Патент на изобретение №2223967
|
||||||||||||||||||||||||||
(54) Ингибиторы фактора VIIa
(57) Реферат: Изобретение относится к группе новых соединений общей формулы I R1-A-B-D-En-R2 (I), в которой R1 представляет собой R12C(O), причем R12 выбран из группы, включающей алкенил, алкенилокси или алкениламино; А представляет собой группу А1-А2-А3, в которой A1 представляет собой NH, А2 представляет собой CHR93, в которой R93 представляет 4-амидинофенилметил; A3 представляет собой C(O); В представляет собой группу В1-В2-В3, в которой B1 представляет собой NH; B2 представляет собой CHR97, где R97 представляет этил, который замещен в положении 2 гидроксикарбонилом или алкилоксикарбонилом; В3 представляет собой C(O); D представляет группу D1-D2-D3, в которой D1 представляет собой NH, D2 представляет собой CR81R82, в которой R81 и R82 независимо выбраны из группы, состоящей из водорода и незамещенных или замещенных остатков алкила, арила, арилалкила, гетероарилалкила; D3 представляет собой С(О); En представляет собой (E1-Е2-Е3)n, в которой n равно 0 или 1; E1 представляет NR70, где R70 представляет H; Е2 представляет CR71R72, где R71 и R72 независимо выбраны из группы, состоящей из водорода и незамещенных или замещенных остатков алкила, арила, арилалкила, гетероарилалкила; Е3 представляет собой C(O); R2 представляет собой NR21R22, где R21 и R22 независимо выбраны из водорода и незамещенных или замещенных остатков алкила, арила, арилалкила, гетероарилалкила и гетероциклоалкилалкила, причем алкил содержит от 1 до 13 атомов углерода, алкенил содержит от 2 до 13 атомов углерода, арил и гетероарил содержат от 5 до 13 кольцевых атомов углерода, где в остатке гетероарила один или несколько атомов углерода замещены гетероатомами, выбранными из группы, состоящей из N, О и S; гетероциклоалкил содержит от 3 до 8 кольцевых атомов углерода, из которых от одного до трех атомов углерода замещены гетероатомами, выбранными из группы, состоящей из N, О и S; в их любых стереоизомерных формах или их смесях в любом соотношении и их фармацевтически приемлемым солям; к способу получения соединений общей формулы I, включающему связывание защищенных аминокислот; к фармацевтической композиции обладающей способностью оказывать антитромботический эффект посредством активированного фактора VII(FVIIa) свертывания крови. 3 с. и 16 з.п. ф-лы, 4 табл. Настоящее изобретение относится к новым соединениям, их получению, их применению и к фармацевтическим композициям, содержащим эти соединения, которые оказывают сильный антитромботический эффект посредством обратимого ингибирования активированного фактора VIIa (FVIIa) свертывания крови. Образование тромба является обычно результатом повреждения ткани, которое инициирует каскад свертывания и оказывает эффект замедления или предотвращения тока крови при заживлении ран. Каскад свертывания могут также инициировать другие факторы, которые косвенно связаны с повреждением ткани, подобные атеросклерозу и воспалению, и они могут привести к патологическим последствиям. Свертывание крови представляет собой сложный процесс, вовлекающий прогрессивно нарастающий каскад реакций активации ферментов, при котором путем ограниченного протеолиза последовательно активируются зимогены плазмы. Каскад свертывания крови был механистически разделен на внутренний и внешний пути, которые сходятся при активации фактора X; последующая выработка тромбина продолжается единым общим путем (см. схему 1). 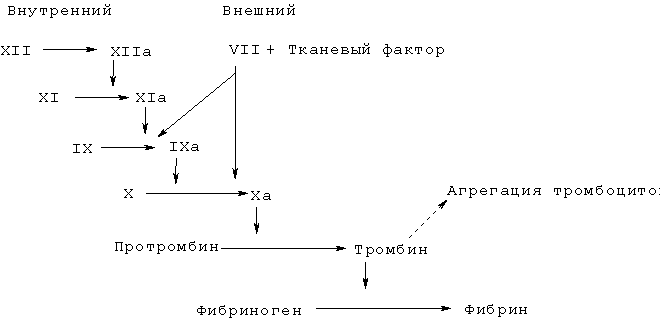 Схема 1: Каскад свертывания крови
Имеющиеся в настоящее время данные свидетельствуют о том, что внутренний путь играет важную роль в поддержании и росте образования фибрина, в то время как внешний путь имеет решающее значение в инициации фазы свертывания крови (H.Cole, Aust.J.Med.Sci. 16 (1995) 87; G.J.Broze, Blood Coagulation and Fibrinolysis 6, Suppl. l (1995) S7-S13). В целом признано, что свертывание крови физически инициируется при образовании комплекса тканевый фактор (TF) / фактор VIIa. Однажды образовавшись, этот комплекс быстро инициирует свертывание путем активации факторов IX и X. Вновь генерированный активированный фактор X, т.е. фактор Ха, затем образует комплекс один к одному с фактором Va и фосфолипидами для образования комплекса протромбиназы, который ответственен за превращение растворимого фибриногена в нерастворимый фибрин посредством активации тромбина из его предшественника протромбина. С течением времени активность комплекса фактор VIIa/тканевый фактор (внешний путь) подавляется белком-ингибитором протеазы типа Kunitz, TFPI, который при образовании комплекса с фактором Ха может непосредственно ингибировать протеолитическую активность фактора VIIa/тканевого фактора. Для поддержания процесса свертывания в присутствии ингибированной экзогенной системы под воздействием опосредованной тромбином активности внутреннего пути вырабатывается дополнительный фактор Ха. Таким образом, тромбин играет двойную автокаталитическую роль, опосредуя свою собственную выработку и превращение фибриногена в фибрин.
Автокаталитическая природа генерирования тромбина представляет собой важный защитный механизм против неконтролируемого кровотечения, и он обеспечивает то, что при наличии данного порогового уровня протромбиназы свертывание крови будет продолжаться до полного завершения. Способность образовывать сгустки крови является жизненно важной для выживания. Однако при определенных патологических состояниях образование сгустков крови внутри системы кровообращения само по себе является источником осложнений. Тем не менее, при таких патологических состояниях нежелательно полностью ингибировать систему свертывания, потому что это бы вызвало угрожающее жизни кровотечение. Таким образом, наиболее желательно разработать средства, которые ингибируют свертывание путем ингибирования фактора VIIa без прямого ингибирования тромбина.
При многих видах клинического применения существует большая необходимость в предотвращении образования внутрисосудистых сгустков крови или в некотором антикоагулянтном лечении. При многих специфических видах клинического применения имеющиеся в настоящее время препараты являются неудовлетворительными. Например, почти у 50% пациентов, которые перенесли полное протезирование тазобедренного сустава, развивается тромбоз глубоких вен (ТГВ). Одобренные в настоящее время методы лечения представляют собой применение фиксированных доз низкомолекулярного гепарина (НМГ) и различных доз гепарина. Даже при использовании этих схем лекарственной терапии у 10-20% пациентов развивается ТГВ, а у 5-10% развиваются осложнения, связанные с кровотечением.
Другая клиническая ситуация, при которой необходимы более совершенные антикоагулянты, касается лиц, подвергающихся транслюминальной коронарной ангиопластике, и лиц с риском инфаркта миокарда или лиц, страдающих нарастающей стенокардией. Принятая в настоящее время обычная терапия, которая состоит во введении гепарина и аспирина, связана с частотой внезапной окклюзии сосудов в пределах 24 ч после выполнения вмешательства, составляющей от 6% до 8%. Частота связанных с кровотечением осложнений, требующих трансфузионной терапии вследствие применения гепарина, также составляет приблизительно 7%. Более того, даже хотя существенны случаи отдаленной окклюзии сосудов, введение гепарина после окончания вмешательства малоэффективно и может оказать вредное воздействие.
Наиболее широко применяемые ингибиторы свертываемости крови представляют собой гепарин и родственные сульфатированные полисахариды, НМГ и гепаринсульфат. Эти молекулы оказывают свои противосвертывающие эффекты, способствуя связыванию естественного регулятора процесса свертывания, антитромбина III, с тромбином и фактором Ха. Ингибирующая активность гепарина в первую очередь направлена на тромбин, который инактивируется приблизительно в 100 раз быстрее, чем фактор Ха. Гирудин и гирулог представляют собой два дополнительных специфических по отношению к тромбину антикоагулянта, которые в настоящее время проходят клинические испытания. Однако эти антикоагулянты, которые ингибируют тромбин, также связаны с осложнениями в виде кровотечения. Преклинические исследования на бабуинах и собаках показали, что прицельно направляющие ферменты, участвующие на ранних стадиях каскада свертывания, такие как фактор Ха или фактор VIIa, предотвращают образование сгустка, не вызывая побочных эффектов в виде кровотечения, наблюдаемых при использовании прямых ингибиторов тромбина (T.Yokoyama, A.B.Kelly, U.M.Marzec, S.R.Hanson, S.Kunitada, L.A.Harker, Ciculation 92 (1995) 485-491; L.A.Harker, S.R.Hanson, A.B.Kelly, Thromb. Hemostas 74 (1995) 464-472; C.R.Benedict, J.Ryan, J.Todd, K.Kuwabara, P.Tyburg, Jr., J.Cartwright, D.Stern, Blood 81 (1993) 2059-2066).
Специфическое ингибирование каталитического комплекса фактор VIIa/TF с использованием моноклонального антитела (международная заявка на патент № WО92/06711) и белка, такого как инактивированный хлорметилкетоном фактор VIIa (международная заявка на патент № WО96/12800 и WО97/47651) представляет собой крайне эффективное средство контроля образования тромба, вызванного острым повреждением артерии, или тромботических осложнений, связанных с бактериальной септицемией. Имеется также экспериментальное доказательство, свидетельствующее о том, что ингибирование активности фактора VIIa/TF ингибирует рестеноз после баллонной ангиопластики (L.A.Harker, S.R.Hanson, J.N.Wilcox, A.B.Kelly, Haemostasis 26 (1996) S1:76-82). Исследования кровотечения были проведены на бабуинах, и они показывают, что ингибирование комплекса фактор VIIa/TF имеет самый широкий диапазон безопасности в отношении терапевтической эффективности и риска кровотечения из всех испытанных подходов к антикоагулянтной терапии, включая ингибирование тромбина, тромбоцитов и фактора Ха (L.A.Harker, S.R.Hanson, A.B.Kelly, Thromb. Hemostas 74 (1995) 464-472).
Специфический ингибитор фактора VIIa имел бы существенную практическую ценность в медицинской практике. В частности, ингибитор фактора VIIa был бы эффективен в условиях, при которых используемые в настоящее время препараты выбора, гепарин и родственные сульфатированные полисахариды, неэффективны или оказывают лишь пограничный эффект. Таким образом, существует необходимость в низкомолекулярном, специфическом для фактора VIIa ингибиторе свертывания, который эффективен, но не вызывает нежелательных побочных эффектов. Настоящее изобретение удовлетворяет этому требованию путем предоставления производных формулы I, ингибирующих активность фактора VIIa, и также с помощью обеспечения связанных с ними преимуществ.
Соединения формулы I представляют собой ингибиторы фермента фактора VIIa свертывания крови. Изобретение также относится к способам получения соединений формулы I, к способам ингибирования активности фактора VIIa и ингибирования свертывания крови, к применению соединений формулы I для лечения и профилактики заболеваний, которые можно излечить или предотвратить с помощью ингибирования активности фактора VIIa, таких как тромбоэмболические заболевания, включая тромбоз, рестеноз, инфаркт и стенокардию, и к применению соединений формулы I для приготовления лекарственных средств, которые предполагается применять при таких заболеваниях. Изобретение, кроме того, относится к композициям, содержащим соединения формулы I, смешанным или связанным другим образом с инертным носителем, в частности, к фармацевтическим композициям, содержащим соединение формулы I, вместе с фармацевтически приемлемыми веществами-носителями или наполнителями и/или дополнительными веществами или добавками.
Сущность изобретения
Настоящее изобретение предоставляет соединения, которые специфически ингибируют активность фактора VIIa. В частности, предметом настоящего изобретения являются соединения формулы I
R1-A-B-D-En-R2 (I),
в которой R1 представляет собой R13, R12C(O) или от 1 до 3 аминокислот, N-конец которых может быть замещен заместителем, выбранным из группы, состоящей из R14C(O), R15S(O)2 и группы, защищающей аминогруппу, в которых
R12 выбрана из ряда, состоящего из алкила, алкенила, алкинила, алкилокси, алкиламино, алкениламино, алкиниламино, алкенилокси, алкинилокси, арила, гетероарила, гетероциклоалкила, арилалкила, гетероарилалкила, гетероциклоалкилалкила, гетероалкила, гетероалкенила и гетероалкинила, причем все эти остатки могут быть замещены,
R13 выбрана из ряда, состоящего из группы, защищающей аминогруппу, водорода, алкила, арила, арилалкила, гетероарила, гетероарилалкила, гетероциклоалкила и гетероциклоалкилалкила,
R14 и R15 независимо выбраны из ряда, состоящего из алкила, арила, арилалкила, гетероарила, гетероарилалкила, гетероциклоалкила и гетероциклоалкилалкила,
А представляет собой группу А1-А2-А3, в которой
А1 представляет собой NH,
А2 представляет собой CHR93, в которой R93 представляет собой 4-амидинфенилметил,
A3 представляет собой С(О),
В представляет собой группу В1-В2-В3, в которой
В1 представляет собой NR95, в которой R95 выбран из ряда, состоящего из водорода и алкила,
B2 представляет собой CHR97, в которой R97 представляет собой этил, который замещен в положении 2 заместителем, выбранным из ряда, состоящего из гидроксикарбонила, алкилоксикарбонила и арилалкилоксикарбонила,
B3 представляет собой С(О),
D представляет собой группу D1-D2-D3, в которой
D1 представляет собой NH,
D2 представляет собой CR81R82, в которой R81 и R82 независимо выбраны из ряда, состоящего из водорода и незамещенных или замещенных остатков алкила, арила, арилалкила, гетероарила, гетероарилалкила, гетероциклоалкила и гетероциклоалкилалкила, D3 представляет собой С(О),
En представляет собой (Е1-Е2-Е3)n, в которой n равно 0, 1, 2 или 3,
E1 представляет собой NR70, в которой R70 выбрана из ряда, состоящего из водорода, алкила, арила, арилалкила, гетероарила, гетероарилалкила, гетероциклоалкила и гетероциклоалкилалкила,
Е2 представляет собой CR71R72, в которой R71 и R72 независимо выбраны из ряда, состоящего из водорода и незамещенных или замещенных остатков алкила, арила, арилалкила, гетероарила, гетероарилалкила, гетероциклоалкила и гетероциклоалкилалкила,
Е3 представляет собой С(О),
R2 выбрана из ряда, состоящего из NR21R22, OR23 и R24, в которой R21, R22, R23 и R24 независимо выбраны из ряда, состоящего из водорода и незамещенных или замещенных остатков алкила, арила, арилалкила, гетероарила, гетероарилалкила, гетероциклоалкила и гетероциклоалкилалкила,
алкил и гетероалкил содержат от 1 до 13 атомов углерода, где в остатке гетероалкила один или несколько атомов углерода замещены гетероатомами, выбранными из ряда, состоящего из N, О и S;
алкенил, алкинил, гетероалкенил и гетероалкинил содержат от 2 до 13 атомов углерода, где в остатке гетероалкенила и гетероалкинила один или несколько атомов углерода замещены гетероатомами, выбранными из ряда, состоящего из N, О и S;
арил и гетероарил содержат от 5 до 13 кольцевых атомов углерода, где в остатке гетероарила один или несколько атомов углерода замещены гетероатомами, выбранными из ряда, состоящего из N, О и S;
гетероциклоалкил содержит от 3 до 8 кольцевых атомов углерода, из которых от одного до трех атомов углерода замещены гетероатомами, выбранными из ряда, состоящего из N, О и S;
в их любых стереоизомерных формах и их смесях в любом соотношении и их фармацевтически приемлемые соли.
Настоящее изобретение предоставляет пептиды формулы I
R1-A-B-D-En-R2 (I),
в которой R1, R2, А, В, D, Е и n определены, как указано выше, и которые представляют собой соединения, ингибирующие активность фактора VIIa, но существенно не ингибирующие активность других протеаз, которые участвуют в пути свертывания крови. В соединениях формулы I, например, в группах А, В, D или Е или в группе R1 в случае, если R1 представляет 1, 2 или 3 аминокислоты, содержатся структурные элементы, которые представляют собой аминокислоты или их производные, или аналоги аминокислот, или миметические структуры, и которые по типу пептидов связаны с прилегающими группами посредством амидных связей С(О)-N, образованных между карбоксильной группой одной такой аминокислоты и т.д. и аминогруппой другой аминокислоты и т.д. Как обычно в пептидной химии, двухвалентный остаток аминокислоты или группы, подобной А, В, D или Е присутствующих в формуле I, получают из соответствующей аминокислоты с помощью формального удаления атома водорода из аминогруппы и гидроксильной группы из карбоксильной группы.
Используемый здесь термин “аминокислота” используется в его самом широком смысле для обозначения 20 естественно встречающихся аминокислот, которые транслируются из генетического кода и содержат строительные блоки белков, включая, если специально не указано иначе, L-аминокислоты и D-аминокислоты, а также химически модифицированные аминокислоты, такие как аналоги аминокислот, естественно встречающиеся аминокислоты, которые обычно не включены в белки, такие как норлейцин, и химически синтезированные соединения, имеющие свойства, которые, как известно в данной области, характерны для аминокислот. Например, аналоги или миметики фенилаланина или пролина, которые обеспечивают возможность такой же конформационной рестрикции пептидных соединений, как естественный Phe или Pro, включены в определение “аминокислот” и известны специалистам в этой области. Такие аналоги и миметики именуются здесь “функциональными эквивалентами” аминокислоты. Другие примеры аминокислот и аналогов аминокислот перечислены в публикации Roberts и Vellaccio (The Peptides: Analysis, Synthesis, Biology, eds. Gross and Meinhofer, vol.5, p.341, Academic Press, Inc., New York, 1983, которая включена сюда в виде ссылки). Аббревиатуры аминокислот, аналогов аминокислот и миметических структур, а также другие аббревиатуры, используемые в заявке, перечислены в табл.1.
Таблица 1
Аббревиатуры, используемые в заявке
Соединение/остаток Аббревиатура
Уксусная кислота АсОН
Ацетиламинометил Асm
Аланин Аlа
Аллилоксикарбонил Alloc
пара-Амидинфенилаланин pAph
2-Аминомасляная кислота 2-Abu
Аргинин Arg
Аспарагин Asn
Аспарагиновая кислота Asp
Бензил Bzl
трет-Бутилоксикарбонил Boc
трет-Бутил tBu
Циклогексилглицин Chg
Циклогексил Chx
Циклогексилаланин Cha
Цистеин Cys
2,4-Диаминмасляная кислота Dab
2,3-Диаминпропионовая кислота Dap
Дихлорметан DCM
Диизопропилкарбодиимид DIC
Диизопропилэтиламин DIEA
N,N-Диметилформамид DMF
Диметилсульфоксид DMSO
9-Фторенилметилоксикарбонил Fmoc
Глутаминовая кислота Glu
Глутамин Gln
Глицин Gly
Гистидин His
N-Гидроксибензотриазол HOBt
4-Гидроксиметилфеноксиуксусная кислота HMPA
Изолейцин Ile
Лейцин Leu
Лизин Lys
Метил Me
N-Метилимидазол NMI
N-Метилморфолин NMM
2,2,5,7,8-Пентаметилхроман-6-сульфонил Pmc
Орнитин Orn
Фенил Ph
Фенилаланин Phe
Фенилглицин Phg
Пролин Pro
Серин Ser
Тетрагидрофуран THF
Тетраметилфторформамидингексафторфосфат TFFH
Треонин Thr
Трифторуксусная кислота TFA
Тритил Trt
Триптофан Trp
Валин Val
При отсутствии других уточнений указанные выше аминокислоты в виде аббревиатур имеют L-конфигурацию. Аминокислоты D-конфигурации обозначены приставкой D с использованием трехбуквенного кода (например, D-Ala, D-Cys, D-Asp, D-Trp, D-pAph). Аббревиатуры, подобные, например, Phe(4-CN) и Phe[4-C(-S-CH2-CH2-S-)-Ph] означают остаток аминокислоты фе-нилаланина, который в положении 4 фениловой группы несет, соответственно, цианозаместитель или заместитель 2-фенил-1,3-дитиолан-2-ил. Аббревиатура, подобная, например. Dap[-С(=NH)-NН2], означает остаток аминокислоты 2,3-диаминпропионовой кислоты, в которой аминогруппа в боковой цепи, т.е. аминогруппа в положении 3, замещена амидиногруппой -C(=NH)-NH2 (карбамимидоильной группой), в результате чего получается элемент, состоящий из гуанидиногруппы -NH-C(=NH)-NH2, прикрепленной к положению 3 пропионовой кислоты. Аббревиатуры, подобные, например, Orn[-С(=NH)-NН2] или Cys(Me), означают остаток аминокислоты орнитина, в которой, соответственно, аминогруппа в боковой цепи несет амидиногруппу или остаток аминокислоты цистеина, в которой меркаптогруппа несет метиловую группу.
Термины TOTU, HATU и ВОР означают, соответственно, O-[циан(этоксикарбонил)метиленамино]-1,1,3,3-тетраметилуронийтетрафторборат, О-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилуронийгексафторфосфат и 1-бензотриазолилокси-трис-(диметиламино)-фосфонийгексафторфосфат.
Используемый здесь термин “специфическое” при использовании со ссылкой на ингибирование активности фактора VIIa означает, что соединение формулы I может ингибировать активность фактора VIIa без существенного ингибирования активности других определенных протеаз, включая плазмин и тромбин (при использовании той же концентрации ингибитора). Такие протеазы участвуют в каскаде свертывания крови и фибринолиза.
Используемый здесь термин “заместитель” относится к любой из различных химических групп, которая замещена на раскрытых здесь каркасе пептида или боковой цепи пептида, аналоге пептида, миметическом или органическом соединении. Заместитель может включать в себя любую из множества различных групп, известных специалистам в данной области (см., например, публикацию Giannis and Kolter, Angew. Chem. Int. Ed. Engl. 32 (1993) 1244-1267, которая включена сюда в виде ссылки).
Используемый здесь термин “алкил” применяется в самом широком смысле для обозначения насыщенных или ненасыщенных, линейных, разветвленных или циклических цепей приблизительно из 1-13 атомов углерода, в которых ненасыщенная алкильная группа, конечно, содержит по меньшей мере 2 атома углерода, а циклическая алкильная группа – по меньшей мере 3 атома углерода. Ненасыщенная группа может содержать одну или несколько двойных связей и/или тройных связей. Таким образом, термин “алкил” включает в себя, например, метильную, этильную, н-пропильную, изопропильную, н-бутильную, изобутильную, втор-бутильную, трет-бутильную, 1-метилбутильную, 2,2-диметилбутильную, 2-метилпентильную, 2,2-диметилпропильную, н-пентильную и н-гексильную группы, алкиленовые группы, циклические цепи атомов углерода, такие как циклопропильная, циклобутильная, циклопентильная, циклогексильная и циклогептильная группы, а также комбинации линейных или разветвленных цепей и циклических цепей атомов углерода, таких как метилциклогексильная, циклогексилметильная, 1-циклогексилэтильная, 2-циклогексилэтильная, циклопентилметильная, 1-циклопентилэтильная, 2-циклопентилэтильная, циклопропилметильная, 1-циклопропилэтильная, 2-циклопропилэтильная или циклопропилметиленовая группы. Таким образом, алкил также включает в себя циклические алкильные группы, которые несут один или несколько алкильных заместителей. Дальнейшие примеры алкила представляют собой упомянутые ниже специфические ненасыщенные группы. Кроме того, следует понимать, что определенный здесь алкил может быть замещен одним или несколькими идентичными или различными заместителями, например одним, двумя, тремя или четырьмя заместителями, которые могут присутствовать в любом желаемом подходящем положении.
Термин “алкил” предпочтительно означает насыщенные, линейные или разветвленные цепи из 1-6 атомов углерода, ненасыщенные линейные или разветвленные цепи из 2-6 атомов углерода или циклические алкильные группы из 3-8 атомов углерода, в частности от 3 до 6 или от 4 до 6 кольцевых атомов углерода. С точки зрения ненасыщенных алкильных цепей предпочтительны (С2-С6)-алкенил и (С2-С6)-алкинил. Примеры ненасыщенных алкильных групп представляют собой алкенильную и алкинильную группы, такие как винил, проп-1-енил, проп-2-енил (=аллил), бут-2-енил, бутен-3-ил, 3-метилбут-2-енил, этинил, проп-2-инил, бут-2-инил и им подобные.
Аналогичным образом термин “ацил” используется в своем самом широком смысле для обозначения насыщенных или ненасыщенных, линейных, разветвленных или циклических цепей из приблизительно 1-13 атомов углерода или арильных групп, имеющих от 5 до 13 кольцевых атомов углерода, которые прикреплены к карбонильной группе -С(О)- и связаны через указанную карбонильную группу. Ацильная группа может рассматриваться как полученная из соответствующего соединения, содержащего карбоксильную группу С(О)-ОН путем формального удаления гидроксильной группы. Таким образом, термин “ацил” охватывает, например, такие группы как формил, ацетил, бензоил и им подобные. Предпочтительная группа из ацильных групп охватывает упомянутые здесь выше насыщенные или ненасыщенные, линейные, разветвленные или циклические цепи, имеющие предпочтительный диапазон атомов углерода, которые дополнительно содержат карбонильную группу, посредством которой они связаны.
Термин “арил” относится к ароматическим группам, содержащим приблизительно от 5 до 13 кольцевых атомов углерода и по меньшей мере одну “кольцевую” группу, имеющую конъюгированную систему pi электрона. Предпочтительно термин “арил” относится к ароматическим группам, имеющим от 6 до 10 кольцевых атомов углерода. Примеры арила включают в себя например, фенильную, нафтильную, такую как 1-нафтильная и 2-нафтильная, фторенильная, бифенильная группы и их аналоги и производные, все из которых могут быть необязательно замещены одним или несколькими, например одним, двумя, тремя или четырьмя, идентичными или различными заместителями, которые могут присутствовать в любом желаемом подходящем положении. Например, однозамещенная фенильная группа может быть замещена в положении 2-, 3- или 4-, двузамещенная фенильная группа – в положении 2,3-, 2,4-, 2,5-, 2,6-, 3,4- или 3,5-.
Термин “арилалкил” относится к алкилу, как определено выше, замещенному одной или несколькими, например одной или двумя, идентичными или различными группами. Подходящие арилалкильные группы включают в себя бензил, фенилэтил, такой как 1-фенилэтил и 2-фенилэтил, дифенилметил, дифенилэтил, такой как 1,2-дифенилэтил и 2,2-дифенилэтил, фенилпропил, такой как 1-фенилпропил, 2-фенилпропил и 3-фенилпропил, дифенилпропил, такой как 2,3-дифенилпропил и 3,3-дифенилпропил, нафтилметил, нафтилэтил, такой как 1-нафтилэтил и 2-нафтилэтил, нафтилпропил, такой как 1-нафтилпропил, 2-нафтилпропил и 3-нафтилпропил, 1,2,3,4-тетрагидро-1-нафтил, 1,2,3,4-тетрагидро-2-нафтил и им подобные, все из которых могут быть необязательно замещены.
