Патент на изобретение №2198185
|
||||||||||||||||||||||||||
(54) РЕАКЦИОННОСПОСОБНЫЕ ГЕТЕРОЦЕПНЫЕ ОЛИГОМЕРЫ НА ОСНОВЕ ПРОТОНСОДЕРЖАЩИХ АКРИЛОВЫХ ПРОИЗВОДНЫХ И СПОСОБ ИХ ПОЛУЧЕНИЯ
(57) Реферат: Описаны реакционноспособные гетероцепные олигомеры на основе протонсодержащих акриловых производных общей формулы СН2= СН-С(О)О-(СН2)n-Х-[CH2-СН2-С(О)О-(СН2)n-Х-] mH, где Х- -О-, -О-С(О)-N(Ph)-; n=2-10; m=2-15, а также способ их получения путем анионной полимеризации акриловых производных в присутствии инициатора, причем в качестве мономеров берут протонсодержащие акриловые производные, анионную полимеризацию ведут в инертной среде в присутствии инициаторов, выбранных из группы щелочные металлы или их производные, смесь  -окиси и третичного амина, мольное отношение компонентов в которой берут в пределах 0,5-1,5 соответственно, причем соотношение мономера и инициатора выбирают из интервала (10-40) -окиси и третичного амина, мольное отношение компонентов в которой берут в пределах 0,5-1,5 соответственно, причем соотношение мономера и инициатора выбирают из интервала (10-40) 1 соответственно. Изобретение может быть применено в области синтеза новых реакционноспособных олигомеров, макромономеров, предназначенных для использования в качестве модифицирующих добавок, поверхностно активных веществ, аппретов, компатибилизаторов при получении полимерных смесей, покрытий в электротехнике, электронике, материаловедении. 2 с. и 2 з.п.ф-лы, 1 табл. 1 соответственно. Изобретение может быть применено в области синтеза новых реакционноспособных олигомеров, макромономеров, предназначенных для использования в качестве модифицирующих добавок, поверхностно активных веществ, аппретов, компатибилизаторов при получении полимерных смесей, покрытий в электротехнике, электронике, материаловедении. 2 с. и 2 з.п.ф-лы, 1 табл.
Изобретение относится к новым гетероцепным реакционноспособным олигомерам и способу и их получения. Синтезированные макромонометры могут быть использованы для получения новых полимеров, а также в качестве модифицирующих добавок, поверхностно активных веществ, аппретов, компатибилизаторов при получении полимерных смесей, покрытий в электротехнике, электронике, материаловедении. Известно, что (мет)акрилаты полимеризуются по радикальному и ионному механизмам с раскрытием двойной связи и образованием карбоцепных высокомолекулярных полимеров [Энциклопедия полимеров. М.: изд. “Советская энциклопедия”. Т. 1. С. 31-33. 1972.] Имеются сведения о полимеризации гидроксиэтил(мет)акрилата или их аналогов по радикальному механизму [Патент США 4157892, 12.06.1979.] при радиационном инициировании [Патент США 4904708, 27.02.1990.] или под действием фотоинициаторов [Патент США 5681871, 28.10.1997] с образованием высокомолекулярных карбоцепных полимеров, используемых в качестве основы контактных линз, а также о применении гидроксилсодержащих (мет)акрилатов в качестве добавок к эпокси(мет)акрилатным смолам с целью снижения вязкости [Патент Германии 3334329, 22.09.1982.] или в качестве компонента в композиции с диаминами [Патент США 5830987, 26.01.1995.]. Из производных (мет)акрилатов только акриламид способен полимеризоваться под действием ионных инициаторов с образованием соответствующих гетероцепных высокомолекулярных полимеров [Энциклопедия полимеров. М.: изд. “Советская энциклопедия” Т. 1. С. 30-31. 1972.]. Таким образом, не обнаружено сведений о получении низкомолекулярных реакционноспособных гетероцепных олигомеров на основе протонсодержащих (мет)акриловых производных. Наиболее близкими к заявляемому техническому решению являются макромономеры на основе производных (мет)акрилатов и способ их получения [Патент США 5147952, 15.09.1992.]