Патент на изобретение №2201919
|
||||||||||||||||||||||||||
(54) СТИРИЛСУЛЬФОНОВЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ, СПОСОБ ИНГИБИРОВАНИЯ РОСТА ОПУХОЛЕВЫХ КЛЕТОК, СПОСОБ ИНДУКЦИИ АПОПТОЗА ОПУХОЛЕВЫХ КЛЕТОК И СПОСОБ ЛЕЧЕНИЯ
(57) Реферат: Изобретение относится к стирилсульфоновым соединениям формулы I, где n представляет собой ноль или единицу; R1 выбирают из группы, включающей хлор, фтор или бром; и R2 выбирают из группы, включающей хлор, фтор и бром; R3 выбирают из группы, включающей водород и фтор; оба радикала R1 и R2 могут не являться хлором в случае, когда R3 представляет собой водород; и R1 может не являться хлором в случае, когда R2 представляет фтор, и R3 представляет водород в том же самом соединении. Описаны также фармацевтические композиции на основе соединений формулы II, III и IV. Стирилсульфоновые соединения, являющиеся предметом настоящего изобретения, селективно ингибируют пролиферацию клеток опухоли молочной железы и опухоли простаты и индуцируют апоптоз таких опухолевых клеток, не затрагивая нормальные клетки. 10 с. и 32 з.п. ф-лы, 9 ил, 5 табл. 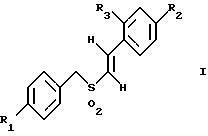 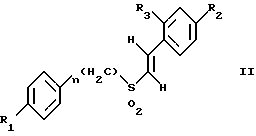 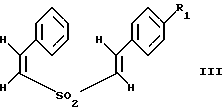 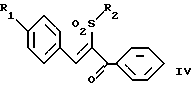
Изобретение относится к композициям для лечения рака и способам лечения рака, в частности рака молочной железы и рака простаты. Уровень техники Внеклеточные сигналы, принимаемые трансмембранными рецепторами, передаются в клетки путем каскада реакций трансдукции сигнала (Pelech et al., Science 257:1335 (1992)), который включает множество физиологических процессов, таких как индукция пролиферации клеток, дифференцировка клеток или апоптоз (Davis et al., J.Biol. Chem. 268:14553 (1993)). Каскад реакций активированной митогеном протеинкиназы (МАРК) является основной сигнальной системой, с помощью которой клетки осуществляют трансдукцию внеклеточных сигналов, вызывающую внутриклеточные ответные реакции (Nishida et al., Trends Biochem. Sci. 18: 128 (1993); Blumer et al. Trends Biochem. Sci. 19:236 (1994)). Продукты многих стадий этого каскада реакций сохраняются, и в различных препаратах обнаруживают гомологи активированных митогеном протеинкиназ. В клетках млекопитающих наиболее типичными и наилучшим образом изученными членами МАРК-семейства являются киназы, регулируемые внеклеточным сигналом (ERK), ERK-1 и ERK-2, причем уникальной особенностью их всех является то, что они активируются при фосфорилировании остатков треонина или тирозина при действии киназы “двойной” специфичности в обратном направлении (Posada et al. . Science 255:212 (1992); Biggs III et al., Proc. Natl. Acad. Sci. USA 89:6295 (1992); Garner et al., Genes Dev. 6:1280 (1992)). Последние исследования позволили идентифицировать дополнительную подгруппу семейства МАРК, в которую входят известные как c-Jun NН2-терминированные киназы 1 и 2 (JNK-1 и JNK-2), которые имеют различную специфичность в отношении субстрата и регулируются действием различных стимулов (Hibi et al., Genes Dev. 7:2135 (1993)). JNK относятся к классу стресс-активируемых протеинкиназ (SPK). Как было показано, JNKs активируются при действии на клетки УФ-излучения, при предвоспалительном цитокинезе или стрессе, вызванном действием окружающей среды (Derijard et al., Cell 1025 (1994)). Активированная JNK связывается с концевой аминогруппой c-Jun белка и повышает транскрипционную активность белка путем фосфорилирования его в положении ser63 и ser73 (Adler et al., Proc. Natl. Acad. Sci. USA 89:5341 (1992); Kwok et al., Nature 370:223 (1994)). Анализ установленной первичной последовательности киназ JNK показывает, что они отдаленно схожи с киназами ERK (Davis. Trends Biochem. Sci. 19:1470 (1994)). Как ERK, так и JNK в ответ на воздействие внешних стимулов фосфорилируются по Туг и Thr, что приводит к их активации (Davis, Trends Biochem. Sci. 19: 1470 (1994)). Участки фосфорилирования (Thr и Туr), которые играют критическую роль в их активации, сохраняются между ERK и JNK (Davis. Trends Biochem. Sci. 19: 1470 (1994)). Однако эти участки фосфорилирования расположены в очевидно двойственных мотивах фосфорилирования: Thr-Pro-Tyr (JNK) и Thr-Glu-Tyr (ERK). Фосфорилирование МАРК и JNK при воздействии внешнего сигнала часто связано с активацией протеинтирозинкиназ (РТК) (Gille et al., Nature 358:414 (1992)), что приводит к возникновению большого семейства белков, включающего некоторые рецепторы фактора роста и другие молекулы, передающие сигнал. Протеинтирозинкиназы представляют собой ферменты, которые катализируют хорошо известные химические реакции: фосфорилирование тирозинового остатка (Hunter et al. Annu. Rev. Biochem. 54:897 (1985)). Тирозинкиназы рецептора представляют собой, в частности, привлекающие мишени для создания лекарственных препаратов, поскольку блокаторы доменов субстрата этих киназ, вероятно, будут являться эффективными и селективными антипролиферативными агентами. Возможное использование блокаторов протеинтирозинкиназы в качестве антипролиферативных средств было выявлено еще в 1981 году, когда в качестве РТК-блокатора был предложен кверцетин (Graziani et al., Eur. J. Biochem. 135: 583-589 (1983)). Наиболее вероятный каскад реакций МАРК связан с внеклеточным сигналом регуляции киназы, который запускает каскад реакций Ras/Raf/MEK/BRK-киназа (Boudewijn et al., Trends Biochem. Sci. 20, 18 (1995)). Когда такой каскад запускается под действием различных сигналов, МАРК фосфорилируют ряд белков, включающий некоторые факторы транскрипции, которые транслоцируются в ядра и активируют ген транскрипции. Возможно такое негативно-регулирующее воздействие, которое может остановить этот каскад реакций. Таким образом, необходимы новые противораковые химио-терапевтические средства, которые направленно воздействуют на тирозинкиназу рецепторов и останавливают каскад реакций Ras/Raf/MEK/BRK-киназа. Онкобелки в целом и белки, передающие сигнал, в частности, вероятно, являются наиболее селективной мишенью для химиотерапии, поскольку они представляют подкласс белков, активность которых имеет существенное значение для пролиферации клеток, а также поскольку их активность значительно возрастает при пролиферативных заболеваниях. Согласно одному из вариантов осуществления изобретения предлагаются новые соединения формулы I 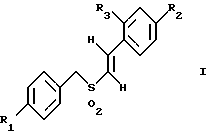 где R1 и R2 независимо выбирают из группы, включающей хлор, фтор или бром; и R3 выбирают из группы, включающей водород и фтор; оба радикала R1 и R2 могут не являться хлором в случае, когда R3 представляет собой водород; и R1 может не являться хлором в случае, когда R2 обозначает фтор, и R3 обозначает водород в том же самом соединении. Согласно другому варианту осуществления изобретения предлагается фармацевтическая композиция, включающая фармацевтически приемлемый носитель и соединение формулы II 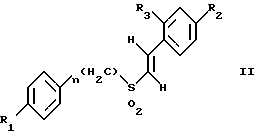 где n представляет ноль или единицу; R1 выбирают из группы, включающей водород, хлор, фтор или бром; и R2 выбирают из группы, включающей водород, хлор, фтор, бром, метил и метокси; и R3 выбирают из группы, включающей водород, хлор и фтор; при условии, что R2 может не являться метилом или метокси в случае, когда оба радикала R1 и R3 представляют собой водород, и n равно нулю или единице; и все радикалы R1, R2 и R3 могут не являться водородом, в случае, когда n представляет собой единицу. Согласно предпочтительному варианту осуществления изобретения предлагается фармацевтическая композиция, включающая фармацевтически приемлемый носитель и соединение формулы II, где R3 представляет собой водород, R1 и R2 независимо выбирают из группы, включающей хлор, фтор и бром. Согласно другому варианту осуществления изобретения предлагается фармацевтическая композиция, включающая фармацевтически приемлемый носитель и соединение формулы III: 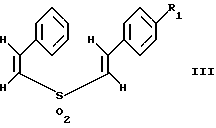 где R1 выбирают из группы, включающей водород, хлор, фтор и бром. Согласно другому варианту осуществления изобретения предлагается способ лечения пациента от рака молочной железы или рака простаты, включающий введение указанному пациенту эффективного количества соединения формулы II или III, одного или в сочетании с фармацевтически приемлемым носителем. Согласно другому варианту осуществления изобретения предлагается способ ингибирования роста клеток опухоли молочной железы или простаты у пациента, пораженного раком молочной железы или простаты, включающий введение указанному пациенту эффективного количества соединения формулы III, одного или в сочетании с фармацевтически приемлемым носителем. Кроме того, предлагается способ индукции апоптоза клеток опухоли молочной железы или простаты у пациента, пораженного раком молочной железы или простаты, включающий введение указанному пациенту эффективного количества соединения формулы III, одного или в сочетании с фармацевтически приемлемым носителем. Изобретение также относится к фармацевтическим композициям и терапевтическим способам, описанным выше, в которых указанное соединение представляет собой соединение формулы IV: 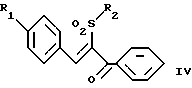 где R1 выбирают из группы, включающей фтор и бром, и R2 выбирают из группы, включающей 2-хлорофенил, 4-хлорофенил, 4-фторофенил и 2-нитрофенил. На фиг. 1А и фиг.1В представлены гистограммы, иллюстрирующие действие соединений: Е-2,4-дифторостирил-4-фторобензилсульфон (FRI-2), Е-4-фторостирил-4-бромо-бензилсульфон (FRI-6), Е-4-бромостирил-4-фторобензилсульфон (FRI-7), Е-4-фторостирил-4-хлоробензилсульфон (FRI-20), и Е-4-хлоростирил-4-хлоробензилсульфон (FRI-22) – на клетки линий NIH3T3, MCF7, ВТ-20 и LnCaP. Клетки