущение от действия лекарства) и/или путем измерения, например, размера зрачка (измеряемого с помощью пупиллометрии). Такие измерения позволяют специалисту в данной области определить дозу антагониста относительно дозы агониста, которая вызывает уменьшение опиатных эффектов агониста. Впоследствии специалист может определить уровень опиоидного антагониста, который вызывает эффект отвращения у лиц с физической зависимостью, а также уровень опиоидного антагониста, который минимизирует “оценки пристрастия” или поддерживающие свойства опиоида у наркоманов без физической зависимости. Если эти уровни опиоидного антагониста один раз определены, становится возможным определить диапазон дозировок антагониста вокруг или ниже того уровня, который следует поддерживать для выполнения условий (i)-(iii), указанных ранее в предшествующем абзаце.
Комбинации опиоидного агониста и опиоидного антагониста могут использоваться в смесях с общепринятыми наполнителями, т.е. фармацевтически приемлемыми веществами – носителями органической или неорганической природы, пригодными для использования орально, известными в данной области. Подходящие фармацевтически пригодные носители включают (но не ограничиваются ими): воду, солевые растворы, спирты, камеди, растительные масла, бензиловые спирты, полиэтиленгликоли, желатин, углеводы, такие как лактоза, амилоза или крахмал, стеарат магния, тальк, кремневую кислоту, вязкий парафин, ароматизированное масло, моноглицериды и диглицериды жирных кислот, эфиры пентаэритритола и жирных кислот, гидроксицеллюлозу, поливинилпирролидон и т.д. Фармацевтические препараты могут быть простерилизованы и, при желании, смешаны со вспомогательными средствами, например лубрикантами, консервантами и стабилизирующими добавками, увлажнителями, эмульгаторами, солями для воздействия на осмотическое давление буферов, веществами, улучшающими цвет, вкус и/или запах, и т.п. Они могут быть также скомбинированы как угодно с другими активными веществами, например другими анальгетиками. Для орального применения пригодны, в частности, таблетки, драже, растворы, капли, суппозитории или капсулы, таблетки с покрытием и желированные пилюли. Композиции, предназначенные для орального применения, могут быть получены любым методом, известным в данной области, и такие композиции могут содержать один или несколько агентов, выбранных из группы, состоящей из инертных фармацевтически нетоксичных наполнителей, которые пригодны для изготовления таблеток. Такие наполнители включают, например, инертный разбавитель, такой как лактоза; гранулирующие и дезинтегрирующие агенты, например кукурузный крахмал; связывающие агенты, например крахмал, средства для смазки, например стеарат магния. Таблетки могут быть без покрытия или с покрытием, нанесенным для придания изысканного вида или для уменьшения высвобождения активных ингредиентов. Составы для орального использования могут также представлять собой твердые желатиновые капсулы, в которых активный ингредиент смешан с инертным разбавителем.
Водные суспензии содержат вышеопределенную комбинацию лекарств, и эта смесь содержит один или несколько наполнителей, пригодных в качестве суспендирующих агентов, например фармацевтически приемлемые синтетические смолы, такие как гидроксипропилметилцеллюлоза или природные смолы (камеди). Масляные суспензии также могут быть составлены путем суспендирования вышеозначенной комбинации лекарств в растительном или минеральном масле. Масляные суспензии могут содержать загущающий агент, например, пчелиный воск или цетиловый спирт. Сироп, эликсир или что-то подобное могут использоваться тогда, когда требуется подслащивающий носитель. Суспензии для инъекций также могут быть приготовлены, в этом случае используются соответствующие жидкие носители, суспендирующие агенты и т.п.
Способ лечения и фармацевтические композиции настоящего изобретения могут далее включать одно или несколько лекарств в дополнение к опиоидному анальгетику и опиоидному антагонисту, причем дополняющие лекарства (лекарство) могут быть или могут не быть к тому же синергически действующими. Таким образом, в некоторых вариантах в состав может быть включена комбинация двух опиоидных анальгетиков в дополнение к опиоидному антагонисту. Например, лекарственная форма может включать два опиоидных анальгетика с разными свойствами, например, период полувыведения, растворимость, активность, и комбинацию из вышеприведенных веществ. В других же вариантах включен один или несколько опиоидных анальгетиков и также в дополнение к опиоидному антагонисту включено еще другое неопиоидное лекарство. Такие неопиоидные лекарства предпочтительно должны обеспечивать дополнительную анальгезию, это, например, аспирин, ацетаминафен, нестероидные противовоспалительные лекарства (“NSAIDS”), например ибупрофен, кетопрофен и т.д.; N-метил-D-аспартатные (NMDA) рецепторные антагонисты, например, морфинан, такой как декстрометорфан или декстрорфан или кетамин, ингибиторы циклооксигеназы – П (“СОХ-П ингибиторы”); и/или глициновые рецепторные антагонисты.
В некоторых предпочтительных вариантах настоящего изобретения предусматривается использование более низких доз опиоидного анальгетика благодаря включению дополнительного неопиоидного анальгетика, такого как NSAID или СОХ-2 ингибитор. При использовании меньших количеств каждого или обоих лекарств побочные эффекты, связанные с эффективным контролем боли у людей, снижаются.
Подходящие нестероидные противовоспалительные средства включают ибупрофен, диклофенак, напроксен, беноксапрофен, флурбипрофен, фенопрофен, флубуфен, кетопрофен, индопрофен, пиропрофен, карпрофен, оксапрозин, прамопрофен, муропрофен, триоксапрофен, супрофен, аминопрофен, тиапрофеновую кислоту, флупрофен, буклоксиновую кислоту, индометацин, сулиндак, толметин, зомепирак, тиопинак, зидометацин, ацеметацин, фентиазак, клиданак, оксипинак, мефенаминовую кислоту, меклофенаминовую кислоту, флуфенаминовую кислоту, нифлуминовую кислоту, толфенаминовую кислоту, дифлуризал, флуфенизал, пироксикам, судоксикам или изоксикам и т.п. Приемлемые дозы этих лекарств хорошо известны специалистам в данной области.
Антагонисты рецепторов на основе N-метиловой соли D-аспарагиновой кислоты (NMDA) хорошо известны в данной области и охватывают морфинаны, например, декстрометорфан или декстрорфан, кетамин, d-метадон или их фармацевтически приемлемые соли. Для целей настоящего изобретения считают, что термин “NMDA-антагонист” охватывает лекарства, которые блокируют главную внутриклеточную последовательность акивации NMDA-рецепторов, например, ганглиозид, такой как GTI или GMIb фенотиазин, например, трифлуоперазин или нафталинсульфонамид, такой как N-(6-аминотексил)-5-хлор-1-нафталинсульфонамид. Установлено, что эти лекарства ингибируют развитие толерантности и/или зависимости к наркотическим веществам, например наркотическим анальгетикам, таким как морфин, кодеин и т.д. (US патент 5321012 и 5556838 – Mayer et al., оба) и облегчают хронические боли (US патент 5502058 – Mayer et al.) (патенты упомянуты здесь в качестве ссылок). NMDA антагонист может быть включен как сам по себе, так и в сочетании о местным анестезирующим средством, например лидокаином, как описано в этих патентах Mayer et al.
Лечение хронической боли применением глициновых рецепторных антагонистов и идентификация подобных лекарственных средств описаны в US патенте 5514680 (Weber et al.), приведенном здесь в качестве ссылки.
Сообщалось о том, что известно, что СОХ-2 ингибиторы и другие химические структуры вызывают ингибирование циклооксигеназы-2. СОХ-2 ингибиторы описаны, например, в US патентах 5616601, 5604260, 5593994, 5550142, 5536752, 5521213, 5475995, 5639780, 5604253, 5552422, 5510368, 5436265, 5409944 и 5130311, каждый из которых упомянут здесь в качестве ссылки. Некоторые предпочтительные СОХ-2 ингибиторы включают целекоксиб (SC-58635), DUP-697, флосулид (CGP-28238), мелоксикам,6-метокси-2-нафтилуксусную кислоту (6-MNA), МК-966, набуметон (предшественник 6-MNA), нимесулид, NS-398, SC-5766, SC-58215. Т-614 или их комбинации. Уровни дозировки СОХ-2 ингибитора порядка от около 0,005 мг до около 140 мг на кг веса пациента в день являются терапевтически эффективными в сочетании с опиоидным анальгетиком. Или же около 0,25 мг до приблизительно 7 г на пациента в день СОХ-2 ингибитора вводят в сочетании с опиоидным анальгетиком.
В других вариантах в состав может включаться неопиоидное лекарство, обеспечивающее не обезболивание, а другой эффект, например противокашлевое, отхаркивающее, противоотечное, антигистаминное лекарство, анестетики местного действия и т.п.
Лекарственная форма для орального применения согласно изобретению может представлять собой гранулы, шарики, зернышки, драже (объединенные здесь под наименованием “мультичастицы”). Некое количество мультичастиц, которое необходимо, чтобы обеспечить желаемую дозу опиоида в течение какого-то периода времени, можно поместить в капсулу или включить в любую другую подходящую оральную твердую форму. Или же лекарственная форма для орального применения может быть в форме таблетки.
Контролируемое выделение лекарства из лекарственной формы
Композиция из опиоидного агониста и опиоидного антагониста может быть приготовлена как состав для орального применения с контролируемым или непрерывным выделением в виде подходящей таблетки, таблетки с покрытием или совокупности мультичастиц, известных специалисту в данной области. Непрерывно выделяющаяся лекарственная форма может необязательно включать носитель, обеспечивающий непрерывное выделение, который включен в матрицу вместе с опиоидным агонистом и опиоидным антагонистом, или же может быть применено покрытие с непрерывным выделением.
В вариантах, в которых опиоидный анальгетик представлен гидрокодоном, лекарственные формы для орального использования с непрерывным выделением могут включать дозы анальгетика от около 8 мг до около 50 мг гидрокодона на единицу дозировки. В лекарственных оральных формах с непрерывным выделением, где терапевтически активный опиоид – гидроморфон, включается от около 2 мг до около 64 мг соляно-кислого гидроморфона. В другом варианте опиоидный анальгетик включает морфин и лекарственные оральные формы с непрерывным выделением включают от около 2,5 мг до около 800 мг морфина по весу. В еще одном варианте опиоидный анальгетик включает оксикодон, и оральные лекарственные формы с непрерывным выделением включают от около 2,5 мг до около 800 мг оксикодона. Опиоидный анальгетик может включать трамадол, и оральные лекарственные формы с непрерывным выделением могут включать от около 20 мг до 800 мг трамадола на единицу дозировки. Лекарственная форма может содержать более одного опиоидного анальгетика, чтобы обеспечить в значительной мере эквивалентный терапевтический эффект. Или же лекарственная форма может содержать молярные эквивалентные количества других солей опиоидов, применяемых в настоящем изобретении.
В одном предпочтительном варианте настоящего изобретения лекарственное средство с непрерывным выделением включает такие частицы, содержащие или предусматривающие активный ингредиент, причем частицы имеют диаметр от около 0,1 мм до около 2,5 мм, предпочтительно от около 0,5 мм до около 2 мм.
Частицы предпочтительно покрывают пленкой из материала, допускающего выход комбинации опиоидного агониста/антагониста с постоянной скоростью выделения в водную среду. Пленочное покрытие выбирают таким образом, чтобы достигнуть в комбинации с другими установленными свойствами желаемой скорости высвобождения in vitro. Составы покрытия, допускающего непрерывное выделение, должны быть способны обеспечивать крепкую сплошную пленку, которая является однородной и качественной, способной удерживать подкрашивающие вещества и др. добавки, нетоксичной, инертной и нелипкой.
В некоторых вариантах частицы предусматривают матрицы, обеспечивающие среднее выделение, содержащие опиоидный анальгетик с опиоидным антагонистом.
Покрытия
Лекарственные формы настоящего изобретения могут необязательно иметь покрытие из одного или нескольких материалов, пригодных для регулирования выхода или для защиты состава. В одном варианте покрытия предусматриваются для того, чтобы обеспечить либо рН-зависимое, либо рН-независимое выделение, например, при выдерживании в гастроинтестинальной среде. рН-зависимое покрытие служит, чтобы обеспечить выделение опиоида в нужных участках желудочно-кишечного тракта (GI), например во рту или тонкой кишке, так что согласно профилю абсорбции оно способно обеспечить обезболивание у пациента в течение по крайней мере около 8 часов и предпочтительно около 12 часов и вплоть до около 24 часов. Когда требуется рН-независимое покрытие, покрытие создается таким, чтобы обеспечить оптимальное выделение независимо от изменений рН в окружающей жидкости, например, GI-тракте. Возможно также составление комбинаций, которые выделяют порцию дозы в одном нужном участке GI-тракта, например во рту, выделение оставшейся дозы в другом участке GI-тракта, например в тонкой кишке.
Составы, согласно изобретению, в которых использованы рН-зависимые покрытия, могут также обуславливать эффект повторного действия при том, что незащищенное лекарство покрывают покрытием для тонкого кишечника, и оно выделяется в желудке, тогда как остаток, будучи защищенным покрытием для тонкого кишечника, выделяется далее в желудочно-кишечном тракте. Покрытия, которые могут использоваться согласно изобретению, включают шеллак, ацетатфталат, целлюлозы (CAP), поливинилацетатфталат (pvap), фталат гидроксипропилметилцеллюлозы и сополимеры эфиров метакриловой кислоты, зеин и т.п.
В некоторых предпочтительных вариантах субстрат (т.е. зерно сердцевины таблетки, частицы матрицы), содержащий опиоидный анальгетик (с или без СОХ-2 ингибитора), покрывают гидрофобным материалом, выбранным из (i) алкилцеллюлозы; (ii) акрилового полимера; или (iii) их смесей. Покрытие может быть нанесено в виде органического или водного раствора или дисперсии. Покрытие может применяться для увеличения веса от около 2 до около 20% субстрата для того, чтобы получить желаемую кривую непрерывного выделения. Покрытия, полученные на основе водных дисперсий, описаны, например, детально в US патентах 5273760 и 5286493, поданных заявителем настоящего изобретения и включены здесь как ссылки.
Другие примеры составов и покрытий, обеспечивающих непрерывное выделение, которые могут использоваться в соответствии с настоящим изобретением, включают US патенты 5324351, 5356467 и 5472712 заявителя, включенные здесь целиком в виде ссылки.
Алкилцеллюлозные полимеры
Целлюлозные материалы и полимеры, включающие алкилцеллюлозы, представляющие собой гидрофобные материалы, очень подходят для покрытия крупинок согласно изобретению. Просто в качестве примера – один предпочтительный алкилцеллюлозный полимер – этилцеллюлоза, хотя любой специалист заметит, что и другие целлюлозные и/или алкилцеллюлозные полимеры можно легко использовать в одиночку или в любом сочетании как основу или часть гидрофобного покрытия согласно изобретению.
Одна коммерчески доступная водная дисперсия этилцеллюлозы – Aquacoat (FMC Corp., Philadelphia, Pennsylvania U.S.A.). Aquacoat готовится растворением этилцеллюлозы в не смешивающемся с водой органическом растворителе и эмульгированием ее в воду в присутствии поверхностноактивного вещества и стабилизатора. После гомогенизации с получением капелек субмикронного размера органический растворитель выпаривают под вакуумом и получают псевдолатекс. Пластификатор не вводится в псевдолатекс во время производства. Так что прежде, чем использовать его в качестве покрытия, необходимо тщательно перемешать Aquacoat с подходящим пластификатором перед использованием.
Другая водная дисперсия этилцеллюлозы коммерчески доступна как Surelease (Colorcon, Inc., West Point, Pennsylvania, U.S.А.). Этот продукт готовится при введении пластификатора в дисперсию во время процесса производства. Горячий расплав полимера, пластификатор (дибутиловый эфир себациновой кислоты) и стабилизатор (олеиновая кислота) подготавливают в качестве гомогенной смеси, которую затем разбавляют щелочным раствором с получением водной дисперсии, которую используют непосредственно в контакте с непокрываемым материалом.
Акриловые полимеры
В других предпочтительных вариантах настоящего изобретения гидрофобный материал, предусматривающий покрытие с контролируемой степенью выделения представляет собой приемлемый акриловый полимер, включая (но не ограничиваясь) сополимеры акриловой и метакриловой кислот, сополимеры метилметакрилата, этоксиэтилметакрилаты, цианоэтилметакрилат, полиакриловую кислоту, полиметакриловую кислоту, сополимер метакриловой кислоты и алкиламида, полиметилметакрилат, полиметакрилат, сополимер полиметилметакрилата, полиакриламид, сополимер аминоалкилметакрилата, ангидрид полиметакриловой кислоты и сополимеры глицидилметакрилата.
В некоторых предпочтительных вариантах акриловый полимер включает один или несколько сополимеров аммонийметакрилата. Сополимеры аммонийметакрилата хорошо известны в данной области и описываются в NF ХVIII как полностью полимеризованные сополимеры эфиров акриловой и метакриловой кислот с низким содержанием групп четвертичного аммония.
Для того чтобы получить желаемую кривую растворения, может быть необходимым включить два или несколько аммонийметакрилатных сополимеров, имеющих различные физические свойства, например, разные молярные соотношения между группами четвертичного аммония и нейтральными метакриловыми эфирами.
Некоторые полимеры типа эфиров метакриловых кислот используются для получения рН-зависимых покрытий, которые могут использоваться в соответствии с настоящим изобретением. Например, существует семейство сополимеров, синтезированных из диэтиламиноэтилметакрилата и других нейтральных метакриловых эфиров, известных также как сополимеры метакриловой кислоты и полимерные метакрилаты, коммерчески доступные как Eudragit от Rohm Tech, Inc. Существует несколько различных типов Eudragit. Например, Eudragifc E представляет собой сополимер метакриловой кислоты, который набухает и растворяется в кислых средах.
