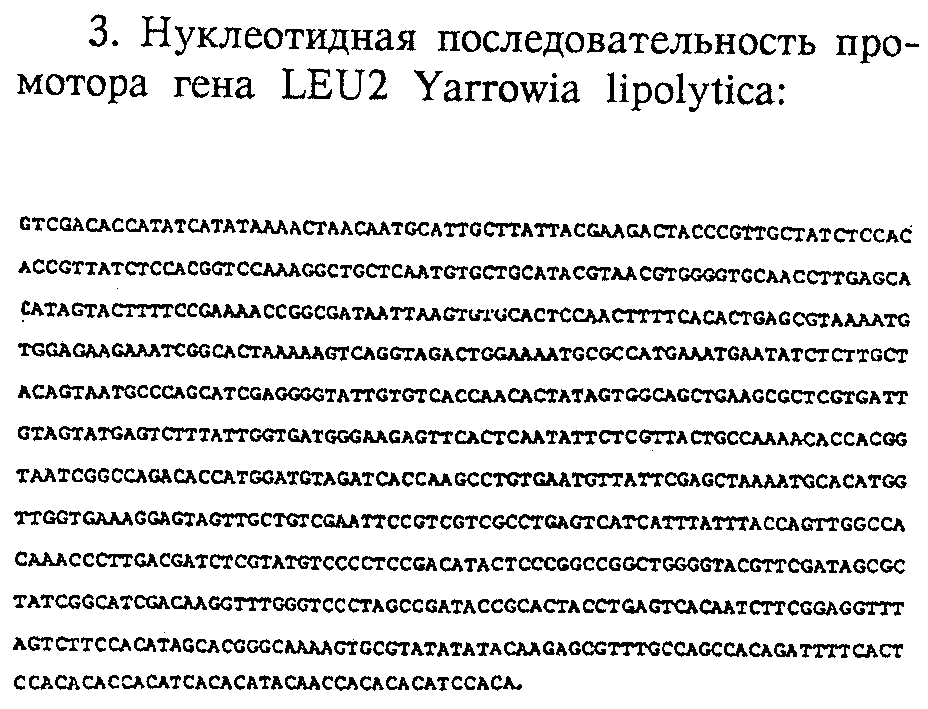Патент на изобретение №2198923
|
||||||||||||||||||||||||||
(54) НУКЛЕОТИДНАЯ ПОСЛЕДОВАТЕЛЬНОСТЬ ГЕНА LEU2 YARROWIA LIPOLYTICA (ВАРИАНТЫ), РЕКОМБИНАНТНЫЙ ДНК-МАТЕРИАЛ (ВАРИАНТЫ), ШТАММ ДРОЖЖЕЙ YARROWIA LIPOLYTICA (ВАРИАНТЫ)
(57) Реферат: Изобретение относится к технологии выделения протеинов дрожжей и касается нуклеотидной последовательности гена LEU2 Yarrowia lipolytica, рекомбинантных ДНК-материалов, каждый из которых содержит, помимо нуклеотидной последовательности гена LEU2 Yarrowia lipolytica, ген прореннина или анафилатоксина человека, а также штаммов Yarrowia lipolytica, трансформированных рекомбинантным ДНК-материалом и способных продуцировать прореннин или анафилатоксин человека. Изобретение обеспечивает надежное получение достаточно высокого количества таких протеинов, как прореннин и анафилатоксин человека, в легко отделяемой функциональной форме. 8 с.п. ф-лы, 8 табл., 15 ил. Заявка является частичным продолжением заявки, серийный номер 789206, поданной 18 октября 1985 г. Данное изобретение относится к технологии выделения протеинов дрожжей. Более конкретно оно относится к рекомбинантным векторам клонирования Yarrowia lipolytica, содержащим чужеродную ДНК, кодирующую экспрессию и выделение протеинов млекопитающих (например, прохимозина) и других полипептидов; к векторам экспрессии, содержащим промотор гена Y. lipolytica (например, XРR2 или LЕU2), ДНК-последовательности, кодирующие сигнальную (или предсигнальную) последовательность щелочной протеазы, область пропептида и участок терминатора XPR2, а также их варианты и функциональные эквиваленты, возникающие при вырождении генетического кода или использовании других генных компонентов Y. lipolytica. Кроме того, изобретение относится к трансформантам дрожжей, несущим указанные векторы экспрессии и выделения, их использованию для получения чужеродных протеинов в их нативной, функциональной форме и методам осуществления вышеуказанного. Экономическая привлекательность как надежного и обеспечивающего достаточно высокий выход разнообразного множества протеинов или полипептидов, представляющих интерес для промышленности (например, прореннина, бычьего гормона роста) или медицинских целей (например, урогастрона, тканевого активатора плазминогена, человеческого анафилатоксина С5а), и в особенности как источника, дающего высококачественный продукт в легко отделяемой функциональной форме, привели многих исследователей к приложению ДНК-рекомбинантной технологии к микроорганизмам как “фабрикам” для производства чужеродных протеинов. Обширные исследования сконцентрированы на секреции протеинов как потенциальном разрешении трудностей, встречающихся при извлечении чужеродных (гетерологичных) или экзогенных протеинов в биологически активной форме из внутриклеточных скоплений их в рекомбинантных клетках хозяина, в особенности из Escherichia coli. В Е. coli чужеродный протеин часто производится в клетке в форме преломляющих тел включения. Эти протеины обычно имеют низкую растворимость в воде и малую биологическую активность или не имеют ее вовсе. Извлечение данных протеинов из преломляющих тел включения обычно требует жесткой химической обработки, которая может быть дорогостоящей и может иметь следствием низкий выход или невозможность получения протеина в желаемой нативной, биологически активной форме. Кроме того, возможность загрязнения протеина нежелательными веществами, производимыми Е. coli, увеличивается ввиду необходимости разрушения клеток для освобождения преломляющих тел. Другие организмы, кроме Е. coli, также производят чужеродные протеины в нерастворимой внутриклеточной форме. Например, патент Великобритании 2091271, опубликованный 28 июля 1982 г., описывает генетическую модификацию S. cerevisiae путем ДНК-рекомбинантной технологии для экспрессии телячьего реннина или химозина; эти термины используются в патенте взаимозаменяемо. Ввиду этих трудностей обратились к секреции упомянутых протеинов с целью получения протеинов в нативной, активной конфигурации. Выделяется ли какой-либо протеин, включая чужеродные протеины, или полипептид данным организмом, предположительно зависит от вида протеина. В большинстве эукариотических клеток часть аппарата синтеза протеинов связана с эндоплазмичной сетчатой мембраной и последовательность аминокислот (называемая “сигнальной последовательностью”) у аминоконца растущей полипептидной цепи служит для управления процессом преодоления протеином мембраны. Сигнальная последовательность затем отщепляется протеолитически, давая активный зрелый протеин. Для ускорения процессов выделения чужеродных протеинов было сделано несколько попыток использовать сигнальные последовательности микроорганизмов, в том числе Bacillus subtilis, Saccharomyces cerevisiae и культур клеток млекопитающих. Однако данные организмы оказались не идеальными. Присущие В. subtilis свойства, например выделение многих протеинов, включая многочисленные протеазы, которые способны деградировать выделяемый чужеродный протеин, нестабильность трансформированных штаммов, возникающая вследствие потери чужеродной ДНК, препятствуют его использованию. Клетки млекопитающих успешно модифицировались генетически для экспрессии и выделения чужеродных протеинов, но эти системы технологически сложны и дорогостоящи для обработки и не имеют практического применения для коммерческого производства большинства протеинов. Хотя исследования выделения протеинов были более успешны с S. cerevisiae, чем с В. subtilis, похоже, что S. cerevisiae тоже имеет некоторые внутренние ограничения, как система выделения протеинов. Европейская патентная заявка 0123544, опубликованная 31 октября 1984 г., описывает выделение  -фактор-генов из S. cerevisiae и использование их промотирующих и/или сигнальных пептидных частей в сочетании с ДНК, кодирующей чужеродные для дрожжей протеины, в плазмиде для трансформации клеток дрожжей, способных производить отдельный зрелый протеин в клеточной культуре. Европейская заявка 0088632, опубликованная 14 сентября 1983 г., описывает способ экспрессии и выделения чужеродного протеина в S. cerevisiae. Однако, как представляется, размер протеинов, которые S. cerevisiae будет эффективно выделять с данной и другими системами выделения, должен быть ограничен величиной примерно 20000 дальтон. Преодоление этой неэффективности S. cerevisiae как организма выделения требует многих мутационных изменений, которые описаны Smith и др. Science 229; 1219-1224 (1985). Единственным исключением в этом отношении является наблюдение, что ферменты Aspergillus величиной более 20000, видимо, могут быть выделены S. cerevisiae, но эти ферменты сильно гликозилируются S. cerevisiae, что и может влиять на эффективность выделения.
Особый интерес представляет Yarrowia lipolytica, промышленно важный вид дрожжей, используемый для производства лимонной кислоты и клеточных протеинов. Он также может быть использован для получения эритрита, маннита и изопропиляблочной кислоты. В противоположность S. cerevisiae Y. lipolytica представляет особый интерес и ценность из-за своей способности эффективно выделять высокомолекулярные протеины (щелочную протеазу, кислую протеазу и ДНК-азу) в питательную среду, предоставляя таким образом потенциальную возможность извлечения чужеродных протеинов в нативном состоянии без необходимости разрушения продуцирующих клеток. Кроме того, Y. lipolytica количественно выделяет очень мало собственных протеинов, таким образом предоставляя возможность получения нужного чужеродного протеина в питательной среде в качестве доминирующей разновидности протеинов и облегчая извлечение указанного чужеродного протеина.
Y. lipolytica производит большое количество внеклеточной протеазы. Это доминирующий протеин, выделяемый Y. lipolytica. Вид протеазы (щелочной, кислый или нейтральный) зависит от используемого штамма Y. lipolytica (Ogrydziak и др. J. Gen. Microbiol. (1982) 128, 1225-1234). Частичный анализ N-концевой аминокислотной последовательности щелочной внеклеточной протеазы описан Ogrydziak и др. (ук. соч.).
Рассматриваемая заявка с серийным 634505, поданная 25 июля 1984 г., описывает методы трансформации Y. lipolytica и клонирования генов Y. lipolytica путем комплементации мутаций. В ней описано клонирование гена XPR2, который кодирует выделяемую щелочную протеазу, путем комплементации хрr2 мутаций Y. lipolytica. Метод включает трансформацию штамма-хозяина Y. lipolytica с частичным расщеплением ВglII из библиотеки генов Y. lipolytica в вектор pLD40, описанный в Европейской заявке 0138508, опубликованной 24 апреля 1985 г. и являющейся аналогом вышеуказанной заявки США.
Данное изобретение предусматривает: рекомбинантные векторы клонирования Y. lipolytica, содержащие чужеродную ДНК, кодирующую протеины млекопитающих и другие полипептиды, в том числе плазмиды, пригодные для трансформации клеток хозяина Y. lipolytica, и в особенности, интегративные векторы экспрессии, содержащие ген-промотор LEU2, ген-промотор XPR2, препрообласти щелочной протеазы и концевой регион XPR2; и плазмиды экспрессии, имеющие чужеродную кодирующую последовательность с сигналами выделения XPR2 далее в направлении считывания от промотора LEU2, которые способны вызывать экспрессию и выделение чужеродного протеина в трансформируемой Y. lipolytica.
Изобретение, таким образом, иллюстрирует процесс экспрессии и выделения готовых чужеродных протеинов, в особенности прореннина и человеческого анафилатоксина С5а, из генетически измененных клеточных культур Y. lipolytica. Установление точной идентичности аминокислотной последовательности, а также последовательности ДНК для внеклеточной щелочной протеазы Y. lipolytica позволило предположить, что чужеродный протеин может быть экспрессирован и выделен с помощью ДНК-рекомбинантной технологии при производстве в клеточной культуре. В случае прореннина экспрессируется и выделяется готовая форма зимогена (предшественник реннина).
