Патент на изобретение №2215288
|
||||||||||||||||||||||||||
(54) СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ФЛАВОНОИДОВ МЕТОДОМ ДИФФЕРЕНЦИАЛЬНОЙ ВОЛЬТАМПЕРОМЕТРИИ
(57) Реферат: Предложенный способ может быть применен при анализе следовых количеств флавоноидов как в чистых растворах, так и для анализа сложных по составу растительных экстрактов, лекарственных форм, фармакологических препаратов. Техническим результатом данного изобретения является повышение чувствительности определения флавоноидов, экспрессности и селективности метода дифференциальной вольтамперометрии. Сущность: флавоноиды переводят в раствор путем растворения навески пробы в 70%-ном этиловом спирте при нагревании. Вольтамперометрическое определение ведут на стеклоуглеродном электроде по пикам восстановления при потенциалах от +0,41 до -0,63 В или по пикам окисления при потециалах от -0,36 до +0,46 В, на фонах 0,2 н. Na2HPO4 или 0,2 н. C6H14O7N2, регистрацию пиков проводят при линейной скорости развертки потенциала 20-30 мВ/с в дифференциальном режиме съемки вольтамперограммы. Концентрации флавоноидов (кверцетина, диквертина, менадиона, байкалина) определяли по высоте анодных или по высоте катодных пиков. 4 з.п.ф-лы, 2 табл. Изобретение относится к области аналитической химии и фармакологии, в частности к инверсионному вольтамперометрическому способу определения флавоноидов кверцетина, менадиона, байкалина, диквертина, структурные формулы которых приведены в таблице 1. Флавоноиды в большем или меньшем количестве содержатся почти во всех высших растениях, реже встречаются у микроорганизмов и насекомых. Флавоноиды имеют широкий спектр действия на организм: высокая Р-витаминная активность, нормализуют проницаемость капилляров, укрепляют и делают более эластичной сосудистую стенку. Обладают диуретическим, гипоазотемическим, гипотензивным, гипогликемическим, эстрогенным, спазмолитическим, желчегонным действием [Блинова К.Ф., Яковлева Г.П., Ботанико-фармакологический словарь. – М.: Высшая школа, 1990]. Некоторые из них являются эффективными антиоксидантами, например кверцетин [Запрометов М.Н., Основы биохимии фенольных соединений. – М.: Высшая школа, 1974]. Исследования последних лет показали, что лекарственные препараты растительного происхождения, содержащие соединения полифенольной структуры, уменьшают ксенобиотическую нагрузку на организм и обладают высокой цитопротекторной активностью, в частности при патологиях, сопровождающихся нарушением функции митохондрий, развитием перекисного окисления липидов. Согласно временной фармакопейной статье 42-1399-80, качество жидких экстрактов флавоноидсодержащих растений оценивают по содержанию в нем сухих веществ [Новые лекарственные препараты из растений Сибири и Дальнего Востока, т. – 2. Тезисы всесоюзной конференции, Томск, 1989]. Очевидно, что только по этому показателю нельзя объективно судить о терапевтической ценности препарата. В связи с этим определение микроколичеств флавоноидов очень важно для оценки качества сырья и биологически активных добавок, идентификации действующих веществ в лекарственных формах. Поэтому аналитическая практика предъявляет повышенные требования к совершенствованию методов определения флавоноидов. Старейшими методами обнаружения флавоноидов являются титрометрические методы в среде неводных растворителей: ацетона, диметилформамида, диметилсульфоксида, с использованием в качестве титрантов гидроксиды тетраэтиламмония или натрия. Для проведения количественной оценки этими методами необходимо предварительное разделение компонентов, что увеличивает длительность методик. Чувствительность определения невелика и составляет 10-2-10-3 моль/л [Георгиевский В. П., Физико-химические методы анализа биологически активных веществ растительного происхождения. – Ашхабад, “Ылым”, 1976].  10-4-110-2 мг/мл [Георгиевский В.П., 1976, Лакович Дж. , Основы флуорисцентной спектроскопии. М.: Мир, 1986, с. 496]. Поэтому такие методы не могут быть использованы для определения исследуемых веществ в многокомпонентных смесях.