обрабатывали соединениями в концентрации 2,5 мкМ (фиг.1А) и 5,0 мкМ (фиг. 1В) и через 48 часов определяли процент выживаемости клеток методом эксклюзии с использованием красителя трипанового синего. Все приведенные данные представляют собой среднее значение для трех независимых экспериментов. Стандартное отклонение не превышает 10%. На фиг.2А представлена гистограмма, иллюстрирующая зависимость подавления роста клеток линий MCF7, ВТ-20, LnCaP и NIH3T3 от концентрации при обработке их FRI-20. Клетки обрабатывают при концентрации FRI-20, равной 0,250 нМ, 500 нМ, 1 мкМ, 2,5 мкМ и 5,0 мкМ в течение 48 часов. Процентное содержание живых клеток определяют методом эксклюзии с использованием красителя трипанового синего. Приведено среднее значение для трех независимых экспериментов. На фиг. 2В представлена гистограмма жизнеспособности клеток линий MCF7, ВТ-20, LnCaP и NIH3T3 после обработки их FRI-20 в течение различных промежутков времени. Все клетки обрабатывали соединением FRI-20 в концентрации 2,5 мкМ, и методом эксклюзии с использованием красителя трипанового синего определяли количество жизнеспособных клеток через 12, 24, 48 и 72 часа. Приведено среднее значение для трех независимых экспериментов. На фиг. 3А представлен график зависимости, иллюстрирующий влияние активности соединения FRI-20 на нормальные клетки линий NIH3T3, HeLa и HFL; позитивно реагирующие на эстроген клетки опухоли молочной железы линий SKBR-3, 435, ВТ-20). Фиг.3В аналогична фиг.3А, с тем исключением, что обработанные клетки включают андрогензависимые клетки опухоли простаты линии LnCaP, а также андрогеннезависимые клетки опухоли простаты линий DU-145 и PC-3, Все клетки обрабатывали соединением FRI-20 в концентрации 2,5 и 5,0 мкМ и анализировали жизнеспособность клеток через 48 часов методом эксклюзии с использованием красителя трипанового синего. Приведено среднее значение для трех независимых экспериментов. Отклонение не превышает 10%. На фиг. 4 приведена серия блот-анализов цикла клеток линии LnCaP, подвергнутых обработке с использованием соединения FRI-20 и контрольной обработке. Клетки линии LnCaP обрабатывали 120 мл ДМСО (контрольные клетки) или 2,5 мкМ FRI-20 в 10 мл ДМСО. Клетки отбирали через 6, 12, 24 и 48 часов после обработки, окрашивали пропидий иодидом и анализировали методом проточной цитометрии. На фиг. 5 представлена авторадиограмма после электрофореза в полиакриламидном геле с использованием додецилсульфата натрия, иллюстрирующий действие соединения FRI-20 на активность ERK/MAPK. Клетки линий LnCaP, MCF-7 и NIH3T3 обрабатывают соединением FRI-20 и также, как и обработанные ДМСО клетки, подвергают иммунокомплексному методу анализа киназной активности ERK/MAPK с использованием в качестве субстрата основного миелинового белка (МBР). Активность ERK-2 в отношении МВР анализировали в присутствии [  –32P]ATP. Фосфорилированный МВР разделяли методом электрофореза в полиакриламидном геле с использованием додецилсульфата натрия при 12% концентрации полиакриламидного геля и визуализировали авторадиографически.
На фиг. 6 представлен блоттинг распределения ERK-2 и JNK/SAPK белков в клетках линий NIH3T3, LnCaP и MCF-7. Лизаты клеточных культур, содержащие 100 мг белков, подвергают электрофорезу. После электрофореза и переноса на поливинилиденовую мембрану белки подвергают блоттингу против поликлональных антител к ERK-2 и JNK-2 и визуализируют с использованием хемилюминесценции.
На фиг. 7 представлена авторадиограмма после электрофореза в полиакриламидном геле с использованием додецилсульфата натрия, иллюстрирующий действие FRI-20 на активность JNK/SAPK. JNK иммунопреципитировали из 100 мг лизатов клеточных культур с поликлональными антителами к JNK и подвергали иммунокомплексному методу анализа киназной активности с использованием в качестве субстрата GST-c-Jun (1-79). Фосфорилированные белки разделяли электрофорезом в полиакриламидном геле с использованием додецилсульфата натрия (SDS-PAGE) и визуализировали авторадиографически. Эксперимент повторяли три раза и получили сходные результаты.
Согласно настоящему изобретению некоторые производные стирилсульфонов оказывают влияние на каскад реакций, связанный с передачей сигнала с участием МАРК, оказывая при этом действие на рост и жизнеспособность опухолевых клеток. Эти соединения ингибируют рост и пролиферацию клеток опухоли молочной железы и опухоли простаты дозозависимым образом, не оказывая влияния на рост нормальных клеток. Ингибирование роста клеток связано с регулированием активности киназ ERK и JNK – разновидностей МАРК. Способность стирилсульфонов регулировать активность этих киназ MAPKs и индуцировать остановку роста клеток определяется природой и положением функциональных групп, имеющихся в соединении.
Обработка клеток опухоли молочной железы и опухоли простаты стирилсульфонами, являющимися предметом настоящего изобретения, приводит к ингибированию пролиферации клеток и индукции гибели клеток, вызванной апоптозом. Этот эффект наблюдается как для клеток, позитивно реагирующих на действие эстрогенового рецептора, так и для клеток, негативно реагирующих на действие эстрогенового рецептора, хотя при тестировании раковых клеток молочной железы, клетки линии 361, показали значительную резистентность к стирилсульфонам. Ингибирование пролиферации клеток и индукция гибели клеток, вызванная апоптозом, также наблюдаются для андрогензависимых, а также андрогеннезависимых клеток опухоли простаты, хотя первые значительно более чувствительны к стирилсульфоновым лекарственым средствам.
Клетки опухоли, подвергнутые действию соединений, являющихся предметом настоящего изобретения, аккумулируются на G2/M фазе цикла клетки. Если клетки проходят С2/М-фазу, по-видимому, они претерпевают апоптоз. Обработка нормальных клеток стирилсульфонами не приводит к такому действию на прогрессию цикла клетки. Нормальные клетки характеризуются обычной прогрессией цикла клетки как в присутствии, так и в отсутствие стирилсульфоновых лекарственных препаратов.
Как клетки, обработанные стирилсульфоновыми лекарственными препаратами, являющимися предметом настоящего изобретения, так и необработанные клетки, характеризуются сходным уровнем внутриклеточной ERK-2, однако биохимическая активность киназы ERK-2, что определяют по способности к фосфорилированию субстрата – основного миелинового белка (МВР), значительно отличается у обработанных лекарственным препаратом клеток по сравнению с необработанными клетками. В случае клеток опухоли простаты обработка соединением FR-20, предпочтительным соединением согласно настоящему изобретению, снижает способность к фосфорилированию МВР более, чем на 80%, по сравнению с обработкой контрольным препаратом, не содержащим соединение FR-20. Вестерн-блоттинг лизатов клеток, обработанных лекарственным препаратом и не содержащим лекарства контролем с антителами к ERK-2 дает то же самое количество белка в обоих лизатах, свидетельствуя о том, что более высокий уровень фосфорилирования МВК в обработанных контролем клетках не является следствием различного количества ERK-2 белка в лизатах. Эти результаты подтверждают, что стирилсульфоны, являющиеся предметом настоящего изобретения, блокируют фосфорилирующую способность ERK-2.
Стирилсульфоны, являющиеся предметом настоящего изобретения, усиливают способность JNK фосфорилировать c-Jun, по сравнению с клетками, обработанными контрольным (не содержащим FR-20) препаратом. Вне связи, с какой-либо теорией, эти результаты указывают на то, что стирилсульфоны могут действовать подобно предвоспалительному цитокинезу или УФ-свету, активируя каскад реакций JNK, который, в свою очередь, может включать в работу гены, ответственные за ингибирование роста клеток и апоптоз.
Синтез стирилсульфонов –32P]ATP. Фосфорилированный МВР разделяли методом электрофореза в полиакриламидном геле с использованием додецилсульфата натрия при 12% концентрации полиакриламидного геля и визуализировали авторадиографически.
На фиг. 6 представлен блоттинг распределения ERK-2 и JNK/SAPK белков в клетках линий NIH3T3, LnCaP и MCF-7. Лизаты клеточных культур, содержащие 100 мг белков, подвергают электрофорезу. После электрофореза и переноса на поливинилиденовую мембрану белки подвергают блоттингу против поликлональных антител к ERK-2 и JNK-2 и визуализируют с использованием хемилюминесценции.
На фиг. 7 представлена авторадиограмма после электрофореза в полиакриламидном геле с использованием додецилсульфата натрия, иллюстрирующий действие FRI-20 на активность JNK/SAPK. JNK иммунопреципитировали из 100 мг лизатов клеточных культур с поликлональными антителами к JNK и подвергали иммунокомплексному методу анализа киназной активности с использованием в качестве субстрата GST-c-Jun (1-79). Фосфорилированные белки разделяли электрофорезом в полиакриламидном геле с использованием додецилсульфата натрия (SDS-PAGE) и визуализировали авторадиографически. Эксперимент повторяли три раза и получили сходные результаты.
Согласно настоящему изобретению некоторые производные стирилсульфонов оказывают влияние на каскад реакций, связанный с передачей сигнала с участием МАРК, оказывая при этом действие на рост и жизнеспособность опухолевых клеток. Эти соединения ингибируют рост и пролиферацию клеток опухоли молочной железы и опухоли простаты дозозависимым образом, не оказывая влияния на рост нормальных клеток. Ингибирование роста клеток связано с регулированием активности киназ ERK и JNK – разновидностей МАРК. Способность стирилсульфонов регулировать активность этих киназ MAPKs и индуцировать остановку роста клеток определяется природой и положением функциональных групп, имеющихся в соединении.