Было обнаружено, что Y. lipolytica может быть генетически модифицирована с помощью ДНК-рекомбинантной технологии с получением трансформантов, способных к экспрессии и выделению чужеродных протеинов в их нативной форме. Это достигается путем конструирования векторов, несущих сигнальную последовательность или сигнальную и первую (про1) или обе последовательности (npo1 + про2) гена XPR2, связанные со структурной генной последовательностью для чужеродного протеина, который необходимо выделить.
Трансформанты, получаемые интеграцией в локусе XPR2 вектора ДНК, содержащего фрагмент гена XPR2 с отсутствующими регуляторными или структурными компонентами на обоих концах гена, не выделяют более щелочной протеазы, характеристика, не только желательная для выделения чужеродного протеина, но и которая может быть использована для скрининга трансформантов.
Кроме того, векторы, несущие промотор XPR2 и последовательности для сигнальной последовательности выделения щелочной протеазы, способны в трансформированной клетке Y. lipolytica вызвать выделение готового чужеродного протеина. Некоторые ДНК-рекомбинантные векторы этого типа способны вызвать экспрессию/выделение независимо от сайта интеграции в геноме дрожжей. Вообще, векторы, содержащие подходящие 5′ и 3′-фланкирующие ДНК, вызывают экспрессию продукта независимо от сайта интеграции.
Кроме того, было неожиданно обнаружено, что интеграция производной от pBR322 плазмиды в хромосомную ДНК Y. lipolytica создает область гомологии, способную благоприятствовать дальнейшей сайт-направленной интегративной трансформации. Интегрированная копия pBR322 служит таким образом “доком” для входящей трансформирующей ДНК. Интеграция резидент-копии pBR322 в хромосомную ДНК Y. lipolytica, несмотря на то, что pBR322 не является ДНК Y. lipolytica, таким образом создает известную мишень для интеграции. Трансформированные реципиенты Y. lipolytica, содержащие такой сайт, обладают двумя главными преимуществами перед реципиентами, у которых подобный сайт отсутствует, а именно, наличием региона с известной последовательностью и известной рестрикционной картой в качестве мишени для сайт-направленной интеграции, и возможность определить, используя pBR322 как мишень для интеграции, содержит ли входящая плазмида полную функциональную единицу или ген, или только часть нужного гена. Например, плазмида, содержащая только 3′-фрагмент гена XPR2, может трансформировать реципиент XPR2-1002, если она содержит кодон дикого типа и интегрирована в локусе XPR2. Однако та же плазмида не будет трансформировать клетку-хозяина XPR2-1002 к положительному фенотипу протеазы, если интегрирована в pBR322, так как в ней отсутствует целая функциональная единица.
Таким образом, в трансформантах Y. lipolytica, содержащих область гомологии к чужеродному вектору ДНК, указанная область, содержащая экзогенную ДНК, служит в качестве принимающего сайта во время интегративной трансформации Y. lipolytica. В дополнение к pBR322 и ее производным для получения трансформантов Y. lipolytica, имеющих “док”, могут быть использованы космиды, бактериофаги, такие как М13 и лямбда, синтетически полученная ДНК и обычные плазмиды, такие как PUC13.
Под промотирующей последовательностью “LEU2” понимается нетранслируемый участок в направлении от ATG, который содержит большинство, если не все черты, требуемые для экспрессии.
Под промотирующей последовательностью “XPR2” понимается нетранслируемый участок, расположенный по направлению считывания перед сигнальной (или предсигнальной) последовательностью, которая необходима для экспрессии. Кроме того, сигнал с или без пропоследовательности из гена XPR2 может быть использован для выделения под контролем промоторов Y.lipolytica иных, чем гена XPR2, векторы, несущие промотор LEU2 и последовательности для сигнала выделения щелочной протеазы, способны в трансформированной клетке Y. lipolytica вызвать выделение готовых чужеродных протеинов.Человеческий анафилатоксин С5а, также известный как человеческий дополнительный протеин С5а (человеческий С5а), представляет собой биоактивный полипептидный фрагмент, генерируемый in vivo в результате комплементарной активации. Он функционирует как иммуномодулятор при регулировании определенных аспектов гуморальной и клеточной иммунореакции. Изучена первичная структура его и других анафилатоксинов. Обзор химических, физических и биологических характеристик представлен Hugli в “Complement”, под редакцией Н.J. Muller-Eberhard и Р.A. Miescher, стр. 73-99, 1985, Springer-Verlag, New-York.
Специалистам в данной области понятно, что в настоящем изобретении может быть использована с соответствующими необходимыми изменениями чужеродная ДНК, кодирующая фактически любую известную аминокислотную последовательность. Методика, описанная здесь, может быть применена с соответствующими необходимыми изменениями для получения и выделения любых известных чужеродных протеинов, представители которых перечислены в патенте США 4532207, опубликованном 30 июля 1985 г. Кроме того, вместо гена XPR2 может быть использован любой другой ген Y. lipolytica для выделения протеинов, такой как гены рибонуклеазы или кислой протеазы, а также гибридные гены, сконструированные путем объединения фрагментов двух или более указанных генов, например, сигнальная последовательность гена XPR2 и промотирующая последовательность гена рибонуклеазы.
В объем данного изобретения также включены функциональные эквиваленты вышеописанных ДНК или нуклеотидных последовательностей. Вырожденность генетического кода дает возможность замещения определенных кодонов другими, которые кодируют ту же самую аминокислоту и, следовательно, приводят к тому же протеину. ДНК или нуклеотидная последовательность могут значительно изменяться, так как за исключением метионина и триптофана известные аминокислоты могут кодироваться более чем одним кодоном. Так, часть или весь ген XPR2 могут быть синтезированы для получения последовательности ДНК, существенно отличающейся от показанной на фиг.3. Кодируемая аминокислотная последовательность для него будет однако сохранена. Подобные функциональные изменения данной ДНК или нуклеотидной последовательности дают возможность промотировать выделение и/или обработку чужеродных протеинов, кодируемых чужеродными ДНК-последовательностями, пришитыми к ним. Все изменения нуклеотидной последовательности гена XPR2 и его фрагментов, разрешенные генетическим кодом, следовательно, включены в данное изобретение. Кроме того, возможно ликвидировать кодоны или заменить один или более кодонов иными, чем вырожденные кодоны, для получения структурно модифицированного полипептида, но по существу имеющего ту же полезность или активность, что и полипептид, полученный с помощью немодифицированной молекулы ДНК. Данные два полипептида функционально эквивалентны, как две молекулы ДНК, вызывающие их появление, даже если разница между этими молекулами ДНК не относится к вырождению генетического кода. Наиболее простой пример этого относится к прореннину А и прореннину В, двум аллельным формам прореннина, которые различаются только присутствием остатка аспарагиновой кислоты в положении 286 прореннина А и остатка глицина в этом положении у прореннина В.
При использовании данной методики достигнута экспрессия и экскреция в Y. lipolytica чужеродных протеинов млекопитающих прореннина и человеческого анафилатоксина С5а при применении сигналов экспрессии и выделения из генов Y. lipolytica XPR2 и/или LEU2. ДНК-последовательности для прореннина и человеческого анафилатоксина С5а были пришиты с помощью синтетических олигонуклеотидов к генной последовательности XPR2 в сайтах, предполагаемых для кодирования сигнального пептида щелочной протеазы, или в сайтах преобразования предшественника протеазы, обозначенных здесь как про1 и про2, и использованы для получения генных конструктов, которые были затем введены в Y. lipolytica путем интегративной трансформации. Рекомбинантные культуры экспрессировали и выделяли в питательную среду чужеродные протеины, имеющие молекулярный вес и иммунореактивность прореннина и человеческого анафилатоксина С5а. Можно полагать, что прореннин, полученный таким образом, имеет нативную конфигурацию, так как после удаления пропептида он обнаруживает полную ферментативную активность.
Термин “рекомбинантный ДНК материал”, используемый здесь, включает любой материал, который содержит по крайней мере один из следующих элементов: ген XPR2 Y. lipolytica, сигнал (или предсигнал), про1- и про2- (которые вместе составляют прорегион) промотор или его концевую последовательность; промотор LEU2 или функциональные эквиваленты вышеуказанных последовательностей, допускаемые вырожденностью генетического кода.
Представителями указанного рекомбинантного ДНК материала являются фрагменты ДНК, плазмиды или векторы или трансформанты, содержащие любую или все из вышеуказанных последовательностей.
Материалы. Рестрикционные эндонуклеазы и Т4 лигаза были получены из New England Biolabs, бактериальная щелочная фосфатаза из Bethesda Research Laboratories, T4 полинуклеотидкиназа из PL-Biochemicals и [гамма-32p]АТФ из New England Nuclear. Все ферменты использовались при условиях, рекомендованных поставщиком.
Среды -фактор-генов из S. cerevisiae и использование их промотирующих и/или сигнальных пептидных частей в сочетании с ДНК, кодирующей чужеродные для дрожжей протеины, в плазмиде для трансформации клеток дрожжей, способных производить отдельный зрелый протеин в клеточной культуре. Европейская заявка 0088632, опубликованная 14 сентября 1983 г., описывает способ экспрессии и выделения чужеродного протеина в S. cerevisiae. Однако, как представляется, размер протеинов, которые S. cerevisiae будет эффективно выделять с данной и другими системами выделения, должен быть ограничен величиной примерно 20000 дальтон. Преодоление этой неэффективности S. cerevisiae как организма выделения требует многих мутационных изменений, которые описаны Smith и др. Science 229; 1219-1224 (1985). Единственным исключением в этом отношении является наблюдение, что ферменты Aspergillus величиной более 20000, видимо, могут быть выделены S. cerevisiae, но эти ферменты сильно гликозилируются S. cerevisiae, что и может влиять на эффективность выделения.
Особый интерес представляет Yarrowia lipolytica, промышленно важный вид дрожжей, используемый для производства лимонной кислоты и клеточных протеинов. Он также может быть использован для получения эритрита, маннита и изопропиляблочной кислоты. В противоположность S. cerevisiae Y. lipolytica представляет особый интерес и ценность из-за своей способности эффективно выделять высокомолекулярные протеины (щелочную протеазу, кислую протеазу и ДНК-азу) в питательную среду, предоставляя таким образом потенциальную возможность извлечения чужеродных протеинов в нативном состоянии без необходимости разрушения продуцирующих клеток. Кроме того, Y. lipolytica количественно выделяет очень мало собственных протеинов, таким образом предоставляя возможность получения нужного чужеродного протеина в питательной среде в качестве доминирующей разновидности протеинов и облегчая извлечение указанного чужеродного протеина.
Y. lipolytica производит большое количество внеклеточной протеазы. Это доминирующий протеин, выделяемый Y. lipolytica. Вид протеазы (щелочной, кислый или нейтральный) зависит от используемого штамма Y. lipolytica (Ogrydziak и др. J. Gen. Microbiol. (1982) 128, 1225-1234). Частичный анализ N-концевой аминокислотной последовательности щелочной внеклеточной протеазы описан Ogrydziak и др. (ук. соч.).
Рассматриваемая заявка с серийным 634505, поданная 25 июля 1984 г., описывает методы трансформации Y. lipolytica и клонирования генов Y. lipolytica путем комплементации мутаций. В ней описано клонирование гена XPR2, который кодирует выделяемую щелочную протеазу, путем комплементации хрr2 мутаций Y. lipolytica. Метод включает трансформацию штамма-хозяина Y. lipolytica с частичным расщеплением ВglII из библиотеки генов Y. lipolytica в вектор pLD40, описанный в Европейской заявке 0138508, опубликованной 24 апреля 1985 г. и являющейся аналогом вышеуказанной заявки США.
Данное изобретение предусматривает: рекомбинантные векторы клонирования Y. lipolytica, содержащие чужеродную ДНК, кодирующую протеины млекопитающих и другие полипептиды, в том числе плазмиды, пригодные для трансформации клеток хозяина Y. lipolytica, и в особенности, интегративные векторы экспрессии, содержащие ген-промотор LEU2, ген-промотор XPR2, препрообласти щелочной протеазы и концевой регион XPR2; и плазмиды экспрессии, имеющие чужеродную кодирующую последовательность с сигналами выделения XPR2 далее в направлении считывания от промотора LEU2, которые способны вызывать экспрессию и выделение чужеродного протеина в трансформируемой Y. lipolytica.
