Патент на изобретение №2206571
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ ОРГАНОМЕТАЛЛОКСИСТАННАНОВ
(57) Реферат: Изобретение относится к усовершенствованному способу получения органометаллоксистаннанов общей формулы YAlk2SnOMnRmYn-m-1 [где М – металл, выбранный из группы Са, Zn, Cu, Al, Si, Ge, Sn, Ti, Fe, Mn, Со, Cr; Y=OR’ (R‘ – алкил с 1 – 4С) или O2CR” (R” – алкил, циклоалкил с 6-11C); R – алкил с 1-4С, винил, фенил; n – валентность металла М, m=0-2, m-n 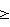 1] путем взаимодействия диалкилоловооксидов с алкоксидами или ацилатами указанных выше металлов М. Реакцию проводят в атмосфере сухого очищенного азота при нагревании реакционной смеси при температуре 80-100oС в присутствии катализатора – алифатической или циклоалифатической карбоновой кислоты, взятой в количестве 2-5% от массы диалкилоловооксида. Способ позволяет получать с высоким выходом и достаточно высоким качеством реакционноспособные, термически и гидролитически малоустойчивые органометаллоксистаннаны. Эффективность способа подтверждается его успешной апробацией в опытных условиях. 1 з.п. ф-лы. 1] путем взаимодействия диалкилоловооксидов с алкоксидами или ацилатами указанных выше металлов М. Реакцию проводят в атмосфере сухого очищенного азота при нагревании реакционной смеси при температуре 80-100oС в присутствии катализатора – алифатической или циклоалифатической карбоновой кислоты, взятой в количестве 2-5% от массы диалкилоловооксида. Способ позволяет получать с высоким выходом и достаточно высоким качеством реакционноспособные, термически и гидролитически малоустойчивые органометаллоксистаннаны. Эффективность способа подтверждается его успешной апробацией в опытных условиях. 1 з.п. ф-лы.
Изобретение относится к способам получения диметаллоорганических соединений, содержащих связь Sn-О-M, где М – металл, выбранный из разных групп Периодической системы элементов, а именно органометаллоксистаннанов. Соединения такого типа рекомендованы к применению как исходные мономеры для получения полимеров, как термо- и светостабилизаторы для полиолефинов, для придания водоотталкивающих свойств цементу, металлам, древесине, тканям, бумаге; в качестве фунгицидов, инсектицидов и антибактериальных средств, как катализаторы или компоненты каталитических систем и инициаторы реакций гомо- и сополимеризации мономеров различной химической природы и для получения композиционных составов (пат. США 3307973, 1967 г., НКИ 428-573; 5461139, 1995 г. , МКИ С 08 G 063/08; 5003015, 1991 г., МКИ С 08 F 004/72; 5128295, 1992 г., МКИ С 08 F 004/64; 5430125, 1995 г., НКИ 528/354; 5034455, 1991 г., МКИ С 08 К 003/26). Существует крайне мало работ с описанием способов получения органометаллоксистаннанов, большинство из которых были разработаны более трех десятилетий назад [М. Dub (bd.) “Organometallic Compounds”, vol. II. 1967; vol. II 1-st Suppl., 1973. Springer Verlag, Berlin-NYj. Высокая реакционная способность, а также гидролитическая и термическая нестабильность подобных соединений являются серьезным препятствием для их препаративного синтеза. В одной из работ (Beckmann J. , a.o. – Organometallics. 1998, v.17, 26, р. 5697-5712) практически значимые результаты были получены по реакции диалкилоловооксидов с органогалогенсиланами лишь в случае использования исходных реагентов с объемными трет-бутильными группами и при проведении синтеза в течение нескольких суток при температуре ~60oС. Известные нам методы имеют ограниченное и в основном теоретическое значение: как правило, в их описаниях отсутствуют условия проведения реакций, нет сведений о технологических приемах и количественных результатах их осуществления. Исключение составляет способ получения органометаллоксистаннанов, защищенный патентом США 3307973 (см. выше), который выбран нами за прототип. Согласно изобретению, вышеназванные соединения, выражаемые общей формулой [R2SnO-]nE (где Е=МХv-n и M(R’m)Xv-n-m, М – металл, выбранный из группы В, Al, Si, Ti, Zr, V, или  VO; X – галоген или ORо, Rо – углеводородный или фторуглеводородный радикал с 1-18С; R’ – алкил с 1-12С: v – валентность М (3 или 4); m – в пределах от 1 до v-1; n – от 1 до v; сумма m+n VO; X – галоген или ORо, Rо – углеводородный или фторуглеводородный радикал с 1-18С; R’ – алкил с 1-12С: v – валентность М (3 или 4); m – в пределах от 1 до v-1; n – от 1 до v; сумма m+n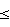 v], получены путем взаимодействия диалкилоловооксида с галогенидом, алкоксидом, алкилгалогенидом или алкилалкоксидом металла при нагревании смеси компонентов реакции в атмосфере сухого инертного газа или в среде инертного органического растворителя. Пределы нагревания реакционной массы от 125 до 185oС. При использовании растворителя с т. кип. <110oС применен автоклав. Выход целевого продукта в примерах, как правило, не указан, за исключением одного, где он составил 92,5%; в другом примере приведен выход продукта в граммах, что позволило рассчитать его в процентах (57%).
