Патент на изобретение №2193040
|
||||||||||||||||||||||||||
(54) НОВЫЕ ПЕНТАСАХАРИДЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
(57) Реферат: Изобретение относится к пентасахариду в кислой форме или его фармацевтически приемлемой соли, анионная форма которого имеет формулу I, где R представляет собой водород или -SO3 –, (C1-С3)алкильную или (С2-С3)ацильную группу, Т представляет собой водород или этильную группу, n представляет собой 1 или 2, а также к фармацевтической композиция на их основе. Заявленные соединения и композиция обладают антитромботической активностью. 2 с. и 6 з.п. ф-лы, 7 ил., 3 табл. 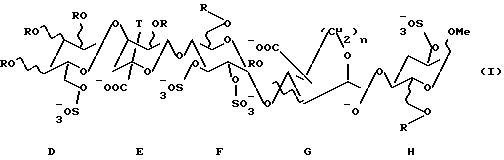
Настоящее изобретение относится к пентасахаридам, способам их получения и содержащим их фармацевтическим композициям. Гепарин – это полисахарид из семейства гликозаминогликанов, известный своими антикоагулянтными свойствами. Известно, что свертывание крови представляет собой сложный физиологический феномен (I.  and U. Lindahl, Molecular and Cell Biochemistry, 1982, Dr. W.Junk Publishers-Netherlands). Определенные стимулы, такие как контактная активация и тканевой фактор, запускают последующую активацию серии факторов свертывания, присутствующих в плазме крови. Независимо от природы стимула конечные стадии идентичны: активированный фактор Х (Ха) активирует фактор II (также известный как протромбин), который в своей активированной форме (фактор IIа, известный как тромбин) вызывает частичный протеолиз растворимого фибриногена с высвобождением нерастворимого фибрина, являющегося одной из основных составляющих кровяного сгустка.
При обычных физиологических условиях активность факторов свертывания регулируется такими белками, как антитромбин III (ATIII) и кофактор гепарина II (НС II), которые также присутствуют в плазме. ATIII проявляет ингибиторную активность в отношении определенного ряда факторов свертывания, и в частности факторов Ха и IIа.
Таким образом, ингибирование фактора Ха или фактора IIа является предпочтительным средством для достижения антикоагулянтной и антитромботической активности, так как эти два фактора вовлечены в две конечные стадии свертывания, которые не зависят от запускающего стимула.
Пентасахарид, такой как описан Синай (Р. and U. Lindahl, Molecular and Cell Biochemistry, 1982, Dr. W.Junk Publishers-Netherlands). Определенные стимулы, такие как контактная активация и тканевой фактор, запускают последующую активацию серии факторов свертывания, присутствующих в плазме крови. Независимо от природы стимула конечные стадии идентичны: активированный фактор Х (Ха) активирует фактор II (также известный как протромбин), который в своей активированной форме (фактор IIа, известный как тромбин) вызывает частичный протеолиз растворимого фибриногена с высвобождением нерастворимого фибрина, являющегося одной из основных составляющих кровяного сгустка.
При обычных физиологических условиях активность факторов свертывания регулируется такими белками, как антитромбин III (ATIII) и кофактор гепарина II (НС II), которые также присутствуют в плазме. ATIII проявляет ингибиторную активность в отношении определенного ряда факторов свертывания, и в частности факторов Ха и IIа.
Таким образом, ингибирование фактора Ха или фактора IIа является предпочтительным средством для достижения антикоагулянтной и антитромботической активности, так как эти два фактора вовлечены в две конечные стадии свертывания, которые не зависят от запускающего стимула.
Пентасахарид, такой как описан Синай (Р.  et al., Carbohydrate Research 1984, 132 С5) представляет собой минимальную гепариновую последовательность, необходимую для связывания с AT III. Это соединение было получено около пятнадцати лет назад посредством полного химического синтеза.
С тех пор в литературе было описано несколько синтетических олигосахаридов, полученных путем полного химического синтеза и обладающих антитромботической антикоагулянтной активностью.
Известны (патент ЕР 0084999) производные, состоящие из моносахаридных звеньев уроновой кислоты (глюкуроновой или идуроновой кислоты) и глюкозамина, которые обладают полезными антитромботическими свойствами. Кроме заместителей, состоящих из гидроксильных групп, эти соединения содержат N-сульфатные группы, N-ацетильные группы и, в некоторых случаях, аномерные гидроксильные группы заменены метоксильными группами.
Известны (заявка на патент ЕР 0165134) синтетические олигосахариды с антитромботической активностью. Эти соединения, состоящие из моносахаридных звеньев уроновой кислоты и глюкозамина и включающие в себя O-сульфатную группу в положении 3 глюкозаминового звена, также описаны в заявке на патент ЕР 0301618. Эти соединения обладают сильными антитромботическими и антикоагулянтными свойствами. Известны (патент ЕР 0454220) производные уроновой кислоты и производные глюкозы, содержащие O-алкильные или O-сульфатные группы в качестве заместителей. Эти последние соединения также обладают антитромботическими и антикоагулянтными свойствами.
Известны (патент ЕР 0529175) также сульфатированные производные гликозаминогликаноидов, в которых N-сульфатные, N-ацетатные или гидроксильные функциональные группы заменены алкоксильной, арилоксильной, аралкилоксильной или O-сульфатной группами. Эти соединения обладают полезными антитромботическими свойствами. Последние соединения также являются ингибиторами пролиферации гладкомышечных клеток.
Известны (Angew. Chem. Int. Ed. Engl. 1993, 32, 3, 434-436) олигосахариды и, в частности, пентасахариды, являющиеся аналогами минимальной гепариновой последовательности, необходимой для связывания с AT-III. Эти соединения содержат глюкуроновую кислоту или звенья глюкозы, чьи гидроксильные функции заменены O-сульфатными или O-метильными группами.
Много исследований проведено по изучению пентасахаридов, и литературные данные указывают на то, что конформация звена G L-идуроновой кислоты играет важную роль в активности продуктов. Описано несколько конформационных состояний для звена G (4С1, 1C4, 2SO) и выдвинуто предположение, что такая конформационная гибкость существенна для биологической активности продуктов, содержащих L-идуроновую кислоту (В. Casu, M. Petitou, A. Provasoli and P. et al., Carbohydrate Research 1984, 132 С5) представляет собой минимальную гепариновую последовательность, необходимую для связывания с AT III. Это соединение было получено около пятнадцати лет назад посредством полного химического синтеза.
С тех пор в литературе было описано несколько синтетических олигосахаридов, полученных путем полного химического синтеза и обладающих антитромботической антикоагулянтной активностью.
Известны (патент ЕР 0084999) производные, состоящие из моносахаридных звеньев уроновой кислоты (глюкуроновой или идуроновой кислоты) и глюкозамина, которые обладают полезными антитромботическими свойствами. Кроме заместителей, состоящих из гидроксильных групп, эти соединения содержат N-сульфатные группы, N-ацетильные группы и, в некоторых случаях, аномерные гидроксильные группы заменены метоксильными группами.
Известны (заявка на патент ЕР 0165134) синтетические олигосахариды с антитромботической активностью. Эти соединения, состоящие из моносахаридных звеньев уроновой кислоты и глюкозамина и включающие в себя O-сульфатную группу в положении 3 глюкозаминового звена, также описаны в заявке на патент ЕР 0301618. Эти соединения обладают сильными антитромботическими и антикоагулянтными свойствами. Известны (патент ЕР 0454220) производные уроновой кислоты и производные глюкозы, содержащие O-алкильные или O-сульфатные группы в качестве заместителей. Эти последние соединения также обладают антитромботическими и антикоагулянтными свойствами.
Известны (патент ЕР 0529175) также сульфатированные производные гликозаминогликаноидов, в которых N-сульфатные, N-ацетатные или гидроксильные функциональные группы заменены алкоксильной, арилоксильной, аралкилоксильной или O-сульфатной группами. Эти соединения обладают полезными антитромботическими свойствами. Последние соединения также являются ингибиторами пролиферации гладкомышечных клеток.
