Патент на изобретение №2201754
|
||||||||||||||||||||||||||
(54) ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, СПОСОБ ЛЕЧЕНИЯ ПСИХОТИЧЕСКИХ СОСТОЯНИЙ И ГИПЕРАКТИВНОСТИ
(57) Реферат: Фармацевтический состав замедленного выделения предназначен для лечения психотических состояний или гиперактивности у теплокровного животного. Состав включает 11-[4-[2-(2-гидроксиэтокси)этил] -1-пиперазинил] дибензо[b,f] [1,4]тиазепин или его фармацевтически приемлемую соль, один или более фармацевтически приемлемых наполнителей и гидроксипропилметилцеллюлозу в качестве гелеобразующего агента. Замедленное выделение обеспечивает одинаковую и постоянную скорость выделения в течение длительного периода времени. За указанный период времени достигается стабильный и желаемый уровень активного ингредиента в плазме крови без необходимости частого приема лекарства. 3 н. и 11 з.п. ф-лы, 2 ил., 11 табл. Настоящее изобретение относится к фармацевтическому составу, в частности к фармацевтическому составу замедленного выделения, включающему 11-[4-[2-(2-гидроксиэтокси)этил] -1-пиперазинил] дибензо[b, f] [1,4] тиазепин или его фармацевтически приемлемую соль. При лечении ряда заболеваний как терапевтически, так и профилактически желательно, чтобы активный фармацевтический ингредиент был замедленного выделения. Желательно, чтобы замедленное выделение в основном обеспечивало одинаковую и постоянную скорость выделения в течение длительного периода времени, за который достигается стабильный и желаемый уровень активного ингредиента в плазме крови без необходимости частого приема лекарства. Несмотря на то, что известно много составов замедленного выделения, в которых используют гелеобраэующие агенты, такие как гидроксипропилметилцеллюлоза, было обнаружено, что по нескольким причинам трудно получить составы замедленного выделения из растворимых лекарств и гелеобразующих агентов, таких как гидроксипропилметилцеллюлоза. Прежде всего, активные ингредиенты, которые растворимы в воде, склонны вырабатывать продукт замедленного выделения, который чувствителен к явлению, известному как “сброс” дозы. То есть, действие активного ингредиента задерживается в течение некоторого времени, но в определенный момент выделение начинается, причем скорость выделения очень высока. Более того, склонны возникать флуктуации концентрации активного ингредиента в плазме крови, которые повышают вероятность токсичности. Кроме того, также была обнаружена некоторая степень ежедневного отклонения концентрации активного ингредиента в плазме крови. Наконец, было обнаружено, что трудно достигнуть желаемых профилей растворения или контролировать скорость выделения растворимого лекарства. Таким образом, существует необходимость в составах замедленного выделения на основе растворимых лекарств, таких как 11-[4-[2-(2-гидроксиэтокси)этил] -1-пиперазинил] дибензo[b, f][1,4]тиазепин или фармацевтически приемлемая соль, которые преодолеют или по крайней мере облегчат (снизят) одну или более из описанных выше трудностей, и которые, кроме того, обеспечат благоприятное свойство: сделают возможным прием (назначение) активного лекарства менее частым, например один раз в день, при достижении уровней активного ингредиента в плазме крови, сходных с уровнями, которых добиваются при назначении меньших доз лекарства более часто, например два или более раз в день. Фиг. 1 показывает профили выделения (растворения) составов замедленного выделения примеров 8, 9 и 10, которые получают путем погружения подходящей таблетки в 750 мл 0,1 N раствора соляной кислоты на 2 ч при 37oС и при перемешивании со скоростью 100 оборотов в минуту и затем путем добавления 250 мл 0,2 М буферного раствора фосфата натрия к среде растворения с тем, чтобы получить рН 6,2. Фиг. 2 показывает профили концентрации активного ингредиента в плазме крови от времени для составов замедленного выделения примеров 1 и 2 и для состава мгновенного выделения примера 12. Соединение 11-[4-[2-(2-гидроксиэтокси)этил] -1-пиперазинил]дибензо[b,f] [1,4] тиазепин (смотри формулу 1) и его фармацевтически приемлемые соли показывают антидопаминергическую активность и могут быть использованы, например, в качестве антипсихотического агента (например, для “управления” проявлений психотических расстройств) или для лечения гиперактивности. Это соединение представляет особый интерес, поскольку оно может быть использовано в качестве антипсихотического агента с существенным снижением возможности вызывать побочные эффекты, такие как острая дистония, острая дискинезия, псевдо-Паркинсонизм и поздняя дискинезия, которые могут возникать при использовании других антипсихотических или нейролептических средств. 