Патент на изобретение №2192413
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ 1-(2-ХЛОРЭТИЛ)-3-ЦИКЛОГЕКСИЛ-1-НИТРОЗОМОЧЕВИНЫ
(57) Реферат: Изобретение касается способа получения 1-(2-хлорэтил)-3-циклогексил-1-нитрозомочевины, известной в фармацевтике, как противоопухолевый препарат ломустин. По предлагаемому способу ломустин получают в несколько стадий: взаимодействие циклогексилизоцианата с моноэтаноламином в среде ацетонитрила с направлением продукта реакции непосредственно на следующую стадию обработки продукта реакции хлористым тионилом в среде толуола в присутствии катализатора – хлористого цинка; взаимодействие полученного хлорэтильного производного с нитритом натрия в 75-85%-ной водной муравьиной кислоте, с последующей обработкой образующейся смеси изомеров соляной кислотой и кристаллизация целевого продукта из смеси ацетон – ацетонитрил. Способ позволяет увеличить выход целевого продукта фармакопейного качества до 51,7% и упростить процесс. Настоящее изобретение относится к органическому синтезу, в частности, оно касается способа получения 1-(2-хлорэтил)-3-циклогексил-1-нитрозомочевины, известной в фармацевтике как противоопухолевый препарат ломустин. Наиболее близким к способу, предлагаемому в настоящем изобретении, по технологической сущности является известный 3-стадийный способ [J. Org. Chem. , т. 46. 12, 2479-2489, 1981 г.], заключающийся в том, что на первой стадии циклогексилизоцианат (I) обрабатывают моноэтаноламином (II) в среде серного эфира в течение 12 часов, затем полученную 1-(2-оксиэтил)-3-циклогексилмочевину (III) 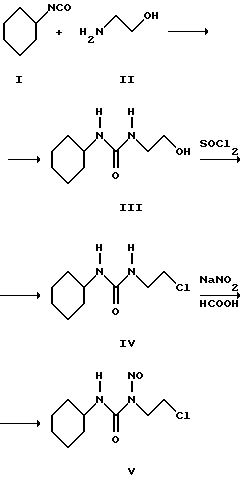 перекристаллизовывают и кипятят в среде хлористого тионила с образованием хлорэтильного производного (IV), которое подвергают нитрозированию сухим нитритом натрия в среде практически безводной (98-99%) муравьиной кислоты. Выход целевого продукта по описанному способу составляет 26,7%. Недостатками известного способа являются: I. На 1-й стадии: 1. Использование взрывоопасного растворителя – серного эфира, что совершенно неприемлемо для производственных условий. 2. Большая продолжительность процесса – 12 часов. 3. Необходимость очистки продукта реакции III, что усложняет технологию, и приводит к увеличению расходных норм сырья. II. На 2-й cтaдии: 1. Кипячение в среде хлористого тионила, приводящее к осмолению, чем обусловлен низкий выход продукта реакции IV – 41%. 2. Сильно загрязненный продукт IV требует сложной очистки (колонка с силикагелем с последующей кристаллизацией). III. На 3-й стадии: 1. Нитрозирование проводят в практически безводной (98-99%-ной) муравьиной кислоте, что обеспечить в производственных условиях весьма затруднительно. 2. Образование изомера целевого продукта – 1-(2-хлорэтил)-3-циклогексил-3-нитрозомочевины, превращение которого в целевой продукт не предусмотрено. IV. На 4-й стадии: 1. Кристаллизацию целевого продукта осуществляют из взрывоопасных растворителей – смеси серного и петролейного эфиров, что крайне нежелательно для производственных условий. Все вышеприведенные недостатки делают описанный процесс технологически сложным и обуславливают довольно низкий выход целевого продукта – 26,7%. Задачей предлагаемого изобретения является разработка такой технологии получения 1-(2-хлорэтил)-3-циклогексил-1-нитрозомочевины по описанной схеме, которая бы обеспечивала увеличение выхода целевого продукта при одновременном упрощении процесса. Поставленная задача решается тем, что в описанном способе получения соединения V по 3-стадийной схеме первую стадию проводят в среде ацетонитрила; продукт III непосредственно направляют на 2 стадию, которую осуществляют в среде толуола в