Изобретение, таким образом, иллюстрирует процесс экспрессии и выделения готовых чужеродных протеинов, в особенности прореннина и человеческого анафилатоксина С5а, из генетически измененных клеточных культур Y. lipolytica. Установление точной идентичности аминокислотной последовательности, а также последовательности ДНК для внеклеточной щелочной протеазы Y. lipolytica позволило предположить, что чужеродный протеин может быть экспрессирован и выделен с помощью ДНК-рекомбинантной технологии при производстве в клеточной культуре. В случае прореннина экспрессируется и выделяется готовая форма зимогена (предшественник реннина).
Было обнаружено, что Y. lipolytica может быть генетически модифицирована с помощью ДНК-рекомбинантной технологии с получением трансформантов, способных к экспрессии и выделению чужеродных протеинов в их нативной форме. Это достигается путем конструирования векторов, несущих сигнальную последовательность или сигнальную и первую (про1) или обе последовательности (npo1 + про2) гена XPR2, связанные со структурной генной последовательностью для чужеродного протеина, который необходимо выделить.
Трансформанты, получаемые интеграцией в локусе XPR2 вектора ДНК, содержащего фрагмент гена XPR2 с отсутствующими регуляторными или структурными компонентами на обоих концах гена, не выделяют более щелочной протеазы, характеристика, не только желательная для выделения чужеродного протеина, но и которая может быть использована для скрининга трансформантов.
Кроме того, векторы, несущие промотор XPR2 и последовательности для сигнальной последовательности выделения щелочной протеазы, способны в трансформированной клетке Y. lipolytica вызвать выделение готового чужеродного протеина. Некоторые ДНК-рекомбинантные векторы этого типа способны вызвать экспрессию/выделение независимо от сайта интеграции в геноме дрожжей. Вообще, векторы, содержащие подходящие 5′ и 3′-фланкирующие ДНК, вызывают экспрессию продукта независимо от сайта интеграции.
Кроме того, было неожиданно обнаружено, что интеграция производной от pBR322 плазмиды в хромосомную ДНК Y. lipolytica создает область гомологии, способную благоприятствовать дальнейшей сайт-направленной интегративной трансформации. Интегрированная копия pBR322 служит таким образом “доком” для входящей трансформирующей ДНК. Интеграция резидент-копии pBR322 в хромосомную ДНК Y. lipolytica, несмотря на то, что pBR322 не является ДНК Y. lipolytica, таким образом создает известную мишень для интеграции. Трансформированные реципиенты Y. lipolytica, содержащие такой сайт, обладают двумя главными преимуществами перед реципиентами, у которых подобный сайт отсутствует, а именно, наличием региона с известной последовательностью и известной рестрикционной картой в качестве мишени для сайт-направленной интеграции, и возможность определить, используя pBR322 как мишень для интеграции, содержит ли входящая плазмида полную функциональную единицу или ген, или только часть нужного гена. Например, плазмида, содержащая только 3′-фрагмент гена XPR2, может трансформировать реципиент XPR2-1002, если она содержит кодон дикого типа и интегрирована в локусе XPR2. Однако та же плазмида не будет трансформировать клетку-хозяина XPR2-1002 к положительному фенотипу протеазы, если интегрирована в pBR322, так как в ней отсутствует целая функциональная единица.
Таким образом, в трансформантах Y. lipolytica, содержащих область гомологии к чужеродному вектору ДНК, указанная область, содержащая экзогенную ДНК, служит в качестве принимающего сайта во время интегративной трансформации Y. lipolytica. В дополнение к pBR322 и ее производным для получения трансформантов Y. lipolytica, имеющих “док”, могут быть использованы космиды, бактериофаги, такие как М13 и лямбда, синтетически полученная ДНК и обычные плазмиды, такие как PUC13.
Под промотирующей последовательностью “LEU2” понимается нетранслируемый участок в направлении от ATG, который содержит большинство, если не все черты, требуемые для экспрессии.
Под промотирующей последовательностью “XPR2” понимается нетранслируемый участок, расположенный по направлению считывания перед сигнальной (или предсигнальной) последовательностью, которая необходима для экспрессии. Кроме того, сигнал с или без пропоследовательности из гена XPR2 может быть использован для выделения под контролем промоторов Y.lipolytica иных, чем гена XPR2, векторы, несущие промотор LEU2 и последовательности для сигнала выделения щелочной протеазы, способны в трансформированной клетке Y. lipolytica вызвать выделение готовых чужеродных протеинов.Человеческий анафилатоксин С5а, также известный как человеческий дополнительный протеин С5а (человеческий С5а), представляет собой биоактивный полипептидный фрагмент, генерируемый in vivo в результате комплементарной активации. Он функционирует как иммуномодулятор при регулировании определенных аспектов гуморальной и клеточной иммунореакции. Изучена первичная структура его и других анафилатоксинов. Обзор химических, физических и биологических характеристик представлен Hugli в “Complement”, под редакцией Н.J. Muller-Eberhard и Р.A. Miescher, стр. 73-99, 1985, Springer-Verlag, New-York.
Специалистам в данной области понятно, что в настоящем изобретении может быть использована с соответствующими необходимыми изменениями чужеродная ДНК, кодирующая фактически любую известную аминокислотную последовательность. Методика, описанная здесь, может быть применена с соответствующими необходимыми изменениями для получения и выделения любых известных чужеродных протеинов, представители которых перечислены в патенте США 4532207, опубликованном 30 июля 1985 г. Кроме того, вместо гена XPR2 может быть использован любой другой ген Y. lipolytica для выделения протеинов, такой как гены рибонуклеазы или кислой протеазы, а также гибридные гены, сконструированные путем объединения фрагментов двух или более указанных генов, например, сигнальная последовательность гена XPR2 и промотирующая последовательность гена рибонуклеазы.
В объем данного изобретения также включены функциональные эквиваленты вышеописанных ДНК или нуклеотидных последовательностей. Вырожденность генетического кода дает возможность замещения определенных кодонов другими, которые кодируют ту же самую аминокислоту и, следовательно, приводят к тому же протеину. ДНК или нуклеотидная последовательность могут значительно изменяться, так как за исключением метионина и триптофана известные аминокислоты могут кодироваться более чем одним кодоном. Так, часть или весь ген XPR2 могут быть синтезированы для получения последовательности ДНК, существенно отличающейся от показанной на фиг.3. Кодируемая аминокислотная последовательность для него будет однако сохранена. Подобные функциональные изменения данной ДНК или нуклеотидной последовательности дают возможность промотировать выделение и/или обработку чужеродных протеинов, кодируемых чужеродными ДНК-последовательностями, пришитыми к ним. Все изменения нуклеотидной последовательности гена XPR2 и его фрагментов, разрешенные генетическим кодом, следовательно, включены в данное изобретение. Кроме того, возможно ликвидировать кодоны или заменить один или более кодонов иными, чем вырожденные кодоны, для получения структурно модифицированного полипептида, но по существу имеющего ту же полезность или активность, что и полипептид, полученный с помощью немодифицированной молекулы ДНК. Данные два полипептида функционально эквивалентны, как две молекулы ДНК, вызывающие их появление, даже если разница между этими молекулами ДНК не относится к вырождению генетического кода. Наиболее простой пример этого относится к прореннину А и прореннину В, двум аллельным формам прореннина, которые различаются только присутствием остатка аспарагиновой кислоты в положении 286 прореннина А и остатка глицина в этом положении у прореннина В.
При использовании данной методики достигнута экспрессия и экскреция в Y. lipolytica чужеродных протеинов млекопитающих прореннина и человеческого анафилатоксина С5а при применении сигналов экспрессии и выделения из генов Y. lipolytica XPR2 и/или LEU2. ДНК-последовательности для прореннина и человеческого анафилатоксина С5а были пришиты с помощью синтетических олигонуклеотидов к генной последовательности XPR2 в сайтах, предполагаемых для кодирования сигнального пептида щелочной протеазы, или в сайтах преобразования предшественника протеазы, обозначенных здесь как про1 и про2, и использованы для получения генных конструктов, которые были затем введены в Y. lipolytica путем интегративной трансформации. Рекомбинантные культуры экспрессировали и выделяли в питательную среду чужеродные протеины, имеющие молекулярный вес и иммунореактивность прореннина и человеческого анафилатоксина С5а. Можно полагать, что прореннин, полученный таким образом, имеет нативную конфигурацию, так как после удаления пропептида он обнаруживает полную ферментативную активность.
Термин “рекомбинантный ДНК материал”, используемый здесь, включает любой материал, который содержит по крайней мере один из следующих элементов: ген XPR2 Y. lipolytica, сигнал (или предсигнал), про1- и про2- (которые вместе составляют прорегион) промотор или его концевую последовательность; промотор LEU2 или функциональные эквиваленты вышеуказанных последовательностей, допускаемые вырожденностью генетического кода.
Представителями указанного рекомбинантного ДНК материала являются фрагменты ДНК, плазмиды или векторы или трансформанты, содержащие любую или все из вышеуказанных последовательностей.
Материалы. Рестрикционные эндонуклеазы и Т4 лигаза были получены из New England Biolabs, бактериальная щелочная фосфатаза из Bethesda Research Laboratories, T4 полинуклеотидкиназа из PL-Biochemicals и [гамма-32p]АТФ из New England Nuclear. Все ферменты использовались при условиях, рекомендованных поставщиком.