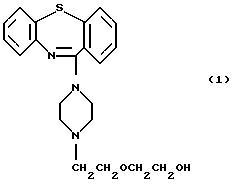 Получение, физические свойства и полезные фармакологические свойства 11-[4-[2-(2-гидроксиэтокси)этил] -1-пиперазинил]дибензо[b,f][1,4]тиазепина и его фармацевтически приемлемых солей описаны в опубликованных Европейских Патентах ЕР 240228 и 282236, а также в Патенте США 4879288, полное содержание которых включено в описание в качестве ссылки. Согласно настоящему изобретению обеспечивается состав замедленного выделения, включающий гелеобразующий агент, предпочтительно гидроксипропилметилцеллюлозу, и 11-[4-[2-(2-гидроксиэтокси)этил]-1-пиперазинил]дибензо[b, f] [1,4] тиазепин или его фармацевтически приемлемую соль вместе с одним или более фармацевтически приемлемыми наполнителями. Предпочтительно, чтобы состав замедленного выделения включал гидрофильную матрицу, содержащую гелеобразующий агент, предпочтительно гидроксипропилметилцеллюлозу, и 11-[4-[2-(2-гидроксиэтокси)этил] -1-пиперазинил] дибензо[b, f] [1,4] тиазепин или его фармацевтически приемлемую соль вместе с одним или более фармацевтически приемлемыми наполнителями. Термин “гелеобразующий агент” означает любое вещество, особенно гидрофильное вещество, которое образует гель при контакте с водой и, следовательно, включает такие вещества, как гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза, гидроксиметилцеллюлоза, гидроксиэтилцеллюлоза, гидроксипропилэтилцеллюлоза, метилцеллюлоза, этилцеллюлоза, карбоксиэтилцеллюлоза, карбоксиметилгидроксиэтилцеллюлоза, карбомер, карбоксиметилцеллюлоза натрия, поливинилпирролидон и тому подобные или их смеси. Гелеобразующий агент – это предпочтительно гидроксипропилметилцеллюлоза. Количество гелеобразующего агента, предпочтительно гидроксипропилметилцеллюлозы, выбирают предпочтительно такое, что, когда активный ингредиент выделяется из состава лекарственного средства регулируемым образом в течение периода времени 4 ч или более, предпочтительно за период времени 8 ч или более и особенно предпочтительно за период времени между 8 и 24 ч, то в конце этого периода выделяется по крайней мере 60% активного ингредиента. Гелеобразующий агент предпочтительно гидроксипропилметилцеллюлоза присутствует в количестве приблизительно от 5 до 50 вес.%, более предпочтительно приблизительно от 5 до 40%, наиболее предпочтительно приблизительно от 8 до 35% и в особенности предпочтительно приблизительно от 10 до 35%. Вообще предпочтительно, чтобы гелеобразующий агент предпочтительно гидроксипропилметилцеллюлоза присутствовал в количестве приблизительно от 10 до 30%, более предпочтительно от приблизительно 15 до 30%. Гидроксипропилметилцеллюлоза может содержать более одного “сорта” полимера и коммерчески доступна под несколькими товарными знаками, например METHOCEL  E, F, J и К от Dow Chemical Co., США, и METALOSEТМ SH от Shin-Etsu, Ltd., Япония. Различные сорта, доступные под данными товарными знаками, имеют различия в содержании метокси- и гидроксипропоксигрупп, а также отличаются по вязкости. Содержание метоксигрупп варьируется от 16,5 до 30 вес.%, содержание гидроксипропоксигрупп варьируется от 4 до 32 вес.%, и вязкость 2%-ного водного раствора при 20oС варьируется от 3 сантипуаз до 100000 сантипуаз. Например, гидроксипропилметилцеллюлоза предпочтительно включает (а) полимер с вязкостью приблизительно от 40 до 60 сантипуаз (в особенности предпочтительно около 50 сантипуаз) с содержанием метоксигрупп приблизительно от 28 до 30 вес.% и содержанием гидроксипропоксигрупп приблизительно от 7 до менее чем 9 вес.%; или (b) полимер с вязкостью приблизительно от 3500 до 5600 сантипуаз (особенно предпочтительно около 4000 сантипуаз) с содержанием метоксигрупп приблизительно от 28 до 30 вес.% и содержанием гидроксипропоксигрупп приблизительно от 7 до 12 вес.%; или (с) полимер с вязкостью приблизительно от 80 до 120 сантипуаз (особенно предпочтительно около 100 сантипуаз) с содержанием метоксигрупп приблизительно от 19 до 24 вес.% и содержанием гидроксипропоксигрупп приблизительно от 7 до менее чем 9 вес. %; или (d) полимер с вязкостью приблизительно от 3500 до 5600 сантипуаз (особенно предпочтительно около 4000 сантипуаз) с содержанием метоксигрупп приблизительно от 19 до 24 вес.% и содержанием гидроксипропоксигрупп приблизительно от 7 до 12 вес.%, или смеси этого. Более предпочтительно, чтобы гидроксипропилметилцеллюлозу выбирали из группы, состоящей из полимеров (а)-(d) или их смесей, описанных выше, с условием, что, если фармацевтический состав содержит гидроксипропилметилцеллюлозу, описанную выше под (d), общее количество гидроксипропилметилцеллюлозы, присутствующей в смеси, должно быть более чем 25,8 вес.%.
В одном варианте настоящего изобретения гидроксипропилметилцеллюлоза включает от 8 до 12% полимера, имеющего вязкость около 4000 сантипуаз, и предпочтительно приблизительно от 5 до 10% полимера. В другом варианте настоящего изобретения гидроксипропилметилцеллюлоза включает от 10 до 35% полимера, имеющего вязкость около 50 сантипуаз, и предпочтительно приблизительно от 10 до 15% полимера.
В предпочтительном варианте настоящего изобретения гидроксипропилметилцеллюлоза включает 15% полимера, имеющего вязкость около 50 сантипуаз, и необязательно около 5% полимера гидроксипропилметилцеллюлозы, имеющего вязкость около 4000 сантипуаз.