Широкое распространение получили при определении флавоноидов различные варианты хроматографии. Методы тонкослойной хроматографии связаны со значительными трудностями при количественной оценке хроматограмм. В последнее время наибольшее применение получил метод высокоэффективной жидкостной хроматографии (ВЭЖХ). Для увеличения чувствительности определения физико-химических методов используют ультрафиолетовое, флуориметрическое и электрохимическое детектирование. Несмотря на высокую чувствительность ВЭЖХ (10-5-10-5
Электрохимическому исследованию флавоноидов посвящены единичные работы. Для определения флавоноидов использовали полярографические методы с ртутным капельным индикаторным электродом. В работе [Георгиевский В.П.,1976] показана способность менадиона и кверцетина восстанавливаться на ртутном капельном электроде. Сделана попытка их количественного определения в растительном сырье. Однако величины потенциалов полуволн (E1/2) различных флавоноидов мало отличались, поэтому полярографическим методом определяли их общее содержание. При этом требовалась тщательная очистка флавоноидов, так как определению мешали сопутствующие вещества. Наиболее близким является метод определения менадиона (прототип) [Мискиджьян С.П., Кравченюк Л.П. Полярография лекарственных препаратов, Киев: Вища школа, 1976, с. 229]. Сущность методики состоит в том, что растворенный менадион предварительно гидролизуют и полученный 2-метил 1,4 нафтохинон окисляют сульфатом Се (IV) или бромом в 20%-ном спиртовом растворе буфера Бриттона – Робинсона (рН 7). Полярографическую волну определяемого вещества регистрируют при потенциале E1/2=-0,28 В. В качестве фона использовали 0,3 моль/л раствор LiCl в безводном метаноле. Классическая постояннотоковая полярография обеспечивает определение менадиона более 510-4 моль/л при относительной области определяемых содержаний от 510-4 до 510-3 моль/л [Мискиджьян С.П., Кравченюк Л.П., 1976]. Этот метод позволяет определять флавоноиды в фармакологических препаратах, но не позволяет определять их в растительных экстрактах. Одним из ограничивающих факторов применения полярографии в биохимических лабораториях является использование больших количеств металлической ртути в качестве электродов в электролитической ячейке.
Условия полярографического определения диквертина и байкалина в литературе практически отсутствуют.
Электороаналитические методы и в первую очередь такие его высокочувствительные варианты, как дифференциальная вольтамперометрия (ДВА), пережили в последние годы своеобразное “возрождение”, и не только по причине невысоких затрат на их реализацию, но главным образом потому, что они отвечают современным требованиям к контролю качества разнообразных и сложных по составу систем. При соблюдении требований ультрамикроанализа вполне реально определение 10-8-10-9 мг/мл. Однако большинство опубликованных работ по анализу электрохимическими методами посвящено определению металлов. Идентификация природных органических веществ, флавоноидов и их метаболитов в лекарственных препаратах, в растительных экстрактах и в пищевых биологически активных добавках становится с каждым годом все более актуальной проблемой.
Информация о применении метода вольтамперометрии (ВА) для определения флавоноидов в литературе не описана. Поэтому разработка экспрессных и высокочувствительных методов определения флавоноидов продолжает представлять интерес.
Задачей заявляемого изобретения является повышение чувствительности, экспрессности и селективности определения флавоноидов методом дифференциальной вольтамперометрии.