Используемые здесь термины “гетероалкил”, “гетероалкенил”, “гетероалкинил”, “гетероарилалкил” и “гетероарил” относятся соответственно к алкильной, арилалкильной и арильной группам, в которых один или несколько атомов углерода, например 1, 2 или 3 атома углерода, замещены идентичными или отличными гетероатомами, такими как N, О или S. Кроме того, термин “гетероциклоалкил” используется при ссылке на циклическую алкильную группу, в которой один или несколько кольцевых атомов углерода замещены гетероатомами. Предпочтительно термин “гетероциклоалкил” означает циклическую алкильную группу, имеющую от 3 до 8 кольцевых атомов углерода, 1, 2 или 3 из которых замещены идентичными или различными гетероатомами, такими как N, О или S.
Все эти группы могут быть связаны посредством любого желаемого положения, включая подходящие кольцевые атомы азота в случае гетероциклов азота. Подходящие гетероарильные группы, гетероарилалкильные группы, гетероалкильные группы и гетероциклоалкильные группы включают в себя, например, 2-пиридил, 3-пиридил, 4-пиридил, 2-тиенил, 3-тиенил, индолил, имидазолил, фурил, пиперонил, 2-пиридилметил, 3-пиридилметил, 4-пиридилметил, 1-(2-пиридил)этил, 1-(3-пиридил)этил, 1-(4-пиридил)этил, 2-(2-пиридил)этил, 2-(3-пиридил)этил, 2-(4-пиридил)этил, пиколил, пирролидинил, пиперидинил, тетрагидрофурил, тетрагидрофуран-2-илметил, морфолинил, 4-морфолинил, 2-(4-морфолинил)этил, пиперазинил, 2-(4-метилпиперазин-1-ил)этил и им подобные, все из которых могут быть необязательно замещены одним или несколькими, например одним, двумя, тремя или четырьмя, идентичными или различными заместителями.
Пептиды согласно изобретению могут изменяться на N-конце и/или С-конце с помощью реакции с подходящими реагентами или с помощью введения (или с помощью присутствия), соответственно, группы, защищающей аминогруппу, или группы, защищающей карбоксильную группу. N-конец пептида или аналога пептида может быть химически изменен так, что N-концевая аминогруппа замещается, например, ацильной группой (например, ацетильной, циклопентилкарбонильной, изохинолилкарбонильной, фуроильной, тозильной, бензоильной, пиразинкарбонильной или другими такими группами) с помощью реакции с изоцианатом, хлорформатом, алкилирующим агентом или с помощью введения другой такой группы, все из которых могут быть замещены заместителем, как описано выше. Следует понимать, что термин “аминогруппа” используется здесь широко для ссылки на любую свободную аминогруппу, включая первичную, вторичную или третичную аминогруппу, присутствующую в пептиде. Для сравнения, термин “N-конец” относится к
Схема 1: Каскад свертывания крови
Имеющиеся в настоящее время данные свидетельствуют о том, что внутренний путь играет важную роль в поддержании и росте образования фибрина, в то время как внешний путь имеет решающее значение в инициации фазы свертывания крови (H.Cole, Aust.J.Med.Sci. 16 (1995) 87; G.J.Broze, Blood Coagulation and Fibrinolysis 6, Suppl. l (1995) S7-S13). В целом признано, что свертывание крови физически инициируется при образовании комплекса тканевый фактор (TF) / фактор VIIa. Однажды образовавшись, этот комплекс быстро инициирует свертывание путем активации факторов IX и X. Вновь генерированный активированный фактор X, т.е. фактор Ха, затем образует комплекс один к одному с фактором Va и фосфолипидами для образования комплекса протромбиназы, который ответственен за превращение растворимого фибриногена в нерастворимый фибрин посредством активации тромбина из его предшественника протромбина. С течением времени активность комплекса фактор VIIa/тканевый фактор (внешний путь) подавляется белком-ингибитором протеазы типа Kunitz, TFPI, который при образовании комплекса с фактором Ха может непосредственно ингибировать протеолитическую активность фактора VIIa/тканевого фактора. Для поддержания процесса свертывания в присутствии ингибированной экзогенной системы под воздействием опосредованной тромбином активности внутреннего пути вырабатывается дополнительный фактор Ха. Таким образом, тромбин играет двойную автокаталитическую роль, опосредуя свою собственную выработку и превращение фибриногена в фибрин.
Автокаталитическая природа генерирования тромбина представляет собой важный защитный механизм против неконтролируемого кровотечения, и он обеспечивает то, что при наличии данного порогового уровня протромбиназы свертывание крови будет продолжаться до полного завершения. Способность образовывать сгустки крови является жизненно важной для выживания. Однако при определенных патологических состояниях образование сгустков крови внутри системы кровообращения само по себе является источником осложнений. Тем не менее, при таких патологических состояниях нежелательно полностью ингибировать систему свертывания, потому что это бы вызвало угрожающее жизни кровотечение. Таким образом, наиболее желательно разработать средства, которые ингибируют свертывание путем ингибирования фактора VIIa без прямого ингибирования тромбина.
При многих видах клинического применения существует большая необходимость в предотвращении образования внутрисосудистых сгустков крови или в некотором антикоагулянтном лечении. При многих специфических видах клинического применения имеющиеся в настоящее время препараты являются неудовлетворительными. Например, почти у 50% пациентов, которые перенесли полное протезирование тазобедренного сустава, развивается тромбоз глубоких вен (ТГВ). Одобренные в настоящее время методы лечения представляют собой применение фиксированных доз низкомолекулярного гепарина (НМГ) и различных доз гепарина. Даже при использовании этих схем лекарственной терапии у 10-20% пациентов развивается ТГВ, а у 5-10% развиваются осложнения, связанные с кровотечением.
Другая клиническая ситуация, при которой необходимы более совершенные антикоагулянты, касается лиц, подвергающихся транслюминальной коронарной ангиопластике, и лиц с риском инфаркта миокарда или лиц, страдающих нарастающей стенокардией. Принятая в настоящее время обычная терапия, которая состоит во введении гепарина и аспирина, связана с частотой внезапной окклюзии сосудов в пределах 24 ч после выполнения вмешательства, составляющей от 6% до 8%. Частота связанных с кровотечением осложнений, требующих трансфузионной терапии вследствие применения гепарина, также составляет приблизительно 7%. Более того, даже хотя существенны случаи отдаленной окклюзии сосудов, введение гепарина после окончания вмешательства малоэффективно и может оказать вредное воздействие.
Наиболее широко применяемые ингибиторы свертываемости крови представляют собой гепарин и родственные сульфатированные полисахариды, НМГ и гепаринсульфат. Эти молекулы оказывают свои противосвертывающие эффекты, способствуя связыванию естественного регулятора процесса свертывания, антитромбина III, с тромбином и фактором Ха. Ингибирующая активность гепарина в первую очередь направлена на тромбин, который инактивируется приблизительно в 100 раз быстрее, чем фактор Ха. Гирудин и гирулог представляют собой два дополнительных специфических по отношению к тромбину антикоагулянта, которые в настоящее время проходят клинические испытания. Однако эти антикоагулянты, которые ингибируют тромбин, также связаны с осложнениями в виде кровотечения. Преклинические исследования на бабуинах и собаках показали, что прицельно направляющие ферменты, участвующие на ранних стадиях каскада свертывания, такие как фактор Ха или фактор VIIa, предотвращают образование сгустка, не вызывая побочных эффектов в виде кровотечения, наблюдаемых при использовании прямых ингибиторов тромбина (T.Yokoyama, A.B.Kelly, U.M.Marzec, S.R.Hanson, S.Kunitada, L.A.Harker, Ciculation 92 (1995) 485-491; L.A.Harker, S.R.Hanson, A.B.Kelly, Thromb. Hemostas 74 (1995) 464-472; C.R.Benedict, J.Ryan, J.Todd, K.Kuwabara, P.Tyburg, Jr., J.Cartwright, D.Stern, Blood 81 (1993) 2059-2066).
Специфическое ингибирование каталитического комплекса фактор VIIa/TF с использованием моноклонального антитела (международная заявка на патент № WО92/06711) и белка, такого как инактивированный хлорметилкетоном фактор VIIa (международная заявка на патент № WО96/12800 и WО97/47651) представляет собой крайне эффективное средство контроля образования тромба, вызванного острым повреждением артерии, или тромботических осложнений, связанных с бактериальной септицемией. Имеется также экспериментальное доказательство, свидетельствующее о том, что ингибирование активности фактора VIIa/TF ингибирует рестеноз после баллонной ангиопластики (L.A.Harker, S.R.Hanson, J.N.Wilcox, A.B.Kelly, Haemostasis 26 (1996) S1:76-82). Исследования кровотечения были проведены на бабуинах, и они показывают, что ингибирование комплекса фактор VIIa/TF имеет самый широкий диапазон безопасности в отношении терапевтической эффективности и риска кровотечения из всех испытанных подходов к антикоагулянтной терапии, включая ингибирование тромбина, тромбоцитов и фактора Ха (L.A.Harker, S.R.Hanson, A.B.Kelly, Thromb. Hemostas 74 (1995) 464-472).
Специфический ингибитор фактора VIIa имел бы существенную практическую ценность в медицинской практике. В частности, ингибитор фактора VIIa был бы эффективен в условиях, при которых используемые в настоящее время препараты выбора, гепарин и родственные сульфатированные полисахариды, неэффективны или оказывают лишь пограничный эффект. Таким образом, существует необходимость в низкомолекулярном, специфическом для фактора VIIa ингибиторе свертывания, который эффективен, но не вызывает нежелательных побочных эффектов. Настоящее изобретение удовлетворяет этому требованию путем предоставления производных формулы I, ингибирующих активность фактора VIIa, и также с помощью обеспечения связанных с ними преимуществ.
Соединения формулы I представляют собой ингибиторы фермента фактора VIIa свертывания крови. Изобретение также относится к способам получения соединений формулы I, к способам ингибирования активности фактора VIIa и ингибирования свертывания крови, к применению соединений формулы I для лечения и профилактики заболеваний, которые можно излечить или предотвратить с помощью ингибирования активности фактора VIIa, таких как тромбоэмболические заболевания, включая тромбоз, рестеноз, инфаркт и стенокардию, и к применению соединений формулы I для приготовления лекарственных средств, которые предполагается применять при таких заболеваниях. Изобретение, кроме того, относится к композициям, содержащим соединения формулы I, смешанным или связанным другим образом с инертным носителем, в частности, к фармацевтическим композициям, содержащим соединение формулы I, вместе с фармацевтически приемлемыми веществами-носителями или наполнителями и/или дополнительными веществами или добавками.
Сущность изобретения
Настоящее изобретение предоставляет соединения, которые специфически ингибируют активность фактора VIIa. В частности, предметом настоящего изобретения являются соединения формулы I
R1-A-B-D-En-R2 (I),
в которой R1 представляет собой R13, R12C(O) или от 1 до 3 аминокислот, N-конец которых может быть замещен заместителем, выбранным из группы, состоящей из R14C(O), R15S(O)2 и группы, защищающей аминогруппу, в которых
R12 выбрана из ряда, состоящего из алкила, алкенила, алкинила, алкилокси, алкиламино, алкениламино, алкиниламино, алкенилокси, алкинилокси, арила, гетероарила, гетероциклоалкила, арилалкила, гетероарилалкила, гетероциклоалкилалкила, гетероалкила, гетероалкенила и гетероалкинила, причем все эти остатки могут быть замещены,
R13 выбрана из ряда, состоящего из группы, защищающей аминогруппу, водорода, алкила, арила, арилалкила, гетероарила, гетероарилалкила, гетероциклоалкила и гетероциклоалкилалкила,
R14 и R15 независимо выбраны из ряда, состоящего из алкила, арила, арилалкила, гетероарила, гетероарилалкила, гетероциклоалкила и гетероциклоалкилалкила,
А представляет собой группу А1-А2-А3, в которой
А1 представляет собой NH,
А2 представляет собой CHR93, в которой R93 представляет собой 4-амидинфенилметил,
A3 представляет собой С(О),
В представляет собой группу В1-В2-В3, в которой
В1 представляет собой NR95, в которой R95 выбран из ряда, состоящего из водорода и алкила,
B2 представляет собой CHR97, в которой R97 представляет собой этил, который замещен в положении 2 заместителем, выбранным из ряда, состоящего из гидроксикарбонила, алкилоксикарбонила и арилалкилоксикарбонила,
B3 представляет собой С(О),
D представляет собой группу D1-D2-D3, в которой
D1 представляет собой NH,
D2 представляет собой CR81R82, в которой R81 и R82 независимо выбраны из ряда, состоящего из водорода и незамещенных или замещенных остатков алкила, арила, арилалкила, гетероарила, гетероарилалкила, гетероциклоалкила и гетероциклоалкилалкила, D3 представляет собой С(О),
En представляет собой (Е1-Е2-Е3)n, в которой n равно 0, 1, 2 или 3,
E1 представляет собой NR70, в которой R70 выбрана из ряда, состоящего из водорода, алкила, арила, арилалкила, гетероарила, гетероарилалкила, гетероциклоалкила и гетероциклоалкилалкила,
Е2 представляет собой CR71R72, в которой R71 и R72 независимо выбраны из ряда, состоящего из водорода и незамещенных или замещенных остатков алкила, арила, арилалкила, гетероарила, гетероарилалкила, гетероциклоалкила и гетероциклоалкилалкила,
Е3 представляет собой С(О),
R2 выбрана из ряда, состоящего из NR21R22, OR23 и R24, в которой R21, R22, R23 и R24 независимо выбраны из ряда, состоящего из водорода и незамещенных или замещенных остатков алкила, арила, арилалкила, гетероарила, гетероарилалкила, гетероциклоалкила и гетероциклоалкилалкила,
алкил и гетероалкил содержат от 1 до 13 атомов углерода, где в остатке гетероалкила один или несколько атомов углерода замещены гетероатомами, выбранными из ряда, состоящего из N, О и S;
алкенил, алкинил, гетероалкенил и гетероалкинил содержат от 2 до 13 атомов углерода, где в остатке гетероалкенила и гетероалкинила один или несколько атомов углерода замещены гетероатомами, выбранными из ряда, состоящего из N, О и S;
арил и гетероарил содержат от 5 до 13 кольцевых атомов углерода, где в остатке гетероарила один или несколько атомов углерода замещены гетероатомами, выбранными из ряда, состоящего из N, О и S;
гетероциклоалкил содержит от 3 до 8 кольцевых атомов углерода, из которых от одного до трех атомов углерода замещены гетероатомами, выбранными из ряда, состоящего из N, О и S;
в их любых стереоизомерных формах и их смесях в любом соотношении и их фармацевтически приемлемые соли.
Настоящее изобретение предоставляет пептиды формулы I
R1-A-B-D-En-R2 (I),
в которой R1, R2, А, В, D, Е и n определены, как указано выше, и которые представляют собой соединения, ингибирующие активность фактора VIIa, но существенно не ингибирующие активность других протеаз, которые участвуют в пути свертывания крови. В соединениях формулы I, например, в группах А, В, D или Е или в группе R1 в случае, если R1 представляет 1, 2 или 3 аминокислоты, содержатся структурные элементы, которые представляют собой аминокислоты или их производные, или аналоги аминокислот, или миметические структуры, и которые по типу пептидов связаны с прилегающими группами посредством амидных связей С(О)-N, образованных между карбоксильной группой одной такой аминокислоты и т.д. и аминогруппой другой аминокислоты и т.д. Как обычно в пептидной химии, двухвалентный остаток аминокислоты или группы, подобной А, В, D или Е присутствующих в формуле I, получают из соответствующей аминокислоты с помощью формального удаления атома водорода из аминогруппы и гидроксильной группы из карбоксильной группы.
Используемый здесь термин “аминокислота” используется в его самом широком смысле для обозначения 20 естественно встречающихся аминокислот, которые транслируются из генетического кода и содержат строительные блоки белков, включая, если специально не указано иначе, L-аминокислоты и D-аминокислоты, а также химически модифицированные аминокислоты, такие как аналоги аминокислот, естественно встречающиеся аминокислоты, которые обычно не включены в белки, такие как норлейцин, и химически синтезированные соединения, имеющие свойства, которые, как известно в данной области, характерны для аминокислот. Например, аналоги или миметики фенилаланина или пролина, которые обеспечивают возможность такой же конформационной рестрикции пептидных соединений, как естественный Phe или Pro, включены в определение “аминокислот” и известны специалистам в этой области. Такие аналоги и миметики именуются здесь “функциональными эквивалентами” аминокислоты. Другие примеры аминокислот и аналогов аминокислот перечислены в публикации Roberts и Vellaccio (The Peptides: Analysis, Synthesis, Biology, eds. Gross and Meinhofer, vol.5, p.341, Academic Press, Inc., New York, 1983, которая включена сюда в виде ссылки). Аббревиатуры аминокислот, аналогов аминокислот и миметических структур, а также другие аббревиатуры, используемые в заявке, перечислены в табл.1.
Таблица 1
Аббревиатуры, используемые в заявке
Соединение/остаток Аббревиатура
Уксусная кислота АсОН
Ацетиламинометил Асm
Аланин Аlа
Аллилоксикарбонил Alloc
пара-Амидинфенилаланин pAph
2-Аминомасляная кислота 2-Abu
Аргинин Arg
Аспарагин Asn
Аспарагиновая кислота Asp
Бензил Bzl
трет-Бутилоксикарбонил Boc
трет-Бутил tBu
Циклогексилглицин Chg
Циклогексил Chx
Циклогексилаланин Cha
Цистеин Cys
2,4-Диаминмасляная кислота Dab
2,3-Диаминпропионовая кислота Dap
Дихлорметан DCM
Диизопропилкарбодиимид DIC
Диизопропилэтиламин DIEA
N,N-Диметилформамид DMF
Диметилсульфоксид DMSO
9-Фторенилметилоксикарбонил Fmoc
Глутаминовая кислота Glu
Глутамин Gln
Глицин Gly
Гистидин His
N-Гидроксибензотриазол HOBt
4-Гидроксиметилфеноксиуксусная кислота HMPA
Изолейцин Ile
Лейцин Leu
Лизин Lys
Метил Me
N-Метилимидазол NMI
N-Метилморфолин NMM
2,2,5,7,8-Пентаметилхроман-6-сульфонил Pmc
Орнитин Orn
Фенил Ph
Фенилаланин Phe
Фенилглицин Phg
Пролин Pro
Серин Ser
Тетрагидрофуран THF
Тетраметилфторформамидингексафторфосфат TFFH
Треонин Thr
Трифторуксусная кислота TFA
Тритил Trt
Триптофан Trp
Валин Val
При отсутствии других уточнений указанные выше аминокислоты в виде аббревиатур имеют L-конфигурацию. Аминокислоты D-конфигурации обозначены приставкой D с использованием трехбуквенного кода (например, D-Ala, D-Cys, D-Asp, D-Trp, D-pAph). Аббревиатуры, подобные, например, Phe(4-CN) и Phe[4-C(-S-CH2-CH2-S-)-Ph] означают остаток аминокислоты фе-нилаланина, который в положении 4 фениловой группы несет, соответственно, цианозаместитель или заместитель 2-фенил-1,3-дитиолан-2-ил. Аббревиатура, подобная, например. Dap[-С(=NH)-NН2], означает остаток аминокислоты 2,3-диаминпропионовой кислоты, в которой аминогруппа в боковой цепи, т.е. аминогруппа в положении 3, замещена амидиногруппой -C(=NH)-NH2 (карбамимидоильной группой), в результате чего получается элемент, состоящий из гуанидиногруппы -NH-C(=NH)-NH2, прикрепленной к положению 3 пропионовой кислоты. Аббревиатуры, подобные, например, Orn[-С(=NH)-NН2] или Cys(Me), означают остаток аминокислоты орнитина, в которой, соответственно, аминогруппа в боковой цепи несет амидиногруппу или остаток аминокислоты цистеина, в которой меркаптогруппа несет метиловую группу.
Термины TOTU, HATU и ВОР означают, соответственно, O-[циан(этоксикарбонил)метиленамино]-1,1,3,3-тетраметилуронийтетрафторборат, О-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилуронийгексафторфосфат и 1-бензотриазолилокси-трис-(диметиламино)-фосфонийгексафторфосфат.
Используемый здесь термин “специфическое” при использовании со ссылкой на ингибирование активности фактора VIIa означает, что соединение формулы I может ингибировать активность фактора VIIa без существенного ингибирования активности других определенных протеаз, включая плазмин и тромбин (при использовании той же концентрации ингибитора). Такие протеазы участвуют в каскаде свертывания крови и фибринолиза.
Используемый здесь термин “заместитель” относится к любой из различных химических групп, которая замещена на раскрытых здесь каркасе пептида или боковой цепи пептида, аналоге пептида, миметическом или органическом соединении. Заместитель может включать в себя любую из множества различных групп, известных специалистам в данной области (см., например, публикацию Giannis and Kolter, Angew. Chem. Int. Ed. Engl. 32 (1993) 1244-1267, которая включена сюда в виде ссылки).
Используемый здесь термин “алкил” применяется в самом широком смысле для обозначения насыщенных или ненасыщенных, линейных, разветвленных или циклических цепей приблизительно из 1-13 атомов углерода, в которых ненасыщенная алкильная группа, конечно, содержит по меньшей мере 2 атома углерода, а циклическая алкильная группа – по меньшей мере 3 атома углерода. Ненасыщенная группа может содержать одну или несколько двойных связей и/или тройных связей. Таким образом, термин “алкил” включает в себя, например, метильную, этильную, н-пропильную, изопропильную, н-бутильную, изобутильную, втор-бутильную, трет-бутильную, 1-метилбутильную, 2,2-диметилбутильную, 2-метилпентильную, 2,2-диметилпропильную, н-пентильную и н-гексильную группы, алкиленовые группы, циклические цепи атомов углерода, такие как циклопропильная, циклобутильная, циклопентильная, циклогексильная и циклогептильная группы, а также комбинации линейных или разветвленных цепей и циклических цепей атомов углерода, таких как метилциклогексильная, циклогексилметильная, 1-циклогексилэтильная, 2-циклогексилэтильная, циклопентилметильная, 1-циклопентилэтильная, 2-циклопентилэтильная, циклопропилметильная, 1-циклопропилэтильная, 2-циклопропилэтильная или циклопропилметиленовая группы. Таким образом, алкил также включает в себя циклические алкильные группы, которые несут один или несколько алкильных заместителей. Дальнейшие примеры алкила представляют собой упомянутые ниже специфические ненасыщенные группы. Кроме того, следует понимать, что определенный здесь алкил может быть замещен одним или несколькими идентичными или различными заместителями, например одним, двумя, тремя или четырьмя заместителями, которые могут присутствовать в любом желаемом подходящем положении.
Термин “алкил” предпочтительно означает насыщенные, линейные или разветвленные цепи из 1-6 атомов углерода, ненасыщенные линейные или разветвленные цепи из 2-6 атомов углерода или циклические алкильные группы из 3-8 атомов углерода, в частности от 3 до 6 или от 4 до 6 кольцевых атомов углерода. С точки зрения ненасыщенных алкильных цепей предпочтительны (С2-С6)-алкенил и (С2-С6)-алкинил. Примеры ненасыщенных алкильных групп представляют собой алкенильную и алкинильную группы, такие как винил, проп-1-енил, проп-2-енил (=аллил), бут-2-енил, бутен-3-ил, 3-метилбут-2-енил, этинил, проп-2-инил, бут-2-инил и им подобные.
Аналогичным образом термин “ацил” используется в своем самом широком смысле для обозначения насыщенных или ненасыщенных, линейных, разветвленных или циклических цепей из приблизительно 1-13 атомов углерода или арильных групп, имеющих от 5 до 13 кольцевых атомов углерода, которые прикреплены к карбонильной группе -С(О)- и связаны через указанную карбонильную группу. Ацильная группа может рассматриваться как полученная из соответствующего соединения, содержащего карбоксильную группу С(О)-ОН путем формального удаления гидроксильной группы. Таким образом, термин “ацил” охватывает, например, такие группы как формил, ацетил, бензоил и им подобные. Предпочтительная группа из ацильных групп охватывает упомянутые здесь выше насыщенные или ненасыщенные, линейные, разветвленные или циклические цепи, имеющие предпочтительный диапазон атомов углерода, которые дополнительно содержат карбонильную группу, посредством которой они связаны.
Термин “арил” относится к ароматическим группам, содержащим приблизительно от 5 до 13 кольцевых атомов углерода и по меньшей мере одну “кольцевую” группу, имеющую конъюгированную систему pi электрона. Предпочтительно термин “арил” относится к ароматическим группам, имеющим от 6 до 10 кольцевых атомов углерода. Примеры арила включают в себя например, фенильную, нафтильную, такую как 1-нафтильная и 2-нафтильная, фторенильная, бифенильная группы и их аналоги и производные, все из которых могут быть необязательно замещены одним или несколькими, например одним, двумя, тремя или четырьмя, идентичными или различными заместителями, которые могут присутствовать в любом желаемом подходящем положении. Например, однозамещенная фенильная группа может быть замещена в положении 2-, 3- или 4-, двузамещенная фенильная группа – в положении 2,3-, 2,4-, 2,5-, 2,6-, 3,4- или 3,5-.
Термин “арилалкил” относится к алкилу, как определено выше, замещенному одной или несколькими, например одной или двумя, идентичными или различными группами. Подходящие арилалкильные группы включают в себя бензил, фенилэтил, такой как 1-фенилэтил и 2-фенилэтил, дифенилметил, дифенилэтил, такой как 1,2-дифенилэтил и 2,2-дифенилэтил, фенилпропил, такой как 1-фенилпропил, 2-фенилпропил и 3-фенилпропил, дифенилпропил, такой как 2,3-дифенилпропил и 3,3-дифенилпропил, нафтилметил, нафтилэтил, такой как 1-нафтилэтил и 2-нафтилэтил, нафтилпропил, такой как 1-нафтилпропил, 2-нафтилпропил и 3-нафтилпропил, 1,2,3,4-тетрагидро-1-нафтил, 1,2,3,4-тетрагидро-2-нафтил и им подобные, все из которых могут быть необязательно замещены.
Используемые здесь термины “гетероалкил”, “гетероалкенил”, “гетероалкинил”, “гетероарилалкил” и “гетероарил” относятся соответственно к алкильной, арилалкильной и арильной группам, в которых один или несколько атомов углерода, например 1, 2 или 3 атома углерода, замещены идентичными или отличными гетероатомами, такими как N, О или S. Кроме того, термин “гетероциклоалкил” используется при ссылке на циклическую алкильную группу, в которой один или несколько кольцевых атомов углерода замещены гетероатомами. Предпочтительно термин “гетероциклоалкил” означает циклическую алкильную группу, имеющую от 3 до 8 кольцевых атомов углерода, 1, 2 или 3 из которых замещены идентичными или различными гетероатомами, такими как N, О или S.
Все эти группы могут быть связаны посредством любого желаемого положения, включая подходящие кольцевые атомы азота в случае гетероциклов азота. Подходящие гетероарильные группы, гетероарилалкильные группы, гетероалкильные группы и гетероциклоалкильные группы включают в себя, например, 2-пиридил, 3-пиридил, 4-пиридил, 2-тиенил, 3-тиенил, индолил, имидазолил, фурил, пиперонил, 2-пиридилметил, 3-пиридилметил, 4-пиридилметил, 1-(2-пиридил)этил, 1-(3-пиридил)этил, 1-(4-пиридил)этил, 2-(2-пиридил)этил, 2-(3-пиридил)этил, 2-(4-пиридил)этил, пиколил, пирролидинил, пиперидинил, тетрагидрофурил, тетрагидрофуран-2-илметил, морфолинил, 4-морфолинил, 2-(4-морфолинил)этил, пиперазинил, 2-(4-метилпиперазин-1-ил)этил и им подобные, все из которых могут быть необязательно замещены одним или несколькими, например одним, двумя, тремя или четырьмя, идентичными или различными заместителями.
