Патент на изобретение №2225859
|
||||||||||||||||||||||||||
(54) СИНТЕЗ 3-АМИНО-3-АРИЛПРОПАНОАТОВ
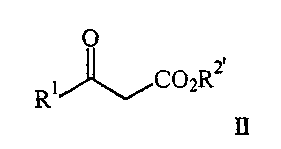
(57) Реферат: Изобретение относится к усовершенствованному способу получения соединения формулы I 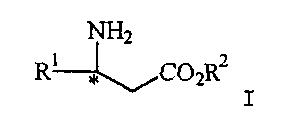 где R1 представляет арил, гетероарил, замещенный арил или замещенный гетероарил, и R2 представляет водород, алкил или аралкил, или его соли, включающий взаимодействие соединения формулы II
где R1 представляет арил, гетероарил, замещенный арил или замещенный гетероарил, и R2 представляет водород, алкил или аралкил, или его соли, включающий взаимодействие соединения формулы II
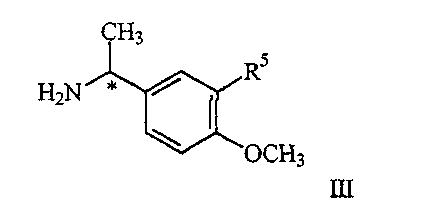
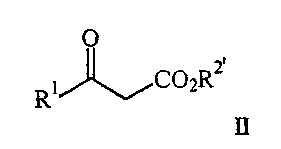 где R1 является таким, как описано выше, и R2′ представляет алкил или аралкил, с соединением формулы III
где R1 является таким, как описано выше, и R2′ представляет алкил или аралкил, с соединением формулы III
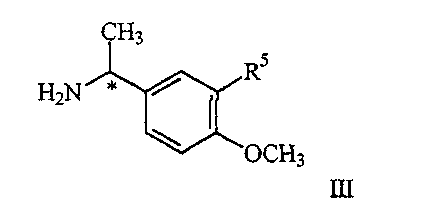 где R5 представляет водород или алкокси, в условиях пониженного давления, при температуре кипения реакционного раствора (от 40 до 65
где R5 представляет водород или алкокси, в условиях пониженного давления, при температуре кипения реакционного раствора (от 40 до 65 С) в инертном растворителе с азеотропной отгонкой воды, полученное соединение IV С) в инертном растворителе с азеотропной отгонкой воды, полученное соединение IV
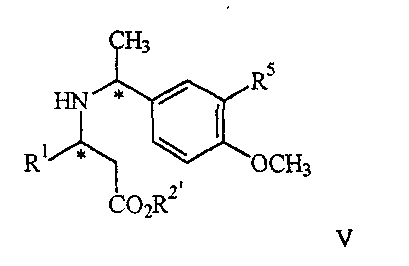
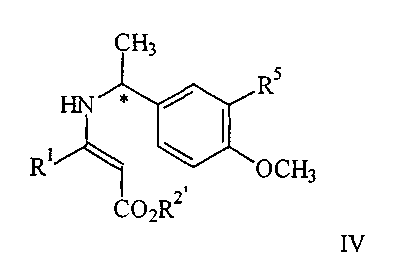 обрабатывают с газообразным водородом в присутствии палладиевого катализатора до получения соединения V
обрабатывают с газообразным водородом в присутствии палладиевого катализатора до получения соединения V
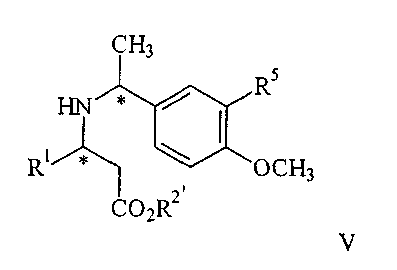 которое при взаимодействии с кислотой дают соединение формулы I;
или путем взаимодействия соединения IV с газообразным водородом в присутствии палладиевого катализатора и переводом полученного соединения V обработкой кислотой в целевой продукт;
промежуточным продуктам для синтеза соединений I, новым соединениям общих формул IV и V.
Предлагаемый способ может быть использован для крупномасштабного производства соединений формулы I с приемлемым уровнем чистоты и выхода. 4 с. и 18 з.п. ф-лы.
Изобретение относится к способу получения соединения формулы
которое при взаимодействии с кислотой дают соединение формулы I;
или путем взаимодействия соединения IV с газообразным водородом в присутствии палладиевого катализатора и переводом полученного соединения V обработкой кислотой в целевой продукт;
промежуточным продуктам для синтеза соединений I, новым соединениям общих формул IV и V.
Предлагаемый способ может быть использован для крупномасштабного производства соединений формулы I с приемлемым уровнем чистоты и выхода. 4 с. и 18 з.п. ф-лы.
Изобретение относится к способу получения соединения формулы
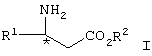 где R1 представляет арил, гетероарил, замещенный арил или замещенный гетероарил, и R2 представляет водород, алкил или аралкил, или его соли.
Соединения формулы I могут использоваться в качестве промежуточных продуктов при синтезе, помимо прочего, соединений, описанных в WO 97/41102. Соединения, описанные в WO 97/41102, представляют собой антагонисты тромбоцитного фибриногенного рецептора (gp IIb/IIIa антагонист) и таким образом могут использоваться для лечения тромбоцитопосредованных тромботических заболеваний, таких как артериальный и венозный тромбоз, острый инфаркт миокарда, повторное сужение после тромболитической терапии и ангиопластики, воспаление, нестабильная стенокардия и заболевания, связанные с закупоркой сосудов.
Известные способы получения соединений формулы I, описанные в патенте США 5254573, включают асимметрическое присоединение по Михаэлю (R)-N-(триметилсилил)-(1)-фенетиламида лития к этил-3-пиридилакрилату с получением сложного этил-
где R1 представляет арил, гетероарил, замещенный арил или замещенный гетероарил, и R2 представляет водород, алкил или аралкил, или его соли.
Соединения формулы I могут использоваться в качестве промежуточных продуктов при синтезе, помимо прочего, соединений, описанных в WO 97/41102. Соединения, описанные в WO 97/41102, представляют собой антагонисты тромбоцитного фибриногенного рецептора (gp IIb/IIIa антагонист) и таким образом могут использоваться для лечения тромбоцитопосредованных тромботических заболеваний, таких как артериальный и венозный тромбоз, острый инфаркт миокарда, повторное сужение после тромболитической терапии и ангиопластики, воспаление, нестабильная стенокардия и заболевания, связанные с закупоркой сосудов.
Известные способы получения соединений формулы I, описанные в патенте США 5254573, включают асимметрическое присоединение по Михаэлю (R)-N-(триметилсилил)-(1)-фенетиламида лития к этил-3-пиридилакрилату с получением сложного этил- -аминоэфира. Данный способ приводит к неэффективному образованию амида лития и трудному удалению N-(метилбензильной) группы.
В J.Org.Chem., vol.61, p.2222 (1996) описан способ, в котором литиевый енолят этилацетата добавляют к энантиомерному сульфинимину, продукт взаимодействия очищают хроматографией и снимают защитную группу в кислых условиях, получая сложный -аминоэфир с более чем 90% ее (энантиомерным избытком). Необходимость хроматографической очистки делает данный способ не привлекательным для крупномасштабного производства.