. В данном случае осуществляется процесс анионной полимеризации с использованием предварительно синтезированного инициатора структуры: СН2=СН-С(О)О-(CН2)n-C–(R1)(R2) M+, где R1 и R2 – электрофильные группы, стабилизирующие карбанион С–, М+ – четвертичный аммониевый ион. Процесс ведут в среде органического растворителя при 0-60oС. В результате получаются макромономеры карбоцепной структуры (гетероцепная только на конце цепи) с одной концевой реакционноспособной группой и молекулярной массой от 2000 до 10000 (в зависимости от количества инициатора). Недостатками данного метода являются получение карбоцепных макромономеров только с одной концевой реакционноспособной группой и необходимость предварительного синтеза дорогостоящего инициатора сложной структуры. Наличие на концах цепи реакционноспособных групп различной природы представляет широкий спектр возможностей для макромолекулярного дизайна и получения таким образом полимерных матриц и модификаторов различного строения, кроме того, гетероцепная структура полимерной молекулы обеспечивает специфические свойства по сравнению с карбоцепной. Задачей изобретения является создание новых реакционноспособных гетероцепных олигомеров – макромонометров с двумя концевыми реакционноспособными группами и средневесовой молекулярной массой не более 3000. Поставленная задача решается олигомеризацией протоносодержащих акриловых производных с получением гетероцепных реакционноспособных макромономеров общей формулы: СН2=СH-С(О)О-(СН2)n-Х-[СН2-СН2-С(О)О-(СН2)n-Х-]mН, где Х- -О-, -О-С(О) -N(Ph)-; n=2-10; m=2-15. Данные химические соединения с полиэфирной или уретановой основной цепью имеют на концах цепи две реакционноспособные группы (двойная акриловая связь и функциональная). Отметим, что наличие функциональной группы в предлагаемых макромономерах существенно расширяет структурную модификацию как самих макромономеров, так и полимеров на их основе. По существу возможно создание нового класса макромономеров на основе акриловых производных с активным протоном в структуре молекулы. Сущность предлагаемого технического решения заключается в том, что при получении реакционноспособных гетероцепных олигомеров путем анионной полимеризации акриловых производных в присутствии инициатора в качестве мономеров используют протонсодержащие акриловые производные, анионную полимеризацию ведут в инертной среде в присутствии инициаторов, выбранных из группы: щелочные металлы или их производные, смесь -окиси и третичного амина, мольное отношение компонентов, в которой берут в пределах 0,5-1,5; причем соотношение мономера и инициатора выбирают из интервала (10-40)1 соответственно.
Например, для получения олигомеров указанной выше структуры могут быть использованы мономеры следующей формулы:СН2=СH С(O)O(CН2)n ОН СН2=CНС(O)O (CH2)nOC(O)NHPh, где n=2-10. Синтез проводят в инертной среде при постоянном перемешивании в температурном диапазоне 40-80oС до получения целевого продукта. При увеличении содержания инициатора в реакционной смеси по отношению к мономеру (>10 мол.% от концентрации мономера) не реализуется рост цепи, при содержании инициатора, меньшем 2,5 мол.%, не обеспечивается количество активных центров, необходимых для получения олигомера. При температуре <40oС существенно замедляется инициирование олигомеризации, а при температуре >80oС увеличивается вклад побочных реакций, ухудшающих молекулярные характеристики олигомера и его распределение по типу функциональности. В случае использования для инициирования смеси -окиси и третичного амина предпочтительным является соотношение 1
При использовании описанного процесса анионной полимеризации образуются растворимые олигомеры со среднечисленной молекулярной массой (MN) в пределах 500-1500 и молекулярно-массовом распределении (ММР), близком к наиболее вероятному (МW/МN= 2). Для подтверждения структуры и определения молекулярных характеристик олигомеров использованы ЯМР-спектроскопия и метод гельпроникающей хроматографии (ГПХ) на приборе фирмы “Waters” с рефрактометрическим (DR 410) и ультрафиолетовым (PDA 996) детекторами, что позволяло получать УФ-спектры каждой фракции олигомера в области 210-290 нм (для тетрагидрофурана (ТГФ)) и идентифицировать наличие двойной связи по пику с длиной волны 210-213 нм. Средние величины Mn и Mw вычислены с помощью калибровочного графика по полистирольным стандартам.
Суть изобретения демонстрируется следующими примерами.