Пептиды согласно изобретению могут изменяться на N-конце и/или С-конце с помощью реакции с подходящими реагентами или с помощью введения (или с помощью присутствия), соответственно, группы, защищающей аминогруппу, или группы, защищающей карбоксильную группу. N-конец пептида или аналога пептида может быть химически изменен так, что N-концевая аминогруппа замещается, например, ацильной группой (например, ацетильной, циклопентилкарбонильной, изохинолилкарбонильной, фуроильной, тозильной, бензоильной, пиразинкарбонильной или другими такими группами) с помощью реакции с изоцианатом, хлорформатом, алкилирующим агентом или с помощью введения другой такой группы, все из которых могут быть замещены заместителем, как описано выше. Следует понимать, что термин “аминогруппа” используется здесь широко для ссылки на любую свободную аминогруппу, включая первичную, вторичную или третичную аминогруппу, присутствующую в пептиде. Для сравнения, термин “N-конец” относится к  -аминогруппе первой аминокислоты, присутствующей в пептиде, написанном обычным образом.
N-конец пептида согласно изобретению может быть защищен с помощью присоединения к нему группы, защищающей аминогруппу. Термин “группа, защищающая аминогруппу”, используется здесь широко для ссылки на химическую группу, которая может вступать в реакцию со свободной аминогруппой, включая, например, -аминогруппу, присутствующую на N-конце пептида изобретения. С помощью реакции с ней группа, защищающая аминогруппу, защищает реактивную в противном случае аминогруппу против нежелательных реакций, которые могут произойти, например, во время процедуры синтеза или вследствие активности экзопептидазы по отношению к конечному соединению. Модификация аминогруппы может также обеспечить дополнительные преимущества, включая, например, увеличение растворимости или активности соединения. Здесь раскрыты или известны другим образом в данной области различные группы, защищающие аминогруппу, и они включают, например, ацильные группы, такие как ацетильная, трет-бутилоксикарбонильная, аллилоксикарбонильная, бензилоксикарбонильная группа или бензоильные группы, а также аминоацильный остаток, который сам может изменяться с помощью группы, защищающей аминогруппу. Другие группы, защищающие аминогруппу, описаны, например, в книге The Peptides, eds. Gross and Meienhofer, vol.3 (Academic Press, Inc., New York, 1981); и в публикации Greene and Wuts Protective Groups in Organic Synthesis, 2nd ed., pages 309-405 (John Wiley&Sons, New York, 1991), каждая из которых включена сюда в качестве ссылки. Продукт любой такой модификации N-концевой аминогруппы пептида или аналога пептида изобретения именуется здесь “N-концевое производное”.
Аналогичным образом, карбоксильная группа, такая как карбоксильная группа, присутствующая на С-конце пептида, может химически модифицироваться с использованием группы, защищающей карбоксильную группу. Термины “карбоксильная группа” и “С-конец” используются согласованным образом с определенными выше терминами “аминогруппа” и “N-конец”. Карбоксильная группа, подобная группе, которая присутствует на С-конце пептида, может модифицироваться с помощью восстановления С-концевой карбоксильной группы до спирта или альдегида или с помощью образования сложного эфира для перорального использования, или с помощью замещения карбоксильной группы таким заместителем как тиазолил, циклогексил или другая группа. Сложные эфиры для перорального использования хорошо известны в этой области и включают в себя, например, алкилоксиметильные группы, такие как метоксиметил, этоксиметил, изопрооксиметил и им подобные; 1-((C1-C4)-алкилокси)этильные группы, такие как метоксиэтил, этоксиэтил, пропоксиэтил, изопропоксиэтил и им подобные; 2-оксо-1,3-диоксолен-4-илметильные группы, такие как 5-метил-2-оксо-1,3-диоксолен-4-илметил, 5-фенил-2-оксо-1,3-диоксолен-4-илметил и им подобные; ((C1-С3)-алкилтио)метильные группы, такие как метилтиометил, этилтиометил, изопропилтиометил и им подобные; ацилоксиметильные группы, такие как пивалоилоксиметил, ацетоксиметил и им подобные; этоксикарбонилметильную группу; 1-ацилокси-1-замещенные метильные группы, такие как 1-ацетоксиэтил; 3-фталидильные или 5,6-диметилфталидильные группы; 1-(((C1-C4)-алкилокси)карбонилокси)этильные группы, такие как 1-(этоксикарбонилокси)этильная группа; и 1-(((C1-C4)-алкиламино)карбонилокси)этильную группу, такую как 1-(метиламинокарбонилокси)этильная группа.
Пептид согласно изобретению может модифицироваться с помощью связывания с ним группы, защищающей карбоксильную группу. Группы, защищающие карбоксильную группу, хорошо известны в этой области, и путем связывания с пептидом они защищают карбоксильную группу против нежелательных реакций (см., например, публикацию Greene and Wuts, supra, pages 224-276 (1991), которая включена сюда в виде ссылки). Специалисту будет понятно, что такие описанные выше модификации, которые могут осуществляться в отношении N-конца аминогруппы или С-конца карбоксильной группы пептида, аналогичным образом могут осуществляться в отношении любой реактивной аминогруппы или карбоксильной группы, присутствующей, например, на боковой цепи аминокислоты или аналога аминокислоты в пептиде согласно изобретению. Способы выполнения таких модификаций раскрыты здесь или известны в данной области из других источников.
Выбор включения L- или D-аминокислоты в соединение согласно изобретению может частично зависеть от желательных характеристик пептида. Например, включение одной или нескольких D-аминокислот может придать соединению повышенную устойчивость in vitro или in vivo. Включение одной или нескольких D-аминокислот может также увеличить или уменьшить фармакологическую активность соединения. В некоторых случаях может быть желательным предоставить соединению возможность оставаться активным в течение лишь короткого периода времени. В таких случаях включение одной или нескольких L-аминокислот в соединение может позволить эндогенным пептидазам у индивидуума переварить соединение in vivo, ограничивая таким образом воздействие на индивидуума активного соединения. Опытный специалист может определить желательные характеристики, требуемые от соединения согласно изобретению, с учетом, например возраста и общего состояния здоровья индивидуума. В целом настоящее изобретение относится к соединениям формулы I во всех их стереоизомерных формах и к смесям двух или более стереоизомеров во всех соотношениях, например, к чистым энантиомерам, чистым диастереомерам, к смесям двух энантиомеров во всех соотношениях, включая рацематы, к смесям диастереомеров, к цисизомерам, трансизомерам, Е-изомерам или Z-изомерам. Изобретение также относится к соединениям формулы I во всех их таутомерных формах. Кроме того, изобретение относится к пролекарствам соединений формулы I, например к сложным эфирам, амидам, альдегидам или спиртам, которые, как уже упоминалось, можно получить из карбоксильных групп, или к ацильным производным, подобным (C1-С6)-алкилкарбонильным, (C1-C6)-алкилоксикарбонильным или арил-(C1-C4)-алкилоксикарбонильным производным, которые можно получить из ацилируемых групп, включая аминогруппы, иминогруппы, гуанидиногруппы и амидиногруппы, и изобретение также относится к активным метаболитам соединений формулы I.
В соединениях формулы I группа R1 предпочтительно представляет собой R12C(O). Особая группа определений R12 образована группами алкила, алкенила, алкинила, алкилокси, алкиламино, алкениламино, алкиниламино, алкенилокси, алкинилокси, арила, гетероарила, гетероциклоалкила, гетероарилалкила, гетероциклоалкилалкила, гетероалкила, гетероалкенила и гетероалкинила, причем все эти остатки могут быть незамещенными или замещенными. R12 предпочтительно представляет собой алкил, алкенил, алкинил, алкилокси, алкенилокси, алкинилокси, алкиламино, алкениламино, алкиниламино, арил, гетероарил, арилалкил или гетероарилалкил, более предпочтительно алкенилокси, алкениламино или арил, причем все эти остатки могут быть незамещенными или замещенными. Особенно предпочтительно R12 представляет собой алкенилокси или алкениламино, подобно (С2-С6)-алкенилокси или (С2-С6)-алкениламино, причем каждый из них содержит одну двойную связь, например, аллилокси или аллиламино. Кроме того, предпочтительно R12 представляет собой (C2-C6)-алкенилокси. Остатки, представляющие R12, могут быть незамещенными или замещенными. В замещенных остатках R12 остатки предпочтительно замещены одним или несколькими идентичными или различными заместителями, выбранными из ряда, состоящего из галогена, т.е. фтора, хлора, брома или иода, трифторметила, гидрокси, нитро, амино, циано, карбокси, аминокарбонила, алкилсульфонила, аминосульфонила, алкилокси, алкилкарбониламино и моно- или диалкиламино. Аналогичным образом остатки, представляющие R13, R14 и R15, могут быть незамещенными или замещенными, например, заместителями, которые могут присутствовать в R12, где R14 и R15 независимы друг от друга и могут быть идентичными или различными.
Группа А в соединениях формулы I, которая представляет собой двухвалентный остаток 4-амидинфенилаланина -NH-CH[-CH2-С6Н4-(4-С(=NH)-NH2)]-С(О)-, предпочтительно представляет собой остаток (L)-4-амидинфенилаланина (=остатку (S)-4-амидинфенилаланина). Группа В, которая представляет собой двухвалентный остаток глутаминовой кислоты -NH-CH[-CH2-CH2-С(О)ОН]-С(О)- или его фармацевтически приемлемую соль или сложный эфир, предпочтительно представляет собой остаток (L)-глутаминовой кислоты (=остатку (S)-глутаминовой кислоты) или его фармацевтически приемлемую соль или сложный эфир.
R95 предпочтительно представляет собой водород или (C1-С4)-алкил, более предпочтительно водород или метил, особенно предпочтительно водород.
Замещенные остатки R81 и R82 могут независимо нести один или несколько, например 1, 2, 3 или 4, идентичных или различных остатков, которые предпочтительно выбраны из ряда, состоящего из амино, аминокарбонила, амидино, гуанидино, аминоалкила, гидрокси, меркапто, все из которых могут быть замещены защитной группой, и ацетимидо (-С(=NH)-СН3), нитро и циано. Что касается нитрогрупп, то в соединениях формулы I в соответствии с изобретением предпочтительно присутствуют лишь до двух нитрогрупп. Подходящие защитные группы для перечисленных групп известны специалисту в этой области и могут быть найдены в упомянутых выше ссылках, подобных публикации Greene and Wuts, Protective Groups in Organic Synthesis, 2nd ed., (John Wiley & Sons, New York, 1991), которая включена сюда в виде ссылки. Примеры защитных групп представляют собой упомянутые выше группы, защищающие аминогруппы, подобные третбутилоксикарбонилу, бензилоксикарбонилу и аллилоксикарбонилу, которые также могут представлять собой защитные группы на амидиногруппах и гуанидиногруппах, нитрогруппу, которая может использоваться для защиты гуанидиногруппы или групп, подобных бензилу, метилу, тритилу или ацетиламинометилу, которые могут использоваться для защиты групп, подобных гидрокси, меркапто и другим. Предпочтительно R81 и R82 выбраны из ряда, состоящего из водорода, алкила, подобного (C1-C6)-алкилу, арила, подобного фенилу, арилалкила, подобного фенил-(C2-C2)-алкилу, и гетероалкила, подобного гетероарил-(C1-C2)-алкилу, все из которых могут быть незамещенными или замещенными и в которых гетероарил предпочтительно представляет собой остаток моноциклической или бициклической ароматической кольцевой системы, содержащей один или два идентичных или различных кольцевых гетероатома, таких как N, О или S. Более предпочтительно R81 представляет собой водород, а R82 представляет собой определенный выше незамещенный или замещенный остаток.
Особенно предпочтительно группа D представляет собой остаток, выбранный из ряда, состоящего из Arg, Dap, Dab, Orn, Lys, Dap[-C(=NH)-NH2], Dab[-C(=NH)-NH2], Lys[-C(=NH)-NH2], Lys[-C(=NH)-СН3], Orn[-C(=NH)-СН3], Dab[-С(=NH)-СН3], Dap[-С(=NН)-СН3], Dab(Alloc), Asn, Gln, Met, Ser, Thr, Ser(Bzl), Thr(Bzl), Cys(Me), Cys(Bzl), Cys(Acm), Arg (NO2), His, Trp, Phg, Gly, Ala, Val, Ile, Leu, Phe, Phe(4-NHO2), Phe(4-NH-C(-NH)-NH2), 2-Abu, Ala(3-CN), Ala[3-C(=NH)-NH2], 2-Abu(4-CN) и 2-Abu[4-C(=NH)-NH2]. Подгруппа остатков, из которых выбраны особенно предпочтительные остатки D, образована рядом, состоящим из Arg, Dap, Dab, Orn, Lys, Dap[-C(=NH)-NH2], Dab[-C(=NH)-NH2], Lys[-C=NH)-NH2], Asn, Ser, Thr, Ser(Bzl), Arg (NO2), Trp, Phg, Ala, Val, Ile, Leu, Phe, 2-Abu, Ala(3-CN), Ala(3C(=NH)-NH2), 2-Abu(4-CN) и 2-Abu(4-C(=NH)-NH2).
Число n предпочтительно равно 0, 1 или 2, более предпочтительно 0 или 1. Если n равно 0, группа R2 непосредственно связана с карбонильной группой, представляющей D3. Если n отлично от нуля, группа R2 связана с карбонильной группой, представляющей концевую группу Е3. Если n равно 2 или 3, все группы Е могут быть идентичными или различными.
Замещенные остатки R71 и R72 могут независимо нести один или несколько, например 1, 2, 3 или 4, идентичных или различных остатков, которые предпочтительно выбраны из ряда, состоящего из алкила, алкилокси, галогена, трифторметила, нитро, циано, алкилсульфонила, алкилкарбонила, фенилкарбонила и 2-фенил-1,3-дитиолан-2-ила, которые могут быть дополнительно замещены. Подгруппа заместителей, которая может присутствовать в R71 и R72, образована рядом, состоящим из алкила, алкилокси, галогена, трифторметила, нитро, циано, алкилсульфонила, алкилкарбонила, которые могут быть дополнительно замещены. Предпочтительно R71 представляет собой водород, а R72 представляет собой определенный выше незамещенный или замещенный остаток. R72 предпочтительно представляет собой алкил, в частности (С3-С8)-алкил, включая циклический алкил, подобный циклоалкилалкилу, такому как циклоалкил-(C1-C2)-алкил, или арил, в частности фенил или арилалкил, в частности фенил-(C1-C2)-алкил, или гетероарилалкил, в частности гетероарил-(C1-C2)-алкил, где все эти остатки могут быть незамещенными или замещенными, и где гетероарил предпочтительно представляет собой моноциклическое пятичленное или шестичленное ароматическое кольцо, содержащее 1 или 2 идентичных или различных кольцевых гетероатома, таких как N, О и S. Группа или группы Е, в частности, в случае, если число n равно 1, предпочтительно выбраны из ряда, состоящего из Phe, который не замещен или замещен в фениловой группе, Cha и Chg. Особенно предпочтительная группа Е выбрана из ряда, состоящего из Cha, Chg и Phe[4-C(S-CH2-CH2-S-)-Ph]. Группа R70, присутствующая в группе Е, представляет собой предпочтительно водород, алкил, в частности (C1-C4)-алкил, включая метил, или арилалкил, в частности фенил-(C1-C4)-алкил, включая бензил и 2-фенилэтил, который может быть незамещенным или замещенным в фениловой группе. Особенно предпочтительно R70 представляет собой водород.
Замещенные остатки R21, R22, R23 и R24 могут независимо нести один или несколько, например 1, 2, 3 или 4, идентичных или различных остатков, которые предпочтительно выбраны из ряда, состоящего из галогена, в частности, F, Cl, Вr, гидрокси, трифторметила, нитро, циано, диалкиламино, алкилокси, подобной метилокси, алкилендиокси, алкилсульфонила, аминсульфонила и оксо(=O), которые могут быть дополнительно замещены. Примерами алкилендиокси являются метилендиокси (O-СН2-О) или 1,2-этилендиокси. Примерами диалкиламино являются диметиламино, диэтиламино или дибутиламино, примерами алкилсульфонила являются метилсульфонил, этилсульфонил или бутилсульфонил. R2 предпочтительно представляет собой NR21R22, в котором R21 и R22 представляют собой определенные выше группы. R21 предпочтительно представляет собой водород, (C1-C4)-алкил или фенил-(C1-C4)-алкил, который не замещен или замещен в фениловой группе. Особенно предпочтительно R21 представляет собой водород, т.е. особенно предпочтительно NR21R22 представляет собой NHR22, и, таким образом, особенно предпочтительно R2 представляет собой NHR22. R22 предпочтительно представляет собой остаток, выбранный из ряда, состоящего из водорода, алкила, в частности (C1-C12)-алкила, включая циклический алкил, подобный циклоалкилалкилу, такому как циклоалкил-(C1-C4)-алкил, арил, в частности (С6-С13)-арил, арилалкил, в частности (C1-C4)-алкил, замещенный одним или двумя остатками (C6-C12)-арила, гетероарилалкил, в частности (C1-C4)-алкил, замещенный моноциклическим или бициклическим остатком гетероарила, содержащим 1 или 2 идентичных или различных гетероатома, таких как N, О или S, и гетероциклоалкилалкил, в частности (C1-C4)-алкил, замещенный моноциклической 4-, 5-, 6- или 7-членной гетероциклоалкиловой группой, содержащей 1 или 2 идентичных или различных гетероатома, таких как N, О или S, причем все эти остатки, как указано выше, могут быть незамещенными или замещенными.
Особенно предпочтительно R22 представляет собой остаток, выбранный из ряда, состоящего из водорода, бензила, нафтилметила, пиридилметила, фенилэтила, нафтилэтила, пиридилэтила, фенилпропила, нафтилпропила, пиридилпропила, фторенила, дифенилметила, дифенилэтила и дифенилпропила, причем эти остатки не замещены или замещены одним или несколькими, например одним, двумя, тремя или четырьмя, идентичными или различными заместителями, которые предпочтительно выбраны из ряда, состоящего из F, Cl, Вr, гидрокси, метокси, метилендиокси, нитро, циано, диалкиламино, алкилсульфонила, аминсульфонила и трифторметила, которые могут быть дополнительно замещены. Ряд особенно предпочтительных соединений формулы I образован соединениями, в которых одновременно число n отлично от 0, R2 представляет собой NHR22, а R22 представляет собой водород. Другой ряд особенно предпочтительных соединений образован соединениями, в которых одновременно n равно 0, R2 представляет собой NHR22, а R22 отличен от водорода, причем в этом ряду соединений предпочтительное обозначение группы D представляет собой Asn.
Предпочтительными соединениями формулы I являются те соединения, в которых одна или несколько групп или остатков имеют предпочтительные обозначения, причем все комбинации предпочтительных обозначений представляют собой предмет настоящего изобретения.
Группа предпочтительных соединений изобретения образована соединениями формулы I, в которой
R1 представляет собой R12C(О), в которой R12 представляет собой определенный выше остаток,
А представляет собой определенный выше остаток,
В представляет собой определенный выше остаток, а предпочтительно В представляет собой NH-CHR97-C(О), в которой
R97 представляет собой этил, который замещен в положении 2 гидроксикарбонилом или его солью или алкилоксикарбонилом, подобным (C1-C4)-алкилоксикарбонилу,
D представляет собой NH-CHR82-C(О), в которой R82 представляет собой определенный выше остаток,
En представляет собой (Е1-Е2-Е3)n, в которой
n равно 0, 1 или 2,
E1 представляет собой NH,
Е2 представляет собой CHR72, в которой остатки R72, которые независимы друг от друга и являются идентичными или различными, представляют собой определенные выше группы,
Е3 представляет собой С (О), а
R2 представляет собой определенный выше остаток,
в любой из их стереоизомерных форм или их смесях в любом соотношении, и их фармацевтически приемлемые соли, амиды и сложные эфиры.
Группа особенно предпочтительных соединений образована соединениями формулы I, в которой
R1 представляет собой аллилоксикарбонил или аллиламинокарбонил,
А представляет собой остаток (L)-4-амидинфенилаланина,
В представляет собой остаток (L)-глутаминовой кислоты или фармацевтически приемлемую соль или сложный эфир (L)-глутаминовой кислоты,
D представляет собой остаток, выбранный из ряда, состоящего из Arg, Dap, Dab, Orn, Lys, Dap[-С(=NH)-NH2], Dab[-C(=NH)-NH2], Lys[-C(=NH)-NH2], Lys[-C(=NH)-CH3], Orn[-C(=NH)-СН3], Dab[-C(=NH)-СН3], Dap[-С(=NH)-СН3], Dab(Alloc), Asn, Gln, Met, Ser, Thr, Ser(Bzl), Thr(Bzl), Cys(Me), Cys(Bzl), Cys (Acm), Arg(NO2), His, Trp, Phg, Gly, Ala, Val, Ile, Leu, Phe, Phe(4-NO2), Phe(4-NH-C(=NH)-NH2), 2-Abu, Ala(3-CN), Ala[3-C(=NH)-NH2], 2-Abu(4-CN) и 2-Abu[4-C(=NH)-NH2],
n равно 0 или 1,
Е представляет собой остаток, выбранный из ряда, состоящего из Cha, Chg и Phe[4-C(-S-CH2-CH2-S-)-Ph],
R2 представляет собой NHR22, R22 представляет собой водород или остаток, выбранный из ряда, состоящего из бензила, нафтилметила, пиридилметила, фенилэтила, нафтилэтила, пиридилэтила, фенилпропила, нафтилпропила, пиридилпропила, фторенила, дифенилметила, дифенилэтила и дифенилпропила, причем эти остатки не замещены или замещены одним или несколькими идентичными или различными заместителями, выбранными из ряда, состоящего из F, Cl, Вr, гидрокси, метокси, метилендиокси, нитро, циано, диалкиламино, алкилсульфонила, аминосульфонила и трифторметила,
в любой из их стереоизомерных форм или их смесей в любом соотношении, и их фармацевтически приемлемые соли, амиды и сложные эфиры.
Особые примеры соединений согласно изобретению включают в себя, например, соединения, перечисленные ниже в табл. 2, и в разделе примеров, и их фармацевтически приемлемые соли, амиды и сложные эфиры.
Соединения формулы I могут быть получены, например, в соответствии со способами твердофазной химии с помощью способа, который включает в себя:
а1) связывание соединения формулы Fmoc-En-OH, в которой n равно 1, 2 или 3, с чувствительным к кислоте линкером, прикрепленным к смоле или в целом к твердой опоре, отщепление защитной группы Fmoc, связывание соединения формулы Fmoc-D1-D2-C(О)ОН с полученной свободной аминогруппой и вновь отщепление защитной группы Fmoc, или для получения соединения формулы I, в которой n равно 0, связывание соединения формулы Fmoc-D1-D2-C(О)ОН с чувствительным к кислоте линкером, прикрепленным к смоле или в целом к твердой опоре, и отщепление защитной группы Fmoc;
а2) связывание соединения формулы Fmoc-B1-B2-C(О)ОН со свободной аминогруппой, полученной на этапе а1), и отщепление защитной группы Fmoc;
а3) связывание соединения формулы R1-A1-A2-C(О)ОН со свободной аминогруппой, полученной на этапе а2);
а4) отщепление соединения, полученного в соответствии с этапами с а1) по а3), от смолы посредством трифторуксусной кислоты.
Смола или линкер, используемые в этом способе, могут относиться к такому типу, в котором карбоксильная группа в соединении, связанном, соответственно, со смолой или с линкером, трансформируется в амидную группу С(O)-NН2, например линкер Knorr или амидная смола Rink. Получение соединения, в котором число n равно 2 или 3, также может осуществляться с помощью поэтапной сборки элемента En следующим образом. На этапе а1) вместо соединения формулы Fmoc-En-OH, в которой n равно 2 или 3, сначала соединение формулы Fmoc-En-OH, в которой n равно 1, связывают с чувствительным к кислоте линкером, прикрепленным к смоле, затем отщепляют защитную группу Fmoc, и второе соединение формулы Fmoc-En-OH, в которой n равно 1, связывают с полученной свободной аминогруппой. Затем для получения соединения, в котором n равно 1, отщепляют защитную группу Fmoc, и третье соединение формулы Fmoc-En-OH, в которой n равно 3, связывают с полученной свободной аминогруппой. Наконец, отщепляют защитную группу Fmoc, и за этим следуют этапы а2) – а4).
Другой способ получения соединений формулы I включает в себя:
b1) связывание карбоновой кислоты боковой цепи соединения формулы Fmoc-B1-CHR97-C(О)OPG, в которой R97 представляет собой 2-гидроксикарбонилэтил, a PG представляет собой защитную группу с чувствительным к кислоте линкером типа бензилового спирта, прикрепленным к смоле с функциональной аминогруппой;
b2) отщепление защитной группы PG;
b3) связывание соединения формулы H2N-D2-D3-En-R2, в котором n равно 0, 1, 2 или 3, со свободной карбоновой кислотой, полученной на этапе b2);
b4) отщепление защитной группы Fmoc;
b5) связывание соединения формулы R1-A1-A2-C(О)ОН со свободной аминогруппой, полученной на этапе b4);
b6) отщепление соединения, полученного в соответствии с этапами bl) – b5), от смолы посредством трифторуксусной кислоты.
Аналогично модификации описанного выше первого способа, в этом способе сборка структурного элемента H2N-D2-D3-En-R2 может поэтапно осуществляться на смоле. В соответствии с еще одним способом, подобным этому способу, соединения формулы I могут быть также получены с помощью связывания сначала группы карбоновой кислоты, которая присутствует в боковой цепи в группе D2 группы D, т.е., которая присутствует в одной из групп R81 и R82, с линкером, прикрепленным к смоле. Аналогично указанному выше соединению формулы Fmoc-B1-CHR97-C(О)OPG, такое соединение может, например, иметь формулу Fmoc-NH-CR81R82-C(О)OPG, в которой R82 представляет собой определенное выше соединение при условии, что оно содержит группу С(О)ОН, а R81 представляет собой определенный выше остаток. Например, R81 может представлять собой водород, а R82 может представлять собой гидроксикарбонилметил, и соединение формулы Fmoc-NH-CR81R82-C(O)OPG может таким образом представлять собой защищенное производное аспарагиновой кислоты. После отсоединения защитной группы C(O)OPG, карбонильная группа которой представляет собой группу D3 в формуле I, полученную свободную группу карбоновой кислоты связывают с соединением, подобным H2N-En-R2 или H-R2. Затем после удаления защитной группы Fmoc полученную аминогруппу связывают с соединением формулы Fmoc-B1-B2-C(О)ОН, и после устранения защиты аминогруппы продукт связывают с соединением формулы R1-A1-A2-С(O)ОН. И снова используемая смола или линкер могут быть такого типа, что карбоксильная группа в соединении, которое связано со смолой или с линкером, трансформируется в амидную группу C(O)-NH2. Например, с помощью использования амидной смолы элемент аспарагиновой кислоты, прикрепленный к смоле, может быть в конечном соединении превращен в аспарагиновый элемент.
Соединение согласно изобретению может быть химически синтезировано с использованием, например, автоматического синтезатора (см. пример 1). Избирательная модификация реактивной группы, такой как группа, присутствующая на боковой цепи аминокислоты, или N-концевая или С-концевая реактивная группа в пептиде, может придать соединению согласно изобретению желаемые характеристики, такие как повышенная растворимость или усиленная ингибирующая функция. При использовании твердофазных способов синтеза химический состав соединения можно изменять, в то время как к смоле прикрепляется образующийся пептид или после того, как пептид был отщеплен от смолы для получения, например, N-концевого производного, такого как соединение, ацилированное на N-конце, например ацетилированное соединение. Аналогичные модификации могут также производиться в карбоксильной группе соединения, включая карбоксильную группу С-конца, которая может быть, например, амидирована.