СредыСреда ГПП (среда глицерин-протеаза-пептон) содержала (на литр): 6,7 г глицерина, 1,6 г Difco протеазы-пептона, 1,7 г Difco азотистого основания дрожжей без аминокислот и сульфата аммония, 30 мг урацила и 0,5 мл/л полипропиленгликоля с мол. в. 2000 (Polysciences) в 40 мМ-фосфатном буфере (рН 6,8). (Полипропиленгликоль не был включен, когда среда использовалась для выращивания культур для анализа ферментной активности реннина). Протеаза-пептон был отдельно обработан в автоклаве в фосфатном буфере. Среда ДЭПД (дрожжевой экстракт-пептон-декстроза) содержала (на литр): 5 г экстракта дрожжей, 10 г пептона и 20 г декстрозы. Е. coli выращивали в среде LB при 37oС. Среда LB содержала (на литр): 10 г бактотриптона, 10 г бактодрожжевого экстракта, 10 г хлорида натрия, доводилась до рН 7,5 гидроксидом натрия. Анализ последовательности ДНК. Фрагменты ДНК из различных описанных здесь плазмид изолировали на геле полиакриламида и секвенировали по методу Маxam и др. Methods in Enzymology, 65, 499 (1980). Процедуры связывания. Фрагменты ДНК, включая расщепленные векторные плазмиды, связывали путем смешения нужных компонентов (фрагменты ДНК с концевыми участками, специально сконструированными для обеспечения правильного спаривания) с Т4 ДНК-лигазой. Добавляли примерно 10 единиц лигазы на мкг количества вектора и вводимых фрагментов. Полученную реакционную смесь трансформировали в компетентные клетки штаммов ММ294 (АТСС-33625) или НВ101 (АТСС-33694) Е. coli K12. Получение химически синтезированной ДНК. Для конструирования гибридных генов для экспрессии и выделения прореннина было синтезировано восемь олигонуклеотидов по модифицированной фосфорамидатной методике (Sinha и др., Tetrahedron Letters 24, 5843 (1983) на Genetic Design 6500 (Watertown, MA) автоматическом ДНК-синтезаторе и очищены на 6М мочевина-20% полиакриламидном геле. Аликвоты комплементарных олигонуклеотидов смешивали и сшивали друг с другом при 4oС в течение ночи в ТЭ (10 мМ трис-HCl, рН 8,0; 1 мМ NaEDTA). Аликвоты (около 2 мкг) двухцепочечных олигонуклеотидов фосфорилировали в 20 мкл реакционной смеси, содержащей 70 мМ трис (рН 7,6), 10 мМ MgCl2, 5 мМ дитиотрейтола, 5 мМ АТФ, при 37oС с использованием Т4 полинуклеотидкиназы. Получение плазмидной ДНК. Плазмидную ДНК в больших количествах получали по методике Holmes и др.. Anal. Biochem., 114, 195-197 (1981), с последующим центрифугированием в градиенте плотности этидиум бромид-CsCl. Миниколичества плазмидной ДНК получали по щелочной SDS методике Birnboim и др., NAR 1, 1513 (1979). Конструирование векторов экспрессии/выделения для прореннина. Была приготовлена серия различных конструкций для получения конечных векторов экспрессии. Все стадии представлены в виде схемы на прилагаемых фигурах. В общем, фрагменты ДНК изолировали путем гель-электрофореза и сшивали с другими фрагментами или расщепленной плазмидной ДНК в 20 мкл 50 мМ трис-НСl (рН 7,5), 10 мМ MgCl2, 20 мМ дитиотрейтола, 1 мМ АТФ, в присутствии 200 единиц Т4 лигазы при 4oС. Если требовалось частичное расщепление ДНК рестрикционной эндонуклеазой, оптимальное время расщепления устанавливалось экспериментально. Идентификация прореннина в культуральной жидкости. Трансформанты дрожжей, содержащие векторы экспрессии, выращивали в течение ночи в среде ГПП (см. выше). После центрифугирования для удаления клеток дрожжей добавляли 1 мл 50% трихлоруксусной кислоты в каждой 5 мл аликвоте культуральной жидкости и выдерживали при 4oС 60 минут. Центрифугированием получали шарики, которые дважды промывали 2 мл холодного ацетона. Осажденный протеин растворяли в 100 мкл стандартного SDS-буфера и аликвоты подвергали электрофорезу на 10% SDS-полиакриламидных гелях (Laemmli, U.K. (1970), Nature 227, 680). Освобожденные из геля протеины электрофоретически переносили на нитроцеллюлозу (Schleicher и Schuell, 0,22 мкм) и прореннин определяли иммуноточечным анализом на пластинке геля (Hawkes, R. и др. (1982) Anal. Biochem. 119, 142). Фильтр покрывали антипрореннин-антителом кролика с последующей инкубацией с конъюгированным пероксидазой антителом козел-кролик (IgG) Cappel, Malvern, Pa). Связанные антитела определяли окрашиванием смесью 4-хлор-1-нафтола и перекиси водорода. Активность прореннина по свертыванию молока в культуральной жидкости. Культуральную жидкость различных трансформантов Y. lipolytica исследовали на активность по свертыванию молока по модифицированному методу Ernstrom, J. Dairy Sci. 41, 1664 (1958). Коротко говоря, исследование включало измерение времени, требуемого для свертывания буферного снятого молока реннином в активированных супернатантах культуры и соотнесения значений со стандартными для очищенного реннина. Культуры дрожжей (25 мл) выращивали в течение ночи в среде ГПП. После удаления клеток центрифугированием 5 мл аликвоты супернатантов культуры высушивали замораживанием под вакуумом. Каждый лиофилизированный супернатант был ресуспендирован в 300 мкл дистиллированной воды. В качестве контрольного стандарта была приготовлена серия разведений очищенного прореннина теленка. Концентраты прореннина в среде и контрольные образцы активировали добавлением примерно 5-10 мкл конц. НСl до получения рН приблизительно 2 и инкубировали один час при 22oС. Снятое молоко готовили добавлением 60 г сухого порошка снятого молока (Difco) к 500 мл 41,5 мМ ацетата натрия (рН 6,3) и 13,6 мМ CaCl2 и перемешиванием в течение 20 минут при 4oС. Субстрат был использован для анализа немедленно после приготовления. Аликвоту 60 мкл (эквивалент 1 мл супернатанта культуры) каждого препарата фермента добавляли к 1 мл аликвоты снятого молока при 37oС и регистрировали время свертывания. Получение синтетических олигонуклеотидов для гена С5а. Олигодезоксинуклеотиды, используемые в синтезе структурного гена С5а, получали по модифицированной фосфорамидатной методике (Sinha и др., ук. соч.) с использованием контролируемой подложки из пористого стекла на Genetic Design 6500 (Watertown, MA) автоматическом ДНК-синтезаторе. При получении использовали 3% (вес/объем) дихлоруксусную кислоту в дихлорметане для удаления тритильной группы, активирование фосфорамидатов насыщенным тетразолом в ацетонитриле, обработку диэтоксифосфинтетразолидом и окисление водным раствором йод/ТГФ (Matteucci и др., 1981, J. Amer. Chem. Soc. 105, 3183). Полное время на цикл добавления составляло 14 минут. Десять 47-мер сегментов A-J (фиг.7) получали с 98,8% средним выходом на стадию (по тритильному анализу), деблокировали по методике Matteucci и др., ук. соч., осаждали этанолом из 0,3 М ацетата натрия и выделяли препаративным гель-электрофорезом на 10% денатурированном геле полиакриламид-мочевина перед сшиванием. Сборка, клонирование и секвенирование человеческого гена С5а. На фиг.7 показана аминокислотная последовательность нужного протеина и расположение синтетических олигонуклеотидов, необходимых для изготовления гена, кодирующего человеческий протеин С5а. Все олигомеры, кроме А и F, фосфорилировали по их 5′-концам Т4 полинуклеотидкиназой. Сборка гена включала две первичные реакции расщепления/сшивания, включающие: олигомеры А, В, I и J и олигомеры С, D, Е, F, G и Н. Образующиеся двухцепочечные фрагменты ДНК размером 94 п.ос. и 141 п. ос. изолировали после электрофореза на 10% полиакриламидном геле, сшивали вместе, и их продукт размером 235 п.ос. изолировали гель-электрофорезом. Этот фрагмент ДНК, содержащий структурный ген, кодирующий С5а, вводили между EcoRI и HindIII-сайтами ДНК вектора pBR322 и использовали для трансформации клеток штамма НВ101 Е. coli К-12. Рестрикционный анализ плазмидной ДНК, изолированной из 6 трансформантов, показал, что 5 из 6 клонов содержали EcoRI/HindIII-фрагмент правильного размера. Нуклеотидную последовательность участка гена С5а каждой из этих плазмид определяли по методу Махаm и др. Method Enzymol. 65, 499 (1980). Конструирование и характеристика плазмиды экспрессии С5а для Е. coli. Методика изолирования фрагментов ДНК и условия реакции сшивания были такими же, как описано Maniatis и др. (1982) Molecular Cloning: A Laboratory Manual, Cold Spring Harbor. E. coli trp промотор-оператор был первоначально получен из ptrpL1 (Edman и др. (1981) Nature 291, 503). Фрагмент EcoRI 360 п. ос. , содержащий промотор-операторную последовательность, используемый в плазмиде экспрессии С5а (рС5а-48), изолировали из плазмиды экспрессии прореннина pPFZ-R2, описанной в Европейской заявке 0147178, опубликованной 3 июля 1985 г. Идентификация С5а в культуральной жидкости Y. lipolytica. Методика была такой же, как описано выше для прореннина, за исключением того, что в иммуноанализе были использованы козел- анти-С5а- и кролик анти-козел IgG (Cappel). Антитело козел-анти-человеческий С5а получали методом Manderino и др., J. Immunol. Methods 53, 41-50 (1982). Векторы pLD40 – описанный в Европейской заявке 0138508, опубликованной 24 апреля 1985 г. Микроорганизмы: АТСС 20774 – Yarrowia lipolytica PC 30869; АТСС 20781 – Yarrowia lipolytica DL112-PC-30869, трансформант с XPR2; АТСС 20776 – Yarrowia lipolytica DL-148. Трансформант Y. Lipolytica ATCC-20688 с SnaBI, расщепленной pLS-3; АТСС 20775 – Yarrowia lipolytica DL-144. Трансформант Y. Lipolytica АТСС 20688 с нерасщепленной pLS-3; АТСС 20777 – Трансформант Y. lipolytica PC-30869 с SnaBI, расщепленной рС5аХ-3; АТСС 20780 – Трансформант Y. lipolytica PC-30869 с SnaBI, расщепленной рХХ-33; АТСС 20794 – Трансформант Y. lipolytica PC-30869 с pLD56; АТСС 20795 – Трансформант Y. lipolytica АТСС 20794 с NruI, расщепленной pLX-34. Они были депонированы в соответствии с Будапештским договором в Американской Коллекции типовых культур, Rockville, Maryland, признанной коллекции, гарантирующей неизменность депозитов и быстрый доступ к ним заинтересованных лиц, если будет выдан патент по данной заявке. В период рассмотрения данной заявки депозиты доступны лицам, которые определяются Комиссионером Ведомства по патентам и товарным знакам США в соответствии с 37 CFR 1.14 и 35 USC 122 и в соответствии с иностранными патентными законодательствами в странах, куда поданы дубликаты данной заявки или основанных на ней последующих заявок. Все ограничения на доступ к депонированным микроорганизмам будут окончательно сняты после выдачи патента. Таксономическое исследование Y. lipolytica АТСС 20774 (идентифицированного в коллекции культур Pfizer Inc. как PC 30869) проведено д-ром J. R. DeZeeuw, который подготовил следующее описание. Использовались методики, рекомендованные J.L. Lodder в “The Yeasts”, второе издание, N. Holland Publishing Co., Амстердам, 1970. CBS 599, типовая культура для вида Candida lipolytica (“The Yeasts”, второе издание, N. Holland Publishing Co., Амстердам, 1970) и CBS 6124, типовая культура для Saccharomycopsis lipolytica в “The Yeasts”, третье издание, были взяты для сравнения. Ранее вид был также сопоставлен с Endomycopsis lipolytica. Его промежуточное обозначение – Candida lipolytica. Таксономическое положение вида было установлено van der Walt и von Arx, Antonie van Leeuwenhoek, 46, 517-521 (1980). Предпочитаемым названием в настоящее время является Yarrowia lipolytica. Культуральные, морфологические и физиологические характеристики штамма PC-30869 согласуются со стандартным описанием вида, указанного как Saccharomycopsis lipolytica в “The Yeasts”, третье издание, под редакцией Kreger-van Rij, стр. 406-408, Elsevier Science Publishers B.V., Амстердам, 1984. Сравнение штаммов Y.lipolytica приведено в таблице 1. PC-30869 был сконструирован с помощью генетически рекомбинированных подходящих мутантов Y. lipolytica PC-22208, Pfizer почвенный изолят и Y. lipolytica PC-30026, субкультура NRRL Y-1094. PC-30869 фенотипически отличается от своих родителей дикого типа следующим: (1) не производит активной внеклеточной щелочной протеазы, (2) требует биотина для роста и (3) требует источник L-лейцина. В течение лог-фазы роста PC-30869 в бульоне дрожжевой экстракт-пептон-глюкоза (ДЭПГ) почкующиеся клетки имеют яйцевидную форму и средний размер 2,6 х 5,5 микрон. На ДЭПГ-агаре доминируют псевдо- и истинные мицелии. Бластоспоры присутствуют в большинстве как одиночки в плевральных положениях. Не обнаруживаются каротиноидные пигменты. Культура ведет себя как “В” спаривающийся гаплоид в скрещивании с аутентичными для вида тестовыми штаммами (Таблица 5). Типичная аскоспоруляция наблюдается на агаре V8. Особенности ассимиляции углерода указаны в Таблице 2. Ферментация отсутствует. Ион аммония и мочевина, но не нитрат, утилизируются как единственные источники азота (Таблица 3). Штамм PC-30869 требует витаминов тиамина и D-биотина (Таблица 4). Только тиамин требуется для родителей культуры дикого типа. Никакого роста не наблюдается при 37oС. В таблице 6 приведены также и другие характеристики штаммов. PC-30869 отличается от других штаммов Y. lipolytica, описанных в патентной литературе, как это следует из сравнения их фенотипов (таблицы 7 и 8). АТСС 20228 (Nubel и др. Патент США 4155811) обнаруживает вид питания дикого типа подобно типовым штаммам для вида CBS 599 и CBS 6124. Конкретно, он не нуждается для роста в урациле, лейцине или биотине и он разжижает желатин. АТСС 20268 (DeZeeuw и др., патент США 4407953) в отличие от АТСС 20228 требует дополнительного лейцина для роста. Подобно АТСС 20228 он не нуждается в урациле или биотине. Он также разжижает желатин. АТСС 20688 (европейская заявка 0138508) растет, только если в среду добавлены как урацил, так и лейцин. Эта необходимость в урациле отличает АТСС 20688 как от АТСС 20228, так и от АТСС 20628. АТСС 20688 не требует биотина и разжижает желатин. Культура PC-30869 отличается от всех вышеуказанных. Она нуждается для роста в биотине и лейцине, но не в урациле. Она не разжижает желатин. Полная среда содержала 16,7 г/л бактодрожжевого основания, свободного от витаминов, плюс 100 мг/л урацила, 100 мг/л L-лейцина, 10 мкг/л D-биотина и 200 мкг/л тиамина  НСl.