Известны (Angew. Chem. Int. Ed. Engl. 1993, 32, 3, 434-436) олигосахариды и, в частности, пентасахариды, являющиеся аналогами минимальной гепариновой последовательности, необходимой для связывания с AT-III. Эти соединения содержат глюкуроновую кислоту или звенья глюкозы, чьи гидроксильные функции заменены O-сульфатными или O-метильными группами.
Много исследований проведено по изучению пентасахаридов, и литературные данные указывают на то, что конформация звена G L-идуроновой кислоты играет важную роль в активности продуктов. Описано несколько конформационных состояний для звена G (4С1, 1C4, 2SO) и выдвинуто предположение, что такая конформационная гибкость существенна для биологической активности продуктов, содержащих L-идуроновую кислоту (В. Casu, M. Petitou, A. Provasoli and P.  , Conformational flexibility: a new concept for explaining binding and biological properties of iduronic acid-containing glycosaminoglycans. Trends Biochem. Sci. 1988,13, 221-225).
Было неожиданно обнаружено, что при замене одной из O-алкильных групп алкиленовым мостиком и, тем самым, фиксации конформации L-идуроновой кислоты, получаются олигосахариды, обладающие полезными биологическими свойствами, хотя и утратившие конформационную гибкость. Причина этого заключается в том, что соединения по настоящему изобретению отличаются от других синтетических гепариноидов, описанных в литературе, благодаря своим новым структурам и сильным и неожиданным биологическим свойствам. Соединения по изобретению являются пентасахаридами, в которых L-идуроновое звено G находится в так называемой “фиксированной” конформации 2SO, а звено Е D-глюкуроновой кислоты возможно имеет этильную группу в положении 5. Эти соединения обладают очень высокой активностью против фактора Ха и сильное сродство к AT III.
Объектом настоящего изобретения является, более конкретно, пентасахарид в кислой форме и его фармацевтически приемлемые соли с одним или более чем одним фармацевтически приемлемым катионом, анионная форма которого имеет формулу (I), приведенную в конце описания, , Conformational flexibility: a new concept for explaining binding and biological properties of iduronic acid-containing glycosaminoglycans. Trends Biochem. Sci. 1988,13, 221-225).
Было неожиданно обнаружено, что при замене одной из O-алкильных групп алкиленовым мостиком и, тем самым, фиксации конформации L-идуроновой кислоты, получаются олигосахариды, обладающие полезными биологическими свойствами, хотя и утратившие конформационную гибкость. Причина этого заключается в том, что соединения по настоящему изобретению отличаются от других синтетических гепариноидов, описанных в литературе, благодаря своим новым структурам и сильным и неожиданным биологическим свойствам. Соединения по изобретению являются пентасахаридами, в которых L-идуроновое звено G находится в так называемой “фиксированной” конформации 2SO, а звено Е D-глюкуроновой кислоты возможно имеет этильную группу в положении 5. Эти соединения обладают очень высокой активностью против фактора Ха и сильное сродство к AT III.
Объектом настоящего изобретения является, более конкретно, пентасахарид в кислой форме и его фармацевтически приемлемые соли с одним или более чем одним фармацевтически приемлемым катионом, анионная форма которого имеет формулу (I), приведенную в конце описания,где R представляет собой водород или -SO-3, (С1-С3)алкил или (C2-С3)ацильную группу; – Т представляет собой водород или этильную группу; – n представляет собой 1 или 2. Изобретение включает в себя пентасахариды в кислой форме или в форме фармацевтически приемлемой соли. В кислой форме функции -СОО– и -SО-3 находятся в форме -СООН и -SО3Н соответственно. Выражение “фармацевтически приемлемая соль пентасахаридов по изобретению” предназначено для обозначения пентасахаридов, в которых одна или более чем одна из функций -СОО– и/или -SO3 – связана ионной связью с фармацевтически приемлемым катионом металла. Предпочтительными солями по изобретению являются такие, в которых катион выбран из катионов щелочных металлов, особое предпочтение отдается солям, в которых катионом является Na+ или К+. Объектом настоящего изобретения также является способ получения пентасахаридов по изобретению, отличающийся тем, что получают предшественник звена G, который сочетают с предшественником звена Н для получения предшественника GH, и наконец получают пентасахарид либо путем сочетания предшественника GH с предшественником DEF, либо путем сочетания предшественника GH с предшественником EF с последующим добавлением D. Может быть использован любой предшественник G, Н, EF или DEF. Это означает, что согласно этим способам можно получить целое семейство пентасахаридов, имеющих совместно звено G фиксированной конфигурации. Способ, описанный выше, является предпочтительным способом по изобретению. Однако пентасахариды по изобретению могут быть получены посредством других известных способов химии сахаров, в частности путем взаимодействия моносахарида, содержащего защитные группы, такие как описаны Грином (Т.W. Green, in Protective Groups in Organic Synthesis (Wiley, N.Y. 1981)), на гидроксильных радикалах и, возможно, на карбоксильных радикалах, если таковые присутствуют, с другим защищенным моносахаридом для получения дисахарида, который затем подвергают взаимодействию с другим защищенным моносахаридом с образованием защищенного трисахарида, из которого можно получить защищенный тетрасахарид и затем защищенный пентасахарид. С защищенных пентасахаридов затем снимают защиту и, возможно, сульфатируют или частично снимают защиту, затем сульфатируют и затем полностью снимают защиту для того, чтобы получить соединения по изобретению. Такие способы известны в химии углеводов и описаны, в частности, Джаурандом (G. Jaurand et al. in Bioorganic and Medical Chemistry Letters 1992, 2, 9, 897-900), Бастеном (J. Basten et al., in Bioorganic and Medicinal Chemistry Letters 1992, 2, 9, 905-910) и Петитоу и Ван Боэкелем (М. Petitou and С. A. A. van Boeckel in “Chemical synthesis of heparin fragment and analogues” 203-210 – Process in the chemistry of organic natural products, Ed. Springer Verlag Vienna – N.Y.1992). Способ, описанный выше, позволяет получать соединения по изобретению в форме солей. Для того чтобы получить соответствующие кислоты, соединения по изобретению в форме солей приводят в контакт с катионообменной смолой в кислой форме. Соединения по изобретению в форме кислот могут быть затем нейтрализованы основанием для получения желаемой соли. Для этого может быть использовано любое неорганическое или органическое основание, которое дает фармацевтически приемлемые соли. Предпочтительно использовать гидроксид натрия, гидроксид калия, гидроксид кальция или гидроксид магния. Предпочтительными солями пентасахаридов по изобретению являются соли натрия и кальция. Соединения, являющиеся объектом настоящего изобретения, обладают полезными фармакологическими и биохимическими свойствами. В частности, они обладают высокой активностью против фактора Ха и сильным сродством к AT III. Как упоминалось выше, в своем каскаде свертывания фактор Ха активирует протромбин в тромбин, который подвергает протеолизу растворимый фибриноген с высвобождением нерастворимого фибрина, являющегося основной составляющей кровяного сгустка. Таким образом, ингибирование фактора Ха является предпочтительным средством для достижения антикоагулянтной и антитромботической активности. Активность против фактора Ха продуктов по изобретению определяли при рН 7 согласно методу, описанному Тейеном и Ли (Teien А. N. and Lie M., in Thrombosis Research 1977, 10, 399-410). Было показано, что продукты по изобретению обладают активностью против Ха, равной или превышающей активность уже известных синтетических гепариноидов. Конкретно, анти-Ха активность соединений по изобретению, определенная по указанной методике, составляла от 900 анти-Ха ед./мг до 1200 анти-Ха ед./мг. Сродство пентасахаридов по изобретению, анион которых имеет формулу (I), к ATIII определяли при помощи спектрофлуориметрии при условиях, описанных Ата (D. Atha et al. in Biochemistry 1987, 26, 6454-6461). Результаты опытов показали, что соединения по изобретению обладают очень сильным сродством к AT III. Более того, общую антитромботическую активность этих соединений определяли в опытах с крысами с помощью модели венозного стаза и индукции тромбопластином согласно методу, описанному Рейерсом (J. Reyers et al. in Thrombosis Research 1980, 18, 669-674). ЭД50 соединений по изобретению по меньшей мере того же порядка или меньше, чем у других