Соединения могут также быть получены с помощью связывания защищенных аминокислот в соответствии со способами традиционной фармацевтической химии или с помощью известных в данной области стандартных процедур жидкофазной органической химии и снятия защиты до целевой молекулы. В целом, подходящие реакции для синтеза соединений формулы I с помощью твердофазных способов или жидкофазных способов, а также экспериментальные детали, подобные подходящим связывающим агентам, таким как карбодиимиды, TOTU или HATU, или растворители и температуры реакции, хорошо известны специалистам в данной области и их можно также найти в стандартных ссылках, включая ссылки, упомянутые здесь, а также приведенные ниже в качестве примеров.
Синтезированное соединение может быть очищено с использованием хорошо известных способов, таких как высокоэффективная жидкостная хроматография с обращенной фазой (RP-HPLC; см. пример 1) или другие способы отделения, основанные, например, на размере, заряде или гидрофобности соединения. Аналогичным образом для характеристики структуры соединения согласно изобретению могут использоваться хорошо известные способы, такие как анализ последовательности аминокислот или масс-спектрометрия (MS), см. пример 1.
Различные соединения, содержащие различные расположения заместителей, проявляют различные уровни ингибирующей активности по отношению к фактору VIIa. Например, выбор заместителей влияет на сродство связывания соединений. Эти соединения были синтезированы в соответствии с процедурами, описанными в примерах. Испытание пептидов на ингибирующую активность осуществлялось с использованием количественного анализа, описанного в примере 22. Используя такие способы, специалист в данной области может синтезировать соединение, как раскрыто здесь, включая его модификацию, и определить ингибирующую активность соединения по отношению к фактору VIIa. Композиция настоящего изобретения может быть представлена в виде однородной композиции или в виде смеси соединений, содержащих различные комбинации заместителей. Гибкость, обеспечиваемая выбором заместителей, обеспечивает возможность существенного управления биологическими и физико-химическими свойствами соединений и композиций согласно изобретению.
Изобретение предоставляет соединения, которые специфически ингибируют активность фактора VII. Такие соединения предпочтительно имеют ингибирующую активность (Ki) -аминогруппе первой аминокислоты, присутствующей в пептиде, написанном обычным образом.
N-конец пептида согласно изобретению может быть защищен с помощью присоединения к нему группы, защищающей аминогруппу. Термин “группа, защищающая аминогруппу”, используется здесь широко для ссылки на химическую группу, которая может вступать в реакцию со свободной аминогруппой, включая, например, -аминогруппу, присутствующую на N-конце пептида изобретения. С помощью реакции с ней группа, защищающая аминогруппу, защищает реактивную в противном случае аминогруппу против нежелательных реакций, которые могут произойти, например, во время процедуры синтеза или вследствие активности экзопептидазы по отношению к конечному соединению. Модификация аминогруппы может также обеспечить дополнительные преимущества, включая, например, увеличение растворимости или активности соединения. Здесь раскрыты или известны другим образом в данной области различные группы, защищающие аминогруппу, и они включают, например, ацильные группы, такие как ацетильная, трет-бутилоксикарбонильная, аллилоксикарбонильная, бензилоксикарбонильная группа или бензоильные группы, а также аминоацильный остаток, который сам может изменяться с помощью группы, защищающей аминогруппу. Другие группы, защищающие аминогруппу, описаны, например, в книге The Peptides, eds. Gross and Meienhofer, vol.3 (Academic Press, Inc., New York, 1981); и в публикации Greene and Wuts Protective Groups in Organic Synthesis, 2nd ed., pages 309-405 (John Wiley&Sons, New York, 1991), каждая из которых включена сюда в качестве ссылки. Продукт любой такой модификации N-концевой аминогруппы пептида или аналога пептида изобретения именуется здесь “N-концевое производное”.
Аналогичным образом, карбоксильная группа, такая как карбоксильная группа, присутствующая на С-конце пептида, может химически модифицироваться с использованием группы, защищающей карбоксильную группу. Термины “карбоксильная группа” и “С-конец” используются согласованным образом с определенными выше терминами “аминогруппа” и “N-конец”. Карбоксильная группа, подобная группе, которая присутствует на С-конце пептида, может модифицироваться с помощью восстановления С-концевой карбоксильной группы до спирта или альдегида или с помощью образования сложного эфира для перорального использования, или с помощью замещения карбоксильной группы таким заместителем как тиазолил, циклогексил или другая группа. Сложные эфиры для перорального использования хорошо известны в этой области и включают в себя, например, алкилоксиметильные группы, такие как метоксиметил, этоксиметил, изопрооксиметил и им подобные; 1-((C1-C4)-алкилокси)этильные группы, такие как метоксиэтил, этоксиэтил, пропоксиэтил, изопропоксиэтил и им подобные; 2-оксо-1,3-диоксолен-4-илметильные группы, такие как 5-метил-2-оксо-1,3-диоксолен-4-илметил, 5-фенил-2-оксо-1,3-диоксолен-4-илметил и им подобные; ((C1-С3)-алкилтио)метильные группы, такие как метилтиометил, этилтиометил, изопропилтиометил и им подобные; ацилоксиметильные группы, такие как пивалоилоксиметил, ацетоксиметил и им подобные; этоксикарбонилметильную группу; 1-ацилокси-1-замещенные метильные группы, такие как 1-ацетоксиэтил; 3-фталидильные или 5,6-диметилфталидильные группы; 1-(((C1-C4)-алкилокси)карбонилокси)этильные группы, такие как 1-(этоксикарбонилокси)этильная группа; и 1-(((C1-C4)-алкиламино)карбонилокси)этильную группу, такую как 1-(метиламинокарбонилокси)этильная группа.
Пептид согласно изобретению может модифицироваться с помощью связывания с ним группы, защищающей карбоксильную группу. Группы, защищающие карбоксильную группу, хорошо известны в этой области, и путем связывания с пептидом они защищают карбоксильную группу против нежелательных реакций (см., например, публикацию Greene and Wuts, supra, pages 224-276 (1991), которая включена сюда в виде ссылки). Специалисту будет понятно, что такие описанные выше модификации, которые могут осуществляться в отношении N-конца аминогруппы или С-конца карбоксильной группы пептида, аналогичным образом могут осуществляться в отношении любой реактивной аминогруппы или карбоксильной группы, присутствующей, например, на боковой цепи аминокислоты или аналога аминокислоты в пептиде согласно изобретению. Способы выполнения таких модификаций раскрыты здесь или известны в данной области из других источников.
Выбор включения L- или D-аминокислоты в соединение согласно изобретению может частично зависеть от желательных характеристик пептида. Например, включение одной или нескольких D-аминокислот может придать соединению повышенную устойчивость in vitro или in vivo. Включение одной или нескольких D-аминокислот может также увеличить или уменьшить фармакологическую активность соединения. В некоторых случаях может быть желательным предоставить соединению возможность оставаться активным в течение лишь короткого периода времени. В таких случаях включение одной или нескольких L-аминокислот в соединение может позволить эндогенным пептидазам у индивидуума переварить соединение in vivo, ограничивая таким образом воздействие на индивидуума активного соединения. Опытный специалист может определить желательные характеристики, требуемые от соединения согласно изобретению, с учетом, например возраста и общего состояния здоровья индивидуума. В целом настоящее изобретение относится к соединениям формулы I во всех их стереоизомерных формах и к смесям двух или более стереоизомеров во всех соотношениях, например, к чистым энантиомерам, чистым диастереомерам, к смесям двух энантиомеров во всех соотношениях, включая рацематы, к смесям диастереомеров, к цисизомерам, трансизомерам, Е-изомерам или Z-изомерам. Изобретение также относится к соединениям формулы I во всех их таутомерных формах. Кроме того, изобретение относится к пролекарствам соединений формулы I, например к сложным эфирам, амидам, альдегидам или спиртам, которые, как уже упоминалось, можно получить из карбоксильных групп, или к ацильным производным, подобным (C1-С6)-алкилкарбонильным, (C1-C6)-алкилоксикарбонильным или арил-(C1-C4)-алкилоксикарбонильным производным, которые можно получить из ацилируемых групп, включая аминогруппы, иминогруппы, гуанидиногруппы и амидиногруппы, и изобретение также относится к активным метаболитам соединений формулы I.
В соединениях формулы I группа R1 предпочтительно представляет собой R12C(O). Особая группа определений R12 образована группами алкила, алкенила, алкинила, алкилокси, алкиламино, алкениламино, алкиниламино, алкенилокси, алкинилокси, арила, гетероарила, гетероциклоалкила, гетероарилалкила, гетероциклоалкилалкила, гетероалкила, гетероалкенила и гетероалкинила, причем все эти остатки могут быть незамещенными или замещенными. R12 предпочтительно представляет собой алкил, алкенил, алкинил, алкилокси, алкенилокси, алкинилокси, алкиламино, алкениламино, алкиниламино, арил, гетероарил, арилалкил или гетероарилалкил, более предпочтительно алкенилокси, алкениламино или арил, причем все эти остатки могут быть незамещенными или замещенными. Особенно предпочтительно R12 представляет собой алкенилокси или алкениламино, подобно (С2-С6)-алкенилокси или (С2-С6)-алкениламино, причем каждый из них содержит одну двойную связь, например, аллилокси или аллиламино. Кроме того, предпочтительно R12 представляет собой (C2-C6)-алкенилокси. Остатки, представляющие R12, могут быть незамещенными или замещенными. В замещенных остатках R12 остатки предпочтительно замещены одним или несколькими идентичными или различными заместителями, выбранными из ряда, состоящего из галогена, т.е. фтора, хлора, брома или иода, трифторметила, гидрокси, нитро, амино, циано, карбокси, аминокарбонила, алкилсульфонила, аминосульфонила, алкилокси, алкилкарбониламино и моно- или диалкиламино. Аналогичным образом остатки, представляющие R13, R14 и R15, могут быть незамещенными или замещенными, например, заместителями, которые могут присутствовать в R12, где R14 и R15 независимы друг от друга и могут быть идентичными или различными.
Группа А в соединениях формулы I, которая представляет собой двухвалентный остаток 4-амидинфенилаланина -NH-CH[-CH2-С6Н4-(4-С(=NH)-NH2)]-С(О)-, предпочтительно представляет собой остаток (L)-4-амидинфенилаланина (=остатку (S)-4-амидинфенилаланина). Группа В, которая представляет собой двухвалентный остаток глутаминовой кислоты -NH-CH[-CH2-CH2-С(О)ОН]-С(О)- или его фармацевтически приемлемую соль или сложный эфир, предпочтительно представляет собой остаток (L)-глутаминовой кислоты (=остатку (S)-глутаминовой кислоты) или его фармацевтически приемлемую соль или сложный эфир.
R95 предпочтительно представляет собой водород или (C1-С4)-алкил, более предпочтительно водород или метил, особенно предпочтительно водород.
Замещенные остатки R81 и R82 могут независимо нести один или несколько, например 1, 2, 3 или 4, идентичных или различных остатков, которые предпочтительно выбраны из ряда, состоящего из амино, аминокарбонила, амидино, гуанидино, аминоалкила, гидрокси, меркапто, все из которых могут быть замещены защитной группой, и ацетимидо (-С(=NH)-СН3), нитро и циано. Что касается нитрогрупп, то в соединениях формулы I в соответствии с изобретением предпочтительно присутствуют лишь до двух нитрогрупп. Подходящие защитные группы для перечисленных групп известны специалисту в этой области и могут быть найдены в упомянутых выше ссылках, подобных публикации Greene and Wuts, Protective Groups in Organic Synthesis, 2nd ed., (John Wiley & Sons, New York, 1991), которая включена сюда в виде ссылки. Примеры защитных групп представляют собой упомянутые выше группы, защищающие аминогруппы, подобные третбутилоксикарбонилу, бензилоксикарбонилу и аллилоксикарбонилу, которые также могут представлять собой защитные группы на амидиногруппах и гуанидиногруппах, нитрогруппу, которая может использоваться для защиты гуанидиногруппы или групп, подобных бензилу, метилу, тритилу или ацетиламинометилу, которые могут использоваться для защиты групп, подобных гидрокси, меркапто и другим. Предпочтительно R81 и R82 выбраны из ряда, состоящего из водорода, алкила, подобного (C1-C6)-алкилу, арила, подобного фенилу, арилалкила, подобного фенил-(C2-C2)-алкилу, и гетероалкила, подобного гетероарил-(C1-C2)-алкилу, все из которых могут быть незамещенными или замещенными и в которых гетероарил предпочтительно представляет собой остаток моноциклической или бициклической ароматической кольцевой системы, содержащей один или два идентичных или различных кольцевых гетероатома, таких как N, О или S. Более предпочтительно R81 представляет собой водород, а R82 представляет собой определенный выше незамещенный или замещенный остаток.
Особенно предпочтительно группа D представляет собой остаток, выбранный из ряда, состоящего из Arg, Dap, Dab, Orn, Lys, Dap[-C(=NH)-NH2], Dab[-C(=NH)-NH2], Lys[-C(=NH)-NH2], Lys[-C(=NH)-СН3], Orn[-C(=NH)-СН3], Dab[-С(=NH)-СН3], Dap[-С(=NН)-СН3], Dab(Alloc), Asn, Gln, Met, Ser, Thr, Ser(Bzl), Thr(Bzl), Cys(Me), Cys(Bzl), Cys(Acm), Arg (NO2), His, Trp, Phg, Gly, Ala, Val, Ile, Leu, Phe, Phe(4-NHO2), Phe(4-NH-C(-NH)-NH2), 2-Abu, Ala(3-CN), Ala[3-C(=NH)-NH2], 2-Abu(4-CN) и 2-Abu[4-C(=NH)-NH2]. Подгруппа остатков, из которых выбраны особенно предпочтительные остатки D, образована рядом, состоящим из Arg, Dap, Dab, Orn, Lys, Dap[-C(=NH)-NH2], Dab[-C(=NH)-NH2], Lys[-C=NH)-NH2], Asn, Ser, Thr, Ser(Bzl), Arg (NO2), Trp, Phg, Ala, Val, Ile, Leu, Phe, 2-Abu, Ala(3-CN), Ala(3C(=NH)-NH2), 2-Abu(4-CN) и 2-Abu(4-C(=NH)-NH2).
Число n предпочтительно равно 0, 1 или 2, более предпочтительно 0 или 1. Если n равно 0, группа R2 непосредственно связана с карбонильной группой, представляющей D3. Если n отлично от нуля, группа R2 связана с карбонильной группой, представляющей концевую группу Е3. Если n равно 2 или 3, все группы Е могут быть идентичными или различными.
Замещенные остатки R71 и R72 могут независимо нести один или несколько, например 1, 2, 3 или 4, идентичных или различных остатков, которые предпочтительно выбраны из ряда, состоящего из алкила, алкилокси, галогена, трифторметила, нитро, циано, алкилсульфонила, алкилкарбонила, фенилкарбонила и 2-фенил-1,3-дитиолан-2-ила, которые могут быть дополнительно замещены. Подгруппа заместителей, которая может присутствовать в R71 и R72, образована рядом, состоящим из алкила, алкилокси, галогена, трифторметила, нитро, циано, алкилсульфонила, алкилкарбонила, которые могут быть дополнительно замещены. Предпочтительно R71 представляет собой водород, а R72 представляет собой определенный выше незамещенный или замещенный остаток. R72 предпочтительно представляет собой алкил, в частности (С3-С8)-алкил, включая циклический алкил, подобный циклоалкилалкилу, такому как циклоалкил-(C1-C2)-алкил, или арил, в частности фенил или арилалкил, в частности фенил-(C1-C2)-алкил, или гетероарилалкил, в частности гетероарил-(C1-C2)-алкил, где все эти остатки могут быть незамещенными или замещенными, и где гетероарил предпочтительно представляет собой моноциклическое пятичленное или шестичленное ароматическое кольцо, содержащее 1 или 2 идентичных или различных кольцевых гетероатома, таких как N, О и S. Группа или группы Е, в частности, в случае, если число n равно 1, предпочтительно выбраны из ряда, состоящего из Phe, который не замещен или замещен в фениловой группе, Cha и Chg. Особенно предпочтительная группа Е выбрана из ряда, состоящего из Cha, Chg и Phe[4-C(S-CH2-CH2-S-)-Ph]. Группа R70, присутствующая в группе Е, представляет собой предпочтительно водород, алкил, в частности (C1-C4)-алкил, включая метил, или арилалкил, в частности фенил-(C1-C4)-алкил, включая бензил и 2-фенилэтил, который может быть незамещенным или замещенным в фениловой группе. Особенно предпочтительно R70 представляет собой водород.
Замещенные остатки R21, R22, R23 и R24 могут независимо нести один или несколько, например 1, 2, 3 или 4, идентичных или различных остатков, которые предпочтительно выбраны из ряда, состоящего из галогена, в частности, F, Cl, Вr, гидрокси, трифторметила, нитро, циано, диалкиламино, алкилокси, подобной метилокси, алкилендиокси, алкилсульфонила, аминсульфонила и оксо(=O), которые могут быть дополнительно замещены. Примерами алкилендиокси являются метилендиокси (O-СН2-О) или 1,2-этилендиокси. Примерами диалкиламино являются диметиламино, диэтиламино или дибутиламино, примерами алкилсульфонила являются метилсульфонил, этилсульфонил или бутилсульфонил. R2 предпочтительно представляет собой NR21R22, в котором R21 и R22 представляют собой определенные выше группы. R21 предпочтительно представляет собой водород, (C1-C4)-алкил или фенил-(C1-C4)-алкил, который не замещен или замещен в фениловой группе. Особенно предпочтительно R21 представляет собой водород, т.е. особенно предпочтительно NR21R22 представляет собой NHR22, и, таким образом, особенно предпочтительно R2 представляет собой NHR22. R22 предпочтительно представляет собой остаток, выбранный из ряда, состоящего из водорода, алкила, в частности (C1-C12)-алкила, включая циклический алкил, подобный циклоалкилалкилу, такому как циклоалкил-(C1-C4)-алкил, арил, в частности (С6-С13)-арил, арилалкил, в частности (C1-C4)-алкил, замещенный одним или двумя остатками (C6-C12)-арила, гетероарилалкил, в частности (C1-C4)-алкил, замещенный моноциклическим или бициклическим остатком гетероарила, содержащим 1 или 2 идентичных или различных гетероатома, таких как N, О или S, и гетероциклоалкилалкил, в частности (C1-C4)-алкил, замещенный моноциклической 4-, 5-, 6- или 7-членной гетероциклоалкиловой группой, содержащей 1 или 2 идентичных или различных гетероатома, таких как N, О или S, причем все эти остатки, как указано выше, могут быть незамещенными или замещенными.
Особенно предпочтительно R22 представляет собой остаток, выбранный из ряда, состоящего из водорода, бензила, нафтилметила, пиридилметила, фенилэтила, нафтилэтила, пиридилэтила, фенилпропила, нафтилпропила, пиридилпропила, фторенила, дифенилметила, дифенилэтила и дифенилпропила, причем эти остатки не замещены или замещены одним или несколькими, например одним, двумя, тремя или четырьмя, идентичными или различными заместителями, которые предпочтительно выбраны из ряда, состоящего из F, Cl, Вr, гидрокси, метокси, метилендиокси, нитро, циано, диалкиламино, алкилсульфонила, аминсульфонила и трифторметила, которые могут быть дополнительно замещены. Ряд особенно предпочтительных соединений формулы I образован соединениями, в которых одновременно число n отлично от 0, R2 представляет собой NHR22, а R22 представляет собой водород. Другой ряд особенно предпочтительных соединений образован соединениями, в которых одновременно n равно 0, R2 представляет собой NHR22, а R22 отличен от водорода, причем в этом ряду соединений предпочтительное обозначение группы D представляет собой Asn.
Предпочтительными соединениями формулы I являются те соединения, в которых одна или несколько групп или остатков имеют предпочтительные обозначения, причем все комбинации предпочтительных обозначений представляют собой предмет настоящего изобретения.
Группа предпочтительных соединений изобретения образована соединениями формулы I, в которой
R1 представляет собой R12C(О), в которой R12 представляет собой определенный выше остаток,
А представляет собой определенный выше остаток,
В представляет собой определенный выше остаток, а предпочтительно В представляет собой NH-CHR97-C(О), в которой
R97 представляет собой этил, который замещен в положении 2 гидроксикарбонилом или его солью или алкилоксикарбонилом, подобным (C1-C4)-алкилоксикарбонилу,
D представляет собой NH-CHR82-C(О), в которой R82 представляет собой определенный выше остаток,
En представляет собой (Е1-Е2-Е3)n, в которой
n равно 0, 1 или 2,
E1 представляет собой NH,
Е2 представляет собой CHR72, в которой остатки R72, которые независимы друг от друга и являются идентичными или различными, представляют собой определенные выше группы,
Е3 представляет собой С (О), а
R2 представляет собой определенный выше остаток,
в любой из их стереоизомерных форм или их смесях в любом соотношении, и их фармацевтически приемлемые соли, амиды и сложные эфиры.
Группа особенно предпочтительных соединений образована соединениями формулы I, в которой
R1 представляет собой аллилоксикарбонил или аллиламинокарбонил,
А представляет собой остаток (L)-4-амидинфенилаланина,
В представляет собой остаток (L)-глутаминовой кислоты или фармацевтически приемлемую соль или сложный эфир (L)-глутаминовой кислоты,
D представляет собой остаток, выбранный из ряда, состоящего из Arg, Dap, Dab, Orn, Lys, Dap[-С(=NH)-NH2], Dab[-C(=NH)-NH2], Lys[-C(=NH)-NH2], Lys[-C(=NH)-CH3], Orn[-C(=NH)-СН3], Dab[-C(=NH)-СН3], Dap[-С(=NH)-СН3], Dab(Alloc), Asn, Gln, Met, Ser, Thr, Ser(Bzl), Thr(Bzl), Cys(Me), Cys(Bzl), Cys (Acm), Arg(NO2), His, Trp, Phg, Gly, Ala, Val, Ile, Leu, Phe, Phe(4-NO2), Phe(4-NH-C(=NH)-NH2), 2-Abu, Ala(3-CN), Ala[3-C(=NH)-NH2], 2-Abu(4-CN) и 2-Abu[4-C(=NH)-NH2],
n равно 0 или 1,
Е представляет собой остаток, выбранный из ряда, состоящего из Cha, Chg и Phe[4-C(-S-CH2-CH2-S-)-Ph],
R2 представляет собой NHR22, R22 представляет собой водород или остаток, выбранный из ряда, состоящего из бензила, нафтилметила, пиридилметила, фенилэтила, нафтилэтила, пиридилэтила, фенилпропила, нафтилпропила, пиридилпропила, фторенила, дифенилметила, дифенилэтила и дифенилпропила, причем эти остатки не замещены или замещены одним или несколькими идентичными или различными заместителями, выбранными из ряда, состоящего из F, Cl, Вr, гидрокси, метокси, метилендиокси, нитро, циано, диалкиламино, алкилсульфонила, аминосульфонила и трифторметила,
в любой из их стереоизомерных форм или их смесей в любом соотношении, и их фармацевтически приемлемые соли, амиды и сложные эфиры.
Особые примеры соединений согласно изобретению включают в себя, например, соединения, перечисленные ниже в табл. 2, и в разделе примеров, и их фармацевтически приемлемые соли, амиды и сложные эфиры.
Соединения формулы I могут быть получены, например, в соответствии со способами твердофазной химии с помощью способа, который включает в себя:
а1) связывание соединения формулы Fmoc-En-OH, в которой n равно 1, 2 или 3, с чувствительным к кислоте линкером, прикрепленным к смоле или в целом к твердой опоре, отщепление защитной группы Fmoc, связывание соединения формулы Fmoc-D1-D2-C(О)ОН с полученной свободной аминогруппой и вновь отщепление защитной группы Fmoc, или для получения соединения формулы I, в которой n равно 0, связывание соединения формулы Fmoc-D1-D2-C(О)ОН с чувствительным к кислоте линкером, прикрепленным к смоле или в целом к твердой опоре, и отщепление защитной группы Fmoc;
а2) связывание соединения формулы Fmoc-B1-B2-C(О)ОН со свободной аминогруппой, полученной на этапе а1), и отщепление защитной группы Fmoc;
а3) связывание соединения формулы R1-A1-A2-C(О)ОН со свободной аминогруппой, полученной на этапе а2);
а4) отщепление соединения, полученного в соответствии с этапами с а1) по а3), от смолы посредством трифторуксусной кислоты.
Смола или линкер, используемые в этом способе, могут относиться к такому типу, в котором карбоксильная группа в соединении, связанном, соответственно, со смолой или с линкером, трансформируется в амидную группу С(O)-NН2, например линкер Knorr или амидная смола Rink. Получение соединения, в котором число n равно 2 или 3, также может осуществляться с помощью поэтапной сборки элемента En следующим образом. На этапе а1) вместо соединения формулы Fmoc-En-OH, в которой n равно 2 или 3, сначала соединение формулы Fmoc-En-OH, в которой n равно 1, связывают с чувствительным к кислоте линкером, прикрепленным к смоле, затем отщепляют защитную группу Fmoc, и второе соединение формулы Fmoc-En-OH, в которой n равно 1, связывают с полученной свободной аминогруппой. Затем для получения соединения, в котором n равно 1, отщепляют защитную группу Fmoc, и третье соединение формулы Fmoc-En-OH, в которой n равно 3, связывают с полученной свободной аминогруппой. Наконец, отщепляют защитную группу Fmoc, и за этим следуют этапы а2) – а4).
Другой способ получения соединений формулы I включает в себя:
b1) связывание карбоновой кислоты боковой цепи соединения формулы Fmoc-B1-CHR97-C(О)OPG, в которой R97 представляет собой 2-гидроксикарбонилэтил, a PG представляет собой защитную группу с чувствительным к кислоте линкером типа бензилового спирта, прикрепленным к смоле с функциональной аминогруппой;
b2) отщепление защитной группы PG;
b3) связывание соединения формулы H2N-D2-D3-En-R2, в котором n равно 0, 1, 2 или 3, со свободной карбоновой кислотой, полученной на этапе b2);
b4) отщепление защитной группы Fmoc;
b5) связывание соединения формулы R1-A1-A2-C(О)ОН со свободной аминогруппой, полученной на этапе b4);
b6) отщепление соединения, полученного в соответствии с этапами bl) – b5), от смолы посредством трифторуксусной кислоты.
Аналогично модификации описанного выше первого способа, в этом способе сборка структурного элемента H2N-D2-D3-En-R2 может поэтапно осуществляться на смоле. В соответствии с еще одним способом, подобным этому способу, соединения формулы I могут быть также получены с помощью связывания сначала группы карбоновой кислоты, которая присутствует в боковой цепи в группе D2 группы D, т.е., которая присутствует в одной из групп R81 и R82, с линкером, прикрепленным к смоле. Аналогично указанному выше соединению формулы Fmoc-B1-CHR97-C(О)OPG, такое соединение может, например, иметь формулу Fmoc-NH-CR81R82-C(О)OPG, в которой R82 представляет собой определенное выше соединение при условии, что оно содержит группу С(О)ОН, а R81 представляет собой определенный выше остаток. Например, R81 может представлять собой водород, а R82 может представлять собой гидроксикарбонилметил, и соединение формулы Fmoc-NH-CR81R82-C(O)OPG может таким образом представлять собой защищенное производное аспарагиновой кислоты. После отсоединения защитной группы C(O)OPG, карбонильная группа которой представляет собой группу D3 в формуле I, полученную свободную группу карбоновой кислоты связывают с соединением, подобным H2N-En-R2 или H-R2. Затем после удаления защитной группы Fmoc полученную аминогруппу связывают с соединением формулы Fmoc-B1-B2-C(О)ОН, и после устранения защиты аминогруппы продукт связывают с соединением формулы R1-A1-A2-С(O)ОН. И снова используемая смола или линкер могут быть такого типа, что карбоксильная группа в соединении, которое связано со смолой или с линкером, трансформируется в амидную группу C(O)-NH2. Например, с помощью использования амидной смолы элемент аспарагиновой кислоты, прикрепленный к смоле, может быть в конечном соединении превращен в аспарагиновый элемент.