В частности, 11-[4-[2-(2-гидроксиэтокси)этил]-1-пиперазинил]дибензо[b,f] [1,4]тиазепин или его фармацевтически приемлемая соль (предпочтительно гемифумарат) присутствует в количестве приблизительно от 10 до 90 вес.%, предпочтительно приблизительно от 20 до 80 вес.%, более предпочтительно приблизительно от 35 до 65 вес.%, наиболее предпочтительно приблизительно от 40 до 60 вес.% и особенно предпочтительно приблизительно от 43,2 до 57,6 вес.%.
В общем состав будет содержать один или более наполнителей. Такие наполнители будут включать разбавители такие, как лактоза, микрокристаллическая целлюлоза, декстроза, маннит, сахароза, сорбит, желатин, смола акации, дикальцийфосфат, трикальцийфосфат, фосфат монокальция, фосфат натрия, карбонат натрия и тому подобное, предпочтительно лактоза и микрокристаллическая целлюлоза; смазочные вещества такие, как стеариновая кислота, стеарат цинка, кальция или магния и тому подобное, предпочтительно стеарат магния; связывающие вещества такие, как сахароза, полиэтиленгликоль, повидон (поливинилпирролидон), кукурузный или маисовый крахмал, предварительно желатинизированный крахмал и тому подобное, предпочтительно повидон (поливинилпирролидон); красящие вещества такие, как оксиды железа, FD&C красители, краски и тому подобное; корригенты, регуляторы рН, которые включают подходящие органические кислоты или соли щелочных металлов (например, лития, натрия или калия) на основе этих кислот таких, как бензойная кислота, лимонная кислота, винная кислота, сукциновая (янтарная) кислота, адипиновая кислота и тому подобные или их соответствующие соли щелочных металлов, предпочтительно соли щелочных металлов на основе таких кислот и, в частности, натриевая соль лимонной кислоты (т.е. цитрат натрия). Наполнитель(и) в общем будет (будут) присутствовать в количестве приблизительно от 10 до 90 вес. %, предпочтительно приблизительно от 20 до 80 вес.%, более предпочтительно приблизительно от 20 до 45 вес.%, наиболее предпочтительно приблизительно от 20 до 40 вес.% и особенно предпочтительно приблизительно от 22,4 до 36,8 вес.%. Фармацевтический состав предпочтительно может содержать один или более фармацевтически приемлемых наполнителей, выбранных из группы, состоящей из микрокристаллической целлюлозы, лактозы, стеарата магния, цитрата натрия и повидона. В частности, состав лекарственного средства может содержать один или более из следующих наполнителей: (а) микрокристаллическую целлюлозу, предпочтительно в количестве приблизительно от 4 до 20 вес.%, (b) лактозу, предпочтительно в количестве приблизительно от 5 до 20 вес.%, (с) стеарат магния, предпочтительно в количестве приблизительно от 1 до 3 вес.%, (d) цитрат натрия в количестве приблизительно от 10 до 30 вес.%, предпочтительно в количестве приблизительно от 12,5 до 25 вес.% и в особенности предпочтительно в количестве около 12,5 вес.%, и (е) повидон (поливинилпирролидон) в количестве приблизительно от 1 до 15 вес.%, предпочтительно в количестве приблизительно от 4 до 6 вес.% и особенно предпочтительно в количестве около 5 вес.% E, F, J и К от Dow Chemical Co., США, и METALOSEТМ SH от Shin-Etsu, Ltd., Япония. Различные сорта, доступные под данными товарными знаками, имеют различия в содержании метокси- и гидроксипропоксигрупп, а также отличаются по вязкости. Содержание метоксигрупп варьируется от 16,5 до 30 вес.%, содержание гидроксипропоксигрупп варьируется от 4 до 32 вес.%, и вязкость 2%-ного водного раствора при 20oС варьируется от 3 сантипуаз до 100000 сантипуаз. Например, гидроксипропилметилцеллюлоза предпочтительно включает (а) полимер с вязкостью приблизительно от 40 до 60 сантипуаз (в особенности предпочтительно около 50 сантипуаз) с содержанием метоксигрупп приблизительно от 28 до 30 вес.% и содержанием гидроксипропоксигрупп приблизительно от 7 до менее чем 9 вес.%; или (b) полимер с вязкостью приблизительно от 3500 до 5600 сантипуаз (особенно предпочтительно около 4000 сантипуаз) с содержанием метоксигрупп приблизительно от 28 до 30 вес.% и содержанием гидроксипропоксигрупп приблизительно от 7 до 12 вес.%; или (с) полимер с вязкостью приблизительно от 80 до 120 сантипуаз (особенно предпочтительно около 100 сантипуаз) с содержанием метоксигрупп приблизительно от 19 до 24 вес.% и содержанием гидроксипропоксигрупп приблизительно от 7 до менее чем 9 вес. %; или (d) полимер с вязкостью приблизительно от 3500 до 5600 сантипуаз (особенно предпочтительно около 4000 сантипуаз) с содержанием метоксигрупп приблизительно от 19 до 24 вес.% и содержанием гидроксипропоксигрупп приблизительно от 7 до 12 вес.%, или смеси этого. Более предпочтительно, чтобы гидроксипропилметилцеллюлозу выбирали из группы, состоящей из полимеров (а)-(d) или их смесей, описанных выше, с условием, что, если фармацевтический состав содержит гидроксипропилметилцеллюлозу, описанную выше под (d), общее количество гидроксипропилметилцеллюлозы, присутствующей в смеси, должно быть более чем 25,8 вес.%.