К недостаткам способа, предложенного автором для получения соединений, защищенных патентом США 3307973, можно отнести необходимость: v], получены путем взаимодействия диалкилоловооксида с галогенидом, алкоксидом, алкилгалогенидом или алкилалкоксидом металла при нагревании смеси компонентов реакции в атмосфере сухого инертного газа или в среде инертного органического растворителя. Пределы нагревания реакционной массы от 125 до 185oС. При использовании растворителя с т. кип. <110oС применен автоклав. Выход целевого продукта в примерах, как правило, не указан, за исключением одного, где он составил 92,5%; в другом примере приведен выход продукта в граммах, что позволило рассчитать его в процентах (57%).
К недостаткам способа, предложенного автором для получения соединений, защищенных патентом США 3307973, можно отнести необходимость:1) использовать органические растворители с предварительной тщательной осушкой их перед синтезом с последующим отделением растворителей (фильтрацией, упариванием) от продуктов реакции после завершения синтеза; 2) применять автоклав при использовании растворителей с т. кип. <110oС, так как в этом случае процесс протекает при повышенном давлении; 3) проводить реакцию при температуре 140-185oС, если в процессе не используется растворитель, что ухудшает качество продуктов (осмоление, появление окраски и пр.). Перед нами возникла задача устранить перечисленные выше технологические трудности и разработать эффективный способ получения органометаллоксистаннанов, соответствующих общей формуле YAlk2SnOMnRmYn-m-1, где М – металл, выбранный из группы Са, Zn, Сu, Al, Si, Ge, Sn, Ti, Fe, Mn, Co, Cr; R – алкил с 1-4 С, винил, фенил; Y=OR’ (R’ – алкил с 1-4С) или О2CR” (R” – алкил, циклоалкил с 6-11 С); Alk – алкил с 1-8С; n – валентность металла М, m=0-2, n-m 1.
Поставленная задача решена тем, что нами предложен способ получения вышеназванных соединений взаимодействием диалкилоловооксидов (Alk2SnO)x с алкоксидами или ацилоксипроизводными металлов MnRmYn-m, взятых в эквимолярном соотношении; процесс проводят при нагревании реакционной смеси до температуры 80-100oС в присутствии катализатора – карбоновой кислоты общей формулы ZCO2H, где Z – алкил или циклоалкил с 3-11 С, в атмосфере сухого очищенного азота. Количество катализатора составляет 2-5 мас.% от количества взятого в реакцию диалкилоловооксида.
Подробности практического осуществления способа могут быть проиллюстрированы следующими примерами. Все реакции проводились в атмосфере сухого очищенного азота.
Пример 1. Получение тетрабутилдикаприлатдистанноксана.
В трехгорлую колбу, снабженную термометром, мешалкой и обратным холодильником, загружают 26,4 г (0,106 моля) дибутилоловооксида, 55 г (0,106 моля) дибутилоловодикаприлата и 0,53 г каприловой кислоты. Реакционную смесь перемешивают при температуре 80-90oС до полного растворения дибутилоловооксида (около 55 мин). В результате получают 81 г (99,5%) тетрабутилдикаприлоксидистанноксана nD 26 1,4872-1,4875, содержание олова 30,7-30,9%.
Пример 2. Получение дибутилбутокси(трибутоксититанокси) станнана.
В условиях, аналогичных описанным в примере 1, из 13,6 г (0,04 моля) тетрабутоксититана и 9,95 г (0,04 моля) дибутилоловооксида в присутствии 0,41 г каприловой кислоты (температура реакции 90oС, время – 35 мин) получают 23 г (97,7%) дибутилбутокси (трибутоксититанокси)станнана. nD 26 1,4959-1,4974, содержание олова 20,0-20,2%.
Пример 3. Получение дибутилэтокси(триэтоксисилокси) станнана.