Среда содержала 120 г/л желатина и 16,7 г/л бактодрожжевого основания, свободного от витаминов, плюс 100 мг/л урацила, 100 мг/л L-лейцина, 10 мкг/л D-биотина и 200 мкг/л тиаминаНСl.
Краткое описание чертежей НСl.
Среда содержала 120 г/л желатина и 16,7 г/л бактодрожжевого основания, свободного от витаминов, плюс 100 мг/л урацила, 100 мг/л L-лейцина, 10 мкг/л D-биотина и 200 мкг/л тиаминаНСl.
Краткое описание чертежейФиг. 1 – Частичная линейная рестрикционная карта перекрывающихся плазмид pLD 57, pLD 58 и pLD 62, изолированных из штамма Y. lipolytica DL 112. Фиг.2 – Пробы синтетических олигонуклеотидов для гена XPR2. Из описанной последовательности для большинства первых 25 аминокислотных остатков активной протеазы (Ogrydziak и др., ук. соч.) два участка, отмеченные I и II, дают возможность конструирования 14-мер-олигонуклеотидных проб с 32-кратным или меньшим вырождением. Эти два участка начинаются соответственно у аминокислот 7 и 18. Четыре различные восьмикратно вырожденные смешанные пробы были приготовлены для каждого участка, они получили номера от 170 до 186, как показано. В изображенной предсказанной последовательности нуклеиновой кислоты “X” означает все 4 основания, “U” означает оба пурина и “Y” означает оба пиримидина. Фиг.3 – Нуклеотидная последовательность гена XPR2 с указанием промотора, пре (от -157 до -136), про1 (от -135 до -98), про2 (от -97 до -1), щелочной внеклеточной протеазы и концевых последовательностей. Фиг. 4 – Сконструированная последовательность для вектора-терминатора pterm 4. Фиг.5 – Сконструированная последовательность и рестрикционная карта плазмиды pLS-3. Фиг.6 – Сконструированная последовательность и рестрикционная карта плазмиды рХХ-33. Фиг. 7 – Аминокислотная последовательность человеческого анафилатоксина С5а. Фиг.8 – Рестрикционная карта плазмиды рС5а-48. Фиг.9 – Сконструированная последовательность и рестрикционная карта плазмиды рС5аХ-3. Фиг.10 – Нуклеотидная последовательность гена LEU2. Фиг. 11 – Сконструированная последовательность и рестрикционная карта плазмиды pLX-34. Анализ последовательности гена XPR2. Анализ ДНК-последовательности клонированного гена XPR2 проводится методом химической деградации (Махаm и др., 1980, Methods Enzymol. 65, 499) на перекрывающихся фрагментах рестрикции, полученных из плазмид pLD 57, pLD 58, pLD 62 (Фиг.1), pLD 84 и pLD 86 (см. ниже). Результаты показали, что клонированные геномные ДНК дрожжей действительно содержат ген для внеклеточной щелочной протеазы. Нуклеотидная последовательность гена XPR2 и аминокислотная последовательность предшественника щелочной протеазы с ее сигнальной последовательностью, выведенная из нуклеотидной последовательности, показаны на фиг.3. Значительная часть аминокислотной последовательности внеклеточной протеазы была неизвестна (Ogrydziak и др., ук. соч.) и представлена здесь впервые. Более того, последовательности, требуемые для экспрессии и выделения внеклеточной протеазы, также описаны здесь в первый раз. Последовательность ДНК, кодирующая щелочную протеазу, ее предшествующие и сигнальные последовательности, состоит из 1362 пар оснований (Фиг.3). Первичная структура этой полипептидной цепи, выводимая из нуклеотидной последовательности, должна содержать 454 аминокислотных остатка. Щелочная протеаза синтезируется в клетке в форме предшественника, который протеолитически переводится в выделяемую или готовую форму. Анализ N-концевой аминокислотной последовательности, выведенной из нуклеотидной последовательности, показал наличие сигнального пептида в молекуле предшественника. Этот сигнальный пептид содержит 22 аминокислотных остатка, и его структурные особенности подобны особенностям сигнальных пептидов высших эукариотов и прокариотов (Perlman и др., 1983, J. Mol. Biol. 167, 391). Области предсказанной аминокислотной последовательности, в целом совпадающей с известными 25 N-концевыми аминокислотами зрелой щелочной протеазы (Ogrydziak и др., 1982, J. Gen. Microbiol. 128, 1225) предшествуют 157 аминокислотных остатков, содержащих сигнальный пептид и два сайта расщепления трипсинового типа (Lys-Arg). Эти сайты расщепления использовались для разделения прорегиона на про1 (от -135 до -98) и про2 (от -97 до -1). См. Фиг.3. Как следует из нуклеотидной последовательности, активная щелочная протеаза имеет 297 аминокислот. Аминокислотные последовательности для различных форм протеазы, выведенные из нуклеотидной последовательности, находятся в соответствии с размерами очищенных форм фермента. В дополнение к структурной последовательности предшественника щелочной протеазы были определены примерно 700 п. ос. 5′-фланкирующей последовательности и 600 п.ос. 3′-фланкирующей последовательности. Анализ этих участков показал, что они содержат последовательности, аналогичные другим эукариотическим промоторам и терминаторам, и, вероятно, они играют существенную роль в экспрессии щелочной протеазы. Как указано выше, методика трансформации Y. lipolytica и клонирования генов Y. lipolytica путем комплементации мутаций, в том числе и клонирование гена XPR2, который кодирует выделяемую щелочную протеазу, путем комплементации мутации хрr2, описаны в Европейской заявке 0138508. Методика, описанная в ней, включает трансформацию клетки-хозяина штамма Y. lipolytica частичным расщеплением BglII библиотеки генов Y. lipolytica в вектор pLD40. Данный вектор характеризуется тем, что он удерживает малый сегмент, содержащий LEU2 участок Y. lipolytica и сайты рестрикции эндонуклеазы 3EcoRI, 4EcoRV, 6AvaI, 1BgII, 1NcoI, 1ApaI, 1XhoI и 1BstXI. Один из трансформантов XPR2 Y. lipolytica был использован для извлечения гена дикого типа (pLD84 и pLD86) из Y. lipolytica NRRL Y-1094 для применения в векторе конструирования экспрессии/выделения, как описано в Примере 1. Анализ последовательности гена LEU2. Анализ последовательности ДНК клонированного гена LEU2 в pLD25 (Европейская заявка 0138508) проводили методом химической деградации (Махаm и др., 1980, Methods Enzymol. 65, 499) на перекрывающихся фрагментах рестрикции. Для определения участка, кодирующего бета-изопропилмалат (ИПМ)-дегидрогеназу, и правильной рамки считывания была использована информация об аминокислотной последовательности, ранее определенной для гена LEU2 S. cerevisiae (Andreadis и др., 1984, J. Biol. Chem. 259, 8059). Участок геномной последовательности Y. lipolytica, который кодирует аминокислотную последовательность, гомологичную к участку протеиновой последовательности S. cerevisiae, был идентифицирован. Нуклеотидная последовательность гена LEU2 размером 2,8 тыс. п. о. и аминокислотная последовательность бета-ИПМ-дегидрогеназы, выведенная из нуклеотидной последовательности, показаны на фиг.10. Кроме того, последовательности, требуемые для экспрессии бета-ИПМ-дегидрогеназы Y. lipolytica, описаны здесь впервые. Последовательность ДНК, кодирующая этот протеин, состоящий из 405 аминокислот, содержит 1215 пар оснований (Фиг.10). В дополнение к последовательности, кодирующей бета-ИПМ-дегидрогеназу, были определены примерно 798 п.ос. 5′-фланкирующей последовательности и 797 п.ос. 3′-фланкирующей последовательности, включая кодон окончания трансляции ТАА). Анализ этих участков показал, что они содержат последовательности, аналогичные другим эукариотическим промоторам и терминаторам, и что они имеют существенное значение для экспрессии. 5′-Участок гена LEU2 Y. lipolytica содержит ТАТАТАТА-последовательность на расстоянии 78 п.ос. перед точкой начала трансляции и на расстоянии 30 п. ос. перед предполагаемой точкой начала образования мРНК. Вторая последовательность, важная для инициации транскрипции в эукариотах, представляет собой бокс СААТ, который в гене LEU2 расположен на расстоянии 74 п.ос. перед предполагаемым сайтом инициации транскрипции, расположенным в -48 п.ос. от ATG (Фиг.10). 3′-Участок вниз по течению имеет последовательность от 72 до 120 нуклеотидов после стоп-кодона (ТАА), гомологичную последовательности 5′-ТАС…. ТА(Т)GT. . . . . ТТТ-3′, предложенной Zaret и др., Cell 28, 563 (1982), как имеющий важное значение для окончания транскрипции в S. cerevisiae. ПРИМЕР 1 Использованным штаммом-хозяином был АТСС 20774 (МАТВ leu2-40 bio-6 xpr2-1002). Трансформант XPR2, Y. lipolytica АТСС 20781 определялся как колония, образующая зону на индикаторных чашках со снятым молоком после посева методом отпечатков на чашки с недостатком лейцина. Хромосомную ДНК получали из трансформанта по методу заявки ЕР 0138508 и использовали для извлечения гена выделяемой протеазы. Хромосомную ДНК частично расщепляли ферментом BglII, сшивали для образования кольцевого фрагмента, содержащего репликон Е. coli и ген устойчивости к ампициллину из вектора и использовали для трансформации Е. coli. Хромосомную ДНК также расщепляли ферментом SalI и использовали в эксперименте Southern, который показал, что нормальный участок LEU2 у трансформанта не был нарушен (520 п.ос. SalI EcoRI-сегмент участка LEU2 в 5′-положении к сегменту LEU2, содержащемуся в pLD40, использовался в качестве пробы). Так как для интеграции библиотечной плазмиды в Y. lipolytica необходима гомология, участок XPR2 должен быть сайтом интеграции. Три перекрывающиеся, но различные плазмиды pLD57, pLD58 и pLD62 были первоначально извлечены из Y. lipolytica АТСС 20781. Они изображены на Фиг.1. Гибридизация синтетическими олигонуклеотидными пробами гена XPR2, основанными на известной последовательности первых 25 аминокислотных остатков выделенной активной протеазы (фиг.2 и 3), показала, что ген выделения протеазы был клонирован. Для определения того, представляет ли извлеченный ген копию дикого типа или мутантную копию, штамм-реципиент Y. lipolytica был трансформирован pLD58. Так как из лейцин-независимых трансформантов не возникло трансформантов, положительных по протеазе, был сделан вывод, что pLD58 содержит мутантный аллель гена. Форма гена XPR2, присутствующая в штамме NRRL Y-1094 дикого типа, получена в эксперименте по гибридизации колонии Е. coli. В качестве пробы использовался фрагмент PvuI-EcoRI размером 2 тыс.п.н, который, как следует из данных по секвенированию, содержит полный структурный ген. Из оригинальной библиотеки Sаu3А частично расщепленных фрагментов, как описано в ЕР 0138508, NRRL Y-1094 ДНК в pLD40 получали несколько колоний, гибридизирующихся пробой. Две из этих колоний содержали очень сходные плазмиды, обозначенные pLD84 и pLD86, которые были использованы для получения векторов экспрессии. Обе плазмиды содержали одинаковый 5′-конец участка XPR2 – сайт Sаu3А (который был присоединен и регенерировал сайт BamHI вектора), от которого начинается последовательность на фиг.3. Каждый содержит целиком структурный ген протеазы и предполагаемый терминатор транскрипции и включает приблизительно 4-5 тыс.п.