В одном варианте настоящего изобретения гидроксипропилметилцеллюлоза включает от 8 до 12% полимера, имеющего вязкость около 4000 сантипуаз, и предпочтительно приблизительно от 5 до 10% полимера. В другом варианте настоящего изобретения гидроксипропилметилцеллюлоза включает от 10 до 35% полимера, имеющего вязкость около 50 сантипуаз, и предпочтительно приблизительно от 10 до 15% полимера.
В предпочтительном варианте настоящего изобретения гидроксипропилметилцеллюлоза включает 15% полимера, имеющего вязкость около 50 сантипуаз, и необязательно около 5% полимера гидроксипропилметилцеллюлозы, имеющего вязкость около 4000 сантипуаз.
В частности, 11-[4-[2-(2-гидроксиэтокси)этил]-1-пиперазинил]дибензо[b,f] [1,4]тиазепин или его фармацевтически приемлемая соль (предпочтительно гемифумарат) присутствует в количестве приблизительно от 10 до 90 вес.%, предпочтительно приблизительно от 20 до 80 вес.%, более предпочтительно приблизительно от 35 до 65 вес.%, наиболее предпочтительно приблизительно от 40 до 60 вес.% и особенно предпочтительно приблизительно от 43,2 до 57,6 вес.%.
В общем состав будет содержать один или более наполнителей. Такие наполнители будут включать разбавители такие, как лактоза, микрокристаллическая целлюлоза, декстроза, маннит, сахароза, сорбит, желатин, смола акации, дикальцийфосфат, трикальцийфосфат, фосфат монокальция, фосфат натрия, карбонат натрия и тому подобное, предпочтительно лактоза и микрокристаллическая целлюлоза; смазочные вещества такие, как стеариновая кислота, стеарат цинка, кальция или магния и тому подобное, предпочтительно стеарат магния; связывающие вещества такие, как сахароза, полиэтиленгликоль, повидон (поливинилпирролидон), кукурузный или маисовый крахмал, предварительно желатинизированный крахмал и тому подобное, предпочтительно повидон (поливинилпирролидон); красящие вещества такие, как оксиды железа, FD&C красители, краски и тому подобное; корригенты, регуляторы рН, которые включают подходящие органические кислоты или соли щелочных металлов (например, лития, натрия или калия) на основе этих кислот таких, как бензойная кислота, лимонная кислота, винная кислота, сукциновая (янтарная) кислота, адипиновая кислота и тому подобные или их соответствующие соли щелочных металлов, предпочтительно соли щелочных металлов на основе таких кислот и, в частности, натриевая соль лимонной кислоты (т.е. цитрат натрия). Наполнитель(и) в общем будет (будут) присутствовать в количестве приблизительно от 10 до 90 вес. %, предпочтительно приблизительно от 20 до 80 вес.%, более предпочтительно приблизительно от 20 до 45 вес.%, наиболее предпочтительно приблизительно от 20 до 40 вес.% и особенно предпочтительно приблизительно от 22,4 до 36,8 вес.%. Фармацевтический состав предпочтительно может содержать один или более фармацевтически приемлемых наполнителей, выбранных из группы, состоящей из микрокристаллической целлюлозы, лактозы, стеарата магния, цитрата натрия и повидона. В частности, состав лекарственного средства может содержать один или более из следующих наполнителей: (а) микрокристаллическую целлюлозу, предпочтительно в количестве приблизительно от 4 до 20 вес.%, (b) лактозу, предпочтительно в количестве приблизительно от 5 до 20 вес.%, (с) стеарат магния, предпочтительно в количестве приблизительно от 1 до 3 вес.%, (d) цитрат натрия в количестве приблизительно от 10 до 30 вес.%, предпочтительно в количестве приблизительно от 12,5 до 25 вес.% и в особенности предпочтительно в количестве около 12,5 вес.%, и (е) повидон (поливинилпирролидон) в количестве приблизительно от 1 до 15 вес.%, предпочтительно в количестве приблизительно от 4 до 6 вес.% и особенно предпочтительно в количестве около 5 вес.%Согласно настоящему изобретению также обеспечивается фармацевтический состав замедленного выделения, включающий гелеобразующий агент, предпочтительно гидроксипропилметилцеллюлозу и 11-[4-[2-(2-гидроксиэтокси)этил]-1-пиперазинил] дибензо[b, f] [1,4]тиазепин или его фармацевтически приемлемую соль вместе с одним или более фармацевтически приемлемыми наполнителями, где один из наполнителей является рН модификатором (регулятором). Согласно настоящему изобретению, также обеспечивается состав лекарственного средства замедленного выделения, включающий 11-[4-[2-(2-гидроксиэтокси)этил] -1-пиперазинил]дибензо[b,f][1,4]тиазепин или его фармацевтически приемлемую соль в качестве активного ингредиента, и от 5 до 40% гидроксипропилметилцеллюлозы вместе с одним или более фармацевтически приемлемыми наполнителями. Согласно настоящему изобретению также обеспечивается состав лекарственного средства замедленного выделения, включающий приблизительно от 35 до 65% 11-[4-[2-(2-гидроксиэтокси)этил]-1-пиперазинил]дибензо[b,f][1,4]тиазепина или его фармацевтически приемлемой соли в качестве активного ингредиента и приблизительно от 5 до 40 вес.% гидроксипропилметилцеллюлозы вместе с одним или более фармацевтически приемлемыми наполнителями. Согласно настоящему изобретению также обеспечивается состав лекарственного средства замедленного выделения, включающий приблизительно от 35 до 65% 11-[4-[2-(2-гидроксиэтокси)этил]-1-пиперазинил]дибензо[b,f][1,4]тиазепина или его фармацевтически приемлемой соли в качестве активного ингредиента и приблизительно от 15 до 30% гидроксипропилметилцеллюлозы вместе с приблизительно от 20 до 45% одного или более фармацевтически приемлемых наполнителей. Согласно настоящему изобретению, также обеспечивается состав лекарственного средства замедленного выделения, включающий приблизительно от 35 до 65% 11-[4-[2-(2-гидроксиэтокси)этил]-1-пиперазинил]дибензо[b,f][1,4]тиазепина в качестве активного ингредиента или его фармацевтически приемлемой соли, приблизительно от 5 до 40 вес.