Патент на изобретение №2213724
|
||||||||||||||||||||||||||
(54) СПОСОБ РАЗДЕЛЕНИЯ НА ЭНАНТИОМЕРЫ РАЦЕМИЧЕСКИХ 3-(2-МЕТОКСИФЕНОКСИ)-1,2-ПРОПАНДИОЛА И 3-(2-МЕТИЛФЕНОКСИ)-1,2-ПРОПАНДИОЛА
(57) Реферат: Изобретение относится к способу разделения на энантиомеры рацемических 3-(2-метоксифенокси)-1,2-пропандиола и 3-(2-метилфенокси)-1,2-пропандиола, который может быть использован в фармацевтической промышленности при получении нерацемических лекарственных препаратов. Способ заключается в том, что рацемический диол слегка обогащают одним из энантиомеров в подходящем растворителе при 40-60oС с получением концентрированного раствора и перемешивают, постепенно охлаждая до 10-40oС, добиваясь его пересыщения, после чего вносят кристаллическую затравку того же энантиомера, который добавлен для обогащения, выдерживают при перемешивании при температуре кристаллизации в интервале 5-33oС в течение 30-60 мин, отфильтровывают твердый осадок скалемического диола, обладающего той же конфигурацией, что и образец внесенной затравки, затем в маточный раствор, обогащенный вторым энантиомером, добавляют рацемическое соединение в количестве, компенсирующем отделенный осадок, нагревают до полного растворения при 40-60oС, затем охлаждают до 10-40oС, вносят кристаллическую затравку преобладающего в растворе энантиомера, выдерживают при перемешивании при температуре кристаллизации в интервале 5-33oС, отделяют осадок, обогащенный вторым энантиомером, с последующим повторением указанного процесса несколько раз. Как правило, в качестве растворителя используют бензол, четыреххлористый углерод или его смесь с хлористым метиленом 1: 1, 40% водный раствор этанола, воду. Способ позволяет получить известные лекарственные средства в нерацемическом виде из дешевых рацемических субстратов. 1 з.п.ф-лы, 1 табл. Изобретение относится к химии органических соединений, а именно к способу разделения на отдельные энантиомеры рацемических 3-(2-метоксифенокси)-1,2-пропандиола (I), международное непатентованное название гвайфенезин (guaiphenesin), и 3-(2-метилфенокси)-1,2-пропандиола (II), международное непатентованное название мефенезин (mephenesin). Изобретение может быть использовано в фармацевтической промышленности при получении нерацемических (скалемических, энантиочистых) лекарственных препаратов. В настоящее время названные соединения зарегистрированы в качестве субстанций лекарственных средств в рацемическом виде.  Гвайфенезин применяется как мышечный релаксант и отхаркивающее средство, мефенезин используется как релаксант скелетных мышц. Зарегистрированными лекарственными средствами являются и простейшие производные рацемических диолов (I) и (II), а именно, их 1-карбаматы, так же являющиеся релаксантами скелетных мышц и спазмолитиками. Химическую структуру (I) и (II) можно модифицировать и в другие физиологически активные производные с хиральным С3 Применительно к получению скалемических 3-арилоксипропандиолов в качестве описанных примеров первого подхода можно привести присоединение фенолов АrОН к скалемическому глицидолу, региоселективно протекающее в присутствии катализаторов, таких как Ti(OAlk)43 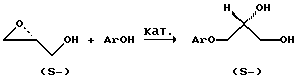 Этот способ достаточно прост и хорошо воспроизводится, однако для реализации он нуждается в наличии дорогостоящего энантиочистого глицидола.  На схеме AD-mix-  – торговое название смеси, содержащей хиральный лиганд (DHQ)2PHAL, К3Fе(СN)6, K2CO3, K2OsO4 – торговое название смеси, содержащей хиральный лиганд (DHQ)2PHAL, К3Fе(СN)6, K2CO3, K2OsO4 2H2O; здесь (DHQ)2PHAL: 2H2O; здесь (DHQ)2PHAL: Для проведения этого процесса используются экологически вредные производные тяжелого металла осмия и дорогостоящие энантиочистые катализаторы. Кроме того, полученные этим способом аналоги (I) и (II) имеют невысокий энантиомерный избыток (в пределах 60%). На первой стадии rас-1,2-диолы с помощью липаз региоселективно ацилируются винилацетатом по первичной гидроксигруппе без проявления энантиоселективности. Последующая стадия катализируемого липазами ацилирования вторичной гидроксигруппы протекает с удовлетворительной энантиоселективностью (для R= OMe, ee= 87%; R= Me, ee=80%). Моноацетат отделяется от диацетата флеш-хроматографией и затем после последовательного удаления ацильных групп выделяются целевые (S)- и (R)-диолы.  