Патент на изобретение №2200061
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ СОДЕРЖАЩИХ СУРЬМУ КАТАЛИЗАТОРОВ ДЛЯ (АММ) ОКСИДИРОВАНИЯ АЛКАНОВ И АЛКЕНОВ
(57) Реферат: Изобретение относится к способу получения катализатора для (АММ)оксидирования пропана или пропилена до акрилонитрила. Описан способ получения катализатора VaSbbMmNnOx, полезного при (АММ)оксидировании (С2-С5) – углеводорода до его соответствующего  , ,  -ненасыщенного нитрила, включающий нагревание водной смеси, содержащей V2O5 и Sb2O3, при температуре свыше примерно 100oС до 250oС, предпочтительно при 110oC-175oC, наиболее предпочтительно при 120oС – 160oС, при аутогенном давлении, при перемешивании, для образования предшественника катализатора, сушку предшественника катализатора и прокаливание предшественника катализатора с образованием конечного катализатора. Технический результат: получен катализатор с более высокой активностью и селективностью по акрилонитрилу. 2 с. и 15 з.п. ф-лы, 3 табл. -ненасыщенного нитрила, включающий нагревание водной смеси, содержащей V2O5 и Sb2O3, при температуре свыше примерно 100oС до 250oС, предпочтительно при 110oC-175oC, наиболее предпочтительно при 120oС – 160oС, при аутогенном давлении, при перемешивании, для образования предшественника катализатора, сушку предшественника катализатора и прокаливание предшественника катализатора с образованием конечного катализатора. Технический результат: получен катализатор с более высокой активностью и селективностью по акрилонитрилу. 2 с. и 15 з.п. ф-лы, 3 табл.
Изобретение относится к каталитическому (АММ) оксидированию парафинов и олефинов, содержащих от двух до пяти атомов углерода, до соответствующих альфа, бета-ненасыщенных нитрилов. В частности, настоящее изобретение относится к способу получения катализатора, полезного при (АММ)оксидировании пропана или пропилена до акрилонитрила. Из-за различия в стоимости между пропиленом и пропаном существует экономический стимул для разработки жизнеспособного катализатора, пригодного для конверсии пропана до акрилонитрила. Разработка способа (АММ)оксидирования пропана до акрилонитрила стала достаточно сложной. Патенты США 5124016, 5008427, 5258543, 4788317, 4746641, 3860534, 3681421 и патенты Великобритании 1136135 и 1336136 относятся к различным, содержащим ванадий и сурьму катализаторам, полезным при (АММ)оксидировании пропана до акрилонитрила, и предлагают различные способы получения таких катализаторов. Настоящее изобретение относится к простой процедуре получения сурьмянованадиевого катализатора, полезного при реакциях окисления и/или (АММ)оксидирования, таких как окисление о-ксилола до фталевого ангидрида или (АММ)оксидирования пропилена и пропана до акрилонитрила. В частности, способ настоящего изобретения относится к получению катализатора, полезного при (АММ)оксидировании пропана. Преимущество способа настоящего изобретения состоит в том, что он представляет очень простую процедуру, по существу исключающую применение редких материалов, используемых в ранее разработанных способах, а также исключающую многие стадии, присущие ранее разработанным способам. Это, конечно, ведет к более экономичному и коммерчески жизнеспособному способу получения катализатора, полезного при (АММ)оксидировании пропана и пропилена. Основной целью настоящего изобретения является способ получения катализатора, полезного при (АММ)оксидировании олефинов и/или парафинов до соответствующих ненасыщенных нитрилов. Другой целью настоящего изобретения является способ получения катализатора, полезного при (АММ)оксидировании пропана или пропилена до акрилонитрила. Еще одной целью настоящего изобретения является способ получения акрилонитрила из пропана или пропилена с применением сурьмянованадиевого промотированного катализатора, полученного специфическим способом. Другие цели, преимущества и новые признаки настоящего изобретения будут формулироваться частично в описании, следующем далее, а частично станут очевидны для специалистов в этой области техники при проверке нижеследующего или могут проясниться при практическом применении изобретения. Цели и преимущества настоящего изобретения можно реализовать и достичь с помощью средств и сочетаний, в особенности, указанных в прилагаемой формуле изобретения. Для достижения вышеуказанных целей в соответствии с целями настоящего изобретения, как они описаны и осуществлены здесь, способ настоящего изобретения включает получение катализатора следующей эмпирической формулы: VaSbbMmNnOx, где а=0,01-2,00; b=0,5-4,00; m=0,01-3,0 и n=0-1, M=Sn, Ti, Fe, Cu, Mn, Ga, или их смеси, N=Li, Mg, Sr, Ca, Ва, Со, Ni, Zn, Ge, Nb, Zr, Mo, W, Cr, Те, Та, Se, Bi, Ce, In, As, В, или их смеси, включающее нагревание водной смеси, содержащей водорастворимый ванадат и Sb2О3, и, необязательно, по меньшей мере, одни М-промотор, при температуре от температуры флегмы водной смеси (т.е. примерно выше 100oС) до 250oС, предпочтительно примерно от 110oС до 250oС, при аутогенном давлении, при перемешивании в течение времени, достаточного для взаимодействия, по меньшей мере, слаборастворимого в воде ванадата и Sb2О3 с образованием предшественника катализатора, сушку предшественника катализатора для удаления воды и прокаливание предшественника катализатора для получения конечного катализатора. В другом своем аспекте настоящее изобретение относится к способу получения промотированного VaSbbOx-катализатора формулы VaSbbMmNnOx, где а=0,01-2,00; b=0,5-4,0; m=0,01-3,0 и n=0-1, M=Sn, Ti, Fe, Cu, Mn, Ga, или их смеси, N=Li, Mg, Sr, Ca, Ва, Со, Ni, Zn, Ge, Nb, Zr, Mo, W, Cr, Те, Та, Se, Bi, Ce, In, As, В, или их смеси, включающему нагревание водной смеси, содержащей V2O5 и Sb2О3 и необязательно, по меньшей мере, один М-промотор, при 125-250oС, при аутогенном давлении, при перемешивании в течение времени, достаточного, по меньшей мере, для