Eudragit L является сополимером метакриловой кислоты, который не набухает при рН приблизительно менее 5,7 и растворим при рН приблизительно более 6. Eudragit S не набухает при рН приблизительно менее 6,5 и растворим при рН приблизительно более 7. Eudragit RL и Eudragit RS набухают в воде, а количество воды, поглощенной этими двумя полимерами, зависит от рН, тем не менее лекарственные формы с покрытием из Eudragit RL и RS являются рН-независимыми.
В некоторых предпочтительных вариантах акриловое покрытие содержит смесь двух полимерных лаков на основе акрилатов, поставляемых Rohm Pharma под торговыми марками Eudragit RL 30D и Eudragit RS 30D соответственно. Eudragit RL 30D и Eudragit RS 30D являются сополимерами эфиров акриловой и метакриловой кислот с низким содержанием групп четвертичного аммония и с молярным соотношением групп аммония к остаткам нейтральных метакриловых эфирных групп как 1:20 в Eudragit RL 30D и 1:40 в Eudragit RS 30D. Молекулярный вес около 150000. Кодовое обозначение RL (высокая проницаемость) и RS (низкая проницаемость) относятся к характеристикам проницаемости этих средств. Смеси Eudragit RL/RS не растворимы в воде и в жидкостях желудочно-кишечного тракта. Однако созданные на их основе покрытия обладают набухаемостью и проницаемостью в водных растворах и жидкостях желудочно-кишечного тракта.
Дисперсии Eudragit RL/RS настоящего изобретения могут быть смешаны между собой в любом соотношении для того, чтобы в конечном счете получить непрерывно выделяющийся состав с желаемой кривой растворения. Желаемые непрерывно выделяющиеся составы могут быть получены, например, на основе покрытия с эффектом задерживания, полученного из 100% Eudragit RL, 50% Eudragit RL и 50% Eudragit RS и 10% Eudragit RL: Eudragit 90% RS. Конечно, любой специалист в данной области сможет определить, что другие полимеры могут быть также использованы, такие как, например, Eudragit L.
Пластификаторы
При осуществлении настоящего изобретения, когда покрытие содержит водную дисперсию гидрофобного вещества, включение эффективного количества пластификатора в водную дисперсию гидрофобного вещества будет еще более улучшать физические свойства покрытия, обеспечивающего непрерывное выделение. Например, поскольку этилцеллюлоза имеет относительно высокую температуру стеклования и не образует эластичных пленок при обычных условиях создания покрытий, предпочтительно в материал покрытия из этилпеллюлозы для непрерывного выделения перед использованием его в этом качестве вводить пластификатор. В целом количество пластификатора, вводимого в растворенный материал покрытия, зависит от концентрации пленкообразующего, материала, например, чаще всего – от около 1 до около 50 вес.% от пленкообразующего материала. Концентрация пластификатора, однако, может быть правильно определена только после кропотливого экспериментирования с раствором конкретного материала, образующего покрытие, и способом использования.
Примеры подходящих пластификаторов для этилцеллюлозы включают водонерастворимые пластификаторы, такие как дибутиловый эфир себациновой кислоты, диэтилфталат, триэтилцитрат, трибутилцитрат и триацетин, хотя возможно, что другие водонерастворимые пластификаторы (как, например, ацетилированные моноглицериды, фталатные эфиры, касторовое масло и т.д.) могут использоваться. Триэтилцитрат является особенно предпочтительным пластификатором для водных дисперсий этилцеллюлозы настоящего изобретения.
Примеры подходящих пластификаторов для акриловых полимеров настоящего изобретения включают (но не ограничиваются ими): эфиры лимонной кислоты, такие как триэтилцитрат NF ХVI, трибутилцитрат, дибутилфталат и, возможно, 1,2-пропиленгликоль. Другие пластификаторы, которые без сомнения пригодны для увеличения эластичности пленок, образованных акрилатами, такими как растворы лакирующих Eudragit RL/RS, включают полиэтиленгликоли, пропиленгликоль, диэтилфталат, касторовое масло и триацетин. Триэтилцитрат является особенно предпочтительным пластификатором для водных дисперсий этилцеллюлозы данного изобретения.
Было также еще установлено, что добавление небольшого количества талька уменьшает возможность слипания водной дисперсии в процессе обработки и действует как полирующее средство.
Способы производства гранул с покрытием
При использовании гидрофобного материала для создания покрытия инертных фармацевтических гранул, например, nu poriel 18/20 гранул, большинство полученных в результате твердых гранул с контролируемой степенью выделения можно затем поместить в желатиновую капсулу в количестве, достаточном для обеспечения эффективной контролируемой выделяющейся дозы при проглатывании и контактировании с окружающей жидкостью, например, желудочным соком или средой растворения.
Гранулированные составы настоящего изобретения с контролируемым выделением медленно выделяют терапевтически активное вещество, например при приеме внутрь (проглатывании) и выдерживании в желудочном соке, а затем в содержимом кишечного тракта. Кривая контролируемого выделения составов данного изобретения может изменяться, например, при изменении количества гидрофобного материала, использованного для покрытия, при изменении способа введения пластификатора в гидрофобный материал, при изменении количества пластификатора по сравнению с количеством гидрофобного материала, при введении дополнительных ингредиентов или наполнителей, при изменении способа производства и т.д. Кривая растворения конкретного продукта также может изменяться, если увеличена или уменьшена толщина задерживающего покрытия.
Шарики или гранулы из терапевтически активного агента готовят, например, растворением терапевтически активного агента в воде и последующим распылением раствора на субстрат, например, nu pariel 18/20 гранул, используя вставку Wuster. Дополнительные ингредиенты необязательно могут добавляться перед нанесением покрытия на гранулы, чтобы способствовать связыванию опиоида с гранулами и/или, например, подцвечивать раствор и т.д. Например, продукт, который включает гидроксипропилметилцеллюлозу и т.д. с красителем ли без него (например, Opadry, поставляемый Colorcon, Inc.) можно добавлять к раствору, раствор перемешивают (например, около 1 часа) до нанесения его на гранулы. Полученный субстрат с нанесенным покрытием, в этом примере – гранулы, могут затем быть покрыты разделяющим, создающим барьер покрытием для того, чтобы разделить терапевтически активный агент от гидрофобного покрытия, обеспечивающего контролируемое выделение. Примером подходящего барьерного средства является такой агент, который содержит гидроксипропилметилцеллюлозу. Однако можно использовать любое вещество, образующее пленку, известное в данной области. Предпочтительно, чтобы агент, создающий барьер, не действовал на скорость растворения конечного продукта.
На гранулы можно потом нанести покрытие обработкой водной дисперсией гидрофобного материала. Водная дисперсия гидрофобного материала предпочтительно включает еще эффективное количество пластификатора, например, триэтилцитрата. Могут использоваться также и предварительно приготовленные водные дисперсии этилцеллюлозы, такие как Aquacoat или Surelease. Если используется Surelease, нет необходимости специально добавлять пластификатор. Или же можно использовать предварительно приготовленные водные дисперсии акриловых полимеров, такие как Eudragit.
Растворы, наносимые для создания покрытий, соответствующие настоящему изобретению, в дополнение к пленкообразователю, пластификатору и системе растворителей (т.е. воде), предпочтительно содержат подкрашивающее средство для улучшения внешнего вида и отличения продукта. Краситель может добавляться к раствору терапевтически активного агента вместо или в дополнение к водной дисперсии гидрофобного материала. Например, краситель может быть добавлен к Aquacoat в результате использования красящих дисперсий на основе спирта или пропиленгликоля, молотого алюминиевого краплака и затемнителей, таких как двуокись титана, при добавлении красителя к раствору водорастворимого полимера с одновременным их смешением и при последующем низкоскоростном смешении с пластифицированным Aquacoat. Альтернативно может использоваться любой подходящий способ подкрашивания составов настоящего изобретения. Подходящими ингредиентами для придания цвета составу, когда используется водная дисперсия акрилового полимера, являются двуокись титана и цветные пигменты, например пигменты оксида железа. Включение пигментов может, однако, увеличивать задерживающий эффект покрытия.
Пластифицированный гидрофобный материал может быть нанесен на субстрат, содержащий терапевтически активный агент, путем распыления с помощью любого подходящего распыляющего устройства, известного в данной области. В предпочтительном варианте способа используют псевдоожижающую систему Wuster, в которой струя воздуха, инжектируемая снизу, поддерживает во взвешенном состоянии частицы сыпучего материала и подсушивает распыляемый на них в виде покрытия акриловый полимер. Чтобы обеспечить заранее определенную контролируемую степень выделения указанного терапевтически активного агента при выдерживании субстрата с нанесенным покрытием в водных растворах, например желудочном соке, основное количество гидрофобного материала наносят, учитывая физические свойства терапевтически активного агента, способа введения пластификатора и т.д. После нанесения покрытия из гидрофобного материала следующий слой покрытия с образованием на гранулах пленки, например из Opadry, может и не применяться. Такое покрытие, если оно есть, обеспечивает существенное уменьшение слипания частиц.
Выделение терапевтически активного агента из состава с контролируемой степенью выделения, соответствующего настоящему изобретению, может также зависеть, т.е. регулироваться в желаемой степени добавлением одного или нескольких средств, обусловливающих изменение выделения, или путем одно- или многократного выделения через покрытие. Отношение гидрофобного материала к водорастворимому материалу определяется, помимо других факторов, требуемой скоростью выделения и параметрами растворимости выбранных веществ.
Средства, обуславливающие изменение выделения, которые играют роль порообразователей, могут быть органической или неорганической природы и включают вещества, которые могут быть нерастворимыми, экстрагируемыми и выщелачиваемыми в среде использования. Порообразователи могут включать один или несколько гидрофильных материалов, таких как гидроксипропилметилцеллюлоза.
Покрытия, обеспечивающие непрерывное выделение, согласно изобретению могут также включать средства, способствующие разрыхлению (эрозии), такие как крахмал и камеди.
Покрытия, обеспечивающие непрерывное выделение, согласно изобретению могут также включать материалы, пригодные для создания в среде окружения микропористых тонкослойных пластинок, такие как поликарбонаты, содержащие в себе линейные полиэфиры карбоновых кислот, в которых карбонатные группы появляются вновь в полимерной цепочке.
Средство, обуславливающее изменение выделения, может также содержать полупроницаемый полимер.
В некоторых предпочтительных вариантах средство, обуславливающее изменение выделения, выбирают из гидроксипропилметилцеллюлозы, лактозы, стеаратов металлов и смесей вышеупомянутых соединений.
Покрытия, обеспечивающие непрерывное выделение, согласно изобретению могут также включать средства для выделения, включающие по крайней мере один канал для протока или отверстие или т.п. Канал может быть создан такими способами, как те, что описаны в US патентах 3845770, 3916889, 4063064 и 4088864 (все приведены здесь в качестве ссылок). Канал может иметь любой профиль, такой как круг, треугольный, квадратный, эллиптический, неправильной формы и т.д.
Составы матрицы гранул
В других вариантах воплощения настоящего изобретения состав с контролируемым выделением создается с помощью матрицы, имеющей покрытие, обеспечивающее контролируемое выделение, как это изложено выше. В настоящем изобретении может также использоваться матрица с контролируемым выделением, которая обеспечивает скорость растворения опиоида in vitro в пределах диапазона предпочтительных значений и которая выделяет опиоид независимо или в зависимости от значений рН. Материалы, пригодные для включения в матрицу с контролируемым выделением, будут зависеть от метода, используемого для создания матрицы.
Например, матрица в дополнение к опиоидному анальгетику и (необязательно) СОХ-2 может включать:
гидрофильные и/или гидрофобные материалы, такие как камеди, эфиры целлюлозы, акриловые смолы, материалы белковой природы; перечень не является исчерпывающим, и любой фармацевтически приемлемый гидрофобный материал или гидрофильный материал, которому можно придать свойство непрерывно выделять активный агент и который плавится (или становится мягче до того предела, который необходим, чтобы его подвергнуть экструзии), может использоваться в соответствии с настоящим изобретением.
Перевариваемые, длинноцепочечные (C8-C50, особенно С12-С40) замещенные или незамещенные углеводороды, такие как жирные кислоты, жирные спирты, эфиры глицерина и жирных кислот, минеральные и растительные масла, и воски, и стеариловый спирт; полиалкиленгликоли.
Из этих полимеров предпочтительны акриловые полимеры, особенно Eudragit RSPO – целлюлозные эфиры, особенно гидроксиалкилцеллюлозы и карбоксиалкилцеллюлозы. Орально применяемая лекарственная форма может содержать по крайней мере один гидрофильный или гидрофобный материал в количестве от 1% до 80% (по весу).
Если гидрофобным материалом является углеводород, он предпочтительно имеет точку плавления между 25
Хотя гидрокодон эффективно побеждает боль, имеет место повышение злоупотребления им со стороны лиц с физической зависимостью от опиоидов или тех, кто злоупотребляет опиоидами в нетерапевтических целях. Предыдущая практика с другими опиоидами показала, что есть возможность уменьшения их использования не по назначению, если вводить опиоиды в сочетании с антагонистом наркотика, особенно пациентам, которые являются бывшими наркоманами.
Weinhold L.L., et al. Buprenorphine Alone and in Combination with Naltrexone in Non-Dependent Humans, Drug and Alcohol Dependence 1992; 30:263-274; Mendelson J., et. al. Buprenorphine and Naloxone Interactions in Opiate-Dependent Volunteers, Clin Pharm Ther 1996; 60:105-114; приведенные здесь в качестве ссылки.
Гидрокодон представляет собой полусинтетический наркотик с обезболивающим и противокашлевым эффектом, оказывающий множественные эффекты на центральную нервную систему и желудочно-кишечный тракт. Химически гидрокодон представляет собой 4,5-эпокси-3-метокси-17-метилморфинан-6-он и известен, кроме того, как дигидрокодеинон. Подобно другим опиоидам, гидрокодон может иметь свойство формировать и может вызывать лекарственную зависимость морфинового типа. При избыточных дозах гидрокодон, как и другие опиаты, будет угнетать дыхание.
Гидрокодон для орального применения доступен также в Европе (Бельгии, Германии, Греции, Италии, Люксембурге, Норвегии и Швейцарии) как противокашлевое средство. Состав для парентерального применения есть также в Германии в качестве противокашлевого средства. Для использования в качестве анальгетика битартрат гидрокодона продается в Соединенных Штатах только в виде связанной композиции с неопиатными лекарствами (т.е. ибупрофен, ацетаминофен, аспирин и т.д.) для облегчения средней или умеренно-сильной боли.
Обычная лекарственная форма гидрокодона представляет собой комбинацию с ацетаминофеном и она продается, например, как Lortab в US от UCB Pharma, Ins. как таблетки 2,5/500 мг, 5/500 мг, 7,5/500 мг и 10/500 мг гидрокодона и ацетаминофена. Доступны также таблетки при соотношении 7,5 мг битартрата гидрокодона и 650 мг ацетаминофена и 7,5 мг битартрата гидрокодона и 750 мг ацетаминофена. Гидрокодон в сочетании с аспирином назначают орально взрослым, в основном, в дозе 1-2 таблетки каждые 4-6 часов, если требуется смягчить боль. Таблетка содержит 5 мг битартрата гидрокодона и 224 мг аспирина вместе с 32 мг кофеина или – 5 мг битартрата гидрокодона и 500 мг аспирина. Относительно новый состав включает битартрат гидрокодона и ибупрофен. Викопрофен, коммерчески доступный в US от Knoll Laboratories, представляет собой таблетку, содержащую 7,5 мг битартрата гидрокодона и 200 мг ибупрофена. Настоящее изобретение следует расценивать как охватывающее все такие составы, в которые включен орально активный опиоидный антагонист в пределах заявленных количеств, указанных ниже.
Возможность неправильного употребления опиоидных анальгетиков, например, гидрокодона, удивительно сокращается за счет предлагаемых композиций согласно настоящему изобретению. В частности, установлено, что можно соединить в отдельную оральную лекарственную форму опиоидный анальгетик вместе с небольшим количеством опиоидного антагониста, чтобы получить продукт, который не только обеспечит обезболивание, но и который значительно снизит возможность того, что человек с физической зависимостью будет продолжать злоупотребление лекарством, принимая больше одной таблетки за 1 раз, например в 2-3 раза больше обычно предназначаемой дозы.
Оральные лекарственные формы изобретения включают орально терапевтически эффективное количество опиоидного агониста с опиоидным антагинистом, таким как налтрексон, в количестве (i), которое не вызывает уменьшения уровня обезболивания, проявляемого лекарственной формой при оральном введении не терапевтического уровня, и (ii), которое обеспечивает по крайней мере неприятное чувство отвращения у лиц с физической зависимостью, например физически зависимых наркоманов (например, с отчетливым синдромом абстиненции) при употреблении за один раз дозы, превышающей предназначенную дозу. Предпочтительно количество антагониста, включенное в оральную лекарственную форму, является (iii) менее решительно используемым, т.е. менее “привлекательным” со стороны лиц с физической зависимостью, например, наркоманом, употребляющим опиаты, чем аналогичная оральная лекарственная форма, в которую антагонист не включен.
Количество антагониста, которое используют для выполнения условий (i)-(iii), приведенных в предшествующем абзаце, можно определить, по крайней мере, частично, с помощью “замещающих” тестов, таких как VAS шкала (когда субъект соотносит по шкале свое ощущение от действия лекарства) и/или путем измерения, например, размера зрачка (измеряемого с помощью пупиллометрии). Такие измерения позволяют специалисту в данной области определить дозу антагониста относительно дозы агониста, которая вызывает уменьшение опиатных эффектов агониста. Впоследствии специалист может определить уровень опиоидного антагониста, который вызывает эффект отвращения у лиц с физической зависимостью, а также уровень опиоидного антагониста, который минимизирует “оценки пристрастия” или поддерживающие свойства опиоида у наркоманов без физической зависимости. Если эти уровни опиоидного антагониста один раз определены, становится возможным определить диапазон дозировок антагониста вокруг или ниже того уровня, который следует поддерживать для выполнения условий (i)-(iii), указанных ранее в предшествующем абзаце.
