Патент на изобретение №2226192
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ 4Н-ТИОПИРАНОВ
(57) Реферат: Изобретение относится к способу получения 4Н-тиопиранов, которые могут применяться в качестве материалов для оптоэлектроники, полупродуктов органического синтеза, лекарственных препаратов и др. Замещенные 4Н-тиопираны общей формулы I, где R = Н; Cl; ОСН3, получают взаимодействием 1,5-дикетонов с сероводородом, источником которого является сульфид цинка, в среде метанола в присутствии эфиров фосфористой кислоты, образующихся непосредственно в реакционной смеси при добавлении в нее треххлористого фосфора. Изобретение позволяет получить 4Н-тиопираны способом, не связанным с применением газообразных сероводорода и хлористого водорода, не имеющим дополнительной стадии очистки, и по которому можно получать соединения, склонные к реакциям диспропорционирования. 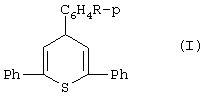 I. Область применения.
II. Предшествующий уровень техники.
Недостатками данного метода являлись
а) невозможность получения тиопиранов, склонных к реакциям диспропорционирования, например триарилзамещенных 4Н-тиопиранов (в прикладном аспекте больший интерес вызывают тиопираны с меньшей степенью замещения, которые легко подвергаются реакциям диспропорционирования);
б) полученные соединения требовали дополнительной очистки, так как часть целевого продукта выпадала из реакционной смеси в виде масла;
в) для проведения реакции требовалось использования газообразного сухого хлористого водорода, которым предварительно насыщали реакционную смесь, контролируя процесс насыщения титрованием.
III. Сущность изобретения.
Целью данного изобретения является создание нового метода синтеза 4Н-тиопиранов, который не был бы связан с применением газообразных сероводорода и хлористого водорода, не имел дополнительной стадии очистки, легко мог быть внедрен в промышленность и по которому можно было получать соединения, склонные к реакциям диспропорционирования.
В связи с этим мы решили провести синтез 4Н-тиопиранов разнообразного строения при действии сероводорода in situ на 1,5-дикетоны в присутствии эфиров фосфористой кислоты и хлористого водорода.
Реакцию проводили в метаноле при комнатной температуре. Насыщение метанола хлороводородом и образование эфиров фосфористой кислоты происходило при взаимодействии РСl3 со спиртом. Источником сероводорода служил сульфид цинка.
I. Область применения.
II. Предшествующий уровень техники.
Недостатками данного метода являлись
а) невозможность получения тиопиранов, склонных к реакциям диспропорционирования, например триарилзамещенных 4Н-тиопиранов (в прикладном аспекте больший интерес вызывают тиопираны с меньшей степенью замещения, которые легко подвергаются реакциям диспропорционирования);
б) полученные соединения требовали дополнительной очистки, так как часть целевого продукта выпадала из реакционной смеси в виде масла;
в) для проведения реакции требовалось использования газообразного сухого хлористого водорода, которым предварительно насыщали реакционную смесь, контролируя процесс насыщения титрованием.
III. Сущность изобретения.
Целью данного изобретения является создание нового метода синтеза 4Н-тиопиранов, который не был бы связан с применением газообразных сероводорода и хлористого водорода, не имел дополнительной стадии очистки, легко мог быть внедрен в промышленность и по которому можно было получать соединения, склонные к реакциям диспропорционирования.
В связи с этим мы решили провести синтез 4Н-тиопиранов разнообразного строения при действии сероводорода in situ на 1,5-дикетоны в присутствии эфиров фосфористой кислоты и хлористого водорода.
Реакцию проводили в метаноле при комнатной температуре. Насыщение метанола хлороводородом и образование эфиров фосфористой кислоты происходило при взаимодействии РСl3 со спиртом. Источником сероводорода служил сульфид цинка.