Обработка клеток опухоли молочной железы и опухоли простаты стирилсульфонами, являющимися предметом настоящего изобретения, приводит к ингибированию пролиферации клеток и индукции гибели клеток, вызванной апоптозом. Этот эффект наблюдается как для клеток, позитивно реагирующих на действие эстрогенового рецептора, так и для клеток, негативно реагирующих на действие эстрогенового рецептора, хотя при тестировании раковых клеток молочной железы, клетки линии 361, показали значительную резистентность к стирилсульфонам. Ингибирование пролиферации клеток и индукция гибели клеток, вызванная апоптозом, также наблюдаются для андрогензависимых, а также андрогеннезависимых клеток опухоли простаты, хотя первые значительно более чувствительны к стирилсульфоновым лекарственым средствам.
Клетки опухоли, подвергнутые действию соединений, являющихся предметом настоящего изобретения, аккумулируются на G2/M фазе цикла клетки. Если клетки проходят С2/М-фазу, по-видимому, они претерпевают апоптоз. Обработка нормальных клеток стирилсульфонами не приводит к такому действию на прогрессию цикла клетки. Нормальные клетки характеризуются обычной прогрессией цикла клетки как в присутствии, так и в отсутствие стирилсульфоновых лекарственных препаратов.
Как клетки, обработанные стирилсульфоновыми лекарственными препаратами, являющимися предметом настоящего изобретения, так и необработанные клетки, характеризуются сходным уровнем внутриклеточной ERK-2, однако биохимическая активность киназы ERK-2, что определяют по способности к фосфорилированию субстрата – основного миелинового белка (МВР), значительно отличается у обработанных лекарственным препаратом клеток по сравнению с необработанными клетками. В случае клеток опухоли простаты обработка соединением FR-20, предпочтительным соединением согласно настоящему изобретению, снижает способность к фосфорилированию МВР более, чем на 80%, по сравнению с обработкой контрольным препаратом, не содержащим соединение FR-20. Вестерн-блоттинг лизатов клеток, обработанных лекарственным препаратом и не содержащим лекарства контролем с антителами к ERK-2 дает то же самое количество белка в обоих лизатах, свидетельствуя о том, что более высокий уровень фосфорилирования МВК в обработанных контролем клетках не является следствием различного количества ERK-2 белка в лизатах. Эти результаты подтверждают, что стирилсульфоны, являющиеся предметом настоящего изобретения, блокируют фосфорилирующую способность ERK-2.
Стирилсульфоны, являющиеся предметом настоящего изобретения, усиливают способность JNK фосфорилировать c-Jun, по сравнению с клетками, обработанными контрольным (не содержащим FR-20) препаратом. Вне связи, с какой-либо теорией, эти результаты указывают на то, что стирилсульфоны могут действовать подобно предвоспалительному цитокинезу или УФ-свету, активируя каскад реакций JNK, который, в свою очередь, может включать в работу гены, ответственные за ингибирование роста клеток и апоптоз.
Синтез стирилсульфоновСоединения, являющиеся предметом настоящего изобретения, характеризуются цис-транс изомерией, являющейся следствием присутствия одной или более двойных связей. Эти соединения называют согласно системе Кана-Ингольда-Прелога, Рекомендации ИЮПАК 1974 года, раздел Е: Стереохимия, в Nomenclature of Organic Chemistry, Pergamon, Elmsford, NY, 1979 (The “Blue Book”). См. также: March, Advanced Organic Chemistry, John Wiley & Sons, Inc., New York. NY, 4th ed., 1992, p. 127-138. Стерическое расположение относительно двойной связи обозначается как “Z” или “Е”. (Е)-стирил- и бензилсульфоны получают конденсацией Кневенагеля ароматических альдегидов с активными метиленовыми компонентами, такими, как арил-, бензил-, стирилсульфонилуксусные кислоты, фенациларилсульфоны и сульфонилдиуксусная кислота. Этот метод описан в: Reddy et. al., Acta. Chim. Hung. 115: 269 (1984); Reddy et. al. , Sulfur Letters 13:83 (1991); Reddy et. al., Synthesis 322 (1984); и Reddy et. al., Sulfur Letters 7:43 (1987), эти сведения включены в настоящее описание в качестве ссылки. (Z)-бензил- и (Z)-стирилсульфоны синтезируют путем нуклеофильного присоединения ароматических или алифатических тиолов к фенилацетилену с последующим окислением полученного продукта 30%-ным пероксидом водорода. Синтез бензилсульфонил- и арилсульфонилуксусных кислот Арил- и бензилсульфонилуксусные кислоты являются исходными соединениями для синтеза (Е)-стириларил- и (Е)-стирилбензилсульфонов. Арилсульфонилуксусные кислоты могут быть получены конденсацией арилсульфината натрия с хлоруксусной кислотой в области щелочных значений рН. Другой метод синтеза тех же самых соединений включает окисление продуктов, полученных при конденсации арилтиолата натрия с хлоруксусной кислотой. Бензилсульфонилуксусные кислоты могут быть синтезированы при окислении продуктов конденсации бензилхлорида с тиогликолятом натрия 30%-ным пероксидом водорода. Кроме того, бензилсульфонилуксусные кислоты могут быть синтезированы при окислении продуктов конденсации натриевой соли бензилтиолов с хлоруксусной кислотой 30%-ным пероксидом водорода. Синтез (Е)-стириларил- и (Е)-бензилсульфонов Для получения (Е)-стирилбензил- и (Е)-бензилсульфонов смесь соответствующей сульфонилуксусной кислоты (например, 10 ммоль), ароматического альдегида (например, 10 ммоль) и каталитическое количество бензиламина в уксусной кислоте (например, 15 мл) нагревают с обратным холодильником в течение 2-3 часов. После охлаждения добавляют осушенный эфир, и охлаждают реакционную смесь в течение ночи. Эфирный раствор последовательно промывают насыщенным раствором гидрокарбоната натрия, бисульфита натрия, разбавленной хлороводородной кислотой и, окончательно, водой. Выпаривание раствора в осушенном эфире, содержащего сульфат натрия, дает твердый продукт – (Е)-стириларил- или бензилсульфон, который может быть затем перекристаллизован из 2-пропанола или из 95%-ного этанола. Синтез (Z)-стириларил- и (Z)-стирилбензилсульфонов (Z)-стириларил- и (Z)-стирилбензилсульфоны могут быть получены путем присоединения арилтиолата натрия или бензилтиолата натрия, полученного из соответствующего тиола (например, 10 ммоль) и гидроксида натрия (например, 20 ммоль), к свежеперегнанному фенилацетилену в метаноле. Смесь нагревают с обратным холодильником в течение 24 часов и выливают на раздробленный лед. (Z)-стириларил- и (Z)-стирилбензилсульфиды окисляют 30%-ным пероксидом водорода и получают (Z)-стириларил- и (Z)-стирилбензилсульфоны. Синтез (Е),(Е)- и (Е),(Z)-бис(стирил)сульфонов (Е), (Е)-бис(стирил)сульфоны могут быть получены конденсацией сульфонилдиуксусной кислоты с ароматическими альдегидами в присутствии бензиламина в качестве катализатора. Реакционную смесь нагревают с обратным холодильником в течение 2 часов в ледяной уксусной кислоте. После охлаждения в реакционную смесь добавляют абсолютный эфир, и последовательно промывают насыщенным раствором гидрокарбоната натрия, бисульфита натрия, разбавленной хлороводородной кислотой и водой. Выпаривание высушенного эфирного слоя дает (Е), (Е)-бис(стирил)сульфоны. (Z), (Е)-бис(стирил)сульфоны могут быть получены путем смешения раствора (Z)-стирилсульфонилуксусной кислоты в ледяной уксусной кислоте с аральдегидом и бензиламином. Раствор кипятят в течение 3 часов. Реакционную смесь охлаждают и добавляют осушенный эфир. Продукты разделяют фильтрованием. Фильтрат разбавляют дополнительным количеством эфира и промывают насыщенным раствором гидрокарбоната натрия, бисульфита натрия, разбавленной хлороводородной кислотой и водой. Эфирный слой отделяют, высушивают и после упаривания получают (Z),(Е)-бис(стирил)сульфоны. Стирилсульфоны, являющиеся предметом настоящего изобретения, могут вводиться в форме фармацевтической композиции, в сочетании с фармацевтически приемлемым носителем. Количество активного ингредиента в такой композиции может составлять от 0,1 до 99,99 весовых процентов. Под термином “фармацевтически приемлемый носитель” понимают любой носитель, разбавитель или наполнитель, который совместим с другими компонентами композиции и не вреден реципиенту. Соединения, являющиеся предметом настоящего изобретения, могут вводиться пациентам (млекопитающим, включая животных и человека), пораженным раком молочной железы или раком простаты. Соединения могут вводиться любым способом, включая оральное и парентеральное введение. Парентеральное введение включает, например, внутривенное, внутримышечное, внутриартериальное, интраперитональное, назальное, ректальное и подкожное введение. Активное средство предпочтительно вводится с фармацевтически приемлемым носителем, выбранным в зависимости от предполагаемого способа введения и общепринятой фармацевтической практики. Активный ингредиент может вводиться в дозированных формах в соответствии с общепринятой практикой в области приготовления фармацевтических препаратов. См.: Gennaro Alphonso, ed. Remington Pharmaceutical Sciences. 