Комбинации опиоидного агониста и опиоидного антагониста могут использоваться в смесях с общепринятыми наполнителями, т.е. фармацевтически приемлемыми веществами – носителями органической или неорганической природы, пригодными для использования орально, известными в данной области. Подходящие фармацевтически пригодные носители включают (но не ограничиваются ими): воду, солевые растворы, спирты, камеди, растительные масла, бензиловые спирты, полиэтиленгликоли, желатин, углеводы, такие как лактоза, амилоза или крахмал, стеарат магния, тальк, кремневую кислоту, вязкий парафин, ароматизированное масло, моноглицериды и диглицериды жирных кислот, эфиры пентаэритритола и жирных кислот, гидроксицеллюлозу, поливинилпирролидон и т.д. Фармацевтические препараты могут быть простерилизованы и, при желании, смешаны со вспомогательными средствами, например лубрикантами, консервантами и стабилизирующими добавками, увлажнителями, эмульгаторами, солями для воздействия на осмотическое давление буферов, веществами, улучшающими цвет, вкус и/или запах, и т.п. Они могут быть также скомбинированы как угодно с другими активными веществами, например другими анальгетиками. Для орального применения пригодны, в частности, таблетки, драже, растворы, капли, суппозитории или капсулы, таблетки с покрытием и желированные пилюли. Композиции, предназначенные для орального применения, могут быть получены любым методом, известным в данной области, и такие композиции могут содержать один или несколько агентов, выбранных из группы, состоящей из инертных фармацевтически нетоксичных наполнителей, которые пригодны для изготовления таблеток. Такие наполнители включают, например, инертный разбавитель, такой как лактоза; гранулирующие и дезинтегрирующие агенты, например кукурузный крахмал; связывающие агенты, например крахмал, средства для смазки, например стеарат магния. Таблетки могут быть без покрытия или с покрытием, нанесенным для придания изысканного вида или для уменьшения высвобождения активных ингредиентов. Составы для орального использования могут также представлять собой твердые желатиновые капсулы, в которых активный ингредиент смешан с инертным разбавителем.
Водные суспензии содержат вышеопределенную комбинацию лекарств, и эта смесь содержит один или несколько наполнителей, пригодных в качестве суспендирующих агентов, например фармацевтически приемлемые синтетические смолы, такие как гидроксипропилметилцеллюлоза или природные смолы (камеди). Масляные суспензии также могут быть составлены путем суспендирования вышеозначенной комбинации лекарств в растительном или минеральном масле. Масляные суспензии могут содержать загущающий агент, например, пчелиный воск или цетиловый спирт. Сироп, эликсир или что-то подобное могут использоваться тогда, когда требуется подслащивающий носитель. Суспензии для инъекций также могут быть приготовлены, в этом случае используются соответствующие жидкие носители, суспендирующие агенты и т.п.
Способ лечения и фармацевтические композиции настоящего изобретения могут далее включать одно или несколько лекарств в дополнение к опиоидному анальгетику и опиоидному антагонисту, причем дополняющие лекарства (лекарство) могут быть или могут не быть к тому же синергически действующими. Таким образом, в некоторых вариантах в состав может быть включена комбинация двух опиоидных анальгетиков в дополнение к опиоидному антагонисту. Например, лекарственная форма может включать два опиоидных анальгетика с разными свойствами, например, период полувыведения, растворимость, активность, и комбинацию из вышеприведенных веществ. В других же вариантах включен один или несколько опиоидных анальгетиков и также в дополнение к опиоидному антагонисту включено еще другое неопиоидное лекарство. Такие неопиоидные лекарства предпочтительно должны обеспечивать дополнительную анальгезию, это, например, аспирин, ацетаминафен, нестероидные противовоспалительные лекарства (“NSAIDS”), например ибупрофен, кетопрофен и т.д.; N-метил-D-аспартатные (NMDA) рецепторные антагонисты, например, морфинан, такой как декстрометорфан или декстрорфан или кетамин, ингибиторы циклооксигеназы – П (“СОХ-П ингибиторы”); и/или глициновые рецепторные антагонисты.
В некоторых предпочтительных вариантах настоящего изобретения предусматривается использование более низких доз опиоидного анальгетика благодаря включению дополнительного неопиоидного анальгетика, такого как NSAID или СОХ-2 ингибитор. При использовании меньших количеств каждого или обоих лекарств побочные эффекты, связанные с эффективным контролем боли у людей, снижаются.
Подходящие нестероидные противовоспалительные средства включают ибупрофен, диклофенак, напроксен, беноксапрофен, флурбипрофен, фенопрофен, флубуфен, кетопрофен, индопрофен, пиропрофен, карпрофен, оксапрозин, прамопрофен, муропрофен, триоксапрофен, супрофен, аминопрофен, тиапрофеновую кислоту, флупрофен, буклоксиновую кислоту, индометацин, сулиндак, толметин, зомепирак, тиопинак, зидометацин, ацеметацин, фентиазак, клиданак, оксипинак, мефенаминовую кислоту, меклофенаминовую кислоту, флуфенаминовую кислоту, нифлуминовую кислоту, толфенаминовую кислоту, дифлуризал, флуфенизал, пироксикам, судоксикам или изоксикам и т.п. Приемлемые дозы этих лекарств хорошо известны специалистам в данной области.
Антагонисты рецепторов на основе N-метиловой соли D-аспарагиновой кислоты (NMDA) хорошо известны в данной области и охватывают морфинаны, например, декстрометорфан или декстрорфан, кетамин, d-метадон или их фармацевтически приемлемые соли. Для целей настоящего изобретения считают, что термин “NMDA-антагонист” охватывает лекарства, которые блокируют главную внутриклеточную последовательность акивации NMDA-рецепторов, например, ганглиозид, такой как GTI или GMIb фенотиазин, например, трифлуоперазин или нафталинсульфонамид, такой как N-(6-аминотексил)-5-хлор-1-нафталинсульфонамид. Установлено, что эти лекарства ингибируют развитие толерантности и/или зависимости к наркотическим веществам, например наркотическим анальгетикам, таким как морфин, кодеин и т.д. (US патент 5321012 и 5556838 – Mayer et al., оба) и облегчают хронические боли (US патент 5502058 – Mayer et al.) (патенты упомянуты здесь в качестве ссылок). NMDA антагонист может быть включен как сам по себе, так и в сочетании о местным анестезирующим средством, например лидокаином, как описано в этих патентах Mayer et al.
Лечение хронической боли применением глициновых рецепторных антагонистов и идентификация подобных лекарственных средств описаны в US патенте 5514680 (Weber et al.), приведенном здесь в качестве ссылки.
Сообщалось о том, что известно, что СОХ-2 ингибиторы и другие химические структуры вызывают ингибирование циклооксигеназы-2. СОХ-2 ингибиторы описаны, например, в US патентах 5616601, 5604260, 5593994, 5550142, 5536752, 5521213, 5475995, 5639780, 5604253, 5552422, 5510368, 5436265, 5409944 и 5130311, каждый из которых упомянут здесь в качестве ссылки. Некоторые предпочтительные СОХ-2 ингибиторы включают целекоксиб (SC-58635), DUP-697, флосулид (CGP-28238), мелоксикам,6-метокси-2-нафтилуксусную кислоту (6-MNA), МК-966, набуметон (предшественник 6-MNA), нимесулид, NS-398, SC-5766, SC-58215. Т-614 или их комбинации. Уровни дозировки СОХ-2 ингибитора порядка от около 0,005 мг до около 140 мг на кг веса пациента в день являются терапевтически эффективными в сочетании с опиоидным анальгетиком. Или же около 0,25 мг до приблизительно 7 г на пациента в день СОХ-2 ингибитора вводят в сочетании с опиоидным анальгетиком.
В других вариантах в состав может включаться неопиоидное лекарство, обеспечивающее не обезболивание, а другой эффект, например противокашлевое, отхаркивающее, противоотечное, антигистаминное лекарство, анестетики местного действия и т.п.
Лекарственная форма для орального применения согласно изобретению может представлять собой гранулы, шарики, зернышки, драже (объединенные здесь под наименованием “мультичастицы”). Некое количество мультичастиц, которое необходимо, чтобы обеспечить желаемую дозу опиоида в течение какого-то периода времени, можно поместить в капсулу или включить в любую другую подходящую оральную твердую форму. Или же лекарственная форма для орального применения может быть в форме таблетки.
Контролируемое выделение лекарства из лекарственной формы
Композиция из опиоидного агониста и опиоидного антагониста может быть приготовлена как состав для орального применения с контролируемым или непрерывным выделением в виде подходящей таблетки, таблетки с покрытием или совокупности мультичастиц, известных специалисту в данной области. Непрерывно выделяющаяся лекарственная форма может необязательно включать носитель, обеспечивающий непрерывное выделение, который включен в матрицу вместе с опиоидным агонистом и опиоидным антагонистом, или же может быть применено покрытие с непрерывным выделением.
В вариантах, в которых опиоидный анальгетик представлен гидрокодоном, лекарственные формы для орального использования с непрерывным выделением могут включать дозы анальгетика от около 8 мг до около 50 мг гидрокодона на единицу дозировки. В лекарственных оральных формах с непрерывным выделением, где терапевтически активный опиоид – гидроморфон, включается от около 2 мг до около 64 мг соляно-кислого гидроморфона. В другом варианте опиоидный анальгетик включает морфин и лекарственные оральные формы с непрерывным выделением включают от около 2,5 мг до около 800 мг морфина по весу. В еще одном варианте опиоидный анальгетик включает оксикодон, и оральные лекарственные формы с непрерывным выделением включают от около 2,5 мг до около 800 мг оксикодона. Опиоидный анальгетик может включать трамадол, и оральные лекарственные формы с непрерывным выделением могут включать от около 20 мг до 800 мг трамадола на единицу дозировки. Лекарственная форма может содержать более одного опиоидного анальгетика, чтобы обеспечить в значительной мере эквивалентный терапевтический эффект. Или же лекарственная форма может содержать молярные эквивалентные количества других солей опиоидов, применяемых в настоящем изобретении.
В одном предпочтительном варианте настоящего изобретения лекарственное средство с непрерывным выделением включает такие частицы, содержащие или предусматривающие активный ингредиент, причем частицы имеют диаметр от около 0,1 мм до около 2,5 мм, предпочтительно от около 0,5 мм до около 2 мм.
Частицы предпочтительно покрывают пленкой из материала, допускающего выход комбинации опиоидного агониста/антагониста с постоянной скоростью выделения в водную среду. Пленочное покрытие выбирают таким образом, чтобы достигнуть в комбинации с другими установленными свойствами желаемой скорости высвобождения in vitro. Составы покрытия, допускающего непрерывное выделение, должны быть способны обеспечивать крепкую сплошную пленку, которая является однородной и качественной, способной удерживать подкрашивающие вещества и др. добавки, нетоксичной, инертной и нелипкой.
В некоторых вариантах частицы предусматривают матрицы, обеспечивающие среднее выделение, содержащие опиоидный анальгетик с опиоидным антагонистом.
Покрытия
Лекарственные формы настоящего изобретения могут необязательно иметь покрытие из одного или нескольких материалов, пригодных для регулирования выхода или для защиты состава. В одном варианте покрытия предусматриваются для того, чтобы обеспечить либо рН-зависимое, либо рН-независимое выделение, например, при выдерживании в гастроинтестинальной среде. рН-зависимое покрытие служит, чтобы обеспечить выделение опиоида в нужных участках желудочно-кишечного тракта (GI), например во рту или тонкой кишке, так что согласно профилю абсорбции оно способно обеспечить обезболивание у пациента в течение по крайней мере около 8 часов и предпочтительно около 12 часов и вплоть до около 24 часов. Когда требуется рН-независимое покрытие, покрытие создается таким, чтобы обеспечить оптимальное выделение независимо от изменений рН в окружающей жидкости, например, GI-тракте. Возможно также составление комбинаций, которые выделяют порцию дозы в одном нужном участке GI-тракта, например во рту, выделение оставшейся дозы в другом участке GI-тракта, например в тонкой кишке.
Составы, согласно изобретению, в которых использованы рН-зависимые покрытия, могут также обуславливать эффект повторного действия при том, что незащищенное лекарство покрывают покрытием для тонкого кишечника, и оно выделяется в желудке, тогда как остаток, будучи защищенным покрытием для тонкого кишечника, выделяется далее в желудочно-кишечном тракте. Покрытия, которые могут использоваться согласно изобретению, включают шеллак, ацетатфталат, целлюлозы (CAP), поливинилацетатфталат (pvap), фталат гидроксипропилметилцеллюлозы и сополимеры эфиров метакриловой кислоты, зеин и т.п.
В некоторых предпочтительных вариантах субстрат (т.е. зерно сердцевины таблетки, частицы матрицы), содержащий опиоидный анальгетик (с или без СОХ-2 ингибитора), покрывают гидрофобным материалом, выбранным из (i) алкилцеллюлозы; (ii) акрилового полимера; или (iii) их смесей. Покрытие может быть нанесено в виде органического или водного раствора или дисперсии. Покрытие может применяться для увеличения веса от около 2 до около 20% субстрата для того, чтобы получить желаемую кривую непрерывного выделения. Покрытия, полученные на основе водных дисперсий, описаны, например, детально в US патентах 5273760 и 5286493, поданных заявителем настоящего изобретения и включены здесь как ссылки.
Другие примеры составов и покрытий, обеспечивающих непрерывное выделение, которые могут использоваться в соответствии с настоящим изобретением, включают US патенты 5324351, 5356467 и 5472712 заявителя, включенные здесь целиком в виде ссылки.
Алкилцеллюлозные полимеры
Целлюлозные материалы и полимеры, включающие алкилцеллюлозы, представляющие собой гидрофобные материалы, очень подходят для покрытия крупинок согласно изобретению. Просто в качестве примера – один предпочтительный алкилцеллюлозный полимер – этилцеллюлоза, хотя любой специалист заметит, что и другие целлюлозные и/или алкилцеллюлозные полимеры можно легко использовать в одиночку или в любом сочетании как основу или часть гидрофобного покрытия согласно изобретению.
Одна коммерчески доступная водная дисперсия этилцеллюлозы – Aquacoat (FMC Corp., Philadelphia, Pennsylvania U.S.A.). Aquacoat готовится растворением этилцеллюлозы в не смешивающемся с водой органическом растворителе и эмульгированием ее в воду в присутствии поверхностноактивного вещества и стабилизатора. После гомогенизации с получением капелек субмикронного размера органический растворитель выпаривают под вакуумом и получают псевдолатекс. Пластификатор не вводится в псевдолатекс во время производства. Так что прежде, чем использовать его в качестве покрытия, необходимо тщательно перемешать Aquacoat с подходящим пластификатором перед использованием.
Другая водная дисперсия этилцеллюлозы коммерчески доступна как Surelease (Colorcon, Inc., West Point, Pennsylvania, U.S.А.). Этот продукт готовится при введении пластификатора в дисперсию во время процесса производства. Горячий расплав полимера, пластификатор (дибутиловый эфир себациновой кислоты) и стабилизатор (олеиновая кислота) подготавливают в качестве гомогенной смеси, которую затем разбавляют щелочным раствором с получением водной дисперсии, которую используют непосредственно в контакте с непокрываемым материалом.
Акриловые полимеры
В других предпочтительных вариантах настоящего изобретения гидрофобный материал, предусматривающий покрытие с контролируемой степенью выделения представляет собой приемлемый акриловый полимер, включая (но не ограничиваясь) сополимеры акриловой и метакриловой кислот, сополимеры метилметакрилата, этоксиэтилметакрилаты, цианоэтилметакрилат, полиакриловую кислоту, полиметакриловую кислоту, сополимер метакриловой кислоты и алкиламида, полиметилметакрилат, полиметакрилат, сополимер полиметилметакрилата, полиакриламид, сополимер аминоалкилметакрилата, ангидрид полиметакриловой кислоты и сополимеры глицидилметакрилата.
В некоторых предпочтительных вариантах акриловый полимер включает один или несколько сополимеров аммонийметакрилата. Сополимеры аммонийметакрилата хорошо известны в данной области и описываются в NF ХVIII как полностью полимеризованные сополимеры эфиров акриловой и метакриловой кислот с низким содержанием групп четвертичного аммония.
Для того чтобы получить желаемую кривую растворения, может быть необходимым включить два или несколько аммонийметакрилатных сополимеров, имеющих различные физические свойства, например, разные молярные соотношения между группами четвертичного аммония и нейтральными метакриловыми эфирами.
Некоторые полимеры типа эфиров метакриловых кислот используются для получения рН-зависимых покрытий, которые могут использоваться в соответствии с настоящим изобретением. Например, существует семейство сополимеров, синтезированных из диэтиламиноэтилметакрилата и других нейтральных метакриловых эфиров, известных также как сополимеры метакриловой кислоты и полимерные метакрилаты, коммерчески доступные как Eudragit от Rohm Tech, Inc. Существует несколько различных типов Eudragit. Например, Eudragifc E представляет собой сополимер метакриловой кислоты, который набухает и растворяется в кислых средах.
Eudragit L является сополимером метакриловой кислоты, который не набухает при рН приблизительно менее 5,7 и растворим при рН приблизительно более 6. Eudragit S не набухает при рН приблизительно менее 6,5 и растворим при рН приблизительно более 7. Eudragit RL и Eudragit RS набухают в воде, а количество воды, поглощенной этими двумя полимерами, зависит от рН, тем не менее лекарственные формы с покрытием из Eudragit RL и RS являются рН-независимыми.
В некоторых предпочтительных вариантах акриловое покрытие содержит смесь двух полимерных лаков на основе акрилатов, поставляемых Rohm Pharma под торговыми марками Eudragit RL 30D и Eudragit RS 30D соответственно. Eudragit RL 30D и Eudragit RS 30D являются сополимерами эфиров акриловой и метакриловой кислот с низким содержанием групп четвертичного аммония и с молярным соотношением групп аммония к остаткам нейтральных метакриловых эфирных групп как 1:20 в Eudragit RL 30D и 1:40 в Eudragit RS 30D. Молекулярный вес около 150000. Кодовое обозначение RL (высокая проницаемость) и RS (низкая проницаемость) относятся к характеристикам проницаемости этих средств. Смеси Eudragit RL/RS не растворимы в воде и в жидкостях желудочно-кишечного тракта. Однако созданные на их основе покрытия обладают набухаемостью и проницаемостью в водных растворах и жидкостях желудочно-кишечного тракта.