Пример 1. Навески гидроксиэтилакрилата (ГЭА) – 4,94 г и раствора третбутилата лития в толуоле (титр 3 10-4 моль/мл) – 6,86 г помещаются в стеклянную ампулу в токе аргона при комнатной температуре. После дополнительного вытеснения воздуха аргоном ампула запаивается, термостатируется при 80oС в течение 70 ч. После охлаждения ампула вскрывается, реакционная смесь нейтрализуется добавлением 0,3 мл ледяной уксусной кислоты и 20 мл 10%-ного раствора кислого сернокислого калия. После встряхивания смесь переливается в делительную воронку, где выдерживается в течение трех суток дo четкого разделения на три слоя: нижний слой – олигомер ГЭА, средний слой – раствор уксуснокислого и сернокислого лития в воде; верхний слой – раствор непрореагировавшего ГЭА в толуоле. После разделения олигомерный слой подвергается вакуумной сушке при температуре не более 50oС в течение трех часов до остаточного давления 0,1 мм рт. ст. Результат ГПХ-анализа полученного олигомера представлен в таблице.
Пример 2. Навески ГЭА – 5,00 г, фенилглицидилового эфира (ФГЭ) – 0,729 г и диметилбензиламина (ДМБА) – 0,505 г заливаются в стеклянную ампулу в токе аргона при комнатной температуре. Процесс ведут в инертной атмосфере в течение 24 ч при 50oС. После окончания процесса продукт реакции анализируется методом ГПХ, результаты представлены в таблице.
Пример 3. Навески: ГЭА – 1,856 г, ФГЭ – 0,246 г и ДМБА – 0,213 г используют для синтеза олигомера аналогично примеру 2. Термостатирование в течение 96 часов при температуре 80oС. После реакции из полученного продукта вымывается ДМБА и растворимые фракции двумя 10-кратными порциями этилового спирта в течение трех суток при комнатной температуре. Олигомер и отмытая этиловым спиртом его фракция высушиваются в вакууме при комнатной температуре и анализируется методоми ГПХ (результаты в таблице) и ЯМР-спектроскопии.
Пример 4. Используют навески фенилуретанэтиленакрилата (ФУЭА) – 0,846 г, ФГЭ – 0,059 г и ДМБА – 0,049 г для получения олигомера ФУЭА аналогично примеру 3. Низкомолекулярные продукты отмываются четырьмя порциями стократного избытка этилового спирта при комнатной температуре в течение десяти суток. Отмытый олигомер анализируется методами ГПХ (результаты в таблице) и ЯМР-спектроскопии.
Пример 5. Олигомер ФУЭА получают как в примере 4 за исключением условия термостатирования: в течение 70 часов при 70oС. Олигомер анализируется термомеханическим методом (прибор УИП-70) и методом ГПХ.
Пример 6. Олигомер ФУЭА синтезируется по условиям примера 5. Олигомер, отмытый от низкомолекулярных фракций, двумя порциями десятикратного избытка этилового спирта в течение трех суток анализируется методами ЯМР и ГПХ.
Пример 7. Олигомер ФУЭА синтезируется по условиям примера 5. После сушки в вакууме олигомер дополнительно выдерживается в течение семи часов при 100oС. Результат ГПХ-анализа представлен в таблице.
Пример 8. Олигомер ФУЭА синтезируется аналогично примеру 4 за исключением соотношения реагентов [С=С]о/[ФГЭ]о=17,1, [ФГЭ]о=[ДМБА]о и режима реакции: при 80oС в течение 24 часов. ГПХ-анализ представлен в таблице.
Пример 9. Олигомер ФУЭА синтезируется аналогично примеру 8 за исключением мольного соотношения реагентов [С=С]о/[ФГЭ]о=36,5, [ФГЭ]о/[ДМБА]о=0,5. Данные ГПХ в таблице.
Пример 10. В качестве мономера берут 3-гидроксипропилакрилат (ГПА), 1,95 г инициатора – трет-бутилат лития 0,06 г. Синтез олигомера проводят без растворителя при 50oС в течение 48 ч при постоянном перемешивании в токе аргона. Реакционную массу растворяют в хлористом метилене и нейтрализуют углекислым газом до рН~7,0, затем фильтруют и удаляют растворитель в вакууме. Выход 83%. Молекулярные характеристики приведены в таблице.