В WO 98/02410 описан способ стереоселективного присоединения реактива Реформатского, полученного из трет-бутилбромацетата, к энантиомерному имину, полученному из 3-пиридинкарбоксальдегида и (R)-2-фенилглицинола. Окислительное расщепление N-(1-фенил-2-гидроксиэтильной) группы NaIO4 в этаноле с последующим кислотным гидролизом дает энантиомерно чистый трет-бутиловый сложный -аминоэфир. Применение окисляющих агентов делает данный способ не привлекательным для крупномасштабного производства.
В WO 97/41102 описано ферментативное разделение (±)-фенилацетамидо кислоты с использованием пенициллинамидазы с получением S-кислоты. Данный способ, в котором используют ферменты, не эффективен и не практичен для крупномасштабного производства.
Таким образом, существует необходимость в способе, который был бы совместим с потребностями крупномасштабного производства и при котором достигался бы приемлемый уровень чистоты и выхода.
Краткое изложение сущности изобретения
Изобретение относится к способу получения соединения формулы I, -аминоэфира. Данный способ приводит к неэффективному образованию амида лития и трудному удалению N-(метилбензильной) группы.
В J.Org.Chem., vol.61, p.2222 (1996) описан способ, в котором литиевый енолят этилацетата добавляют к энантиомерному сульфинимину, продукт взаимодействия очищают хроматографией и снимают защитную группу в кислых условиях, получая сложный -аминоэфир с более чем 90% ее (энантиомерным избытком). Необходимость хроматографической очистки делает данный способ не привлекательным для крупномасштабного производства.
В WO 98/02410 описан способ стереоселективного присоединения реактива Реформатского, полученного из трет-бутилбромацетата, к энантиомерному имину, полученному из 3-пиридинкарбоксальдегида и (R)-2-фенилглицинола. Окислительное расщепление N-(1-фенил-2-гидроксиэтильной) группы NaIO4 в этаноле с последующим кислотным гидролизом дает энантиомерно чистый трет-бутиловый сложный -аминоэфир. Применение окисляющих агентов делает данный способ не привлекательным для крупномасштабного производства.
В WO 97/41102 описано ферментативное разделение (±)-фенилацетамидо кислоты с использованием пенициллинамидазы с получением S-кислоты. Данный способ, в котором используют ферменты, не эффективен и не практичен для крупномасштабного производства.
Таким образом, существует необходимость в способе, который был бы совместим с потребностями крупномасштабного производства и при котором достигался бы приемлемый уровень чистоты и выхода.
Краткое изложение сущности изобретения
Изобретение относится к способу получения соединения формулы I,
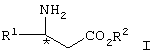 где R1 представляет арил, гетероарил, замещенный арил или замещенный гетероарил, и R2 представляет водород, алкил или аралкил, или его соли, включающему взаимодействие соединения формулы II
где R1 представляет арил, гетероарил, замещенный арил или замещенный гетероарил, и R2 представляет водород, алкил или аралкил, или его соли, включающему взаимодействие соединения формулы II
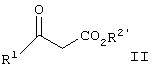 где R1 является таким, как описано выше, и R2′ представляет алкил или аралкил, с соединением формулы III
где R1 является таким, как описано выше, и R2′ представляет алкил или аралкил, с соединением формулы III
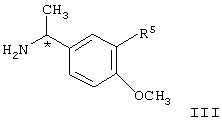 где R5 представляет водород или алкокси, в условиях пониженного давления, так что реакционный раствор кипит при температуре в диапазоне от примерно 40 до примерно 65С, в инертном растворителе, при этом указанный растворитель при пониженном давлении способен к азеотропной отгонке воды с образованием соединения формулы IV,
где R5 представляет водород или алкокси, в условиях пониженного давления, так что реакционный раствор кипит при температуре в диапазоне от примерно 40 до примерно 65С, в инертном растворителе, при этом указанный растворитель при пониженном давлении способен к азеотропной отгонке воды с образованием соединения формулы IV,
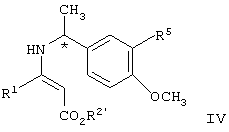 взаимодействие соединения формулы IV с газообразным водородом в присутствии палладиевого катализатора с образованием соединения формулы V
взаимодействие соединения формулы IV с газообразным водородом в присутствии палладиевого катализатора с образованием соединения формулы V
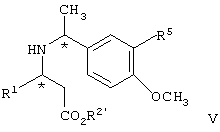 и взаимодействие соединения формулы V с образованием соединения формулы Iа или его соли
и взаимодействие соединения формулы V с образованием соединения формулы Iа или его соли
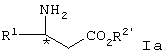 где R2′ представляет алкил или аралкил.
При желании, соединение Iа может быть дополнительно превращено в соединение формулы Ib или его соль
где R2′ представляет алкил или аралкил.
При желании, соединение Iа может быть дополнительно превращено в соединение формулы Ib или его соль
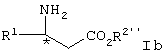 где R2” представляет водород, омылением сложноэфирной группы.
Способ данного изобретения, как он представлен в данном описании, имеет преимущество по сравнению с ранее описанными способами в том, что он является эффективным по объему, что делает его подходящим для крупномасштабного производства.
Подробное описание изобретения
Как использовано в данном описании, если не указано другого, алкил, там, где используется по отдельности или как часть замещающей группы, включает прямые и разветвленные цепи. Например, алкильные радикалы включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, н-гексил и тому подобные. Если не указано другого, “низший” при использовании с алкилом означает состав углеродной цепи из 1-4 атомов углерода.
Как использовано в данном описании, если не указано другого, “алкокси” будет означать простой эфирный радикал с атомом кислорода описанных выше прямых или разветвленных алкильных групп. Например, метокси, этокси, н-пропокси, втор-бутокси, трет-бутокси, н-гексилокси и тому подобные.
Как использовано в данном описании по отдельности или в виде части замещающей группы, если не указано другого, “арил” будет относиться к незамещенным карбоциклическим ароматическим группам, таким как фенил, нафтил и тому подобные. Арильная группа может быть замещена по крайней мере одним заместителем. Подходящие заместители арильной группы выбирают независимо из группы, состоящей из галогена, гидрокси низшего алкила, низшего алкокси, низшего аралкила, -NR32, где R3 представляет низший алкил; R4CONH, где R4 представляет фенил или низший алкил; и -OC(О)R6, где R6 представляет водород, алкил или аралкил.