Соединение согласно изобретению может быть химически синтезировано с использованием, например, автоматического синтезатора (см. пример 1). Избирательная модификация реактивной группы, такой как группа, присутствующая на боковой цепи аминокислоты, или N-концевая или С-концевая реактивная группа в пептиде, может придать соединению согласно изобретению желаемые характеристики, такие как повышенная растворимость или усиленная ингибирующая функция. При использовании твердофазных способов синтеза химический состав соединения можно изменять, в то время как к смоле прикрепляется образующийся пептид или после того, как пептид был отщеплен от смолы для получения, например, N-концевого производного, такого как соединение, ацилированное на N-конце, например ацетилированное соединение. Аналогичные модификации могут также производиться в карбоксильной группе соединения, включая карбоксильную группу С-конца, которая может быть, например, амидирована.
Соединения могут также быть получены с помощью связывания защищенных аминокислот в соответствии со способами традиционной фармацевтической химии или с помощью известных в данной области стандартных процедур жидкофазной органической химии и снятия защиты до целевой молекулы. В целом, подходящие реакции для синтеза соединений формулы I с помощью твердофазных способов или жидкофазных способов, а также экспериментальные детали, подобные подходящим связывающим агентам, таким как карбодиимиды, TOTU или HATU, или растворители и температуры реакции, хорошо известны специалистам в данной области и их можно также найти в стандартных ссылках, включая ссылки, упомянутые здесь, а также приведенные ниже в качестве примеров.
Синтезированное соединение может быть очищено с использованием хорошо известных способов, таких как высокоэффективная жидкостная хроматография с обращенной фазой (RP-HPLC; см. пример 1) или другие способы отделения, основанные, например, на размере, заряде или гидрофобности соединения. Аналогичным образом для характеристики структуры соединения согласно изобретению могут использоваться хорошо известные способы, такие как анализ последовательности аминокислот или масс-спектрометрия (MS), см. пример 1.
Различные соединения, содержащие различные расположения заместителей, проявляют различные уровни ингибирующей активности по отношению к фактору VIIa. Например, выбор заместителей влияет на сродство связывания соединений. Эти соединения были синтезированы в соответствии с процедурами, описанными в примерах. Испытание пептидов на ингибирующую активность осуществлялось с использованием количественного анализа, описанного в примере 22. Используя такие способы, специалист в данной области может синтезировать соединение, как раскрыто здесь, включая его модификацию, и определить ингибирующую активность соединения по отношению к фактору VIIa. Композиция настоящего изобретения может быть представлена в виде однородной композиции или в виде смеси соединений, содержащих различные комбинации заместителей. Гибкость, обеспечиваемая выбором заместителей, обеспечивает возможность существенного управления биологическими и физико-химическими свойствами соединений и композиций согласно изобретению.
Изобретение предоставляет соединения, которые специфически ингибируют активность фактора VII. Такие соединения предпочтительно имеют ингибирующую активность (Ki) 500 нмоль, более предпочтительно 50 нмоль для активности фактора VIIa и не оказывают существенного ингибирующего действия на активность других протеаз, участвующих в каскаде свертывания и фибринолиза, относительно ингибирования фактора VIIa (при использовании такой же концентрации ингибитора). Такие другие протеазы включают в себя, например, фактор Ха, тромбин и плазмин.
В следующей ниже табл.2 показана ингибирующая активность по отношению к фактору VIIa (см. пример 22 по способу определения Ki) отобранных соединений формулы I, которые также иллюстрируют изобретение. 500 нмоль, более предпочтительно 50 нмоль для активности фактора VIIa и не оказывают существенного ингибирующего действия на активность других протеаз, участвующих в каскаде свертывания и фибринолиза, относительно ингибирования фактора VIIa (при использовании такой же концентрации ингибитора). Такие другие протеазы включают в себя, например, фактор Ха, тромбин и плазмин.
В следующей ниже табл.2 показана ингибирующая активность по отношению к фактору VIIa (см. пример 22 по способу определения Ki) отобранных соединений формулы I, которые также иллюстрируют изобретение.
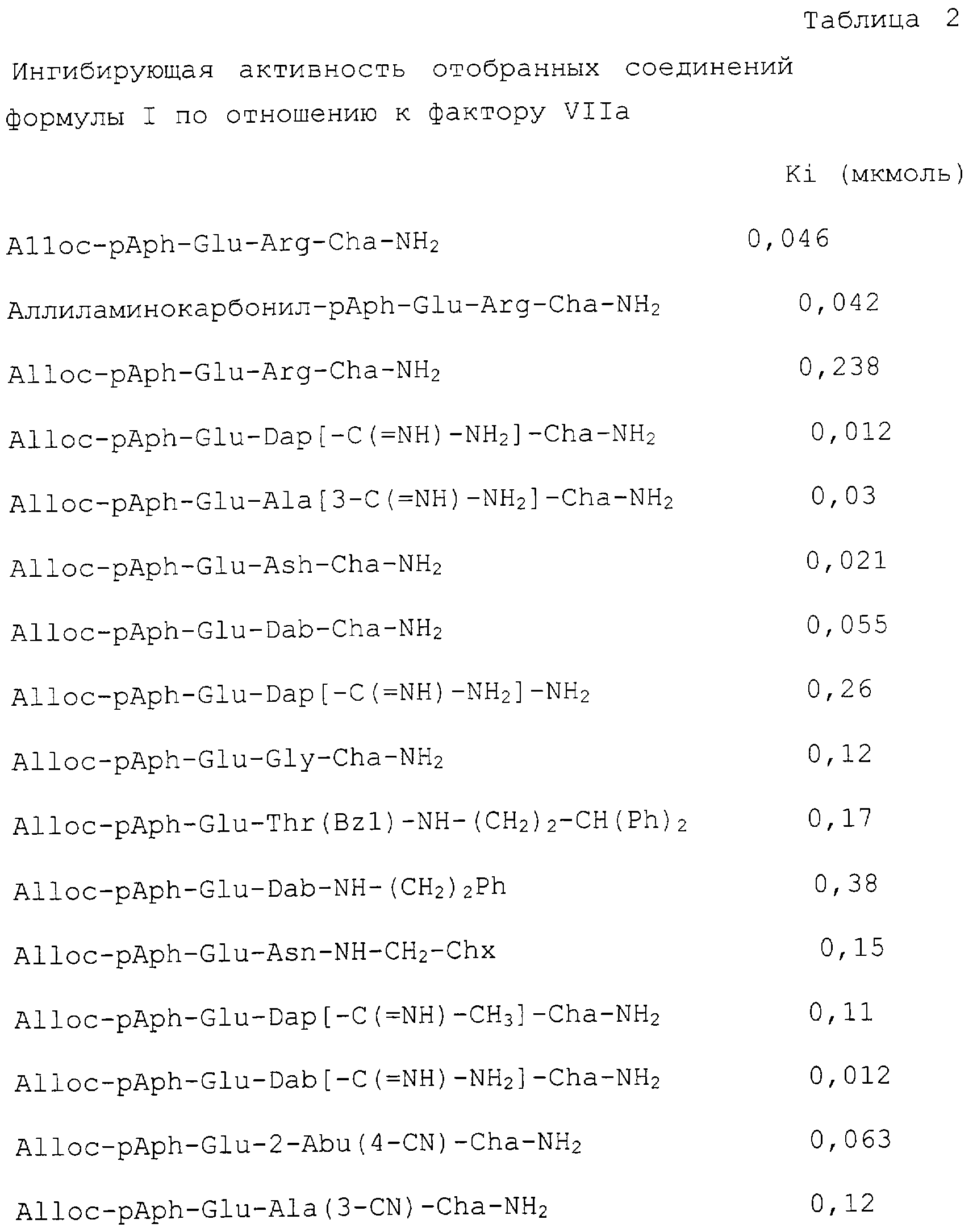
 Ингибирующая тромбин активность указанных выше соединений может быть выражена в величинах Кi, которые в целом значительно выше, чем указанная выше ингибирующая активность по отношению к фактору VIIa, например, приблизительно в 200 раз или приблизительно в 500 раз, или приблизительно в 1000 раз выше, чем ингибирующая активность по отношению к фактору VIIa. Ингибирующая активность указанных выше соединений в отношении фактора Ха также может быть выражена в величинах Ki, которые в целом значительно выше, чем указанная выше ингибирующая активность в отношении фактора VIIa, например, приблизительно в 100 раз выше, чем ингибирующая активность в отношении фактора VIIa.
Эти данные показывают, что соединения формулы I могут использоваться в качестве ингибиторов фактора VIIa, но не оказывают существенного ингибирующего воздействия на активность фактора Ха или сериновых протеаз, таких как тромбин, которые участвуют в процессе свертывания крови и фибринолизе.
Соединение согласно изобретению может применяться преимущественно в качестве антикоагулянта, который может вступать в контакт с образцом крови для предотвращения свертывания. Например, эффективное количество соединения может вступать в контакт со свежим образцом взятой крови для предотвращения свертывания образца крови. Используемый здесь термин “эффективное количество” при использовании со ссылкой на соединение согласно изобретению означает количество соединения, которое ингибирует активность фактора VIIa. Опытному специалисту должно быть понятно, что эффективное количество соединения согласно изобретению может быть определено с использованием раскрытых здесь способов (см. пример 22) или других способов, известных в данной области. С точки зрения возможности применения соединения согласно изобретению, опытному специалисту будет понятно, что такое средство как гепарин может быть заменено на соединение согласно изобретению. Такое применение соединения данного изобретения может, например, привести к экономии затрат по сравнению с другими антикоагулянтами.
Кроме того, соединение согласно изобретению может вводиться индивидууму для лечения различных клинических состояний, включая, например, лечение сердечно-сосудистых расстройств или осложнения, связанного, например, с инфекцией или с хирургической операцией. Примеры сердечно-сосудистых расстройств включают в себя рестеноз после ангиопластики, респираторный дистресс-синдром взрослых, полиорганную недостаточность, инсульт и синдром диссеминированного внутрисосудистого свертывания. Примеры родственных осложнений, связанных с хирургическими вмешательствами, включают в себя, например, тромбоз глубоких вен и проксимальных вен, который может произойти после операции. Таким образом, соединение согласно изобретению может применяться в качестве лекарственного средства для уменьшения или ингибирования нежелательного свертывания крови у индивидуума.
Поскольку соединение согласно изобретению может ингибировать активность фактора VIIa, такое соединение может в целом применяться для уменьшения или ингибирования свертывания крови у индивидуума. Используемый здесь термин “индивидуум” означает позвоночное животное, включая млекопитающее, такое как человек, у которого фактор VIIa участвует в каскаде свертывания.
Свертывание крови у индивидуума может быть уменьшено или ингибировано с помощью введения индивидууму терапевтически эффективного количества соединения согласно изобретению. Используемый здесь термин “терапевтически эффективное количество” означает дозу соединения, которую необходимо ввести индивидууму для ингибирования активности фактора VIIa у индивидуума. Более конкретно, терапевтически эффективное количество соединения согласно изобретению ингибирует каталитическую активность фактора VIIa или непосредственно в пределах комплекса протромбиназы, или в виде растворимой субъединицы, или косвенно путем ингибирования сборки фактора VIIa в комплекс протромбиназы. Предпочтительные соединения могут ингибировать активность фактора VIIa при Ki500 нмоль, а более предпочтительные соединения – при Ki50 нмоль. Терапевтически эффективное количество можно определить с использованием описанных способов, например в примере 22, или с помощью других способов, известных в данной области.
При использовании способа лечения согласно изобретению конкретная дозировка для получения терапевтически эффективного количества фармацевтической композиции, которое предстоит ввести индивидууму, будет зависеть от множества факторов, которые следует учитывать, включая, например, природу или тяжесть заболевания, схему введения, возраст и физические характеристики индивидуума. Соответствующую дозировку можно установить с использованием клинических подходов, хорошо известных в области медицины. Таким образом, изобретение представляет способ специфического ингибирования активности фактора VIIa с помощью обеспечения контакта фактора VIIa с соединением, имеющим формулу R1-A-B-D-En-R2. Изобретение, кроме того, представляет способ уменьшения или ингибирования образования сгустка крови у индивидуума с помощью введения терапевтически эффективного количества соединения согласно изобретению.
Соединение согласно изобретению в целом будет вводиться индивидууму в виде композиции, содержащей одно или несколько соединений формулы I, и фармацевтически приемлемый носитель. Термин “фармацевтически приемлемый носитель” относится к среде или композиции, которая является нетоксичной для индивидуума или имеет приемлемую токсичность по определению соответствующего регулирующего ведомства. Используемый здесь термин “фармацевтически приемлемый носитель” охватывает любой из стандартных фармацевтических носителей, включающих в себя твердые вещества-носители, подобные кукурузному крахмалу, лактозе, жирам, воскам и т.д., или жидкости, такие, например как солевой раствор с фосфатным буфером, вода, эмульсии, такие как эмульсии масло-в-воде или вода-в-масле, и/или обычные добавки, например, любые из различных типов смачивающих средств. Подходящие фармацевтические носители и их композиции описаны Martin в руководстве Remington’s Pharmaceutical Sciences, 15th ed. (Mack Publishing Co., Easton, 1975), которое включено сюда в виде ссылки. Такие композиции будут в целом содержать терапевтически эффективное количество соединения согласно изобретению вместе с подходящим количеством носителя, с тем, чтобы содержать соответствующую дозировку для введения индивидууму. Таким образом, заявляемые соединения могут применяться в качестве лекарственных средств для ингибирования активности фактора VIIa и свертывания крови у индивидуума.
Фармацевтические композиции или лекарственные препараты согласно изобретению могут вводиться перорально, например, в форме пилюль, таблеток, глянцованных таблеток, покрытых таблеток, гранул, твердых и мягких желатиновых капсул, растворов, сиропов, эмульсий, суспензий или аэрозольных смесей. Однако введение может также осуществляться ректально, например, в форме суппозиториев, или парентерально, например внутривенно, внутримышечно или подкожно, в виде растворов для инъекций или растворов для вливания, в виде микрокапсул, имплантатов или стержней, или чрескожно или местно, например, в форме мазей, растворов или настоек, или другими путями, например в форме аэрозолей или аэрозольных композиций для интраназального распыления. Количество активного ингредиента формулы I или его фармацевтически приемлемой соли или производного в стандартной дозе фармацевтической композиции обычно составляет приблизительно от 0,5 мг до 1000 мг, предпочтительно приблизительно от 1 мг до 500 мг, но, в зависимости от типа фармацевтической композиции, количество может также быть более высоким. Дневная доза соединений формулы I, которую предстоит ввести, может представлять собой однократную дневную дозу или может быть разделена на несколько, например, 2, 3 или 4 дробных введения.
Фармацевтически приемлемые носители также могут включать в себя, например, другие среды, соединения или модификации в дополнение к ингибирующему фактор VIIa соединению формулы I, которые усиливают его фармакологическую функцию. Фармацевтически приемлемая среда может включать в себя, например, фармацевтически приемлемую соль. Соль присоединения кислот соединения формулы I может быть образована, например, с неорганической кислотой, такой как хлористоводородная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота или перхлорная кислота, или с органической карбоновой кислотой, такой как уксусная кислота, щавелевая кислота, малеиновая кислота, оксиянтарная кислота, муравьиная кислота, молочная кислота, винная кислота, лимонная кислота, янтарная кислота или малоновая кислота, с органической сульфокислотой, такой как метансульфоновая кислота или пара-толуолсульфоновая кислота. Кислотная группа в соединении формулы I, например карбоксильная группа, может присутствовать в виде соли металла, катион которой основан на щелочных и щелочноземельных металлах, таких как натрий, литий, калий, кальций или магний, или также нетоксичных аммониевых солей, включая соли четвертичного аммония и соли присоединения кислот с аминами, например аммониевая, метиламмониевая, диметиламмониевая, триметиламмониевая, тетраметиламмониевая, этиламмониевая, триэтиламмониевая или тетраэтиламмониевая соль.
Примеры модификаций, которые усиливают фармакологическую функцию соединения, включают в себя, например, эстерификацию, такую как образование (C1-С6)-алкиловых эфиров, предпочтительно (C1-C4)-алкиловых эфиров, в которых алкильная группа представляет собой прямую или разветвленную цепь. Другие приемлемые сложные эфиры включают в себя, например, (С5-С7)-циклоалкиловые эфиры и арилалкиловые эфиры, такие как бензиловые эфиры. Такие сложные эфиры могут быть получены из описанных здесь соединений с использованием обычных способов, хорошо известных в области пептидной химии.
Фармацевтически приемлемые модификации также могут включать в себя, например, образование пептидных амидов. Такие амидные модификации, которые могут производиться в отношении соединений согласно изобретению включают в себя, например, модификации, полученные из аммониев, первичных (C1-C6)-алкиламинов и вторичных ди-(C1-C6)-алкиламинов, где алкильные группы представляют собой прямую или разветвленную цепь, или ариламины, имеющие различные замещения. В случае вторичных аминов амин может также быть в форме 5- или 6-членного гетероцикла, который, в дополнение к амидному атому азота, может содержать незамещенный или замещенный атом азота, атом кислорода или атом серы. Способы получения таких амидов хорошо известны в данной области.
В другом варианте реализации изобретения соединение согласно изобретению может применяться в количественном анализе для выявления присутствия фактора VIIa или для выделения фактора VIIa по существу в очищенной форме. Предпочтительно соединение согласно изобретению метят, например, радиоизотопом, и меченое соединение выявляют с использованием обычного способа, применяемого для выявления определенной метки. Кроме того, соединение согласно изобретению может применяться преимущественно в качестве зонда для выявления локализации или количества активности фактора VIIa in vivo, in vitro или ex vivo.
Понятно, что в раскрытое здесь изобретение включены модификации, которые существенно не влияют на активность различных вариантов реализации этого изобретения. Соответственно, следующие примеры предназначены для иллюстрации, а не для ограничения настоящего изобретения.
Пример 1. Процедуры пептидного синтеза и общие процедуры синтеза
Исходные материалы, используемые при синтезе, получают в компаниях, торгующих химическими товарами, таких как Aldrich, Sigma, Fluka, Nova Biochem и Advanced Chemtech. Во время синтеза функциональные группы используемых производных аминокислот защищают с помощью блокирующих групп для предотвращения побочной реакции во время этапов связывания. Примеры подходящих защитных групп и их применения описаны в публикации The Peptides, supra, 1981, и в vol.9, Udenfriend and Meienhofer (eds.), 1987, которые включены сюда в виде ссылки.
Общий твердофазный пептидный синтез используют для получения соединений согласно изобретению. Такие способы описаны, например, в руководстве Stewart and Young, Solid Phase Peptide Syntesis (Freeman&Co., San Francisco, 1969, которое включено сюда в качестве ссылки.
При отсутствии других указаний пептиды синтезируют на смоле TentaGel S NH2 (Rapp Polymere, Tubingen, Germany). Чувствительное к кислоте связывающее вещество пара-[(R,S)--[1-(9Н-фторен-9-ил)метоксиформамидо]-2,4-диметоксибензил]феноксиуксусную кислоту (связывающее вещество Knorr) связывают с твердым носителем (публикация Bernatowicz et al., Tetr. Lett. 30 (1989) 4645, которая включена сюда в виде ссылки). Альтернативно, пептиды синтезируют на полистироловой смоле, поперечно сшитой 1% дивинилбензолом, модифицированным чувствительным к кислоте линкером (смолой Rink) (Rink, Tetr. Lett. 28(1987)3787; Sieber, Tetr. Lett. 28(1987)2107, каждая из которых включена сюда в ссылки). Когда пептиды синтезируют сначала с помощью связывания карбоновой кислоты боковой цепи соединения формулы Fmoc-B1-CHR97-C(О)OPG со смолой, используют смолу TentaGel S NH2, модифицированную прикреплением связывающего вещества НМРА. Связывание выполняют с использованием N,N’-диизопропилкарбодиимида (DIC) в присутствии эквивалентного количества HOBt, за исключением Alloc-pAph-OH, где используют 2 эквивалента (экв.) HOBt. Все связывания проводят или в N,N-диметилформамиде (DMF), или DMF/DMSO (смесь 1/1) при комнатной температуре (КТ). Завершение связывания контролируют с помощью нингидриновой пробы. Второе (двойное) связывание выполняют, когда в первом случае происходит неполное связывание.
Удаление группы, защищающей группу Fmoc, осуществляют с использованием 50% пиперидина в DMF в течение 2-10 мин. Количество освобождаемой Fmoc определяют по спектральной поглощательной способности при 300 нм раствором после удаления защитной группы, по объему промываний и массе смолы, использованной при синтезе.
Цикл каждого связывания был следующим:
Ингибирующая тромбин активность указанных выше соединений может быть выражена в величинах Кi, которые в целом значительно выше, чем указанная выше ингибирующая активность по отношению к фактору VIIa, например, приблизительно в 200 раз или приблизительно в 500 раз, или приблизительно в 1000 раз выше, чем ингибирующая активность по отношению к фактору VIIa. Ингибирующая активность указанных выше соединений в отношении фактора Ха также может быть выражена в величинах Ki, которые в целом значительно выше, чем указанная выше ингибирующая активность в отношении фактора VIIa, например, приблизительно в 100 раз выше, чем ингибирующая активность в отношении фактора VIIa.
Эти данные показывают, что соединения формулы I могут использоваться в качестве ингибиторов фактора VIIa, но не оказывают существенного ингибирующего воздействия на активность фактора Ха или сериновых протеаз, таких как тромбин, которые участвуют в процессе свертывания крови и фибринолизе.
Соединение согласно изобретению может применяться преимущественно в качестве антикоагулянта, который может вступать в контакт с образцом крови для предотвращения свертывания. Например, эффективное количество соединения может вступать в контакт со свежим образцом взятой крови для предотвращения свертывания образца крови. Используемый здесь термин “эффективное количество” при использовании со ссылкой на соединение согласно изобретению означает количество соединения, которое ингибирует активность фактора VIIa. Опытному специалисту должно быть понятно, что эффективное количество соединения согласно изобретению может быть определено с использованием раскрытых здесь способов (см. пример 22) или других способов, известных в данной области. С точки зрения возможности применения соединения согласно изобретению, опытному специалисту будет понятно, что такое средство как гепарин может быть заменено на соединение согласно изобретению. Такое применение соединения данного изобретения может, например, привести к экономии затрат по сравнению с другими антикоагулянтами.
Кроме того, соединение согласно изобретению может вводиться индивидууму для лечения различных клинических состояний, включая, например, лечение сердечно-сосудистых расстройств или осложнения, связанного, например, с инфекцией или с хирургической операцией. Примеры сердечно-сосудистых расстройств включают в себя рестеноз после ангиопластики, респираторный дистресс-синдром взрослых, полиорганную недостаточность, инсульт и синдром диссеминированного внутрисосудистого свертывания. Примеры родственных осложнений, связанных с хирургическими вмешательствами, включают в себя, например, тромбоз глубоких вен и проксимальных вен, который может произойти после операции. Таким образом, соединение согласно изобретению может применяться в качестве лекарственного средства для уменьшения или ингибирования нежелательного свертывания крови у индивидуума.
Поскольку соединение согласно изобретению может ингибировать активность фактора VIIa, такое соединение может в целом применяться для уменьшения или ингибирования свертывания крови у индивидуума. Используемый здесь термин “индивидуум” означает позвоночное животное, включая млекопитающее, такое как человек, у которого фактор VIIa участвует в каскаде свертывания.
Свертывание крови у индивидуума может быть уменьшено или ингибировано с помощью введения индивидууму терапевтически эффективного количества соединения согласно изобретению. Используемый здесь термин “терапевтически эффективное количество” означает дозу соединения, которую необходимо ввести индивидууму для ингибирования активности фактора VIIa у индивидуума. Более конкретно, терапевтически эффективное количество соединения согласно изобретению ингибирует каталитическую активность фактора VIIa или непосредственно в пределах комплекса протромбиназы, или в виде растворимой субъединицы, или косвенно путем ингибирования сборки фактора VIIa в комплекс протромбиназы. Предпочтительные соединения могут ингибировать активность фактора VIIa при Ki500 нмоль, а более предпочтительные соединения – при Ki50 нмоль. Терапевтически эффективное количество можно определить с использованием описанных способов, например в примере 22, или с помощью других способов, известных в данной области.
При использовании способа лечения согласно изобретению конкретная дозировка для получения терапевтически эффективного количества фармацевтической композиции, которое предстоит ввести индивидууму, будет зависеть от множества факторов, которые следует учитывать, включая, например, природу или тяжесть заболевания, схему введения, возраст и физические характеристики индивидуума. Соответствующую дозировку можно установить с использованием клинических подходов, хорошо известных в области медицины. Таким образом, изобретение представляет способ специфического ингибирования активности фактора VIIa с помощью обеспечения контакта фактора VIIa с соединением, имеющим формулу R1-A-B-D-En-R2. Изобретение, кроме того, представляет способ уменьшения или ингибирования образования сгустка крови у индивидуума с помощью введения терапевтически эффективного количества соединения согласно изобретению.
Соединение согласно изобретению в целом будет вводиться индивидууму в виде композиции, содержащей одно или несколько соединений формулы I, и фармацевтически приемлемый носитель. Термин “фармацевтически приемлемый носитель” относится к среде или композиции, которая является нетоксичной для индивидуума или имеет приемлемую токсичность по определению соответствующего регулирующего ведомства. Используемый здесь термин “фармацевтически приемлемый носитель” охватывает любой из стандартных фармацевтических носителей, включающих в себя твердые вещества-носители, подобные кукурузному крахмалу, лактозе, жирам, воскам и т.д., или жидкости, такие, например как солевой раствор с фосфатным буфером, вода, эмульсии, такие как эмульсии масло-в-воде или вода-в-масле, и/или обычные добавки, например, любые из различных типов смачивающих средств. Подходящие фармацевтические носители и их композиции описаны Martin в руководстве Remington’s Pharmaceutical Sciences, 15th ed. (Mack Publishing Co., Easton, 1975), которое включено сюда в виде ссылки. Такие композиции будут в целом содержать терапевтически эффективное количество соединения согласно изобретению вместе с подходящим количеством носителя, с тем, чтобы содержать соответствующую дозировку для введения индивидууму. Таким образом, заявляемые соединения могут применяться в качестве лекарственных средств для ингибирования активности фактора VIIa и свертывания крови у индивидуума.
Фармацевтические композиции или лекарственные препараты согласно изобретению могут вводиться перорально, например, в форме пилюль, таблеток, глянцованных таблеток, покрытых таблеток, гранул, твердых и мягких желатиновых капсул, растворов, сиропов, эмульсий, суспензий или аэрозольных смесей. Однако введение может также осуществляться ректально, например, в форме суппозиториев, или парентерально, например внутривенно, внутримышечно или подкожно, в виде растворов для инъекций или растворов для вливания, в виде микрокапсул, имплантатов или стержней, или чрескожно или местно, например, в форме мазей, растворов или настоек, или другими путями, например в форме аэрозолей или аэрозольных композиций для интраназального распыления. Количество активного ингредиента формулы I или его фармацевтически приемлемой соли или производного в стандартной дозе фармацевтической композиции обычно составляет приблизительно от 0,5 мг до 1000 мг, предпочтительно приблизительно от 1 мг до 500 мг, но, в зависимости от типа фармацевтической композиции, количество может также быть более высоким. Дневная доза соединений формулы I, которую предстоит ввести, может представлять собой однократную дневную дозу или может быть разделена на несколько, например, 2, 3 или 4 дробных введения.