Пример 11. Берут навески: ГЭА – 4,66 г, тетрагидрофуран (ТГФ) – 18,66 г и калий металлический – 0,157 г. Синтез олигомера проводят в колбе с обратным холодильником при постоянном перемешивании в токе аргона при температуре кипения ТГФ ( 10-4 моль/мл) – 6,86 г помещаются в стеклянную ампулу в токе аргона при комнатной температуре. После дополнительного вытеснения воздуха аргоном ампула запаивается, термостатируется при 80oС в течение 70 ч. После охлаждения ампула вскрывается, реакционная смесь нейтрализуется добавлением 0,3 мл ледяной уксусной кислоты и 20 мл 10%-ного раствора кислого сернокислого калия. После встряхивания смесь переливается в делительную воронку, где выдерживается в течение трех суток дo четкого разделения на три слоя: нижний слой – олигомер ГЭА, средний слой – раствор уксуснокислого и сернокислого лития в воде; верхний слой – раствор непрореагировавшего ГЭА в толуоле. После разделения олигомерный слой подвергается вакуумной сушке при температуре не более 50oС в течение трех часов до остаточного давления 0,1 мм рт. ст. Результат ГПХ-анализа полученного олигомера представлен в таблице.
Пример 2. Навески ГЭА – 5,00 г, фенилглицидилового эфира (ФГЭ) – 0,729 г и диметилбензиламина (ДМБА) – 0,505 г заливаются в стеклянную ампулу в токе аргона при комнатной температуре. Процесс ведут в инертной атмосфере в течение 24 ч при 50oС. После окончания процесса продукт реакции анализируется методом ГПХ, результаты представлены в таблице.
Пример 3. Навески: ГЭА – 1,856 г, ФГЭ – 0,246 г и ДМБА – 0,213 г используют для синтеза олигомера аналогично примеру 2. Термостатирование в течение 96 часов при температуре 80oС. После реакции из полученного продукта вымывается ДМБА и растворимые фракции двумя 10-кратными порциями этилового спирта в течение трех суток при комнатной температуре. Олигомер и отмытая этиловым спиртом его фракция высушиваются в вакууме при комнатной температуре и анализируется методоми ГПХ (результаты в таблице) и ЯМР-спектроскопии.
Пример 4. Используют навески фенилуретанэтиленакрилата (ФУЭА) – 0,846 г, ФГЭ – 0,059 г и ДМБА – 0,049 г для получения олигомера ФУЭА аналогично примеру 3. Низкомолекулярные продукты отмываются четырьмя порциями стократного избытка этилового спирта при комнатной температуре в течение десяти суток. Отмытый олигомер анализируется методами ГПХ (результаты в таблице) и ЯМР-спектроскопии.
Пример 5. Олигомер ФУЭА получают как в примере 4 за исключением условия термостатирования: в течение 70 часов при 70oС. Олигомер анализируется термомеханическим методом (прибор УИП-70) и методом ГПХ.
Пример 6. Олигомер ФУЭА синтезируется по условиям примера 5. Олигомер, отмытый от низкомолекулярных фракций, двумя порциями десятикратного избытка этилового спирта в течение трех суток анализируется методами ЯМР и ГПХ.
Пример 7. Олигомер ФУЭА синтезируется по условиям примера 5. После сушки в вакууме олигомер дополнительно выдерживается в течение семи часов при 100oС. Результат ГПХ-анализа представлен в таблице.
Пример 8. Олигомер ФУЭА синтезируется аналогично примеру 4 за исключением соотношения реагентов [С=С]о/[ФГЭ]о=17,1, [ФГЭ]о=[ДМБА]о и режима реакции: при 80oС в течение 24 часов. ГПХ-анализ представлен в таблице.
Пример 9. Олигомер ФУЭА синтезируется аналогично примеру 8 за исключением мольного соотношения реагентов [С=С]о/[ФГЭ]о=36,5, [ФГЭ]о/[ДМБА]о=0,5. Данные ГПХ в таблице.
Пример 10. В качестве мономера берут 3-гидроксипропилакрилат (ГПА), 1,95 г инициатора – трет-бутилат лития 0,06 г. Синтез олигомера проводят без растворителя при 50oС в течение 48 ч при постоянном перемешивании в токе аргона. Реакционную массу растворяют в хлористом метилене и нейтрализуют углекислым газом до рН~7,0, затем фильтруют и удаляют растворитель в вакууме. Выход 83%. Молекулярные характеристики приведены в таблице.