Как использовано в данном описании, если не указано другого, “гетероарил” будет означать любую пяти- или шестичленную моноциклическую ароматическую кольцевую структуру, содержащую по крайней мере один гетероатом, выбранный из О, N и S, или бициклическую систему, где моноциклический гетероарил сконденсирован с арилом или моноциклическим гетероарилом. Примеры подходящих гетероарильных групп включают, но не ограничиваются ими, пирролил, пиридил, пиразинил, пиримидинил, пиразолил, пиридазинил, фуранил, пиранил, имидазолил, тиенил, оксазолил, изотиазолил, изоксазолил, фуразанил, бензотиенил, бензофуранил, индолил, изоиндолил, индолизинил, индазолил, пуринил, изохинолил, хинолил, изотиазолил и тому подобные. Гетероарил может быть замещен по крайней мере одним заместителем. Подходящие заместители гетероарильной группы выбирают независимо из группы, состоящей из галогена, гидрокси, низшего алкила, низшего алкокси, низшего аралкила, -NR32, где R3 представляет низший алкил; R4CONH, где R4 представляет фенил или низший алкил и -OC(O)R6, где R6 представляет водород, алкил или аралкил; предпочтительно из галогена или низшего алкила. Гетероарильная группа может быть присоединена посредством любого гетероатома или атома углерода кольца так, чтобы приводить к стабильной структуре.
Предпочтительно гетероарил выбирают из группы, состоящей из пиридила, пиримидинила, фуранила и тиенила.
Как использовано в данном описании, если не указано другого, “аралкил” будет означать любую низшую алкильную группу, замещенную арильной группой, такой как фенил, нафтил и тому подобные.
Как использовано в данном описании, “галоген” будет означать хлор, бром, фтор и иод.
Как использовано в данном описании, обозначение “
где R2” представляет водород, омылением сложноэфирной группы.
Способ данного изобретения, как он представлен в данном описании, имеет преимущество по сравнению с ранее описанными способами в том, что он является эффективным по объему, что делает его подходящим для крупномасштабного производства.
Подробное описание изобретения
Как использовано в данном описании, если не указано другого, алкил, там, где используется по отдельности или как часть замещающей группы, включает прямые и разветвленные цепи. Например, алкильные радикалы включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, н-гексил и тому подобные. Если не указано другого, “низший” при использовании с алкилом означает состав углеродной цепи из 1-4 атомов углерода.
Как использовано в данном описании, если не указано другого, “алкокси” будет означать простой эфирный радикал с атомом кислорода описанных выше прямых или разветвленных алкильных групп. Например, метокси, этокси, н-пропокси, втор-бутокси, трет-бутокси, н-гексилокси и тому подобные.
Как использовано в данном описании по отдельности или в виде части замещающей группы, если не указано другого, “арил” будет относиться к незамещенным карбоциклическим ароматическим группам, таким как фенил, нафтил и тому подобные. Арильная группа может быть замещена по крайней мере одним заместителем. Подходящие заместители арильной группы выбирают независимо из группы, состоящей из галогена, гидрокси низшего алкила, низшего алкокси, низшего аралкила, -NR32, где R3 представляет низший алкил; R4CONH, где R4 представляет фенил или низший алкил; и -OC(О)R6, где R6 представляет водород, алкил или аралкил.
Как использовано в данном описании, если не указано другого, “гетероарил” будет означать любую пяти- или шестичленную моноциклическую ароматическую кольцевую структуру, содержащую по крайней мере один гетероатом, выбранный из О, N и S, или бициклическую систему, где моноциклический гетероарил сконденсирован с арилом или моноциклическим гетероарилом. Примеры подходящих гетероарильных групп включают, но не ограничиваются ими, пирролил, пиридил, пиразинил, пиримидинил, пиразолил, пиридазинил, фуранил, пиранил, имидазолил, тиенил, оксазолил, изотиазолил, изоксазолил, фуразанил, бензотиенил, бензофуранил, индолил, изоиндолил, индолизинил, индазолил, пуринил, изохинолил, хинолил, изотиазолил и тому подобные. Гетероарил может быть замещен по крайней мере одним заместителем. Подходящие заместители гетероарильной группы выбирают независимо из группы, состоящей из галогена, гидрокси, низшего алкила, низшего алкокси, низшего аралкила, -NR32, где R3 представляет низший алкил; R4CONH, где R4 представляет фенил или низший алкил и -OC(O)R6, где R6 представляет водород, алкил или аралкил; предпочтительно из галогена или низшего алкила. Гетероарильная группа может быть присоединена посредством любого гетероатома или атома углерода кольца так, чтобы приводить к стабильной структуре.
Предпочтительно гетероарил выбирают из группы, состоящей из пиридила, пиримидинила, фуранила и тиенила.
Как использовано в данном описании, если не указано другого, “аралкил” будет означать любую низшую алкильную группу, замещенную арильной группой, такой как фенил, нафтил и тому подобные.
Как использовано в данном описании, “галоген” будет означать хлор, бром, фтор и иод.
Как использовано в данном описании, обозначение “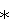 ” будет означать присутствие стереогенного центра.
Соединение формулы IV благодаря присутствию двойной связи может существовать или в виде цис- или в виде трансконфигурации или в виде смеси двух конфигураций.
Соединение формулы V благодаря присутствию двух стереогенных центров может существовать в виде любого из четырех диастереомеров или их смеси.
Как использовано в данном описании, в отношении реагентов и продуктов реакций, термин “энантиомерный избыток или ее” будет означать избыточное количество одного энантиомера по сравнению с другим энантиомером. Энантиомерный избыток (выраженный в процентах) рассчитывают как
[(Количество энантиомера(1)-Количество энантиомера(2))/общее количество обоих энантиомеров)]100% Применение настоящего изобретения для смеси энантиомеров формулы III, по существу не содержащей R энантиомера, будет приводить к получению смеси энантиомеров формулы I, по существу не содержащей R энантиомера. Аналогично, применение настоящего изобретения для смеси энантиомеров формулы III, по существу не содержащей S энантиомера, будет приводить к получению смеси энантиомеров формулы I, по существу не содержащей S энантиомера. Предпочтительно энантиомерный избыток требуемого энантиомера формулы III составляет по крайней мере 90% ее, более предпочтительно по крайней мере 98% ее, наиболее предпочтительно 99% ее.
В предпочтительном воплощении изобретения в соединении формулы I R1 представляет фенил, пиримидил, незамещенный или замещенный пиридил, нафтил или 3,5-дихлорфенил, более предпочтительно 2-пиридил, 3-пиридил или 4-пиридил, наиболее предпочтительно 3-пиридил. R2 предпочтительно представляет низший алкил, более предпочтительно метил или этил.
Изобретение относится к способу получения соединения формулы I ” будет означать присутствие стереогенного центра.
Соединение формулы IV благодаря присутствию двойной связи может существовать или в виде цис- или в виде трансконфигурации или в виде смеси двух конфигураций.
Соединение формулы V благодаря присутствию двух стереогенных центров может существовать в виде любого из четырех диастереомеров или их смеси.
Как использовано в данном описании, в отношении реагентов и продуктов реакций, термин “энантиомерный избыток или ее” будет означать избыточное количество одного энантиомера по сравнению с другим энантиомером. Энантиомерный избыток (выраженный в процентах) рассчитывают как
[(Количество энантиомера(1)-Количество энантиомера(2))/общее количество обоих энантиомеров)]100% Применение настоящего изобретения для смеси энантиомеров формулы III, по существу не содержащей R энантиомера, будет приводить к получению смеси энантиомеров формулы I, по существу не содержащей R энантиомера. Аналогично, применение настоящего изобретения для смеси энантиомеров формулы III, по существу не содержащей S энантиомера, будет приводить к получению смеси энантиомеров формулы I, по существу не содержащей S энантиомера. Предпочтительно энантиомерный избыток требуемого энантиомера формулы III составляет по крайней мере 90% ее, более предпочтительно по крайней мере 98% ее, наиболее предпочтительно 99% ее.