Дисперсии Eudragit RL/RS настоящего изобретения могут быть смешаны между собой в любом соотношении для того, чтобы в конечном счете получить непрерывно выделяющийся состав с желаемой кривой растворения. Желаемые непрерывно выделяющиеся составы могут быть получены, например, на основе покрытия с эффектом задерживания, полученного из 100% Eudragit RL, 50% Eudragit RL и 50% Eudragit RS и 10% Eudragit RL: Eudragit 90% RS. Конечно, любой специалист в данной области сможет определить, что другие полимеры могут быть также использованы, такие как, например, Eudragit L.
Пластификаторы
При осуществлении настоящего изобретения, когда покрытие содержит водную дисперсию гидрофобного вещества, включение эффективного количества пластификатора в водную дисперсию гидрофобного вещества будет еще более улучшать физические свойства покрытия, обеспечивающего непрерывное выделение. Например, поскольку этилцеллюлоза имеет относительно высокую температуру стеклования и не образует эластичных пленок при обычных условиях создания покрытий, предпочтительно в материал покрытия из этилпеллюлозы для непрерывного выделения перед использованием его в этом качестве вводить пластификатор. В целом количество пластификатора, вводимого в растворенный материал покрытия, зависит от концентрации пленкообразующего, материала, например, чаще всего – от около 1 до около 50 вес.% от пленкообразующего материала. Концентрация пластификатора, однако, может быть правильно определена только после кропотливого экспериментирования с раствором конкретного материала, образующего покрытие, и способом использования.
Примеры подходящих пластификаторов для этилцеллюлозы включают водонерастворимые пластификаторы, такие как дибутиловый эфир себациновой кислоты, диэтилфталат, триэтилцитрат, трибутилцитрат и триацетин, хотя возможно, что другие водонерастворимые пластификаторы (как, например, ацетилированные моноглицериды, фталатные эфиры, касторовое масло и т.д.) могут использоваться. Триэтилцитрат является особенно предпочтительным пластификатором для водных дисперсий этилцеллюлозы настоящего изобретения.
Примеры подходящих пластификаторов для акриловых полимеров настоящего изобретения включают (но не ограничиваются ими): эфиры лимонной кислоты, такие как триэтилцитрат NF ХVI, трибутилцитрат, дибутилфталат и, возможно, 1,2-пропиленгликоль. Другие пластификаторы, которые без сомнения пригодны для увеличения эластичности пленок, образованных акрилатами, такими как растворы лакирующих Eudragit RL/RS, включают полиэтиленгликоли, пропиленгликоль, диэтилфталат, касторовое масло и триацетин. Триэтилцитрат является особенно предпочтительным пластификатором для водных дисперсий этилцеллюлозы данного изобретения.
Было также еще установлено, что добавление небольшого количества талька уменьшает возможность слипания водной дисперсии в процессе обработки и действует как полирующее средство.
Способы производства гранул с покрытием
При использовании гидрофобного материала для создания покрытия инертных фармацевтических гранул, например, nu poriel 18/20 гранул, большинство полученных в результате твердых гранул с контролируемой степенью выделения можно затем поместить в желатиновую капсулу в количестве, достаточном для обеспечения эффективной контролируемой выделяющейся дозы при проглатывании и контактировании с окружающей жидкостью, например, желудочным соком или средой растворения.
Гранулированные составы настоящего изобретения с контролируемым выделением медленно выделяют терапевтически активное вещество, например при приеме внутрь (проглатывании) и выдерживании в желудочном соке, а затем в содержимом кишечного тракта. Кривая контролируемого выделения составов данного изобретения может изменяться, например, при изменении количества гидрофобного материала, использованного для покрытия, при изменении способа введения пластификатора в гидрофобный материал, при изменении количества пластификатора по сравнению с количеством гидрофобного материала, при введении дополнительных ингредиентов или наполнителей, при изменении способа производства и т.д. Кривая растворения конкретного продукта также может изменяться, если увеличена или уменьшена толщина задерживающего покрытия.
Шарики или гранулы из терапевтически активного агента готовят, например, растворением терапевтически активного агента в воде и последующим распылением раствора на субстрат, например, nu pariel 18/20 гранул, используя вставку Wuster. Дополнительные ингредиенты необязательно могут добавляться перед нанесением покрытия на гранулы, чтобы способствовать связыванию опиоида с гранулами и/или, например, подцвечивать раствор и т.д. Например, продукт, который включает гидроксипропилметилцеллюлозу и т.д. с красителем ли без него (например, Opadry, поставляемый Colorcon, Inc.) можно добавлять к раствору, раствор перемешивают (например, около 1 часа) до нанесения его на гранулы. Полученный субстрат с нанесенным покрытием, в этом примере – гранулы, могут затем быть покрыты разделяющим, создающим барьер покрытием для того, чтобы разделить терапевтически активный агент от гидрофобного покрытия, обеспечивающего контролируемое выделение. Примером подходящего барьерного средства является такой агент, который содержит гидроксипропилметилцеллюлозу. Однако можно использовать любое вещество, образующее пленку, известное в данной области. Предпочтительно, чтобы агент, создающий барьер, не действовал на скорость растворения конечного продукта.
На гранулы можно потом нанести покрытие обработкой водной дисперсией гидрофобного материала. Водная дисперсия гидрофобного материала предпочтительно включает еще эффективное количество пластификатора, например, триэтилцитрата. Могут использоваться также и предварительно приготовленные водные дисперсии этилцеллюлозы, такие как Aquacoat или Surelease. Если используется Surelease, нет необходимости специально добавлять пластификатор. Или же можно использовать предварительно приготовленные водные дисперсии акриловых полимеров, такие как Eudragit.
Растворы, наносимые для создания покрытий, соответствующие настоящему изобретению, в дополнение к пленкообразователю, пластификатору и системе растворителей (т.е. воде), предпочтительно содержат подкрашивающее средство для улучшения внешнего вида и отличения продукта. Краситель может добавляться к раствору терапевтически активного агента вместо или в дополнение к водной дисперсии гидрофобного материала. Например, краситель может быть добавлен к Aquacoat в результате использования красящих дисперсий на основе спирта или пропиленгликоля, молотого алюминиевого краплака и затемнителей, таких как двуокись титана, при добавлении красителя к раствору водорастворимого полимера с одновременным их смешением и при последующем низкоскоростном смешении с пластифицированным Aquacoat. Альтернативно может использоваться любой подходящий способ подкрашивания составов настоящего изобретения. Подходящими ингредиентами для придания цвета составу, когда используется водная дисперсия акрилового полимера, являются двуокись титана и цветные пигменты, например пигменты оксида железа. Включение пигментов может, однако, увеличивать задерживающий эффект покрытия.
Пластифицированный гидрофобный материал может быть нанесен на субстрат, содержащий терапевтически активный агент, путем распыления с помощью любого подходящего распыляющего устройства, известного в данной области. В предпочтительном варианте способа используют псевдоожижающую систему Wuster, в которой струя воздуха, инжектируемая снизу, поддерживает во взвешенном состоянии частицы сыпучего материала и подсушивает распыляемый на них в виде покрытия акриловый полимер. Чтобы обеспечить заранее определенную контролируемую степень выделения указанного терапевтически активного агента при выдерживании субстрата с нанесенным покрытием в водных растворах, например желудочном соке, основное количество гидрофобного материала наносят, учитывая физические свойства терапевтически активного агента, способа введения пластификатора и т.д. После нанесения покрытия из гидрофобного материала следующий слой покрытия с образованием на гранулах пленки, например из Opadry, может и не применяться. Такое покрытие, если оно есть, обеспечивает существенное уменьшение слипания частиц.
Выделение терапевтически активного агента из состава с контролируемой степенью выделения, соответствующего настоящему изобретению, может также зависеть, т.е. регулироваться в желаемой степени добавлением одного или нескольких средств, обусловливающих изменение выделения, или путем одно- или многократного выделения через покрытие. Отношение гидрофобного материала к водорастворимому материалу определяется, помимо других факторов, требуемой скоростью выделения и параметрами растворимости выбранных веществ.
Средства, обуславливающие изменение выделения, которые играют роль порообразователей, могут быть органической или неорганической природы и включают вещества, которые могут быть нерастворимыми, экстрагируемыми и выщелачиваемыми в среде использования. Порообразователи могут включать один или несколько гидрофильных материалов, таких как гидроксипропилметилцеллюлоза.
Покрытия, обеспечивающие непрерывное выделение, согласно изобретению могут также включать средства, способствующие разрыхлению (эрозии), такие как крахмал и камеди.
Покрытия, обеспечивающие непрерывное выделение, согласно изобретению могут также включать материалы, пригодные для создания в среде окружения микропористых тонкослойных пластинок, такие как поликарбонаты, содержащие в себе линейные полиэфиры карбоновых кислот, в которых карбонатные группы появляются вновь в полимерной цепочке.
Средство, обуславливающее изменение выделения, может также содержать полупроницаемый полимер.
В некоторых предпочтительных вариантах средство, обуславливающее изменение выделения, выбирают из гидроксипропилметилцеллюлозы, лактозы, стеаратов металлов и смесей вышеупомянутых соединений.
Покрытия, обеспечивающие непрерывное выделение, согласно изобретению могут также включать средства для выделения, включающие по крайней мере один канал для протока или отверстие или т.п. Канал может быть создан такими способами, как те, что описаны в US патентах 3845770, 3916889, 4063064 и 4088864 (все приведены здесь в качестве ссылок). Канал может иметь любой профиль, такой как круг, треугольный, квадратный, эллиптический, неправильной формы и т.д.
Составы матрицы гранул
В других вариантах воплощения настоящего изобретения состав с контролируемым выделением создается с помощью матрицы, имеющей покрытие, обеспечивающее контролируемое выделение, как это изложено выше. В настоящем изобретении может также использоваться матрица с контролируемым выделением, которая обеспечивает скорость растворения опиоида in vitro в пределах диапазона предпочтительных значений и которая выделяет опиоид независимо или в зависимости от значений рН. Материалы, пригодные для включения в матрицу с контролируемым выделением, будут зависеть от метода, используемого для создания матрицы.
Например, матрица в дополнение к опиоидному анальгетику и (необязательно) СОХ-2 может включать:
гидрофильные и/или гидрофобные материалы, такие как камеди, эфиры целлюлозы, акриловые смолы, материалы белковой природы; перечень не является исчерпывающим, и любой фармацевтически приемлемый гидрофобный материал или гидрофильный материал, которому можно придать свойство непрерывно выделять активный агент и который плавится (или становится мягче до того предела, который необходим, чтобы его подвергнуть экструзии), может использоваться в соответствии с настоящим изобретением.
Перевариваемые, длинноцепочечные (C8-C50, особенно С12-С40) замещенные или незамещенные углеводороды, такие как жирные кислоты, жирные спирты, эфиры глицерина и жирных кислот, минеральные и растительные масла, и воски, и стеариловый спирт; полиалкиленгликоли.
Из этих полимеров предпочтительны акриловые полимеры, особенно Eudragit RSPO – целлюлозные эфиры, особенно гидроксиалкилцеллюлозы и карбоксиалкилцеллюлозы. Орально применяемая лекарственная форма может содержать по крайней мере один гидрофильный или гидрофобный материал в количестве от 1% до 80% (по весу).
Если гидрофобным материалом является углеводород, он предпочтительно имеет точку плавления между 25 С и 90С. Длинноцепочечные углеводороды, жирные (алифатические) спирты являются предпочтительными. Орально применяемая лекарственная форма может содержать до 60% (по весу) по крайней мере одного перевариваемого длинноцепочечного углеводорода.
Предпочтительно орально применяемая лекарственная форма содержит до 60% (по весу), по крайней мере, одного полиалкиленгликоля.
Гидрофобный материал предпочтительно выбирают из группы, состоящей из алкилцеллюлоз, полимеров и сополимеров акриловой и метакриловой кислот, шеллака, зеина, гидрогенированного касторового масла, гидрогенизированного растительного масла или их смесей. В некоторых предпочтительных вариантах воплощения настоящего изобретения гидрофобный материал представляет собой фармацевтически приемлемый акриловый полимер, включая (но без ограничения) сополимеры акриловой и метакриловой кислот, метилметакрилат, сополимеры метилметакрилата, этоксиэтилметакрилаты, цианоэтилметакрилат, сополимер аминоалкилметакрилата, полиакриловую кислоту, полиметакриловую кислоту, сополимер алкиламина метакриловой кислоты, полиметилметакрилат, ангидрид полиметакриловой кислоты, полиметакрилат, полиакриламид, полиангидрид метакриловой кислоты и сополимеры глицидилметакрилата. В других вариантах гидрофобный материал выбирают из таких материалов, как гидроксиалкилцеллюлозы, например, гидроксипропилметилцеллюлоза, и их смеси. Предпочтительные гидрофобные материалы являются водонерастворимыми с более или менее выраженными гидрофильными и/или гидрофобными тенденциями.
Предпочтительно гидрофобные материалы, используемые в изобретении, имеют точку плавления от около 30 до около 200С, предпочтительно от около 45 до около 90С. В частности, гидрофобный материал может содержать натуральные или синтетические воски, жирные спирты (такие как лауриловый, миристиловый, стеариловый, цетиловый или предпочтительно цетостеариловый спирт), жирные кислоты, включая (без ограничения) эфиры жирных кислот, глицериды жирных кислот (мо- но-, ди- и триглицериды), гидрогенизированные жиры, углеводороды, обычные воска, стеариновое вспомогательное средство, стеариловый спирт и гидрофобные и гидрофильные материалы с углеводородной цепочкой. Подходящие воски включают, например, пчелиный воск, гликовоск, касторовый воск и карнаубский воск. Для целей настоящего изобретения воскоподобная субстанция подразумевает любой материал, который обычно является твердым при комнатной температуре и имеет точку плавления от около 30 до около 100С. Подходящие гидрофобные материалы, которые можно использовать в соответствии с настоящим изобретением, включают перевариваемые длинноцепочечные (С8-С50, особенно C12-C40) замещенные или незамещенные углеводороды, такие как жирные кислоты, жирные спирты, эфиры глицерина и жирных кислот, минеральные и растительные масла и природные и синтетические воска. Предпочтительны углеводороды с точкой плавления между 25 и 90С. Из длинноцепочечных углеводородов в некоторых вариантах предпочтительны жирные (алифатические) спирты. Орально применяемая лекарственная форма может содержать до 60% (по весу) по крайней мере одного перевариваемого длинноцепочечного углеводорода.
Предпочтительно в состав матрицы включают два или более гидрофобных материала, его предпочтительно выбирают из натуральных или синтетических восков, жирных кислот, жирных спиртов и их смесей. Примерами являются пчелиный воск, стеариновая кислота и стеариновый спирт. Этот перечень не является исчерпывающим.
Конкретная подходящая матрица содержит по крайней мере одну водорастворимую гидроксиалкилцеллюлозу, по крайней мере, один С12-С36 предпочтительно C14-C22 алифатический спирт и, необязательно, по крайней мере один полиалкиленгликоль. По крайней мере одна гидроксиалкилцеллюлоза представляет собой предпочтительно гидрокси(C1-C6)алкилцеллюлозу, например, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу и особенно гидроксиэтилцеллюлозу. Количество по меньшей мере одной гидроксиалкилцеллюлозы в данной оральной лекарственной форме будет определяться, между прочим, определенной требуемой скоростью выделения опиоида. По крайней мере, один алифатический спирт может быть, к примеру, лауриловым, миристиловым или стеариловым спиртом. В особенно предпочтительных вариантах конкретной оральной лекарственной формы, однако, по крайней мере один алифатический спирт представляет собой цетиловый спирт или цетостеариловый спирт. Количество, по крайней мере, одного алифатического спирта в конкретной оральной лекарственной форме будет определяться, как указано выше, определенной требуемой скоростью выделения опиоида. Оно будет также зависеть от того, присутствует или отсутствует в оральной лекарственной форме, по крайней, мере, один полиалкиленгликоль. В отсутствие, по крайней мере, одного полиалкиленгликоля оральная лекарственная форма предпочтительно содержит от 20 до 50% (по весу), по крайней мере, одного алифатического спирта. Если, по крайней мере, один полиалкиленгликоль содержится в оральной лекарственной форде, тогда общий вес по крайней мере одного алифатического спирта и по крайней мере одного полиалкиленгликоля предпочтительно составляет от 20 до 50% (по весу) от всей лекарственной формы.
В одном варианте отношение, например, по крайней мере, одной гидроксиалкилцеллюлозы или акриловой смолы к смеси по крайней мере одного алифатического спирта и полиалкиленгликоля в значительной степени определяет скорость выделения опиоида из лекарственной формы. Отношение, по крайней мере, одной гидроксиалкилцеллюлозы к смеси по крайней мере одного алифатического спирта и от полиалкиленгликоля предпочтительна 1:2 до 1:4, причем особенно предпочтительно от 1:3 до 1:4.
По крайней мере, один полиалкиленгликоль может быть, например, полипропиленгликолем или, что предпочтительно, полиэтиленгликолем. Средняя молекулярная масса, по крайней мере, одного полиалкиленгликоля составляет предпочтительно от 1000 до 15000, особенно – от 1500 до 12000.
Другая подходящая матрица, обеспечивающая контролируемое выделение, должна содержать алкилцеллюлозу (особенно этилцеллюлозу), С12-С36 алифатический спирт и, необязательно, полиалкиленгликоль.
В другом предпочтительном варианте матрица включает фармацевтически приемлемую комбинацию из, по крайней мере, двух гидрофобных материалов.