% гидроксипропилметилцеллюлозы, приблизительно от 4 до 12% микрокристаллической целлюлозы, приблизительно от 8 до 20% лактозы и остаток, представляющий собой один или более дополнительных фармацевтически приемлемых наполнителей. Такие дополнительные наполнители могут включать компоненты, которые действуют как смазывающие агенты (например, стеарат магния) во время изготовления фармацевтического состава или изготовления лекарственных форм. Согласно настоящему изобретению также обеспечивается состав лекарственного средства замедленного выделения, включающий приблизительно от 5 до 40 вес.% гидроксипропилметилцеллюлозы, выбранной из группы, состоящей из (а) гидроксипропилметилцеллюлозы, имеющей вязкость приблизительно от 40 до 60 сантипуаз, с содержанием метоксигрупп приблизительно от 28 до 30 вес.% и содержанием гидроксипропоксигрупп приблизительно от 7 до менее чем 9 вес.%, (b) гидроксипропилметилцеллюлозы, имеющей вязкость приблизительно от 3500 до 5600 сантипуаз, с содержанием метоксигрупп приблизительно от 28 до 30 вес.% и содержанием гидроксипропоксигрупп приблизительно от 7 до 12 вес.%, (с) гидроксипропилметилцеллюлозы, имеющей вязкость приблизительно от 80 до 120 сантипуаз, с содержанием метоксигрупп приблизительно от 19 до 24 вес.% и содержанием гидроксипропоксигрупп приблизительно от 7 до менее чем 9 вес.% и (d) гидроксипропилметилцеллюлозы, имеющей вязкость приблизительно от 3500 до 5600 сантипуаз, с содержанием метоксигрупп приблизительно от 19 до 24 вес.% и содержанием гидроксипропоксигрупп приблизительно от 7 до 12 вес.% или их смесей; приблизительно от 35 до 65 вес.% 11-[4-[2-(2-гидроксиэтокси)этил]-1-пиперазинил]дибензо[b,f]-[1,4]тиазепина или его фармацевтически приемлемой соли; и приблизительно от 20 до 45 вес.% одного или более одного фармацевтически приемлемых наполнителей, при условии, что, если состав содержит гидроксипропилметилцеллюлозу, описанную выше под (d), то общее количество гидроксипропилметилцеллюлозы, присутствующей в составе, должно быть более чем 25,8 вес.%. Другие фармацевтические составы в пределах указанной последней группы – это составы, включающие приблизительно от 8 до 35 вес.% гидроксипропилметилцеллюлозы, выбранной из группы, состоящей из (а) гидроксипропилметилцеллюлозы, имеющей вязкость около 40-60 сантипуаз, с содержанием метоксигрупп приблизительно от 28 до 30 вес.% и содержанием гидроксипропоксигрупп приблизительно от 7 до менее чем 9 вес.%, (b) гидроксипропилметилцеллюлозы, имеющей вязкость приблизительно от 3500 до 5600 сантипуаз, с содержанием метоксигрупп приблизительно от 28 до 30 вес.% и содержанием гидроксипропоксигрупп приблизительно от 7 до 12 вес.%, (с) гидроксипропилметилцеллюлозы, имеющей вязкость приблизительно от 80 до 120 сантипуаз, с содержанием метоксигрупп приблизительно от 19 до 24 вес.% и содержанием гидроксипропоксигрупп приблизительно от 7 до менее 9 вес.% и (d) гидроксипропилметилцеллюлозы, имеющей вязкость приблизительно от 3500 до 5600 сантипуаз с содержанием метоксигрупп приблизительно от 19 до 24 вес.% и содержанием гидроксипропоксигрупп приблизительно от 7 до 12 вес.% или их смесей; приблизительно от 35 до 65 вес.% 11-[4-[2-(2-гидроксиэтокси)этил]-1-пиперазинил]дибензо[b,f] [1,4] тиазепина или его фармацевтически приемлемой соли; и приблизительно от 20 до 45 вес.% одного или более одного фармацевтически приемлемых наполнителей. Другие фармацевтические составы в пределах этой последней группы представляют собой составы, включающие приблизительно от 10 до 30 вес.% гидроксипропилметилцеллюлозы, выбранной из группы, состоящей из соединений (а)-(d) или их смесей, как описано выше; приблизительно от 40 до 60 вес.% 11-[4-[2-(2-гидроксиэтокси)этил]-1-пиперазинил]дибензо[b,f]-[1,4]тиазепина или его фармацевтически приемлемой соли и приблизительно от 20 до 40 вес.% одного или более одного фармацевтически приемлемых наполнителей. Предпочтительными фармацевтическими составами в пределах этой последней группы являются составы, включающие приблизительно от 15 до 30 вес.% гидроксипропилметилцеллюлозы, выбранной из групп (а)-(d) или их смесей, описанных выше; приблизительно от 43,2 до 57,6 вес.% 11-[4-[2-(2-гидроксиэтокси)этил]-1-пиперазинил]дибензо[b,f][1,4]тиазепина или его фармацевтически приемлемой соли и приблизительно от 22,4 до 36,8 вес.% одного или более одного фармацевтически приемлемых наполнителей. Особенно предпочтительными фармацевтическими составами в пределах этой последней группы являются составы, включающие приблизительно от 15 до 30 вес. % гидроксипропилметилцеллюлозы, выбранной из групп (а)-(d) или их смесей, описанных выше; приблизительно от 43,2 до 57,6 вес.% 11-[4-[2-(2-гидроксиэтокси)этил] -1-пиперазинил] дибензо[b,f]-[1,4]тиазепина или его фармацевтически приемлемой соли и приблизительно от 22,4 до 36,8 вес.% одного или более одного фармацевтически приемлемых наполнителей, выбранных из группы, состоящей из (а) приблизительно от 4 до 12 вес.% микрокристаллической целлюлозы, (b) приблизительно от 5 до 20 вес.