известных синтетических гепариноидов. Таким образом, соединения по изобретению обладают полезной специфичностью действия и полезной антикоагулянтной и антитромботической активностью. Соединения по изобретению пригодны для получения фармацевтических композиций для парентерального введения. Соединения по изобретению имеют очень низкую токсичность; их токсичность полностью совместима с их использованием в качестве лекарственных препаратов. Соединения по изобретению очень устойчивы и поэтому особенно пригодны для того, чтобы являться активным началом лекарственного препарата. Изобретение также включает в себя фармацевтические композиции, содержащие в качестве активного начала соединение по изобретению или одну из его фармацевтически приемлемых солей, возможно объединенную с одним или более чем одним фармацевтически приемлемым нетоксичным инертным эксципиентом. В каждой единице дозирования активное начало присутствует в количествах, соответствующих предусмотренными суточными дозами. Каждая единица дозирования содержит от 0,1 до 100 мг активного начала, предпочтительно от 0,5 до 50 мг. Соединения по изобретению могут также быть использованы в сочетании с одним или более чем одним другим активным началом, подходящим для желаемой терапии, таким как, например, антитромботические агенты, антикоагулянты, агенты, препятствующие агрегации тромбоцитов, такие как, например, дипиридамол, аспирин, тиклопидин, клопидогрель или антагонисты гликопротеинового комплекса IIb/IIIa. Фармацевтические композиции готовят в виде препаратов для введения млекопитающим, включая человека, для лечения вышеупомянутых заболеваний. Фармацевтические композиции, полученные таким образом, полезны в различных формах, таких как, например, растворы для инъекций или для питья, таблетки с сахарным покрытием, обычные таблетки или желатиновые капсулы. Предпочтительные фармацевтические формы – это растворы для инъекций. Фармацевтические композиции по настоящему изобретению полезны, в частности, для профилактики и лечения заболеваний сосудистой стенки, таких как атеросклероз, состояния гиперкоагуляции, наблюдаемые, например, после операции по поводу опухоли или нарушения свертывания, вызванного бактериальными, вирусными или ферментативными активаторами. Более широко пентасахариды по изобретению могут быть использованы в лечении патологий, связанных с дисфункцией свертывания. Дозировка может варьировать в широком диапазоне в зависимости от возраста пациента, массы и состояния здоровья, природы и тяжести жалобы и способа введения. Эта дозировка охватывает введение одной или более чем одной дозы от примерно 0,5 мг до примерно 1000 мг в день, предпочтительно от примерно 1 до примерно 100 мг в день, и лучше от примерно 0,5 до примерно 50 мг в день, например примерно 20 мг в день внутримышечно или подкожно при дозированных введениях или введениях через регулярные интервалы времени, или суточной дозы от примерно 200 мг до примерно 1000 мг в день перорально. Естественно, эти дозы могут быть уточнены для каждого пациента в зависимости от наблюдаемых результатов и анализов крови, сделанных заранее. Предпочтительный способ введения – это подкожная инъекция. Таким образом, объектом настоящего изобретения являются также фармацевтические композиции, которые содержат в качестве активного начала одно из вышеупомянутых соединений, возможно объединенное с другим активным началом. Эти композиции приготовлены так, чтобы их можно было вводить через пищеварительный тракт или парентерально. В фармацевтических композициях по настоящему изобретению для перорального, сублингвального, подкожного, внутримышечного, внутривенного, трансдермального введения, введения через слизистую, местного или ректального введения активный ингредиент может быть введен животным и людям в стандартных формах введения, в смеси со стандартными фармацевтическими вспомогательными средствами. Подходящие стандартные формы введения включают в себя пероральные формы, такие как пероральные суспензии, растворы, гранулы и порошки, желатиновые капсулы и таблетки, сублингвальные и трансбуккальные формы введения, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения. Когда твердую композицию готовят в форме таблеток, основной активный ингредиент смешивают с фармацевтическим носителем, таким как желатин, крахмал, лактоза, стеарат магния, тальк, гуммиарабик или подобными. Таблетки могут быть покрыты оболочкой из сахарозы или других подходящих материалов или, с другой стороны, они могут быть обработаны так, чтобы иметь отсроченную или замедленную активность, и так, чтобы они непрерывно высвобождали предопределенное количество активного начала. Препарат в форме желатиновых капсул получают путем смешивания активного ингредиента с разбавителем и розлива полученной смеси в мягкие или твердые желатиновые капсулы. Вододиспергируемые порошки или гранулы могут содержать активный ингредиент, смешанный с диспергирующими агентами или смачивающими агентами, или суспендирующими агентами, такими как поливинилпирролидон, а также с подсластителями или усилителями вкуса. Для ректального введения используют суппозитории, приготовленные со связывающими веществами, плавящимися при ректальной температуре, например кокосовым маслом или полиэтиленгликолями. Для парентерального, интраназального или внутриглазного введения используют водные суспензии, изотонические физиологические растворы или стерильные растворы и растворы для инъекций, содержащие фармакологически совместимые диспергирующие агенты и/или смачивающие агенты, например пропиленгликоль или бутиленгликоль. Для введения через слизистые активное начало может быть приготовлено в виде препарата с наличием активатора, такого как соль желчной кислоты, гидрофильный полимер, такой как, например, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза, этилцеллюлоза, карбоксиметилцеллюлоза, декстран, поливинилпирролидон, пектины, крахмалы, желатин, казеин, акриловые кислоты, эфиры акриловых кислот и их сополимеры, полимеры виниловых соединений или сополимеры, виниловые спирты, алкоксиполимеры, полиэтиленоксидные полимеры, полиэфиры или их смеси. Активное начало может также быть приготовлено в виде препарата в форме микрокапсул, возможно с одним или более чем одним вспомогательным веществом или добавкой. Активное начало также может быть в форме комплекса с циклодекстрином, например  -, -,  – или – или  -циклодекстрином, 2-гидроксипропил--циклодекстрином или метил--циклодекстрином.
Активное начало также может высвобождаться баллоном, в котором оно содержится, или через эндоваскулярный удлинитель, введенный в кровеносные сосуды. Так на фармакологическую эффективность активного начала не оказывается влияния.
Подкожное введение является предпочтительным способом.
Нижеследующие методы, способы получения и схемы иллюстрируют синтез различных промежуточных продуктов, которые необходимы для получения пентасахаридов по изобретению.
Используют следующие аббревиатуры: -циклодекстрином, 2-гидроксипропил--циклодекстрином или метил--циклодекстрином.
Активное начало также может высвобождаться баллоном, в котором оно содержится, или через эндоваскулярный удлинитель, введенный в кровеносные сосуды. Так на фармакологическую эффективность активного начала не оказывается влияния.
Подкожное введение является предпочтительным способом.
Нижеследующие методы, способы получения и схемы иллюстрируют синтез различных промежуточных продуктов, которые необходимы для получения пентасахаридов по изобретению.
Используют следующие аббревиатуры:TBDMS: mpem-бутилдиметилсилил; Lev: левулинил; Вn: бензил; Bz: бензоил; ТСХ: тонкослойная хроматография; Olm: трихлорацетимидил, LSIMS: жидкостная масс-спектрометрия вторичных ионов; ESIMS: масс-спектрометрия с ионизацией распылением электронов; TMS: триметилсилил; TSP: триметилсилилтетрадейтеропропионат натрия; Tf: трифлат; MS: молекулярные сита; All: аллил; РМВ: п-метоксибензил; SE: триметилсилилэтил. Dowex  , Sephadex, Chelex и Toyopearl – зарегистрированные товарные знаки.
В методах, способах получения и примерах, приведенных ниже, обычные процедуры, относящиеся к каталитическому сочетанию имидатов, расщеплению эфиров левулиновой кислоты, каталитическому сочетанию тиогликозидов, омылению, метилированию и избирательному снятию защиты с п-метоксибензильной группы, снятию защиты и сульфатированию олиго- и полисахаридов путем гидрогенолиза сложных или простых эфиров бензила, омылению сложных эфиров или сульфат-ионов, могут быть выполнены с использованием обычных методов для подходящих промежуточных продуктов, рассмотренных ниже.