Пример 11. Берут навески: ГЭА – 4,66 г, тетрагидрофуран (ТГФ) – 18,66 г и калий металлический – 0,157 г. Синтез олигомера проводят в колбе с обратным холодильником при постоянном перемешивании в токе аргона при температуре кипения ТГФ (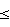 66oС) в течение 6 ч. После нейтрализации реакционной среды углекислым газом до рН~7,0 и фильтрования выделяют олигомер. Выход 72%. Молекулярные характеристики приведены в таблице.
Из таблицы видно, что по предлагаемому способу образуются олигомеры со средневесовой молекулярной массой ~3000 и ММР, близком к наиболее вероятному. Исключением являются данные примеров 2,8 и 9, в которых из-за незавершенности процесса (более низкие температуры, концентрации инициатора и меньшее время термостатирования) образуются олигомеры с ММР>2.
При инициировании олигомеризации гидроксилсодержащих акрилатов щелочными металлами в реакционной среде образуются алкоголяты, алкоксианионы которых способны атаковать на -атом углерода двойной связи мономера и вызывать рост цепи, что приводит к наличию одной концевой С=С-связи в макромолекуле при гетероцепной структуре олигомера. При инициировании трет-бутилатом лития из-за существенного различия кислотных свойств первичного и третичного спиртов (соответственно основности их алкоксианионов, увеличивающейся с уменьшением кислотности спирта R3СОН>R2OH>RCH2O) происходит быстрое смещение равновесия в сторону образования алкоксианионов гидроксилсодержащего акрилата. Третбутоксианион из-за низкой основности не способен атаковать С=С-связь мономера и инициировать процесс олигомеризации. Отсутствие концевой третбутоксигруппы в образующихся олигомерах установлено экспериментально.
Известно, что третичные амины не способны вызывать полимеризацию эпоксидных соединений типа глицидиловых эфиров при отсутствии протонодонорного соединения (ПС), а при его наличии рост цепи начинается с алкоксианиона алкоголята тетраалкиламмония, образующегося из используемого ПС, -окиси и третичного амина приводит к аналогичному механизму зарождения цепи соответственно и к наличию концевой акрилатной С=С-связи в макромономере.
Сделанные заключения подтверждаются экспериментально. Одновременный анализ на двух детекторах (рефрактометрическом и ультрафиолетовом) олигомеров, отмытых от низкомолекулярных компонентов реакционных систем и высушенных в вакууме, позволил идентифицировать по пику с длиной волны 210-213 нм наличие С=С-связи независимо от природы инициатора.
Гетероцепная структура получаемых предлагаемым способом олигомеров подтверждается ЯМР-спектроскопическим исследованием. Спектры ЯМР записывали на спетрометре ЯМР AC-200 P фирмы “Bruker” при 25oС с накоплением сигналов. Для измерений использовали насыщенные растворы олигомеров в четыреххлористом углероде.
Действительно, 1Н ЯМР-спектры растворимой и нерастворимой в этиловом спирте фракций олигомера ГЭА (пример 3) [ 66oС) в течение 6 ч. После нейтрализации реакционной среды углекислым газом до рН~7,0 и фильтрования выделяют олигомер. Выход 72%. Молекулярные характеристики приведены в таблице.
Из таблицы видно, что по предлагаемому способу образуются олигомеры со средневесовой молекулярной массой ~3000 и ММР, близком к наиболее вероятному. Исключением являются данные примеров 2,8 и 9, в которых из-за незавершенности процесса (более низкие температуры, концентрации инициатора и меньшее время термостатирования) образуются олигомеры с ММР>2.
При инициировании олигомеризации гидроксилсодержащих акрилатов щелочными металлами в реакционной среде образуются алкоголяты, алкоксианионы которых способны атаковать на -атом углерода двойной связи мономера и вызывать рост цепи, что приводит к наличию одной концевой С=С-связи в макромолекуле при гетероцепной структуре олигомера. При инициировании трет-бутилатом лития из-за существенного различия кислотных свойств первичного и третичного спиртов (соответственно основности их алкоксианионов, увеличивающейся с уменьшением кислотности спирта R3СОН>R2OH>RCH2O) происходит быстрое смещение равновесия в сторону образования алкоксианионов гидроксилсодержащего акрилата. Третбутоксианион из-за низкой основности не способен атаковать С=С-связь мономера и инициировать процесс олигомеризации. Отсутствие концевой третбутоксигруппы в образующихся олигомерах установлено экспериментально.