В условиях, аналогичных описанным в примере 1, из 35,8 г (0,144 моля) дибутилоловооксида и 30 г (0,144 моля) тетраэтоксисилана в присутствии 1,79 г каприловой кислоты (температура реакции 100oС, время – 50 минут) получают 64,9 г (98,6%) дибутилэтокси(триэтоксисилокси)станнана. Содержание олова 25,8-26,1%.
Пример 4. Получение дибутилметокси(метилвинилметоксисилокси) станнанаВ условиях, аналогичных описанным в примере 1, из 94 г (0,378 моля) дибутилоловооксида и 50 г (0,378 моля) метилвинилдиметоксисилана в присутствии 4,7 г каприловой кислоты (температура реакции 100oС, время – 4 часа) получают 143 г (99,3%) дибутилметокси(метилвинилметоксисилокси)станнана, содержание олова 30,9-31,2%. Пример 5. Получение дибутилэтокси(метилфенилэтоксисилокси) станнана В условиях, аналогичных описанным в примере 1, из 94 г (0,378 моля) дибутилоловооксида и 77,6 г (0,378 моля) метилфенилдиэтоксисилана в присутствии 4,7 г лауриновой кислоты (температура реакции 85-90oС, время – 4 часа) получают 171 г (97%) дибутилэтокси(метилфенилэтоксисилокси)станнана, содержание олова 25,9- 26,2%. Пример 6. Получение дибутилкаприлат (дикаприлатферроокси) станнана В условиях, аналогичных описанным в примере 2, из 2,38 г (0,01 моля) дибутилоловооксида и 4,65 г (0,01 моля) трикаприлата железа в присутствии 0,1 г каприловой кислоты (температура реакции 95-100oС, время – 50 мин) получают 6,58 г (93,6%) дибутилкаприлат (дикаприлатферроокси)станнана. Содержание олова 15,9-16,3%. Пример 7. Получение диметил(2-этилгексанат)(2-этилгексанаткальцийокси)станнана В условиях, аналогичных описанным в примере 2, из 3,29 г (0,02 моля) диметилоловооксида и 6,53 г (0,02 моля) октоата кальция в присутствии 0,07 г 2-этилгексановой кислоты (температура реакции 85-95oС, время – 40 мин) получают 9,43 г (96%) диметил (2-этилгексанат)(2-этилгексанаткальцийокси)станнана. Содержание олова 24,1-24,3%. Пример 8. Получение диметил(2-этилгексанат)(2-этилгексанатцинкокси)станнана В условиях, аналогичных описанным в примере 2, из 3,29 г (0,02 моля) диметилоловооксида и 7,04 г (0,02 моля) октоата цинка в присутствии 0,07 г 2-этилгексановой кислоты (температура реакции 95-100oС, время – 50 мин) получают 9,81 г (95%) диметил (2-этилгексанат)(2-этилгексанатцинкокси)станнана. Содержание олова 22,7-23,1%. Пример 9. Получение диметилизобутокси(диизобутоксиалюмоокси) станнана В условиях, аналогичных описанным в примере 3, из 3,29 г (0,02 моля) диметилоловооксида и 4,93 г (0,02 моля) триизобутоксиалюминия в присутствии 0,07 г масляной кислоты (температура реакции 94-98oС, время – 60 мин) получают 7,64 г (93%) диметилизобутокси (диизобутоксиалюмоокси)станнана. Содержание олова 28,7-29,1%. Пример 10. Получение дибутилизобутокси(диизобутоксихромокси) станнана В условиях, аналогичных описанным в примере 2, из 2,38 г (0,01 моля) дибутилоловооксида и 2,71 г (0,01 моля) триизобутоксиалюминия в присутствии 0,07 г масляной кислоты (температура реакции 80-90oС, время – 50 мин) получают 4,64 г (91,2%) дибутилизобутокси (диизобутоксихромокси)станнана. Содержание олова 23,1-23,5%. Пример 11. Получение диметилциклопентилацетат(циклопентил-ацетатмарганецокси)станнана В условиях, аналогичных описанным в примере 2, из 3,29 г (0,02 моля) диметилоловооксида и 6,19 г (0,02 моля) циклопентилацетата марганца в присутствии 0,07 г циклопентилуксусной кислоты (температура реакции 85-95oС, время – 55 мин) получают 9,0 г (95%) диметилциклопентилацетат(циклопентилацетатмарганецокси)станнана. Содержание олова 24,9-25,2%. Пример 12. Получение дибутилциклопентилацетат(циклопентил-ацетаткобальтокси)станнана В условиях, аналогичных описанным в примере 1, из 2,38 г (0,01 моля) дибутилоловооксида и 3,13 г (0,01 моля) циклопентилацетата кобальта в присутствии 0,07 г циклопентилуксусной кислоты (температура реакции 95-100oС, время – 40 мин) получают 5,26 г (95,5%) дибутилциклопентилацетат(циклопентилацетаткобальтокси) станнана. Содержание олова 21,3-21,6%. Пример 13. Получение диоктилциклопентилацетат(циклопентил-ацетатмедьокси)станнана В условиях, аналогичных описанным в примере 1, из 3,61 г (0,01 моля) диоктилоловооксида и 3,18 г (0,01 моля) циклопентилацетата меди в присутствии 0,07 г циклопентилуксусной кислоты (температура реакции 94-100oС, время – 40 мин) получают 6,52 г (96%) диоктилциклопентилацетат(циклопентилацетатмедьокси) станнана. Содержание олова 17,3-17,6%. Пример 14. Получение тетрабутилдикаприлатдистанноксана В реактор емкостью 400 л с рубашкой, снабженный термометром, мешалкой и обратным холодильником, загружают 77,26 кг дибутилоловооксида, 160,98 кг дибутилстаннандикаприлата и 1,76 кг каприловой кислоты. Реакционную смесь перемешивают при температуре 90  10oС до полного растворения дибутилоловооксида (около 2 часов). В результате получают 228,47 кг (95,9%) тетрабутилдикаприлатдистанноксана. nD 26 1,4874, содержание олова 30,8%.