н., введенных из участка XPR2 штамма NRRL Y-1094. Включение в pLD86 содержит дополнительно несколько сотен пар оснований на 3′-конце. Так как использовали 3′-цепь до сайта BglII (2655 пар оснований) для конструирования вектора экспрессии, две плазмиды передают одинаковую ДНК, которая функционально идентична последовательности на фиг.3. Конструирование векторов экспрессии/выделения. План, разработанный для получения экспрессии и выделения прореннина в Y. lipolytica, предусматривает конструирование различных гибридных генов в интегративном векторе клонирования. Подобный подход требует нескольких различных плазмид, которые содержат обширные участки общих последовательностей ДНК. Фактически была использована модулированная конструкционная схема для объединения векторов с геном прореннина, введенным в положение 3′ предсказанного сайта XPR2, ответственного за сигнальный пептид, перед предполагаемым про-1-перерабатывающим сайтом и расщепляющим сайтом – известным генератором активной щелочной протеазы. Вообще, желательно, чтобы чужеродный ген был введен между последовательностями дрожжей для промотора и терминатора экспрессии. Ясно, что N-концевая часть последовательностей гибридного гена будет изменяться в различных конструкциях плазмид, однако последовательность структурного гена прореннина, последовательность терминатора XPR2 и челночный вектор ДНК будут одинаковы в каждой конструкции плазмиды экспрессии. Предполагалось, что одни и те же фрагменты структурного гена прореннина и терминатор/вектор плазмиды будут использованы в каждой конструкции плазмиды экспрессии, как описано ниже. Различные конструкции плазмид экспрессии/выделения прореннина различаются в участке сразу за промотирующей последовательностью гена XPR2 длиной N-концевой последовательности предшественника щелочной протеазы, которая предшествует последовательности гена прореннина. Поэтому промотирующий фрагмент каждой плазмиды экспрессии конструировался как изменяемая последовательность в участке связи ХРR2-прореннин. Все векторы экспрессии/выделения собирались схожими реакциями связывания, содержащими три компонента. Экспериментальные стадии, использованные для конструирования вектора-терминатора pterm 4, изображены на фиг.4. Сначала синтетический связующий присоединяли к фрагменту, содержащему 3′-конец гена XPR2, включающему сигналы окончания транскрипции и полиаденилирования. Коротко говоря, плазмиду pLD84 расщепляли эндонуклеазой KpnI и связывали синтетической двухцепочечной связующей ДНК, изображенной на фиг.4. Продукт связывания расщепляли эндонуклеазами HindIII и BglII и фрагмент из 760 п.ос. вводили в плазмиду pLD41, линеализированную теми же двумя эндонуклеазами до образования pterm 4. Плазмиду pterm 4 идентифицировали по ее рестрикционной карте. Результаты серии рестрикционных расщеплений эндонуклеазой анализировали при использовании EcoRV, EcoRI, KpnI, BglII-HindIII и BglII-BclI. Расщепления дают пригодные фрагменты, подтверждающие присутствие синтетического связующего и “полного” 3′-конца гена XPR2 в челночной плазмиде pLD41, как описано в ЕР 0138508. Частичная карта этого 7,3 тыс. п.о. вектора-терминатора показана на фиг.4. Конструирование плазмиды экспрессии/выделения pLS-3. На фиг.5 показано конструирование первоначальной плазмиды, использованной для выделения прореннина в Y. lipolytica. Ее рестрикционная карта представлена на фиг.5. Конструирование плазмиды выделения прореннина было инициировано приготовлением фрагмента, содержащего большую часть последовательности структурного гена прореннина. Фрагмент ДНК BclI-BamHI (частичный) из 1080 п.ос., содержащий кодирующую последовательность для остатков прореннина 6-365, изолировали из плазмиды экспрессии прореннина Е. coli pPFZ-84A. (Плазмида pPFZ-84A является производным плазмиды экспрессии прореннина pPFZ-R2, конструкция которой описана в заявке ЕР 0147178, опубликованной 3 июля 1985 г., и была получена сайт-специфическим мутагенезом при использовании замещения фрагментов рестрикции. Конкретно, pPFZ-84A отличается от pPFZ-R2 только двумя парами оснований для аминокислотных остатков прореннина 214 (Asn –> Asp) и 286 (Asp –> Gly), чтобы кодировать так называемый аллель А прореннина, однако обе плазмиды содержат необходимую последовательность для прореннина и функционально эквивалентны в данном примере). Фрагмент промотора XPR2, содержащий кодирующую последовательность для предшественника щелочной протеазы 1-157 и прореннина 1-5, получали следующим образом. Фрагмент HindIII-AvaI ДНК из 870 п.ос., содержащий участок промотора и 5′-конец гена щелочной протеазы, изолировали из субклона XPR2 плазмиды pLD90. Этот фрагмент связывали с синтетическим фрагментом, имеющим структуру: 5′ CCGAGATTCCTGCTTCTTCTAATGCCAAGCGAGCTGAGATCACTAG 3′ 3′ CTAAGGACGAAGAAGATTACGGTTCGCTCGACTCTAGTGATCCTAG 5′ направление считывания –> Эта последовательность содержит “липкий” конец AvaI, за которым следует последовательность, кодирующая последние девять аминокислот пропептида щелочной протеазы, за которой следует последовательность, кодирующая первые четыре аминокислоты прореннина и оканчивающаяся в сайте BamHI. Фрагмент промотора получали по стандартной реакции связывания, используя синтетический фрагмент, фрагмент HindIII-AvaI из 870 п..ос. и Т4-лигазу с последующим расщеплением HindIII и BamHI. Образовавшиеся связанные последовательности очищали электрофорезом на геле полиакриламида, выделяя соответствующий фрагмент ДНК HindIII-BamHI из 916 п.ос. 3′-Конец гибридного гена получали из плазмиды терминатор/вектора pterm 4, описанной выше. Плазмиду pterm 4 расщепляли HindIII и BclI и приблизительно 7,3 тыс.п.н. HindIII-BclI фрагмент ДНК терминатор/вектора, содержащего терминатор XPR2, селективный маркер LEU2 и pBR322, выделяли на геле агарозы. Плазмиду экспрессии/выделения прореннина pLS-3 собирали путем инкубирования трех фрагментов ДНК (HindIII-BclI-расщепленной плазмиды pterm 4, промотора HindIII-BamHI из 916 п.ос. и фрагментов, содержащих ген прореннина BamHI-BclI из 1080 п.ос.), сконструированных, как описано выше, в присутствии T4-лигазы (см. фиг.5). Лигирующую смесь использовали для трансформации штамма ММ294 Е. coli K12 по CaCl2 – методу Dagert и др., Gene 6, 23-28 (1979). Плазмиды изолировали от выделенных трансформантов, устойчивых к ампициллину, и плазмиду pLS-3 идентифицировали по ее рестрикционной карте (фиг.6А). Участок ХРR2-прореннин этой плазмиды секвенировали для подтверждения наличия нужной последовательности синтетической ДНК и правильного соединения нужных фрагментов. Получение pLD90. Эта плазмида содержит субклон из pLD84. Участок ДНК от сайта Pvul в участке промотора XPR2 до сайта EcoRI в концевом участке субклонировали в сайт HindIII pBR322 по следующей методике. Несколько микрограмм pLD84 расщепляли двумя вышеупомянутыми ферментами рестрикции. Затем в “липкие” концы расщепленных молекул ДНК вводили Klenow-фрагмент ДНК-полимеразы I. Затем киназные HindIII-линкеры (CAAGCTTG из New England Biolabs) добавляли к концам с помощью Т4-лигазы. Избыток линкеров удаляли и генерировали “липкие” концы HindIII последующим расщеплением ферментом HindIII. Смесь молекул ДНК разделяли на препаративном агарозном геле, отделяли нужную группу с размером 2 тыс.п.н., очищали и вводили в реакцию связывания с HindIII-расщепленным вектором pBR322, обработанным бактериальной щелочной фосфатазой. Продукт связывания использовали для трансформации компетентных Е. coli. Ориентация с сайтом EcoRI терминатора XPR2, более близкая к сайту EcoRI pBR322, была названа pLD90, а обратная ориентация была названа pLD91. 5′ Конец участка промотора XPR2, который был включен в pLS-3, представляет собой сайт PvuI, приблизительно в 280 п.ос. от начала области, последовательность которой указана на фиг.3. Было обнаружено, что плазмиды, содержащие ген протеазы дикого типа под контролем только этой части промотора, при интеграции в геном в сайте, удаленном от резидент-локуса хрr2, не делают трансформант способным производить большие количества протеазы (определено по зонам пояснения на чашках снятого молока;. Было обнаружено, что, когда pLS-3 содержит укороченный и поэтому “недостаточный” промотор, интегрант, возникающий при рекомбинации между плазмидой и резидент-геном XPR2 дикого типа, будет образовывать полный промотор, направляющий экспрессию продукта плавления прохимозина, но недостаточный, однако, для экспрессии протеазы. Аналогичный эксперимент типа дезинтеграции гена выполняли с актин-геном S. cerevisiae Shortle и др. (Science 217:371-373, 1982). В соответствии с ожиданиями некоторые лейцин-независимые трансформанты с pLS-3 были действительно недостаточны по протеазе. Трансформанты, недостаточные по протеазе, представляли собой скорее нужные интегранты в локусе XPR2, чем нежелательные побочные продукты, такие как преобразователи гена в leu2. Со штаммом-реципиентом АТСС 20638 было обнаружено, что нерасщепленная pLS-3 генерировала 6,5% псотеаза-недостаточных трансформантов, тогда как SnaBI-расщепленная плазмида давала приблизительно 70% протеаза-недостаточных трансформантов. Аспект дезинтеграции гена при этой трансформации использовали для того, чтобы избежать необходимости в большом количестве Southern точечных экспериментов для нахождения правильного интегранта среди всех трансформантов. Плазмиды, содержащие структурный ген протеазы дикого типа под контролем промотора XPR2, начинающегося, как указано на фиг.3, дают возможность экспрессии значительного количества протеазы, если интегрированы в клетки Y. lipolytica в сайте, ином, чем локус xpr2. Однако для эффективной экспрессии чужеродных генов этим видом интегрантов может потребоваться дальнейшая модификация этого контрольного участка ДНК. Выделение прореннина. Штамм АТСС 20688 Y. lipolytica трансформировали нерасщепленной pLS-3 ДНК и SnaBI-расщепленной pLS-3 ДНК для получения xpr–leu+ трансформантов АТСС 20775 (DL144) и АТСС 20776 (DL148) соответственно. Эти трансформированные штаммы высевали в тест-пробирку, содержащую среду ДЭПД. Клетки выращивали в течение ночи при 28oС. Аликвоту (250 мкл) этих культур разводили 1: 100 в 25 мл среды ГПП. Клетки выращивали во встряхиваемой колбе при 28oС в течение 16-18 часов до возникающей абсорбции 5,0-7,0 при 600 нм и отбирали центрифугированием. Образующуюся культуральную жидкость, или супернатант, исследовали на присутствие прореннина концентрированием супернатанта и нанесением концентрата на SDS-PAGE. Пластину геля электрофоретически переносили на нитроцеллюлозную бумагу в присутствии 20 мМ трис-основания, 150 мМ глицина, 20% метанола при 500 m amp в течение 2 часов при 4oС. Удаление протеина из пластины геля проверяли окрашиванием с Coomassie blue. Нитроцеллюлозную бумагу сушили при 37oС и обжигали при 65oС в течение 1 часа, затем промывали в TBS (200 мМ NaCl, 50 мМ трис-HCl, рН 7,5). Бумагу затем инкубировали при комнатной температуре 30 минут в TBS, содержащем 10% лошадиной сыворотки (Gibco, Chagrin Falls, Ohio), с последующей инкубацией в TBS и соответствующим разведением антитела прореннина в течение 16 часов при комнатной температуре. Бумагу затем три раза промывали в TBS с последующей инкубацией в TBS, содержащем 10% лошадиной сыворотки, с последующей инкубацией в течение 2 часов в TBS, содержащем 10% лошадиной сыворотки, и соответствующим разведением антитела козел-антикролик IgG, сопряженного с пероксидазой ложечницы приморской. Бумагу затем промывали три раза в течение 10 минут в TBS и проявляли в присутствии 4-хлор-1-нафтола (3 мг/мл в метаноле), добавленного в концентрации 0,5 мг/мл в TBS, содержащем 0,01% перекиси водорода. Присутствие прореннина с молекулярным весом 40000 было подтверждено в обоих супернатантах. После кислотной активации концентрированных супернатантов культуры (см. выше) значительная активность по свертыванию молока присутствовала в образцах, приготовленных из трансформированных культур АТСС 20775 и 20776, содержащих pLS-3. Как ожидалось, активность по свертыванию молока не обнаружена в супернатантах контрольной культуры штамма-реципиента Y. lipolytica АТСС 20688. Конструирование плазмиды экспрессии/выделения рХХ-33. Модификация для превращения pLS-3 в усовершенствованную плазмиду экспрессии рХХ-33 изображена на фиг.6. Такая модификация увеличивает участок промотора XPR2 на 280 п.