% лактозы, (с) приблизительно от 1 до 3 вес. % стеарата магния, (d) приблизительно от 10 до 30 вес.% цитрата натрия и (е) приблизительно от 1 до 15 вес.% повидона (поливинилпирролидона). В вышеописанных фармацевтических составах 11-[4-[2-(2-гидроксиэтокси)этил]-1-пиперазинил]дибензо[b,f][1,4]тиазепин находится предпочтительно в виде гемифумаратной соли, которая имеет равновесную растворимость в воде 3,29 мг/мл при 20oС. Фармацевтические составы, представляющие особый интерес, включают составы, описанные в примерах, и, таким образом, составы, определенные в примерах, по существу обеспечивают дополнительный признак настоящего изобретения. Как указано выше, соединение 11-[4-[2-(2-гидроксиэтокси)этил]-1-пиперазинил]дибензо[b,f][1,4]тиазепин и его фармацевтически приемлемые соли показывают антидопаминергическую активность и могут быть использованы, например, в качестве антипсихотического агента (например, для “управления” проявлениями психотических расстройств) или для лечения гиперактивности. Таким образом, настоящее изобретение также обеспечивает способ лечения психотических состояний, например психоз у теплокровного животного, как, например, у человека, который включает введение эффективного количества фармацевтического состава настоящего изобретения указанному теплокровному животному. Настоящее изобретение также обеспечивает способ лечения гиперактивности у теплокровного животного, который включает введение указанному теплокровному животному эффективного количества фармацевтического состава настоящего изобретения. Фармацевтические составы настоящего изобретения могут быть приготовлены согласно общепринятой технологии, хорошо известной специалистам в данной области исследований, такой как мокрое гранулирование, непосредственное прессование, сухое плотное прессование (путем ударов) и тому подобное. Так, например, активный ингредиент 11-[4-[2-(2-гидроксиэтокси)этил]-1-пиперазинил] дибензо[b, f][1,4]тиазепин или его фармацевтически приемлемую соль, гелеобразующий агент, предпочтительно гидроксипропилметилцеллюлозу и другие наполнители смешивают вместе с тем, чтобы получить фармацевтические составы настоящего изобретения. Предпочтительно, чтобы активный ингредиент 11-[4-[2-(2-гидроксиэтокси)этил] -1-пиперазинил] дибензо[b,f][1,4]тиазепин или его фармацевтически приемлемую соль, гелеобразующий агент, предпочтительно гидроксипропилметилцеллюлозу и другие наполнители смешивают вместе, чтобы получить смесь, пригодную для прессования в таблетки, которую затем прессуют для формирования таблеток или которой заполняют капсулы. Процесс смешивания предпочтительно выполняют путем смешивания компонентов, мокрого гранулирования смешанных компонентов, высушивания смеси, измельчения высушенной смеси, смешивания смеси со смазывающим агентом, таким как стеарат магния, и прессования смеси для формования таблеток или заполнения капсул полученной смесью. Предпочтительный способ получения фармацевтических составов настоящего изобретения включает следующие стадии: (a) смешивание 11-[4-[2-(2-гидроксиэтокси)этил]-1-пиперазинил]дибензо[b, f] [1,4] тиазепина или его фармацевтически приемлемой соли, гелеобразующего агента, предпочтительно гидроксипропилметилцеллюлозы, и других наполнителей; (b) мокрое гранулирование смешанных компонентов; (c) высушивание смеси; (d) измельчение высушенной смеси; (e) смешивание смеси со смазывающим агентом, таким как стеарат магния; и (f) прессование высушенной смеси для формирования таблеток. Дозированные формы могут быть покрыты одной или более оболочками, хорошо известными в данной области, такими как, например, шеллак, зеин, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, этилцеллюлоза, полиметакрилаты, поливинилацетатфталат, ацетатфталат целлюлозы, триацетин, дибутилсебацат, смесь полиэтиленгликоля, диоксида титана и гидроксипропилметилцеллюлозы и тому подобное. Такое свойство, как замедленное выделение фармацевтического состава настоящего изобретения может быть продемонстрировано путем наблюдения за растворением активного ингредиента. Растворение активного ингредиента можно проконтролировать, используя стандартные методики, хорошо известные специалистам данной области исследований (например, тестовые методики растворения: такие, как метод вращающейся корзины (Прибор I) или метод гребка (Прибор II), раскрытые в U.S. Pharmacopeia (USP)). Такие методики включают способы, в которых фармацевтический состав погружают в водную среду, такую как вода или соляная кислота, и в различные моменты времени в течение 24 ч равные количества среды изымаются. Эти равные количества анализируют, используя жидкостную хроматографию высокого давления (HPLC) с УФ детектированием для определения концентрации растворенного активного ингредиента при использовании стандартной методики. В одном примере таблетку погружают приблизительно в 900 мл воды и определяют профиль растворения. В другом примере профиль растворения определяют с помощью метода вращающейся корзины путем погружения таблетки в 750 мл 0,1 N раствора соляной кислоты на 2 ч при вращении со скоростью 100 оборотов в минуту и затем добавления к среде растворения 250 мл 0,2 М фосфатного буфера для достижения рН 6,2. Фармацевтический состав предпочтительно выделяет активный ингредиент регулируемым образом в течение 8 ч или более. Например, состав, описанный в примере 2 выделял около 90% активного ингредиента за 16 ч, и