Поставленная задача достигается тем, что флавоноиды переводят в раствор путем растворения навески пробы в 70%-ном этиловом спирте при нагревании с последующей регистрацией катодных или анодных пиков. Новым в способе является то, что вольтамперометрическое определение ведут на стеклоуглеродном электроде по пикам восстановления при потенциалах от +0,41 до -0,63 В (относительно насыщенного хлоридсеребрянного электрода, нас. х.с.) или по пикам окисления при потециалах от -0,36 до +0,46 В, на фонах 0,2 н. Na2HPO4 или 0,2 н. аммония лимоннокислого двузамещенного (С6Н14O7N2), регистрацию пиков проводят при линейной скорости развертки потенциала 20-30 мВ/с в дифференциальном режиме съемки вольтамперограммы и концентрации флавоноидов: кверцетина, диквертина, менадиона, байкалина, определяли по высоте пиков восстановления при потенциалах +0,37 В; (+0,14 10-4-110-2 мг/мл [Георгиевский В.П., 1976, Лакович Дж. , Основы флуорисцентной спектроскопии. М.: Мир, 1986, с. 496]. Поэтому такие методы не могут быть использованы для определения исследуемых веществ в многокомпонентных смесях.
Широкое распространение получили при определении флавоноидов различные варианты хроматографии. Методы тонкослойной хроматографии связаны со значительными трудностями при количественной оценке хроматограмм. В последнее время наибольшее применение получил метод высокоэффективной жидкостной хроматографии (ВЭЖХ). Для увеличения чувствительности определения физико-химических методов используют ультрафиолетовое, флуориметрическое и электрохимическое детектирование. Несмотря на высокую чувствительность ВЭЖХ (10-5-10-5
Электрохимическому исследованию флавоноидов посвящены единичные работы. Для определения флавоноидов использовали полярографические методы с ртутным капельным индикаторным электродом. В работе [Георгиевский В.П.,1976] показана способность менадиона и кверцетина восстанавливаться на ртутном капельном электроде. Сделана попытка их количественного определения в растительном сырье. Однако величины потенциалов полуволн (E1/2) различных флавоноидов мало отличались, поэтому полярографическим методом определяли их общее содержание. При этом требовалась тщательная очистка флавоноидов, так как определению мешали сопутствующие вещества. Наиболее близким является метод определения менадиона (прототип) [Мискиджьян С.П., Кравченюк Л.П. Полярография лекарственных препаратов, Киев: Вища школа, 1976, с. 229]. Сущность методики состоит в том, что растворенный менадион предварительно гидролизуют и полученный 2-метил 1,4 нафтохинон окисляют сульфатом Се (IV) или бромом в 20%-ном спиртовом растворе буфера Бриттона – Робинсона (рН 7). Полярографическую волну определяемого вещества регистрируют при потенциале E1/2=-0,28 В. В качестве фона использовали 0,3 моль/л раствор LiCl в безводном метаноле. Классическая постояннотоковая полярография обеспечивает определение менадиона более 510-4 моль/л при относительной области определяемых содержаний от 510-4 до 510-3 моль/л [Мискиджьян С.П., Кравченюк Л.П., 1976]. Этот метод позволяет определять флавоноиды в фармакологических препаратах, но не позволяет определять их в растительных экстрактах. Одним из ограничивающих факторов применения полярографии в биохимических лабораториях является использование больших количеств металлической ртути в качестве электродов в электролитической ячейке.
Условия полярографического определения диквертина и байкалина в литературе практически отсутствуют.
Электороаналитические методы и в первую очередь такие его высокочувствительные варианты, как дифференциальная вольтамперометрия (ДВА), пережили в последние годы своеобразное “возрождение”, и не только по причине невысоких затрат на их реализацию, но главным образом потому, что они отвечают современным требованиям к контролю качества разнообразных и сложных по составу систем. При соблюдении требований ультрамикроанализа вполне реально определение 10-8-10-9 мг/мл. Однако большинство опубликованных работ по анализу электрохимическими методами посвящено определению металлов. Идентификация природных органических веществ, флавоноидов и их метаболитов в лекарственных препаратах, в растительных экстрактах и в пищевых биологически активных добавках становится с каждым годом все более актуальной проблемой.
Информация о применении метода вольтамперометрии (ВА) для определения флавоноидов в литературе не описана. Поэтому разработка экспрессных и высокочувствительных методов определения флавоноидов продолжает представлять интерес.