взаимодействия 2O5 и Sb2О3 с образованием предшественника катализатора, сушку предшественника катализатора для удаления воды и прокаливание предшественника катализатора для получения катализатора. В предпочтительном варианте осуществления способа настоящего изобретения для образования предшественника катализатора к водной смеси перед нагреванием добавляют, по меньшей мере, один промотор N, определение которого дается выше. В другом предпочтительном варианте осуществления способа настоящего изобретения, по меньшей мере, один промоторный элемент М добавляют к водной смеси для образования предшественника катализатора после нагревания. Еще в одном предпочтительном варианте осуществления способа настоящего изобретения в качестве М-промотора выбирают олово и олово-промотор добавляют к водной смеси в форме коллоидного раствора оксида олова. В другом предпочтительном варианте осуществления способа настоящего изобретения поддерживают рН водной смеси менее 7 посредством добавления кислоты. Кислоту можно выбрать из числа либо неорганических, либо органических кислот. Еще в одном предпочтительном варианте осуществления способа настоящего изобретения поддерживают рН водной смеси выше 7 посредством добавления основания. Основание предпочтительно представляет либо органический, либо неорганический амин (например, алканоламин, гидроксид тетраэтиламмония). Теперь способ настоящего изобретения будет описан подробно. Настоящее изобретение относится к способу получения катализатора на основе VaSbbOx для (АММ)оксидирования углеводородов, особенно (С3-С5)-алканов и олефинов, до соответствующих альфа, бета-ненасыщенных нитрилов. В частности, настоящее изобретение относится к получению катализаторов на основе антимоната ванадия для (АММ)оксидирования пропана и пропилена до акрилонитрила. Способ настоящего изобретения включает получение катализатора эмпирической формулы VaSbbMmNnOx, где а=0,01-2,00; b=0,5-4,0; m=0,01-3,0 и n=0-1, M=Sn, Ti, Fe, Cu, Mn, Ga, или их смеси, N=Li, Mg, Sr, Ca, Ва, Со, Ni, Zn, Ge, Nb, Zr, Mo, W, Cr, Те, Та, Se, Bi, Ce, In, As, В, или их смеси, включающее нагревание водной смеси, содержащей водорастворимые ванадаты (например, VO4 -3, VО3 -1, V2O5), и Sb2О3, и необязательно, по меньшей мере, один М-промотор, при температуре выше температуры флегмы водной смеси и до 250oС, при аутогенном давлении, при перемешивании в течение времени, достаточного для взаимодействия, по меньшей мере, слаборастворимых в воде ванадатов и Sb2О3 с образованием предшественника катализатора, сушку предшественника катализатора для удаления воды и прокаливание предшественника катализатора для получения конечного катализатора. В другом варианте осуществления способа настоящего изобретения М-промотор добавляют в водную смесь после смешивания и взаимодействия водораствороимых ванадатов и Sb2О3. В другом аспекте настоящего.изобретения способ получения катализатора VaSbbMmNnOx указанной выше формулы включает нагревание водной смеси, содержащей V2O5 и Sb2О3, при 110-250oС, при аутогенном давлении, при перемешивании в течение времени, достаточного для взаимодействия оксидов металлов с образованием предшественника катализатора, добавление, по меньшей мере, одного М-промотора к водной смеси, сушку предшественника катализатора для удаления воды и прокаливание предшественника катализатора для получения катализатора. Гидротермальная реакция оксидов металлов в водном растворе длится в течение времени, достаточного для соответствующего взаимодействия оксидов металлов с образоваием предшественника катализатора. Требуемое время реакции определяют в конечном счете по каталитическим и физическим свойствам конечного материала, полученного после прокаливания. Как правило, реакция продолжается 0,5-100,0 ч, предпочтительно 1-50 ч, причем особенно предпочтительно время 1-10 ч. Отмечено, что более короткое время реакции требуется, если увеличивают температуру в процессе образования предшественника катализатора. Водная смесь, содержащая ванадиевые и сурьмяные компоненты, может также содержать кислоту или основание, предпочтительно основание, присутствующее в коллоидном растворе оксида олова. Для доведения рН до величины ниже 7 можно использовать любую неорганическую или органическую кислоту. Желательнее использовать кислоты, которые могут разлагаться во время прокаливания, чтобы они не оставались после этого в виде нелетучих компонентов, как, например, хлориды или сульфаты, которые могут модифицировать композицию за исключением того случая, когда это желательно. Можно использовать неорганические кислоты, такие как азотная кислота, однако такие кислоты могут выделять в окружающую среду нежелательные газы, например NOx. Представляется, что предпочтительными кислотами являются органические кислоты, такие как уксусная кислота, лимонная кислота или щавелевая кислота, которые при прокаливании разлагаются с образованием СОх и воды. Количество кислоты, которое используют при этих синтезах, существенно ниже, чем то количество, которое требуется для растворения всех неорганических компонентов (например, V2O5) перед реакцией. Для достижения величины рН выше 7 можно использовать любое неорганическое основание (например, гидроксид аммония) или органический амин (в том числе диэтоксиэтиламин, гидроксид тетраметилалкиламмония, алканоламины, гидроксид трет-бутиламмония). Однако желательнее использовать основания, которые могут разлагаться во время прокаливания, после которых не остается нелетучих компонентов, таких как щелочной или щелочноземельный металл, если только их присутствие нежелательно в конечной композиции катализатора. Подходящими основаниями, которые можно использовать, являются гидроксид аммония и органические основания, такие как алкиламины и алканоламины. Наиболее предпочтительно, если основание может присутствовать уже в водной смеси как стабилизатор/диспергатор для гидратированного неорганического оксида металла (например, коллоидного раствора оксида олова), который использован в качестве источника сырья для М-промотора в процессе образования предшественника катализатора и поэтому не будет необходимости добавлять его отдельно. Процедура настоящего изобретения может включать взаимодействие в соответствующих количествах V2O5 и Sb2О3 (так же, как необязательно оксидов других металлов, известных для промотирования катализа (АММ)оксидрования, например (но их число не ограничивается этими примерами) оксид олова (коллоидный раствор), оксиды титана, теллура, рения, кремния, алюминия, железа, ниобия, хрома, кобальта, меди, висмута, вольфрама и т.