присутствии хлористого цинка, а процесс нитрозирования сухим нитритом натрия проводят в 75-85%-ной водной муравьиной кислоте; образующуюся смесь изомеров обрабатывают концентрированной НС1; кристаллизацию технического продукта V проводят из смеси растворителей – ацетон – ацетонитрил. Выход целевого продукта V по описанной технологии составляет 51,7%, качество его соответствует требованиям к фармакопейному продукту. Нижеследующий пример характеризует предлагаемое изобретение. Пример. Стадия 1. Получение 1-(2-оксиэтил)-3-циклогексилмочевины. К охлаждаемой до 0-5oС смеси 31,16 г 98% (30,54 г 100%; 0,5 Мол) моноэтаноламина в 750 мл ацетонитрила при перемешивании прикапывают 64,5 мл (63,21 г – 99% или 62,59 г 100%; 0,5 Мол) циклогексилизоцианата. По окончании прибавления реакционную смесь выдерживают 4 часа, выпавший осадок отфильтровывают, промывают 50 мл ацетонитрила и сушат. Получают 88,7 г (95%) 1-(2-оксиэтил)-3-циклогексилмочевины в виде кристаллов белого цвета с т. пл. 99-102oС. Стадия 2. Получение 1-(2-хлорэтил)-3 -циклогексилмочевины. К перемешиваемой смеси 84 г (0,45 Мол) 1-(2-оксиэтил)-3-циклогексилмочевины, 9,0 г 98% (8,82 г 100%) ZnCl2; и 1350 мл толуола при температуре 20oС прикапывают 50,3 мл (0,675 Мол) хлористого тионила. Реакционную массу нагревают до 80oС и выдерживают при этой температуре 1 час. Затем в вакууме отгоняют смесь хлористого тионила и толуола до прекращения погона. К остатку добавляют 300 мл воды и перемешивают 1 час. Выделившийся осадок отфильтровывают, промывают водой и сушат. Получают 73,79 г (80%) 1-(2-хлорэтил)-3-циклогексилмочевины в виде кристаллов белого цвета с т. пл. 116-118oС. Стадия 3. Получение 1-(2-хлорэтил)-3-циклогексил-1-нитрозомочевины. К перемешиваемой смеси 73,44 г 1-(2-хлорэтил)-3-циклогексилмочевины и 1075 мл 85%-ной водной муравьиной кислоты порциями добавляют 73,44 г сухого нитрита натрия, поддерживая температуру не выше 5oС. Реакционную массу, представляющую собой смесь изомеров: 1-(2-хлорэтил)-3-циклогексил-1-нитрозомочевины и 1-(2-хлорэтил)-3-циклогексил-3-нитрозомочевины, выдерживают при этой температуре 2 часа, добавляют 537 мл концентрированной НСl, выдерживают 1 час и разбавляют 1075 мл воды. Осадок отфильтровывают, промывают водой и сушат. Получают 93,5 г (85%) технического ломустина с т. пл. 89-90,5oС. Кристаллизацией из смеси ацетон – ацетонитрил (1:2) с выходом 80% получают фармакопейный продукт (температура плавления – 90,0-91,0oС, хлориды – менее 0,05%, содержание основного вещества – 98,85%, что соответствует требованиям, предьявляемым к субстанции “Ломустин” Британской Фармакопеей 2000 г.). Таким образом, предлагаемый способ позволяет: 1. На 1-й стадии: исключить использование взрывоопасного растворителя, значительно сократить продолжительность процесса с 12 до 4 часов, исключить очистку продукта реакции III. 2. На 2-й стадии: исключить кипячение в среде хлористого тионила, повысить выход продукта IV с 41% до 80%, исключить необходимость сложной очистки. 3. На 3-й стадии: сделать стадию более технологичной за счет проведения процесса в 75-85%-ной водной муравьиной кислоте, исключить 2-й изомер – 1-(2-хлорэтил)-3-циклогексил-3-нитрозомочевину – за счет применения концентрированной НСl. 4. Исключить на стадии кристаллизации целевого продукта применение взрывоопасной смеси растворителей. Все вышеприведенные усовершенствования позволяют повысить в два раза выход целевого продукта при значительном упрощении технологии. Формула изобретения
MM4A Досрочное прекращение действия патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 13.03.2005
Извещение опубликовано: 7.01.2006 БИ: 03/2006
|
||||||||||||||||||||||||||