Задачей заявляемого изобретения является повышение чувствительности, экспрессности и селективности определения флавоноидов методом дифференциальной вольтамперометрии.
Поставленная задача достигается тем, что флавоноиды переводят в раствор путем растворения навески пробы в 70%-ном этиловом спирте при нагревании с последующей регистрацией катодных или анодных пиков. Новым в способе является то, что вольтамперометрическое определение ведут на стеклоуглеродном электроде по пикам восстановления при потенциалах от +0,41 до -0,63 В (относительно насыщенного хлоридсеребрянного электрода, нас. х.с.) или по пикам окисления при потециалах от -0,36 до +0,46 В, на фонах 0,2 н. Na2HPO4 или 0,2 н. аммония лимоннокислого двузамещенного (С6Н14O7N2), регистрацию пиков проводят при линейной скорости развертки потенциала 20-30 мВ/с в дифференциальном режиме съемки вольтамперограммы и концентрации флавоноидов: кверцетина, диквертина, менадиона, байкалина, определяли по высоте пиков восстановления при потенциалах +0,37 В; (+0,14 0,02) В; (-0,220,02) В; -(0,600,03) В соответственно на фоне 0,2 н. Na2HPO4 или по высоте пиков восстановления при потенциалах (+0,330,01) В; (-0,400,02) В; (-0,150,02) В; -0,60 В; соответственно на фоне 0,2 н. C6H14O7N2 или по высоте пиков окисления при потенциалах (+0,390,02) В; (+0,400,02) В; -0,34 В; (+0,200,02)В соответственно на фоне 0,2 н Na2HPO4 или по высоте пиков окисления при потенциалах (+0,360,02) В; +0,45 В; (-0,150,01) В; (+0,350,01) В соответственно на фоне 0,2 н. С6Н14O7N2.
В прототипе описано использование в качестве фона 0,3 моль/л раствор LiCl в безводном метаноле. Определение менадиона в этих условиях затруднено из-за низкой чувствительности определения, связанной, по-видимому, с плохой растворимостью флавоноидов в метаноле. Использование высокотоксичного метанола ограничивает применение методики в серийных анализах. Предлагаемые в заявленном изобретении фоны 0,2 н. Na2HPО4, pH 8,6 и 0,2 н. C6H14О7N2, pH 7 позволяют определять флавоноиды на уровне нанограммовых содержаний с хорошей воспроизводимостью. Относительное стандартное отклонение (Sr) для концентрации флавоноидов 110-8 мг/мл не превышает 0,2. Все фоны подобраны экспериментально. Фон 0,2 н. Na2HPО4 использовался в вольтамперометрических исследованиях, однако абсолютной новизной является использование подобранных значений pH раствора Na2HPO4 и использование в качестве фона 0,2 н. аммония лимоннокислого двузамещенного, от чего зависит количественное определение флавоноидов. Кроме того, использование цитратного фона приводит к лучшей воспроизводимости результатов при анализе биологических и природных объектов, так как раствор аммония лимоннокислого двузамещенного является хорошим осадителем растворимых белковых примесей в таких пробах. Оптимальный диапазон pH фоновых растворов электролитов 7-9 определяется хорошей воспроизводимостью и фиксированием практически одного пика при регистрации вольтамперограмм. Более низкие значения pH (pH<7) и более высокие (pH>9) нежелательны, так как изменяется форма нахождения флавоноида в растворе, что связано со смещением протеолитического равновесия органического вещества в растворе. Это приводит к регистрации дополнительных предпиков, увеличению остаточного тока и снижает чувствительность, экспрессность и селективность определения. На фонах 0,2 н. Na2HPO4 и 0,2 н/ C6H14O7N2 впервые получены пики окисления и восстановления на стеклоуглеродном электроде (в прототипе исследовались только пики восстановления с использованием ртутного капельного электрода). Что позволило проводить определение флавоноидов на уровне 10-8-10-6 мг/мл методом ДВА.