п.), в присутствии воды и необязательно кислоты или основания для подгонки рН, при 100 (температура флегмы) – 250oС, при аутогенном давлении и энергичном перемешивании. В случаях, когда нельзя получить сам оксид промоторного металла для гидротермальной реакции (или нет легкодоступного в подходящей форме), можно необязательно использовать другие источники металлов-промоторов (например, нитраты, ацетаты, гидроксиды, комплексы с ионом аммония, карбонилы и т.п.). Гидротермальная реакция этих оксидов металлов продолжается в течение времени (как правило, 0,5-100,0 ч), достаточного для подходящего диспергирования оксидов металлов с образованием предшественника катализатора. Некоторые оксиды металлов или другие источники можно добавлять после стадии гидротермальной реакции, и они могут находиться в форме мелкодисперсных металлических порошков, коллоидных растворов или комплексов. Примерами являются высокодисперсный TiO2, коллоидный раствор оксида олова, коллоидный раствор диоксида кремния и коллоидный раствор оксида алюминия. После охлаждения полученный материал (как правило, довольно однородная суспензия, хотя может происходить некоторое расслоение, если смесь стоит без перемешивания) можно смешать/ввести в дальнейшую реакцию с дополнительными оксидами металлов (или с другими источниками металлов, такими как нитраты, ацетаты и т.п.), упарить досуха (или сушить распылением) и прокалить в одну или несколько стадий для получения катализатора. Величину рН полученной гидротермально суспензии можно подогнать перед добавлением других реагентов для оптимального получения катализатора. Требуемое время реакции, температура, содержание твердых веществ и порядок добавления других реагирующих оксидов металлов определяются в конечном счете каталитическими и физическими свойствами конечного полученного материала. Дополнительные стадии промывки и прокаливания, применяемые для полученного катализатора при других путях его получения, также можно использовать для улучшения рабочих характеристик этих катализаторов. Например, для улучшения рабочих характеристик катализатора можно использовать стадии промывки и прокаливания, описанные в патентах США 5214016, 5008427 и 5258543, включенных в настоящее в качестве ссылок. Добавление щелочи, например гидроксида лития, для уменьшения вязкости суспензии перед сушкой распылением также приемлемо для суспензий, полученных таким способом. Обнаружено, что выгодно присутствие промотора-олова в форме коллоидного раствора оксида олова/стабилизаторов во время гидротермального синтеза предшественника промотированного катализатора, особенно при температуре выше температуры флегмы ( 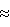 100oС) и до 250oС (например, от 110oC до 250oС), предпочтительно при 110-175oС, причем наиболее предпочтительна температура 120-160oС, при аутогенном давлении.
Кроме того, получение водных суспензий с высоким содержанием твердых веществ представляется особенно благоприятным для практического осуществления настоящего изобретения. Применение стабилизированного гидроксидом тетраметиламмония коллоидного раствора SnO2 можно использовать для получения водных суспензий с содержанием твердых веществ примерно 70%, подходящих для практического осуществления настоящего изобретения.
Проведение стадии гидротермальной реакции при повышенных температурах снижает время, необходимое для получения суспензий, которые в конечном счете дают на выходе катализаторы, которые также хороши или лучше катализаторов, полученных при температуре флегмы (приблизительно 100oС). Также представляется, что при операции при повышенных температурах (температурах выше температуры флегмы, т.е. выше 100oС) усиливается включение других промоторных элементов в основную композицию для улучшения общих характеристик. Кроме того, способ настоящего изобретения легко масштабируется до коммерческого производства катализатора, поскольку нет необходимости, чтобы все исходные материалы находились в растворе в любой момент времени. Поэтому можно достичь более высокого содержания твердых веществ в суспензиях без затрат на их концентрирование, например, путем выпаривания перед распылительной сушкой. Это увеличивает эффективность использования реакторов и другого оборудования, используемого при получении катализатора. Способ настоящего изобретения также снижает количество воды, которую надо выпарить из суспензии, чтобы достичь соответствующего содержания твердых веществ для распылительной сушки, за счет чего в результате сберегаются время и энергия.
Также отмечено, что скорость реакции оксидов металлов при более высоких температурах и при аутогенном давлении существенно увеличивается при присутствии промотора-олова в форме коллоидного раствора оксида олова. В частности, существенно усиливается взаимодействие оксидов металлов в присутствии коллоидного раствора оксида олова, т. е. Nalco 88-SN-123 и Nalco TX7684, поставляемых Nalco Chemical Co., Haпервилль, Иллинойс. Это позволяет получать катализаторы (предшественники) при меньшем времени реакции и более низких температурах реакции, чем время и температуры, используемые ранее.