ос. Как в случае pLS-3, плазмида экспрессии рХХ-33 содержит гибридный ген, кодирующий весь препропептид (157 аминокислотных остатков) щелочной протеазы, присоединенный к полной структурной генной последовательности прореннина. Перед конструированием плазмид экспрессии/выделения прореннина с последовательностью промотора XPR2 на 280 п.ос. больше, чем в pLS-3, было необходимо субклонировать фрагмент рестрикции, содержащий полный ген щелочной протеазы, в сайт HindIII. Этот субклон был собран добавлением синтетических линкеров к фрагменту рестрикции, выделенному из клона XPR2-геномной библиотеки pLD86. Конструирование этого субклона XPR2 с сайтом HindIII в сторону 5′ инициировали приготовлением фрагмента ДНК, содержащего весь ген щелочной протеазы. 2,3 тыс.п.н. фрагмент (частичный) EcoRI-BamHI из геномного участка клона XPR2 pLD-86 очищали электрофорезом на агарозном геле и лигировали с синтетическим фрагментом, имеющим последовательность 5′ GATCGAAGCTTG 3′ 3′ TTCGAACTTAA 5′ Эта линкерная последовательность содержит “липкий” конец BamHI (но не регенерирует сайт BamHI), за которым следует сайт HindIII и за которым следует “липкий” конец EcoRI. Лигированный продукт расщепляли HindIII и вставляли в HindIII сайт рВ322. Плазмиду рХНР-24 идентифицировали по ее рестрикционной карте и превращали в источник фрагментов промотора XPR2 для будущих конструкций экспрессии. В плазмиде рХНР-24 субклонированный ген XPR2 содержит приблизительно на 280 п. ос. больше 5′ последовательности промотора XPR2, чем последовательность промотора XPR2, содержащаяся в pLS-3. Первый фрагмент промотора получали стандартной реакцией связывания синтетического фрагмента ДНК (описанного выше для pLS-3) и фрагмента HindIII-AvaI размером 1150 п.ос. из рХНР-24 с Т4-лигазой с последующим расщеплением HindIII и BamHI. Образующиеся связанные последовательности очищали гель-электрофорезом, выделяя фрагмент HindIII-BamHI приблизительно из 1196 п.ос. Второй фрагмент, содержащий последовательности, кодирующие аминокислотные остатки прореннина 6-151, получали из pLS-3 расщеплением BamHI и XmaI и очисткой на геле образующегося фрагмента ДНК BamHI-XmaI из 440 п.о. Третий фрагмент, содержащий остальную часть гена прореннина, терминатор XPR2 и последовательности вектора получали из pLS-3 расщеплением HindIII и Xmal и очисткой на геле фрагмента вектора HindIII- Xmal приблизительно из 8,0 тыс.п.н. Эти три фрагмента затем лигировали, используя стандартную методику, описанную выше. Реакцию лигирования использовали для трансформации штамма ММ294 Е. coli K12. Плазмиды изолировали из трансформантов, отобранных на основе устойчивости к ампициллину, и плазмиду рХХ-33 идентифицировали по ее рестрикционной карте (фиг.6). Протеаза-прореннин-участок этой плазмиды секвенировали для подтверждения правильного соединения нужных фрагментов. Y. lipolytica ATCC 20774 затем трансформировали рХХ-33, расщепленной SnaBI, для получения Y. lipolytica ATCC 20780, и прореннин, выделенный трансформированными культурами в культуральный бульон, анализировали, как описано выше в случае pLS-3. Было подтверждено присутствие прореннина в культуральном супернатанте. После кислотной активации концентрированных культуральных супернатантов (см. выше) значительная активность по свертыванию молока наблюдалась в образцах, приготовленных из трансформированной культуры Y. lipolytica ATCC 20780. ПРИМЕР 2 Ген BIO дикого типа, соответствующий bio-6 аллелю в ATCC 20774, клонировали путем комплементации следующим образом. Была сконструирована библиотека генов частично Sаu3А-расщепленной хромосомной ДНК Y. lipolytica, введенной в BamHI сайт pLD40 (который представляет собой pBR322 плюс LEU2 в сайте EcoRI), и приготовлено большое количество библиотечной ДНК как плазмидного препарата смешанной культуры Е. coli (Это та же самая библиотека, что была использована для клонирования гена XPR2). Несколько микрограмм библиотечной ДНК расщепляли ферментом ApaI (который расщепляет одну связь в участке LEU2). Затем эту ДНК использовали для трансформации ATCC 20774 (leu2 xpr2 bio), и трансформированные смеси высевали на синтетической среде с недостатком лейцина. Были получены десятки тысяч лейцин-независимых трансформантов. Чтобы обнаружить колонии, если они есть, содержащие библиотечные плазмиды, включающие ген BIO, лейцин-независимые трансформанты методом отпечатков высевали на агаровые чашки, содержащие биотин-селективную среду (рецептура на литр: 25 мг дестибиотина, 20 г глюкозы, 5 г сульфата аммония, 1 г КН2РO4, 0,5 мг MgSO4 7H2O, 0,1 г CaCl2, 0,1 г NaCl, 500 мкг борной кислоты, 400 мкг тиаминНСl, 400 мкг ZnSO47H2O, 400 мкг MnSO4H2O, 200 мкг Na2MoO42H2O, 200 мкг FеСl36Н2O, 100 мкг KI и 40 мкг CuSO45H2O).
Один из нескольких Y. lipolytica BIO-трансформантов, растущий на биотин-селективной среде, был назван DL31. Затем приступили к извлечению “библиотечной” плазмиды, содержащей ген BIO из штамма DL31 Y. lipolytica. Приготовляли хромосомную ДНК из культуры штамма DL31. Несколько микрограммов этой хромосомной ДНК расщепляли ферментом рестрикции ApaI для того, чтобы вырезать “библиотечную” плазмиду. Аликвоту расщепленной ДНК использовали в реакции связывания для замыкания неизвестной библиотечной плазмиды. Концевой продукт затем использовали для трансформации культуры Е. coli в направлении устойчивости к ампициллину введением неизвестной BIO-содержащей плазмиды в Е. coli. Получали немного трансформантов Е. coli, устойчивых к ампициллину. На трансформантах Е. coli приготовляли немного препаратов плазмид. Рестрикционные расщепления полученной таким образом плазмидной ДНК показывают, что неизвестная BIO-содержащая плазмида, как ожидалось, эквивалентна pLD40 с введением в сайт BamHI. Эта плазмида должна была первоначально происходить из библиотеки генов заявителя и была названа pLD51.
Плазмида pLD56 была образована как субклон pLD51 удалением гена LEU2 из pLD51 следующим образом. Аликвоту плазмиды pLD51 расщепляли ферментом EcoRI для удаления участка LEU2. Расщепленную ДНК использовали в реакции связывания ДНК для замыкания плазмиды. Затем для клонирования выполняли трансформацию Е. coli меньшей ВIO-содержащей плазмиды. Один из ампициллин-устойчивых трансформантов Е. coli, как показано, содержал ожидаемую меньшую плазмиду, которая была названа pLD56. Было выполнено несколько рестрикционных расщеплений pLD56. ВIO-содержащий сегмент pLD56, который обнаруживается как вставка в BamHI сайт pBR322, имеет длину приблизительно 3,6 тыс.п. н.
Очень грубая рестрикционная карта вставки ДНК Y. lipolytica из 3,6 тыс. п. н. в BamHI сайт pBR322 (включая pLD56) описана ниже с указанием в скобках примерного расстояния в парах оснований от начала вставки. Оценка расстояний сделана из немногих анализов на агарозных гелях и, возможно, содержит относительно большие количественные погрешности: PvuII (800), PvuII (1200), PstI (1800), MluI (2000), PstI (2300), EcoRV (2700), NcoI (3200) (Для ориентации SalI сайт pBR322 будет предшествовать описанным сайтам и сайт HindIII будет следовать за ними).
Штамм АТСС 20774 (МАТВ leu2-40 bio-6 xpr2-1002) трансформировали интактной pLD56 (pBR322 плюс приблизительно 3,6 тыс.п.н. хромосомной ДНК Y. lipolytica, содержащей ген ВIO). Три различных биотин-независимых трансформанта испытывали на высокочастотную трансформацию NruI-расщепленной (мишень для pBR322) pLD40 (LEU2 на pBR322) относительно родительского штамма для определения, который из них содержал резидент pBR322, интегрированный в участок BIO. Все три показали способность к высокочастотной трансформации ввиду интеграции pLD40 в резидент pBR322. Это было подтверждено Southern точечным экспериментом по гибридизации. Один из трех исходных ВIO-трансформантов Y. lipolytica был назван DL118 и использован далее как реципиент ДНК. Данная рестрикционная карта требовалась для установления: а) что использовать в качестве ВIO-специфичной пробы гибридизации (область NcoI-PvuII), б) какой фермент требуется, чтобы правильно вырезать плазмиду pLD56 (MluI), в) какой фермент расщепляет только одну связь в области pBR322 (ClaI) и г) какой фермент не расщепляет плазмиду вообще (ApaI). Southern гибридизация ClaI и ApaI дайджестов ДНК из АТСС 20774 и DL118 (испытанная с BIO-фрагментом) показала, что, как и ожидалось, биотин-участок DL118 (при сравнении с ВIO-участком АТСС 20774) был разрушен добавлением ДНК размером, приблизительно равным размеру pLD56. Mlul дайджесты ДНК DL118 (проба с pBR322) показали далее, что добавка была того же размера, как интактная pLD56.
Конструирование плазмиды экспрессии/выделения pLX-34. Была сконструирована плазмида экспрессии, которая содержит последовательность, кодирующую прореннин, с сигналами выделения XPR2 (предпропоследовательности из 157 аминокислот) вниз по направлению считывания от промотора LEU2. Эта плазмида экспрессии демонстрирует, что может быть использован иной, чем XPR2, промотор для достижения выделения чужеродных протеинов. Кроме того, этот вектор экспрессии способен вызвать экспрессию/выделение независимо от сайта интеграции в геноме Y. lipolytica. Успешное выделение прореннина с промотором, иным, чем промотор XPR2, демонстрирует возможность конструкции вектора экспрессии для идентификации альтернативных новых сильных промоторов в Y. lipolytica. В дополнение к этому данный подход может быть использован для получения культуры экспрессии с двумя отдельными гибридными генами прореннина (один, экспрессируемый промотором LEU2, а другой – промотором XPR2), интегрированными в различных сайтах генома хозяина.