Другим отличительным признаком является использование в качестве индикаторного электрода стеклоуглеродных электродов. Впервые установлена возможность электроокисления и электровосстановления флавоноидов с использованием угольных электродов. В качестве индикаторных применяли три типа углеродных электродов – графитовый, пропитанный полиэтиленом с парафином в вакууме или эпоксидной смолой, пирографитовый (ПГ) и стеклоуглеродный (СУ). Использование таких электродов обусловлено высокой химической и электрохимической устойчивостью графита, широкой областью рабочих потенциалов как в водной, так и в неводных средах, а также простотой механического обновления поверхности [Брайнина Х.З., Нейман Е.Я., Слепушкин В.В., Инверсионные электроаналитические методы. – М.: Химия, 1998, c. 239]. Такие электроды являются нетоксичными.
Величина потенциалов окисления и восстановления флавоноидов определяется строением, структурой и степенью адсорбируемости на гексагонах графита, имеющих 0,02) В; (-0,220,02) В; -(0,600,03) В соответственно на фоне 0,2 н. Na2HPO4 или по высоте пиков восстановления при потенциалах (+0,330,01) В; (-0,400,02) В; (-0,150,02) В; -0,60 В; соответственно на фоне 0,2 н. C6H14O7N2 или по высоте пиков окисления при потенциалах (+0,390,02) В; (+0,400,02) В; -0,34 В; (+0,200,02)В соответственно на фоне 0,2 н Na2HPO4 или по высоте пиков окисления при потенциалах (+0,360,02) В; +0,45 В; (-0,150,01) В; (+0,350,01) В соответственно на фоне 0,2 н. С6Н14O7N2.
В прототипе описано использование в качестве фона 0,3 моль/л раствор LiCl в безводном метаноле. Определение менадиона в этих условиях затруднено из-за низкой чувствительности определения, связанной, по-видимому, с плохой растворимостью флавоноидов в метаноле. Использование высокотоксичного метанола ограничивает применение методики в серийных анализах. Предлагаемые в заявленном изобретении фоны 0,2 н. Na2HPО4, pH 8,6 и 0,2 н. C6H14О7N2, pH 7 позволяют определять флавоноиды на уровне нанограммовых содержаний с хорошей воспроизводимостью. Относительное стандартное отклонение (Sr) для концентрации флавоноидов 110-8 мг/мл не превышает 0,2. Все фоны подобраны экспериментально. Фон 0,2 н. Na2HPО4 использовался в вольтамперометрических исследованиях, однако абсолютной новизной является использование подобранных значений pH раствора Na2HPO4 и использование в качестве фона 0,2 н. аммония лимоннокислого двузамещенного, от чего зависит количественное определение флавоноидов. Кроме того, использование цитратного фона приводит к лучшей воспроизводимости результатов при анализе биологических и природных объектов, так как раствор аммония лимоннокислого двузамещенного является хорошим осадителем растворимых белковых примесей в таких пробах. Оптимальный диапазон pH фоновых растворов электролитов 7-9 определяется хорошей воспроизводимостью и фиксированием практически одного пика при регистрации вольтамперограмм. Более низкие значения pH (pH<7) и более высокие (pH>9) нежелательны, так как изменяется форма нахождения флавоноида в растворе, что связано со смещением протеолитического равновесия органического вещества в растворе. Это приводит к регистрации дополнительных предпиков, увеличению остаточного тока и снижает чувствительность, экспрессность и селективность определения. На фонах 0,2 н. Na2HPO4 и 0,2 н/ C6H14O7N2 впервые получены пики окисления и восстановления на стеклоуглеродном электроде (в прототипе исследовались только пики восстановления с использованием ртутного капельного электрода). Что позволило проводить определение флавоноидов на уровне 10-8-10-6 мг/мл методом ДВА.