состав, описанный в примере 1, выделял около 90% активного ингредиента за 8 ч. Профили концентрации активного ингредиента в плазме крови от времени, приведенные на фиг. 2, были получены с использованием следующей методики. Тридцать два пациента были распределены либо в группу А, либо в группу Б по 16 пациентов в каждой группе. После 2-дневного периода без принятия лекарств (дни 1 и 2) всем пациентам давали оральные дозы состава мгновенного выделения примера 12 дважды в день в течение 9-дневного периода (дни с 3 по 11) с точно установленными ступенчатыми увеличениями дозы от 25 до 200 мг. Начиная с 12-го дня, пациенты начинали следовать выбранному наугад порядку лечения в пределах их соответственных групп (группа А или Б). Пациенты группы А следовали порядку (последовательности) лечения, который включал один из каждых следующих фармацевтических составов активного ингредиента, назначенных согласно выбранной наугад последовательности: две таблетки по 100 мг состава мгновенного выделения примера 12 натощак принимали каждые 12 ч (лечение 1), одну таблетку 400 мг состава примера 2 – натощак (лечение 2) и одну таблетку 400 мг состава примера 2 – во время приема пищи (Лечение 3). Пациентов группы Б подвергали лечению в последовательности, которая включала один из каждых следующих составов активного ингредиента, назначенных в соответствии с выбранной наугад последовательностью: две таблетки по 100 мг состава мгновенного выделения примера 12 натощак принимали каждые 12 часов (лечение 1), одну таблетку 400 мг состава примера 1 – натощак (лечение 4) и одну таблетку 400 мг состава примера 1 – во время приема пищи (лечение 5). На 12, 16 и 20 дни пациентам назначали лечение согласно предписанному им порядку лечения. Вечером на 13 и 17 дни пациенты получали 200 мг-овые дозы состава мгновенного выделения примера 12 и на 14, 15, 18 и 19 дни пациенты получали 200 мг-овую дозу состава мгновенного выделения примера 12 дважды в день. У каждого субъекта были взяты образцы крови на 3, 10, 11, 14, 15, 18 и 19 дни до получения утренней дозы. На 12, 16 и 20 дни были взяты образцы крови у каждого субъекта непосредственно до приема дозы и через интервалы времени, точно определенные от момента непосредственно после приема дозы до момента через 36 ч после приема дозы. Концентрацию активного ингредиента в образцах крови определяли, используя экстракцию жидкость-жидкость и жидкостную хроматографию высокого разрешения с детектированием ультрафиолетового поглощения. На фиг.2 приведены профили концентрации активного ингредиента в плазме крови от времени для составов примеров 1 (n=11), 2 (n=10) и 12 (n=10 для группы А и 12 для группы Б), и табл. А суммирует значения средней площади под кривой (AUC) для 24 часового интервала приема лекарства дозами и значения среднего максимума концентрации активного ингредиента в крови (Сmах) для каждого из примеров. Дозу соединения настоящего изобретения, которую назначают, необходимо будет варьировать согласно принципам, хорошо известным в данной области, принимая во внимание путь введения лекарства, длительность лечения, тяжесть психотического состояния, вес и возраст пациента, эффективность (силу) активного компонента и, кроме того, реакцию пациента. Следовательно, эффективное дозированное количество активного компонента можно легко определить клинически после рассмотрения всех критериев и путем применения самого лучшего решения (заключения) в пользу пациента. Соединение следует назначать теплокровному животному (такому, как человек) так, чтобы получить эффективную дозу, как правило суточную дозу, в диапазоне от приблизительно 0,01 до приблизительно 40 мг/кг веса тела. Например, при назначении орально его обычно назначают в диапазоне приблизительно от 0,1 до 40 мг/кг веса человека. Предпочтительно соединение настоящего изобретения назначают в количестве около 25, 50, 200, 300 или 400 мг. Фармацевтический состав настоящего изобретения представлен в виде стандартной дозированной формы, и, в частности, фармацевтический состав представлен в виде таблетки. Для специалиста в данной области будет очевидно то, что фармацевтический состав может быть назначен в сочетании с другими терапевтическими или профилактическими агентами и/или лекарствами, с которыми он терапевтически совместим. Фармацевтический состав настоящего изобретения обычно не дает какого-либо показания на явную токсичность у лабораторных тестовых животных при получении дозы, превышающей в несколько раз минимальную эффективную дозу активного ингредиента. Далее изобретение проиллюстрировано следующими не ограничивающими изобретение примерами, в которых температуры выражены в градусах Цельсия. Соединение 11-[4-[2-(2-гидроксиэтокси)этил]-1-пиперазинил]дибензо[b,f][1,4]тиазепин и его фармацевтически приемлемые соли могут быть получены так, как описано в опубликованных Европейских Патентах 240228 или 282236, а также в Патенте США 4879288, полное содержание которых включено в описание в качестве ссылки. Пример 1. Для изготовления таблеток, имеющих состав, определенный в табл. 1, был использован следующий технологический процесс. Гемифумарат 11-[4-[2-(2-гидроксиэтокси)этил]-1-пиперазинил]дибензо[b,f] [1,4] тиазепина (3453,8 г), лактозу (1144,7 г), микрокристаллическую целлюлозу (381,5 г) и METHOCEL E50LV (900 г) смешивали в планетарном смесителе (мешалке, миксере) в течение приблизительно 3 мин.