Существуют качественные факторы, которые указывают, что присутствие коллоидного раствора оксида олова повышает скорость реакции оксидов ванадия и сурьмы. В отсутствии коллоидного раствора оксида олова окраска флегмы смеси существенно не темнеет (и наблюдают желтые прожилки, когда каплю суспензии наносят на лист фильтровальной бумаги, свидетельствующие о компонентах, содержащих свободный и растворимый ванадий) после 3-6 ч реакции. В присутствии коллоидного раствора оксида олова окраска становится черной в пределах ~1/2 ч, что указывает на существенное взаимодействие. Рентгеновский анализ промежуточных прокаленных материалов (650oС) показывает, что оксид олова и оксид титана лучше диспергируются, если на стадии гидротермальной реакции присутствует коллоидный раствор оксида олова. Присутствие органических стабилизаторов в коллоидном растворе оксида олова может оказаться пагубным для свойств конечного катализатора, если не принять специальных мер предосторожности для удаления стабилизатора перед последним прокаливанием. Соответственно будет существовать некоторая выгода использования низких концентраций стабилизатора и/или стабилизаторов, которые легко улетучиваются. Летучесть стабилизаторов не является проблемой во время синтеза при использовании способа настоящего изобретения, который использует герметизированную систему при аутогенном давлении.
При наиболее простом и удобном способе использования процедуры по настоящему изобретению в щелочных условиях на стадии гидротермальной реакции оксидов ванадия и сурьмы должен присутствовать коллоидный оловосодержащий раствор и сопутствующие стабилизаторы/диспергаторы. Это может быть выгодным в отношении минимизации миграции частиц олова к поверхности частиц, образовавшихся во время распылительной сушки, для получения катализатора в форме псевдоожиженного слоя.
Следующие далее примеры приводятся только в иллюстративных целях.
(АММ)оксидирование пропилена с неподвижным слоем катализатора.
ПРИМЕР 1.
Катализатор состава 60% VSb1,2Fe0,08Sn0,5Ti1Ox – 40% SiO2 получают следующим образом. В тефлоновую гильзу, соответствующую 125 мл автоклаву Парра, снабженному стержнем магнитной мешалки, помещают 4,55 г V2O5 (Stratcore), 8,75 г Sb2О3, 0,32 г F2О3 (Baker) и 40 г воды. После перемешивания приблизительно в течение 15 мин добавляют 1,58 г щавелевой кислоты (Baker) и дополнительные 20 г воды, продолжая перемешивание. Затем гильзу герметизируют в корпусе автоклава и помещают в печь с температурой 175oС. Суспензию, полученную через 93 ч, переносят в 800 мл химический стакан, снабженный перемешивающим стержнем, и разбавляют водой приблизительно до 150 мл. Добавляют к суспензии 4 г TiO2 (Degussa Р25) и перемешивают приблизительно в течение 1 ч. Затем смесь нагревают до кипения. Затем добавляют 18,84 г коллоидного раствора SnО2 (20% твердого вещества) и 47,50 г коллоидного раствора SiO2 (30% твердого вещества) и выпаривают воду до тех пор, пока гель не загустеет настолько, что стержень остановится. Полученное вещество затем сушат в печи при 120oС в течение ночи. Полученное твердое вещество затем прокаливают на воздухе при 290oС в течение 3 ч, при 425oС в течение 3 ч и при 650oС в течение 8 ч. Затем материал измельчают, и просеивают, и некоторую часть, составляющую 20-40 меш, прокаливают при 900oС в течение 3 ч, промывают горячим метанолом, затем прокаливают при 650oС в течение 3 ч и затем промывают горячим метанолом.
ПРИМЕР 2.
Получают катализатор состава 60% VSb1,2Fe0,08Sn0,5Ti1Ox – 40% SiO2 так же, как описано выше, за исключением того, что TiO2 присутствует на стадии 93-часовой гидротермальной реакции при 175oС.
ПРИМЕР 3.
Катализатор состава 60% VSb1,2Fe0,025Nb0,025Sn0,5Ti1Ox – 40% SiO2 получают таким же способом, как в примере 1, но с 0,10 г Fе2О3 и 0,17 г Nb2O5 (Alfa), присутствующими на стадии гидротермальной реакции (175oС, 138 ч), и без щавелевой кислоты.
ПРИМЕР 4.
Катализатор состава 60% VSb1,2Cu0,04Sn0,5Ti1Ox – 40% SiO2 получают способом, подобным способу примера 1, но с 0,14 г Cu2O (Alfa), присутствующими на стадии гидротермальной реакции (175oС, 91 ч).
ПРИМЕР 5.
Катализатор состава 60% VSb1,2Cu0,04Sn0,5Ti1Ox – 40% SiO2 получают способом, подобным способу примера 4, но с 0,16 г СuО (Alfa), присутствующими на стадии гидротермальной реакции.
ПРИМЕР 6.
Катализатор состава 60% VSb1,2Cu0,04Sn0,5Ti1Ox – 40% SiO2 получают способом, подобным способу примера 4, но с 0,16 г CuO (Alfa), присутствующими на стадии гидротермальной реакции.
ПРИМЕР 7.
Катализатор состава 60% VSb1,2Cu0,06Sn0,5Ti1Ox – 40% SiO2 получают способом, подобным способу примера 4, но с 0,24 г CuO (Alfa), и на стадии гидротермальной реакции присутствуют только 0,79 г щавелевой кислоты.
ПРИМЕР 8. 60% VSb1,2Fe0,08Sn0,5Ti1Ox – 40% SiO2 100oС) и до 250oС (например, от 110oC до 250oС), предпочтительно при 110-175oС, причем наиболее предпочтительна температура 120-160oС, при аутогенном давлении.
Кроме того, получение водных суспензий с высоким содержанием твердых веществ представляется особенно благоприятным для практического осуществления настоящего изобретения. Применение стабилизированного гидроксидом тетраметиламмония коллоидного раствора SnO2 можно использовать для получения водных суспензий с содержанием твердых веществ примерно 70%, подходящих для практического осуществления настоящего изобретения.