Другим отличительным признаком является использование в качестве индикаторного электрода стеклоуглеродных электродов. Впервые установлена возможность электроокисления и электровосстановления флавоноидов с использованием угольных электродов. В качестве индикаторных применяли три типа углеродных электродов – графитовый, пропитанный полиэтиленом с парафином в вакууме или эпоксидной смолой, пирографитовый (ПГ) и стеклоуглеродный (СУ). Использование таких электродов обусловлено высокой химической и электрохимической устойчивостью графита, широкой областью рабочих потенциалов как в водной, так и в неводных средах, а также простотой механического обновления поверхности [Брайнина Х.З., Нейман Е.Я., Слепушкин В.В., Инверсионные электроаналитические методы. – М.: Химия, 1998, c. 239]. Такие электроды являются нетоксичными.
Величина потенциалов окисления и восстановления флавоноидов определяется строением, структурой и степенью адсорбируемости на гексагонах графита, имеющих  -зонную структуру. Структурное подобие материла электрода и адсорбция плоскостью молекулы (стереоспецифическая адсорбция), по- видимому, благоприятствует переходу электронов при меньшем значении потенциала и способствует обратимому окислению и восстановлению флавоноидов на графите, чем на СУ и ПГ. Однако из-за большого остаточного тока графитовый электрод оказался менее удобным в работе, чем СУ, особенно при количественном определении флавоноидов в растительных экстрактах.
Из табл. 2 видно, что легче всего восстанавливается кверцетин, а окисляется менадион.
Важным для определения флавоноидов методом ДВА является выбор скорости развертки потенциала. Оптимальной является скорость 20-30 мВ/с. Увеличение скорости более 20 мВ/с увеличивает чувствительность, но при этом растет остаточный ток и уменьшается разрешающая способность способа. При скорости менее 20 мВ/с снижается величина анодного и катодного токов и понижается чувствительность определения флавоноидов. Значение используемой скорости в прототипе не приведено.
Таким образом, подобранные условия впервые позволили количественно определять флавоноиды на уровне 3,510-8 мг/мл без предварительного выделения их из проб. Диапазон определяемых содержаний 3,510-8-110-6 мг/мл. Определению не мешают тысячекратные избытки лимонной, щавелевой, аскорбиновой кислот.
Пример 1. Определение менадиона на уровне 10-8-10-6 мг/мл по пикам восстановления на фоне 0,2 н. Na2HPO4 -зонную структуру. Структурное подобие материла электрода и адсорбция плоскостью молекулы (стереоспецифическая адсорбция), по- видимому, благоприятствует переходу электронов при меньшем значении потенциала и способствует обратимому окислению и восстановлению флавоноидов на графите, чем на СУ и ПГ. Однако из-за большого остаточного тока графитовый электрод оказался менее удобным в работе, чем СУ, особенно при количественном определении флавоноидов в растительных экстрактах.
Из табл. 2 видно, что легче всего восстанавливается кверцетин, а окисляется менадион.
Важным для определения флавоноидов методом ДВА является выбор скорости развертки потенциала. Оптимальной является скорость 20-30 мВ/с. Увеличение скорости более 20 мВ/с увеличивает чувствительность, но при этом растет остаточный ток и уменьшается разрешающая способность способа. При скорости менее 20 мВ/с снижается величина анодного и катодного токов и понижается чувствительность определения флавоноидов. Значение используемой скорости в прототипе не приведено.
Таким образом, подобранные условия впервые позволили количественно определять флавоноиды на уровне 3,510-8 мг/мл без предварительного выделения их из проб. Диапазон определяемых содержаний 3,510-8-110-6 мг/мл. Определению не мешают тысячекратные избытки лимонной, щавелевой, аскорбиновой кислот.