Смесь была подвергнута мокрому гранулированию в планетарном смесителе с использованием очищенной воды. Мокрую массу высушивали в сушильном аппарате с псевдоожиженным слоем при температуре около 65oС до тех пор, пока потеря воды при сушке не составила менее чем приблизительно 3%, что было измерено по оставшейся влажности.
Высушенную гранулированную массу измельчали, используя мельницу типа “молот” или подобную мельницу, действующую с высокой скоростью, с ножами, идущими впереди сита (например, от 20 до 40 меш).
Через соответствующее сито (например, от 20 до 40 меш) был пропущен стеарат магния.
Сухой гранулированный материал смешивали в течение приблизительно 3 мин в обычном смесителе (например, Patterson – Kelley с двойной оболочкой (кожухом)) с отсортированным стеаратом магния.
Изготовленная смесь была подвергнута прессованию в таблетки с использованием обычного вращательного пресса для изготовления таблеток (например, Killan LX-21).
Профиль концентрации активного ингредиента в плазме крови в зависимости от времени для фармацевтического состава примера 1 показан на фиг.2.
Пример 2.
Технологический процесс, описанный в примере 1, был воспроизведен с использованием METHOCEL E50LV и METHOCEL E4M вместо METHOCEL E50LV с тем, чтобы получить таблетки следующего состава (см. табл. 2).
Профиль концентрации активного ингредиента в плазме крови от времени для фармацевтического состава примера 2 показан на фиг.2.
Пример 3.
Следуя методике проведения, описанной в примере 1, можно приготовить таблетки следующего состава (см. табл. 3).
Пример 4.
Следуя методике проведения, описанной в примере 1, можно изготовить таблетки следующего состава (см. табл. 4).
Пример 5.
Следуя методике проведения, описанной в примере 1, можно сделать таблетки следующего состава (см. табл. 5).
Пример 6.
Следуя методике проведения, описанной в примере 1, можно получить таблетки следующего состава (см. табл. 6).
Пример 7.
Следуя методике проведения, описанной в примере 1, можно изготовить таблетки следующего состава (см. табл. 7).
Следуя методике проведения, описанной в примере 1, были изготовлены таблетки следующих составов (см. табл. 8).
На фиг.1 показаны профили растворения фармацевтических составов примеров 8, 9 и 10.
Пример 11.
Следуя методике проведения, описанной в примере 1, были приготовлены таблетки следующего состава (см. табл. 9).
Пример 12 (см. табл. 10).
Вышеописанный состав мгновенного выделения был получен следующим способом.
Активный ингредиент, повидон, дигидрат дикальцийфосфата, части микрокристаллической целлюлозы и крахмалгликолята натрия смешивали в смесителе-грануляторе (например, Littleford MGT) в течение приблизительно 5 мин. При перемешивании добавляли очищенную воду до тех пор, пока не получилась подходящая масса. Мокрые гранулы пропустили через конусную дробилку, снабженную соответствующим ситом (например, 6,35 мм), и затем высушили в сушильном аппарате с псевдоожиженным слоем при установленной впускной температуре приблизительно 65oС до потери при сушке менее чем 2,5 вес.%. Высушенные гранулы затем пропустили через соответствующую дробилку, снабженную соответствующим ситом (например, 20 меш в дробилке типа “молот”). Гранулированную массу соединили в смесителе (например, V-смесителе) с лактозой и оставшейся частью микрокристаллической целлюлозы и крахмалгликолята натрия и смешивали в течение приблизительно 5 мин. Стеарат магния пропустили через соответствующую дробилку, снабженную подходящим ситом (например, 40 меш), затем добавили к сухому гранулированному материалу и смешивали в течение приблизительно 3 мин. Изготовленную смесь затем подвергли прессованию в таблетки, используя традиционное вращательное прессовочное оборудование. Затем таблетки были покрыты пленочной оболочкой с использованием традиционного оборудования для покрытия лекарств водной суспензией пленкообразующих компонентов (т.е. гидроксипропилметилцеллюлозы, полиэтиленгликоля 400, желтого оксида железа и диоксида титана) при впускной температуре приблизительно 80oС.
Формула изобретения
РИСУНКИ
|
||||||||||||||||||||||||||