Пример 15. Получение дибутилэтокси(триэтоксигерманийокси) станнана.
В условиях, аналогичных описанным в примере 1, из 23,8 г (0,1 моля) дибутилоловооксида и 23,7 г (0,1 моля) тетраэтоксигермана в присутствии 0,95 г каприловой кислоты (температура реакции 85-95oС, время – 50 минут) получают 46,3 г (97,5%) дибутилэтокси (триэтоксигерманийокси)станнана, содержание олова 24,8-25,1%.
В результате изучения патентной документации и научно-технической литературы нами не обнаружено материалов, которые бы порочили новизну предлагаемого способа. Последний обладает рядом преимуществ по сравнению с прототипом, представляет собой шаг вперед по сравнению с известным способом и соответствует изобретательскому уровню. Способ успешно реализован в условиях опытного производства.
Таким образом, предложенное техническое решение соответствует критериям патентоспособности. 10oС до полного растворения дибутилоловооксида (около 2 часов). В результате получают 228,47 кг (95,9%) тетрабутилдикаприлатдистанноксана. nD 26 1,4874, содержание олова 30,8%.
Пример 15. Получение дибутилэтокси(триэтоксигерманийокси) станнана.
В условиях, аналогичных описанным в примере 1, из 23,8 г (0,1 моля) дибутилоловооксида и 23,7 г (0,1 моля) тетраэтоксигермана в присутствии 0,95 г каприловой кислоты (температура реакции 85-95oС, время – 50 минут) получают 46,3 г (97,5%) дибутилэтокси (триэтоксигерманийокси)станнана, содержание олова 24,8-25,1%.
В результате изучения патентной документации и научно-технической литературы нами не обнаружено материалов, которые бы порочили новизну предлагаемого способа. Последний обладает рядом преимуществ по сравнению с прототипом, представляет собой шаг вперед по сравнению с известным способом и соответствует изобретательскому уровню. Способ успешно реализован в условиях опытного производства.
Таким образом, предложенное техническое решение соответствует критериям патентоспособности.
Формула изобретения
YAlk2SnOMnRmYn-m-1, где М представляет собой металл, выбранный из группы Са, Zn, Cu, Al, Si, Ge, Sn, Ti, Fe, Mn, Co, Cr; Y – OR’ (R’ означает алкил с 1-4C) или O2CR” (R” означает алкил, циклоалкил с 6-11С); Alk – алкил с 1-8С; R – алкил с 1-4С, винил, фенил; n – валентность металла М; m=0-2; n-m 1,отличающийся тем, что взаимодействию подвергают диалкилоловооксид с металлоорганическими соединениями формулы MnRmYn-m, где М, R, Y, m, n имеют указанные выше значения, при температуре 80-100oС в присутствии катализатора – карбоновой кислоты общей формулы ZCO2H, где Z – алкил или циклоалкил с 3-11С, в атмосфере сухого очищенного азота. 2. Способ по п.1, отличающийся тем, что катализатор добавляют в реакционную смесь в количестве от 2 до 5% от массы диалкилоловооксида. MM4A Досрочное прекращение действия патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 17.01.2004
Извещение опубликовано: 10.03.2005 БИ: 07/2005
NF4A Восстановление действия патента Российской Федерации на изобретение
Извещение опубликовано: 27.05.2005 БИ: 15/2005
|
||||||||||||||||||||||||||