Фармацевтически приемлемые носители также могут включать в себя, например, другие среды, соединения или модификации в дополнение к ингибирующему фактор VIIa соединению формулы I, которые усиливают его фармакологическую функцию. Фармацевтически приемлемая среда может включать в себя, например, фармацевтически приемлемую соль. Соль присоединения кислот соединения формулы I может быть образована, например, с неорганической кислотой, такой как хлористоводородная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота или перхлорная кислота, или с органической карбоновой кислотой, такой как уксусная кислота, щавелевая кислота, малеиновая кислота, оксиянтарная кислота, муравьиная кислота, молочная кислота, винная кислота, лимонная кислота, янтарная кислота или малоновая кислота, с органической сульфокислотой, такой как метансульфоновая кислота или пара-толуолсульфоновая кислота. Кислотная группа в соединении формулы I, например карбоксильная группа, может присутствовать в виде соли металла, катион которой основан на щелочных и щелочноземельных металлах, таких как натрий, литий, калий, кальций или магний, или также нетоксичных аммониевых солей, включая соли четвертичного аммония и соли присоединения кислот с аминами, например аммониевая, метиламмониевая, диметиламмониевая, триметиламмониевая, тетраметиламмониевая, этиламмониевая, триэтиламмониевая или тетраэтиламмониевая соль.
Примеры модификаций, которые усиливают фармакологическую функцию соединения, включают в себя, например, эстерификацию, такую как образование (C1-С6)-алкиловых эфиров, предпочтительно (C1-C4)-алкиловых эфиров, в которых алкильная группа представляет собой прямую или разветвленную цепь. Другие приемлемые сложные эфиры включают в себя, например, (С5-С7)-циклоалкиловые эфиры и арилалкиловые эфиры, такие как бензиловые эфиры. Такие сложные эфиры могут быть получены из описанных здесь соединений с использованием обычных способов, хорошо известных в области пептидной химии.
Фармацевтически приемлемые модификации также могут включать в себя, например, образование пептидных амидов. Такие амидные модификации, которые могут производиться в отношении соединений согласно изобретению включают в себя, например, модификации, полученные из аммониев, первичных (C1-C6)-алкиламинов и вторичных ди-(C1-C6)-алкиламинов, где алкильные группы представляют собой прямую или разветвленную цепь, или ариламины, имеющие различные замещения. В случае вторичных аминов амин может также быть в форме 5- или 6-членного гетероцикла, который, в дополнение к амидному атому азота, может содержать незамещенный или замещенный атом азота, атом кислорода или атом серы. Способы получения таких амидов хорошо известны в данной области.
В другом варианте реализации изобретения соединение согласно изобретению может применяться в количественном анализе для выявления присутствия фактора VIIa или для выделения фактора VIIa по существу в очищенной форме. Предпочтительно соединение согласно изобретению метят, например, радиоизотопом, и меченое соединение выявляют с использованием обычного способа, применяемого для выявления определенной метки. Кроме того, соединение согласно изобретению может применяться преимущественно в качестве зонда для выявления локализации или количества активности фактора VIIa in vivo, in vitro или ex vivo.
Понятно, что в раскрытое здесь изобретение включены модификации, которые существенно не влияют на активность различных вариантов реализации этого изобретения. Соответственно, следующие примеры предназначены для иллюстрации, а не для ограничения настоящего изобретения.
Пример 1. Процедуры пептидного синтеза и общие процедуры синтеза
Исходные материалы, используемые при синтезе, получают в компаниях, торгующих химическими товарами, таких как Aldrich, Sigma, Fluka, Nova Biochem и Advanced Chemtech. Во время синтеза функциональные группы используемых производных аминокислот защищают с помощью блокирующих групп для предотвращения побочной реакции во время этапов связывания. Примеры подходящих защитных групп и их применения описаны в публикации The Peptides, supra, 1981, и в vol.9, Udenfriend and Meienhofer (eds.), 1987, которые включены сюда в виде ссылки.
Общий твердофазный пептидный синтез используют для получения соединений согласно изобретению. Такие способы описаны, например, в руководстве Stewart and Young, Solid Phase Peptide Syntesis (Freeman&Co., San Francisco, 1969, которое включено сюда в качестве ссылки.
При отсутствии других указаний пептиды синтезируют на смоле TentaGel S NH2 (Rapp Polymere, Tubingen, Germany). Чувствительное к кислоте связывающее вещество пара-[(R,S)--[1-(9Н-фторен-9-ил)метоксиформамидо]-2,4-диметоксибензил]феноксиуксусную кислоту (связывающее вещество Knorr) связывают с твердым носителем (публикация Bernatowicz et al., Tetr. Lett. 30 (1989) 4645, которая включена сюда в виде ссылки). Альтернативно, пептиды синтезируют на полистироловой смоле, поперечно сшитой 1% дивинилбензолом, модифицированным чувствительным к кислоте линкером (смолой Rink) (Rink, Tetr. Lett. 28(1987)3787; Sieber, Tetr. Lett. 28(1987)2107, каждая из которых включена сюда в ссылки). Когда пептиды синтезируют сначала с помощью связывания карбоновой кислоты боковой цепи соединения формулы Fmoc-B1-CHR97-C(О)OPG со смолой, используют смолу TentaGel S NH2, модифицированную прикреплением связывающего вещества НМРА. Связывание выполняют с использованием N,N’-диизопропилкарбодиимида (DIC) в присутствии эквивалентного количества HOBt, за исключением Alloc-pAph-OH, где используют 2 эквивалента (экв.) HOBt. Все связывания проводят или в N,N-диметилформамиде (DMF), или DMF/DMSO (смесь 1/1) при комнатной температуре (КТ). Завершение связывания контролируют с помощью нингидриновой пробы. Второе (двойное) связывание выполняют, когда в первом случае происходит неполное связывание.
Удаление группы, защищающей группу Fmoc, осуществляют с использованием 50% пиперидина в DMF в течение 2-10 мин. Количество освобождаемой Fmoc определяют по спектральной поглощательной способности при 300 нм раствором после удаления защитной группы, по объему промываний и массе смолы, использованной при синтезе.
Цикл каждого связывания был следующим:
 После завершения сборки пептида на смоле, если необходимо, выполняют окончательное удаление группы, защищающей Fmoc. Затем пептидную смолу последовательно промывают DMF и DCM, и затем, при отсутствии других указаний, пептид расщепляют и удаляют защитную группу с помощью смеси TFA/тиоанизола (95/5) в течение 1,5 ч. Смолу промывают DCM, и промывочный DCM комбинируют с освобождающимся при промывке TFA. Раствор выпаривают, продукт осаждают с помощью безводного простого диэтилового эфира, и твердый осадок отделяют с помощью фильтрации или центрифугирования и сушат в вакууме над гранулами твердого КОН. Твердое вещество повторно растворяют в смеси воды и ацетонитрила и лиофилизируют.
Высушенный пептид подвергают очистке с помощью HPLC с использованием соответствующего градиента 0,1% TFA в воде и ацетонитриле (ACN). После сбора пика, содержащего предлагаемый синтетический продукт, раствор пептида лиофилизируют и пептид подвергают процессу идентификации, который включает в себя электроаэрозольный масс-спектр (MS) и/или ЯМР, и/или аминокислотный анализ для подтверждения того, что синтезировано правильное соединение.
Для HPLC-анализа образец продукта анализируют с использованием устройства HPLC Beckman (состоящего из устройства подачи растворителя 126, устройства для автоматического взятия проб 507е с программируемым детекторным модулем 166, управляемого блоком управления Data Station с программным обеспечением Gold Nouveau) и колонки YMC ODS-AM 4,6х250 мм при 230 нм и скорости потока 1 мл/мин.
Для очистки продукта образец неочищенного лиофилизированного пептида растворяют в смеси 0,1% водного TFA, содержащего от 10% до 50% ACN. Раствор пептида обычно фильтруют через шприц, соединенный с фильтром “ACRODISC”13CR PTFE (Gelman Sciences; Ann Arbor MI) с размером пор 0,45 мкм. Соответствующий объем профильтрованного раствора пептида инжектируют в полупрепаративную колонку С18 (Vydac Protein and Peptide С18, 218ТР1022 (22
После завершения сборки пептида на смоле, если необходимо, выполняют окончательное удаление группы, защищающей Fmoc. Затем пептидную смолу последовательно промывают DMF и DCM, и затем, при отсутствии других указаний, пептид расщепляют и удаляют защитную группу с помощью смеси TFA/тиоанизола (95/5) в течение 1,5 ч. Смолу промывают DCM, и промывочный DCM комбинируют с освобождающимся при промывке TFA. Раствор выпаривают, продукт осаждают с помощью безводного простого диэтилового эфира, и твердый осадок отделяют с помощью фильтрации или центрифугирования и сушат в вакууме над гранулами твердого КОН. Твердое вещество повторно растворяют в смеси воды и ацетонитрила и лиофилизируют.
Высушенный пептид подвергают очистке с помощью HPLC с использованием соответствующего градиента 0,1% TFA в воде и ацетонитриле (ACN). После сбора пика, содержащего предлагаемый синтетический продукт, раствор пептида лиофилизируют и пептид подвергают процессу идентификации, который включает в себя электроаэрозольный масс-спектр (MS) и/или ЯМР, и/или аминокислотный анализ для подтверждения того, что синтезировано правильное соединение.
Для HPLC-анализа образец продукта анализируют с использованием устройства HPLC Beckman (состоящего из устройства подачи растворителя 126, устройства для автоматического взятия проб 507е с программируемым детекторным модулем 166, управляемого блоком управления Data Station с программным обеспечением Gold Nouveau) и колонки YMC ODS-AM 4,6х250 мм при 230 нм и скорости потока 1 мл/мин.
Для очистки продукта образец неочищенного лиофилизированного пептида растворяют в смеси 0,1% водного TFA, содержащего от 10% до 50% ACN. Раствор пептида обычно фильтруют через шприц, соединенный с фильтром “ACRODISC”13CR PTFE (Gelman Sciences; Ann Arbor MI) с размером пор 0,45 мкм. Соответствующий объем профильтрованного раствора пептида инжектируют в полупрепаративную колонку С18 (Vydac Protein and Peptide С18, 218ТР1022 (22 250 мм); The Separation Group; Hesperia CA, или колонку YMC ODS-A (20250 мм), YMC, Inc., Wilmington, NC). Скорость потока градиента или изократической смеси 0,1% TFA буфера и ACN (сорта HPLC) в качестве элюента поддерживают с использованием программируемого модуля растворителя 126 и программируемого модуля детектора 166, управляемого с помощью программного обеспечения “SYSTEM GOLD” устройства Beckman “SYSTEM GOLD” HPLC (Beckman, System Gold). Элюирование пептида контролируют с помощью УФ-детекции при 230 нм. После выявления с использованием MS-пика, соответствующего синтезируемому соединению, соединение собирают, лиофилизируют и проводят его биологическое испытание. MS выполняют с использованием прибора VG Piatforn (Fisons Instruments) в режиме ES+. Для ЯМР образцы обычно измеряют в DMSO-d6 (Aldrich) с использованием прибора Bruker Avance DPX300.
Пример 2. Синтез Alloc-pAph-OH
Аналогичная процедура применима для Alloc-pAph-OH.
Alloc-Phe(4-CN)-ОН
5,7 г (30 ммоль) H-Phe (4-CN)-ОН растворяют в 100 мл 1М NaOH с добавлением 2М NaOH до рН 10 с ледяным охлаждением. При энергичном перемешивании медленно добавляют аллилхлорформат (7,5 мл) (рН поддерживают на уровне 10 с помощью 2М NaOH). Реакционную смесь перемешивают при 0°С в течение 15 мин и при КТ в течение 30 мин, подкисляют НСl до рН 2, экстрагируют этилацетатом (3 раза), сушат с MgSO4 и выпаривают. Остаток подвергают перекристаллизации из этилацетата/гексана для получения белого твердого вещества. Выход: 7,0 г (85%).
Alloc-Phe[4-С(=S)-NH2]-ОН
2,74 г Alloc-Phe(4-CN)-ОН растворяют в смеси пиридина (50 мл) и триэтиламина (20 мл) и в течение 30 мин пропускают H2S. Реакционную смесь держат в течение ночи при КТ и выпаривают. Сушка в высоком вакууме дает 3,21 г сплошной пены неочищенного тиоамида, который непосредственно превращается в метилтиоимидат.
Alloc-Phe[4-С(=NH)-SCH3]-OH·HI
1 г Alloc-Phe[4-С(=S)-NH2]-ОН растворяют в ацетоне (50 мл) и добавляют метилиодид (5 мл). Реакционную смесь держат в течение ночи при КТ, летучие растворители выпаривают (быстро, максимум 35°С), остаток обрабатывают простым диэтиловым эфиром. Через 1 ч при 0°С эфир декантируют, продукт промывают простым диэтиловым эфиром и сушат в вакууме. Получают желтую плотную пену, которая непосредственно превращается в амидин.
Alloc-pAph-OH
Весь указанный выше Alloc-Phe(4-С(=NH)-SСН3)-OH·HI растворяют в 50 мл метанола с 300 мкл уксусной кислоты и добавляют 0,5 г ацетата аммония. Смесь нагревают в течение 3 ч при 55°С, выпаривают и добавляют 10 мл ацетона. Через 2 ч при 0°С твердый продукт фильтруют, промывают небольшим количеством холодного ацетона, небольшим количеством холодного метанола и простым диэтиловым эфиром и сушат в вакууме для получения желтоватого твердого вещества. Выход: 0,53г.
Пример 3. Синтез Alloc-pAph-Glu-Arg-Cha-NH2
К 1 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют амидный линкер Knorr. В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Cha-OH, Fmoc-Arg(Pmc)-ОН, Fmoc-Glu(OtBu)-ОН и Alloc-pAph. Пептид расщепляют и с помощью воздействия в течение 3 ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 729,1, по расчетам 729,4.
Пример 4. Синтез аллил-NH-C(O)-pAph-Glu-Arg-Cha-NH2
К 0,5 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют амидный линкер Knorr. В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Cha-OH, Fmoc-Arg(Pmc)-ОН, Fmoc-Glu(OtBu)-ОН и Fmoc-Phe(4-CN). После снятия защиты N-концевой Fmoc смолу в течение 2 ч обрабатывают раствором 1 ммоль аллилизоцианата в 3 мл DMF. Затем смолу промывают DMF и триэтиламином/пиридином (1/2) и в течение ночи обрабатывают насыщенным раствором H2S в пиридине/триэтиламине. Смолу промывают ацетоном и в течение 6 ч проводят реакцию тиоамидной смолы с метилиодидом (3 мл 10% раствора метилиодида в ацетоне). Метилтиоимидатную смолу промывают ацетоном, метанолом и обрабатывают раствором 0,2 г ацетата аммония, 100 мкл уксусной кислоты в 3 мл метанола при 55°С в течение 3 ч. Смолу промывают метанолом, DMF и DCM и пептид расщепляют и снимают защиту с помощью воздействия в течение 3 ч TFA/тиоанизола (95/5) и обрабатывают, как описано в примере 1. Неочищенный материал очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 728,3, по расчетам 728,4.
Пример 5. Синтез Alloc-pAph-Glu-Arff-Chg-NH2
К 1 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют амидный линкер Knorr. В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Chg-OH, Fmoc-Arg(Pmc)-ОН, Fmoc-Glu(OtBu)-ОН и Alloc-pAph. Пептид расщепляют и с помощью воздействия в течение 3 ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 715,8, по расчетам 715,4.
Пример 6. Синтез Alloc-D-pAph-Glu-Arg-Cha-NH2
К 1 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют амидный линкер Knorr. В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Cha-OH, Fmoc-Arg (Pmc)-ОН, Fmoc-Glu(OtBu)-ОН и Alloc-D-pAph-ОН (синтезированные в соответствии с той же процедурой, что и Alloc-pAph-ОН в примере 2). Пептид расщепляют и с помощью воздействия в течение 3 ч TFA/тиоанизола (95/5) удаляют защитную группу и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 729,2, по расчетам 729, 4.
Пример 7. Синтез Alloc-pAph-Glu-Phe(4-гуанидино)-Cha-NH2
К 0,25 г смолы TentaGel S NH2 (замещение 0,23 ммоль/г) прикрепляют амидный линкер Knorr. В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Cha-OH, Fmoc-Phe(4-NH-C(=NBoc)-NH-Boc)-OH, Fmoc-Glu(OtBu)-ОН и Alloc-pAph-OH. Пептид расщепляют и с помощью воздействия в течение 1 ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 777,1, по расчетам 777,4.
Пример 8. Синтез Alloc-pAph-Glu-Dap[-C(=NH)-NH2]-Cha-NH2
К 0,25 г смолы TentaGel S NH2 (замещение 0,23 ммоль/г) прикрепляют амидный линкер Knorr. В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Cha-OH, Fmoc-Dap[-С(=NBoc)-NH-Boc]-OH, Fmoc-Glu(OtBu)-ОН и Alloc-pAph-OH. Пептид расщепляют и с помощью воздействия в течение 1ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 729,1, по расчетам 729,4.
Пример 9. Синтез Alloc-pAph-Glu-Dap[-С(=NН-СН3]-Cha-NH2
К 0,25 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют амидный линкер Knorr. В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Cha-OH, Fmoc-Dap(Alloc)-ОН и Fmoc-Glu(OtBu)-ОН. С прикрепленной N-концевой группой, защищающей Fmoc, смолу промывают смесью DMF/NMM/AcOH (5/0,5/1) и при постоянном смешивании струёй аргона снимают защиту группы Alloc с помощью добавления 100 мг Рd(Р(Рh)3)4 в течение 3 ч. Смолу промывают DMF и обрабатывают раствором 150 мг 2-метилнафтилацеттиоимидата в 4 мл этанола/DMSO (3/1) в течение 1 ч. После промывания DMF снимают защиту группы Fmoc (1-5мин) и связывают N-концевую Alloc-pAph-OH. Пептид расщепляют и с помощью воздействия в течение 1 ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 700,1, по расчетам 700,4.
Пример 10. Синтез Alloc-pAph-Glu-Ala[3-C(=NH)-NH2]-Cha-NH2
К 0,25 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют амидный линкер Knorr. В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Cha-OH, Fmoc-Ala(3-CN)-ОН, Fmoc-Glu(OtBu)-OH и Alloc-Phe(4-CN)-ОН. Смесь пиридина и триэтиламина (2/1) насыщают Н2S (КТ, 15-30 мин), и этот раствор добавляют в смолу, предварительно промытую пиридином/триэтиламином (2/1). После хранения в течение ночи смолу промывают ацетоном и в течение ночи обрабатывают 20% метилиодидом в ацетоне. Затем смолу промывают ацетоном и метанолом. Связанный со смолой метилтиоимидат затем превращают в амидин с помощью нагревания (водяная баня, 55°С, 3 ч) смолы с раствором 10 экв. ацетата аммония в метаноле, содержащем 5% уксусную кислоту. После этого окончательного превращения смолу промывают метанолом, DMF, DCM. Пептид расщепляют и с помощью воздействия в течение 1ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 685,9, по расчетам 686,4.
Пример 11. Синтез Alloc-pAph-Glu-Asn-Cha-NH2
С 0,125 г смолы Rink (замещение 0,78 ммоль/г) после снятия защиты Fmoc в соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Cha-OH, Fmoc-Asn-OH, Fmoc-Glu(OtBu)-ОН и Alloc-pAph-ОН. Пептид расщепляют и с помощью воздействия в течение 1ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 686,9, по расчетам 687,3.
Пример 12. Синтез Alloc-pAph-Glu-Dab-Cha-NH2
К 0,25 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют амидный линкер Knorr. В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Cha-OH, Fmoc-Dab(Воc)-ОН, Fmoc-Glu(OtBu)-ОН и Alloc-pAph. Пептид расщепляют и с помощью воздействия в течение 1 ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 673,2, по расчетам 673,4.
Пример 13. Синтез Alloc-pAph-Glu-Ala[3-C(=NH)-NH2]-NH2
К 0,25 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют амидный линкер Knorr. В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Ala(3-CN)-ОН, Fmoc-Glu(OtBu)-ОН и Alloc-Phe(4-CN)-ОН. Смесь пиридина и триэтиламина (2/1) насыщают H2S (КТ, 15-30 мин), и этот раствор добавляют в смолу, предварительно промытую пиридином/триэтиламином (2/1). После хранения в течение ночи смолу промывают ацетоном и в течение ночи обрабатывают раствором 20% метилиодида в ацетоне. Затем смолу промывают ацетоном и метанолом. Связанный со смолой метилтиоимидат затем превращают в амидин с помощью нагревания (водяная баня, 55°С, 3 ч) смолы с раствором 10 экв. ацетата аммония в метаноле, содержащем 5% уксусную кислоту. После этого окончательного превращения смолу промывают метанолом, DMF, DCM. Пептид расщепляют и с помощью воздействия в течение 1 ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (М+Н)+: обнаружено 533,3, по расчетам 533,2.
Пример 14. Синтез Alloc-pAph-Glu-Gly-Cha-NH2
С 0,150 г смолы Rink (замещение 0,78 ммоль/г) после снятия защиты Fmoc в соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Cha-OH, Fmoc-Gly-OH, Fmoc-Glu(OtBu)-ОН и Alloc-pAph-OH. Пептид расщепляют и с помощью воздействия в течение 1 ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 630,1, по расчетам 630,3.
Пример 15. Синтез Alloc-pAph-Glu-Asn-(Ph-CH2-CH2-)Gly-NH2
Для N-замещенных глицинов используют методику Zuckermann et al. (J.Am. Chem. Soc.ll4 (1992) 10646, которая включена сюда в виде ссылки). С 0,1 г смолы Rink (замещение 0,78 ммоль/г) после снятия защиты Fmoc связывают бромуксусную кислоту посредством симметричного ангидрида в DCM/DMF. Через 10 мин смолу промывают DCM и еще раз повторяют связывание. После промывания DCM и DMF смолу в течение ночи обрабатывают 1М раствором 2-фенилэтиламина в DMSO. После промывания DMF смолу, несущую теперь остаток (Ph-CH2-CH2-)NH-CH2-C(О), прикрепляют к связывающему веществу и проводят реакцию с симметричным ангидридом Fmoc-Asn(Trt)-ОН в DCM/DMF. После снятия защиты Fmoc в соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Glu(OtBu)-ОН и Alloc-pAph-OH. Пептид расщепляют и с помощью воздействия в течение 1 ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 694,9, по расчетам 695,3.
Пример 16. Синтез Alloc-pAph-Glu-Thr(Bzl)-NH-CH2-CH2-CH(Ph)2
H-Thr(Bzl)-NH-CH2-CH2-CH(Ph)2·HCl
0,62 г (2 ммоль) Вос-Thr (Bzl)-ОН растворяют в 10 мл DCM, добавляют 2 ммоль триэтиламина, и раствор охлаждают до 0°С. При перемешивании медленно добавляют 2 ммоль изобутилхлороформата. Охлаждающую баню удаляют, раствор перемешивают в течение 15 мин и добавляют 2,5 ммоль 3,3-дифенилпропиламина в 2 мл DMF и перемешивают при комнатной температуре в течение 1 ч. Раствор выпаривают, растворяют в этилацетате и экстрагируют раствором 0,5 М KHSO4, насыщенным раствором NaHCО3 и соляным раствором, сушат с МgSО4 и выпаривают. Маслянистый продукт растворяют в 10 мл DCM и добавляют 10 мл 4М раствора хлористоводородной кислоты в диоксане. Через 10 мин растворы выпаривают, гидрохлорид продукта осаждают простым диэтиловым эфиром, фильтруют, промывают простым диэтиловым эфиром и сушат в вакууме для получения белого твердого вещества. МS-анализ: (М+Н)+: обнаружено 403,1, по расчетам 403,2.
Alloc-pAph-Glu-Thr(Bzl)-NH-CH2-CH2-CH(Ph)2
К 0,5 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют 4-гидроксиметилфеноксиуксусную кислоту (3 экв., активированную в течение 1,5 ч DIC/HOBt). К смоле посредством боковой цепи прикрепляют Fmoc-Glu(ОН)-O-аллил с использованием DIC/HOBt/NMI в DMF в течение ночи. Группу, защищающую аллильную группу, удаляют с помощью встряхивания смолы с Рd(РРh3)4 в DMF/AcOH/NMM (10/2/1) в течение 4 ч под аргоном. Лишенную защиты карбоксильную группу активируют раствором 0,5 ммоль ВОР, 0,5 ммоль HOBt, 1,5 ммоль DIEA и 0,5 ммоль Н-Thr(Bzl)-NH-CH2-СН2-СН (Ph)2·HCl в 1,5 мл DMF в течение 2 ч. После снятия защиты Fmoc Alloc-pAph-OH связывают в соответствии с общей процедурой, описанной в примере 1. Пептид расщепляют и с помощью воздействия в течение 1,5 ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 805,0, по расчетам 805,4.
Пример 17. Синтез Alloc-pAph-Glu-Dab-NH-CH2-CH2-Ph
К 0,2 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют 4-гидроксиметилфеноксиуксусную кислоту (2,5 экв., активированную в течение 4 ч DIC/HOBt). Гидроксильную группу замещают бромом с помощью обработки смолы СВr4 (5 экв.)/РРh3 (5 экв.) в DCM в течение 4 ч. Полученную в результате замещения бромом смолу в течение ночи обрабатывают 2М раствором фенилэтиламина в DCM. Fmoc-Dab(Воc)-ОН связывают со смолой с использованием TFFH/DIEA (ацилфторид, генерированный in situ). В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Glu(OtBu)-ОН и Alloc-pAph-OH. Пептид расщепляют и с помощью воздействия в течение 2 ч TFA/триизопропилсилана (99/1) снимают защиту. TFA выпаривают, пептид растворяют в H2O/ACN и лиофилизируют. Неочищенный материал очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (М+Н)+: обнаружено 624,2, по расчетам 624,3.
Пример 18. Синтез Alloc-pAph-Glu-NH-CH2-CH2-CH
К 0,2 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют 4-гидроксиметилфеноксиуксусную кислоту (3 экв., активированную в течение 1,5 ч DIC/HOBt). К смоле посредством боковой цепи прикрепляют Fmoc-Glu(ОН)-O-аллил с использованием DIC/HOBt/NMI в DMF в течение ночи. Группу, защищающую аллильную группу, удаляют с помощью встряхивания смолы с Рd(РРh3)4 в DMF/AcOH/NMM (10/2/1) в течение 4 ч под аргоном. Лишенную защиты карбоксильную группу активируют DIC (3 экв.)/HOBt (3 экв.) в течение 10 мин, и в смолу в течение 3 ч добавляют 2-цианоэтиламин (3 экв.) в DMF. После снятия защиты Fmoc Alloc-pAph-OH связывают в соответствии с общей процедурой, описанной в примере 1. Пептид расщепляют и путем воздействия в течение 2 ч TFA/триизопропилсилана (99/1) снимают защиту. TFA выпаривают, пептид растворяют в 2O/ACN и лиофилизируют. Неочищенный материал очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (М+Н)+: обнаружено 473,1, по расчетам 473,2.