Известно, что третичные амины не способны вызывать полимеризацию эпоксидных соединений типа глицидиловых эфиров при отсутствии протонодонорного соединения (ПС), а при его наличии рост цепи начинается с алкоксианиона алкоголята тетраалкиламмония, образующегося из используемого ПС, -окиси и третичного амина приводит к аналогичному механизму зарождения цепи соответственно и к наличию концевой акрилатной С=С-связи в макромономере.
Сделанные заключения подтверждаются экспериментально. Одновременный анализ на двух детекторах (рефрактометрическом и ультрафиолетовом) олигомеров, отмытых от низкомолекулярных компонентов реакционных систем и высушенных в вакууме, позволил идентифицировать по пику с длиной волны 210-213 нм наличие С=С-связи независимо от природы инициатора.
Гетероцепная структура получаемых предлагаемым способом олигомеров подтверждается ЯМР-спектроскопическим исследованием. Спектры ЯМР записывали на спетрометре ЯМР AC-200 P фирмы “Bruker” при 25oС с накоплением сигналов. Для измерений использовали насыщенные растворы олигомеров в четыреххлористом углероде.
Действительно, 1Н ЯМР-спектры растворимой и нерастворимой в этиловом спирте фракций олигомера ГЭА (пример 3) [ , м. д.: 4,12 (уш. м., 2Н, СН2-О); 3,58 (уш. м. , 2Н, СН2-О); 3,47 (уш. м., 2Н, СН2-O-); 2,42 (уш. м, 2Н, -СН2-СО)] и олигомера ФУЭА (пример 4) [ м. д.: 7,06 (уш. м., 5Н, С6Н5): 4,01 (уш. м. 4Н. О-СН2СН2-О); 3,79 (уш. м. , 2Н, СН2-О), 2,42 (уш. м. , 2Н -СН2-СО-)], а также 13С ЯМР-спектры олигомера ФУЭА в растворе диметилформамида-Д7[, м. д. : 32,3 (-СН2СО); 45,7 (СН2-О); 61,7 (СН2-O); 62,8 (СН2-O); 126,1, 126,7, 128,4, 141,0 (С6Н5); 154,2 (C=N); 170,2 (С=O)] показывают наличие в олигомере четырех неэквивалентных СН2-групп соответствующих гетероцепной природе олигомеров.
Исключением является олигомер ФУЭА, полученный в примере 6. По сравнению с 4-м примером в 1Н ЯМР-спектре этого олигомера появляются дополнительные сигналы: 2,16 (уш. м., 2Н, СН2-СН) и 1,96 (уш. м., 1Н, СН2-СН). Оценка показала, что в этом образце имеется ~ 70% гетероцепной и ~30% карбоцепной структур. Обусловлено это следующим. ФУЭА синтезируется путем некаталитического взаимодействия ГЭА с фенилизоцианатом при эквифункциональном соотношении и при 60oС в течение 8 с. Присутствующий в ГЭА в качестве ингибитора 4-метоксифенол также взаимодействует с фениизоиианатом с образованием соответствующего уретансодержащего производного, причем его относительная концентрация снижается практически в два раза. В связи с этим при олигомеризации ФУЭА, несмотря на тщательную изоляцию от влаги и кислорода воздуха (все операции проводятся в токе аргона), частично процесс идет по карбоцепному пути. В каждом звене макромолекулы карбоцепной структуры имеется содержащий активный протон алкил-арильный заместитель. Растворимость таких олигомеров в этиловом спирте существенно выше, чем олигомеров гетероцепной структуры. Поэтому менее жесткие условия отмывки низкомолекулярных продуктов, как в примере 6 по сравнению с примером 4, становятся недостаточными для полного удаления карбоцепной фракции олигомера, что и подтверждается экспериментально.
Термомеханические испытания макромономера ФУЭА показали, что его температура стеклования равна 10oС (пример 5), а дополнительная термообработка при 100oС в течение 7 ч не привела к существенному изменению молекулярных характеристик (пример 7), что свидетельствует об отсутствиии термоинициируемой сшивки при нагревании до 100oС. Отметим, что введение дополнительного количества ингибитора в мономер ФУЭА позволяет полностью предотвратить карбоцепной путь его олигомеризации.