Проведение стадии гидротермальной реакции при повышенных температурах снижает время, необходимое для получения суспензий, которые в конечном счете дают на выходе катализаторы, которые также хороши или лучше катализаторов, полученных при температуре флегмы (приблизительно 100oС). Также представляется, что при операции при повышенных температурах (температурах выше температуры флегмы, т.е. выше 100oС) усиливается включение других промоторных элементов в основную композицию для улучшения общих характеристик. Кроме того, способ настоящего изобретения легко масштабируется до коммерческого производства катализатора, поскольку нет необходимости, чтобы все исходные материалы находились в растворе в любой момент времени. Поэтому можно достичь более высокого содержания твердых веществ в суспензиях без затрат на их концентрирование, например, путем выпаривания перед распылительной сушкой. Это увеличивает эффективность использования реакторов и другого оборудования, используемого при получении катализатора. Способ настоящего изобретения также снижает количество воды, которую надо выпарить из суспензии, чтобы достичь соответствующего содержания твердых веществ для распылительной сушки, за счет чего в результате сберегаются время и энергия.
Также отмечено, что скорость реакции оксидов металлов при более высоких температурах и при аутогенном давлении существенно увеличивается при присутствии промотора-олова в форме коллоидного раствора оксида олова. В частности, существенно усиливается взаимодействие оксидов металлов в присутствии коллоидного раствора оксида олова, т. е. Nalco 88-SN-123 и Nalco TX7684, поставляемых Nalco Chemical Co., Haпервилль, Иллинойс. Это позволяет получать катализаторы (предшественники) при меньшем времени реакции и более низких температурах реакции, чем время и температуры, используемые ранее.
Существуют качественные факторы, которые указывают, что присутствие коллоидного раствора оксида олова повышает скорость реакции оксидов ванадия и сурьмы. В отсутствии коллоидного раствора оксида олова окраска флегмы смеси существенно не темнеет (и наблюдают желтые прожилки, когда каплю суспензии наносят на лист фильтровальной бумаги, свидетельствующие о компонентах, содержащих свободный и растворимый ванадий) после 3-6 ч реакции. В присутствии коллоидного раствора оксида олова окраска становится черной в пределах ~1/2 ч, что указывает на существенное взаимодействие. Рентгеновский анализ промежуточных прокаленных материалов (650oС) показывает, что оксид олова и оксид титана лучше диспергируются, если на стадии гидротермальной реакции присутствует коллоидный раствор оксида олова. Присутствие органических стабилизаторов в коллоидном растворе оксида олова может оказаться пагубным для свойств конечного катализатора, если не принять специальных мер предосторожности для удаления стабилизатора перед последним прокаливанием. Соответственно будет существовать некоторая выгода использования низких концентраций стабилизатора и/или стабилизаторов, которые легко улетучиваются. Летучесть стабилизаторов не является проблемой во время синтеза при использовании способа настоящего изобретения, который использует герметизированную систему при аутогенном давлении.
При наиболее простом и удобном способе использования процедуры по настоящему изобретению в щелочных условиях на стадии гидротермальной реакции оксидов ванадия и сурьмы должен присутствовать коллоидный оловосодержащий раствор и сопутствующие стабилизаторы/диспергаторы. Это может быть выгодным в отношении минимизации миграции частиц олова к поверхности частиц, образовавшихся во время распылительной сушки, для получения катализатора в форме псевдоожиженного слоя.
Следующие далее примеры приводятся только в иллюстративных целях.
(АММ)оксидирование пропилена с неподвижным слоем катализатора.
ПРИМЕР 1.
Катализатор состава 60% VSb1,2Fe0,08Sn0,5Ti1Ox – 40% SiO2 получают следующим образом. В тефлоновую гильзу, соответствующую 125 мл автоклаву Парра, снабженному стержнем магнитной мешалки, помещают 4,55 г V2O5 (Stratcore), 8,75 г Sb2О3, 0,32 г F2О3 (Baker) и 40 г воды. После перемешивания приблизительно в течение 15 мин добавляют 1,58 г щавелевой кислоты (Baker) и дополнительные 20 г воды, продолжая перемешивание. Затем гильзу герметизируют в корпусе автоклава и помещают в печь с температурой 175oС. Суспензию, полученную через 93 ч, переносят в 800 мл химический стакан, снабженный перемешивающим стержнем, и разбавляют водой приблизительно до 150 мл. Добавляют к суспензии 4 г TiO2 (Degussa Р25) и перемешивают приблизительно в течение 1 ч. Затем смесь нагревают до кипения. Затем добавляют 18,84 г коллоидного раствора SnО2 (20% твердого вещества) и 47,50 г коллоидного раствора SiO2 (30% твердого вещества) и выпаривают воду до тех пор, пока гель не загустеет настолько, что стержень остановится. Полученное вещество затем сушат в печи при 120oС в течение ночи. Полученное твердое вещество затем прокаливают на воздухе при 290oС в течение 3 ч, при 425oС в течение 3 ч и при 650oС в течение 8 ч. Затем материал измельчают, и просеивают, и некоторую часть, составляющую 20-40 меш, прокаливают при 900oС в течение 3 ч, промывают горячим метанолом, затем прокаливают при 650oС в течение 3 ч и затем промывают горячим метанолом.
ПРИМЕР 2.
Получают катализатор состава 60% VSb1,2Fe0,08Sn0,5Ti1Ox – 40% SiO2 так же, как описано выше, за исключением того, что TiO2 присутствует на стадии 93-часовой гидротермальной реакции при 175oС.
ПРИМЕР 3.
Катализатор состава 60% VSb1,2Fe0,025Nb0,025Sn0,5Ti1Ox – 40% SiO2 получают таким же способом, как в примере 1, но с 0,10 г Fе2О3 и 0,17 г Nb2O5 (Alfa), присутствующими на стадии гидротермальной реакции (175oС, 138 ч), и без щавелевой кислоты.