В дополнение к вышеперечисленным ингредиентам матрица с регулируемым выделением может также содержать подходящие количества других веществ, например разбавителей, лубрикантов, связующих, добавок для гранулирования, красителей, отдушек, глидантов, известных в фармацевтической отрасли.
Способы получения связанных с матрицей гранул
Для того чтобы облегчить получение твердой, применяемой орально лекарственной формы с контролируемым выделением, согласно данному изобретению может использоваться любой способ производства составов, связанных с матрицей, известный специалистам в данной области. Например, включение в матрицу может быть осуществлено путем: а) образования гранул, содержащих по крайней мере одну водорастворимую гидроксиалкилцеллюлозу и опиоид или соль опиоида; b) смешивания гранул, содержащих гидроксиалкилцеллюлозу с по крайней мере одним C12-С36 алифатическим спиртом; и с) необязательно – прессования и формирования гранул. Предпочтительно гранулы формируют влажным гранулированием смеси гидроксиалкилцеллюлозы и опиоида с водой. В особенно предпочтительном варианте этого способа количество воды, добавляемое в процессе стадии влажного гранулирования, является предпочтительно 1,5-5-кратным, особенно 1,75-3,5-кратным по отношению к сухому весу опиоида.
В других альтернативных вариантах окатывающий агент для придания гранулам округленной формы может быть подвергнут скатыванию вместе с активным ингредиентом. Предпочтительна микрокристаллическая целлюлоза. Подходящей микрокристаллической целлюлозой является, например, материал, продаваемый как Avicel РН 101 (Trade Mark, FMC Corporation). В таких вариантах в дополнение к активному ингредиенту и окатывающему агенту сфероиды могут также включать связующее. Подходящие связующие, например водорастворимые полимеры с низкой вязкостью, хорошо известны специалистам-фармацевтам. Однако предпочтительной является водорастворимая гидрокси-низший алкилцеллюлоза, такая как гидроксипропилцеллюлоза. Дополнительно (или альтернативно) сфероиды могут содержать водонерастворимый полимер, особенно – акриловый полимер, акриловый сополимер, например, сополимер метакриловой кислоты и этилакрилата или этилцеллюлозу. В таких вариантах покрытие, обеспечивающее непрерывное выделение, должно вообще включать гидрофобный материал, такой как (а) воск, или сам по себе, или в смеси с жирным спиртом; или b) шеллак или зеин.
Матрица, полученная экструзией из расплава
Матрицы, обеспечивающие непрерывное выделение, могут быть также изготовлены методами грануляции расплава или экструзии расплава. В общем, методы грануляции расплава включают расплавление твердого в нормальных условиях гидрофобного материала, например воска, и включение в него порошкообразного лекарства. Чтобы получить лекарственную форму с непрерывным выделением, может быть необходимым включение дополнительно еще одного гидрофобного вещества, например этилцеллюлозы или водонерастворимого акрилового полимера в расплавленный восковой гидрофобный материал. Примеры составов с непрерывным выделением, полученных методами гранулирования расплава, содержатся в US патенте 4861598, поданном заявителем и включенном здесь в качестве ссылки.
Дополнительный гидрофобный материал может включать одну или несколько водонерастворимых воскоподобных термопластичных субстанций, возможно смешанных с одной или несколькими воскоподобными термопластичными субстанциями, менее гидрофобными, чем упомянутые одна или несколько водонерастворимых воскоподобных субстанций. Чтобы добиться непрерывного выделения, индивидуальные воскоподобные субстанции, в составе должны быть в значительной мере недеградируемыми и не растворимыми в гастроинтестинальных жидкостях во время начальных фаз выделения. Полезными здесь водонерастворимыми воскоподобными веществами могут быть такие, у которых растворимость в воде менее 1:5000 (вес/вес).
В дополнение к вышеперечисленным ингредиентам матрица с непрерывным выделением может также содержать приемлемые количества других материалов, например разбавителей, лубрикантов, связующих, добавок для гранулирования, красителей, отдушек и глидантов, которые известны фармацевтам. Количества этих дополнительных материалов должны быть достаточными, чтобы обеспечить желаемый эффект в выбранном составе.
В дополнение к вышеперечисленным ингредиентам матрица с непрерывным выделением, включающая мультичастицы, полученные экструзией расплава, может также содержать приемлемые количества других материалов, например разбавителей, лубрикантов, связующих, добавок для гранулирования, красителей, отдушек и глидантов, которые являются общеизвестными для фармацевтов, в количествах, по желанию до 50 вес.% от веса частиц.
Специальные примеры фармацевтически приемлемых носителей и наполнителей, которые могут использоваться для составления композиций применяемых орально лекарственных форм, описываются в Haundbook of Pharmaceutical Excipients, American Pharmaceutical Association (1986), включенной здесь в качестве ссылки.
Частицы, полученные экструзией расплава
Получение подходящей экструдированной из расплава матрицы, в соответствии с настоящим изобретением, может, например, включать стадии смешивания опиоидного анальгетика вместе с по крайней мере одним гидрофобным материалом и предпочтительно с еще одним гидрофобным материалом до получения гомогенной смеси. Гомогенную смесь затем нагревают до температуры, достаточной для того, чтобы, по крайней мере, сделать смесь достаточно мягкой, чтобы подвергнуть ее экструзии. Получившуюся гомогенную смесь затем экструдируют с получением тяжей. Экструдат предпочтительно охлаждают и режут с помощью любых известных средств с получением мультичастиц. Тяжи охлаждают и режут, получая мультичастицы. Мультичастицы потом разделяют на единичные дозы. Экструдат предпочтительно имеет диаметр от около 0,1 до около 5 мм и обеспечивает непрерывное выделение терапевтически активного агента в течение времени от около 8 до около 24 часов.
Произвольный процесс экструзии из расплавов по настоящему изобретению включает непосредственно измерение внутри экструдера количеств гидрофобного материала, терапевтически активного агента и связующего, которое является необязательным компонентом; нагрев гомогенной смеси; экструдирование гомогенной смеси с образованием тяжей; охлаждение тяжей, содержащих гомогенную смесь; резку тяжей на частицы, имеющие размер от около 0,1 мм до около 12 мм; и разделение указанных частиц на единичные дозировки. В этом аспекте изобретения отражена относительно длительная процедура производства.
Диаметр отверстия экструдера или выходного отверстия может быть также отрегулирован, чтобы варьировать толщину экструдируемых тяжей. К тому же выпускное отверстие экструдера может не быть круглым, оно может быть продолговатым, прямоугольным и т.д. Выходящие тяжи можно уменьшить до частиц с помощью горячих кусачек, гильотины и т.д.
Система из мультичастиц, полученных при экструзии расплава, может быть, например, в форме гранул, сфероидов или пеллет, в зависимости от конфигурации выпускного отверстия экструдера. Для целей настоящего изобретения термины “мультичастица (-цы), полученные экструзией расплава” и “система(-ы) мультичастиц, полученная экструзией расплава” и “частицы, полученные экструзией расплава” будут относиться к совокупности частиц, предпочтительно имеющих приблизительно одинаковые размеры и/или форму и содержащих один или несколько активных агентов и один или несколько наполнителей, предпочтительно включающих гидрофобный материал, согласно описанному здесь. В этом плане мультичастицы, полученные экструзией расплава, будут иметь размеры от около 0,1 до около 12 мм в длину и диаметр от около 0,1 до около 5 мм. Кроме того, следует понимать, что мультичастицы, полученные экструдированием расплава, могут иметь любую геометрическую форму вместе с указанными размерами. Альтернативно, экструдат можно просто нарезать на куски желаемой длины и разделить на единичные дозы терапевтически активного агента без необходимости проведения стадии окатывания.
В одном предпочтительном варианте оральные лекарственные формы готовят таким образом, чтобы включить эффективное количество мультичастиц, полученных экструзией расплава, внутрь капсулы. Например, совокупность таких мультичастиц может быть помещена в желатиновую капсулу в количестве, достаточном для обеспечения эффективной непрерывно выделяющейся дозы при проглатывании их и контактировании с желудочным соком.
В другом предпочтительном варианте нужное количество экструдата мультичастиц таблетируют известным способом с помощью обычно применяемого таблетирующего устройства. Способы и композиции для изготовления таблеток (таблетирование и формирование), капсул (твердых и мягких желатиновых) и пилюль описаны также в Remington’s Pharmaceutical Sciences, (Arthur Osol, editor), 1553-1593 (1980), включенной здесь в качестве ссылки.
Еще в одном предпочтительном варианте экструдату может придаваться форма таблеток, как указано в US патенте №4957681 (Klimesch, et al.), детально описано выше и включено здесь в качестве ссылки.
Необязательно на систему мультичастиц, полученных экструзией расплава, с непрерывным выделением или на таблетки может быть нанесено покрытие, или желатиновая капсула может затем быть снабжена покрытием с непрерывным выделением, например, покрытием с непрерывным выделением, описанные выше. Такие покрытия предпочтительно включают достаточное количество гидрофобного материала, чтобы получить увеличение веса от около 2 до около 30%, хотя материал для покрытия может больше зависеть от физических свойств конкретного используемого соединения опиоидного анальгетика и желаемой скорости выделения, помимо всего прочего.
Единичные лекарственные формы настоящего изобретения, полученные экструдированием, могут также включать комбинации мультичастиц из экструдированного расплава, содержащие один или более описанных выше терапевтически активных агентов перед инкапсулированием. Кроме того, единичные лекарственные формы могут также включать какое-то количество сразу же выделяющегося активного агента для осуществления немедленного терапевтического действия. Немедленно выделяющийся терапевтически активный агент может быть включен, например, как отдельная частица внутрь желатиновой капсулы или может быть нанесен в виде покрытия на поверхность мультичастиц по завершении процесса изготовления лекарственных форм (например, покрытие с контролируемым выделением или связанное с матрицей). Единичные лекарственные формы настоящего изобретения могут также содержать комбинацию гранул с контролируемым выделением и матрицы для мультичастиц для достижения желаемого эффекта.
Составы настоящего изобретения с непрерывным выделением предпочтительно обеспечивают медленное выделение терапевтически активного агента, например, при проглатывании и выдержке в желудочном соке, а затем в интестинальных жидкостях. Кривая непрерывного выделения составов данного изобретения, полученных экструзией расплава, может быть различной в зависимости от количества задерживающего агента, например гидрофобного материала, в зависимости от изменения количества пластификатора по сравнению с количеством гидрофобного материала, в зависимости от включения дополнительных ингредиентов или наполнителей, при изменении способа производства и т.д.
В другом варианте изобретения экструдированный из расплава материал готовят, не включая терапевтически активного агента, который добавляют потом, к экструдату. В таких составах обычно терапевтически активный агент смешан с материалом экструдированной матрицы, затем смесь следует таблетировать с получением состава с медленным выделением. Такие составы хороши, например, когда терапевтически активный агент, включенный в состав, чувствителен к температурам, требующимся для размягчения гидрофобного материала и/или материала, задерживающего выделение.
Подробное описание предпочтительных вариантов
Следующие примеры иллюстрируют различные аспекты настоящего изобретения. Их не следует рассматривать как ограничивающие каким-либо образом объем изобретения.
Пример 1.
Проверяют отделяемость гидрохлорида налтрексона от битартрата гидрокодона путем экстракции, подражая тому, как это делает лицо, злоупотребляющее лекарствами. Проверка структур и оценка значений рН позволяют предположить, что оба соединения будут растворимы в кислоте. Однако налтрексон должен быть также более растворимым при более высоких рН с минимумом растворимости между рН 8,4 и 10,3. Хотели проверить гипотезу о том, что оба соединения можно экстрагировать из таблетки в кислоте и затем осадить гидрокодон повышением рН.
Так как таблетки с контролируемым выделением гидрокодона (HYCR) и таблетки налтрексона не годятся для этого исследования, были приготовлены “поддельные” образцы добавлением известных количеств лекарственных субстанций битартрата гидрокодона и гидрохлорида налтрексона к HYCR AcroContin 15 мг таблеткам плацебо (“AcroContin”- запатентованная основа, обеспечивающая контролируемое выделение, состоящая из аммонийметакрилатного полимера вместе с высшим алифатическим спиртом, как описано в примере (US патента 4861598, упомянутого здесь в качестве ссылки)). Различные растворители, меняющие значения рН, использовались для экстракции битартрата гидрокодона и/или солянокислого налтрексона при концентрациях 4 таблетки/25 мл (раздел 2.1) и 5 таблеток/5 мл (раздел 2.2) растворителя. Выделенные продукты определялись с помощью ВЭЖХ.
2.1. Экстракция при концентрации 4 таблетки на 25 мл растворителя.
2.1.1. Около 60 мг битартрата гидрокодона/ 25 мг гидрохлорида налтрексона и 400 мг HYCR 15 мг AcroContin таблеток плацебо (эквивалентных 4 таблеткам) добавили в 25 мл мерную колбу. Добавили около 15 мл воды в мерную колбу и раствор озвучивали в течение 10 мин. Раствор довели до объема водой и хорошо перемешали. Это был основной раствор для опыта. Таким образом приготовили 13 образцов основных растворов.
2.1.2. Регулировали рН растворов либо ледяной уксусной кислотой, либо 0,2 н NaOH до значений рН: 2,0, 4,0, 5,1, 6,0, 6,5, 7,0, 7,4, 8,0, 8,5, 9,0, 9,4 и 10,0. Однако при получении раствора с рН 1,1 использовали соляную кислоту. Затем следовала стадия 2.1.4.
2.1.3. Повторяли стадию 2.1.1., чтобы вместо воды приготовить основные испытуемые растворы в этаноле, метаноле и ацетоне.
2.1.4. Каждый раствор фильтровали, используя 5 мл шприц с 0,45 мкм фильтровальным блоком Millex-HV. 1,0 мл чистого фильтрата отбирали пипеткой в 25 мл мерную колбу, доводили до объема водой и хорошо взбалтывали. Испытуемые растворы наносили на колонку ВЭЖХ, результаты показаны в таблице 1.
2.2. Экстракция при концентрациях 5 таблеток на 5 мл растворителя.
2.2.1. Около 75 мг битартрата гидрокодона и 32 мг гидрохлорида налтрексона добавили в сцинтилляционный сосуд, который содержал 475 мг HYCR 15 мг AcroContin таблеток плацебо (эквивалентных 5 таблеткам). В сцинтилляционный сосуд добавили 5,0 мл воды и раствор озвучивали в течение 10 минут. Это был основной раствор опыта.
2.2.2. рН растворов довели с помощью 50 вес.%/вес. NaOH до 7,1. После того, как растворы отстоялись в течение 1 часа, весь раствор пропустили через 5 мл, имеющийся в распоряжении шприц с 0,45 мкм фильтровальным элементом Millex-HV. 1,0 мл чистого раствора было отмерено пипеткой в 25 мл мерную колбу, содержимое довели водой до объема и хорошо перемешали. Получился основной раствор для опыта с рН 7,1.
2.2.3. Стадии 2.2.1. и 2.2.2. повторили для получения опытных растворов с рН 8,0, 9,0, 10,0, 11,0, 12,0 и 12,7. Образцы растворов потом вводили в систему ВЭЖХ, результаты показаны в таблице 2.
3. Результаты.
Результаты представлены в таблицах 4 и 5. В таблице 5 отмечается, что и гидрокодон, и налтрексон полностью растворимы во всех растворителях, кроме ацетона. В таблице 5 отмечается, что количество налтрексона оставалось в растворе уменьшенных при рН 8 и увеличенным при рН 10,0, а количество налтрексона оставалось в растворе уменьшенным при более высоких значениях рН.
В таблице 4 показана “поддельная” экстрагируемость гидрохлорида налтрексона из содержащих битартрат гидрокодона CR, 15 мг AcroContin таблеток при концентрации 4 таблетки на 25 мл растворителя. С и 90С. Длинноцепочечные углеводороды, жирные (алифатические) спирты являются предпочтительными. Орально применяемая лекарственная форма может содержать до 60% (по весу) по крайней мере одного перевариваемого длинноцепочечного углеводорода.
Предпочтительно орально применяемая лекарственная форма содержит до 60% (по весу), по крайней мере, одного полиалкиленгликоля.
Гидрофобный материал предпочтительно выбирают из группы, состоящей из алкилцеллюлоз, полимеров и сополимеров акриловой и метакриловой кислот, шеллака, зеина, гидрогенированного касторового масла, гидрогенизированного растительного масла или их смесей. В некоторых предпочтительных вариантах воплощения настоящего изобретения гидрофобный материал представляет собой фармацевтически приемлемый акриловый полимер, включая (но без ограничения) сополимеры акриловой и метакриловой кислот, метилметакрилат, сополимеры метилметакрилата, этоксиэтилметакрилаты, цианоэтилметакрилат, сополимер аминоалкилметакрилата, полиакриловую кислоту, полиметакриловую кислоту, сополимер алкиламина метакриловой кислоты, полиметилметакрилат, ангидрид полиметакриловой кислоты, полиметакрилат, полиакриламид, полиангидрид метакриловой кислоты и сополимеры глицидилметакрилата. В других вариантах гидрофобный материал выбирают из таких материалов, как гидроксиалкилцеллюлозы, например, гидроксипропилметилцеллюлоза, и их смеси. Предпочтительные гидрофобные материалы являются водонерастворимыми с более или менее выраженными гидрофильными и/или гидрофобными тенденциями.