В предпочтительном воплощении изобретения в соединении формулы I R1 представляет фенил, пиримидил, незамещенный или замещенный пиридил, нафтил или 3,5-дихлорфенил, более предпочтительно 2-пиридил, 3-пиридил или 4-пиридил, наиболее предпочтительно 3-пиридил. R2 предпочтительно представляет низший алкил, более предпочтительно метил или этил.
Изобретение относится к способу получения соединения формулы I
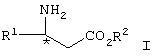 где R1 представляет арил, гетероарил, замещенный арил или замещенный гетероарил, и R2 представляет водород, алкил или аралкил, или его соли, включающему взаимодействие соединения формулы II
где R1 представляет арил, гетероарил, замещенный арил или замещенный гетероарил, и R2 представляет водород, алкил или аралкил, или его соли, включающему взаимодействие соединения формулы II
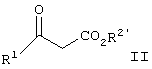 где R1 является таким, как описано выше, и R2′ представляет алкил или аралкил, с соединением формулы III
где R1 является таким, как описано выше, и R2′ представляет алкил или аралкил, с соединением формулы III
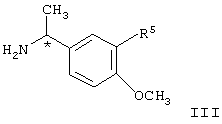 где R5 представляет водород или алкокси, предпочтительно водород или метокси, в условиях пониженного давления, так что реакционный раствор кипит при температуре в диапазоне от примерно 40 до примерно 65С, в инертном растворителе, при этом указанный растворитель при пониженном давлении способен к азеотропной отгонке воды, с образованием соединения формулы IV,
где R5 представляет водород или алкокси, предпочтительно водород или метокси, в условиях пониженного давления, так что реакционный раствор кипит при температуре в диапазоне от примерно 40 до примерно 65С, в инертном растворителе, при этом указанный растворитель при пониженном давлении способен к азеотропной отгонке воды, с образованием соединения формулы IV,
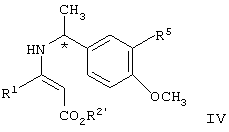 взаимодействие соединения формулы IV с газообразным водородом в присутствии палладиевого катализатора с образованием соединения формулы V
взаимодействие соединения формулы IV с газообразным водородом в присутствии палладиевого катализатора с образованием соединения формулы V
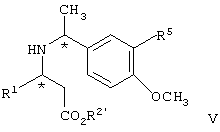 и взаимодействие соединения формулы V с образованием соединения формулы Iа, где R2′ представляет алкил или аралкил или его соли.
и взаимодействие соединения формулы V с образованием соединения формулы Iа, где R2′ представляет алкил или аралкил или его соли.
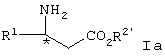 При желании соединение Iа может быть дополнительно превращено в соединение формулы Ib или его соль, где R2” представляет водород, путем омыления сложноэфирной группы.
При желании соединение Iа может быть дополнительно превращено в соединение формулы Ib или его соль, где R2” представляет водород, путем омыления сложноэфирной группы.
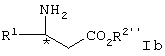 В соответствии с изобретением соединение формулы II, представляющее собой известное соединение или соединение, полученное известными способами (J.Org.Chem., 1975, 40, 532; J.Org.Chem., 1983, 48, 5006), подвергают взаимодействию с соединением формулы III, являющимся известным соединением или соединением, полученным известными способами (Вест. Москов. Универ., Сер.2, Хим. 1977, 18, 446; CAN 88:62074) в присутствии кислоты, предпочтительно карбоновой кислоты, наиболее предпочтительно уксусной кислоты, в вакууме, предпочтительно в вакууме, отрегулированном таким образом, что температура кипения смеси составляет от примерно 40 до примерно 65С в инертном растворителе, указанный растворитель при пониженном давлении способен к азеотропной отгонке воды, таком как ксилол, гептан или толуол, предпочтительно толуол, с образованием соединения формулы IV.
Когда способ применяют к соединению формулы II, где R1 представляет собой азотсодержащий гетероарил, взаимодействие осуществляют в присутствии по крайней мере двух эквивалентов карбоновой кислоты, предпочтительно уксусной кислоты.
Предпочтительно, когда R1 представляет собой N-содержащий гетероарил, реакционный раствор дополнительно промывают водным основанием, таким как бикарбонат натрия, карбонат натрия и тому подобными, для удаления избытка кислоты.
Соединение формулы IV взаимодействует с газообразным водородом в присутствии палладиевого катализатора, такого как гидроксид палладия на угле, палладий-на-угле и тому подобного, предпочтительно по крайней мере 10 вес.%, 20% гидроксида палладия на угле, предпочтительно при атмосферном давлении в спиртовом растворителе, таком как низший алкиловый спирт, предпочтительно метанол, предпочтительно при температуре от примерно 0°C до примерно 40°С, наиболее предпочтительно при комнатной температуре, с образованием соответствующего соединения формулы V.
Требуемый диастереомер соединения формулы V предпочтительно выделяют обычными способами, известными специалистам в данной области, такими как перекристаллизация из органического растворителя, такого как этилацетат, метанол, метил-трет-бутиловый эфир и тому подобное, ВЭЖХ или флэш-хроматографией.
Соединение формулы V взаимодействует в кислоте, такой как уксусная кислота, муравьиная кислота, пропионовая кислота, трифторуксусная кислота (ТФУК), хлористоводородная кислота или их смеси, предпочтительно в муравьиной кислоте, предпочтительно в присутствии гидросилана, такого как ди(низший)алкилсилан или три(низший)алкилсилан, предпочтительно триэтилсилана, при температуре в диапазоне от примерно 40 до примерно 100С, предпочтительно от примерно 80 до примерно 100С, с образованием соответствующего соединения формулы 1а, где R2′ представляет алкил или аралкил.
При желании, соединение формулы Iа или его соль, где R2′ представляет алкил или аралкил, дополнительно может быть преобразовано в соединение формулы Ib или его соль, где R2” представляет водород, с помощью омыления сложноэфирной группы обычными способами, такими как взаимодействие соединения формулы Iа или его соли с гидроксидом лития, гидроксидом натрия или гидроксидом калия в растворителе, таком как тетрагидрофуран (ТГФ), диметилформамид (ДМФ) или метанол.
Следующие примеры более подробно описывают изобретение и предназначены только для иллюстрации изобретения, а не для его ограничения.
Пример 1
Метил N-[(S)-1-(4-метоксифенил)этил]-3-амино-3-(3-пиридил)пропаноат.