Соединения по настоящему изобретению синтезируют различными способами получения, описанными ниже.
Получение 1.
6-O-трет-бутилдиметилсилил-1,2-O-изопропилиден-3-O-метил--D-глюкофураноза (2) , Sephadex, Chelex и Toyopearl – зарегистрированные товарные знаки.
В методах, способах получения и примерах, приведенных ниже, обычные процедуры, относящиеся к каталитическому сочетанию имидатов, расщеплению эфиров левулиновой кислоты, каталитическому сочетанию тиогликозидов, омылению, метилированию и избирательному снятию защиты с п-метоксибензильной группы, снятию защиты и сульфатированию олиго- и полисахаридов путем гидрогенолиза сложных или простых эфиров бензила, омылению сложных эфиров или сульфат-ионов, могут быть выполнены с использованием обычных методов для подходящих промежуточных продуктов, рассмотренных ниже.
Соединения по настоящему изобретению синтезируют различными способами получения, описанными ниже.
Получение 1.
6-O-трет-бутилдиметилсилил-1,2-O-изопропилиден-3-O-метил--D-глюкофураноза (2)Диол 1 (10 г; 42,7 ммоль) разводят в безводном дихлрометане (100 мл) и добавляют трет-бутилдиметилсилилхлорид (7,1 г; 47,3 ммоль) и имидазол (5,8 г; 85,3 ммоль). Реакционную смесь перемешивают при комнатной температуре. Через 2 часа смесь разводят в дихлорметане и промывают водой. Органическую фазу высушивают над сульфатом магния и концентрируют, остаток очищают, посредством хроматографии на колонке с силикагелем (1/9 о/о этилацетат/циклогексан) с получением желаемого продукта 2 (11,9 г; 80%) в форме сиропа. [ ]D-34 (c 1,9; CHCl3). (c 1,9; CHCl3).Получение 2 6-O-трет-бутилдиметилсилил-1,2-O-изопропилиден-3-O-метил-5-С-винил- -D-глюкофураноза (4)Оксалил хлорид (3,2 мл; 36,8 ммоль) и диметилсульфоксид (5,2 мл; 73,4 ммоль) добавляют к безводному дихлорметану (40 мл) при температуре -78oС и перемешивают смесь в течение 30 минут. Затем добавляют соединение 2 (6,4 г; 18,4 ммоль) и полученную смесь перемешиваю в течение часа. Затем добавляют триэтиламин (15,3 мл; 110,0 ммоль) и через 30 минут реакционную смесь разводят в дихлорметане. Стандартная обработка дает пентулозное соединение (3), которое используют непосредственно для следующей реакции. Неочищенный кетон 3 разводят в безводном тетрагидрофуране (100 мл) и добавляют 1 М раствор винилмагнийбромида в тетрагидрофуране (28 мл; 27,6 ммоль) при температуре 0oС. Через час реакционную смесь разбавляют, но не хлоридом аммония, и промывают водой. Органическую фазу высушивают над сульфатом магния и концентрируют, остаток очищают хроматографией на колонке с силикагелем (1/9 о/о этилацетат/циклогексан) с получением желаемого соединения 4 (70%; 4,8 г) в форме сиропа. [ ]D-40(c 1,3; CHCl3).Аналитически вычислено: С, 57,72; Н, 9,15. Найдено: С, 57,77; Н, 9,23. Получение 3 1,2,4,6-тетра-O-ацетил-3-O-метил-5-С-винил- -D-глюкопираноза (6)Соединение 4 (3,5 г; 9,4 ммоль) растворяют в воде (50 мл); добавляют смолу IR-120 (1 г) и нагревают смесь в течение 6 часов при 80oС. Смолу отфильтровывают и фильтрат концентрируют. Неочищенный продукт 5 ацетилируют, используя ангидрид уксусной кислоты (12 мл) и пиридин (13 мл). Излишек ангидрида уксусной кислоты разрушают метанолом, а растворители концентрируют. Остаток экстрагируют водой и дихлорметаном. Органическую фазу высушивают над сульфатом магния и концентрируют, и после очистки хроматографией на колонке с силикагелем (3/2 о/о этилацетат/циклогексан), получают тетраацетат 6 в форме твердого вещества (75%; 2,7 г). Точка плавления = 50oС. [ ]D-84(c 1,6; CHCl3).Аналитически вычислено: С, 52,47; Н, 6,19. Найдено: С, 52,51; Н, 6,19. ХИ-МС: 406 (М + NH4), 389 (М + 1). Получение 4 Метил-2,3,6-три-O-бензил-4-O-(2,4,6-три-O-ацетил-3-O-метил-5-С-винил- -D-глюкопиранозил)--D-глюканозид (8)Соединение 6 (1,6 г; 4,1 ммоль) и соединение 7 (2,1 г; 4,5 ммоль) (P.J. Garegg and Н. Hultberg, Carbohydr. Res. 1981, 93, C10) растворяют в безводном дихлорметане (50 мл) и добавляют молекулярные сита (4,0 г). Реакционную смесь перемешивают при комнатной температуре в течение часа и затем добавляют TMSOTf (триметилсилил-О-трифлат) (0,95 мл; 5,2 ммоль) при -78oС. Реакционную смесь затем оставляют постепенно нагреваться до комнатной температуры. Через 2 часа реакционную смесь нейтрализуют триэтиламином и фильтруют через целит; фильтрат промывают водой. Органическую фазу высушивают над сульфатом магния и концентрируют, остаток очищают хроматографией на колонке с силйкагелем (4/1 о/о этилацетат/циклогексан) с получением желаемого соединения 8 (2,77 г; 85%) в форме твердого вещества. Точка плавления = 47oС. [ ]D-36(c 0,6; CHCl3).Аналитически вычислено: С, 65,14; Н, 6,61. Найдено: С, 65,09; Н, 6,70. Получение 5 Метил-2,3,6-O-три-O-бензил-4-O-(4,6-O-изопропилиден-3-O-метил-5-С-винил- -D-глюкопиранозил)--D-глюкопиранозид (10)Соединение 8 (2,7 г; 3,4 ммоль) растворяют в метаноле (40 мл). Натрий (каталитический) добавляют при 0oС и смесь перемешивают в течение 3-х часов при комнатной температуре. Растворитель концентрируют, а остаток 9 растворяют в безводном ацетоне (40 мл) и добавляют 2,2-диметоксипропан (2 мл) и п-толуолсульфоновую кислоту (каталитическую). Реакционную смесь перемешивают в течение ночи при комнатной температуре. Растворитель выпаривают, а остаток растворяют в хлороформе и промывают водой. Органическую фазу высушивают над сульфатом магния и концентрируют, остаток очищают хроматографией на колонке с диоксидом кремния (1/1 o/o этилацетат/циклогексан) с получением 4′,6′-изопропилиден-O-производного 10 (1,7 г; 70%) в форме твердого вещества. Точка плавления = 55oС. [ ]D-13(c 0,8; CHCl3).Аналитически вычислено: С, 67,97; Н, 7,13. Найдено: С, 67,87; Н, 7,16. ХИ-МС: 707 (М + 1), 724 (М + NH4). Получение 6 Метил-2,3,6-три-O-бензил-4-O-(4,6-O-изопропилиден-3-O-метил-5-С-винил- -D-маннопиранозил)--D-глюкопиранозид (12)Оксалилхлорид (0,35 мл; 4,0 ммоль) и безводный DMSO (диметилсульфоксид) (0,57 мл; 8,0 ммоль) перемешивают в безводном дихлорметане (10 мл) при -78oС в течение 30 минут. Соединение 10 (1,4 г; 2,0 ммоль) в безводном дихлорметане (10 мл) добавляют к полученному раствору и продолжают перемешивать в течение следующих 45 минут. Реакционную смесь нейтрализуют добавлением безводного триэтиламина (1,7 мл; 12,0 ммоль) и затем разбавляют дихлорметаном. После промывания водой органическую фазу высушивают над сульфатом магния и концентрируют, а остаток 11 используют непосредственно для следующей реакции без очистки. Кетон 11 растворяют в безводном тетрагидрофуране (15 мл) и добавляют 1 N раствор супергидрида в тетрагидрофуране (4 мл; 4,0 ммоль) при -78oС. Реакционную смесь перемешивают в течение часа при комнатной температуре и затем добавляют 5%-ный гидроксид натрия (2 мл) и перекись водорода (1 мл). Растворитель выпаривают, а остаток растворяют в этилацетате и промывают водой. Органическую фазу высушивают над сульфатом магния и концентрируют, а остаток очищают хроматографией (2/1 о/о этилацетат/циклогексан) с получением соединения 12 (1,0 г; 70%). [ ]D-11(c 0,5; CHCl3).