18 th Ed. , (1990) Mack Publishing Co., Easton, PA. Подходящие дозированные формы могут включать, например, таблетки, капсулы, растворы, парентеральные растворы, пастилки, суппозитории или суспензии. Для парентерального введения активное средство может быть смешано с подходящим носителем или разбавителем, таким как вода, масло, солевой раствор, водный раствор декстрозы (глюкозы) и других сахаров, гликоль, например, такой, как пропиленгликоль или полиэтиленгликоль. Растворы для парентерального введения предпочтительно содержат водорастворимую соль активного средства. Также могут быть добавлены подходящие антиокислительные средства и консерванты. Подходящие антиокислительные средства включают сульфит, аскорбиновую кислоту, лимонную кислоту и ее соли, натриевую соль ЭДТА. Подходящие консерванты включают бензалконий хлорид, метил- или пропилпарабен и хлорбутанол. Для орального введения активное средство может быть смешано с одним или более твердым неактивным ингредиентом для приготовления таблеток, капсул, или других подходящих дозированных форм для орального введения. Например, активный ингредиент может быть смешан с карбоксиметилцеллюлозой кальция, стеаратом магния, маннитом или крахмалом, а затем сформован в таблетки обычными методами таблетирования. Конкретная доза соединения, являющегося предметом настоящего изобретения, необходимая для получения терапевтического эффекта, конечно, будет зависеть от индивидуальных особенностей пациента, которые включают величину, вес, возраст и пол пациента, природу и стадию заболевания, степень выраженности заболевания и способ введения лекарства. Например, может быть использована суточная доза от около 0,05 до около 50 мг/кг в день. Также возможны более высокие или более низкие дозы. Изобретение иллюстрируется следующими примерами, которые не ограничивают объем изобретения. Методика 1 Общая методика синтеза стирил- и бензиларилсульфонов К раствору гидроксида натрия (8 г, 0,2 моль) в метаноле (200 мл) медленно добавляют соответствующий тиофенол или бензилмеркаптан (0,1 моль). Затем частями добавляют хлоруксусную кислоту (0,1 моль), и реакционную смесь нагревают с обратным холодильником в течение 2-3 часов. Охлажденный продукт выливают на измельченный лед и нейтрализуют разбавленной хлороводородной кислотой (200 мл). Полученные арил- и бензилтиоуксусные кислоты (0,1 моль) подвергают окислению 30%-ным раствором пероксида водорода (0,12 моль) в ледяной уксусной кислоте (25 мл) при нагревании с обратным холодильником в течение 1-2 часов. Содержимое охлаждают и выливают на измельченный лед. Отделенное твердое вещество перекристаллизовывают из горячей воды и получают арил- и бензилсульфонилуксусные кислоты. Смесь соответствующей арил- или бензилсульфонилуксусной кислоты (0,001 моль), ароматического альдегида (0,001 моль) и бензиламина (1 мл) в ледяной уксусной кислоте (15 мл) нагревают с обратным холодильником в течение 2-3 часов. Продукт охлаждают и обрабатывают осушенным эфиром (50 мл). Разделенный продукт собирают с помощью фильтрации. Фильтрат разбавляют дополнительным количеством эфира и последовательно промывают насыщенным раствором бикарбоната натрия (20 мл), бисульфита натрия (20 мл), разбавленной хлороводородной кислотой (20 мл) и окончательно водой (35 мл). Выпаривание высушенного эфирного слоя в большинстве случаев позволяет получить твердое вещество. Однако в некоторых случаях отделяется сиропообразный продукт, который переходит в твердое состояние при обработке 2-пропанолом. Чистоту продукта контролируют ТСХ (силикагель BDH, гексан/этилацетат в соотношении 3:1). Методика 2 Общая методика синтеза (Е)(Е)- и (Е)(Z)-бис-(стирил)сульфонов К свежеперегнанному фенилацетилену (51,07 г, 0,5 моль) добавляют тиогликолят натрия, полученый из тиогликолевой кислоты (46 г, 0,5 моль) и гидроксида натрия (40 г, 1 моль) в метаноле (250 мл). Смесь нагревают с обратным холодильником в течение 24 часов и выливают на измельченный лед (500 мл) после охлаждения. Стирилтиоуксусную кислоту, образующуюся после нейтрализации разбавленной хлороводородной кислотой (250 мл), фильтруют и высушивают; выход 88 г (90%); т.пл. 84-86oС. Затем стирилтиоуксусную кислоту окисляют до стирилсульфонилуксусной кислоты следующим образом. Смесь стирилтиоуксусной кислоты (5 г, 25 ммоль) в ледяной уксусной кислоте (35 мл) и 30%-ный пероксид водорода (15 мл) нагревают с обратным холодильником в течение 60 мин, после охлаждения смесь выливают на измельченный лед (200 мл). Отделенное соединение фильтруют и перекристаллизовывают из горячей воды, получают белые кристаллические хлопья (Z)-стирилсульфонилуксусной кислоты; выход 2,4 г (41%); т.пл. 150-151oС. Раствор (Z)-стирилсульфонилуксусной кислоты (2,263 г, 10 ммоль) в ледяной уксусной кислоте (6 мл) смешивают с ароматическим альдегидом (10 ммоль) и бензиламином (0,2 мл) и нагревают с обратным холодильником в течение 3 часов. Реакционную смесь охлаждают, обрабатывают осушенным эфиром (50 мл), и продукты разделяют фильтрацией. Полученный фильтрат разбавляют дополнительным количеством эфира и последовательно промывают насыщенным раствором гидрокарбоната натрия (15 мл), бисульфита натрия (15 мл), разбавленной хлороводородной кислотой (20 мл) и окончательно водой (30 мл). Упаривание эфирного слоя дает (Е)(Z)-бис-(стирил)сульфоны. (Е), (Е)-бис-(стирил)сульфоны получают согласно той же самой методике, которая описана выше, с тем исключением, что вместо (Z)-стирилсульфонилуксусной кислоты используют сульфонилдиуксусную кислоту, и используют вдвое большее количество ароматического альдегида (20 ммоль). Методика 3 Общая методика синтеза 2-(арилсульфонил)-1-фенил-3-арил-2-пропен-1-онов Эти соединения синтезируют двумя способами, согласно которым используют различные условия реакции, различные растворители и катализаторы. Способ 1: Фенациларилсульфоны получают при нагревании с обратным холодильником в течение 6-8 часов  -бромацетофенонов (0,05 моль) и арилсульфинатов натрия (0,05 моль) в абсолютном этаноле (200 мл). Продукт, который отделяют после охлаждения, фильтруют и промывают несколько раз водой до удаления бромида натрия. Затем продукт перекристаллизовывают из этанола: фенацилфенилсульфон, т. пл. 90-91oС; фенацил-п-фторофенилсульфон, т.пл. 148-149oС; фенацил-п-бромофенилсульфон, т. пл. 121-122oС; фенацил-п-метоксифенилсульфон, т.пл. 104-105oС; п-нитрофенацилфенилсульфон, т.пл. 136-137oС.
Раствор фенациларилсульфона (0,01 моль) в уксусной кислоте (10 мл) смешивают с аральдегидом (0,01 моль) и бензиламином (0,02 мл), нагревают с обратным холодильником в течение 3 часов. Раствор охлаждают и добавляют осушенный эфир (50 мл). Эфирный раствор последовательно промывают разбавленной хлороводородной кислотой, водным 10%-ным раствором NaOH, насыщенным раствором NаНSО3 и водой. Упаривание высушенного эфирного слоя дает твердый продукт, который очищают перекристаллизацией.
Способ 2: В коническую колбу объемом 500 мл, снабженную вводом азота, помещают сухой тетрагидрофуран (200 мл). К содержимому колбы добавляют по каплям при непрерывном перемешивании раствор хлорида титана (IV) (11 мл, 0,01 моль) в абсолютном четыреххлористом углероде. В течение всего времени добавления температуру колбы поддерживают равной -20oС. К реакционной смеси добавляют смесь фенациларилсульфона (0,01 моль) и ароматического альдегида (0,01 моль), а также медленно добавляют пиридин (4 мл, 0,04 моль) в тетрагидрофуране (8 мл), в течение 1 часа. Содержимое колбы перемешивают в течение 10-12 часов, обрабатывают водой (50 мл) и затем добавляют эфир (50 мл). Эфирный слой отделяют и промывают 15 мл насыщенных растворов 10%-ного гидроксида натрия, бисульфита натрия и насыщенного солевого раствора. Упаривание высушенного эфирного слоя дает 2-(арилсульфонил)-1-фенил-3-арил-2-пропен-1-оны.
Пример 1 -бромацетофенонов (0,05 моль) и арилсульфинатов натрия (0,05 моль) в абсолютном этаноле (200 мл). Продукт, который отделяют после охлаждения, фильтруют и промывают несколько раз водой до удаления бромида натрия. Затем продукт перекристаллизовывают из этанола: фенацилфенилсульфон, т. пл. 90-91oС; фенацил-п-фторофенилсульфон, т.пл. 148-149oС; фенацил-п-бромофенилсульфон, т. пл. 121-122oС; фенацил-п-метоксифенилсульфон, т.пл. 104-105oС; п-нитрофенацилфенилсульфон, т.пл. 136-137oС.
Раствор фенациларилсульфона (0,01 моль) в уксусной кислоте (10 мл) смешивают с аральдегидом (0,01 моль) и бензиламином (0,02 мл), нагревают с обратным холодильником в течение 3 часов. Раствор охлаждают и добавляют осушенный эфир (50 мл). Эфирный раствор последовательно промывают разбавленной хлороводородной кислотой, водным 10%-ным раствором NaOH, насыщенным раствором NаНSО3 и водой. Упаривание высушенного эфирного слоя дает твердый продукт, который очищают перекристаллизацией.
Способ 2: В коническую колбу объемом 500 мл, снабженную вводом азота, помещают сухой тетрагидрофуран (200 мл). К содержимому колбы добавляют по каплям при непрерывном перемешивании раствор хлорида титана (IV) (11 мл, 0,01 моль) в абсолютном четыреххлористом углероде. В течение всего времени добавления температуру колбы поддерживают равной -20oС. К реакционной смеси добавляют смесь фенациларилсульфона (0,01 моль) и ароматического альдегида (0,01 моль), а также медленно добавляют пиридин (4 мл, 0,04 моль) в тетрагидрофуране (8 мл), в течение 1 часа. Содержимое колбы перемешивают в течение 10-12 часов, обрабатывают водой (50 мл) и затем добавляют эфир (50 мл). Эфирный слой отделяют и промывают 15 мл насыщенных растворов 10%-ного гидроксида натрия, бисульфита натрия и насыщенного солевого раствора. Упаривание высушенного эфирного слоя дает 2-(арилсульфонил)-1-фенил-3-арил-2-пропен-1-оны.