Экспериментальные стадии, использованные для конструирования вектора экспрессии, который содержит ген прореннина с сигналами выделения щелочной протеазы (препропоследовательность XPR2 157 аминокислот), экспрессированными последовательностями промотора LEU2, изображены на фиг.11. Конструирование этой плазмиды было инициировано приготовлением фрагмента промотора LEU2, содержащего около 300 пар оснований 5′-нетранслируемой последовательности, предшествующей кодону инициации трансляции ATG гена бета-изопропилмалатдегидрогеназы (Фиг.10). Фрагмент ДНК HindIII-FokI из 300 п.ос., кодирующий участок из 270 п.ос. последовательности промотора LEU2, был выделен из челночного вектора pLD40. Этот фрагмент лигировали с синтетическим линкером из 54 п.ос., имеющими последовательность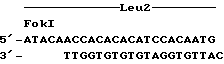 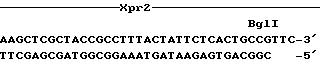 T4-лигазой с последующим расщеплением HindIII. Образующиеся связанные последовательности очищали гель-электрофорезом, выделяя фрагмент ДНК HindIII-BglI приблизительно из 360 п.ос. Второй составляющий фрагмент, кодирующий остальную часть препропоследовательности XPR2 и первые 152 аминокислоты прореннина, изолировали из плазмиды экспрессии рХХ-33 (Фиг.6) расщеплением BglI и XmaI и очисткой на геле образующегося фрагмента ДНК из 887 п. ос. Третий фрагмент, содержащий остальную часть гена прореннина, терминатор XPR2 и последовательности вектора, приготовляли расщеплением рХХ-33 HindIII и XmaI и очисткой на геле, выделяя приблизительно 8,5 тыс.п.н. фрагмента вектора. Три фрагмента ДНК лигировали при использовании стандартной методики, описанной выше. Реакцию лигирования использовали для трансформации штамма НВ101 Е. coli K12. Плазмиды изолировали из трансформантов, отобранных на основе устойчивости к ампициллину, и плазмиду pLX-34 идентифицировали по ее рестрикционной карте (фиг.11). Y. lipolytica ATCC 20794 (DL118) трансформировали NruI-расщепленной ДНК pLX-34 для получения Y. lipolytica ATCC 20795 (DL251), и прореннин, выделенный в культуральную жидкость лейцин-независимыми трансформантами культуры, анализировали, как описано выше в случае pLS-3. Данная методика трансформации обеспечивала интеграцию pLX-34 в последовательность pBR322, предварительно введенную в bio-локус в хромосоме хозяина (описано выше). Интеграцию pLX-34 в этот сайт подтверждали Southern-анализом. Использование DL118 в качестве реципиента. Southern-эксперименты по гибридизации выполнялись следующим образом: NruI дайджесты ДНК из трансформантов DL118 (гибридизованных прохимозиновой пробой, например, когда входящая плазмида представляла плазмиду экспрессии прохимозина) точно вырезали вошедшую плазмиду. Несколько нанограмм NruI-расщепленной трансформирующей плазмиды служили для проверки правильности размера гибридизирующейся полосы. Также MluI дайджесты (MluI не расщепляют трансформирующую плазмиду) ДНК из этих трансформантов (проба с 32р-помеченной pBR322) показали, что резидентная pBR322 последовательность DL118 разрушалась при добавлении одной или более молекул трансформирующей плазмиды. Это показывает, что интеграция произошла в нужный сайт. Трансформантные культуры Y. lipolytica ATCC 20795 (DL251) выращивали в среде ДЭНД при 22oС, чтобы благоприятствовать экспрессии промотора LEU2. Присутствие прореннина в культуральных супернатантах подтверждали анализом на свертываемость молока (см. выше) культуральных супернатантов, активированных кислотой, и проверяли иммуноточечным анализом (см. выше). Эти результаты показывают, что этот гибридный ген представляет собой независимую единицу экспрессии, способную к экспрессии/выделению при интеграции в сайт, иной, чем XPR2 или LEU2. Это позволяет сконструировать культуру экспрессии с множественными гибридными генами, потенциально способными увеличивать уровень внеклеточного прореннина. ПРИМЕР 3 Последовательность синтетического гена для человеческого С5а. План, разработанный для достижения бактериальной продукции человеческого анафилатоксина С5а, был аналогичен предыдущим методикам, использованным для синтеза и экспрессии EGF, как описано в ЕР заявке 00147178. В нем применялось конструирование гена, в котором кодирующую последовательность для активированного дополнительного компонента С5а получали синтетически. Зная аминокислотную последовательность человеческого С5а, сконструировали фрагмент ДНК, кодирующий информацию для его 74 аминокислот (фиг.7). Синтетическую генную последовательность выбирали так, чтобы максимизировать утилизацию предпочтительного кодона Е. coli и S. cerevisiae и обеспечить несколько сайтов рестрикции эндонуклеазой для облегчения определения. Данный подход дает возможность прямой экспрессии анафилатоксина в Е. coli путем введения кодона инициации ATG для синтеза протеина перед триплетом, кодирующим первую аминокислоту полипептида С5а. Для облегчения его введения в нужной ориентации в плазмиду pBR322 был сконструирован синтетический ген С5а, содержащий сайты узнавания эндонуклеаз рестрикции EcoRI и HindIII на своих концах. Для получения конечной генной последовательности С5а синтезировали фосфорамидатным методом десять 47-мер и соединяли в двухцепочечный фрагмент ДНК из 235 п.ос. Фрагмент гена С5а вводили в соответствующим образом расщепленную pBR322, и клонированный ген идентифицировали рестрикционным анализом плазмидной ДНК из произвольно выбранных трансформантов. Несколько клонов С5а затем анализировали секвенированием ДНК для идентификации клона с правильной последовательностью. Нужная нуклеотидная последовательность для участка гена С5а была найдена в 2 из 5 исследованных клонов. Бактериальная экспрессия человеческого С5а. Конструирование плазмиды экспрессии С5а инициировали расщеплением субклона С5а эндонуклеазой рестрикции EcoRI с последующим дефосфорилированием обработкой бактериальной щелочной фосфатазой. Используя фрагмент ДНК EcoRI из pPFZ-R2 размером 360 п. ос. , содержащий trp промотор-оператор и последовательности сайта, связывающего рибосому, была сконструирована плазмида экспрессии С5а. Компетентные клетки штамма НВ101 Е. coli трансформировали реакцией лигирования. Несколько устойчивых к лекарствам колоний от каждой трансформации очищали и их плазмидные ДНК подвергали анализу с помощью рестрикционной эндонуклеазы для идентификации ДНК с trp промотором в ориентации, которая будет проявляться в транскрипции гена С5а. Множественные изоляты из этой реакции связывания идентифицировали с помощью плазмид, содержащих ген анафилатоксина, примыкающий к последовательности бактериального промотора в конфигурации, требуемой для прямой экспрессии С5а. Рестрикционная карта плазмиды экспрессии С5а рС5а-48 представлена на фиг.8. Экспрессия и выделение человеческого анафилатоксина в Y. lipolytica Вектор экспрессии/выделения рС5аХ-3, кодирующий выделение человеческого анафилатоксина С5а, приготавливали по методике, изложенной в Примере 1 для рХХ-33. Y. lipolytica ATCC 20774 затем трансформировали этим вектором выделения, и человеческий С5а, продуцированный трансформированными культурами, анализировали, как описано выше, за исключением того, что в иммуноточечной методике использовали козел-анти-С5а и кролик-анти-козел IgG. Для плазмиды, описанной в данном примере, было подтверждено наличие С5а в культуральном супернатанте. Конструирование плазмиды экспрессии/выделения рС5аХ-3 Экспериментальные стадии для конструирования плазмиды экспрессии/выделения анафилатоксина рС5аХ-3 показаны на фиг.9. Эта плазмида содержит последовательность для “полного” промотора XPR2 и сигнала пропептида из 157 аминокислотных остатков, присоединенную к синтетической последовательности, кодирующей 74 аминокислотных остатка С5а. План конструирования, использованный для рС5аХ-3, был подобен использованному для рХХ-33. Сначала плазмиду рХНР-24 (или другую плазмиду, содержащую нужную последовательность) расщепляли HindIII и AvaI и фрагмент из 1150 п.ос., содержащий промотор XPR2, очищали на геле. Второй фрагмент, содержащий 3′ конец пропептида XPR2 и последовательность структурного гена С5а, получали стандартной реакцией лигирования при использовании фрагмента ДНК HindI-HindIII размером приблизительно 220 п. ос. из плазмиды экспрессии Е. coli pC5a-48 синтетического фрагмента с последовательностью: 5′ CCGAGATTCCTGCTTCTTCTAATGCCAAGCGA 3′ 3′ CTAAGGACGAAGAAGATTACGGTTCGCTTGA 5′ и Т4-лигазы с последующим расщеплением AvaI и HindIII. Образующиеся связанные последовательности очищали гель-электрофорезом, отбирая фрагмент AvaI-HindIII около 250 п.ос. Фрагмент HindIII-AvaI, содержащий промотор, и фрагмент AvaI-HindIII, кодирующий С5а, затем лигировали Т4-лигазой с последующим расщеплением HindIII. Фрагмент приблизительно в 1,4 тыс.п.н. очищали на геле и использовали для лигирования с HindIII-расщепленным pterm4 (описанным выше). Продукт реакции лигирования использовали для трансформации штамма ММ294 Е. coli K12. Плазмиды, отобранные по устойчивости к ампициллину, изолировали из отобранных трансформантов и плазмиду рС5аХ-3 идентифицировали по ее рестрикционной карте. Штамм Y. lipolytica PC-30869, ATCC 20774 затем трансформировали SnaBI-расщепленной рС5аХ-3, и анафилатоксин, выделенный в культуральную среду трансформированными культурами, анализировали, как описано выше. Присутствие С5а в культуральном супернатанте подтверждали по вышеописанной методике. Известно, что многие протеины, которые синтезируются рибосомами, связанными с эндоплазмической сетью, производятся в виде гликопротеинов. Действительно, гликозилирование может оказывать влияние на выделение данного протеина. N-связанное гликозилирование эукариотических протеинов происходит в трипептидных последовательностях аспарагин-Х-треонин и аспарагин-Х-серин, где Х может быть любой аминокислотой, кроме, возможно, аспарата (Hubbard, S. и др. 1981, Ann. Rev. Biochem. 50; 555). Аминокислотная последовательность прореннина включает две такие трипептидные последовательности, однако гель-электрофоретический анализ прореннина, выделяемого в культурах Y. lipolytica, не показывает наличия гликозилирования. В других выделяемых эукариотических протеинах не все аспарагин-Х-треонин/серин сайты гликозилированы. Видимо, определенные аспарагины в трипептидных последовательностях не гликозилируются, так как они недоступны соответствующим ферментам. В случае человеческого С5а аминокислотная последовательность включает отдельный сайт гликозилирования, или трипептидную последовательность (Asn-Ile-Ser), которая обычно содержит сложный олигосахарид, присоединенный к аспарагину (Fernandez, Н. и др. 1976, J. Immunol. 117, 1688). Часть молекул С5а, выделяемых в культуральную среду Y. lipolytica, как представляется, гликозилирована, так как широкая область антигенной активности наблюдается в высокомолекулярной части иммуноточечного анализа. Данная гетерогенная электрофоретическая мобильность аналогична наблюдаемой для других выделяемых протеинов и, вероятно, является следствием различий в степени добавления углеводов. По настоящему изобретению очевидное гликозилирование определенных выделяемых чужеродных протеинов наводит на мысль, что экспрессия и выделение Y. lipolytica будут полезны для получения многих обычно гликозилированных эукариотических протеинов. Формула изобретения
используемая в экспрессии гетерологичных белков. 2. Кодирующая последовательность гена LEU2 Yarrowia lipolytica: (см. графическую часть), используемая в экспрессии гетерологичных белков. 3. Нуклеотидная последовательность промотора гена LEU2 Yarrowia lipolytica: (см. графическую часть), 4. Нуклеотидная последовательность терминатора гена LEU2 Yarrowia lipolytica: (см. графическую часть), 5. Рекомбинантный ДНК-материал, содержащий нуклеотидную последовательность, охарактеризованную в любом из пп.1-4, и нуклеотидную последовательность гена прореннина. 6. Рекомбинантный ДНК-материал, содержащий нуклеотидную последовательность, охарактеризованную в любом из пп.1-4, и нуклеотидную последовательность гена анафилатоксина человека С5а. 7. Штамм дрожжей Yarrowia lipolytica, трансформированный рекомбинантным ДНК-материалом, охарактеризованным в п.5, и способный продуцировать прореннин. 8. Штамм дрожжей Yarrowia lipolytica, трансформированный рекомбинантным ДНК-материалом, охарактеризованным в п.6, и способный продуцировать анафилатоксин человека С5а. РИСУНКИ
Изменения:
Извещение опубликовано: 10.03.2005 БИ: 07/2005
|
||||||||||||||||||||||||||