Предпочтительно гидрофобные материалы, используемые в изобретении, имеют точку плавления от около 30 до около 200С, предпочтительно от около 45 до около 90С. В частности, гидрофобный материал может содержать натуральные или синтетические воски, жирные спирты (такие как лауриловый, миристиловый, стеариловый, цетиловый или предпочтительно цетостеариловый спирт), жирные кислоты, включая (без ограничения) эфиры жирных кислот, глицериды жирных кислот (мо- но-, ди- и триглицериды), гидрогенизированные жиры, углеводороды, обычные воска, стеариновое вспомогательное средство, стеариловый спирт и гидрофобные и гидрофильные материалы с углеводородной цепочкой. Подходящие воски включают, например, пчелиный воск, гликовоск, касторовый воск и карнаубский воск. Для целей настоящего изобретения воскоподобная субстанция подразумевает любой материал, который обычно является твердым при комнатной температуре и имеет точку плавления от около 30 до около 100С. Подходящие гидрофобные материалы, которые можно использовать в соответствии с настоящим изобретением, включают перевариваемые длинноцепочечные (С8-С50, особенно C12-C40) замещенные или незамещенные углеводороды, такие как жирные кислоты, жирные спирты, эфиры глицерина и жирных кислот, минеральные и растительные масла и природные и синтетические воска. Предпочтительны углеводороды с точкой плавления между 25 и 90С. Из длинноцепочечных углеводородов в некоторых вариантах предпочтительны жирные (алифатические) спирты. Орально применяемая лекарственная форма может содержать до 60% (по весу) по крайней мере одного перевариваемого длинноцепочечного углеводорода.
Предпочтительно в состав матрицы включают два или более гидрофобных материала, его предпочтительно выбирают из натуральных или синтетических восков, жирных кислот, жирных спиртов и их смесей. Примерами являются пчелиный воск, стеариновая кислота и стеариновый спирт. Этот перечень не является исчерпывающим.
Конкретная подходящая матрица содержит по крайней мере одну водорастворимую гидроксиалкилцеллюлозу, по крайней мере, один С12-С36 предпочтительно C14-C22 алифатический спирт и, необязательно, по крайней мере один полиалкиленгликоль. По крайней мере одна гидроксиалкилцеллюлоза представляет собой предпочтительно гидрокси(C1-C6)алкилцеллюлозу, например, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу и особенно гидроксиэтилцеллюлозу. Количество по меньшей мере одной гидроксиалкилцеллюлозы в данной оральной лекарственной форме будет определяться, между прочим, определенной требуемой скоростью выделения опиоида. По крайней мере, один алифатический спирт может быть, к примеру, лауриловым, миристиловым или стеариловым спиртом. В особенно предпочтительных вариантах конкретной оральной лекарственной формы, однако, по крайней мере один алифатический спирт представляет собой цетиловый спирт или цетостеариловый спирт. Количество, по крайней мере, одного алифатического спирта в конкретной оральной лекарственной форме будет определяться, как указано выше, определенной требуемой скоростью выделения опиоида. Оно будет также зависеть от того, присутствует или отсутствует в оральной лекарственной форме, по крайней, мере, один полиалкиленгликоль. В отсутствие, по крайней мере, одного полиалкиленгликоля оральная лекарственная форма предпочтительно содержит от 20 до 50% (по весу), по крайней мере, одного алифатического спирта. Если, по крайней мере, один полиалкиленгликоль содержится в оральной лекарственной форде, тогда общий вес по крайней мере одного алифатического спирта и по крайней мере одного полиалкиленгликоля предпочтительно составляет от 20 до 50% (по весу) от всей лекарственной формы.
В одном варианте отношение, например, по крайней мере, одной гидроксиалкилцеллюлозы или акриловой смолы к смеси по крайней мере одного алифатического спирта и полиалкиленгликоля в значительной степени определяет скорость выделения опиоида из лекарственной формы. Отношение, по крайней мере, одной гидроксиалкилцеллюлозы к смеси по крайней мере одного алифатического спирта и от полиалкиленгликоля предпочтительна 1:2 до 1:4, причем особенно предпочтительно от 1:3 до 1:4.
По крайней мере, один полиалкиленгликоль может быть, например, полипропиленгликолем или, что предпочтительно, полиэтиленгликолем. Средняя молекулярная масса, по крайней мере, одного полиалкиленгликоля составляет предпочтительно от 1000 до 15000, особенно – от 1500 до 12000.
Другая подходящая матрица, обеспечивающая контролируемое выделение, должна содержать алкилцеллюлозу (особенно этилцеллюлозу), С12-С36 алифатический спирт и, необязательно, полиалкиленгликоль.
В другом предпочтительном варианте матрица включает фармацевтически приемлемую комбинацию из, по крайней мере, двух гидрофобных материалов.
В дополнение к вышеперечисленным ингредиентам матрица с регулируемым выделением может также содержать подходящие количества других веществ, например разбавителей, лубрикантов, связующих, добавок для гранулирования, красителей, отдушек, глидантов, известных в фармацевтической отрасли.
Способы получения связанных с матрицей гранул
Для того чтобы облегчить получение твердой, применяемой орально лекарственной формы с контролируемым выделением, согласно данному изобретению может использоваться любой способ производства составов, связанных с матрицей, известный специалистам в данной области. Например, включение в матрицу может быть осуществлено путем: а) образования гранул, содержащих по крайней мере одну водорастворимую гидроксиалкилцеллюлозу и опиоид или соль опиоида; b) смешивания гранул, содержащих гидроксиалкилцеллюлозу с по крайней мере одним C12-С36 алифатическим спиртом; и с) необязательно – прессования и формирования гранул. Предпочтительно гранулы формируют влажным гранулированием смеси гидроксиалкилцеллюлозы и опиоида с водой. В особенно предпочтительном варианте этого способа количество воды, добавляемое в процессе стадии влажного гранулирования, является предпочтительно 1,5-5-кратным, особенно 1,75-3,5-кратным по отношению к сухому весу опиоида.
В других альтернативных вариантах окатывающий агент для придания гранулам округленной формы может быть подвергнут скатыванию вместе с активным ингредиентом. Предпочтительна микрокристаллическая целлюлоза. Подходящей микрокристаллической целлюлозой является, например, материал, продаваемый как Avicel РН 101 (Trade Mark, FMC Corporation). В таких вариантах в дополнение к активному ингредиенту и окатывающему агенту сфероиды могут также включать связующее. Подходящие связующие, например водорастворимые полимеры с низкой вязкостью, хорошо известны специалистам-фармацевтам. Однако предпочтительной является водорастворимая гидрокси-низший алкилцеллюлоза, такая как гидроксипропилцеллюлоза. Дополнительно (или альтернативно) сфероиды могут содержать водонерастворимый полимер, особенно – акриловый полимер, акриловый сополимер, например, сополимер метакриловой кислоты и этилакрилата или этилцеллюлозу. В таких вариантах покрытие, обеспечивающее непрерывное выделение, должно вообще включать гидрофобный материал, такой как (а) воск, или сам по себе, или в смеси с жирным спиртом; или b) шеллак или зеин.
Матрица, полученная экструзией из расплава
Матрицы, обеспечивающие непрерывное выделение, могут быть также изготовлены методами грануляции расплава или экструзии расплава. В общем, методы грануляции расплава включают расплавление твердого в нормальных условиях гидрофобного материала, например воска, и включение в него порошкообразного лекарства. Чтобы получить лекарственную форму с непрерывным выделением, может быть необходимым включение дополнительно еще одного гидрофобного вещества, например этилцеллюлозы или водонерастворимого акрилового полимера в расплавленный восковой гидрофобный материал. Примеры составов с непрерывным выделением, полученных методами гранулирования расплава, содержатся в US патенте 4861598, поданном заявителем и включенном здесь в качестве ссылки.
Дополнительный гидрофобный материал может включать одну или несколько водонерастворимых воскоподобных термопластичных субстанций, возможно смешанных с одной или несколькими воскоподобными термопластичными субстанциями, менее гидрофобными, чем упомянутые одна или несколько водонерастворимых воскоподобных субстанций. Чтобы добиться непрерывного выделения, индивидуальные воскоподобные субстанции, в составе должны быть в значительной мере недеградируемыми и не растворимыми в гастроинтестинальных жидкостях во время начальных фаз выделения. Полезными здесь водонерастворимыми воскоподобными веществами могут быть такие, у которых растворимость в воде менее 1:5000 (вес/вес).
В дополнение к вышеперечисленным ингредиентам матрица с непрерывным выделением может также содержать приемлемые количества других материалов, например разбавителей, лубрикантов, связующих, добавок для гранулирования, красителей, отдушек и глидантов, которые известны фармацевтам. Количества этих дополнительных материалов должны быть достаточными, чтобы обеспечить желаемый эффект в выбранном составе.
В дополнение к вышеперечисленным ингредиентам матрица с непрерывным выделением, включающая мультичастицы, полученные экструзией расплава, может также содержать приемлемые количества других материалов, например разбавителей, лубрикантов, связующих, добавок для гранулирования, красителей, отдушек и глидантов, которые являются общеизвестными для фармацевтов, в количествах, по желанию до 50 вес.% от веса частиц.
Специальные примеры фармацевтически приемлемых носителей и наполнителей, которые могут использоваться для составления композиций применяемых орально лекарственных форм, описываются в Haundbook of Pharmaceutical Excipients, American Pharmaceutical Association (1986), включенной здесь в качестве ссылки.
Частицы, полученные экструзией расплава
Получение подходящей экструдированной из расплава матрицы, в соответствии с настоящим изобретением, может, например, включать стадии смешивания опиоидного анальгетика вместе с по крайней мере одним гидрофобным материалом и предпочтительно с еще одним гидрофобным материалом до получения гомогенной смеси. Гомогенную смесь затем нагревают до температуры, достаточной для того, чтобы, по крайней мере, сделать смесь достаточно мягкой, чтобы подвергнуть ее экструзии. Получившуюся гомогенную смесь затем экструдируют с получением тяжей. Экструдат предпочтительно охлаждают и режут с помощью любых известных средств с получением мультичастиц. Тяжи охлаждают и режут, получая мультичастицы. Мультичастицы потом разделяют на единичные дозы. Экструдат предпочтительно имеет диаметр от около 0,1 до около 5 мм и обеспечивает непрерывное выделение терапевтически активного агента в течение времени от около 8 до около 24 часов.
Произвольный процесс экструзии из расплавов по настоящему изобретению включает непосредственно измерение внутри экструдера количеств гидрофобного материала, терапевтически активного агента и связующего, которое является необязательным компонентом; нагрев гомогенной смеси; экструдирование гомогенной смеси с образованием тяжей; охлаждение тяжей, содержащих гомогенную смесь; резку тяжей на частицы, имеющие размер от около 0,1 мм до около 12 мм; и разделение указанных частиц на единичные дозировки. В этом аспекте изобретения отражена относительно длительная процедура производства.
Диаметр отверстия экструдера или выходного отверстия может быть также отрегулирован, чтобы варьировать толщину экструдируемых тяжей. К тому же выпускное отверстие экструдера может не быть круглым, оно может быть продолговатым, прямоугольным и т.д. Выходящие тяжи можно уменьшить до частиц с помощью горячих кусачек, гильотины и т.д.
Система из мультичастиц, полученных при экструзии расплава, может быть, например, в форме гранул, сфероидов или пеллет, в зависимости от конфигурации выпускного отверстия экструдера. Для целей настоящего изобретения термины “мультичастица (-цы), полученные экструзией расплава” и “система(-ы) мультичастиц, полученная экструзией расплава” и “частицы, полученные экструзией расплава” будут относиться к совокупности частиц, предпочтительно имеющих приблизительно одинаковые размеры и/или форму и содержащих один или несколько активных агентов и один или несколько наполнителей, предпочтительно включающих гидрофобный материал, согласно описанному здесь. В этом плане мультичастицы, полученные экструзией расплава, будут иметь размеры от около 0,1 до около 12 мм в длину и диаметр от около 0,1 до около 5 мм. Кроме того, следует понимать, что мультичастицы, полученные экструдированием расплава, могут иметь любую геометрическую форму вместе с указанными размерами. Альтернативно, экструдат можно просто нарезать на куски желаемой длины и разделить на единичные дозы терапевтически активного агента без необходимости проведения стадии окатывания.
В одном предпочтительном варианте оральные лекарственные формы готовят таким образом, чтобы включить эффективное количество мультичастиц, полученных экструзией расплава, внутрь капсулы. Например, совокупность таких мультичастиц может быть помещена в желатиновую капсулу в количестве, достаточном для обеспечения эффективной непрерывно выделяющейся дозы при проглатывании их и контактировании с желудочным соком.
В другом предпочтительном варианте нужное количество экструдата мультичастиц таблетируют известным способом с помощью обычно применяемого таблетирующего устройства. Способы и композиции для изготовления таблеток (таблетирование и формирование), капсул (твердых и мягких желатиновых) и пилюль описаны также в Remington’s Pharmaceutical Sciences, (Arthur Osol, editor), 1553-1593 (1980), включенной здесь в качестве ссылки.
Еще в одном предпочтительном варианте экструдату может придаваться форма таблеток, как указано в US патенте №4957681 (Klimesch, et al.), детально описано выше и включено здесь в качестве ссылки.
Необязательно на систему мультичастиц, полученных экструзией расплава, с непрерывным выделением или на таблетки может быть нанесено покрытие, или желатиновая капсула может затем быть снабжена покрытием с непрерывным выделением, например, покрытием с непрерывным выделением, описанные выше. Такие покрытия предпочтительно включают достаточное количество гидрофобного материала, чтобы получить увеличение веса от около 2 до около 30%, хотя материал для покрытия может больше зависеть от физических свойств конкретного используемого соединения опиоидного анальгетика и желаемой скорости выделения, помимо всего прочего.
Единичные лекарственные формы настоящего изобретения, полученные экструдированием, могут также включать комбинации мультичастиц из экструдированного расплава, содержащие один или более описанных выше терапевтически активных агентов перед инкапсулированием. Кроме того, единичные лекарственные формы могут также включать какое-то количество сразу же выделяющегося активного агента для осуществления немедленного терапевтического действия. Немедленно выделяющийся терапевтически активный агент может быть включен, например, как отдельная частица внутрь желатиновой капсулы или может быть нанесен в виде покрытия на поверхность мультичастиц по завершении процесса изготовления лекарственных форм (например, покрытие с контролируемым выделением или связанное с матрицей). Единичные лекарственные формы настоящего изобретения могут также содержать комбинацию гранул с контролируемым выделением и матрицы для мультичастиц для достижения желаемого эффекта.
Составы настоящего изобретения с непрерывным выделением предпочтительно обеспечивают медленное выделение терапевтически активного агента, например, при проглатывании и выдержке в желудочном соке, а затем в интестинальных жидкостях. Кривая непрерывного выделения составов данного изобретения, полученных экструзией расплава, может быть различной в зависимости от количества задерживающего агента, например гидрофобного материала, в зависимости от изменения количества пластификатора по сравнению с количеством гидрофобного материала, в зависимости от включения дополнительных ингредиентов или наполнителей, при изменении способа производства и т.д.
В другом варианте изобретения экструдированный из расплава материал готовят, не включая терапевтически активного агента, который добавляют потом, к экструдату. В таких составах обычно терапевтически активный агент смешан с материалом экструдированной матрицы, затем смесь следует таблетировать с получением состава с медленным выделением. Такие составы хороши, например, когда терапевтически активный агент, включенный в состав, чувствителен к температурам, требующимся для размягчения гидрофобного материала и/или материала, задерживающего выделение.
Подробное описание предпочтительных вариантов
Следующие примеры иллюстрируют различные аспекты настоящего изобретения. Их не следует рассматривать как ограничивающие каким-либо образом объем изобретения.
Пример 1.
Проверяют отделяемость гидрохлорида налтрексона от битартрата гидрокодона путем экстракции, подражая тому, как это делает лицо, злоупотребляющее лекарствами. Проверка структур и оценка значений рН позволяют предположить, что оба соединения будут растворимы в кислоте. Однако налтрексон должен быть также более растворимым при более высоких рН с минимумом растворимости между рН 8,4 и 10,3. Хотели проверить гипотезу о том, что оба соединения можно экстрагировать из таблетки в кислоте и затем осадить гидрокодон повышением рН.
Так как таблетки с контролируемым выделением гидрокодона (HYCR) и таблетки налтрексона не годятся для этого исследования, были приготовлены “поддельные” образцы добавлением известных количеств лекарственных субстанций битартрата гидрокодона и гидрохлорида налтрексона к HYCR AcroContin 15 мг таблеткам плацебо (“AcroContin”- запатентованная основа, обеспечивающая контролируемое выделение, состоящая из аммонийметакрилатного полимера вместе с высшим алифатическим спиртом, как описано в примере (US патента 4861598, упомянутого здесь в качестве ссылки)). Различные растворители, меняющие значения рН, использовались для экстракции битартрата гидрокодона и/или солянокислого налтрексона при концентрациях 4 таблетки/25 мл (раздел 2.1) и 5 таблеток/5 мл (раздел 2.2) растворителя. Выделенные продукты определялись с помощью ВЭЖХ.
2.1. Экстракция при концентрации 4 таблетки на 25 мл растворителя.
2.1.1. Около 60 мг битартрата гидрокодона/ 25 мг гидрохлорида налтрексона и 400 мг HYCR 15 мг AcroContin таблеток плацебо (эквивалентных 4 таблеткам) добавили в 25 мл мерную колбу. Добавили около 15 мл воды в мерную колбу и раствор озвучивали в течение 10 мин. Раствор довели до объема водой и хорошо перемешали. Это был основной раствор для опыта. Таким образом приготовили 13 образцов основных растворов.
2.1.2. Регулировали рН растворов либо ледяной уксусной кислотой, либо 0,2 н NaOH до значений рН: 2,0, 4,0, 5,1, 6,0, 6,5, 7,0, 7,4, 8,0, 8,5, 9,0, 9,4 и 10,0. Однако при получении раствора с рН 1,1 использовали соляную кислоту. Затем следовала стадия 2.1.4.
2.1.3. Повторяли стадию 2.1.1., чтобы вместо воды приготовить основные испытуемые растворы в этаноле, метаноле и ацетоне.
2.1.4. Каждый раствор фильтровали, используя 5 мл шприц с 0,45 мкм фильтровальным блоком Millex-HV. 1,0 мл чистого фильтрата отбирали пипеткой в 25 мл мерную колбу, доводили до объема водой и хорошо взбалтывали. Испытуемые растворы наносили на колонку ВЭЖХ, результаты показаны в таблице 1.
2.2. Экстракция при концентрациях 5 таблеток на 5 мл растворителя.