ПРИМЕР 4.
Катализатор состава 60% VSb1,2Cu0,04Sn0,5Ti1Ox – 40% SiO2 получают способом, подобным способу примера 1, но с 0,14 г Cu2O (Alfa), присутствующими на стадии гидротермальной реакции (175oС, 91 ч).
ПРИМЕР 5.
Катализатор состава 60% VSb1,2Cu0,04Sn0,5Ti1Ox – 40% SiO2 получают способом, подобным способу примера 4, но с 0,16 г СuО (Alfa), присутствующими на стадии гидротермальной реакции.
ПРИМЕР 6.
Катализатор состава 60% VSb1,2Cu0,04Sn0,5Ti1Ox – 40% SiO2 получают способом, подобным способу примера 4, но с 0,16 г CuO (Alfa), присутствующими на стадии гидротермальной реакции.
ПРИМЕР 7.
Катализатор состава 60% VSb1,2Cu0,06Sn0,5Ti1Ox – 40% SiO2 получают способом, подобным способу примера 4, но с 0,24 г CuO (Alfa), и на стадии гидротермальной реакции присутствуют только 0,79 г щавелевой кислоты.
ПРИМЕР 8. 60% VSb1,2Fe0,08Sn0,5Ti1Ox – 40% SiO2Смесь V2O5 (4,55 г), Sb2О3 (8,75 г), Fe2О3 (Nanocat (R), далее называемый здесь “Nano”, от Mach I Company, расположенной в Пенсильвании, 0,32 г), 18,84 г коллоидного раствора оксида олова (3,77 г SnO2) и 125 мл воды реагирует при кипячении с обратным холодильником в течение ~24 ч. После охлаждения добавляют 4,00 г ТiO2 и 47,50 г коллоидного раствора диоксида кремния (30% твердого вещества) и концентрируют суспензию на электроплитке до тех пор, пока стержень мешалки не остановится. Высокотемпературное прокаливание проводят при 885oС в течение 3 ч. ПРИМЕР ДЛЯ СРАВНЕНИЯ 9. 60% VSb1,3Fe0,2Sn0,5Ti1Ox – 40% SiO2 Катализатор эмпирической формулы 60% VSb1,3Fe0,2Sn0,5Ti1-Ох – 40% SiO2 получают следующим образом. К смеси, состоящей из 900 см3 H2O и 100 г 30% Н2О2, в 2-литровом лабораторном стакане добавляют 12,08 г V2О5 и перемешивают при комнатной температуре примерно 15 мин, пока не образуется темно-красный пероксидный комплекс. Затем добавляют 25,17 г Sb2О3, 2,12 г Fе2О3 (Nano) и 10,61 г TiO2 (Degussa, P-25), устанавливают регулятор температуры электроплитки на “высокая” и накрывают стакан часовым стеклом. При нагревании цвет суспензии изменяется от желтого до зеленого, а затем черного. Смесь варят приблизительно 4,5 ч; время от времени добавляют воду, чтобы объем оставался постоянным. Добавляют 50,05 г 20% коллоидного раствора SnO2 (Naico 88SN123), затем 133,33 г 30% коллоидного раствора диоксида кремния (Nissan). Суспензию катализатора упаривают на электроплитке при непрерывном перемешивании, пока она не загустеет. Остальная часть процедуры такая же, как в примере 8. ПРИМЕР 10. 60% VSb1,2Fe0,025Ni0,025Sn0,5Ti1Ox – 40% SiO2 Получают катализатор состава 60% VSb1,2Fe0,025Ni0,025Sn0,5-Ti10x – 40% SiO2 с 0,093 г NiO и 0,01 г Fе2O3 и 0,79 г щавелевой кислоты, присутствующими на стадии гидротермальной реакции (175oС, 16 ч), по способу, подобному способу примера 4. ПРИМЕР 11. 60% VSb1,2Mn0,025Sn0,5Ti1Ox – 40% SiO2 Катализатор состава 60% VSb1,2Mn0,025Sn0,5Ti1Ox – 40% SiO2 получают тем же способом, что в примере 10, за исключением того, что присутствует 0,11 г МnO2. ПРИМЕР 12. 60% VSb1,2Fe0,025Mn0,025Sn0,5Ti1Ox – 40% SiO2 Катализатор состава 60% VSb1,2Fe0,025Mn0,025Sn0,5Ti1Ox – 40% SiO2 получают тем же способом, что в примере 10, за исключением того, что присутствуют 0,12 г Fе2O3 (Baker) и 0,11 г МnO2. ПРИМЕР 13. 60% VSb1,2Fe0,12Sn0,5Ti1Ox – 40% SiO2 Катализатор состава 60% VSb1,2Fe0,12Sn0,5Ti1Ox – 40% SiO2 получают путем реакции смеси V2O5 (4,55 г), Sb2О3 (8,75 г), Fе2О3 (Nano, 0,479 г), 18,84 г коллоидного раствора оксида олова (3,77 г SnО2) и 125 мл воды при кипячении с обратным холодильником в течение ~24 ч. После охлаждения добавляют 4,00 г TiO2 и 47,86 г коллоидного раствора диоксида кремния (30% твердого вещества) и концентрируют суспензию на электроплитке до тех пор, пока перемешивающий стержень не остановится. Высокотемпературное прокаливание проводят при 900oС в течение 3 ч. ПРИМЕР 14. 60% VSb1,2Fe0,12Sn0,5Ti1Ox – 40% SiO2 Катализатор состава 60% VSb1,2Fe0,12Sn0,5Ti1Ox – 40% SiO2 получают путем реакции смеси V2O5 (4,55 г), Sb2O3 (8,75 г), Fе2O3 (Nano, 0,479 г), 18,84 г коллоидного раствора оксида олова (3,77 г SnO2) и 120 мл воды при 150oС при аутогенном давлении в автоклаве с мешалкой в течение 21 ч. После охлаждения добавляют 4,00 г TiO2 и 47,86 г коллоидного раствора диоксида кремния (30% твердого вещества) и концентрируют суспензию на электроплитке до тех пор, пока перемешивающий стержень не остановится. Высокотемпературное прокаливание проводят при 900oС в течение 2,5 ч. ПРИМЕР 15. 