Смесь метилникотиноилацетата (23,6 г, 0,13 моль) и (S)-1-(4-метоксифенил)этиламина (20,0 г, 0,13 моль) растворяли в толуоле (60 мл), получая гомогенный раствор. Добавляли ледяную уксусную кислоту (19,5 г, 0,33 моль), что приводило к образованию осадка. Реакционную смесь нагревали до 62С при пониженном давлении, смесь опять принимала вид прозрачного раствора. Пониженное давление регулировали таким образом, чтобы дать возможность раствору кипеть с обратным холодильником при 62С с постоянной скоростью для азеотропного удаления воды. Реакцию останавливали через 24 часа; 1Н ЯМР указывал, что реакция на >90% завершилась. Растворитель упаривали при пониженном давлении при 60С, получая коричневое масло. Сырое масло повторно растворяли в толуоле (50 мл) и промывали насыщенным раствором NaHCO3 (2
В соответствии с изобретением соединение формулы II, представляющее собой известное соединение или соединение, полученное известными способами (J.Org.Chem., 1975, 40, 532; J.Org.Chem., 1983, 48, 5006), подвергают взаимодействию с соединением формулы III, являющимся известным соединением или соединением, полученным известными способами (Вест. Москов. Универ., Сер.2, Хим. 1977, 18, 446; CAN 88:62074) в присутствии кислоты, предпочтительно карбоновой кислоты, наиболее предпочтительно уксусной кислоты, в вакууме, предпочтительно в вакууме, отрегулированном таким образом, что температура кипения смеси составляет от примерно 40 до примерно 65С в инертном растворителе, указанный растворитель при пониженном давлении способен к азеотропной отгонке воды, таком как ксилол, гептан или толуол, предпочтительно толуол, с образованием соединения формулы IV.
Когда способ применяют к соединению формулы II, где R1 представляет собой азотсодержащий гетероарил, взаимодействие осуществляют в присутствии по крайней мере двух эквивалентов карбоновой кислоты, предпочтительно уксусной кислоты.
Предпочтительно, когда R1 представляет собой N-содержащий гетероарил, реакционный раствор дополнительно промывают водным основанием, таким как бикарбонат натрия, карбонат натрия и тому подобными, для удаления избытка кислоты.
Соединение формулы IV взаимодействует с газообразным водородом в присутствии палладиевого катализатора, такого как гидроксид палладия на угле, палладий-на-угле и тому подобного, предпочтительно по крайней мере 10 вес.%, 20% гидроксида палладия на угле, предпочтительно при атмосферном давлении в спиртовом растворителе, таком как низший алкиловый спирт, предпочтительно метанол, предпочтительно при температуре от примерно 0°C до примерно 40°С, наиболее предпочтительно при комнатной температуре, с образованием соответствующего соединения формулы V.
Требуемый диастереомер соединения формулы V предпочтительно выделяют обычными способами, известными специалистам в данной области, такими как перекристаллизация из органического растворителя, такого как этилацетат, метанол, метил-трет-бутиловый эфир и тому подобное, ВЭЖХ или флэш-хроматографией.
Соединение формулы V взаимодействует в кислоте, такой как уксусная кислота, муравьиная кислота, пропионовая кислота, трифторуксусная кислота (ТФУК), хлористоводородная кислота или их смеси, предпочтительно в муравьиной кислоте, предпочтительно в присутствии гидросилана, такого как ди(низший)алкилсилан или три(низший)алкилсилан, предпочтительно триэтилсилана, при температуре в диапазоне от примерно 40 до примерно 100С, предпочтительно от примерно 80 до примерно 100С, с образованием соответствующего соединения формулы 1а, где R2′ представляет алкил или аралкил.
При желании, соединение формулы Iа или его соль, где R2′ представляет алкил или аралкил, дополнительно может быть преобразовано в соединение формулы Ib или его соль, где R2” представляет водород, с помощью омыления сложноэфирной группы обычными способами, такими как взаимодействие соединения формулы Iа или его соли с гидроксидом лития, гидроксидом натрия или гидроксидом калия в растворителе, таком как тетрагидрофуран (ТГФ), диметилформамид (ДМФ) или метанол.
Следующие примеры более подробно описывают изобретение и предназначены только для иллюстрации изобретения, а не для его ограничения.
Пример 1
Метил N-[(S)-1-(4-метоксифенил)этил]-3-амино-3-(3-пиридил)пропаноат.
Смесь метилникотиноилацетата (23,6 г, 0,13 моль) и (S)-1-(4-метоксифенил)этиламина (20,0 г, 0,13 моль) растворяли в толуоле (60 мл), получая гомогенный раствор. Добавляли ледяную уксусную кислоту (19,5 г, 0,33 моль), что приводило к образованию осадка. Реакционную смесь нагревали до 62С при пониженном давлении, смесь опять принимала вид прозрачного раствора. Пониженное давление регулировали таким образом, чтобы дать возможность раствору кипеть с обратным холодильником при 62С с постоянной скоростью для азеотропного удаления воды. Реакцию останавливали через 24 часа; 1Н ЯМР указывал, что реакция на >90% завершилась. Растворитель упаривали при пониженном давлении при 60С, получая коричневое масло. Сырое масло повторно растворяли в толуоле (50 мл) и промывали насыщенным раствором NaHCO3 (2 100 мл), затем насыщенным раствором соли (75 мл). Органический слой отделяли и упаривали досуха в вакууме при 60С, получая 41,1 г светло-коричневого масла. 1Н ЯМР показал, что сырое масло содержало целевой продукт, загрязненный 4,3% толуола и 0,86% воды по весу. Данное сырое масло использовали непосредственно для последующего восстановления без дополнительной очистки.
Аналитический образец получали перекристаллизацией из этилацетата.
Масс-спектр m/z (относительная интенсивность): 281,20(50), 313,22(МН+, 100), 354,24 (MH++MeCN, 40) , 432,27 (<5). 100 мл), затем насыщенным раствором соли (75 мл). Органический слой отделяли и упаривали досуха в вакууме при 60С, получая 41,1 г светло-коричневого масла. 1Н ЯМР показал, что сырое масло содержало целевой продукт, загрязненный 4,3% толуола и 0,86% воды по весу. Данное сырое масло использовали непосредственно для последующего восстановления без дополнительной очистки.
Аналитический образец получали перекристаллизацией из этилацетата.
Масс-спектр m/z (относительная интенсивность): 281,20(50), 313,22(МН+, 100), 354,24 (MH++MeCN, 40) , 432,27 (<5).
Элементный анализ, вычислено для C18H20N2O3:С, 69,21; Н, 6,45; N, 8,97%. Найдено: С, 69,27; Н, 6,59; N, 8,92%. Пример 2 Метил N-[(S)-1-(4-метоксифенил)этил]-(S)-3-амино-3-(3-пиридил)пропаноат Неочищенный метил N-[(S)-1-(4-метоксифенил)этил]-3-амино-3-(3-пиридил)пропаноат (40,6 г, 130 ммоль) растворяли в метаноле (200 мл) и смешивали с 20% Pd(OH)2/C (4,1 г). Смесь гидрировали при атмосферном давлении в течение 30 часов. Смесь разбавляли этилацетатом (50 мл) и фильтровали через целит (20 г) для удаления катализатора. Целит промывали горячим (70С) этилацетатом (200 мл). Объединенный фильтрат концентрировали при пониженном давлении почти досуха, получая маслянистый остаток. Сырой амин (27,5 г) растворяли в горячем этилацетате (40 мл). Полученный горячий желтый раствор фильтровали и прополаскивали 15 мл горячего этилацетата. Прозрачный раствор концентрировали до объема <50 мл и оставляли при комнатной температуре в течение ночи. Полученное белое кристаллическое твердое вещество собирали фильтрованием и промывали холодным этилацетатом (EtOAc) (~15-20 мл), затем сушили на воздухе. Выход выделенного твердого вещества: 15,5 г. ВЭЖХ анализ показал >99% целевого диастереомера. Вторую порцию получали из фильтрата после концентрирования до объема примерно 20 мл. Выход: 1,7 г, по ВЭЖХ идентично первой порции; общий выход: 17,2 г (62,5%).