ХИ-МС: 724 (М + 18), 707 (М + 1). Получение 7 Метил-2,3,6-три-O-бензил-4-O-(2-O-ацетил-3-O-метил-5-С-винил -D-маннопиранозил)--D-глюкопиранозид (14)Соединение 12 (940 мг; 1,3 ммоль) растворяют в пиридине (3 мл) и добавляют уксусный ангидрид (0,3 мл). Реакционную смесь перемешивают в течение 3-х часов при комнатной температуре. Излишек пиридина и ангидрида уксусной кислоты концентрируют, а остаток 13 используют непосредственно для снятия защиты с изопропилидена, используя 80%-ную уксусную кислоту (5 мл) при 60oС в течение 2 часов. Излишек уксусной кислоты выпаривают, а остаток очищают хроматографией на колонке с силикагелем (4/1 о/о этилацетат/циклогексан) с получением диола 14 (660 мг, 70%) в форме твердого вещества. Точка плавления = 53oС. [ ]D-10(c 0,8; CHCl3).ХИ-МС: 709 (М + 1), 726 (М + 18). Получение 8 Метил-2,3,6-три-O-бензил-4-O-(2-O-ацетил-3-O-метил-6-O-тозил-5-С-винил- –D-маннопиранозил)--D-глюкопираноза (15)Соединение 14 (600 мг; 0,9 ммоль) растворяют в пиридине (3 мл) и добавляют тозилхлорид (240 мг; 1,3 ммоль). Реакционную смесь перемешивают в течение 3 часов при комнатной температуре. Растворитель выпаривают, а остаток разводят хлороформом и промывают водой. Органическую фазу высушивают над сульфатом магния и концентрируют, а остаток очищают хроматографией на колонке с силикагелем (1/1 о/о этилацетат/циклогексан) с получением соединения тозила 15 (297 мг, 80%) в форме сиропа. [ ]D-26(c 0,8; CHCl3).Получение 9 Метил-2,3,6-три-O-бензил-4-O-(2,6-ангидро-3-O-метил-5-С-винил- -D-маннопиранозил)--D-глюкопиранозид (16)Соединение 15 (550 мг; 0,6 ммоль) растворяют в этаноле (3 мл) и затем добавляют 0,1 N этанольный раствор гидроксида натрия (5 мл). Реакционную смесь нагревают при температуре 70oС в течение 3 часов и затем нейтрализуют смолой IR-120 (форма Н+) и фильтруют через целит. После концентрации остаток очищают хроматографией на колонке с силикагелем (1/1 о/о этилацетат/циклогексан) с получением соединения 16 (292 мг, 70%) в форме сиропа. [ ]D-13(c 0,5; CHCl3).ХИ-МС: 666 (М+18). Получение 10 Метил-2,3,6-три-O-бензил-4-O-(бензил-3-O-метил-2-O-5-С-метилиден- -L-идопирануронат)--D-глюкопиранозид (17)Соединение 16 (260 мг; 0,4 ммоль) растворяют в дихлорметане (20 мл), раствор перемешивают при -78oС и затем барботируют через него озон в течение 30 секунд. Раствор становится бледно-желтого цвета. В раствор добавляют диметилсульфид, и реакционную смесь затем промывают водой. Органическую фазу высушивают над сульфатом магния и концентрируют, а последующую реакцию проводят непосредственно без дальнейшей очистки. Неочищенный альдегид разводят в mpem-бутаноле (16 мл) и добавляют 2-метил-2-бутен (5 мл) и воду (16 мл). Затем к смеси последовательно добавляют NaH2PО4 (700 мг) и NaClО2 (700 мг). Суспензию энергично перемешивают при комнатной температуре в течение ночи, разбавляют водой и экстрагируют этилацетатом. Органическую фазу высушивают над сульфатом магния и концентрируют, и непосредственно проводят следующую реакцию. Неочищенную кислоту растворяют в диметилформамиде (25 мл) и иодиде тетрабутиламмония (0,7 г; 2,0 ммоль) и добавляют бикарбонат калия (0,25 г; 2,5 ммоль) и бензилбромид (0,250 мл; 2,1 ммоль). Реакционную смесь перемешивают в течение 5 часов при комнатной температуре. Реакционную смесь экстрагируют водой и простым диэтиловым эфиром. Эфирную фазу высушивают над сульфатом магния и концентрируют, а остаток очищают хроматографией на колонке с силикагелем (2/1 о/о этилацетат/циклогексан) с получением производного 17 (236 мг, 80%) в форме сиропа. ХИ-МС:774(М+18). Получение 11 Метил-O-(6-O-ацетил-2,3,4-три-O-метил- -D-глюкопиранозил)-(1–>4)-O-(бензил-2,3-ди-O-метил--D-глюкопиранозилуронат)-(1–>4)-O-(3,6-ди-O-ацетил-2-O-бензил--D-глюкопиранозил)-(1–>4)-O-(2,6-ангидро-5-С-бензилоксикарбонил-3-O-метил--D-маннопиранозил)-(1–>4)-2,3,6-три-O-бензил--D-глюкопиранозид (19)Имидат 18 (Van der Heijden et al., abstr. 9th Eur. Carbohydr. Symp. Utrecht, July 6-11, 1997; A74, p. 154) (81 мг; 78,2 мкмоль), акцептор 17 (65 мг; 86,0 мкмоль) и молекулярные сита с мелкими отверстиями (70 мг, 4  ) перемешивают в смеси 1/2 (о/о) дихлорметан / диэтиловый эфир (2,4 мл) в инертной атмосфере.
Реакционную смесь перемешивают в течение 30 минут и затем добавляют МаНСО3 до точки нейтрализации. После фильтрации и концентрации остаток очищают на колонке с гелем Sephadex LH 20 (1/1 о/о дихлорметан/этанол), а затем на колонке с силикагелем (1/1 (о/о) этилацетат/циклогексан) с получением соединения 19 (86 мг, 67%). ) перемешивают в смеси 1/2 (о/о) дихлорметан / диэтиловый эфир (2,4 мл) в инертной атмосфере.
Реакционную смесь перемешивают в течение 30 минут и затем добавляют МаНСО3 до точки нейтрализации. После фильтрации и концентрации остаток очищают на колонке с гелем Sephadex LH 20 (1/1 о/о дихлорметан/этанол), а затем на колонке с силикагелем (1/1 (о/о) этилацетат/циклогексан) с получением соединения 19 (86 мг, 67%).
 1H ЯМР представлен в табл.1. Получение 12 Метил-O-(2,3,4-три-O-метил- -D-глюкопиранозил)-(1–>4)-O-(2,3-ди-O-метил--D-глюкопиранозилуроноваякислота)-(1–>4)-O-(--D-глюкопиранозил)-(1–>4)-O-(2,6-ангидро-5-С-карбокси-3-O-метил--D-маннопиранозил)-(1–>4)--D-глюкопиранозид (20)По Петитоу и Ван Боэкелю (М.Petitou and C.A.A. van Boeckel, Progress in the Chemistry of Organic Natural Products, published by W.Herz et al. Vienna, Springer-Verlag, New-York, 1992, 143-210). Раствор соединения 19 (49 мг; 30,0 мкмоль) в уксусной кислоте (3 мл) перемешивают под водородом (3,5 МПа) в течение 12 часов при 40oС в присутствии 5%-ного палладия на угле (73 мг, 35 бар). Смесь фильтруют через целит, концентрируют и перегоняют совместно с водой (4х5 мл). Остаток растворяют в водном 1 М растворе NaOH (3 мл) и нагревают при 55oС в течение 3 часов. Раствор охлаждают и пропускают через колонку Sephadex 625 F (170 мл) и элюируют водой. Фракции, содержащие соединение 20, пропускают через колонку со смолой Доуэкс Н+. Элюат концентрируют, чтобы получить соединение 20 (25 мг, 86%).