60% VSb1,2Fe0,12Sn0,5Ti1Ox – 40% SiO2 Катализатор состава 60% VSb1,2Fe0,12Sn0,5Ti1Ox – 40% SiO2 получают путем реакции смеси V2O5 (7,00 г), Sb2O3 (13,46 г), Fе2О3 (Nano, 0,74 г), 28,98 г коллоидного раствора оксида олова (5,80 г SnO2) и 112 мл воды при 150oС при аутогенном давлении в автоклаве с мешалкой в течение 3 ч. После охлаждения добавляют 6,15 г TiO2 и 73,62 г коллоидного раствора диоксида кремния (30% твердого вещества) и концентрируют суспензию на электроплитке до тех пор, пока перемешивающий стержень не остановится. Высокотемпературное прокаливание проводят при 900oС в течение 2,5 ч. Катализаторы приведенных выше примеров 1-15 испытывают при следующих условиях. Загрузочное соотношение С3 =/NН3/-O2/N2/H2O в каждом примере составляет 1,8/2,2/3,9/2,4/6,0 соответственно. Результаты приводятся ниже в табл. I. (АММ)оксидирование пропилена на псевдоожиженном слое ПРИМЕР 1. Получают крупную партию псевдоожиженного катализатора состава 60% V1Sb1,2Fe0,12Sn0,5Ti1Ox – 40% SiO2, следуя процедуре, использованной для получения катализатора в приведенном выше примере 13. В данном случае оксид титана и коллоидный раствор диоксида кремния добавляют к суспензии, полученной со стадии гидротермальной реакции. Затем перед распылительной сушкой суспензию концентрируют примерно до 32-33% твердого вещества посредством испарения воды. ПРИМЕР 2. Получают крупную партию псевдоожиженного катализатора состава 60% V1Sb1,2Fe0,12Sn0,5Ti1Ox – 40% SiO2, следуя процедуре, использованной выше, за исключением того, что суспензию, полученную со стадии гидротермальной реакции, концентрируют до 18-20% твердого вещества перед добавлением оксида титана и диоксида кремния. Затем суспензию концентрируют примерно до ~35% твердого вещества перед распылительной сушкой посредством испарения воды. Проводят испытания катализаторов примеров 1 и 2 в 40 см3 реакторе с псевдоожиженным слоем. Загрузочное соотношение CH3 =/NH3/O2/N2/H2O в случае катализаторов примеров 1 и 2 составляет 1,8/2,2/3,9/2,4/16 соответственно. Результаты приводятся ниже в табл.II. (АММ)оксидирование пропана ПРИМЕР ДЛЯ СРАВНЕНИЯ 1. Катализатор состава VSb1,4Sn0,2Ti0,1Ox получают способом, подобным способу, описанному выше в примере 9, за исключением того, что не добавляют ни оксид железа, ни SiO2. На конечной стадии катализатор прокаливают при 820oС в течение 3 ч, прокаливают при 650oС в течение 3 ч и промывают изобутанолом. ПРИМЕР 2. Катализатор состава VSb1,4Sn0,2Ti0,1Ox получают следующим образом. В круглодонную 250 мл колбу, снабженную обратным холодильником, помещают 7,58 г V2O5, 17,01 г Sb2О3 и 25,57 г коллоидного раствора SnO2, стабилизированного NH4OH, содержащего 2,51 г SnO2, и приблизительно 125 г воды. Эту смесь кипятят с обратным хролодильником при перемешивании в течение 22 ч. После охлаждения полученную суспензию при перемешивании добавляют в химический стакан, содержащий 0,67 г высокодисперсного TiO2. Через 1 ч суспензию концентрируют путем выпаривания пока смесь не загустеет и помещают на ночь в печь при 125oС для сушки. Высушенное твердое вещество греют на воздухе при 290oС в течение 3 ч, затем при 425oС в течение 3 ч и затем в течение 8 ч при 650oС. Затем материал измельчают и просеивают, и часть вещества 20-35 меш прокаливают при 820oС в течение 3 ч, затем при 650oС в течение 3 часов и затем промывают изобутанолом. ПРИМЕР 3. Получают ту же композицию, какая указана в примере 2, за исключением того, что выскодисперсный TiO2 присутствует на стадии кипячения с обратным холодильником в течение 24 ч. ПРИМЕР 4. Катализатор состава VSb1,4Sn0,2Ti0,1Ox получают следующим образом. В кварцевую гильзу помещают 7,58 г V2O5, 17,01 г Sb2О3, 0,67 г высокодисперсного TiO2 и 27,57 г коллоидного раствора SnO2, стабилизированного NH4OH, содержащего 2,51 г SnO2, и приблизительно 125 г воды. Эту гильзу затем размещают и плотно закрывают в 300 см3 автоклаве, снабженном подвесной мешалкой. Затем смесь нагревают до 125oС и выдерживают в течение 6 ч при перемешивании. Затем нагреватель удаляют от автоклава и оставляют смесь охлаждаться при перемешивании в течение ночи. Остальную часть процедуры проводят так же, как в примерах 2 и 3. ПРИМЕР 5. Для получения суспензии, содержащей компоненты для получения катализатора состава VSb1,4Sn0,2Ti0,1Ox, используют ту же процедуру, что в примере 4. После извлечения из охлажденного автоклава суспензию концентрируют в химическом стакане на электроплитке до содержания твердого вещества 29-30%. Добавляют гидроксид лития в воде для снижения вязкости суспензии. Продолжают испарять воду и перед тем, как стержень мешалки остановится, получают содержание твердого вещества приблизительно 46%. Остальная часть процедуры подобна описанному выше за исключением того, что катализатор прокаливают в токе воздуха при 810 и 650oС перед единственной стадией промывки изобутанолом после прокаливания при 650oС. Состав полученного в результате катализатора Li0,05VSb1,4Sn0,2-Ti0,1Ox. ПРИМЕР 6. (6 ч) Осуществляют ту же процедуру, что в примере 4, за исключением