2.2.1. Около 75 мг битартрата гидрокодона и 32 мг гидрохлорида налтрексона добавили в сцинтилляционный сосуд, который содержал 475 мг HYCR 15 мг AcroContin таблеток плацебо (эквивалентных 5 таблеткам). В сцинтилляционный сосуд добавили 5,0 мл воды и раствор озвучивали в течение 10 минут. Это был основной раствор опыта.
2.2.2. рН растворов довели с помощью 50 вес.%/вес. NaOH до 7,1. После того, как растворы отстоялись в течение 1 часа, весь раствор пропустили через 5 мл, имеющийся в распоряжении шприц с 0,45 мкм фильтровальным элементом Millex-HV. 1,0 мл чистого раствора было отмерено пипеткой в 25 мл мерную колбу, содержимое довели водой до объема и хорошо перемешали. Получился основной раствор для опыта с рН 7,1.
2.2.3. Стадии 2.2.1. и 2.2.2. повторили для получения опытных растворов с рН 8,0, 9,0, 10,0, 11,0, 12,0 и 12,7. Образцы растворов потом вводили в систему ВЭЖХ, результаты показаны в таблице 2.
3. Результаты.
Результаты представлены в таблицах 4 и 5. В таблице 5 отмечается, что и гидрокодон, и налтрексон полностью растворимы во всех растворителях, кроме ацетона. В таблице 5 отмечается, что количество налтрексона оставалось в растворе уменьшенных при рН 8 и увеличенным при рН 10,0, а количество налтрексона оставалось в растворе уменьшенным при более высоких значениях рН.
В таблице 4 показана “поддельная” экстрагируемость гидрохлорида налтрексона из содержащих битартрат гидрокодона CR, 15 мг AcroContin таблеток при концентрации 4 таблетки на 25 мл растворителя.
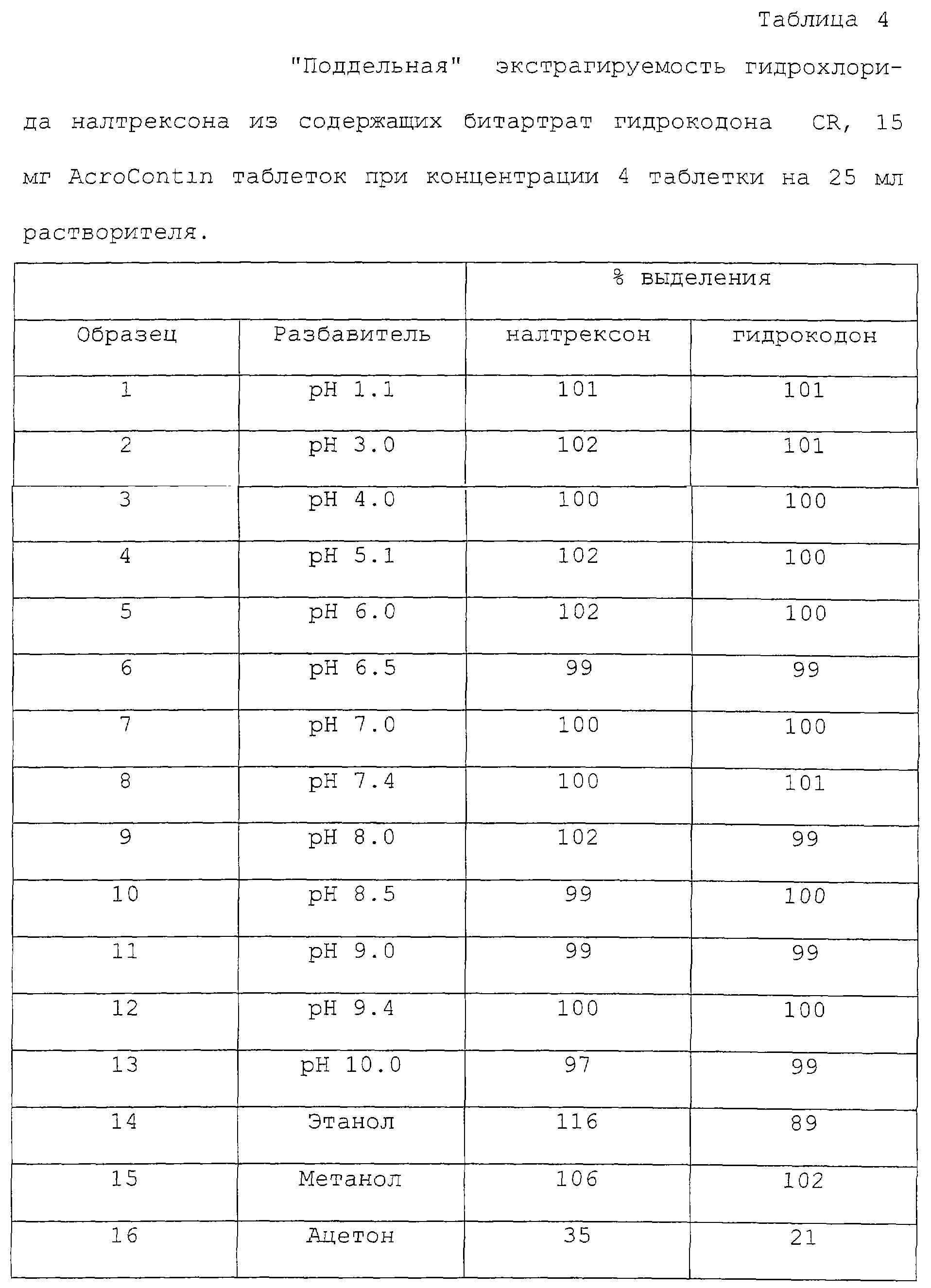 В таблице 5 показана “поддельная” экстрагируемость гидрохлорида налтрексона из содержащего битартрат гидрокодона CR 15 мг, AcroContin таблеток при концентрации 5 таблеток на 5 мл растворителя.
В таблице 5 показана “поддельная” экстрагируемость гидрохлорида налтрексона из содержащего битартрат гидрокодона CR 15 мг, AcroContin таблеток при концентрации 5 таблеток на 5 мл растворителя.
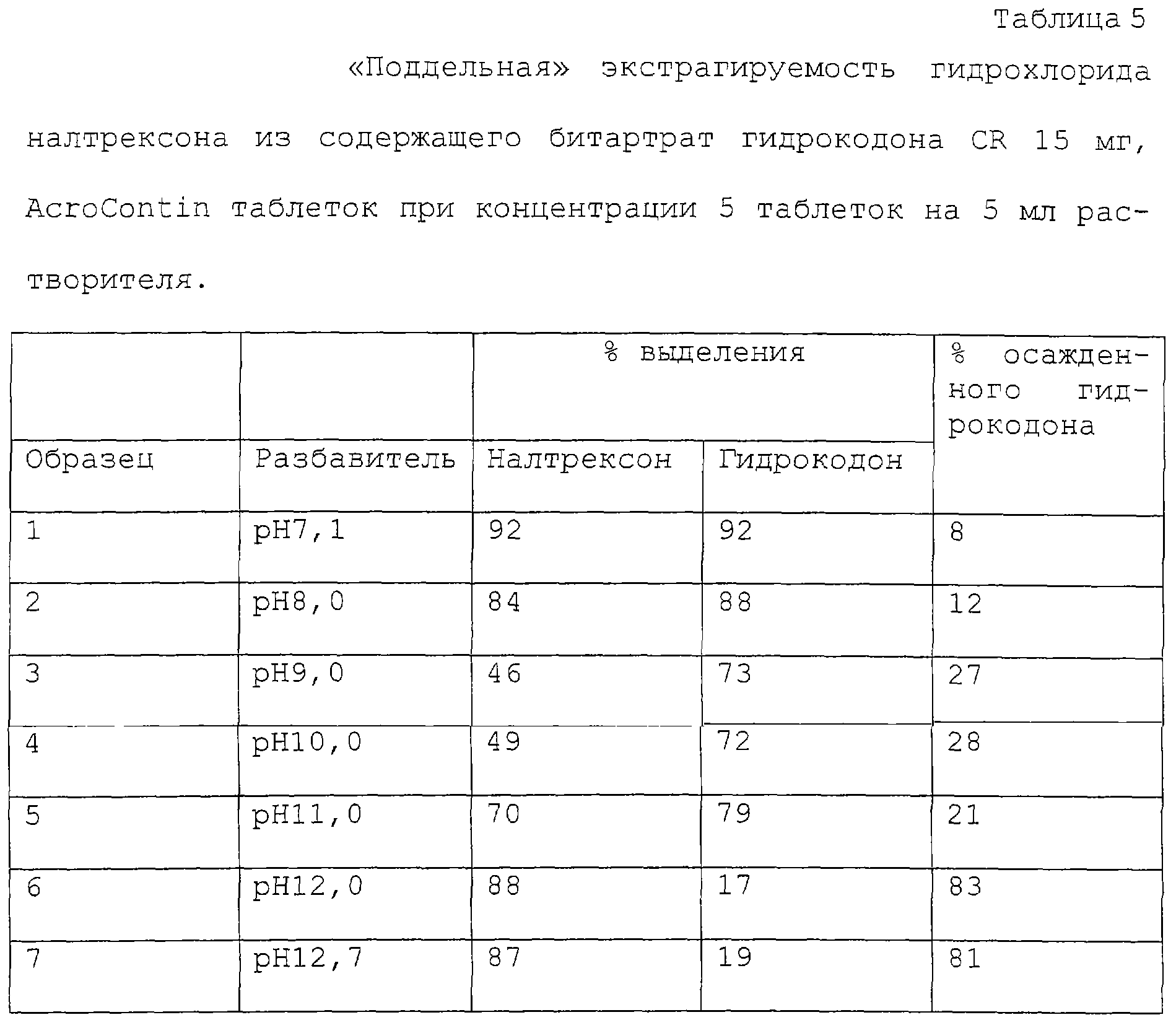 4. Выводы
В таблице 4 показано, что концентрации гидрокодона и налтрексона были слишком низкими в 25 мл растворителей и они почти полностью растворяются при различных рН как в этаноле, так и в метаноле. В ацетоне гидрокодон и налтрексон менее растворимы и получаются небольшие осадки.
В таблице 5 результаты можно объяснить оценкой величин рН лекарственных субстанций. Величины рН для гидрохлорида налтрексона, которые получены в PRC, Yonkers, составляют 8,4 (при функциональной аминогруппе) и 10,3 (при функциональной фенольной группе), а величина рН битартрата гидрокодона (при аминогруппе) составляет 9,2.
Для гидрохлорида налтрексона: так как рН достигает 8,4, налтрексон превращается в свободное основание и начинает осаждаться из раствора, и когда рН достигает 10,3, фенольная ОН- функциональная группа деионизируется и соединение снова растворяется, переходя в раствор. Для битартрата гидрокодона: гидрокодон становится свободным основанием при рН выше 9,2 и начинает осаждаться из раствора.
В таблице 4 показано, что 80% битартрата гидрокодона и 10% соляно-кислого налтрексона могут экстрагироваться из таблетки при более высоких значениях рН.
Эту процедуру нелегко осуществить на улице. Требуются и сильная кислота, и сильное основание плюс стадии размалывания и фильтрования. Кроме того, выделенный гидрокодон смачивают сильной щелочью, любая попытка отмыть каустик приведет к некоторой потере гидрокодона.
Однако важно отметить, что в этом опыте по влажному выделению ни одно лекарство не было включено в матрицу таблеток в процессе производства (горячий воск). Наиболее вероятным является то, что выделения из данной таблетки могли быть хуже. Кроме того, добавление желирующего агента или других наполнителей могло сделать его даже еще более трудным.
Пример 2.
Изучали отделение гидрохлорида налтрексона (1,5 мг) от гидрохлорида гидроморфона (15 мг) экстракцией при концентрации 5 таблеток, 5 мл растворителя с помощью методов, описанных в примере 1. Результаты даны в таблице 6 ниже:
4. Выводы
В таблице 4 показано, что концентрации гидрокодона и налтрексона были слишком низкими в 25 мл растворителей и они почти полностью растворяются при различных рН как в этаноле, так и в метаноле. В ацетоне гидрокодон и налтрексон менее растворимы и получаются небольшие осадки.
В таблице 5 результаты можно объяснить оценкой величин рН лекарственных субстанций. Величины рН для гидрохлорида налтрексона, которые получены в PRC, Yonkers, составляют 8,4 (при функциональной аминогруппе) и 10,3 (при функциональной фенольной группе), а величина рН битартрата гидрокодона (при аминогруппе) составляет 9,2.
Для гидрохлорида налтрексона: так как рН достигает 8,4, налтрексон превращается в свободное основание и начинает осаждаться из раствора, и когда рН достигает 10,3, фенольная ОН- функциональная группа деионизируется и соединение снова растворяется, переходя в раствор. Для битартрата гидрокодона: гидрокодон становится свободным основанием при рН выше 9,2 и начинает осаждаться из раствора.
В таблице 4 показано, что 80% битартрата гидрокодона и 10% соляно-кислого налтрексона могут экстрагироваться из таблетки при более высоких значениях рН.
Эту процедуру нелегко осуществить на улице. Требуются и сильная кислота, и сильное основание плюс стадии размалывания и фильтрования. Кроме того, выделенный гидрокодон смачивают сильной щелочью, любая попытка отмыть каустик приведет к некоторой потере гидрокодона.
Однако важно отметить, что в этом опыте по влажному выделению ни одно лекарство не было включено в матрицу таблеток в процессе производства (горячий воск). Наиболее вероятным является то, что выделения из данной таблетки могли быть хуже. Кроме того, добавление желирующего агента или других наполнителей могло сделать его даже еще более трудным.
Пример 2.
Изучали отделение гидрохлорида налтрексона (1,5 мг) от гидрохлорида гидроморфона (15 мг) экстракцией при концентрации 5 таблеток, 5 мл растворителя с помощью методов, описанных в примере 1. Результаты даны в таблице 6 ниже:
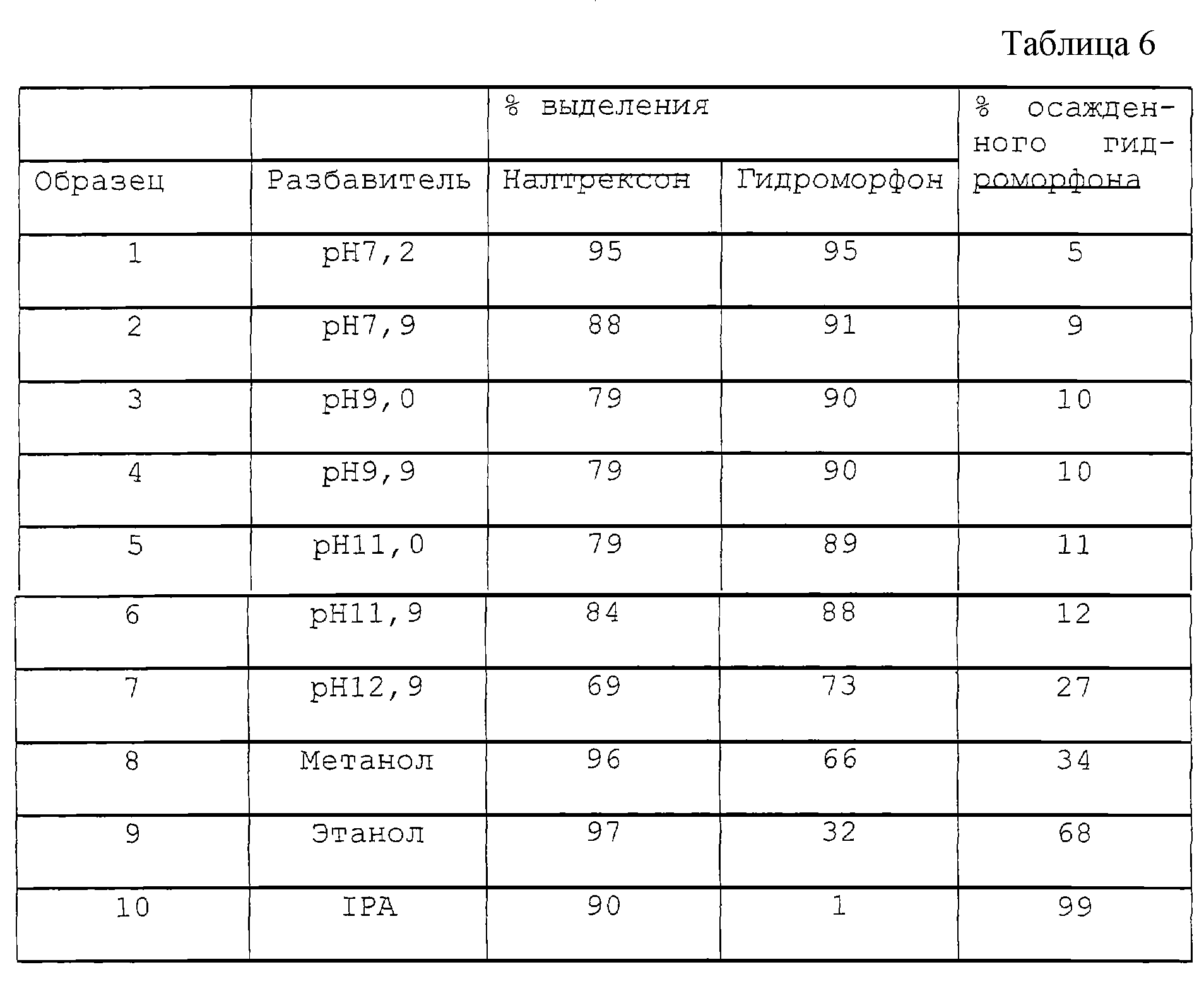 Пример 3.
Изучалось отделение гидрохлорида налтрексона (1,5 мг) от гидрохлорида оксикодона (15 мг) экстракцией при концентрации 5 таблеток/ 5 мл растворителя с помощью способов, описанных в примере 1. Результаты даны в таблице 7 ниже.