Белое кристаллическое твердое вещество, т.пл. 117,3-119,0С.
Масс-спектр(ES), 315 МН+.
Элементный анализ, вычислено для C18H22N2O3: С, 68,77; Н, 7,05; N, 8,91%. Найдено: С, 68,66; Н, 6,95; N, 8,86%.
Пример 3
Дигидрохлорид метил (S)-3-амино-3-(3-пиридил)пропаноата.
Смесь метил N-[(S)-1-(4-метоксифенил)этил]-(S)-3-амино-3-(3-пиридил)пропаноата (57,35 г, 0,148 моль) и триэтилсилана (25,81 г, 0,222 моль) в муравьиной кислоте (120 мл) нагревали до 90С (масляная баня) при перемешивании. Первоначальная суспензия превращалась в прозрачный раствор через 5 минут при 90С. Нагревание и перемешивание продолжали в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, и растворитель удаляли при пониженном давлении при 50С, получая в остатке масло. Оставшееся масло растворяли в этилацетате (300 мл) и метаноле (100 мл). Раствор фильтровали. Фильтрат медленно обрабатывали 9,8М НСl в метаноле (30,2 мл, 0,296 моль) при перемешивании. Полученный раствор разбавляли этилацетатом (200 мл) и нагревали на паровой бане для удаления избытка метанола до помутнения раствора. Затем смесь перемешивали при комнатной температуре в течение ночи, и из раствора выкристаллизовывался твердый продукт. К суспензии добавляли этилацетат (150 мл) и смесь перемешивали еще в течение 2 часов. Твердое вещество собирали фильтрованием и промывали 200 мл этилацетата, затем сушили в вакууме в течение 1 часа. Целевой продукт получали в виде белого порошка (27,05 г, 72% выход).
ВЭЖХ область чистота=98%, ее=99,4%.
т.пл=197,5-199С.
Вторую порцию получали концентрированном фильтрата и растворением оставшегося масла в 200 мл этилацетата и 20 мл метанола. После внесения затравки раствор перемешивали в течение 6 часов, вторую порцию собирали фильтрованием и промывали 100 мл этилацетата, затем сушили в вакууме в течение 1 часа (4,9 г, 13% выход).
Общий выход выделенного продукта: 31,95 г, 85%
ВЭЖХ область чистота = 90%, ее=99,8%.
т.пл=197,5-199,5С.
Масс-спектр (Esl) m/z 181,2 (МН+), 222,2 (MH++MeCN)
Элементный анализ, вычислено для C9H14N2O2Cl2: С, 42,71; Н, 5,57; N, 11,07; Cl, 28,01. Найдено: С, 42,68; Н, 5,64; N, 11,05; Cl, 28,00.
Пример арила
Этил N-[(S)-1-(4-метоксифенил)этил]-3-амино-3-(3-фенил)пропаноат гидрохлорид
Смесь этилбензоилацетата (3,8 г, 20 ммоль) и (s)-l-(4-метоксифенил)этиламина (3,0 г, 20 ммоль) растворяли в толуоле (30 мл), получая гомогенный раствор. Добавляли ледяную уксусную кислоту (2,4 г, 40 ммоль), что приводило к образованию осадка. Реакционную смесь нагревали до 65С при пониженном давлении, смесь опять принимала вид прозрачного раствора. Пониженное давление регулировали таким образом, чтобы дать раствору возможность кипеть с обратным холодильником при 60-65С с постоянной скоростью для азеотропного удаления воды. Реакцию останавливали через 18 ч; 1H-ЯМР указывал, что реакция на >95% завершилась. Растворитель выпаривали при пониженном давлении при 60С с получением масла золотистого цвета. Сырое масло повторно растворяли в толуоле (50 мл) и промывали насыщенным раствором NaHCO3 (2х50 мл), а затем насыщенным солевым раствором (75 мл). Органический слой отделяли и упаривали до сухости в вакууме при 60С с получением 6,4 г светло-золотистого масла. Сырой енамин (1,0 г, 3,0 ммоль) растворяли в 20 мл ТГФ и к раствору добавляли ВF3·Et2О (0,75 мл, 6,0 ммоль) с последующим добавлением 10 мас.% катализаторов (20% Pd(OH)2/C). Смесь гидрировали в условиях атмосферного давления и температуры окружающей среды при перемешивании. Через 2 часа реакционную смесь фильтровали через целит для удаления катализатора и промывали EtOAc. Фильтрат промывали насыщенным раствором NaHCO3 (2х50 мл) и насыщенным солевым раствором (1х50 мл). Сушили над MgSO4 и выпаривали растворитель с получением бесцветного масла. Сырое масло растворяли в гексане, фильтровали для удаления нерастворимых веществ и затем обрабатывали 1,1 экв. 1н. НСl в Et2O. Осажденное твердое вещество собирали фильтрацией, промывали Et2O и затем сушили на воздухе. Сырой продукт растворяли в 5 мл EtOAc и добавляли небольшое количество (~1 мл) гексана с получением прозрачного раствора. Кристаллизовавшийся продукт выделяли фильтрацией и сушили на воздухе с получением 0,74 г продукта (общий выход 74%).
MS (ES+), 328 МН+.
Этил (S)-3-амино-3-(3-фенил)пропаноат фумарат
Этил N-[(S)-1-(4-метоксифенил)этил]-3-амино-3-(3-фенил)пропаноат гидрохлорид (0,5 г, 1,37 ммоль) растворяли в 10 мл НСООН. Добавляли Et3SiH (0,44 мл, 2,75 ммоль) и реакционную смесь нагревали до 90С. Через 20 час растворитель выпаривали с получением масла золотистого цвета. Сырое масло растворяли в 25 мл EtOAc, промывали насыщенным раствором NaHCO3 (1х50 мл) и насыщенным солевым раствором (1х50 мл) и сушили над MgSO4. После удаления осушителя фильтрованием фильтрат обрабатывали 1 экв. фумаровой кислоты (0,16 г, 1,37 ммоль) в 3 мл МеОН и концентрировали до половины объема. Осажденное белое твердое вещество собирали фильтрованием и промывали EtOAc с получением 0,37 г (87%) указанного в заголовке соединения.
MS(ES+), 194 MH+.