Недостатком этого способа является то, что результаты использования биоэнзимов сильно зависят от условий проведения процесса и от характера заместителей в ароматическом кольце. Метод нуждается в значительном ассортименте вспомогательных реактивов, при разработке используется хроматография, процесс трудно масштабируется. Нами впервые обнаружено, что рацемические диолы (I) и (II) кристаллизуются в виде рацемических конгломератов без какого-либо предварительного превращения их в другие химические производные, например соли, как в большинстве известных случаев, и мы предлагаем способ их разделения на отдельные энантиомеры по схеме избирательной кристаллизации. Ранее свойство самопроизвольного расщепления при кристаллизации для соединений (I) и (II) не было известно, и таким способом нерацемические соединения (I) и (II) не получались. К тому же было неочевидно, что использование этого свойства приведет к получению нерацемических соединений (I) и (II). Цель предлагаемого изобретения – создание способа, позволяющего получить известные лекарственные средства – гвайфенезин и мефенезин в нерацемическом виде из дешевых рацемических субстратов путем их разделения при кристаллизации методом вовлечения. Поставленная цель достигается тем, что рацемических диол слегка обогащают одним из энантиомеров в подходящем растворителе при температуре 40-60oС с получением концентрированного раствора и перемешивают, постепенно охлаждая до 10-40oС, добиваясь его пересыщения, после чего вносят кристаллическую затравку того же энантиомера, который добавлен для обогащения, выдерживают при перемешивании при температуре кристаллизации в интервале 5-33oС в течение 30-60 мин, затем отфильтровывают твердый осадок скалемического диола, обладающего той же конфигурацией, что и образец внесенной затравки. В маточный раствор, обогащенный вторым энантиомером, добавляют рацемическое соединение в количестве, компенсирующем отделенный осадок, нагревают до полного растворения при 40-60oС, затем охлаждают до 10-40oС, вносят кристаллическую затравку преобладающего в растворе энантиомера, выдерживают при перемешивании при температуре кристаллизации в интервале 5-33oС, отделяют осадок, обогащенный вторым энантиомером, с последующим повторением указанного процесса несколько раз, тщательно анализируя оптическую чистоту и количество выделенного осадка. Разделение на энантиомеры проводили для соединений (I) и (II) в бензоле, водном этаноле, воде, четыреххлористом углероде или его смеси с хлористым метиленом (1: 1). Заявляемый способ разделения диолов (I) и (II) иллюстрируется следующими примерами. ПРИМЕР 1 4.56 г (R, S)-гвайфенезина (I) (Т. пл. 76-78oС) и 0.50 г (S)-гвайфенезина (Т. пл. 97oС, [ ]25D+9.4 (с 1.0, МеОН), ор=99.5%) растворяют в 135 мл бензола при 50oС и охлаждают при перемешивании до 30oС, затем вносят мелкоизмельченную затравку (S)-гвайфенезина (0.02 г) с той же оптической чистотой и в течение 60 мин перемешивают раствор при 29oС. После фильтрования и высушивания собирают 0.90 г (S)-гвайфенезина с []25D+6.5 (с 2.0, МеОН), ор= 69.1%. К фильтрату, оставшемуся после отделения осадка, добавляют 0.90 г (R, S)-гвайфенезина, растворяют его при 50oС, охлаждают раствор до 30oС, вносят затравку измельченного (R)-гвайфенезина (0.02 г), op>99%, и перемешивают раствор при 29oС 60 мин. Отделяют 0.80 г (R)-гвайфенезина с []25D-7.2 (с 1.6, МеОН), ор=76.6%. Таким же образом цикл повторяют несколько раз, добавляя каждый раз недостающее количество рацемического диола. После 2-го цикла выделено 0.8 г (S)-гвайфенезина с ор=64.5% и 0.70 г (R)-гвайфенезина с ор= 66.7%. Поcле 3-го цикла выделено 0.70 г (S)-гвайфенезина с ор=60.4% и 0.65 г (R)-гвайфенезина с ор=61.2%.