Пример 1. Определение менадиона на уровне 10-8-10-6 мг/мл по пикам восстановления на фоне 0,2 н. Na2HPO4В кварцевый стаканчик емкостью 20 мл наливают 10 мл 0,2 н. раствора Na2HPO4. Раствор деаэрируют азотом с содержанием кислорода менее 0,001% (ос. ч. ) в течение 5 мин. Отключают газ и фиксируют катодную вольтамперограмму при скорости развертки потенциала 20 мВ/с, начиная с потенциала Енач=1,0 В. Отсутствие пиков свидетельствует о чистоте фона. Затем добавляют 0,01 мл стандартного раствора менадиона в концентрации 1,0 10-5-1,010-3 моль/л, перемешивают 10 с. Съемку вольтамперограммы начинают с потенциала Енач=1,0 В. Катодный пик для указанного флавоноида регистрируют в диапазоне потенциала -0,220,02 В при чувствительности прибора 110-9 А/мм. Время единичного анализа не превышает 15 мин.
Аналогичные измерения проводили с растворами кверцетина, байкалина, диквертина. Катодные пики соответствующих веществ регистрируют при потенциалах (+0,410,04) В, (-0,600,03) В, (+0,140,02) В.
Пример 2. Определение байкалина на уровне 10-8-10-6 мг/мл по пикам окисления на фоне 0,2 н. Nа2НРO4В кварцевый стаканчик емкостью 20 мл наливают 10 мл 0,2 н раствора Na2HPO4. Раствор деаэрируют азотом с содержанием кислорода менее 0,001% (ос. ч.) в течение 5 мин. Отключают газ и фиксируют анодную вольтамперограмму при скорости развертки потенциала 20 мВ/с, начиная с потенциала Енач.=-1,0 В. Отсутствие пиков свидетельствует о чистоте фона. Затем добавляют 0,01 мл стандартного раствора байкалина в концентрации 1,0 10-5-1,010-3 моль/л, перемешивают 10 с. Съемку вольтамперограммы вновь начинают с потенциала Енач.=-1,0 В. Анодный пик для указанного флавоноида регистрируют в диапазоне потенциалов (+0,200,02) В при чувствительности прибора 110-9 А/мм. Время единичного анализа не превышает 15 мин.
Аналогично регистрируют анодные пики кверцетина, менадиона, диквертина. Анодные пики соответствующих веществ регистрируют при потенциалах (+0,390,02) В, (-0,360,02) В, (+0,400,02) В.
Пример 3. Определение диквертина на уровне 10-8-10-6 мг/мл по пикам окисления на фоне 0,2 н. С6Н14O7N2В кварцевый стаканчик емкостью 20 мл наливают 10 мл 0,2 н. 6H14О7N2. Раствор деаэрируют азотом с содержанием кислорода менее 0,001% (ос.ч.) в течение 5 мин. Отключают газ и фиксируют анодную вольтамперограмму при скорости развертки потенциала 30 мВ/с, начиная с потенциала Енач.=-1,0 В. Отсутствие пиков свидетельствует о чистоте фона. Затем добавляют 0,01мл стандартного раствора диквертина 1,0 10-5-1,010-3 моль/л, перемешивают 10 с. Съемку вольтамперограммы начинают с потенциала Енач.=-1,0 В. Анодный пик для указанного флавоноида регистрируют при потенциале (+0,460,01) В при чувствительности прибора 110-9 А/мм. Время единичного анализа не превышает 15 мин.
Аналогичные измерения проводили с растворами кверцетина, менадиона, байкалина. Анодные пики соответствующих веществ регистрируют при потенциалах (+0,360,02) В, (-0,150,02) В, (+0,350,01) В.
Пример 4. Определение кверцетина в экстракте водяники черной по пикам окисления на уровне 10-8-10-6 мг/млВ кварцевый стаканчик емкостью 20 мл наливают 10 мл 0,2 н. раствора C6H14O7N2. Раствор деаэрируют азотом с содержанием кислорода менее 0,001% (ос. ч. ) в течение 5 мин. Отключают газ и фиксируют анодную вольтамперограмму при скорости развертки потенциала 20 мВ/с, начиная с потенциала Енач.= -1,0 В. Отсутствие пиков свидетельствует о чистоте фона. Затем добавляют 0,01 мл раствора экстракта водяники, перемешивают 10 с. Съемку вольтамперограммы начинают с потенциала Енач.=-1,0 В. Анодные пики для указанного флавоноида регистрируют в диапазоне потенциалов (+0,36 0,02) В при чувствительности прибора 110-9 А/мм. Концентрацию определяемого вещества оценивали методом добавок аттестованных смесей по общепринятой методике.