Пример 3.
Изучалось отделение гидрохлорида налтрексона (1,5 мг) от гидрохлорида оксикодона (15 мг) экстракцией при концентрации 5 таблеток/ 5 мл растворителя с помощью способов, описанных в примере 1. Результаты даны в таблице 7 ниже.
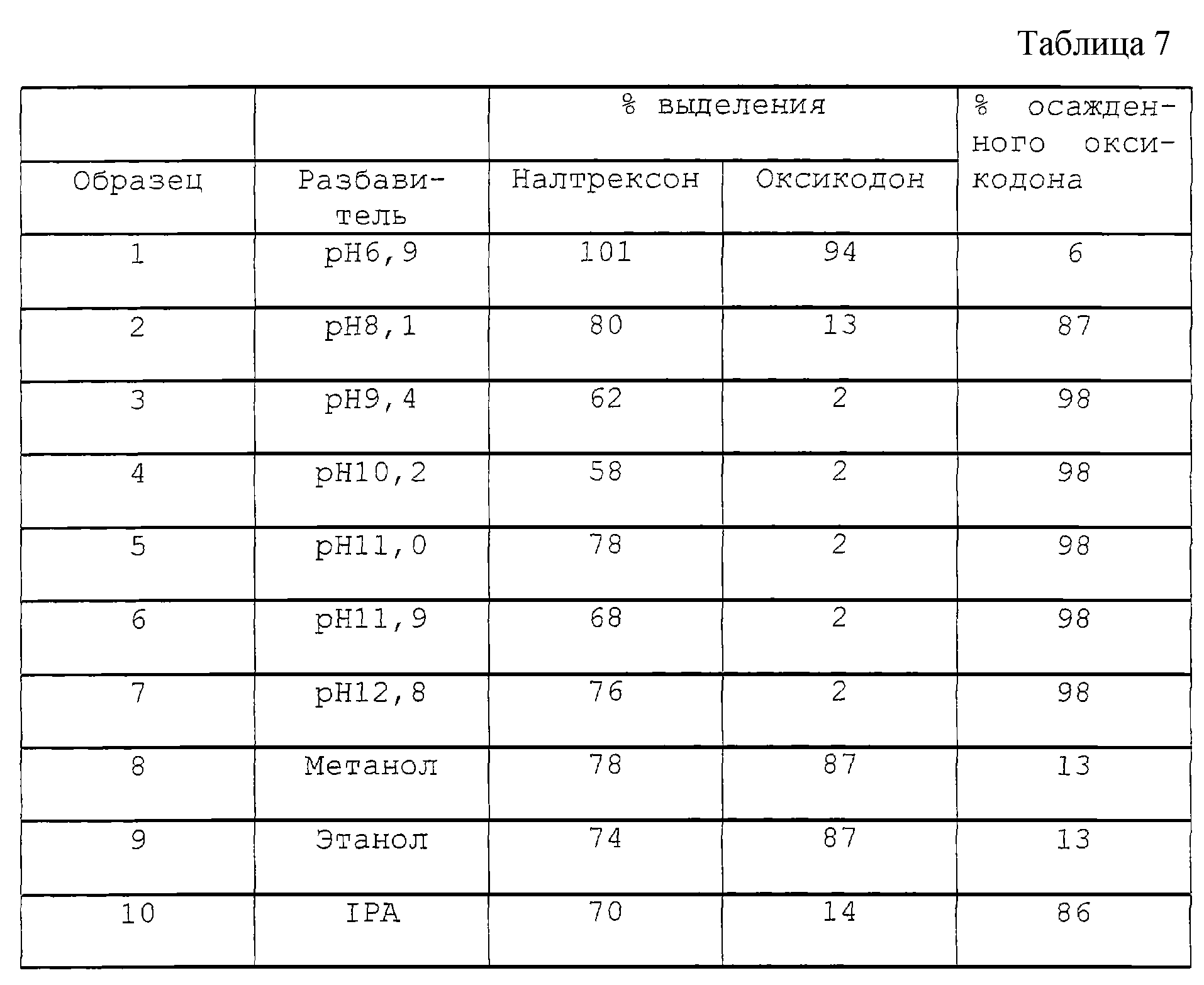 Хотя изобретение описано и проиллюстрировано с акцентом на некоторые предпочтительные варианты, специалисту в данной области будет понятно, что могут быть сделаны очевидные модификации без искажения идеи и объема изобретения. Такие изменения рассматриваются как подпадающие под формулу изобретения.
Хотя изобретение описано и проиллюстрировано с акцентом на некоторые предпочтительные варианты, специалисту в данной области будет понятно, что могут быть сделаны очевидные модификации без искажения идеи и объема изобретения. Такие изменения рассматриваются как подпадающие под формулу изобретения.
Формула изобретения 1. Способ уменьшения возможности неправильного употребления (злоупотребления) лекарственной оральной формы опиоидного анальгетика, включающий объединение анальгетически эффективного количества активного опиоидного агоиста с опиоидным антагонистом в лекарственную оральную форму, причем указанная комбинация опиоидных агониста и антагониста выбрана таким образом, что опиоидные агонист и антагонист могут быть экстрагированы из лекарственной формы только совместно, и для отделения опиоидного антагониста от опиоидного агониста требуется, по меньшей мере, двухэтапная экстракция, а количество опиоидного антагониста достаточно, чтобы противодействовать опиодному действию агониста при их совместной экстракции из оральной лекарственной формы и парентеральном введении. 2. Способ по п.1, в котором доза указанного антагониста обусловливает то, что комбинация опиоидных агониста и антагониста обеспечивает создание отвращения у физически зависимого человека при оральном введении лекарственной формы. 3. Способ по п.2, в котором указанная комбинация указанных опиоидных агониста и антагониста такова, что она требует только совместной экстракции из лекарственной формы, а затем они должны отделяться друг от друга на отдельном этапе экстракции. 4. Способ по п.3, в котором и указанные опиоидные агонист и антагонист растворимы в кислоте, и должны разделяться с использованием раствора с высоким значением рН. 5. Способ по п.4, в котором указанный опиоидный агонист представляет собой битартрат гидрокодона, а указанный опиоидный антагонист представляет собой гидрохлорид налтрексона, причем и гидрокодон, и налтрексон растворяются при значении рН менее 8, и около 80% указанного гидрокодона и около 10% указанного налтрексона могут быть экстрагированы при высоком значении рН. 6. Способ по пп.1 и 2, в котором опиоидный агонист представляет собой гидрохлорид гидроморфона, а опиоидный антагонист представляет собой гидрохлорид налтрексона. 7. Способ по пп.1 и 2, в котором опиоидный агонист представляет собой гидрохлорид оксикодона, а опиоидный антагонист представляет собой гидрохлорид налтрексона. 8. Способ по пп.1 и 2, в котором опиоидный агонист представляет собой сульфат морфина, а опиоидный антагонист представляет собой гидрохлорид налтрексона. 9. Способ по п.4, включающий введение в лекарственную форму дополнительного ингредиента, который затрудняет отделение опиоидного агониста от опиоидного антагониста. 10. Способ по п.9, в котором указанный дополнительный ингредиент выбран из группы, состоящей из желатинирующих агентов, восков и их смесей. 11. Способ по п.9, дополнительно включающий в процесс получения лекарственной формы один или несколько этапов обработки, которые дополнительно препятствуют разделению опиоидного агониста от опиоидного антагониста. 12. Способ по п.1, в котором опиоидный агонист выбран из группы, состоящей из гидрокодона, гидроморфона, оксикодона, морфина, кодеина, леворфанола, меперидина, метадона и их солей, а доза опиоидного антагониста или его фармацевтически приемлемой соли достаточна для того, чтобы противодействовать опиоидному действию при совместной экстракции их с опиоидным агонистом из оральной лекарственной формы и парентеральном введении. 13. Способ по п.12, в котором указанная комбинация опиоидных агониста и антагониста такова, что они могут быть только совместно экстрагированы из лекарственной формы, а затем они должны отделяться друг от друга на отдельном этапе экстракции. 14. Способ по п.13, в котором указанные опиоидный агонист и антагонист растворимы в кислоте и должны разделяться с использованием раствора с высоким значением рН. 15. Способ по п.14, в котором указанный опиоидный агонист представляет собой битартрат гидрокодона, а указанный опиоидный антагонист представляет собой гидрохлорид налтрексона, причем и гидрокодон, и налтрексон растворяются при значении рН менее 8, и около 80% указанного гидрокодона и около 10% указанного налтрексона могут быть экстрагированы при высоком значении рН. 16. Способ по п.12, в котором опиоидный агонист представляет собой гидрохлорид гидроморфона. 17. Способ по п.12, в котором опиоидный агонист представляет собой гидрохлорид оксикодона. 18. Способ по п.12, в котором опиоидный агонист представляет собой сульфат морфина. 19. Способ по п.12, в котором опиоидный агонист представляет собой битартрат гидрокодона. 20. Способ по п.14, включающий введение в лекарственную форму дополнительного ингредиента, который затрудняет отделение опиоидного агониста от антагониста. 21. Способ по п.20, в котором указанный дополнительный ингредиент выбран из группы, состоящей из желатинирующих агентов, восков и их смесей. 22. Способ по п.12, в котором указанный опиоидный антагонист представляет собой налтрексон или его фармацевтически приемлемую соль. 23. Способ по п.16, в котором указанный опиоидный антагонист представляет собой налтрексон или его фармацевтически приемлемую соль, и соотношение между указанным налтрексоном и указанным гидроморфоном составляет от около 0,148:1 до около 1,185:1 (мас.). 24. Способ по п.16, в котором указанный опиоидный антагонист представляет собой налтрексон или его фармацевтически приемлемую соль, и соотношение между указанным налтрексоном и указанным гидроморфоном составляет от около 0,222:1 до около 0,889:1 (мас.). 25. Способ по п.17, в котором указанный опиоидный антагонист представляет собой налтрексон или его фармацевтически приемлемую соль, и соотношение между указанным налтрексоном и указанным оксикодоном составляет от около 0,037:1 до около 0,296:1 (мас.). 26. Способ по п.17, в котором указанный опиоидный антагонист представляет собой налтрексон или его фармацевтически приемлемую соль, и соотношение между указанным налтрексоном и указанным оксикодоном составляет от около 0,056:1 до около 0,222:1 (мас.). 27. Способ по п.18, в котором указанный опиоидный антагонист представляет собой налтрексон или его фармацевтически приемлемую соль, и соотношение между указанным налтрексоном и указанным морфином составляет от около 0,018:1 до около 1,148:1 (мас.). 28. Способ по п.18, в котором указанный опиоидный антагонист представляет собой налтрексон или его фармацевтически приемлемую соль, и соотношение между указанным налтрексоном и указанным морфином составляет от около 0,028:1 до около 0,111:1 (мас.). 29. Способ по п.19, в котором указанный опиоидный антагонист представляет собой налтрексон или его фармацевтически приемлемую соль, и соотношение между указанным налтрексоном и указанным гидрокодоном составляет от около 0,03:1 до около 0,27:1 (мас.). 30. Способ по п.19, в котором указанный опиоидный антагонист представляет собой налтрексон или его фармацевтически приемлемую соль, и соотношение между указанным налтрексоном и указанным гидрокодоном составляет от около 0,05:1 до около 0,20:1 (мас.). 31. Способ по п.1, в котором опиоидный агонист выбран из группы, состоящей из гидрокодона, гидроморфона, оксикодона, морфина, кодеина, леворфанола, меперидина, метадона и их солей, а доза опиоидного антагониста или его фармацевтически приемлемой соли достаточна для того, чтобы противодействовать анальгетическому действию указанного опиоида при их совместной экстракции с опиоидным агонистом из оральной лекарственной формы и парентеральном введении. 32. Способ по п.31, в котором указанный опиоидный агонист представляет собой битартрат гидрокодона, а указанный опиоидный антагонист представляет собой гидрохлорид налтрексона. 33. Способ по п.32, включающий введение в лекарственную форму дополнительного ингредиента, который затрудняет отделение опиоидного агониста от антагониста, причем указанный дополнительный ингредиент выбран из группы, состоящей из желатинирующих агентов, восков и их смесей. 34. Способ по п.31, в котором указанный опиоидный антагонист представляет собой налтрексон или его фармацевтически приемлемую соль. 35. Способ по п.31, в котором доза указанного антагониста обусловливает то, что комбинация опиоидного агониста и антагониста обеспечивает создание отвращения действие у физически зависимого человека при оральном введении лекарственной формы. 36. Способ по п.1, в котором оральная лекарственная форма опиоидного анальгетика является формой непрерывного высвобождения, включающий объединение анальгетически эффективного количества активного при оральном применении опиоидного агониста с опиоидным антагонистом и носителем непрерывного высвобождения в лекарственную форму, которая обеспечивает высвобождение указанного опиоидного анальгетика в течение от около 12 до около 24 ч. 37. Способ по п.36, в котором указанная комбинация указанных опиоидных агониста и антагониста такова, что они могут быть только совместно экстрагированы из лекарственной формы, а затем должны отделяться друг от друга на отдельном этапе экстракции. 38. Способ по п.37, в котором и указанные опиоидный агонист и антагонист растворимы в кислоте, и должны разделяться с использованием раствора с высоким значением рН. 39. Способ по п.38, в котором указанный опиоидный агонист представляет собой битартрат гидрокодона, а указанный опиоидный антагонист представляет собой гидрохлорид налтрексона, причем и гидрокодон, и налтрексон растворяются при значении рН менее 8, и около 80% указанного гидрокодона и около 10% указанного налтрексона могут быть экстрагированы при высоком значении рН. 40. Способ по п.36, в котором опиоидный агонист представляет собой гидрохлорид гидроморфона, а опиоидный антагонист представляет собой гидрохлорид налтрексона. 41. Способ по п.36, в котором опиоидный агонист представляет собой гидрохлорид оксикодона, а опиоидный антагонист представляет собой гидрохлорид налтрексона. 42. Способ по п.36, в котором опиоидный агонист представляет собой сульфат морфина, а опиоидный антагонист представляет собой гидрохлорид налтрексона. 43. Способ по п.38, включающий введение в лекарственную форму дополнительного ингредиента, который затрудняет отделение опиоидного агониста от опиоидного антагониста. 44. Способ по п.43, в котором указанный дополнительный ингредиент выбран из группы, состоящей из желатинирующих агентов, восков и их смесей. 45. Способ по п.43, дополнительно включающий в процесс получения лекарственной формы один или несколько этапов обработки, которые дополнительно препятствуют отделению опиоидного агониста от опиоидного антогониста. 46. Способ по п.36, в котором указанный опиоидный антагонист представляет собой налтрексон или его фармацевтически приемлемую соль, указанный опиоидный агонист представляет собой гидроморфон или его фармацевтически приемлемую соль, и соотношение между указанным налтрексоном и указанным гидроморфоном составляет от около 0,222:1 до около 0,889:1 (мас.). 47. Способ по п.36, в котором указанный опиоидный антагонист представляет собой налтрексон или его фармацевтически приемлемую соль, указанный опиоидный агонист представляет собой оксикодон или его фармацевтически приемлемую соль, и соотношение между указанным налтрексоном и указанным оксикодоном составляет от около 0,037:1 до около 0,296:1 (мас.). 48. Способ по п.36, в котором указанный опиоидный антагонист представляет собой налтрексон или его фармацевтически приемлемую соль, указанный опиоидный агонист представляет собой оксикодон или его фармацевтически приемлемую соль, и соотношение между указанным налтрексоном и указанным оксикодоном составляет от около 0,056:1 до около 0,222:1 (мас.). 49. Способ по п.36, в котором указанный опиоидный антагонист представляет собой налтрексон или его фармацевтически приемлемую соль, указанный опиоидный агонист представляет собой морфин или его фармацевтически приемлемую соль, и соотношение между указанным налтрексоном и указанным морфином составляет от около 0,018:1 до около 0,148:1 (мас.). 50. Способ по п.36, в котором указанный опиоидный антагонист представляет собой налтрексон или его фармацевтически приемлемую соль, указанный опиоидный агонист представляет собой морфин или его фармацевтически приемлемую соль, и соотношение между указанным налтрексоном и указанным морфином составляет от около 0,028:1 до около 0,111:1 (мас.). 51. Способ по п.36, в котором указанный опиоидный антагонист представляет собой налтрексон или его фармацевтически приемлемую соль, указанный опиоидный агонист представляет собой гидрокодон или его фармацевтически приемлемую соль, и соотношение между указанным налтрексоном и указанным гидрокодоном составляет от около 0,03:1 до около 0,27:1 (мас.). 52. Способ по п.36, в котором указанный опиоидный антагонист представляет собой налтрексон или его фармацевтически приемлемую соль, указанный опиоидный агонист представляет собой гидрокодон или его фармацевтически приемлемую соль, и соотношение между указанным налтрексоном и указанным гидрокодоном составляет от около 0,05:1 до около 0,20:1 (мас.). 53. Способ по п.36, в котором указанный опиоидный антагонист представляет собой налтрексон или его фармацевтически приемлемую соль. 54. Способ по п.36, в котором указанный опиоидный антагонист обусловливает то, что комбинация опиоидных агониста и антагониста обеспечивает создание отвращения у физически зависимого человека при оральном введении лекарственной формы непрерывного высвобождения. 55. Способ по п.36, в котором указанные опиоидные агонист и антагонист включены в матричную композицию. 56. Способ по п.55, в котором указанный носитель непрерывного высвобождения включен в указанную матричную композицию с указанными опиоидными агонистом и антагонистом. 57. Способ по п.55, в котором указанный носитель непрерывного высвобождения нанесен в виде покрытия на указанную матричную композицию. 58. Способ по п.55, в котором указанный носитель непрерывного высвобождения включен в указанную матричную композицию с указанным опиоидным агонистом и нанесен в виде покрытия на указанную матричную композицию. MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 23.12.2005
Извещение опубликовано: 20.12.2007 БИ: 35/2007
|
||||||||||||||||||||||||||