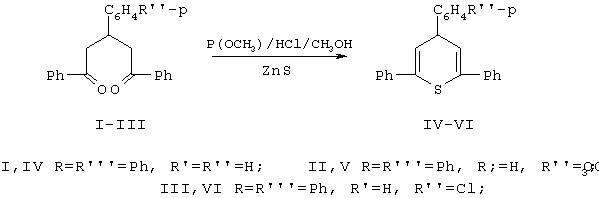 Чистота полученных соединений контролировалась методом КГХ с масс-селективным детектором HP 5890/5972. Условия хроматографии: Tинж=200
Чистота полученных соединений контролировалась методом КГХ с масс-селективным детектором HP 5890/5972. Условия хроматографии: Tинж=200 С; tнaч=3 мин; Ткон=280С; С; tнaч=3 мин; Ткон=280С;  Т=10С/мин; газ-носитель гелий, Т=10С/мин; газ-носитель гелий,  =1 мл/мин; капиллярная колонка HP-5MS.
Для полной иллюстрации изобретения приводим конкретные методики выполнения синтеза.
Пример 1.
Получение 2,6-дифенил-4-(п-метоксифенил)-4Н-тиопирана.
К 19 мл метанола по каплям при интенсивном перемешивании и охлаждении добавляют 3,25 мл треххлористого фосфора. Затем нагревают содержимое колбы до 30С и засыпают небольшими порциями при перемешивании 0,0025М (0,815 г) 1,5-дифенил-3-(п-метоксифенил)-пентандиона-1,5. После полного растворения дикетона (~15 мин) в колбу добавляют небольшими порциями 0,003М (0,29 г) сульфида цинка. Реакционную смесь перемешивают при комнатной температуре до полного исчезновения исходного дикетона по ТСХ (~3 ч). Полученные светло-желтые кристаллы отфильтровывают, промывают на фильтре 10 мл этилового спирта. Сушат в темноте на воздухе. Получают 0.734 г (90%) 2,6-дифенил-4-(п-метоксифенил)-4Н-тиопирана. Т.пл. 94-96С (без дополнительной очистки). Лит. Т.пл. 94-96С [SU 225216, опубл. 16.12.1968]. Данные хроматомасс-спектрометрии показали одинаковые время удерживания и масс-спектры для стандартного и опытного образца.
Реакционную колбу споласкивают 10 мл эфира и выливают содержимое в маточный раствор, добавляют 20 мл воды. Эфирный слой отделяют, промывают водой, сушат, анализируют методом хроматомасс-спектрометрии. Эфирный раствор содержит 2,6-дифенил-4-(п-метоксифенил)-4Н-тиопиран с примесью 2,6-дифенил-4-(п-метоксифенил)-2,3-дигидро-4Н-тиопирана.
Пример 2. Получение 2,4,6-трифенил-4Н-тиопирана.
Получают аналогично вышеприведенной методике, исходя из 0,0025М (0,82 г) 1,3,5-трифенил-пентандиона-1,5. Выход целевого продукта 0,755 г (92%). Т.пл. 103-105С (без дополнительной очистки). Лит. Т.пл. 104-106С [SU 225216, опубл. 16.12.1968]. Данные хроматомасс-спектрометрии показали одинаковые результаты для опытного и контрольного образцов (время удерживания, масс-спектры).
Маточный раствор содержал 2,4,6-трифенил-4Н-тиопиран с примесью 2,4,6-трифенил-2,3-дигидро-4Н-тиопирана.
Пример 3. Получение 2,6-дифенил-4-(п-хлорфенил)-4Н-тиопирана.
Получают аналогично вышеприведенной методике исходя из 0,0025М (0,83 г) 1,5-дифенил-3-(п-хлорфенил)-пентандиона-1,5.
Выход целевого продукта 0,73 г (88%). Т.пл. 95-97С (без дополнительной очистки). Лит. Т.пл. 95-97С [SU 225216, опубл. 16.12.1968]. Данные хроматомасс-спектрометрии показали одинаковые результаты для опытного и контрольного образцов (время удерживания, масс-спектры).
Маточный раствор содержал 2,6-дифенил-4-(п-хлорфенил)-4Н-тиопиран с примесью 2,6-дифенил-4-(п-хлорфенил)-2,3-дигидро-4Н-тиопирана. =1 мл/мин; капиллярная колонка HP-5MS.
Для полной иллюстрации изобретения приводим конкретные методики выполнения синтеза.
Пример 1.