того, что конечные две стадии прокаливания проводят так, как в примере 5. ПРИМЕР 7. (4 ч) Осуществляют ту же процедуру, что в примере 6, за исключением того, что гидротермальную стадию проводят только в течение 4 ч. ПРИМЕР 8. Осуществляют ту же процедуру, что в примере 4, за исключением того, что стадию гидротермальной реакции проводят в течение 4 ч при 150oС. Результаты испытаний в реакторе с неподвижным слоем в случае (АММ)оксидирования пропана приводятся ниже в табл.III. Приведенное выше описание не предназначено предоставить исчерпывающее осуществление и комбинации способа настоящего изобретения. Хотя композиции, полученные до настоящего времени, намечены для катализатора (АММ)оксидирования в случае пропана и пропилена, представляется, что способ будет подходящим для получения катализаторов окисления другого типа. Имеется в виду, что способ настоящего изобретения определяется формулой изобретения, составляющей предмет изобретения. Формула изобретения
VaSbbMmNnOx, где а= 0,01-2,00; b= 0,5-4,0; m= 0,01-3,00; n= 0-1; х – число атомов кислорода, которое определяется степенью окисления остальных элементов, М= Sn, Ti, Fe, Сu, Mn, Ga или их смеси; N= Li, Mg, Sr, Ca, Ва, Со, Ni, Zn, Ge, Nb, Zr, Mo, W, Cr, Те, Та, Se, Bi, Ce, In, As, В или их смеси, включающий нагревание водной смеси, содержащей водорастворимые ванадаты и Sb2О3 и по меньшей мере один М-промотор, при 100 – 250oС с образованием предшественника катализатора, сушку указанного предшественника катализатора для удаления воды и прокаливание предшественника катализатора, отличающийся тем, что взаимодействие между ванадием и сурьмой происходит при аутогенном давлении и перемешивании в течение времени, достаточного, чтобы прореагировать по меньшей мере водорастворимым ванадатам и Sb2О3 с образованием предшественника катализатора. 2. Способ по п. 1, отличающийся тем, что добавляют по меньшей мере один N-промотор к водной смеси перед нагреванием водной смеси для образования предшественника катализатора. 3. Способ по п. 1, отличающийся тем, что используют Sn в качестве М-промотора. 4. Способ по п. 3, отличающийся тем, что Sn-промотор добавляют к водной смеси в форме коллоидного раствора оксида олова. 5. Способ по п. 1, отличающийся тем, что рН водной смеси поддерживают выше 7 добавлением основания. 6. Способ по п. 1, отличающийся тем, что рН водной смеси поддерживают ниже 7 добавлением кислоты. 7. Способ по п. 2, отличающийся тем, что N-промотор добавляют к катализатору после того, как водорастворимый ванадат и Sb2О3 диспергированы и прореагировали с образованием предшественника катализатора. 8. Способ по п. 5, отличающийся тем, что основание выбирают из группы, состоящей из неорганического или органического амина. 9. Способ по п. 8, отличающийся тем, что основание представляет органический амин, выбранный из группы, состоящей из алкиламинов, алканоламинов или их смесей. 10. Способ по п. 6, отличающийся тем, что кислота является органической кислотой. 11. Способ по п. 10, отличающийся тем, что органическую кислоту выбирают из группы, состоящей из уксусной, щавелевой кислоты или их смесей. 12. Способ по п. 6, отличающийся тем, что кислота является неорганической кислотой. 13. Способ получения содержащих сурьму катализаторов для (АММ) оксидирования алканов и алкенов эмпирической формулы VaSbbMmNnOx, где а= 0,01-2,00; b= 0,5-4,0; m= 0,01-3,00; n= 0-1; х – число атомов кислорода, которое определяется степенью окисления остальных элементов; М= Sn, Ti, Fe, Сu, Mn, Ga или их смеси; N= Li, Mg, Sr, Ca, Ва, Со, Ni, Zn, Ge, Nb, Zr, Mo, W, Cr, Те, Та, Se, Bi, Ce, In, As, В или их смеси, включающий нагревание водной смеси, содержащей V2O5 и Sb2О3 и по меньшей мере один М-промотор, при 110-250oC c образованием предшественника катализатора, сушку предшественника катализатора для удаления воды и прокаливание предшественника катализатора, отличающийся тем, что взаимодействие между ванадием и сурьмой протекает при аутогенном давлении и перемешивании в течение времени, достаточного, чтобы прореагировать по меньшей мере V2O5 и Sb2О3 с образованием предшественника катализатора. 14. Способ по п. 13, отличающийся тем, что М-промотором является Sn. 15. Способ по п. 13, отличающийся тем, что Sn добавляют к водной смеси в форме коллоидного раствора оксида олова. 16. Способ по п. 13, отличающийся тем, что по меньшей мере один М-промотор добавляют к водной смеси перед образованием предшественника катализатора. 17. Способ по п. 13, отличающийся тем, что N-промотор добавляют к водной смеси после образования предшественника катализатора. РИСУНКИ
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 27.03.2008
Извещение опубликовано: 20.07.2010 БИ: 20/2010
|
||||||||||||||||||||||||||