[]D+105(c 1,0; H2).1H ЯМР представлен в таблице 1. Получение 13 Метил-O-(4,6-O-изопропилиден-2,3-ди-O-метил-5-С-винил- -D-глюкопиранозил)-(1–>4)-2,3,6-три-O-бензил--D-глюкопиранозид (22)Гидрид натрия (0,31 г; 13,0 ммоль) добавляют при температуре 0oС к раствору метил-O-(4,6-O-изопропилиден-3-O-метил-5-С-винил- -D глюкопиранозил)-(1–>4)-2,3,6-три-O-бензил--D-глюкопиранозида (10) (6,11 г; 8,65 ммоль) и метилиодида (0,80 мл; 13,0 ммоль) в N,N-диметилформамиде (9,00 мл). Смесь перемешивают в течение 2 часов (ТСХ), добавляют метанол и выливают реакционную смесь в воду. Смесь экстрагируют этилацетатом, а экстракты промывают водой, высушивают и концентрируют. Остаток очищают на колонке с диоксидом кремния (3/2 (о/о) циклогексан/простой диэтиловый эфир), чтобы получить соединение 22 (5,92 г; 88%). ТСХ: Rf = 0,28, (3,2 (о/о) циклогексан / простой диэтиловый эфир.
Получение 14Метил-O-(4,6-O-изопропилиден-2,3-ди-O-метил-5-С-этил- -D-глюкопиранозил)-(1–>4)-2,3,6-три-O-бензил--D-глюкопиранозид (23)Оксид платины (160 мг) добавляют к раствору 22 (5,80 г; 8,04 ммоль) в этилацетате (400 мл). Вводят водород. Смесь перемешивают в течение 40 минут (ТСХ), фильтруют и выпаривают, чтобы получить соединение 23. ТСХ: Rf = 0,60 (4/1 (о/о) толуол/этилацетат). Получение 15 Метил-O-(2,3-ди-O-метил-5-С-этил- -D-глюкопиранозил)-(1–>4)-2,3,6-три-O-бензил--D-глюкопиранозид (24)Неочищенное соединение 23 растворяют в 70%-ной уксусной кислоте (60 мл) и перемешивают при 80oС в течение 2 часов. Смесь концентрируют под вакуумом и выпаривают совместно с толуолом, чтобы получить соединение 24. ТСХ: Rf = 0,52 (4/1 (о/о) дихлорметан/метанол). Получение 16 Метил-O-(бензил-2,3-ди-O-метил-5-С-этил- -D-глюкопиранозилуронат)-(1–>4)-2,3,6-три-O-бензил--D-глюкопиранозид (25)2,2,6,6-тетраметил-1-пиперидилокси (17,5 мг), раствор гидрокарбоната натрия (17,5 мл), бромид калия (87 мг) и хлорид тетрабутиламмония (115,5 мг) добавляют к раствору соединения 24 (5, 75 г) в тетрагидрофуране (28 мл). Смесь охлаждают до 0oС и в течение 15 минут добавляют смесь насыщенного раствора хлорида натрия (17 мл), насыщенного раствора гидрокарбоната натрия (8,70 мл) и гипохлорита натрия (1,3 М; 20 мл). После перемешивания в течение 1 часа смесь разводят водой и экстрагируют (3 раза) дихлорметаном. Органическую фазу промывают водным раствором хлорида натрия, высушивают над сульфатом магния, фильтруют и выпаривают до сухого состояния, чтобы получить неочищенное производное кислоты. Производное кислоты растворяют в N,N-диметилформамиде (107 мл) в азотной атмосфере. Добавляют гидрокарбонат калия (4,10 г) и бензилбромид (9,70 мл) и перемешивают смесь в течение 16 часов. Добавляют этилацетат и воду, и после экстракции концентрируют органическую фазу. Очисткой с помощью хроматографии через колонку с силикагелем получают 3,81 г соединения 25 (60% выхода от соединения 23). [ ]D+24o(с=0,15; дихлорметан)Получение 17 Метил-O-(бензил-5-С-этил-4-O-левулинил-2,3-ди-O-метил- -D-глюкопиранозилуронат)-(1–>4)-2,3,6-три-O-бензил--D-глюкопиранозид (26)Соединение 25 (3,51 г; 4,46 ммоль) растворяют в безводном диоксане (45 мл). Добавляют левулиновую кислоту (1,00 г; 8,93 ммоль), гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (1,70 г; 8,93 ммоль) и 4-диметиламинопиридин (0,11 г; 8,93 ммоль). Смесь оставляют перемешиваться в течение 16 часов и экстрагируют этилацетатом. Экстракты промывают последовательно 5%-ным водным раствором гидросульфата калия, водой, насыщенным водным раствором гидрокарбоната натрия и водой, затем высушивают и концентрируют. Остаток очищают, пропуская через колонку с диоксидом кремния (2/1 и затем 3/2 (о/о) циклогексан/этилацетат), чтобы получить чистое соединение 26 (3,64 г; 85%). [ ]D+26o(c=0,9; дихлорметан)Получение 18 O-(Бензил-5-С-этил-4-O-левулинил-2,3-ди-O-метил- -D-глюкопиранозилуронат)-(1–>4)-1,3,6-три-O-ацетил-2-O-бензил-D-глюкопираноза (27)Соединение 26 (3,35 г; 3,78 ммоль) растворяют в ангидриде уксусной кислоты (22 мл). Раствор охлаждают до -20oС и добавляют 22 мл холодного раствора серной кислоты в ангидриде уксусной кислоты (1 мл серной кислоты в 10 мл ангидрида уксусной кислоты). Смесь разводят этилацетатом, промывают насыщенным водным раствором гидрокарбоната натрия, затем водой, высушивают и концентрируют. Остаток очищают, пропуская через колонку с диоксидом кремния (1/1 (о/о) циклогексан/этилацетат), чтобы получить соединение 27 (2,20 г; 65,5%). ТСХ: Rf = 0,24, (1/1 (о/о) циклогексан/этилацетат). Получение 19 O-(Бензил-5-С-этил-4-O-левулинил-2,3-ди-O-метил- -D-глюкопиранозилуронат)-(1–>4)-3,6-ди-O-ацетил-2-O-бензил-D-глюкопираноза (28)Бензиламин (11 мл; 101,4 ммоль) добавляют к раствору соединения 27 (2,18 г; 2,67 ммоль) в тетрагидрофуране (50 мл). Смесь оставляют перемешиваться в течение 4 часов. Смесь экстрагируют этилацетатом, а экстракты промывают водным раствором 1 М соляной кислоты (102 мл), затем водой, высушивают и концентрируют. Остаток очищают, пропуская через колонку с диоксидом кремния (1/1 (о/о) толуол/этилацетат), чтобы получить смесь ( / = 50/50) соединения 28 (1,33 г; 65%). ТСХ: Rf = 0,24, (1/1 (о/о) толуол/этилацетат).
Получение 20O-(Бензил-5-С-этил-4-O-левулинил-2,3-ди-O-метил- -D-глюкопиранозилуронат)-(1–>4)-3,6-ди-O-ацетил-2-O-бензил-D-глюкопиранозила трихлорацетимидат (29)Соединение 28 (1,32 г; 1,71 ммоль) растворяют в дихлорметане (34 мл) и добавляют под аргоном трихлороацетонитрил (0,87 мл; 8,50 ммоль) и карбонат цезия (0,90 г; 2,73 ммоль). Смесь оставляют перемешиваться в течение 2 часов (ТСХ) и фильтруют. Остаток очищают, пропуская через колонку с диоксидом кремния (3/2 (o/o) циклогексан/этилацетат), чтобы получить смесь ( / = 85/15) имидатов 29 (1,20 г; 77%). ТСХ: Rf = 0,36, (1/2 (о/о) циклогексан/этилацетат).
