Патент на изобретение №2210565
|
||||||||||||||||||||||||||
(54) НОВЫЙ СПОСОБ ПРОИЗВОДСТВА МЕТОПРОЛОЛА
(57) Реферат: Изобретение относится к улучшенному способу получения метопролола, который является известным лекарственным средством для лечения аритмии и инфаркта миокарда. Способ заключается в том, что на первой стадии п-(2-метоксиэтил)фенол и эпихлоргидрин подвергают взаимодействию в воде в качестве растворителя во время добавления основания и при температуре 50 – 70oС, избыток эпихлоргидрина выпаривают, а полученный 1-(2,3-эпоксипропокси)-4-(2-метоксиэтил)бензол затем дистиллируют при пониженном давлении и на второй стадии подвергают взаимодействию полученный 1-(2,3-эпоксипропокси)-4-(2-метоксиэтил)бензол и изопропиламин в присутствии изопропилового спирта с образованием метопролола-основания, который при необходимости превращают в метопролола тартрат или метопролола сукцинат. Обычно первую стадию осуществляют в присутствии гидроксида натрия или гидроксида калия. Полученный метопролол-основание можно очистить путем растворения метопролола-основания в растворителе, выбранном из толуола, изобутилметилкетона и бутилацетата, с экстракцией раствором либо соляной, либо серной кислоты. Способ экологически безопасен и позволяет быстро с хорошим выходом и высокой степенью чистоты получить целевой продукт. 5 з.п ф-лы. Настоящее изобретение относится к усовершенствованному способу производства метопролола-основания, 1-(изопропиламино)-3-[пара-(2-метоксиэтил)-фенокси] -2-пропанола, путем взаимодействия п-(2-метоксиэтил)-фенола (А) и эпихлоргидрина (В) с последующим взаимодействием полученного 1-(2,3-эпоксипропокси)-4-(2-метоксиэтил)-бензола (АВ) с изопропиламином (С). Неочищенный метопролол-основание далее подвергают очистке. В Chemical Abstracts, vol. 112 (1990), реферат 197820 описано взаимодействие п-(2-метоксиэтил)-фенола и эпихлоргидрина в двухфазной системе, состоящей из воды и органического растворителя. В патентах Швеции 354851 и 368004 описано взаимодействие п-(2-метоксиэтил)-фенола и эпихлоргидрина, при котором эпихлоргидрин используют не только в качестве промежуточного соединения, но также и в качестве растворителя. Описание изобретения В настоящее время установлено, что метопролол может быть получен быстрым, экологически чистым способом с хорошим выходом и высокой степенью чистоты с использованием реактивов, известных per se. Отличие от прототипа состоит в том, что в новом способе для взаимодействия А и В не используется никаких других растворителей кроме воды. С экологической точки зрения, равно как и с точки зрения профессионального риска, возможность замены опасного органического растворителя невредным растворителем, таким как вода, является большим преимуществом. Способ по изобретению иллюстрируется приведенной в конце текста реакционной схемой. п-(2-Метоксиэтил)фенол (А) и эпихлоргидрин (В; 1,4-2,0 экв.) подвергают взаимодействию в воде, по меньшей мере 1,5 кг, предпочтительно примерно 2 кг воды на кг фенола, во время добавления раствора гидроксида натрия (или калия) (1,3-1,7 экв. ) с образованием 1-(2,3-эпоксипропокси)-4-(2-метоксиэтил)бензола; (п-метоксиэтил-эпоксипропоксибензола или просто более кратко эпоксида). Взаимодействие осуществляют при температуре 50-70oС. Осуществляют разделение двух фаз и п-метоксиэтилэпоксипропоксибензол выделяют дистилляцией при пониженном давлении. Более конкретно, избыток эпихлоргидрина выпаривают и полученный п-метоксиэтилэпоксипропоксибензол, затем дистиллируют при пониженном давлении, получая продукт с чистотой приблизительно 96-98%. При желании перед дистилляцией основной фракции эпоксида можно отогнать его префракцию/головной погон (2-8%, предпочтительно 4-6%). Выделение эпоксида дистилляцией при пониженном давлении является важной частью процесса и существенно для качества конечного продукта. Эпоксид подвергают взаимодействию с изопропиламином в изопропиловом спирте с образованием метопролола-основания. Количество изопропиламина по отношению к эпоксиду составляет по меньшей мере 1 эквивалент, предпочтительно 3-6 эквивалентов. Затем реакционную смесь обрабатывают для того, чтобы удалить избыток изопропиламина. Альтернативно, аминирование изопропиламином осуществляют в герметичной системе без изопропилового спирта при 70  10oС и давлении 2,8-3,2 кг/275-315 кПа.
Полученный метопролол растворяют в толуоле, изобутилметилкетоне или бутилацетате и экстрагируют разбавленной соляной или серной кислотой, предпочтительно при рН 4-6. Производят разделение фаз, и выбранный растворитель с раствором гидроксида натрия или калия для доведения рН до 11-13 добавляют к водной фазе. Осуществляют разделение двух фаз, и органическую фазу выпаривают в вакууме до маслянистого остатка метопролола-основания, который растворяют в ацетоне. Очищенный метопролол-основание далее получают традиционными способами.
Возможно также получение солей метопролола. Так, метопролол-основание можно превратить в метопролола тартрат или метопролола сукцинат путем, например, обработки в водном растворе метопролол-основания соответствующей кислотой и последующего выпаривания воды или при помощи смешивания соответствующей кислоты и метопролола-основания в ацетоне или водном растворе ацетона, с последующей кристаллизацией целевого продукта – соли метопролола.