Пример 1Е-стирилфенилсульфон Синтез осуществляют согласно методике 1, используя раствор фенилсульфонилуксусной кислоты (0,01 моль) и бензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 68-72%. Пример 2 Е-4-хлорстирилфенилсульфон Синтез осуществляют согласно методике 1, используя раствор фенилсульфонилуксусной кислоты (0,01 моль) и 4-хлоробензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 78-80%. Пример 3 Е-2,4-дихлорстирилфенилсульфон Синтез осуществляют согласно методике 1, используя раствор фенилсульфонилуксусной кислоты (0,01 моль) и 2,4-дихлоробензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 60-65%. Пример 4 Е-4-бромстирилфенилсульфон Синтез осуществляют согласно методике 1, используя раствор фенилсульфонилуксусной кислоты (0,01 моль) и 4-бромбензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 78-80%. Пример 5 Е-4-хлорстирил-4-хлорфенилсульфон Синтез осуществляют согласно методике 1, используя раствор 4-хлорфенилсульфонилуксусной кислоты (0,01 моль) и 4-хлорбензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 70-72%. Пример 6 Е-4-метилстирил-4-хлорфенилсульфон Синтез осуществляют согласно методике 1, используя раствор 4-хлорфенилсульфонилуксусной кислоты (0,01 моль) и 4-метилбензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 60-64%. Пример 7 Е-4-метоксистирил-4-хлорфенилсульфон Синтез осуществляют согласно методике 1, используя раствор 4-хлорофенилсульфонилуксусной кислоты (0,01 моль) и 4-метоксибензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 68-70%. Пример 8 Е-4-бромстирил-4-хлорфенилсульфон Синтез осуществляют согласно методике 1, используя раствор 4-хлорофенилсульфонилуксусной кислоты (0,01 моль) и 4-бромобензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 80%. Пример 9 Е-2-хлорстирилбензилсульфон Синтез осуществляют согласно методике 1, используя раствор бензилсульфонилуксусной кислоты (0,01 моль) и 2-хлоробензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 72%. Пример 10 Е-4-хлорстирилбензилсульфон Синтез осуществляют согласно методике 1, используя раствор бензилсульфонилуксусной кислоты (0,01 моль) и 4-хлорбензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 78%. Пример 11 Е-4-фторстирил-4-хлорбензилсульфон Синтез осуществляют согласно методике 1, используя раствор 4-хлорбензилсульфонилуксусной кислоты (0,01 моль) и 4-фторбензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 72%. Пример 12 Е-4-хлорстирил-4-хлорбензилсульфон Синтез осуществляют согласно методике 1, используя раствор 4-хлорбензилсульфонилуксусной кислоты (0,01 моль) и 4-хлорбензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 80%. Пример 13 Е-4-фторстирил-4-фторбензилсульфон Синтез осуществляют согласно методике 1, используя раствор 4-фторбензилсульфонилуксусной кислоты (0,01 моль) и 4-фторбензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 73%. Пример 14 Е-2,4-дифторстирил-4-фторбензилсульфон Синтез осуществляют согласно методике 1, используя раствор 4-фторбензилсульфонилуксусной кислоты (0,01 моль) и 2,4-дифторбензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 68%. Пример 15 Е-4-фторстирил-4-бромбензилсульфон Синтез осуществляют согласно методике 1, используя раствор 4-бромбензилсульфонилуксусной кислоты (0,01 моль) и 4-фторбензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 82%. Пример 16 Е-4-бромстирил-4-бромбензилсульфон Синтез осуществляют согласно методике 1, используя раствор 4-бромбензилсульфонилуксусной кислоты (0,01 моль) и 4-бромбензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 88%. Пример 17 Е-4-бромстирил-4-фторбензилсульфон Синтез осуществляют согласно методике 1, используя раствор 4-фторбензилсульфонилуксусной кислоты (0,01 моль) и 4-бромбензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 82%. Пример 18 Е-4-хлорстирил-4-бромбензилсульфон Синтез осуществляют согласно методике 1, используя раствор 4-бромбензилсульфонилуксусной кислоты (0,01 моль) и 4-хлорбензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 88%. Пример 19 Е-4-бромстирил-4-хлорбензилсульфон Синтез осуществляют согласно методике 1, используя раствор 4-хлоробензилсульфонилуксусной кислоты (0,01 моль) и 4-бромбензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 92%. Пример 20 (Z)-стирил-(Е)-4-фторстирилсульфон Синтез осуществляют согласно методике 2, используя раствор (Z)-стирилсульфонилуксусной кислоты (0,01 моль) и 4-4-фторбензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 68%. Пример 21 (Z)-стирил-(Е)-4-бромстирилсульфон Синтез осуществляют согласно методике 2, используя раствор (Z)-стирилсульфонилуксусной кислоты (0,01 моль) и 4-бромбензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 70%. Пример 22 (Z)-стирил-(Е)-4-хлорстирилсульфон Синтез осуществляют согласно методике 2, используя раствор (Z)-стирилсульфонилуксусной кислоты (0,01 моль) и 4-хлорбензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 64%. Пример 23 2-[(4-фторфенил)-сульфонил]-1-фенил-3-(4-фторфенил)-2-пропен-1-он Синтез осуществляют согласно способу 1 методики 3, используя раствор фенацил-4-фторфенилсульфона (0,01 моль) и 4-фторбензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 63%. Пример 24 2-[(2-хлорфенил)-сульфонил]-1-фенил-3-(4-фторофенил)-2-пропен-1-он Синтез осуществляют согласно способу 1 методики 3, используя раствор фенацил-2-хлорфенилсульфона (0,01 моль) и 4-фторбензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 58%. Пример 25 2-[(2-хлорфенил)-сульфонил]-1-фенил-3-(4-бромфенил)-2-пропен-1-он Синтез осуществляют согласно способу 1 методики 3, используя раствор фенацил-2-хлорфенилсульфона (0,01 моль) и 4-бромобензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 66%. Пример 26 2-[(4-хлорфенил)-сульфонил]-1-фенил-3-(4-бромфенил)-2-пропен-1-он Синтез осуществляют согласно способу 1 методики 3, используя раствор фенацил-4-хлорфенилсульфона (0,01 моль) и 4-бромбензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 60%. Пример 27 2-[(2-нитрофенил)-сульфонил]-1-фенил-3-(4-бромфенил)-2-пропен-1-он Синтез осуществляют согласно способу 1 методики 3, используя раствор фенацил-2-нитрофенилсульфона (0,01 моль) и 4-бромбензальдегида (0,01 моль). Указанное в заглавии соединение получают с выходом 56%. Пример 28 Ингибирование роста опухолевых клеток стирилсульфонами А. Клетки Влияние стирилсульфонов на рост нормальных клеток и клеток опухоли молочной железы и простаты исследовали, используя четыре линии клеток, NIH3T3, MCF-7, ВТ-20 и LnCap. Клетки линии NIH3T3 относятся к нормальным фибробластам, в то время как клетки LnCap представляют собой линию андрогензависимых клеток опухоли простаты. Клетки MCF-7 относятся к линии эстрогензависимых клеток опухоли молочной железы, в то время как клетки ВТ-20 относятся к линии эстрогеннезависимых клеток опухоли молочной железы. Клетки линий MCF-7 и ВТ-20 выращивают в модифицированной Дульбекко среде Игла (DMEM), содержащей 10% эмбриональной бычьей сыворотки, с добавками пенициллина и стрептомицина. Клетки линии LnCaP культивируют в RPMI с 10% эмбриональной бычьей сыворотки, содержащей пенициллин и стрептомицин. Клетки линии NIH3T3 выращивают в DMEM, содержащей 10% сыворотки теленка, с добавками пенициллина и стрептомицина. Все клеточные культуры выращивают при температуре 37oС и влажной атмосфере, содержащей 5% СO2. В. Обработка стирилсульфонами и оценка выживаемости клеток Клетки обрабатывают растворами исследуемых соединений с концентрацией 2,5 мкМ или 5,0 мкМ и определяют выживаемость клеток через 48 часов методом эксклюзии с использованием красителя трипанового синего. Соединения, приведенные в таблицах 1, 2 и 3, ингибируют рост клеток и индуцируют гибель клеток в различной степени. В таблицах показан процент выживаемости клеток для клеток линий LnCap и MCF-7, обработанных 5,0 мкМ соединения. Пять из числа более активных соединений, которые проявляют наибольшую активность, были обозначены как FRI-2 (Е-2,4-дифторстирил-4-фторбензилсульфон), FRI-6 (Е-4-фторостирил-4-бромобензилсульфон), FRI-7 (Е-4-бромостирил-4-фторобензилсульфон), FRI-20 (Е-4-фторстирил-4-хлоробензилсульфон), FRI-22 (Е-4-хлоростирил-4-хлорбензилсульфон). Как было обнаружено, эти соединения существенно ингибируют рост и индуцируют гибель клеток линии LnCaP, ВТ-20 и МСР-7 при концентрации соединений 2,5 мМ (фигура 1А) и 5 мМ (фигура 1В), через 48 часов после обработки указанными соединениями. В идентичных условиях более 80% клеток линии NIH3T3 выживают после инкубации в течение 48 часов (фигуры 1А и 1В). Е-4-хлорстирил-4-бромбензилсульфон и Е-4-бромстирил-4-хлорбензилсульфон также являются высокоактивными соединениями. С. Исследование влияния используемой дозы Исследование влияния используемой дозы при обработке клеток стирилсульфоном проводили при обработке клеток соединением FRI-20, одним из пяти наиболее активных соединений. Клетки линий NIH3T3, MCF-7, ВТ-20 и LnCaP обрабатывали соединением FRI-20, растворенным в ДМСО в концентрации 