Получение 21Метил-O-(бензил-5-С-этил-4-O-левулинил-2,3-ди-O-метил- -D-глюкопиранозилуронат)-(1–>4)-O-(3,6-ди-O-ацетил-2-O-бензил--D-глюкопиранозил)-(1–>4)-O-(2,6-ангидро-5-С-бензилоксикарбонил-3-O-метил--D-маннопиранозил)-(1–>4)-2,3,6-три-O-бензил--D-глюкопиранозид (30)Раствор трифлата трет-бутилдиметилсилила в дихлорметане (1М; 0,19 мл) добавляют под аргоном при -20oС к раствору имидата 29 (1,19 г; 1,29 ммоль) и метил-2,3,6-три-O-бензил-4-(бензил-3-O-метил-2-O-5-С-метилиден- -L-идопирануронат)--D-глюкопиранозида 17 (1,02 г; 1,35 ммоль) в толуоле (40 мл)в присутствии молекулярных сит 4 А (1,93 г). Через 30 минут (ТСХ) снова добавляют раствор трифлата трет-бутилдиметилсилила в дихлорметане (1М; 0,19 мл). Через 30 минут (ТСХ) добавляют твердый гидрокарбонат натрия. Раствор фильтруют, промывают водой, высушивают и выпаривают до сухого состояния. Остаток очищают посредством колоночной хроматографии на Sephadex LH20, а затем на колонке с диоксидом кремния (2/1 (о/о) толуол/этилацетат), чтобы получить чистый тетрасахарид 30- (1,14 г; 58%).
[]D+47o (с = 0,21; дихлорметан).
Получение 22Метил-O-(бензил-5-С-этил-2,3-ди-O-метил- -D-глюкопиранозилуронат)-(1–>4)-O-(3,6-ди-O-ацетил-2-O-бензил--D-глюкопиранозил)-(1–>4)-O-(2,6-ангидро-5-С-бензилоксикарбонил-3-O-метил--D-маннопиранозил)-(1–>4)-2,3,6-три-O-бензил--D-глюкопиранозид (31)Соединение 30 (1,13 г; 0,75 ммоль) растворяют в смеси 2/1 (о/о) этанол / толуол (150 мл) и добавляют гидразинацетат (0,35 г; 3,73 ммоль). Смесь оставляют перемешиваться в течение 1 часа (ТСХ) и концентрируют. Остаток очищают на колонке с диоксидом кремния (3/2 (о/о) толуол/этилацетат), чтобы получить соединение 31 (0,816 г; 83%). [ ]D+35o (с = 1,01; дихлорметан).
Получение 23 Метил-O-(6-O-ацетил-2,3,4-три-O-метил--D-глюкопиранозил)-(1–>4)-O-(бензил-5-С-этил-2,3-ди-O-метил-D-глюкопиранозилуронат)-(1–>4)-O-(3,6-ди-O-ацетил-2-O-бензил--D-глюкопиранозил)-(1–>4)-O-(2,6-ангидро-5-С-бензилоксикарбонил-3-O-метил--D-маннопиранозил)-(1–>4)-2,3,6-три-O-бензил--D-глюкопиранозид (33)Трихлорацетимидат 6-O-ацетил-2,3,4-три-O-метил-O-глюкопиранозы 32 (34,7 мг; 0,0245 ммоль) (Р. Westerduin, et al. BioOrg. Med. Chem., 1994, 2, 1267) и акцептор гликозила 31 (80 мг; 0,056 ммоль) подвергают обработке, описанной в Получении 21. Соединение очищают на хроматографической колонке Sephadex LH20 (1/1 (о/о) дихлорметан/этанол), а затем на колонке с диоксидом кремния (3/2 (о/о) диизопропиловый эфир/этилацетат), чтобы получить производное 33 (54,6 мг; 58%).
[]D+55o (с = 1; дихлорметан).
Получение 24Метил-O-(6-O-ацетил-2,3,4-три-O-метил- -D-глюкопиранозил)-(1–>4)-O-(5-С-этил-2,3-ди-O-метил--D-глюкопиранозилуроновая кислота)-(1–>4)-O-(3,6-ди-O-ацетил--D-глюкопиранозил)-(1–>4)-O-(2,6-ангидро-5-С-карбокси-3-O-метил--D-маннопиранозил)-(1–>4)--D-глюкопиранозид (34)Раствор соединения 33 (40 мг; 0,024 ммоль) в уксусной кислоте (2 мл) перемешивают в атмосфере водорода в присутствии 10%-ного палладия на угле (80 мг) в течение 16 часов и фильтруют. Фильтрат концентрируют, чтобы получить соединение 34. Получение 25 Метил-O-(2,3,4-три-O-метил- -D-глюкопиранозил)-(1–>4)-O-(5-С-этил-2,3-ди-О-метил--D-глюкопиранозилуроновая кислота)-(1–>4)-O-(--D-глюкопиранозил)-(1–>4)-O-(2,6-ангидро-5-С-карбокси-3-O-метил--D-маннопиранозил)-(1–>4)--D-глюкопиранозид (35)Водный 5 М раствор гидроксида натрия (216 мкл) добавляют к раствору неочищенного соединения 34 в метаноле (866 мкл). Через 25 часов добавляют воду, и реакционную смесь пропускают через колонку с гелем Sephadex G-25 (2 х 3,8 см) и элюируют водой. Элюат концентрируют, пропускают через колонку Dowex 50 H+ (2 мл) и сушат вымораживанием. На этой стадии используют 1H ЯМР, чтобы проверить, все ли защитные группы были удалены.
Пример 1Метил-O-(2,3,4-три-O-метил-6-O-сульфо- -D-глюкопиранозил)-(1–>4)-O-(2,3-ди-О-метил--D-глюкопиранозилуроновая кислота)-(1–>4)-O-(2,3,4-три-O-сульфо--D-глюкопиранозил)-(1–>4)-O-(2,6-ангидро-5-С-карбокси-3-O-метил--D-маннопиранозил)-(1–>4)-2,3,6-три-O-сульфо--D-глюкопиранозида натриевая соль (21)По Ван Боэкелю и Петитоу (С.А.А. van Boeckel and M. Petitou, Angew. Chem. Int. Ed. Engl., 1993, 32,1671-1690). Раствор соединения 20 (20 мг; 20,7 мкмоль) и комплекса триэтиламин/триоксид серы (164 мг; 0,90 мкмоль) в диметилформамиде (2 мл) нагревают до 55oС, защищая от света, в течение 18 часов 30 минут. Реакционную смесь охлаждают до комнатной температуры и затем разбавляют водным 0,2 М раствором NaCl. Раствор затем наносят на вершину колонки Sephadex G25F (170 мл), элюируют водным 0,2 М раствором NaCl.
Фракции, содержащие пентасахариды, концентрируют и обессоливают, используя ту же колонку, элюируя водой. После сушки вымораживанием получают соединение 21 (30,5 мг; 85%).
[]D+49 (с = 0,63; Н2О).
1H ЯМР приведен в табл.2.
Пример 2Метил-O-(2,3,4-три-O-метил-6-О-сульфо- -D-глюкопиранозил)-(1–>4)-О-(5-C-этил-2,3-ди-O-метил--D-глюкопиранозилуроновая кислота)-(1–>4)-O-{ 2,3,6-три-O-сульфо--D-глюкопиранозил)-(1–>4)-O-(2,6-ангидро-5-С-карбокси-3-O-метил--D-маннопиранозил)-(1–>4)-2,3,6-три-O-сульфо--D-глюкопиранозид, натриевая соль (36)Комплекс триэтиламин/триоксид серы (91 мг) добавляют к раствору неочищенного соединения 35 в диметилформамиде (1,3 мл). Через 20 часов нагрева при 55oС, раствор наносят на вершину колонки Sephadex G-25 (2 х 38 см), элюируют 0,2 М хлоридом натрия. Фракции, содержащие продукт, концентрируют и обессоливают, используя ту же колонку, элюируя водой.
Соединение 36 получают после сушки вымораживанием (21,9 мг; 52% от соединения 33).
1H ЯМР приведен в табл.3.