Получение 2,6-дифенил-4-(п-метоксифенил)-4Н-тиопирана.
К 19 мл метанола по каплям при интенсивном перемешивании и охлаждении добавляют 3,25 мл треххлористого фосфора. Затем нагревают содержимое колбы до 30С и засыпают небольшими порциями при перемешивании 0,0025М (0,815 г) 1,5-дифенил-3-(п-метоксифенил)-пентандиона-1,5. После полного растворения дикетона (~15 мин) в колбу добавляют небольшими порциями 0,003М (0,29 г) сульфида цинка. Реакционную смесь перемешивают при комнатной температуре до полного исчезновения исходного дикетона по ТСХ (~3 ч). Полученные светло-желтые кристаллы отфильтровывают, промывают на фильтре 10 мл этилового спирта. Сушат в темноте на воздухе. Получают 0.734 г (90%) 2,6-дифенил-4-(п-метоксифенил)-4Н-тиопирана. Т.пл. 94-96С (без дополнительной очистки). Лит. Т.пл. 94-96С [SU 225216, опубл. 16.12.1968]. Данные хроматомасс-спектрометрии показали одинаковые время удерживания и масс-спектры для стандартного и опытного образца.
Реакционную колбу споласкивают 10 мл эфира и выливают содержимое в маточный раствор, добавляют 20 мл воды. Эфирный слой отделяют, промывают водой, сушат, анализируют методом хроматомасс-спектрометрии. Эфирный раствор содержит 2,6-дифенил-4-(п-метоксифенил)-4Н-тиопиран с примесью 2,6-дифенил-4-(п-метоксифенил)-2,3-дигидро-4Н-тиопирана.
Пример 2. Получение 2,4,6-трифенил-4Н-тиопирана.
Получают аналогично вышеприведенной методике, исходя из 0,0025М (0,82 г) 1,3,5-трифенил-пентандиона-1,5. Выход целевого продукта 0,755 г (92%). Т.пл. 103-105С (без дополнительной очистки). Лит. Т.пл. 104-106С [SU 225216, опубл. 16.12.1968]. Данные хроматомасс-спектрометрии показали одинаковые результаты для опытного и контрольного образцов (время удерживания, масс-спектры).
Маточный раствор содержал 2,4,6-трифенил-4Н-тиопиран с примесью 2,4,6-трифенил-2,3-дигидро-4Н-тиопирана.
Пример 3. Получение 2,6-дифенил-4-(п-хлорфенил)-4Н-тиопирана.
Получают аналогично вышеприведенной методике исходя из 0,0025М (0,83 г) 1,5-дифенил-3-(п-хлорфенил)-пентандиона-1,5.
Выход целевого продукта 0,73 г (88%). Т.пл. 95-97С (без дополнительной очистки). Лит. Т.пл. 95-97С [SU 225216, опубл. 16.12.1968]. Данные хроматомасс-спектрометрии показали одинаковые результаты для опытного и контрольного образцов (время удерживания, масс-спектры).
Маточный раствор содержал 2,6-дифенил-4-(п-хлорфенил)-4Н-тиопиран с примесью 2,6-дифенил-4-(п-хлорфенил)-2,3-дигидро-4Н-тиопирана.
Формула изобретения Способ получения замещенных 4Н-тиопиранов общей формулы 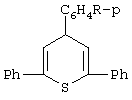 где R=Н, Сl, ОСН3,
включающий взаимодействие 1,5-дикетонов с сероводородом, источником которого является сульфид цинка, в среде метанола, отличающийся тем, что процесс проводят в присутствии эфиров фосфористой кислоты, образующихся непосредственно в реакционной смеси при добавлении в нее треххлористого фосфора.
где R=Н, Сl, ОСН3,
включающий взаимодействие 1,5-дикетонов с сероводородом, источником которого является сульфид цинка, в среде метанола, отличающийся тем, что процесс проводят в присутствии эфиров фосфористой кислоты, образующихся непосредственно в реакционной смеси при добавлении в нее треххлористого фосфора.
MM4A Досрочное прекращение действия патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 10.10.2003
Извещение опубликовано: 20.05.2006 БИ: 14/2006
|
||||||||||||||||||||||||||