Повышение энантиомерной чистоты полученных таким образом скалемических (R)- и (S)-гвайфенезина возможно путем дополнительной перекристаллизации из органических или смешанных растворителей. Например, 0.8 г (S)-гвайфенезина (ор=69.1%) растворяют в 3.5 мл 40% (по объему) водного этанола при 50oС. После охлаждения раствора до 10oС и внесения затравки (S)-гвайфенезина (0.008 г) смесь перемешивают при 5oС 5 ч. После фильтрования и высушивания собирают (5)-гвайфенезин: выход 0.45 г, []25D+8.9 (с 0.5, МеОН), ор=94.3% {ср. для (R)-гвайфенезина []25D
ПРИМЕР 25.00 г (R,S)-гвайфенезина (I) и 0.20 г (R)- гвайфенезина растворяют в 20 мл 40% водного этанола при 40oС и охлаждают при перемешивании до 10oС, затем вносят мелкоизмельченную затравку (R)-гвайфенезина (0.05 г, op>99%) и в течение 60 мин перемешивают раствор при 5oС. После фильтрования и высушивания собирают 0.45 г (R)-гвайфенезина с [ ]25D-8.5 (с 1.0, МеОН), ор=89.1%. К фильтрату, оставшемуся после отделения осадка, добавляют 0.45 г (R, S)-гвайфенезина, растворяют его при 40oС, охлаждают раствор до 10oС, вносят затравку измельченного (S)-гвайфенезина (0.05 г), op>99%, и перемешивают раствор при 5oС 60 мин. Отделяют 0.41 г (S)-гвайфенезина с []25D+8.4 (с 0.8, МеОН), ор=88.5%. Цикл повторяют несколько раз.
ПРИМЕР 32.55 г (R, S)-гвайфенезина (I) и 0.25 г (R)-гвайфенезина (ор=90.0%) растворяют в 20 мл воды при 60oС и охлаждают при перемешивании до 30oС, затем вносят мелкоизмельченную затравку (R)-гвайфенезина (0.02 г, op>99%) и в течение 60 мин перемешивают раствор при 20oС. После фильтрования и высушивания собирают 0.44 г (R)-гвайфенезина с [ ]25D-8.6 (с 1.0, МеОН), ор= 90.5%. К фильтрату, оставшемуся после отделения осадка, добавляют 0.44 г (R, S)-гвайфенезина, растворяют его при 60oС, охлаждают раствор до 30oС, вносят затравку измельченного (S)-гвайфенезина (0.02 г), op>99%, и перемешивают раствор при 20oС 60 мин. Отделяют 0.41 г (S)-гвайфенезина с []25D+7.8 (с 0.8, МеОН), ор=82.1%. Цикл повторяют несколько раз.
ПРИМЕР 40.4 г (R, S)-мефенезина (II) (Т. пл. 70-71 oС) и 0.04 г (S)-мефенезина (Т. пл. 90-92 oС, [ ]25D-19.3 (с 0.9, гексан-изопропиловый спирт 4:1), ор= 99.5%) растворяют в 15 мл бензола при 50oС и охлаждают при перемешивании до 30oС, затем вносят мелкоизмельченную затравку (S)-мефенезина той же оптической чистоты (0.004 г) и в течение 60 минут перемешивают раствор при 30oС. Затем отфильтровывают 0.09 г (S)-мефенезина с []20D-17.1 (с 2.0, с 0.9, гексан-изопропиловый спирт 4: 1), ор=86.0% {ср. []20DoС, охлаждают до 30oС, вносят затравку измельченного (R)-мефенезина (0.004 г), op>99%, и перемешивают раствор при 30oС 60 мин. Отфильтровывают 0.08г (R)-мефенезина с []20D+15.5 (с 0.5, гексан-изопропиловый спирт 4:1), ор=78.0% {ср. []20D
ПРИМЕР 52.00 г (R, S)-мефенезина (II) и 0.20 г (R)- мефенезина растворяют в 24 мл 40% (по объему) водного этанола при 40oС и охлаждают при перемешивании до 10oС, затем вносят мелкоизмельченную затравку (R)-мефенезина (0.01 г), op>99%, и в течение 60 мин перемешивают раствор при +5oС. Затем отфильтровывают 0.50 г (R)- мефенезина с [ ]20D+16.3 (с 1.15, гексан-изопропиловый спирт 4: 1), ор=81.8%. К фильтрату, оставшемуся после отделения осадка, добавляют 0.50 г (R,S)-мефенезина, растворяют твердую компоненту при 40oС, охлаждают до 10oС, вносят затравку измельченного (S)-мефенезина (0.01 г), op>99%, и перемешивают раствор при +5oС 60 мин. Отфильтровывают 0.31 г (S)-мефенезина с []20D-17.1 (с 0.49, гексан-изопропиловый спирт 4:1), ор= 85.5%.