Пример 19. Синтез Alloc-pAph-Glu-Asn-NH-CH2-Chx
К 0,1 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют амидный линкер Knorr. К линкеру посредством боковой цепи прикрепляют Fmoc-Asp(ОН)-O-аллил, и группу, защищающую аллиловую группу, удаляют, как описано в примере 18. Лишенную защиты карбоксильную группу активируют DIC (5 экв.)/ HOBt (5 экв.) и в течение 2,5 ч добавляют циклогексилметиламин (5 экв.) в DMF. После снятия защиты Fmoc Fmoc-Glu(OtBu)-ОН и Alloc-pAph-OH связывают в соответствии с общей процедурой, описанной в примере 1. Пептид расщепляют и путем воздействия в течение 2ч TFA/триизопропилсилана (99/1) снимают защиту. TFA выпаривают, пептид растворяют в H2O/ACN и лиофилизируют. Неочищенный материал очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 629,9, по расчетам 630,3.
Пример 20. Синтез Alloc-pAph-Glu-Asn-NH-CH2-CH2-Ph
Гидрохлорид 5-трет-бутил-1-метилового эфира 2-(S)-[2-(S)-Аллилоксикарбониламино-3-(4-карбамимидоил-фенил)-пропиониламино]-пентандиовой кислоты
К гидрохлориду 2-(S)-аллилоксикарбониламино-3-(4-карбамимидоил-фенил)-пропионовой кислоты (3,48 г, 10,6 ммоль) и гидрохлориду 5-трет-бутил-1-метилового эфира 2-(S)-аминопентандиовой кислоты (2,7 г, 10,6 ммоль) в 20 мл DMF при -15° С добавляют TOTU (3,83 г, 11,67 ммоль) и N-этилморфин (2,7 мл, 21,2 ммоль). Смесь перемешивают в течение 1ч, а затем дают согреться до комнатной температуры. После выпаривания в остаток добавляют этилацетат и органический слой экстрагируют водным раствором гидрокарбоната натрия, раствором гидросульфата калия и водой. Органический слой выпаривают. Выход: 2,8 г (50%). MS: m/z = 491,3 (М+Н)+.
5-трет-бутиловый эфир 2-(S)-[2-(S)-Аллилоксикарбониламино-3-(4-карбамимидоил-фенил)-пропиониламино]-пентандиовой кислоты
К гидрохлориду 5-трет-бутил-1-метилового эфира 2-(S)-[2-(S)-аллилоксикарбониламино-3-(4-карбамимидоил-фенил)-пропиониламино]-пентандиовой кислоты (3,06 г, 5,8 ммоль) в 100 мл воды и 30 THF добавляют гидрат гидроксида лития (0,49 г, 11,6 ммоль). Раствор перемешивают в течение 12 ч при комнатной температуре, выпаривают и лиофилизируют. Остаток очищают с помощью хроматографии на колонке Sephadex LH20, используя в качестве элюента н-бутанол /ледяную уксусную кислоту/ воду (17/1/2). Чистые фракции комбинируют. Растворитель выпаривают, остаток собирают в воде, и водный раствор лиофилизируют. Выход: 2,7 г (97%). MS: m/z=477,4 (М+Н)+.
Гидрохлорид 4-(S)-[2-(S)-Аллилоксикарбониламино-3-(4-карбамимидоилфенил)-пропиониламино]-4-(2-карбамоил-1(S)-(2-фенилэтилкарбамоил)-этилкарбамоил)-масляной кислоты (Alloc-pAph-Glu-Asn-NH-CH2-CH2-Ph)
К 5-трет-бутиловому эфиру 2-(S)-[2-(S)-аллилоксикарбониламино-3-(4-карбамимидоил-фенил)-пропиониламино]-пентандиовой кислоты (48 мг, 0,1 ммоль) и гидрохлориду 2-(S)-амино-N1-фенилэтил-сукцинамида (27 мг, 0,1 ммоль) в 5 мл DMF при 0°С добавляют HATU (39 мг, 0,1 ммоль) и коллидин (24,2 мг, 0,2 ммоль). Смесь перемешивают в течение 1 ч и дают согреться до комнатной температуры. После выпаривания остаток очищают с помощью хроматографии на колонке Sephadex LH20, используя в качестве элюента н-бутанол /ледяную уксусную кислоту/ воду (17/1/2). Чистые фракции комбинируют. Растворитель выпаривают, остаток собирают в воде и водный раствор лиофилизируют. Выход: 45 мг (66%). MS: m/z = 638,4 (М+Н)+.
Пример 21. Синтез Alloc-pAph-Glu-Asn-NH-(3-хлорбензила)
К 5-трет-бутиловому эфиру 2-(S)-[2-(S)-Аллилоксикарбониламино-3-(4-карбамимидоил-фенил)-пропиониламино]-пентандиовой кислоты (50 мг, 0,105 ммоль) и 2-(S)-амино-N1-(3-хлорбензил)сукцинамид трифторацетату (61 мг, 0,16 ммоль) в 5 мл DMF при 0°С добавляют TOTU (36 мг, 0,11 ммоль) и N-этилморфолин (57 мкл, 0,4 ммоль). Смесь перемешивают в течение 1ч, и затем дают согреться до комнатной температуры. После выпаривания остаток очищают с помощью хроматографии на колонке. Sephadex LH20, используя в качестве элюента н-бутанол /ледяную уксусную кислоту/ воду (17/1/2). Чистые фракции комбинируют. Растворитель выпаривают, остаток собирают в воде, и водный раствор лиофилизируют. Выход 4-(S)-[2-(S)-аллилоксикарбониламино-3-(4-карбамимидоил-фенил)-пропиониламино]-4-(2-карбамоил-1(S)-(3-хлорбензилкарбамоил)-этилкарбамоил)-масляной кислоты (Alloc-pAph-Glu-Asn-NH-(3-хлорбензила) или Alloc-pAph-Glu-Asn-3-хлорбензиламида): 28 мг (41%). MS: m/z = 658,3 (М+Н)+.
Другие иллюстративные соединения, полученные аналогично приведенным выше примерам, перечислены выше в табл.2.
Пример 22. Определение Ki ингибирования фактора VIIa
Ингибирующую активность (Ki) каждого соединения по отношению к активности фактора VIIa/тканевого фактора определяют с использованием хромогенного количественного анализа, по существу, в соответствии с описанной ранее процедурой (J.A. Ostrem, F.Al-Obeidi, P. Safar, A. Safarova, S.К. Stringer, M. Patek, M.T. Cross, J. Spoonamore, J.C. LoCascio, P. Kasireddy, D.S. Thorpe, N. Sepetov, M. Lebi, P. Wildgoose, P. Strop, Discovery of a novel, potent, and specific family of factor Xa inhibitors via combinatorial chemistry. Biochemistry 37(1998)1053-1059). Кинетические количественные анализы проводят при 25°С в половинных микротитровочных планшетах (Costar Corp., Cambridge, MA), используя кинетический планшет-ридер (Molecular Devices Spectromax 250). Типичный количественный анализ состоит из 25 мкл человеческого фактора VIIa и TF (5 нмоль и 10 нмоль относительно конечной концентрации), в комбинации с 40 мкл разведений ингибитора в 10% буфере DMSO/TBS-PEG (50 ммоль Tris, 15 ммоль NaCl, 5 ммоль СаСl2; 0,05% PEG 8k, pH 8,15). После преинкубационного периода длительностью 15 мин количественный анализ начинают добавлением 35 мкл хромогенного субстрата S-2288 (D-Ile-Pro-Arg-pNa, Pharmacia Hepar Inc., конечная концентрация 500 мкмоль). Константы видимого ингибирования рассчитывают по наклону кривых динамики в их линейной части, обычно с 1 по 5 мин после добавления субстрата. Фактическую Ki в последствии определяют для каждого соединения с помощью коррекции на концентрацию субстрата (S) и Km с использованием формулы Ki=Ki арр/(1+(S)/Km) (I.H. Segal, Enzyme Kinetics, pp 100-125(John Wiley & Sons, New York, 1975)).
Определение характеристик соединений примеров
Для определения времени удерживания ВЭЖХ (Rt) образец продукта анализировали с использованием системы ВЭЖХ Beckman (включающей систему подачи растворителя 126 (126 Solvent Deliver System), модуль программируемого детектора 166 (166 Programmable Programmable Detector Module), автоколлектор (дозатор, пробоотборник) 507е, регулируемый пунктом сбора и предварительной обработки данных с программным обеспечением Gold Nouveau) и колонку YMC ODS-AM 4,6250 мм при 230 нм и скорости потока 1 мл/мин бинарного градиента, образованного из воды (0,1% ТФУ) и ацетонитрила, начиная от 0% воды (0,1% ТФУ), повышаемого линейно на 2% ацетонитрила в минуту. Время удерживания, отмеченное как “DA”, определяли с помощью той же системы, за исключением того, что в качестве детектора использовали детектор с диодной матрицей Beckman Diode Array 168. Время удерживания, отмеченное как VY”, определяли при тех же условиях, за исключением того, что использовали аналитическую колонку Vydac 218TP54 (С18, 5 пм, 4,6 250 мм).
Масс-спектрометрию (MS) проводили методом ES+ с использованием прибора VG Platform (от Fisons Instruments).
Время удерживания Rt соединений по примерам описания представлено в таблице 3. 250 мм); The Separation Group; Hesperia CA, или колонку YMC ODS-A (20250 мм), YMC, Inc., Wilmington, NC). Скорость потока градиента или изократической смеси 0,1% TFA буфера и ACN (сорта HPLC) в качестве элюента поддерживают с использованием программируемого модуля растворителя 126 и программируемого модуля детектора 166, управляемого с помощью программного обеспечения “SYSTEM GOLD” устройства Beckman “SYSTEM GOLD” HPLC (Beckman, System Gold). Элюирование пептида контролируют с помощью УФ-детекции при 230 нм. После выявления с использованием MS-пика, соответствующего синтезируемому соединению, соединение собирают, лиофилизируют и проводят его биологическое испытание. MS выполняют с использованием прибора VG Piatforn (Fisons Instruments) в режиме ES+. Для ЯМР образцы обычно измеряют в DMSO-d6 (Aldrich) с использованием прибора Bruker Avance DPX300.
Пример 2. Синтез Alloc-pAph-OH
Аналогичная процедура применима для Alloc-pAph-OH.
Alloc-Phe(4-CN)-ОН
5,7 г (30 ммоль) H-Phe (4-CN)-ОН растворяют в 100 мл 1М NaOH с добавлением 2М NaOH до рН 10 с ледяным охлаждением. При энергичном перемешивании медленно добавляют аллилхлорформат (7,5 мл) (рН поддерживают на уровне 10 с помощью 2М NaOH). Реакционную смесь перемешивают при 0°С в течение 15 мин и при КТ в течение 30 мин, подкисляют НСl до рН 2, экстрагируют этилацетатом (3 раза), сушат с MgSO4 и выпаривают. Остаток подвергают перекристаллизации из этилацетата/гексана для получения белого твердого вещества. Выход: 7,0 г (85%).
Alloc-Phe[4-С(=S)-NH2]-ОН
2,74 г Alloc-Phe(4-CN)-ОН растворяют в смеси пиридина (50 мл) и триэтиламина (20 мл) и в течение 30 мин пропускают H2S. Реакционную смесь держат в течение ночи при КТ и выпаривают. Сушка в высоком вакууме дает 3,21 г сплошной пены неочищенного тиоамида, который непосредственно превращается в метилтиоимидат.
Alloc-Phe[4-С(=NH)-SCH3]-OH·HI
1 г Alloc-Phe[4-С(=S)-NH2]-ОН растворяют в ацетоне (50 мл) и добавляют метилиодид (5 мл). Реакционную смесь держат в течение ночи при КТ, летучие растворители выпаривают (быстро, максимум 35°С), остаток обрабатывают простым диэтиловым эфиром. Через 1 ч при 0°С эфир декантируют, продукт промывают простым диэтиловым эфиром и сушат в вакууме. Получают желтую плотную пену, которая непосредственно превращается в амидин.
Alloc-pAph-OH
Весь указанный выше Alloc-Phe(4-С(=NH)-SСН3)-OH·HI растворяют в 50 мл метанола с 300 мкл уксусной кислоты и добавляют 0,5 г ацетата аммония. Смесь нагревают в течение 3 ч при 55°С, выпаривают и добавляют 10 мл ацетона. Через 2 ч при 0°С твердый продукт фильтруют, промывают небольшим количеством холодного ацетона, небольшим количеством холодного метанола и простым диэтиловым эфиром и сушат в вакууме для получения желтоватого твердого вещества. Выход: 0,53г.
Пример 3. Синтез Alloc-pAph-Glu-Arg-Cha-NH2
К 1 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют амидный линкер Knorr. В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Cha-OH, Fmoc-Arg(Pmc)-ОН, Fmoc-Glu(OtBu)-ОН и Alloc-pAph. Пептид расщепляют и с помощью воздействия в течение 3 ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 729,1, по расчетам 729,4.
Пример 4. Синтез аллил-NH-C(O)-pAph-Glu-Arg-Cha-NH2
К 0,5 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют амидный линкер Knorr. В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Cha-OH, Fmoc-Arg(Pmc)-ОН, Fmoc-Glu(OtBu)-ОН и Fmoc-Phe(4-CN). После снятия защиты N-концевой Fmoc смолу в течение 2 ч обрабатывают раствором 1 ммоль аллилизоцианата в 3 мл DMF. Затем смолу промывают DMF и триэтиламином/пиридином (1/2) и в течение ночи обрабатывают насыщенным раствором H2S в пиридине/триэтиламине. Смолу промывают ацетоном и в течение 6 ч проводят реакцию тиоамидной смолы с метилиодидом (3 мл 10% раствора метилиодида в ацетоне). Метилтиоимидатную смолу промывают ацетоном, метанолом и обрабатывают раствором 0,2 г ацетата аммония, 100 мкл уксусной кислоты в 3 мл метанола при 55°С в течение 3 ч. Смолу промывают метанолом, DMF и DCM и пептид расщепляют и снимают защиту с помощью воздействия в течение 3 ч TFA/тиоанизола (95/5) и обрабатывают, как описано в примере 1. Неочищенный материал очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 728,3, по расчетам 728,4.
Пример 5. Синтез Alloc-pAph-Glu-Arff-Chg-NH2
К 1 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют амидный линкер Knorr. В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Chg-OH, Fmoc-Arg(Pmc)-ОН, Fmoc-Glu(OtBu)-ОН и Alloc-pAph. Пептид расщепляют и с помощью воздействия в течение 3 ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 715,8, по расчетам 715,4.
Пример 6. Синтез Alloc-D-pAph-Glu-Arg-Cha-NH2
К 1 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют амидный линкер Knorr. В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Cha-OH, Fmoc-Arg (Pmc)-ОН, Fmoc-Glu(OtBu)-ОН и Alloc-D-pAph-ОН (синтезированные в соответствии с той же процедурой, что и Alloc-pAph-ОН в примере 2). Пептид расщепляют и с помощью воздействия в течение 3 ч TFA/тиоанизола (95/5) удаляют защитную группу и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 729,2, по расчетам 729, 4.
Пример 7. Синтез Alloc-pAph-Glu-Phe(4-гуанидино)-Cha-NH2
К 0,25 г смолы TentaGel S NH2 (замещение 0,23 ммоль/г) прикрепляют амидный линкер Knorr. В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Cha-OH, Fmoc-Phe(4-NH-C(=NBoc)-NH-Boc)-OH, Fmoc-Glu(OtBu)-ОН и Alloc-pAph-OH. Пептид расщепляют и с помощью воздействия в течение 1 ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 777,1, по расчетам 777,4.
Пример 8. Синтез Alloc-pAph-Glu-Dap[-C(=NH)-NH2]-Cha-NH2
К 0,25 г смолы TentaGel S NH2 (замещение 0,23 ммоль/г) прикрепляют амидный линкер Knorr. В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Cha-OH, Fmoc-Dap[-С(=NBoc)-NH-Boc]-OH, Fmoc-Glu(OtBu)-ОН и Alloc-pAph-OH. Пептид расщепляют и с помощью воздействия в течение 1ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 729,1, по расчетам 729,4.
Пример 9. Синтез Alloc-pAph-Glu-Dap[-С(=NН-СН3]-Cha-NH2
К 0,25 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют амидный линкер Knorr. В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Cha-OH, Fmoc-Dap(Alloc)-ОН и Fmoc-Glu(OtBu)-ОН. С прикрепленной N-концевой группой, защищающей Fmoc, смолу промывают смесью DMF/NMM/AcOH (5/0,5/1) и при постоянном смешивании струёй аргона снимают защиту группы Alloc с помощью добавления 100 мг Рd(Р(Рh)3)4 в течение 3 ч. Смолу промывают DMF и обрабатывают раствором 150 мг 2-метилнафтилацеттиоимидата в 4 мл этанола/DMSO (3/1) в течение 1 ч. После промывания DMF снимают защиту группы Fmoc (1-5мин) и связывают N-концевую Alloc-pAph-OH. Пептид расщепляют и с помощью воздействия в течение 1 ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 700,1, по расчетам 700,4.
Пример 10. Синтез Alloc-pAph-Glu-Ala[3-C(=NH)-NH2]-Cha-NH2
К 0,25 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют амидный линкер Knorr. В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Cha-OH, Fmoc-Ala(3-CN)-ОН, Fmoc-Glu(OtBu)-OH и Alloc-Phe(4-CN)-ОН. Смесь пиридина и триэтиламина (2/1) насыщают Н2S (КТ, 15-30 мин), и этот раствор добавляют в смолу, предварительно промытую пиридином/триэтиламином (2/1). После хранения в течение ночи смолу промывают ацетоном и в течение ночи обрабатывают 20% метилиодидом в ацетоне. Затем смолу промывают ацетоном и метанолом. Связанный со смолой метилтиоимидат затем превращают в амидин с помощью нагревания (водяная баня, 55°С, 3 ч) смолы с раствором 10 экв. ацетата аммония в метаноле, содержащем 5% уксусную кислоту. После этого окончательного превращения смолу промывают метанолом, DMF, DCM. Пептид расщепляют и с помощью воздействия в течение 1ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 685,9, по расчетам 686,4.
Пример 11. Синтез Alloc-pAph-Glu-Asn-Cha-NH2
С 0,125 г смолы Rink (замещение 0,78 ммоль/г) после снятия защиты Fmoc в соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Cha-OH, Fmoc-Asn-OH, Fmoc-Glu(OtBu)-ОН и Alloc-pAph-ОН. Пептид расщепляют и с помощью воздействия в течение 1ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 686,9, по расчетам 687,3.
Пример 12. Синтез Alloc-pAph-Glu-Dab-Cha-NH2
К 0,25 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют амидный линкер Knorr. В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Cha-OH, Fmoc-Dab(Воc)-ОН, Fmoc-Glu(OtBu)-ОН и Alloc-pAph. Пептид расщепляют и с помощью воздействия в течение 1 ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 673,2, по расчетам 673,4.
Пример 13. Синтез Alloc-pAph-Glu-Ala[3-C(=NH)-NH2]-NH2
К 0,25 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют амидный линкер Knorr. В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Ala(3-CN)-ОН, Fmoc-Glu(OtBu)-ОН и Alloc-Phe(4-CN)-ОН. Смесь пиридина и триэтиламина (2/1) насыщают H2S (КТ, 15-30 мин), и этот раствор добавляют в смолу, предварительно промытую пиридином/триэтиламином (2/1). После хранения в течение ночи смолу промывают ацетоном и в течение ночи обрабатывают раствором 20% метилиодида в ацетоне. Затем смолу промывают ацетоном и метанолом. Связанный со смолой метилтиоимидат затем превращают в амидин с помощью нагревания (водяная баня, 55°С, 3 ч) смолы с раствором 10 экв. ацетата аммония в метаноле, содержащем 5% уксусную кислоту. После этого окончательного превращения смолу промывают метанолом, DMF, DCM. Пептид расщепляют и с помощью воздействия в течение 1 ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (М+Н)+: обнаружено 533,3, по расчетам 533,2.
Пример 14. Синтез Alloc-pAph-Glu-Gly-Cha-NH2
С 0,150 г смолы Rink (замещение 0,78 ммоль/г) после снятия защиты Fmoc в соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Cha-OH, Fmoc-Gly-OH, Fmoc-Glu(OtBu)-ОН и Alloc-pAph-OH. Пептид расщепляют и с помощью воздействия в течение 1 ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 630,1, по расчетам 630,3.
Пример 15. Синтез Alloc-pAph-Glu-Asn-(Ph-CH2-CH2-)Gly-NH2
Для N-замещенных глицинов используют методику Zuckermann et al. (J.Am. Chem. Soc.ll4 (1992) 10646, которая включена сюда в виде ссылки). С 0,1 г смолы Rink (замещение 0,78 ммоль/г) после снятия защиты Fmoc связывают бромуксусную кислоту посредством симметричного ангидрида в DCM/DMF. Через 10 мин смолу промывают DCM и еще раз повторяют связывание. После промывания DCM и DMF смолу в течение ночи обрабатывают 1М раствором 2-фенилэтиламина в DMSO. После промывания DMF смолу, несущую теперь остаток (Ph-CH2-CH2-)NH-CH2-C(О), прикрепляют к связывающему веществу и проводят реакцию с симметричным ангидридом Fmoc-Asn(Trt)-ОН в DCM/DMF. После снятия защиты Fmoc в соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Glu(OtBu)-ОН и Alloc-pAph-OH. Пептид расщепляют и с помощью воздействия в течение 1 ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 694,9, по расчетам 695,3.
Пример 16. Синтез Alloc-pAph-Glu-Thr(Bzl)-NH-CH2-CH2-CH(Ph)2
H-Thr(Bzl)-NH-CH2-CH2-CH(Ph)2·HCl
0,62 г (2 ммоль) Вос-Thr (Bzl)-ОН растворяют в 10 мл DCM, добавляют 2 ммоль триэтиламина, и раствор охлаждают до 0°С. При перемешивании медленно добавляют 2 ммоль изобутилхлороформата. Охлаждающую баню удаляют, раствор перемешивают в течение 15 мин и добавляют 2,5 ммоль 3,3-дифенилпропиламина в 2 мл DMF и перемешивают при комнатной температуре в течение 1 ч. Раствор выпаривают, растворяют в этилацетате и экстрагируют раствором 0,5 М KHSO4, насыщенным раствором NaHCО3 и соляным раствором, сушат с МgSО4 и выпаривают. Маслянистый продукт растворяют в 10 мл DCM и добавляют 10 мл 4М раствора хлористоводородной кислоты в диоксане. Через 10 мин растворы выпаривают, гидрохлорид продукта осаждают простым диэтиловым эфиром, фильтруют, промывают простым диэтиловым эфиром и сушат в вакууме для получения белого твердого вещества. МS-анализ: (М+Н)+: обнаружено 403,1, по расчетам 403,2.
Alloc-pAph-Glu-Thr(Bzl)-NH-CH2-CH2-CH(Ph)2
К 0,5 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют 4-гидроксиметилфеноксиуксусную кислоту (3 экв., активированную в течение 1,5 ч DIC/HOBt). К смоле посредством боковой цепи прикрепляют Fmoc-Glu(ОН)-O-аллил с использованием DIC/HOBt/NMI в DMF в течение ночи. Группу, защищающую аллильную группу, удаляют с помощью встряхивания смолы с Рd(РРh3)4 в DMF/AcOH/NMM (10/2/1) в течение 4 ч под аргоном. Лишенную защиты карбоксильную группу активируют раствором 0,5 ммоль ВОР, 0,5 ммоль HOBt, 1,5 ммоль DIEA и 0,5 ммоль Н-Thr(Bzl)-NH-CH2-СН2-СН (Ph)2·HCl в 1,5 мл DMF в течение 2 ч. После снятия защиты Fmoc Alloc-pAph-OH связывают в соответствии с общей процедурой, описанной в примере 1. Пептид расщепляют и с помощью воздействия в течение 1,5 ч TFA/тиоанизола (95/5) снимают защиту и обрабатывают, как описано в примере 1. Неочищенное соединение очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 805,0, по расчетам 805,4.
Пример 17. Синтез Alloc-pAph-Glu-Dab-NH-CH2-CH2-Ph
К 0,2 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют 4-гидроксиметилфеноксиуксусную кислоту (2,5 экв., активированную в течение 4 ч DIC/HOBt). Гидроксильную группу замещают бромом с помощью обработки смолы СВr4 (5 экв.)/РРh3 (5 экв.) в DCM в течение 4 ч. Полученную в результате замещения бромом смолу в течение ночи обрабатывают 2М раствором фенилэтиламина в DCM. Fmoc-Dab(Воc)-ОН связывают со смолой с использованием TFFH/DIEA (ацилфторид, генерированный in situ). В соответствии с общими процедурами, описанными в примере 1, связывают следующие защищенные аминокислоты: Fmoc-Glu(OtBu)-ОН и Alloc-pAph-OH. Пептид расщепляют и с помощью воздействия в течение 2 ч TFA/триизопропилсилана (99/1) снимают защиту. TFA выпаривают, пептид растворяют в H2O/ACN и лиофилизируют. Неочищенный материал очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (М+Н)+: обнаружено 624,2, по расчетам 624,3.
Пример 18. Синтез Alloc-pAph-Glu-NH-CH2-CH2-CH
К 0,2 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют 4-гидроксиметилфеноксиуксусную кислоту (3 экв., активированную в течение 1,5 ч DIC/HOBt). К смоле посредством боковой цепи прикрепляют Fmoc-Glu(ОН)-O-аллил с использованием DIC/HOBt/NMI в DMF в течение ночи. Группу, защищающую аллильную группу, удаляют с помощью встряхивания смолы с Рd(РРh3)4 в DMF/AcOH/NMM (10/2/1) в течение 4 ч под аргоном. Лишенную защиты карбоксильную группу активируют DIC (3 экв.)/HOBt (3 экв.) в течение 10 мин, и в смолу в течение 3 ч добавляют 2-цианоэтиламин (3 экв.) в DMF. После снятия защиты Fmoc Alloc-pAph-OH связывают в соответствии с общей процедурой, описанной в примере 1. Пептид расщепляют и путем воздействия в течение 2 ч TFA/триизопропилсилана (99/1) снимают защиту. TFA выпаривают, пептид растворяют в 2O/ACN и лиофилизируют. Неочищенный материал очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (М+Н)+: обнаружено 473,1, по расчетам 473,2.
Пример 19. Синтез Alloc-pAph-Glu-Asn-NH-CH2-Chx
К 0,1 г смолы TentaGel S NH2 (замещение 0,26 ммоль/г) прикрепляют амидный линкер Knorr. К линкеру посредством боковой цепи прикрепляют Fmoc-Asp(ОН)-O-аллил, и группу, защищающую аллиловую группу, удаляют, как описано в примере 18. Лишенную защиты карбоксильную группу активируют DIC (5 экв.)/ HOBt (5 экв.) и в течение 2,5 ч добавляют циклогексилметиламин (5 экв.) в DMF. После снятия защиты Fmoc Fmoc-Glu(OtBu)-ОН и Alloc-pAph-OH связывают в соответствии с общей процедурой, описанной в примере 1. Пептид расщепляют и путем воздействия в течение 2ч TFA/триизопропилсилана (99/1) снимают защиту. TFA выпаривают, пептид растворяют в H2O/ACN и лиофилизируют. Неочищенный материал очищают с использованием HPLC, как описано в примере 1, и характеризуют с помощью MS. (M+H)+: обнаружено 629,9, по расчетам 630,3.