Замещенный арил
Этил N-[(S)-1-(4-метоксифенил)этил]-3-амино-3-[3-(4-гидрокси)фенил]пропаноат фумарат
Смесь метил 4-гидроксибензоилацетата (0,45 г, 2,30 ммоль) и (s)-l-(4-метоксифенил)этиламина (0,31 г, 2,30 ммоль) растворяли в толуоле (10 мл) с получением гомогенного раствора. Добавляли ледяную уксусную кислоту (0,28 г, 4,6 ммоль), что приводило к образованию осадка. Реакционную смесь нагревали до 65°С при пониженном давлении, смесь опять принимала вид прозрачного раствора. Пониженное давление регулировали таким образом, чтобы дать раствору возможность кипеть с обратным холодильником при 60-65С с постоянной скоростью для азеотропного удаления воды. Реакцию останавливали через 18 ч; 1Н-ЯМР указывал, что реакция на >95% завершилась. Растворитель выпаривали при пониженном давлении при 60С с получением масла золотистого цвета. Сырое масло повторно растворяли в толуоле (20 мл) и промывали насыщенным раствором NaHCO3 (2х10 мл), а затем насыщенным солевым раствором (15 мл). Органический слой отделяли и упаривали до сухости в вакууме при 60С с получением 0,75 г светло-золотистого масла. Сырой енамин (0,75 г, 2,3 ммоль) растворяли в 20 мл ТГФ и к раствору добавляли ВF3·Еt3O (0,6 мл, 4,6 ммоль) с последующим добавлением 10 мас.% катализаторов (20% Pd(OH)2/C). Смесь гидрировали в условиях атмосферного давления и температуры окружающей среды при перемешивании. Через 2,5 ч реакционную смесь фильтровали через целит для удаления катализатора и промывали ЕtoАс. Фильтрат промывали насыщенным раствором МаНСО3 (2х25 мл) и насыщенным солевым раствором (1х25 мл). Сушили над MgSO4 и выпаривали растворитель с получением бесцветного масла. Сырое масло растворяли в 10 мл ЕtOАс и затем обрабатывали 1 экв. (0,26 г) фумаровой кислоты в 2 мл МеОН. Осажденное твердое вещество собирали фильтрацией, промывали ЕtOАс, затем сушили на воздухе с получением 0,67 г продукта (общий выход 89%).
MS (ES+), 330 МН+
Метил (S)-3-амино-3-[З-(4-гидрокси)фенил]пропаноат ацетат
Метил N- [(S)-1 -(4-метоксифенил)этил]-3-амино-3-[3-(4-гидрокси)фенил]пропаноат фумарат (0,5 г, 1,12 ммоль) растворяли в 10 мл НСООН. Добавляли Et3SiH (0,36 мл, 2,24 ммоль) и реакционную смесь нагревали до 90С. Через 20 ч растворитель выпаривали с получением масла золотистого цвета. Сырое масло растворяли в 25 мл ЕtOАс, промывали насыщенным раствором NaHCO3 (1х50 мл) и насыщенным солевым раствором (1х50 мл) и сушили над MgSO4. После удаления осушителя фильтрованием фильтрат обрабатывали 1 экв. уксусной кислоты (0,06 г, 1,12 ммоль) и концентрировали до половины объема. Осажденное белое твердое вещество собирали фильтрованием и промывали ЕtOАс с получением 0,22 г (79%) указанного в заголовке соединения.
МS(ЕS+), 196 MH+.
Формула изобретения 1. Способ получения соединения формулы I 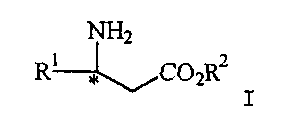 где R1 представляет арил, гетероарил, замещенный арил или замещенный гетероарил;
R2 представляет водород, алкил или аралкил,
или его соли, включающий взаимодействие соединения формулы II
где R1 представляет арил, гетероарил, замещенный арил или замещенный гетероарил;
R2 представляет водород, алкил или аралкил,
или его соли, включающий взаимодействие соединения формулы II
 где R1 является таким, как описано выше,
R2′ представляет алкил или аралкил,
с соединением формулы III
где R1 является таким, как описано выше,
R2′ представляет алкил или аралкил,
с соединением формулы III
 где R5 представляет водород или алкокси,
в условиях пониженного давления, так что реакционный раствор кипит при температуре в диапазоне примерно 40 – 65°С, в инертном растворителе, при этом указанный растворитель при пониженном давлении способен к азеотропной отгонке воды, с образованием соединения формулы IV
где R5 представляет водород или алкокси,
в условиях пониженного давления, так что реакционный раствор кипит при температуре в диапазоне примерно 40 – 65°С, в инертном растворителе, при этом указанный растворитель при пониженном давлении способен к азеотропной отгонке воды, с образованием соединения формулы IV
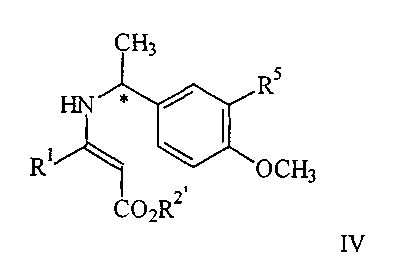 взаимодействие соединения формулы IV с газообразным водородом в присутствии палладиевого катализатора с образованием соединения формулы V
взаимодействие соединения формулы IV с газообразным водородом в присутствии палладиевого катализатора с образованием соединения формулы V
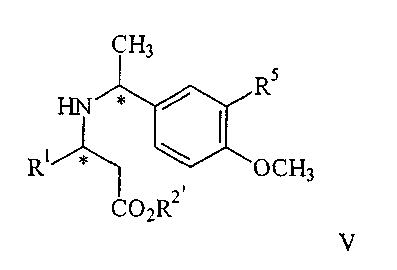 и взаимодействие соединения формулы V в кислоте с образованием соединения формулы I.
2. Способ по п.1, где соединения формулы I и II присутствуют при энантиомерном избытке одного энантиомера.
3. Способ по п.2, где соединения формулы I и III присутствуют при энантиомерном избытке одного энантиомера, составляющем по крайней мере 90% ее.
4. Способ по п.3, где соединения формулы I и III присутствуют при энантиомерном избытке одного энантиомера, составляющем по крайней мере 98% ее.
5. Способ по п.1, где R1 представляет арил, гетероарил, моно- или дизамещенный арил или моно- или дизамещенный гетероарил, где заместители арила или гетероарила независимо выбирают из группы, состоящей из галогена, гидрокси, низшего алкила, низшего алкокси, низшего аралкила, -NR32, где R3 представляет низший алкил; R4-CONH, где R4 представляет фенил или низший алкил; и -OC(О)R6, где R6 представляет водород, алкил или аралкил.
6. Способ по п.5, где R1 представляет фенил, пиримидил, незамещенный или замещенный пиридил, нафтил или 3,5-дихлорфенил.
7. Способ по п.6, где R1 представляет 2-пиридил, 3-пиридил или 4-пиридил.
8. Способ по п.7, где R1 представляет 3-пиридил.
9. Способ по п.1, где R2 представляет низший алкил.
10. Способ по п. 9, где R2 представляет метил.
11. Способ по п.1, где R1 представляет 3-пиридил, a R2 представляет метил.