250 нМ, 500 нМ, 1 мкМ, 2,5 мкМ и 5 мкМ, и исследовали пролиферацию клеток и жизнеспособность через 48 часов (фигура 2А). Процентное содержание жизнеспособных клеток определяли методом эксклюзии с использованием красителя трипанового синего. Контрольные образцы клеток обрабатывали растворителем ДМСО, для того, чтобы определить действие растворителя на клетки. При концентрации 250 нМ через 48 часов наблюдалась примерно 10%-ная гибель клеток линий MCF-7, ВТ-20 и LnCaP и примерно 15-20%-ное ингибирование деления клеток по сравнению с необработанными клетками. При концентрации 500 нМ наблюдалось примерно 30-50%-ное ингибирование пролиферации клеток и 25-30%-ная гибель клеток линий LnCaP, ВТ-20 и MCF-7. В этих условиях только 2-3% клеток линии NIH3T3 при указанных концентрациях оказывались нежизнеспособными. Рост клеток линий LnCaP, ВТ-20 и MCF-7 значительно ингибируется при концентрации соединения FRI-20, равной 1 мкМ, с одновременной потерей клетками жизнеспособности. После 48 часов инкубации при концентрации соединения FRI-20, равной 2,5 мМ, погибают 60-75% клеток линий LnCaP, BT-20 и MCF-7, в то время как более 90% клеток линии NIH3T3 остаются жизнеспособными (фигура 2А). В случае клеток линий LnCaP, BT-20 и MCF-7, обработанных 5 мкМ соединения FRI-20 (фигура 2А), наблюдается примерно 90%-ная гибель клеток. Способность к росту клеток линии NIH3T3 не изменяется или изменяется незначительно, и жизнеспособность этих клеток в присутствии FRI-2, -6, -7, -20 или -22 в концентрации 5 мкМ составляет более 80%. D. Исследование влияния времени Влияние времени на активность FRI-20 было показано следующим образом. Клетки линий NIH3T3, MCF-7, BT-20 и LnCap обрабатывали FRI-20 в концентрации 2,5 мкМ и методом эксклюзии с использованием красителя трипанового синего определяли число жизнеспособных клеток через 12, 24, 48 и 72 часа. Среднее значение для трех независимых экспериментов приведено на фиг.2В. Исследование влияния времени показало, что более 95% клеток линий МСР-7, LnCaP и BT-20 погибают через 72 часа после обработки FRI-20 при концентрации 2,5 мкМ (фигура 2В). Пример 29 Ингибирование роста опухолевых клеток с использованием FRI-20 А. Клетки Влияние FR-20 на рост нормальных клеток и клеток опухоли молочной железы и простаты исследовали, используя девять линий клеток, NIH/3T3 и HFL (линия клеток нормальных фибробластов); MCF-7 и 361 (линии клеток опухоли молочной железы, негативно реагирующих на эстрогеновые рецепторы); ВТ-20, 435 и SKBR-3 (линии клеток опухоли молочной железы, позитивно реагирующих на эстрогеновые рецепторы); LnCaP (линия андрогенчувствительных клеток опухоли простаты); РС-3 и DU-145 (линия не чувствительных к андрогену клеток опухоли простаты). В. Обработка FRI-20 и оценка выживаемости клеток Клетки выращивали по методике, описанной в примере 22.А. FR-20 растворяли в ДМСО и добавляли к клеткам в концентрации 2,5 мкМ и 5,0 мкМ. К контрольным образцам клеток добавляли ДМСО в количестве, эквивалентном количеству растворителя (ДМСО) при добавлении соединения в наибольшей концентрации. Активность соединения определяли через 48 часов методом эксклюзии с использованием красителя трипанового синего. Как было обнаружено, клетки линий NIH3T3 и HFL при обработке соединением с концентрацией, равной 2,5 мкМ и 5,0 мкМ, характеризуются 85-90%-ной жизнеспособностью. Что касается обработки соединением FRI-20 семи линий клеток опухоли молочной железы, то для клеток линий MCF-7, HTB126, Т470 и 435 характерна очень высокая гибель в сочетании с жизнеспособностью, равной менее чем 25 и 10% при концентрации лекарства, равной 2,5 мкМ и 5,0 мкМ., соответственно (фигура 3А). Примерно 50% клеток линий SKBR-3 и ВТ-20 погибают при концентрации соединения, равной 2,5 мкМ, и 75% – при концентрации соединения, равной 5,0 мкМ. С другой стороны, клетки опухоли молочной железы линии 361 проявляют значительную устойчивость к действию соединения FRI-20, 50-75% клеток остаются жизнеспособными при концентрации 2,5 мкМ и 5,0 мкМ. По сравнению с действием на андрогеннезависимые клетки простаты линий DU-145 и РС-3 соединение FRI-20 оказывает сильное действие на жизнеспособность андрогензависимых клеток опухоли простаты линии LnCaP. При концентрации соединения FRI-20, равной 2,5 мМ, погибают 80% клеток линии LnCaP, 40% клеток линии РС-3 и 20% клеток линии DU-145. При концентрации соединения FRI-20, равной 5,0 мМ, погибают 72% клеток линии LnCaP, 47% клеток линии РС-3 и 40% клеток линии DU-145 (фигура 3В). Пример 30 Действие соединения FRI-20 на регуляцию цикла клетки Андрогензависимые клетки опухоли простаты LnCaP обрабатывают 2,0 мкМ соединения FRI-20, растворенного в ДМСО или эквивалентным количеством (10 мл) только одного растворителя. Клетки отбирают через 6, 12, 24 и 48 часов после обработки, окрашивают пропидий иодидом и подвергают анализу методом поточной цитометрии (FACS) для анализа содержания ДНК. Как показано на фиг. 4, добавление FR-20 к культуральной среде приводит к аккумуляции клеток на фазе G2/M цикла клетки, и поскольку клетки проходят эту фазу цикла, они, по-видимому, претерпевают апоптоз. Клетки, которые обработаны только ДМСО, не претерпевают такую блокировку в G2/M фазе цикла клетки, и можно предположить, что наблюдаемый эффект связан с добавлением FRI-20. Обработка нормальных клеток линий NIH3T3 или HFL соединением FRI-20 не оказывает такого эффекта на прогрессию цикла клетки. Клетки линий NIH3T3 и HFL и в присутствии, и в отсутствие лекарства характеризуются обычной прогрессией цикла клетки. Пример 31 Действие соединения FRI-20 на каскад реакций, вызываемый МАРК А. Иммунокомплексное исследование ERK-2 Для изучения действия соединения FRI-20 на каскад реакций, вызываемый МАРК, клетки линий NIH3T3, LnCaP и MCF-7 инкубировали с FRI-20 в концентрации 2,5 мМ в течение 48 часов. После инкубирования клеток в присутствии или в отсутствие соединения FRI-20 клетки лизируют, используя лизисный буфер ERK, содержащий 20 мМ HEPES (N-2-гидроксиэтилпиперазин-N’-2-этансульфоновая кислота) (рН 7,4), 50 мМ  -глицерофосфата, 0,5% тритона Х-100, 2 мМ МgСl2, 1 мМ EGTA, 1 мМ дитиотреитола, 2 мкг/мл лейпептина, 2 мкг/мл апротинина, 100 мкМ фенилметилсульфонилфторида и 1 мМ бензамидина. ERK-2 в 100 мг клеточного лизата иммунопреципитируют путем инкубации лизатного белка с 1 мг поликлональных антител к ERK-2 (антитела sс-154 к ERK-2 получают от Santa Cruz Biotechnology, Inc. ) в течение 1 часа, после чего осуществляют дополнительную инкубацию в течение 1 часа с 20 мкл белок А-сефарозы (Pharmacia). Частицы белка А-сефарозы, связанные с иммунным комплексом, дважды промывают лизисным буфером и дважды ERK/MAPK-буфером, содержащим 20 мМ HEPES (N-2-гидроксиэтилпиперазин-N’-2-этансульфоновая кислота) (рН 7,4), 50 мМ -глицерофосфата, 10 мМ МgСl2, 1 мМ EGTA, 1 мМ дитиотреитола и 100 MM Nа3VO4.
После этого иммунопреципитаты тестируют на МАР-киназную активность in vitro по методике, которая включает использование в качестве субстрата для ERK-2 основного миелинового белка (МВР) в присутствии [–32P]ATP. Затем частицы ресуспендируют в 40 мкл МАРК-буфера, содержащего 100 мкМ [–32P]ATP (5000 распад. мин/пмоль) и проводят анализ киназной активности в течение 20 минут при 30oС, используя в качестве субстрата 5 мкг МВР. Реакцию останавливают добавлением буфера Лэммли с последующим кипячением образцов в течение 3 минут. Белки разделяют электрофорезом в полиакридамидном геле с использованием додецилсульфата натрия при 12% концентрации полиакриламидного геля (SDS-PAGE); гель высушивают и исследуют авторадиограмму. Полученные результаты свидетельствуют о том, что как обработанные, так и необработанные лекарственным препаратом клетки характеризуются одинаковым содержанием внутриклеточной ERK-2, но биохимическая активность ERK-2, которую определяют по способности к фосфорилированию МВР, значительно снижена у обработанных лекарством клеток по сравнению с клетками, обработанными только ДМСО. В случае клеток опухоли простаты обработка соединением FRI-20 снижает способность к фосфорилированию МБР более, чем на 80% по сравнению с контрольными образцами клеток (фигура 5).
В. Вестерн-блоттинг -глицерофосфата, 0,5% тритона Х-100, 2 мМ МgСl2, 1 мМ EGTA, 1 мМ дитиотреитола, 2 мкг/мл лейпептина, 2 мкг/мл апротинина, 100 мкМ фенилметилсульфонилфторида и 1 мМ бензамидина. ERK-2 в 100 мг клеточного лизата иммунопреципитируют путем инкубации лизатного белка с 1 мг поликлональных антител к ERK-2 (антитела sс-154 к ERK-2 получают от Santa Cruz Biotechnology, Inc. ) в течение 1 часа, после чего осуществляют дополнительную инкубацию в течение 1 часа с 20 мкл белок А-сефарозы (Pharmacia). Частицы белка А-сефарозы, связанные с иммунным комплексом, дважды промывают лизисным буфером и дважды ERK/MAPK-буфером, содержащим 20 мМ HEPES (N-2-гидроксиэтилпиперазин-N’-2-этансульфоновая кислота) (рН 7,4), 50 мМ -глицерофосфата, 10 мМ МgСl2, 1 мМ EGTA, 1 мМ дитиотреитола и 100 MM Nа3VO4.