Пример 20. Синтез Alloc-pAph-Glu-Asn-NH-CH2-CH2-Ph
Гидрохлорид 5-трет-бутил-1-метилового эфира 2-(S)-[2-(S)-Аллилоксикарбониламино-3-(4-карбамимидоил-фенил)-пропиониламино]-пентандиовой кислоты
К гидрохлориду 2-(S)-аллилоксикарбониламино-3-(4-карбамимидоил-фенил)-пропионовой кислоты (3,48 г, 10,6 ммоль) и гидрохлориду 5-трет-бутил-1-метилового эфира 2-(S)-аминопентандиовой кислоты (2,7 г, 10,6 ммоль) в 20 мл DMF при -15° С добавляют TOTU (3,83 г, 11,67 ммоль) и N-этилморфин (2,7 мл, 21,2 ммоль). Смесь перемешивают в течение 1ч, а затем дают согреться до комнатной температуры. После выпаривания в остаток добавляют этилацетат и органический слой экстрагируют водным раствором гидрокарбоната натрия, раствором гидросульфата калия и водой. Органический слой выпаривают. Выход: 2,8 г (50%). MS: m/z = 491,3 (М+Н)+.
5-трет-бутиловый эфир 2-(S)-[2-(S)-Аллилоксикарбониламино-3-(4-карбамимидоил-фенил)-пропиониламино]-пентандиовой кислоты
К гидрохлориду 5-трет-бутил-1-метилового эфира 2-(S)-[2-(S)-аллилоксикарбониламино-3-(4-карбамимидоил-фенил)-пропиониламино]-пентандиовой кислоты (3,06 г, 5,8 ммоль) в 100 мл воды и 30 THF добавляют гидрат гидроксида лития (0,49 г, 11,6 ммоль). Раствор перемешивают в течение 12 ч при комнатной температуре, выпаривают и лиофилизируют. Остаток очищают с помощью хроматографии на колонке Sephadex LH20, используя в качестве элюента н-бутанол /ледяную уксусную кислоту/ воду (17/1/2). Чистые фракции комбинируют. Растворитель выпаривают, остаток собирают в воде, и водный раствор лиофилизируют. Выход: 2,7 г (97%). MS: m/z=477,4 (М+Н)+.
Гидрохлорид 4-(S)-[2-(S)-Аллилоксикарбониламино-3-(4-карбамимидоилфенил)-пропиониламино]-4-(2-карбамоил-1(S)-(2-фенилэтилкарбамоил)-этилкарбамоил)-масляной кислоты (Alloc-pAph-Glu-Asn-NH-CH2-CH2-Ph)
К 5-трет-бутиловому эфиру 2-(S)-[2-(S)-аллилоксикарбониламино-3-(4-карбамимидоил-фенил)-пропиониламино]-пентандиовой кислоты (48 мг, 0,1 ммоль) и гидрохлориду 2-(S)-амино-N1-фенилэтил-сукцинамида (27 мг, 0,1 ммоль) в 5 мл DMF при 0°С добавляют HATU (39 мг, 0,1 ммоль) и коллидин (24,2 мг, 0,2 ммоль). Смесь перемешивают в течение 1 ч и дают согреться до комнатной температуры. После выпаривания остаток очищают с помощью хроматографии на колонке Sephadex LH20, используя в качестве элюента н-бутанол /ледяную уксусную кислоту/ воду (17/1/2). Чистые фракции комбинируют. Растворитель выпаривают, остаток собирают в воде и водный раствор лиофилизируют. Выход: 45 мг (66%). MS: m/z = 638,4 (М+Н)+.
Пример 21. Синтез Alloc-pAph-Glu-Asn-NH-(3-хлорбензила)
К 5-трет-бутиловому эфиру 2-(S)-[2-(S)-Аллилоксикарбониламино-3-(4-карбамимидоил-фенил)-пропиониламино]-пентандиовой кислоты (50 мг, 0,105 ммоль) и 2-(S)-амино-N1-(3-хлорбензил)сукцинамид трифторацетату (61 мг, 0,16 ммоль) в 5 мл DMF при 0°С добавляют TOTU (36 мг, 0,11 ммоль) и N-этилморфолин (57 мкл, 0,4 ммоль). Смесь перемешивают в течение 1ч, и затем дают согреться до комнатной температуры. После выпаривания остаток очищают с помощью хроматографии на колонке. Sephadex LH20, используя в качестве элюента н-бутанол /ледяную уксусную кислоту/ воду (17/1/2). Чистые фракции комбинируют. Растворитель выпаривают, остаток собирают в воде, и водный раствор лиофилизируют. Выход 4-(S)-[2-(S)-аллилоксикарбониламино-3-(4-карбамимидоил-фенил)-пропиониламино]-4-(2-карбамоил-1(S)-(3-хлорбензилкарбамоил)-этилкарбамоил)-масляной кислоты (Alloc-pAph-Glu-Asn-NH-(3-хлорбензила) или Alloc-pAph-Glu-Asn-3-хлорбензиламида): 28 мг (41%). MS: m/z = 658,3 (М+Н)+.
Другие иллюстративные соединения, полученные аналогично приведенным выше примерам, перечислены выше в табл.2.
Пример 22. Определение Ki ингибирования фактора VIIa
Ингибирующую активность (Ki) каждого соединения по отношению к активности фактора VIIa/тканевого фактора определяют с использованием хромогенного количественного анализа, по существу, в соответствии с описанной ранее процедурой (J.A. Ostrem, F.Al-Obeidi, P. Safar, A. Safarova, S.К. Stringer, M. Patek, M.T. Cross, J. Spoonamore, J.C. LoCascio, P. Kasireddy, D.S. Thorpe, N. Sepetov, M. Lebi, P. Wildgoose, P. Strop, Discovery of a novel, potent, and specific family of factor Xa inhibitors via combinatorial chemistry. Biochemistry 37(1998)1053-1059). Кинетические количественные анализы проводят при 25°С в половинных микротитровочных планшетах (Costar Corp., Cambridge, MA), используя кинетический планшет-ридер (Molecular Devices Spectromax 250). Типичный количественный анализ состоит из 25 мкл человеческого фактора VIIa и TF (5 нмоль и 10 нмоль относительно конечной концентрации), в комбинации с 40 мкл разведений ингибитора в 10% буфере DMSO/TBS-PEG (50 ммоль Tris, 15 ммоль NaCl, 5 ммоль СаСl2; 0,05% PEG 8k, pH 8,15). После преинкубационного периода длительностью 15 мин количественный анализ начинают добавлением 35 мкл хромогенного субстрата S-2288 (D-Ile-Pro-Arg-pNa, Pharmacia Hepar Inc., конечная концентрация 500 мкмоль). Константы видимого ингибирования рассчитывают по наклону кривых динамики в их линейной части, обычно с 1 по 5 мин после добавления субстрата. Фактическую Ki в последствии определяют для каждого соединения с помощью коррекции на концентрацию субстрата (S) и Km с использованием формулы Ki=Ki арр/(1+(S)/Km) (I.H. Segal, Enzyme Kinetics, pp 100-125(John Wiley & Sons, New York, 1975)).
Определение характеристик соединений примеров
Для определения времени удерживания ВЭЖХ (Rt) образец продукта анализировали с использованием системы ВЭЖХ Beckman (включающей систему подачи растворителя 126 (126 Solvent Deliver System), модуль программируемого детектора 166 (166 Programmable Programmable Detector Module), автоколлектор (дозатор, пробоотборник) 507е, регулируемый пунктом сбора и предварительной обработки данных с программным обеспечением Gold Nouveau) и колонку YMC ODS-AM 4,6250 мм при 230 нм и скорости потока 1 мл/мин бинарного градиента, образованного из воды (0,1% ТФУ) и ацетонитрила, начиная от 0% воды (0,1% ТФУ), повышаемого линейно на 2% ацетонитрила в минуту. Время удерживания, отмеченное как “DA”, определяли с помощью той же системы, за исключением того, что в качестве детектора использовали детектор с диодной матрицей Beckman Diode Array 168. Время удерживания, отмеченное как VY”, определяли при тех же условиях, за исключением того, что использовали аналитическую колонку Vydac 218TP54 (С18, 5 пм, 4,6 250 мм).
Масс-спектрометрию (MS) проводили методом ES+ с использованием прибора VG Platform (от Fisons Instruments).
Время удерживания Rt соединений по примерам описания представлено в таблице 3.
 Характеристики соединений Таблицы 2 представлены в Таблице 4.
Характеристики соединений Таблицы 2 представлены в Таблице 4.
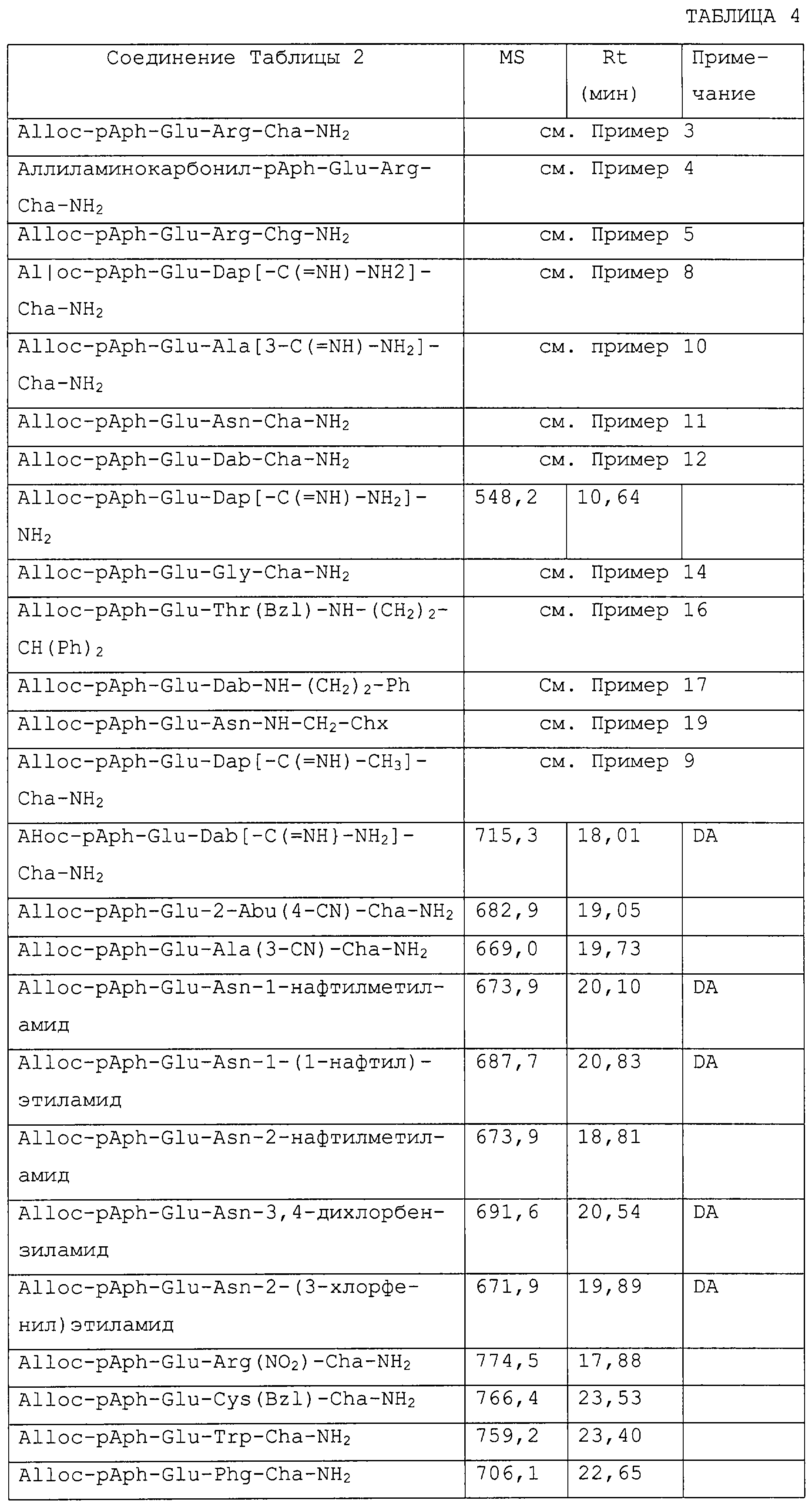
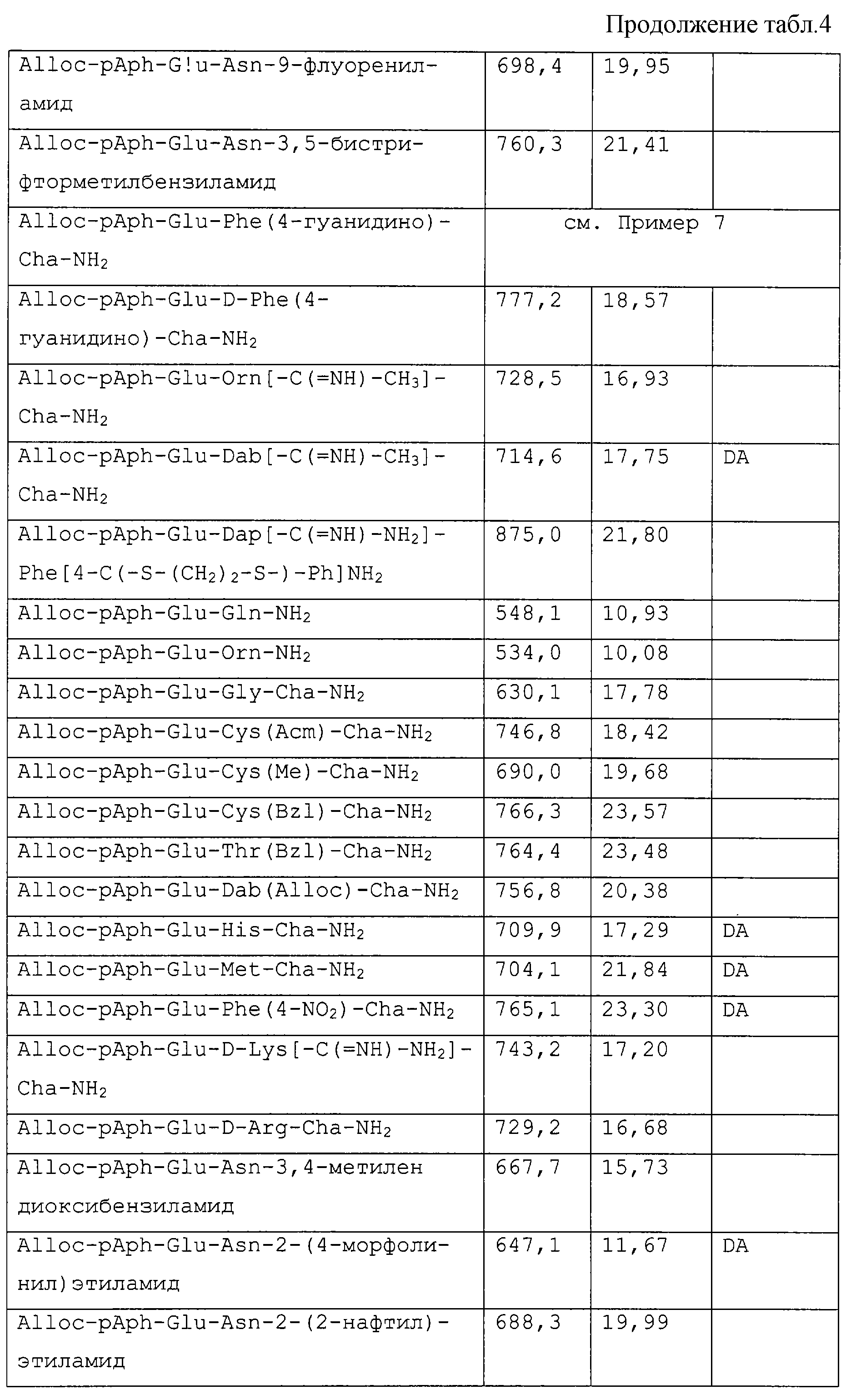

Формула изобретения 1. Соединение формулы I R1-A-B-D-En-R2 (I), в которой R1 представляет собой R12C(O), причем R12 выбран из группы, включающей алкенил, алкенилокси или алкениламино; А представляет собой группу А1-А2-АЗ, в которой A1 представляет собой NH; А2 представляет собой CHR93, в которой R93 представляет 4-амидинофенилметил; A3 представляет собой C(O); В представляет собой группу В1-В2-В3, в которой B1 представляет собой NH; B2 представляет собой CHR97, где R97 представляет этил, который замещен в положении 2 гидроксикарбонилом или алкилоксикарбонилом, В3 представляет собой C(O); D представляет группу D1-D2-D3, в которой D1 представляет собой NH; D2 представляет собой CR81R82, в которой R81 и R82 независимо выбраны из группы, состоящей из водорода и незамещенных или замещенных остатков алкила, арила, арилалкила, гетероарилалкила, D3 представляет собой С(О); En представляет собой (E1-Е2-ЕЗ)n, в которой n равно 0 или 1; E1 представляет NR70, где R70 представляет H, Е2 представляет CR71R72, где R71 и R72 независимо выбраны из группы, состоящей из водорода и незамещенных или замещенных остатков алкила, арила, арилалкила, гетероарилалкила, Е3 представляет собой C(O); R2 представляет собой NR21R22, где R21 и R22 независимо выбраны из водорода и незамещенных или замещенных остатков алкила, арила, арилалкила, гетероарилалкила и гетероциклоалкилалкила, причем алкил содержит от 1 до 13 атомов углерода, алкенил содержит от 2 до 13 атомов углерода, арил и гетероарил содержат от 5 до 13 кольцевых атомов углерода, где в остатке гетероарила один или несколько атомов углерода замещены гетероатомами, выбранными из группы, состоящей из N, О и S, гетероциклоалкил содержит от 3 до 8 кольцевых атомов углерода, из которых от одного до трех атомов углерода замещены гетероатомами, выбранными из группы, состоящей из N, О и S, в их любых стереоизомерных формах или их смесях в любом соотношении и их фармацевтически приемлемые соли. 2. Соединение по п.1, в котором остатки, представляющие R81 и R82, могут быть независимо замещены заместителями, выбранными из ряда, состоящего из амино, аминокарбонила, амидино, гуанидино, аминоалкила, гидрокси, меркапто, все из которых могут быть замещены защитной группой и ацетимидо, нитро и циано. 3. Соединение по п.1 и/или 2, в котором остатки, представляющие R71 и R72, могут быть независимо замещены заместителями, выбранными из ряда, состоящего из алкила, алкилокси, галогена, трифторметила, нитро, циано, алкилсульфонила, алкилкарбонила, фенилкарбонила и 2-фенил-1,3-дитиолан-2-ила. 4. Соединение по одному или нескольким из пп. 1-3, в котором остатки, представляющие R21 и R22, могут быть независимо замещены заместителями, выбранными из ряда, состоящего из галогена, трифторметила, гидрокси, нитро, циано, алкилокси, алкилендиокси, диалкиламино, алкилсульфонила, аминосульфонила, и 5. Соединение по одному или нескольким из пп.1-4, в котором насыщенные линейные или разветвленные алкильные цепи имеют от 1 до 6 атомов углерода, ненасыщенные линейные или разветвленные алкенильные цепи имеют от 2 до 6 атомов углерода и циклические алкильные группы имеют от 3 до 8 атомов углерода. 6. Соединение по одному или нескольким из пп.1-5, в котором R1 представляет собой R12C(О), где R12 – такой, как определено выше, D представляет собой NH-CHR82-C(О), где R82 – такой, как определено выше, En представляет собой (Е1-Е2-Е3)n, где n такой, как определено выше, E1 представляет собой NH, Е2 представляет собой CHR72, где R72 – такой, как определено выше, Е3 представляет собой C(O). 7. Соединение по одному или нескольким из пп.1-6, в котором n – такой, как определено выше, R2 представляет собой NHR22, где R22, такой, как определено выше. 8. Соединение по одному или нескольким из пп.1-7, в котором R1 представляет собой аллилоксикарбонил или аллиламинокарбонил. 9. Соединение по одному или нескольким из пп.1-8, в котором А представляет собой остаток (L)-4-амидинофенилаланина. 10. Соединение по одному или нескольким из пп.1-9, в котором В представляет собой остаток (L)-глутаминовой кислоты, или ее фармацевтически приемлемую соль, или сложный эфир. 11. Соединение по одному или нескольким из пп.1-10, в котором D представляет собой остаток, выбранный из ряда, состоящего из Arg, Dap, Dab, Orn, Lys, Dap[-C (=NH)-NH2], Dab[-C(=NH)-NH2], Lys[-C(=NH)-NH2], Lys[-C(=NH)-CH3], Orn[-C(=NH)-CH3], Dab[-C(=NH)-CH3], Dap[-C(=NH)-CH3], Dab(Alloc), Asn, Gln, Met, Ser, Thr, Ser(Bzl), Thr(Bzl), Cys(Me), Cys(Bzl), Cys(Acm), Arg(NO2), His, Trp, Phg, GIy, Ala, Val, Ile, Leu, Phe, Phe(4-NH-C(=NH)-NH2), Phe(4-NO2), 2-Abu, Ala(3-CN), Ala[3-C(=NH)-NH2], 2-Abu(4-CN) и 2-Abu[4-C(=NH)-NH2]. 12. Соединение по одному или нескольким из пп.1-11, в котором E представляет собой остаток, выбранный из ряда, состоящего из Cha, Chg и Phe[4-C(-S-CH2-CH2-S-)-Ph]. 13. Соединение по одному или нескольким из пп.1-12, в котором R22 представляет собой остаток, выбранный из ряда, состоящего из водорода, алкила, арила, арилалкила, гетероарилалкила и гетероциклоалкилалкила, причем все эти остатки могут быть замещены заместителями, выбранными из ряда, состоящего из галогена, гидрокси, алкилокси, алкилендиокси, нитро, циано, диалкиламино, алкилсульфонила, аминосульфонила и трифторметила, которые могут быть дополнительно замещены. 14. Соединение формулы I по одному или нескольким из пп.1-13, в котором R1 представляет собой аллилоксикарбонил или аллиламинокарбонил, А представляет собой остаток (L)-4-амидинофенилаланина, В представляет собой остаток (L)-глутаминовой кислоты, или фармацевтически приемлемую соль, или сложный эфир (L)-глутаминовой кислоты, D представляет собой остаток, выбранный из ряда, состоящего из Arg, Dap, Dab, Orn, Lys, Dap[-С(=NH)-NH2], Dab[-C(=NH)-NH2], Lys[-C(=NH)-NH2], Lys[-C(=NH)-CH3], Orn[-C(=NH)-CH3], Dab[-C(=NH)-CH3], Dap[-C(=NH)-CH3], Dab(Alloc), Asn, Gln, Met, Ser, Thr, Ser(Bzl), Thr(Bzl), Cys(Me), Cys(Bzl), Cys(Acm), Arg(NO2), His, Trp, Phg, GIy, Ala, Val, Ile, Leu, Phe, Phe(4-NО2), Phe(4-NH-C(=NH)-NH2), 2-Abu, Ala(3-CN), AIa[3-C(=NH)-NH2], 2-Abu(4-CN) и 2-Abu[4-C(=NH)-NH2], n = 0 или 1, E представляет собой остаток, выбранный из ряда, состоящего из Cha, Chg и Phe[4-C(-S-CH2-CH2-S-)-Ph], R2 представляет собой NHR22, R22 представляет собой водород или остаток, выбранный из ряда, состоящего из бензила, нафтилметила, пиридилметила, фенилэтила, нафтилэтила, пиридилэтила, фенилпропила, нафтилпропила, пиридилпропила, фторенила, дифенилметила, дифенилэтила и дифенилпропила, причем эти остатки не замещены или замещены заместителями, выбранными из ряда, состоящего из F, Cl, Br, гидрокси, метокси, метилендиокси, нитро, циано, диалкиламино, алкилсульфонила, аминосульфонила и трифторметила, в любой из его стереоизомерных форм или их смеси в любом соотношении или его фармацевтически приемлемая соль. 15. Соединение формулы I по одному или нескольким из пп.1-13, которое представляет собой Alloc-pAph-Glu-Arg-Cha-NH2, Аллиламинокарбонил-pAph-Glu-Arg-Cha-NH2, Alloc-pAph-Glu-Arg-Chg-NH2, Alloc-pAph-Glu-Dap[-C(=NH)-NH2]-Cha-NH2, Alloc-pAph-Glu-Ala[3-C(=NH)-NH2]-Cha-NH2, Alloc-pAph-Glu-Asn-Cha-NH2, Alloc-pAph-Glu-Dab-Cha-NH2, Alloc-pAph-Glu-Dap[-C(=NH)-NH2]-NH2, Alloc-pAph-Glu-Gly-Cha-NH2, Alloc-pAph-Glu-Thr(Bzl)-NH-(CH2)2-CH(Ph)2, Alloc-pAph-Glu-Dab-NH-(CH2)2-Ph, Alloc-pAph-Glu-Asn-NH-CH2-Chx, Alloc-pAph-Glu-Dap[-C(=NH)-CH3]-Cha-NH2, Alloc-pAph-Glu-Dab[-C(=NH)-NH2]-Cha-NH2, Alloc-pAph-Glu-2-Abu(4-CN)-Cha-NH2, Alloc-pAph-Glu-Ala(3-CN)-Cha-NH2, Alloc-pAph-Glu-Asn-1-нафтилметиламид, Alloc-pAph-Glu-Asn-1-(1-нафтил)этиламид, Alloc-pAph-Glu-Asn-2-нафтилметиламид, Alloc-pAph-Glu-Asn-3,4-дихлорбензиламид, Alloc-pAph-Glu-Asn-2-(3-хлорфенил)этиламид, Alloc-pAph-Glu-Arg(NO2)-Cha-NH2, Alloc-pAph-Glu-Cys(Bzl)-Cha-NH2, Alloc-pAph-Glu-Trp-Cha-NH2, Alloc-pAph-Glu-Phg-Cha-NH2, Alloc-pAph-Glu-Asn-9-флуорениламид, Alloc-pAph-Glu-Asn-3,5-бистрифторметилбензиламид, Alloc-pAph-Glu-Dap[-C (=NH)-NH2]-Phe[4-C(-S-(CH2)2-S-)-Ph]-NH2, Alloc-pAph-Glu-Cys(Bzl)-Cha-NH2, Alloc-pAph-Glu-Thr(Bzl)-Cha-NH2, Alloc-pAph-Glu-Phe(4-NO2)-Cha-NH2, Alloc-pAph-Glu-Asn-3,4-метилендиоксибензиламид, Alloc-pAph-Glu-Asn-2-(2-нафтил)этиламид, Alloc-pAph-Glu-Asn-2-(1-нафтил)этиламид, Alloc-pAph-Glu-Asn-2-(2-пиридил)этиламид, Alloc-pAph-Glu-Asn-2,2-дифенилэтиламид, Alloc-pAph-Glu-Asn-2,4-дифторбензиламид или Alloc-pAph-Glu-Asn-4-диметиламинобензиламид или их фармацевтически приемлемая соль, амид или сложный эфир. 16. Способ получения соединения по одному или нескольким из пп.1-15, который включает в себя связывание защищенных аминокислот с помощью методов традиционной медицинской химии и снятие защиты из целевой молекулы с помощью стандартных процедур, известных в данной области. 17. Фармацевтическая композиция, обладающая способностью оказывать антитромботический эффект посредством обратимого ингибирования активированного фактора VIIa (FVIIa) свертывания крови, содержащая эффективное количество соединения по одному из пп.1-15 или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель. 18. Соединение по одному или нескольким из пп.1-15 или его фармацевтически приемлемая соль для применения в качестве ингибитора фактора VIIa. 19. Соединение по одному или нескольким из пп. 1-15 или его фармацевтически приемлемая соль для применения при ингибировании или уменьшении свертывания крови, воспалительной реакции, тромбоэмболических заболеваний или рестеноза сосудов. PD4A – Изменение наименования обладателя патента Российской Федерации на изобретение
(73) Новое наименование патентообладателя:
Извещение опубликовано: 27.12.2005 БИ: 36/2005
|
||||||||||||||||||||||||||