ПРИМЕР 64.50 г (R, S)-мефенезина (II) и 0.50 г (R)- мефенезина растворяют в 165 мл воды при 60oС и охлаждают при перемешивании до 40oС, затем вносят мелкоизмельченную затравку (R)-мефенезина (0.025 г), op>99%, охлаждают раствор до 33oС (7 мин) и в течение 60 мин перемешивают раствор при этой температуре. Затем отфильтровывают 0.86 г (R)- мефенезина с ]20D+17.1 (с 1.15, гексан-изопропиловый спирт 4: 1), ор=87.7% (операция 1 в табл.). К фильтрату, оставшемуся после отделения осадка, добавляют 0.86 г (R, S)-мефенезина, растворяют твердую компоненту при 60oС, охлаждают до 40oС, вносят затравку измельченного (S)-мефенезина (0.025 г), op>99%, охлаждают раствор до 33oС и перемешивают раствор при этой температуре в течение 40 мин. Отфильтровывают 0.82 г (S)-мефенезина с []20D-16.8 (с 1.10, гексан-изопропиловый спирт 4:1), ор=86.1% (операция 2 в табл.).Таким же образом цикл повторяют несколько раз, добавляя каждый раз недостающее количество рацемического диола. Детали описаны в таблице под операциями 3-6.
Повышение энантиомерной чистоты полученных таким образом скалемических (R)- и (S)-мефенезина возможно путем дополнительной перекристаллизации из органических или смешанных растворителей. Например, 0.47 г (R)-мефенезина (ор 81.8%) растворяют в 7.0 мл 40%-ного водного этанола при 50oС, затем охлаждают до 20oС, вносят затравку (0.004 г) (R)-мефенезина (oр>99%), охлаждают до 8oС, перемешивают 30 мин, отфильтровывают 0.21 г (R)-мефенезина []20D+18.6 (с 1.0, гексан-изопропиловый спирт 4:1), ор=95.4%ПРИМЕР 7. 0.53 г (R,S)-гвайфенезина (I) и 0.06 г (S)-гвайфенезина растворяют в 14 мл смеси четыреххлористого углерода и хлористого метилена 1:1 при 40oС и охлаждают при перемешивании до 25oС, затем вносят мелкоизмельченную затравку (S)-гвайфенезина (0.002 г) ор=99.5%) и в течение 30 мин перемешивают раствор при 20oС. После фильтрования и высушивания собирают 0.08 г (S)-гвайфенезина с [ ]25D+7.7 (с 1.0, МеОН), ор=82.3%.
Формула изобретения 1. Способ разделения рацемических 3-(2-метоксифенокси)- или 3-(2метилфенокси)-1,2-пропандиолов на энантиомеры, заключающийся в том, что рацемический диол слегка обогащают одним из энантиомеров в подходящем растворителе при 40-60oС с получением концентрированного раствора и перемешивают, постепенно охлаждая до 10-40oС, добиваясь его пересыщения, после чего вносят кристаллическую затравку того же энантиомера, который добавлен для обогащения, выдерживают при перемешивании при температуре кристаллизации в интервале 5-33oС в течение 30-60 мин, отфильтровывают твердый осадок скалемического диола, обладающего той же конфигурацией, что и образец внесенной затравки, затем в маточный раствор, обогащенный вторым энантиомером, добавляют рацемическое соединение в количестве, компенсирующем отделенный осадок, нагревают до полного растворения при 40-60oС, затем охлаждают до 10-40oС, вносят кристаллическую затравку преобладающего в растворе энантиомера, выдерживают при перемешивании при температуре кристаллизации в интервале 5-33oС, отделяют осадок, обогащенный вторым энантиомером, с последующим повторением указанного процесса несколько раз. 2. Способ по п. 1, отличающийся тем, что в качестве растворителя используют бензол, четыреххлористый углерод или его смесь с хлористым метиленом 1:1, 40% водный раствор этанола, воду. РИСУНКИ
|
||||||||||||||||||||||||||