Рабочий пример 10oС и давлении 2,8-3,2 кг/275-315 кПа.
Полученный метопролол растворяют в толуоле, изобутилметилкетоне или бутилацетате и экстрагируют разбавленной соляной или серной кислотой, предпочтительно при рН 4-6. Производят разделение фаз, и выбранный растворитель с раствором гидроксида натрия или калия для доведения рН до 11-13 добавляют к водной фазе. Осуществляют разделение двух фаз, и органическую фазу выпаривают в вакууме до маслянистого остатка метопролола-основания, который растворяют в ацетоне. Очищенный метопролол-основание далее получают традиционными способами.
Возможно также получение солей метопролола. Так, метопролол-основание можно превратить в метопролола тартрат или метопролола сукцинат путем, например, обработки в водном растворе метопролол-основания соответствующей кислотой и последующего выпаривания воды или при помощи смешивания соответствующей кислоты и метопролола-основания в ацетоне или водном растворе ацетона, с последующей кристаллизацией целевого продукта – соли метопролола.
Рабочий пример1-(2,3-Эпоксипропокси)-4-(2-метоксиэтил)бензол п-(2-Метоксиэтил)фенол (А, ~ 6,6 моль), эпихлоргидрин (В; 1,45 экв.) и воду (~ 2 кг) объединяют и смесь нагревают до ~50oС. В течение 3 часов добавляют раствор гидроксида натрия (50%-ный; 1,4 экв.), температуру в продолжение добавления поднимают до достижения приблизительно 60oС. В течение этого периода времени происходит образование указанного в заголовке соединения. Раствор перемешивают в течение следующего часа при температуре приблизительно 60oС, затем охлаждают приблизительно до 50oС, разделяют фазы и продукт промывают водой. Остаток (п-метоксиэтилэпоксипропоксибензол) дистиллируют при температуре 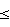 190oС и давлении 20 мм ртутного столба (2,666 кПа) и дистиллят собирают. Выход указанного в заголовке соединения составляет 80% от теоретического, а чистота составляет 98% по данным ГХ-анализа (газовохроматографический анализ).
Метопролол-основание 190oС и давлении 20 мм ртутного столба (2,666 кПа) и дистиллят собирают. Выход указанного в заголовке соединения составляет 80% от теоретического, а чистота составляет 98% по данным ГХ-анализа (газовохроматографический анализ).
Метопролол-основание1-(2,3-Эпоксипропокси)-4-(2-метоксиэтил)бензол (1 кг; 4,8 моль), изопропиловый спирт (~0,9 кг) и изопропиламин (0,8-1,7 кг, 3-6 экв.) смешивают и подвергают взаимодействию в течение 2-5 часов при температуре дефлегмации. В продолжение этого периода времени происходит образование метопролола-основания. Затем реакционную смесь концентрируют при атмосферном давлении до тех пор, пока внутренняя температура не достигнет ~100oС. Воду добавляют в эту загрузку, а затем отгоняют в вакууме до тех пор, пока внутренняя температура не достигнет ~100oС, с образованием концентрата. Полученный концентрат разбавляют изобутилметилкетоном (~0,6 кг) и водой (~ 2,2 кг) и добавляют концентрированную серную кислоту, чтобы довести рН до 4-6. После разделения к водному слою добавляют изобутилметилкетон (~1 кг) и концентрированный раствор гидроксида натрия, чтобы довести рН до 13. Органический слой концентрируют в вакууме при температуре 80oС до тех пор, пока дистилляция не прекращается, и концентрированную загрузку заново растворяют в ацетоне (~1,6 кг) и фильтруют с получением раствора метопролола-основания. Содержание метопролола-основания в растворе определяют титрованием. Выход: ~1,2 кг метопролола-основания (100%) ~95% от теоретического. Чистота метопролола-основания составляет 96%.
Формула изобретения 1. Способ производства метопролола, отличающийся тем, что на первой стадии п-(2-метоксиэтил)фенол и эпихлоргидрин подвергают взаимодействию в воде в качестве растворителя во время добавления основания и при температуре 50-70oС, избыток эпихлоргидрина выпаривают, а полученный 1-(2,3-эпоксипропокси)-4-(2-метоксиэтил)бензол затем дистиллируют при пониженном давлении и на второй стадии подвергают взаимодействию полученный 1-(2,3-эпоксипропокси)-4-(2-метоксиэтил)бензол и изопропиламин в присутствии изопропилового спирта с образованием метопролола-основания, который возможно превращают в метопролола тартрат или метопролола сукцинат. 2. Способ по п. 1, отличающийся тем, что первую стадию осуществляют в присутствии гидроксида натрия. 3. Способ по п. 1, отличающийся тем, что первую стадию осуществляют в присутствии гидроксида калия. 4. Способ по любому из пп. 1-3, отличающийся тем, что полученный метопролол-основание очищают путем растворения метопролола-основания в растворителе, выбранном из толуола, изобутилметилкетона и бутилацетата, и экстрагируют раствором либо соляной, либо серной кислоты. 5. Способ по любому из пп. 1-4, отличающийся тем, что полученный метопролол превращают в метопролола тартрат. 6. Способ по любому из пп. 1-4, отличающийся тем, что полученный метопролол превращают в метопролола сукцинат. РИСУНКИ
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 19.11.2009
Извещение опубликовано: 20.09.2010 БИ: 26/2010
|
||||||||||||||||||||||||||