12. Способ по п.11, где соединения формулы I и III присутствуют при энантиомерном избытке одного энантиомера, составляющем по крайней мере 90% ее.
13. Способ по п.11, где инертный растворитель, способный к азеотропной отгонке воды, представляет ксилол, гептан или толуол.
14. Способ по п.13, дополнительно включающий взаимодействие соединения формулы II с соединением формулы III с образованием соединения формулы IV в присутствии по крайней мере двух эквивалентов карбоновой кислоты.
15. Способ по п.14, где карбоновая кислота представляет уксусную кислоту.
16. Способ по п.15, дополнительно включающий промывку соединения формулы IV водным основанием для удаления избытка кислоты.
17. Способ по п.15, где палладиевый катализатор присутствует в количестве, составляющем по крайней мере 10 вес.% 20%-ного гидроксида палладия на угле.
18. Способ по п.1, дополнительно включающий отделение и выделение соединения формулы V в виде требуемого диастереомера.
19. Способ по п.15, дополнительно включающий отделение и выделение соединения формулы V в виде требуемого диастереомера.
20. Способ по п.15, дополнительно включающий взаимодействие соединения формулы V в муравьиной кислоте в присутствии гидросилана с образованием соединения формулы I.
21. Соединение формулы IV
и взаимодействие соединения формулы V в кислоте с образованием соединения формулы I.
2. Способ по п.1, где соединения формулы I и II присутствуют при энантиомерном избытке одного энантиомера.
3. Способ по п.2, где соединения формулы I и III присутствуют при энантиомерном избытке одного энантиомера, составляющем по крайней мере 90% ее.
4. Способ по п.3, где соединения формулы I и III присутствуют при энантиомерном избытке одного энантиомера, составляющем по крайней мере 98% ее.
5. Способ по п.1, где R1 представляет арил, гетероарил, моно- или дизамещенный арил или моно- или дизамещенный гетероарил, где заместители арила или гетероарила независимо выбирают из группы, состоящей из галогена, гидрокси, низшего алкила, низшего алкокси, низшего аралкила, -NR32, где R3 представляет низший алкил; R4-CONH, где R4 представляет фенил или низший алкил; и -OC(О)R6, где R6 представляет водород, алкил или аралкил.
6. Способ по п.5, где R1 представляет фенил, пиримидил, незамещенный или замещенный пиридил, нафтил или 3,5-дихлорфенил.
7. Способ по п.6, где R1 представляет 2-пиридил, 3-пиридил или 4-пиридил.
8. Способ по п.7, где R1 представляет 3-пиридил.
9. Способ по п.1, где R2 представляет низший алкил.
10. Способ по п. 9, где R2 представляет метил.
11. Способ по п.1, где R1 представляет 3-пиридил, a R2 представляет метил.
12. Способ по п.11, где соединения формулы I и III присутствуют при энантиомерном избытке одного энантиомера, составляющем по крайней мере 90% ее.
13. Способ по п.11, где инертный растворитель, способный к азеотропной отгонке воды, представляет ксилол, гептан или толуол.
14. Способ по п.13, дополнительно включающий взаимодействие соединения формулы II с соединением формулы III с образованием соединения формулы IV в присутствии по крайней мере двух эквивалентов карбоновой кислоты.
15. Способ по п.14, где карбоновая кислота представляет уксусную кислоту.
16. Способ по п.15, дополнительно включающий промывку соединения формулы IV водным основанием для удаления избытка кислоты.
17. Способ по п.15, где палладиевый катализатор присутствует в количестве, составляющем по крайней мере 10 вес.% 20%-ного гидроксида палладия на угле.
18. Способ по п.1, дополнительно включающий отделение и выделение соединения формулы V в виде требуемого диастереомера.
19. Способ по п.15, дополнительно включающий отделение и выделение соединения формулы V в виде требуемого диастереомера.
20. Способ по п.15, дополнительно включающий взаимодействие соединения формулы V в муравьиной кислоте в присутствии гидросилана с образованием соединения формулы I.
21. Соединение формулы IV
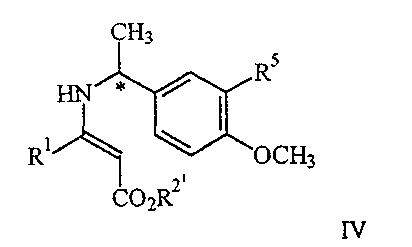 где R1 представляет арил, гетероарил, замещенный арил или замещенный гетероарил;
R2′ представляет алкил или аралкил;
R5 представляет водород или гидрокси,
или его соль.
22. Соединение формулы V
где R1 представляет арил, гетероарил, замещенный арил или замещенный гетероарил;
R2′ представляет алкил или аралкил;
R5 представляет водород или гидрокси,
или его соль.
22. Соединение формулы V
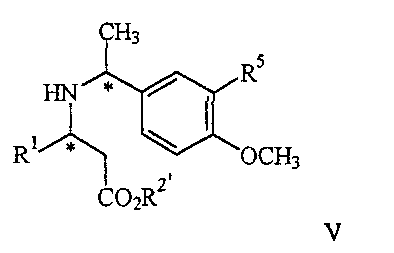 где R1 представляет арил, гетероарил, замещенный арил или замещенный гетероарил;
R2′ представляет алкил или аралкил;
R5 представляет водород или гидрокси, или его соль.
23. Способ получения соединения формулы I
где R1 представляет арил, гетероарил, замещенный арил или замещенный гетероарил;
R2′ представляет алкил или аралкил;
R5 представляет водород или гидрокси, или его соль.
23. Способ получения соединения формулы I
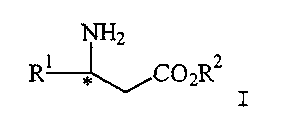 где R1 представляет арил, гетероарил, замещенный арил или замещенный гетероарил;
R2 представляет водород, алкил или аралкил,
или его соли, включающий взаимодействие соединения формулы IV
где R1 представляет арил, гетероарил, замещенный арил или замещенный гетероарил;
R2 представляет водород, алкил или аралкил,
или его соли, включающий взаимодействие соединения формулы IV
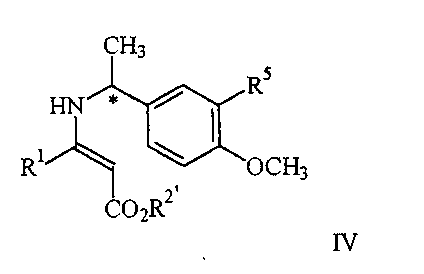 где R1 является таким, как описано выше;
R2′ представляет алкил или аралкил;
R5 представляет водород или алкокси,
с газообразным водородом в присутствии палладиевого катализатора с образованием соединения формулы V
где R1 является таким, как описано выше;
R2′ представляет алкил или аралкил;
R5 представляет водород или алкокси,
с газообразным водородом в присутствии палладиевого катализатора с образованием соединения формулы V
 и взаимодействие соединения формулы V в кислоте с образованием соединения формулы I.
и взаимодействие соединения формулы V в кислоте с образованием соединения формулы I.
|
||||||||||||||||||||||||||