Таким образом, получены новые гетероцепные реакционноспособные олигомеры с широкими возможностями их дальнейшего преобразования и использования. , м. д.: 4,12 (уш. м., 2Н, СН2-О); 3,58 (уш. м. , 2Н, СН2-О); 3,47 (уш. м., 2Н, СН2-O-); 2,42 (уш. м, 2Н, -СН2-СО)] и олигомера ФУЭА (пример 4) [ м. д.: 7,06 (уш. м., 5Н, С6Н5): 4,01 (уш. м. 4Н. О-СН2СН2-О); 3,79 (уш. м. , 2Н, СН2-О), 2,42 (уш. м. , 2Н -СН2-СО-)], а также 13С ЯМР-спектры олигомера ФУЭА в растворе диметилформамида-Д7[, м. д. : 32,3 (-СН2СО); 45,7 (СН2-О); 61,7 (СН2-O); 62,8 (СН2-O); 126,1, 126,7, 128,4, 141,0 (С6Н5); 154,2 (C=N); 170,2 (С=O)] показывают наличие в олигомере четырех неэквивалентных СН2-групп соответствующих гетероцепной природе олигомеров.
Исключением является олигомер ФУЭА, полученный в примере 6. По сравнению с 4-м примером в 1Н ЯМР-спектре этого олигомера появляются дополнительные сигналы: 2,16 (уш. м., 2Н, СН2-СН) и 1,96 (уш. м., 1Н, СН2-СН). Оценка показала, что в этом образце имеется ~ 70% гетероцепной и ~30% карбоцепной структур. Обусловлено это следующим. ФУЭА синтезируется путем некаталитического взаимодействия ГЭА с фенилизоцианатом при эквифункциональном соотношении и при 60oС в течение 8 с. Присутствующий в ГЭА в качестве ингибитора 4-метоксифенол также взаимодействует с фениизоиианатом с образованием соответствующего уретансодержащего производного, причем его относительная концентрация снижается практически в два раза. В связи с этим при олигомеризации ФУЭА, несмотря на тщательную изоляцию от влаги и кислорода воздуха (все операции проводятся в токе аргона), частично процесс идет по карбоцепному пути. В каждом звене макромолекулы карбоцепной структуры имеется содержащий активный протон алкил-арильный заместитель. Растворимость таких олигомеров в этиловом спирте существенно выше, чем олигомеров гетероцепной структуры. Поэтому менее жесткие условия отмывки низкомолекулярных продуктов, как в примере 6 по сравнению с примером 4, становятся недостаточными для полного удаления карбоцепной фракции олигомера, что и подтверждается экспериментально.
Термомеханические испытания макромономера ФУЭА показали, что его температура стеклования равна 10oС (пример 5), а дополнительная термообработка при 100oС в течение 7 ч не привела к существенному изменению молекулярных характеристик (пример 7), что свидетельствует об отсутствиии термоинициируемой сшивки при нагревании до 100oС. Отметим, что введение дополнительного количества ингибитора в мономер ФУЭА позволяет полностью предотвратить карбоцепной путь его олигомеризации.
Таким образом, получены новые гетероцепные реакционноспособные олигомеры с широкими возможностями их дальнейшего преобразования и использования.
Формула изобретения
СН2=СН-С(О)О-(СН2)n-Х-[CH2-СН2-С(О)О-(СН2)n-Х-]m Н, где Х – -О-, -О-С(О)-N(Ph)-; n=2-10; m=2-15. 2. Способ получения реакционноспособных гетероцепных олигомеров по п.1 путем анионной полимеризации акриловых производных в присутствии инициатора, отличающийся тем, что в качестве мономеров берут протонсодержащие акриловые производные, анионную полимеризацию ведут в инертной среде в присутствии инициаторов, выбранных из группы щелочные металлы или их производные, смесь -окиси и третичного амина, мольное отношение компонентов в которой берут в пределах 0,5-1,5 соответственно, причем соотношение мономера и инициатора выбирают из интервала (10-40)1 соответственно.
3. Способ по п.2, отличающийся тем, что в качестве производных щелочных металлов берут их алкоголяты.
4. Способ по п.2, отличающийся тем, что полимеризацию ведут при 40-80oС.
РИСУНКИ
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 31.03.2008
Извещение опубликовано: 10.04.2010 БИ: 10/2010
|
||||||||||||||||||||||||||