После этого иммунопреципитаты тестируют на МАР-киназную активность in vitro по методике, которая включает использование в качестве субстрата для ERK-2 основного миелинового белка (МВР) в присутствии [–32P]ATP. Затем частицы ресуспендируют в 40 мкл МАРК-буфера, содержащего 100 мкМ [–32P]ATP (5000 распад. мин/пмоль) и проводят анализ киназной активности в течение 20 минут при 30oС, используя в качестве субстрата 5 мкг МВР. Реакцию останавливают добавлением буфера Лэммли с последующим кипячением образцов в течение 3 минут. Белки разделяют электрофорезом в полиакридамидном геле с использованием додецилсульфата натрия при 12% концентрации полиакриламидного геля (SDS-PAGE); гель высушивают и исследуют авторадиограмму. Полученные результаты свидетельствуют о том, что как обработанные, так и необработанные лекарственным препаратом клетки характеризуются одинаковым содержанием внутриклеточной ERK-2, но биохимическая активность ERK-2, которую определяют по способности к фосфорилированию МВР, значительно снижена у обработанных лекарством клеток по сравнению с клетками, обработанными только ДМСО. В случае клеток опухоли простаты обработка соединением FRI-20 снижает способность к фосфорилированию МБР более, чем на 80% по сравнению с контрольными образцами клеток (фигура 5).
В. Вестерн-блоттингЛизаты клеток, обработанных соединением FRI-20, подготавливали для вестерн-блоттинга следующим образом. Клетки линий NIH3T3, LnCaP и MCF-7 высевали с плотностью 2 х 105 клеток на лунку в шестилуночный планшет и выращивали в течение 24 часов. За 2 часа до обработки в каждую лунку добавляли свежую среду. Соединение растворяли в ДМСО для приготовления 2 мМ исходного раствора, который затем добавляли к среде (2 мл) для получения окончательной концентрации, равной 2,5 мкМ и 5,0 мкМ. После выдержки в течение 48 часов при 37oС клетки дважды промывали 10 мл охлажденного на льду фосфатно-солевого буфера и помещали в 400 мкл лизисного буфера, содержащего 25 мМ HEPES (N-2-гидроксиэтилпиперазин-N’-2-этансульфоновая кислота) (рН 7,6), 0,1% тритона Х-100, 30 мМ NaCl, 1,5 мМ МgСl2, 20 мМ -глицерофосфата, 100 мкМ Nа3VO4, 0,2 мМ EDTA, 0,5 мМ дитиотреитола, 2 мкг/мл апротинина, 2 мкг/мл лейпептина, 100 мкМ фенилметилсульфонилхлорида и 1 мМ бензамидина. Лизаты клеток выдерживали на льду в течение 30 минут и центрифугировали в течение 10 минут на микроцентрифуге (16000 х g). Клеточные лизаты отделяли от осадка и нормализовали по содержанию белка.
Вестерн-блоттинг лизатов клеток, обработанных лекарством или обработанных контролем (растворитель без лекарственного препарата), проводили с антителами к ERK-2. Одинаковые количества по суммарному белку (100 мкг) образцов разделяли на отдельных дорожках электрофорезом в 10-12% полиакриламидном геле (SDS-PAGE) и переносили на мембрану Immobilon-P (Millipore, USA). После переноса мембраны блокируют в 3% молоке, затем помечают поликлональными антителами кролика против BRK-2 и Jnk-1 (Santa Cruz Biotechnology Inc., Santa Cruz, CA), после чего вводят зонд – меченные пероксидазой хрена вторые антитела – антитела осла против Ig кролика (Amersham) (разбавление 1:1000). Антитела определяют, используя набор для вестерн-блоттинга “ECL Western blotting analysis kit” (Amersham), следуя представленным изготовителем инструкциям. Вестерн-блоттинг лизатов клеток, обработанных лекарством или обработанных контролем (растворитель без лекарственного препарата), с антителами к ERK-2 свидетельствует об одинаковом количестве белка в обоих лизатах (фигура 6), что говорит о том, что более высокий уровень фосфорилирования МБР в обработанных контролем клетках не является следствием неодинакового содержания белка ERK-2 в лизатах. Эти данные указывают на то, что PRI-20 блокирует фосфорилирующую активность ERK-2.
Пример 32Действие соединения FRI-20 на активируемую действием стресса активность белка Дополнительно для выяснения того, зависит ли активируемая действием стресса активность протеинкиназ (SAPKs), к числу которых относится JNK, от присутствия соединения FRI-20, клетки (линии NIH3T3, MCF-7 или LnCaP) обрабатывали соединением FRI-20, растворенным в ДМСО, или только одним ДМСО. Через 48 часов клетки лизировали с киназным буфером, и использовали лизаты для оценки количества киназы JNK, присутствующей в каждом лизате, вестерн-блоттингом с использованием поликлональных тел к JNK. Биохимическую активность JNK, присутствующей в обработанных соединением FRI-20 и в контрольно обработанных клеточных лизатах, также определяют по иммунопреципитации JNK с последующим инкубированием с GST-c-Jun белком в качестве субстрата для JNK в присутствии [ –32Р]АТР.
Аналогично этому JNK-1 в 100 мг клеточного экстракта иммунопреципитировали путем инкубирования лизата с 1 мг поликлональных антител к JNK-1 (от Santa Cruz Biotechnology) в течение одного часа с последующим дополнительным инкубированием с 20 мкл белка А-сефарозы (“Pharmacia”) в течение одного часа. Частички дважды промывали JNK лизисным буфером (как описано выше) и затем дважды JNK реакционным буфером. Частички ресуспендировали в 40 мкл JNK буфера, содержащего 20 мМ [–32P]ATP (5000 распад мин/пмоль), и проводили киназную реакцию в течение 20 минут при 30oС, используя в качестве субстрата очищенный GST-c-Jun (1-79). Реакцию останавливали и оценивали радиоактивность фосфорилированного GST-c-Jun белка. Результаты свидетельствуют о том, что обработка соединением FRI-20 увеличивает способность JNK к фосфорилированию рекомбинантного GST-c-Jun белка на 60-80% по сравнению с контрольно обработанными клетками (фигура 7).
Как было показано, JNK активируется при действии на клетки УФ-излучения, про-воспалительных цитокинов и стресса, вызванного действием факторов окружающей среды (Derijard et al., Cell 1025 (1994)). Активированная киназа JNK связывается с аминоконцов c-Jun белка и повышает его транскрипционную активность при фосфорилировании в положения ser63 и ser73 (Adler et al., Proc. Natl. Acad. Sci. USA 89:5341 (1992); Kwok et al., Nature 370:223 (1994)). Вне связи с какой-либо теорией, эти результаты указывают на то, что FRI-20 может действовать подобно про-воспалительному цитокину или УФ-свету, активируя каскад реакций JNK, который, в свою очередь, может включать в работу гены, ответственные за ингибирование роста клеток и апоптоз.
Пример 33 мин/пмоль), и проводили киназную реакцию в течение 20 минут при 30oС, используя в качестве субстрата очищенный GST-c-Jun (1-79). Реакцию останавливали и оценивали радиоактивность фосфорилированного GST-c-Jun белка. Результаты свидетельствуют о том, что обработка соединением FRI-20 увеличивает способность JNK к фосфорилированию рекомбинантного GST-c-Jun белка на 60-80% по сравнению с контрольно обработанными клетками (фигура 7).
Как было показано, JNK активируется при действии на клетки УФ-излучения, про-воспалительных цитокинов и стресса, вызванного действием факторов окружающей среды (Derijard et al., Cell 1025 (1994)). Активированная киназа JNK связывается с аминоконцов c-Jun белка и повышает его транскрипционную активность при фосфорилировании в положения ser63 и ser73 (Adler et al., Proc. Natl. Acad. Sci. USA 89:5341 (1992); Kwok et al., Nature 370:223 (1994)). Вне связи с какой-либо теорией, эти результаты указывают на то, что FRI-20 может действовать подобно про-воспалительному цитокину или УФ-свету, активируя каскад реакций JNK, который, в свою очередь, может включать в работу гены, ответственные за ингибирование роста клеток и апоптоз.