Соединения 37-40 в Примерах 3-6, данных ниже, получают, следуя вышеприведенным Примерам 1 и 2. Спектр 1H ЯМР, полученный для этих соединений, согласуется с конфигурациями, указанными ниже.
Пример 3Метил-O-(2,3,4-три-O-метил-6-O-сульфо- -D-глюкопиранозил)-(1–>4)-O-(2,3-ди-О-метил--D-глюкопиранозилуроновая кислота)-(1–>4)-O-(2,3,6-три-O-сульфо--D-глюкопиранозил)-(1–>4)-O-(2,6-ангидро-5-С-карбокси-3-O-метил--D-маннопиранозил)-(1–>4)-2-O-сульфо-3,6-ди-O-метил--D-глюкопиранозид, натриевая соль (37)[ ]D+51 (с = 0,48; вода).
Пример 4Метил-O-(2,3,4-три-O-метил-6-O-сульфо- -D-глюкопиранозил)-(1–>4)-O-(2,3-ди-О-метил--D-глюкопиранозилуроновая кислота)-(1–>4)-O-(2,3,6-три-O-сульфо D-глюкопиранозил)-(1–>4)-O-(2,6-ангидро-5-С-карбокси-3-O-метил--D-маннопиранозил)-(1–>4)-2,3-ди-O-сульфо-6-O-метил--D-глюкопиранозид, натриевая соль (38) D-глюкопиранозил)-(1–>4)-O-(2,6-ангидро-5-С-карбокси-3-O-метил--D-маннопиранозил)-(1–>4)-2,3-ди-O-сульфо-6-O-метил--D-глюкопиранозид, натриевая соль (38)[ ]D+57 (с = 0,28; вода).
Пример 5Метил-O-(2,3,4-три-O-метил-6-O-сульфо- -D-глюкопиранозил)-(1–>4)-O-(2,3-ди-О-метил--D-глюкопиранозилуроновая кислота)-(1–>4)-O-(2,3,6-три-O-сульфо--D-глюкопиранозил)-(1–>4)-O-(2,6-ангидро-5-С-карбокси-3-O-метил--D-маннопиранозил)-(1–>4)-2,6-ди-O-сульфо-3-O-метил--D-глюкопиранозид, натриевая соль (39)[ ]D+53 (с = 0,3; вода).
Пример 6Метил-O-(2,3,4-три-O-метил-6-O-сульфо- -D-глюкопиранозил)-(1–>4)-O-(2,3-ди-О-метил--D-глюкопиранозилуроновая кислота)-(1–>4)-O-(2,3,6-три-O-сульфо--D-глюкопиранозил)-(1–>4)-O-(2,7-ангидро-5-С-карбокси-6-дезокси-3-O-метил--D-манногептопиранозил)-(1–>4)-2,3,6-три-O-сульфо--D-глюкопиранозид, натриевая соль (40)[ ]D+49 (с = 0,25; вода).
Примером фармацевтической композиции по изобретению является раствор для инъекций следующего состава:соединение из примера 1: 2,5 мг NaOH или HCl официнальные, сколько требуется до рН 6-7,5 вода для инъекций, сколько требуется до 0,5 мл Соединение по изобретению растворяют в воде при перемешивании. Затем доводят рН полученного раствора до указанного значения с помощью HCl или NaOH. Раствор стерилизуют фильтрацией через стерильные фильтры на 0,22 мкм и заполняют им стерильные флаконы для инъекций. Формула изобретения
где R представляет собой водород или -SО3 –, (С1-С3)алкильную или (С2-С3)ацильную группу; Т представляет собой водород или этильную группу; n представляет собой 1 или 2. 2. Пентасахарид по п. 1 в форме натриевой соли или калиевой соли. 3. Пентасахарид по любому из пп. 1 и 2, выбранный из метил-O-(2,3,4-три-O-метил-6-O-сульфо- -D-глюкопиранозил)-(1–>4)-O-(2,3-ди-O-метил--D-глюкопиранозилуроновая кислота)-(1–>4)-O-(2,3,4-три-O-сульфо--D-глюкопиранозил)-(1–>4)-O-(2,6-ангидро-5-С-карбокси-3-O-метил--D-маннопиранозил)-(1–>4)-(2,3,6-три-O-сульфо--D-глюкопиранозида натриевой соли;метил-O-(2,3,4-три-O-метил-6-O-сульфо- -D-глюкопиранозил)-(1–>4)-O-(5-С-этил-2,3-ди-O-метил--D-глюкопиранозилуроновая кислота)-(1–>4)-O-(2,3,6-три-O-сульфо--D-глюкопиранозил)-(1–>4)-O-(2,6-ангидро-5-С-карбокси-3-O-метил--D-маннопиранозил)-(1–>4)-2,3,6-три-O-сульфо--D-глюкопиранозида натриевой соли;метил-O-(2,3,4-три-O-метил-6-O-сульфо- -D-глюкопиранозил)-(1–>4)-O-(2,3-ди-O-метил--D-глюкопиранозилуроновая кислота)-(1–>4)-O-(2,3,6-три-O-сульфо--D-глюкопиранозил)-(1–>4)-O-(2,6-ангидро-5-С-карбокси-3-O-метил--D-маннопиранозил)-(1–>4)-2-O-сульфо-3,6-ди-O-метил--D-глюкопиранозида натриевой соли;метил-O-(2,3,4-три-O-метил-6-O-сульфо- -D-глюкопиранозил)-(1–>4)-O-(2,3-ди-O-метил--D-глюкопиранозилуроновая кислота)-(1–>4)-O-(2,3,6-три-O-сульфо--D-глюкопиранозил)-(1–>4)-O-(2,6-ангидро-5-С-карбокси-3-O-метил--D-маннопиранозил)-(1–>4)-2,3-ди-O-сульфо-6-O-метил--D-глюкопиранозида натриевой соли;метил-O-(2,3,4-три-O-метил-6-O-сульфо- -D-глюкопиранозил)-(1–>4)-O-(2,3-ди-O-метил--D-глюкопиранозилуроновая кислота)-(1–>4)-O-(2,3,6-три-O-сульфо--D-глюкопиранозил)-(1–>4)-O-(2,6-ангидро-5-С-карбокси-3-O-метил--D-маннопиранозил)-(1–>4)-2,6-ди-O-сульфо-3-О-метил--D-глюкопиранозида натриевой соли;метил-O-(2,3,4-три-O-метил-6-O-сульфо- -D-глюкопиранозил)-(1–>4)-O-(2,3-ди-O-метил--D-глюкопиранозилуроновая кислота)-(1–>4)-O-(2,3,6-три-O-сульфо--D-глюкопиранозил)-(1–>4)-O-(2,7-ангидро-5-С-карбокси-6-дезокси-3-O-метил--D-манногептопиранозил)-(1–>4)-2,3,6-три-O-сульфо--D-глюкопиранозида натриевой соли.
4. Фармацевтическая композиция, обладающая антитромботической активностью, содержащая в качестве активного начала пентасахарид по любому из пп. 1-3 в форме соли с фармацевтически приемлемым основанием или в кислой форме, объединенный или смешанный с фармацевтически приемлемым нетоксичным инертным эксципиентом.
5. Фармацевтическая композиция по п. 4 в форме единиц дозирования, где активное начало смешано с по меньшей мере одним фармацевтическим эксципиентом.
6. Композиция по п. 5, где каждая единица дозирования содержит от 0,1 до 100 мг активного начала.
7. Композиция по п. 6, где каждая единица дозирования содержит от 0,5 до 50 мг активного начала.
8. Полисахарид по пп. 1-3 для приготовления лекарства, которое полезно при патологиях, связанных с дисфункцией свертывания крови.
РИСУНКИ
PC4A – Регистрация договора об уступке патента Российской Федерации на изобретение
Прежний патентообладатель:
(73) Патентообладатель:
(73) Патентообладатель:
Дата и номер государственной регистрации перехода исключительного права: 14.09.2004 № 20078
Извещение опубликовано: 27.10.2004 БИ: 30/2004
PD4A – Изменение наименования обладателя патента Российской Федерации на изобретение
(73) Новое наименование патентообладателя:
Извещение опубликовано: 7.01.2006 БИ: 03/2006
|
||||||||||||||||||||||||||