Таким образом, дифференциальная вольтамперометрия с использованием стеклоуглеродного электрода позволила существенно улучшить метрологические характеристики анализа флавоноидов. На основании проведенных исследований была показана возможность определения анодных и катодных пиков флавоноидов и разработана экспрессная методика количественного их определения в водных средах. Предел обнаружения, рассчитанный по 3 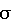 -критерию, равен 1,010-8 моль/л. Минимально определяемая концентрация флавоноидов 3,510-8 моль/л (Sr -критерию, равен 1,010-8 моль/л. Минимально определяемая концентрация флавоноидов 3,510-8 моль/л (Sr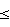 0,20). По сравнению с прототипом чувствительность повышается на 3-4 порядка.
Предложенный способ может быть применен при анализе следовых количеств флавоноидов как в чистых растворах, так и для анализа сложных по составу растительных экстрактов, лекарственных форм, фармакологических препаратов. Вольтамперограммы хорошо воспроизводимы, условия съемки легко могут быть автоматизированы. Методики отличаются простотой исполнения и могут быть использованы в любой лаборатории, имеющей полярограф, особенно в настоящее время, когда налажен выпуск отечественной и зарубежной электроаппаратуры с компьютерным управлением и обработкой данных. 0,20). По сравнению с прототипом чувствительность повышается на 3-4 порядка.
Предложенный способ может быть применен при анализе следовых количеств флавоноидов как в чистых растворах, так и для анализа сложных по составу растительных экстрактов, лекарственных форм, фармакологических препаратов. Вольтамперограммы хорошо воспроизводимы, условия съемки легко могут быть автоматизированы. Методики отличаются простотой исполнения и могут быть использованы в любой лаборатории, имеющей полярограф, особенно в настоящее время, когда налажен выпуск отечественной и зарубежной электроаппаратуры с компьютерным управлением и обработкой данных.
Формула изобретения
0,01) В на фоне 0,2 н C6H14O7N2 или по высоте пиков окисления при потенциалах (+0,390,02) В, или при (+0,360,02) В соответственно на фонах 0,2 н Na2HPO4 и 0,2 н C6H14O7N2.
3. Способ по п.1, отличающийся тем, что концентрацию диквертина определяют по высоте пиков восстановления при потенциалах (+0,140,02) В и (-0,400,02) В соответственно на фонах 0,2 н Na2HPO4 и 0,2 н C6H14O7N2 или по высоте пиков окисления при потенциалах (+0,400,02) В и +0,45 В соответственно на фонах 0,2 н Na2HPO4 и 0,2 н C6H14O7N2.
4. Способ по п.1, отличающийся тем, что концентрацию менадиона определяют по высоте пиков восстановления при потенциалах (-0,220,02) В и (-0,150,02) В соответственно на фонах 0,2 н Na2HPO4 и 0,2 н C6H14O7N2 или по высоте пиков окисления при потенциалах -0,34 В и (-0,150,01) В соответственно на фонах 0,2 н Na2HPO4 и 0,2 н C6H14O7N2.
5. Способ по п.1, отличающийся тем, что концентрацию байкалина определяют по высоте пиков восстановления при потенциалах (-0,600,02) В и -0,60 В соответственно на фонах 0,2 н Na2HPO4 и 0,2 н СбН14О7К2 или по высоте пиков окисления при потенциалах (+0,200,02) В и (+0,350,01) В соответственно на фонах 0,2 н Na2HPO4 и 0,2 н C6H14O7N2.
РИСУНКИ
MM4A Досрочное прекращение действия патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 11.01.2004
Извещение опубликовано: 27.04.2006 БИ: 12/2006
|
||||||||||||||||||||||||||