Пример 33Сравнение соединения FRI-20 и цисплатина Противоопухолевая активность Вызывающее гибель клеток противоопухолевое действие соединения FRI-20 на чувствительные к андрогену (LnCaP) и нечувствительные к андрогену (DU 145) клетки опухоли простаты сравнивали с аналогичным действием цисплатина (цис-диаминодихлорплатина II), широко используемого средства против опухоли простаты. Клетки выращивали таким образом, как описано в примере 26. Соединение FRI-20 или цисплатин растворяли в ДМСО и добавляли к клеткам в различной концентрации. Жизнеспособность клеток оценивали через 72 часа методом эксклюзии с использованием красителя трипанового синего. Концентрация соединения FRI-20, необходимая для полной гибели клеток линий LnCaP и DU 145, составляет 2,5 мкМ и 5,0 мкМ, соответственно. В аналогичных условиях для полной гибели клеток линий LnCaP и DU 145 под действием цисплатина требуется концентрация 25 мкМ и 15 мкМ, соответственно. Таким образом, соединение FRI-20, по меньшей мере, в 10 раз более активно, чем цисплатин в отношении гибели как гормонзависимых, так и гормоннезависимых клеток опухоли простаты. Все ссылки, приведенные при описании синтетических, препаративных и аналитических методов, даются только в качестве ссылок. Настоящее изобретение может быть использовано в других вариантах, находящихся в пределах существенных признаков изобретения, изложенных в формуле изобретения и в описании изобретения и определяющих объем изобретения. Протокол испытания на голых мышах и результаты I. Получение раковых клеток В проведенных экспериментах были использованы клетки карциномы молочной железы человека линии ВТ-20, эстроген-отрицательные, прогестрон-отрицательные клетки аденокарциномы. Раковые клетки выращивали в соответствии с общепринятой методикой культивирования ткани в термостатируемых вентилируемых колбах объемом 225 см до тех пор, пока не образовывалось количество клеток, достаточное для введения мышам. В указанный момент клетки промывали забуференным фосфатом – физиологическим раствором (PBS) и трипсинизировали 0,25% раствором трипсин-ЭДТА. Клетки трижды промывали PBS и подсчитывали с помощью гемацитометра. Полученную суспензию клеток ресуспендировали в стерильном PBS до концентрации 5,0х107 клеток/мл. Указанная концентрация требуется для введения каждой мыши 5,0х106 клеток в объеме инъекции 0,1 мл. II. Инъекция голым мышам и анализ объема опухоли Голые мыши (ncr/ncr) были приобретены у фирмы Taconic в возрасте 8-10 недель. Мышей содержали в клетках с фильтрующей крышкой при постоянном притоке воздуха. Перед подачей в клетки пищу, воду и подстилку автоклавировали. Мышам в заднюю часть правого бока подкожно вводили 5,0х106 клеток в объеме 0,1 мл с использованием туберкулинового шприца с иглой 27,5. Мышей наблюдали до появления пальпируемых опухолей. В указанный момент определяли объем каждой опухоли по трем измерениям с помощью штангенциркуля. Объем опухоли вычисляли по опубликованной формуле 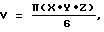 где X, Y и Z – три измеренных диаметра минус 1 мм на толщину кожи (Х-1, Y-1, Z-1). Когда объемы опухолей стали измеряться 2-40 мм3, мышей сгруппировали таким образом, чтобы каждая группа состояла из 7-10 мышей, имеющих как большие, так и малые опухоли, дающие при усреднении приблизительно одинаковый средний объем опухоли. Семи мышам в испытуемой группе каждые два дня (Q2D) внутрибрюшинно вводили 100 мкг Е-4-фторстирил-4-хлорбензилсульфона (пример 11) в объеме 0,1 мл. Это соответствует дозировке соединения 5 мг/кг. Мыши из контрольной группы получали 0,1 мл ДМСО, который является растворителем, применяемом для растворения соединения из примера 11. Массу тела каждой мыши определяли вначале инъекций и затем каждую неделю после начала инъекций. Измерения опухолей проводили дважды в неделю вплоть до 14 дней. Результаты эксперимента представляли как средний объем опухоли плюс или минус стандартная ошибка среднего (SEM). Результаты Фиг.8 показывает, что Q2D внутрибрюшинные инъекции 100 мкг соединения из примера 11 способны в значительной степени уменьшить рост карциномы молочной железы человека. Объем опухоли в контрольной группе увеличивался экспоненциально, тогда как у животных, получавших соединение из примера 11, вначале наблюдалось некоторое снижение объема опухоли с последующим значительно более медленным ростом среднего объема опухоли. Общее уменьшение объема опухоли составило более чем 3 раза за период лечения 14 дней. Введение Q2D 5 мг/кг соединения из примера 11 не вызывало видимых признаков его токсичности. В течение всего времени эксперимента контролировали массу тела мышей, и среднюю массу тела вычисляли и наносили на график. Фиг. 9 показывает, что введение лекарства не оказывает нежелательного эффекта на массу тела животного. У мышей также не проявлялись никакие признаки интоксикации, как то: летаргия, взъерошенность шерсти и сутулость спины. Приведенные данные подтверждают, что заявляемые соединения в соответствии с данным изобретением обладают активностью уничтожать раковые клетки in vivo без связанной с этим токсичностью. Формула изобретения
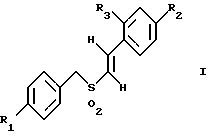 где R1 и R2 независимо выбирают из группы, включающей хлор, фтор и бром; R3 выбирают из группы, включающей водород и фтор; оба радикала R1 и R2 могут не являться хлором в случае, когда R3 представляет собой водород; и R1 может не являться хлором в случае, когда R2 представляет фтор, и R3 представляет водород в том же самом соединении. 2. Соединение по п.1, в котором указанное соединение представляет Е-4-фторстирил 4-фторбензилсульфон. 3. Соединение по п.1, в котором указанное соединение представляет Е-2,4-дифторстирил 4-фторбензилсульфон. 4. Соединение по п.1, в котором указанное соединение представляет Е-4-фторстирил-4-бромбензилсульфон. 5. Соединение по п.1, в котором указанное соединение представляет Е-4-бромстирил-4-бромбензилсульфон. 6. Соединение по п.1, в котором указанное соединение представляет Е-4-хлорстирил-4-бромбензилсульфон. 7. Соединение по п.1, в котором указанное соединение представляет Е-4-бромстирил-4-хлорбензилсульфон. 8. Соединение по п.1, в котором указанное соединение представляет Е-4-бромстирил-4-фторбензилсульфон. 9. Фармацевтическая композиция, обладающая способностью ингибировать рост опухолевых клеток или индуцировать апоптоз опухолевых клеток, отличающаяся тем, что она содержит фармацевтически приемлемый носитель и соединение формулы II 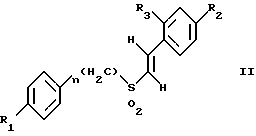 где n = 0 или 1; R1 выбирают из группы, включающей водород, хлор, фтор и бром; R2 выбирают из группы, включающей водород, хлор, фтор, бром, метил и метокси; R3 выбирают из группы, включающей водород, хлор и фтор; при условии, что R2 может не являться метилом или метокси в случае, когда оба радикала R1 и R3 представляют водород, и n = 0 или 1; и все радикалы R1, R2 и R3 могут не являться водородом в случае, когда n = 1. 10. Композиция по п.9, в которой указанное соединение представляет собой Е-4-бромстирилфенилсульфон. 11. Композиция по п.9, в которой указанное соединение представляет собой Е-2,4-дифторстирил-4-фторбензилсульфон. 12. Композиция по п.9, в которой R3 представляет водород, и R1 и R2 независимо выбирают из группы, включающей хлор, фтор и бром. 13. Композиция по п.12, в которой указанное соединение представляет Е-4-хлорстирил-4-хлорфенилсульфон. 14. Композиция по п.12, в которой указанное соединение представляет Е-4-бромстирил-4-хлорфенилсульфон. 15. Композиция по п.12, в которой указанное соединение представляет Е-4-фторстирил-4-хлорбензилсульфон. 16. Композиция по п.12, в которой указанное соединение представляет Е-4-хлорстирил-4-хлорбензилсульфон. 17. Композиция по п.12, в которой указанное соединение представляет Е-4-фторстирил-4-фторбензилсульфон. 18. Композиция по п.12, в которой указанное соединение представляет Е-4-фторстирил-4-бромбензилсульфон. 19. Композиция по п.12, в которой указанное соединение представляет Е-4-бромстирил-4-бромбензилсульфон. 20. Композиция по п.12, в которой указанное соединение представляет Е-4-бромстирил-4-фторбензилсульфон. 21. Композиция по п. 12, в которой указанное соединение представляет собой Е-4-хлорстирил-4-бромбензилсульфон. 22. Композиция по п.12, в которой указанное соединение представляет Е-4-бромстирил-4-хлорбензилсульфон. 23. Фармацевтическая композиция, обладающая способностью ингибировать рост опухолевых клеток или индуцировать апоптоз опухолевых клеток, отличающаяся тем, что она содержит фармацевтически приемлемый носитель и соединения формулы III 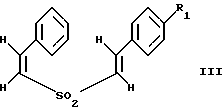 где R1 выбирают из группы, включающей водород, хлор, фтор и бром. 24. Фармацевтическая композиция, обладающая способностью ингибировать рост опухолевых клеток или индуцировать апоптоз опухолевых клеток, отличающаяся тем, что она содержит фармацевтически приемлемый носитель и соединение формулы IV 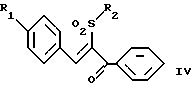 где R1 выбирают из группы, включающей фтор или бром; R2 выбирают из группы, включающей 2-хлорофенил, 4-хлорофенил, 4-фторофенил и 2-нитрофенил. 25. Способ лечения пациента от рака или других пролиферативных заболеваний, включающий введение указанному пациенту эффективного количества фармацевтической композиции по п.9. 26. Способ ингибирования роста опухолевых клеток путем воздействия на них фармацевтической композицией по п.9. 27. Способ индукции апоптоза опухолевых клеток путем воздействия на них фармацевтической композицией по п.9. 28. Способ лечения пациента от рака или других пролиферативных заболеваний, включающий введение указанному пациенту эффективного количества фармацевтической композиции по п.23 или 24. 29. Способ ингибирования роста опухолевых клеток путем воздействия на них фармацевтической композицией по п.23 или 24. 30. Способ индукции апоптоза опухолевых клеток путем воздействия на них фармацевтической композицией по п.23 или 24. 31. Способ по п.25, используемый для лечения рака молочной железы или простаты. 32. Способ по п.26, отличающийся тем, что для ингибирования роста клеток используют клетки опухоли молочной железы или простаты. 33. Способ по п.27, отличающийся тем, что для индукции апоптоза клеток используют клетки опухоли молочной железы или простаты. 34. Способ по п.28, используемый для лечения рака молочной железы или простаты. 35. Способ по п.29, отличающийся тем, что для ингибирования роста клеток используют клетки опухоли молочной железы или простаты. 36. Способ по п.30, отличающийся тем, что для индукции апоптоза клеток используют клетки опухоли молочной железы или простаты. 37. Способ по п.25, отличающийся тем, что композиция содержит соединение, выбранное из (Е)-4-фторстирил-4-хлорбензилсульфона, (Е)-4-фторстирил-4-бромбензилсульфона и (Е)-4-хлорстирил-4-бромбензилсульфона. 38. Способ по п.26, отличающийся тем, что композиция содержит соединение, выбранное из (Е)-4-фторстирил-4-хлорбензилсульфона, (Е)-4-фторстирил-4-бромбензилсульфона и (Е)-4-хлорстирил-4-бромбензилсульфона. 39. Способ по п.27, отличающийся тем, что композиция содержит соединение, выбранное из (Е)-4-фторстирил-4-хлорбензилсульфона, (Е)-4-фторстирил-4-бромбензилсульфона и (Е)-4-хлорстирил-4-бромбензилсульфона. 40. Способ по п.37, предназначенный для лечения рака молочной железы или простаты. 41. Способ по п.38, предназначенный для индукции апоптоза клеток опухоли молочной железы или простаты у пациента, пораженного раком молочной железы или простаты. 42. Способ по п.39, предназначенный для индукции апоптоза клеток опухоли молочной железы или простаты у пациента, пораженного раком молочной железы или простаты. РИСУНКИ
|
||||||||||||||||||||||||||