Патент на изобретение №2186073
|
||||||||||||||||||||||||||
(54) КОМПЛЕКСЫ МЕТАЛЛОВ, СОДЕРЖАЩИЕ ЛИГАНДЫ 3-АРИЛЗАМЕЩЕННОГО ИНДЕНИЛА, КАТАЛИЗАТОР ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ И СПОСОБ ПОЛИМЕРИЗАЦИИ
(57) Реферат: Изобретение относится к комплексам металлов формулы (I), где М – титан, цирконий или гафний в формальной степени окисления +2,+3 или +4; R’ – фенил, бифенил или нафтил; R* – водород или гидрокарбил; Х – галоген или метил, к катализаторам полимеризации олефинов, содержащих эти лиганды, и способу полимеризации С2-С100000–  -олефинов, особенно этилена и стирола, с использованием этих катализаторов. Технический результат – новый катализатор полимеризации олефинов, использование которого приводит к высокоэффективному образованию высокомолекулярных полиолефинов в широком интервале условий полимеризации. 3 с. и 4 з.п.ф-лы, 1 табл. -олефинов, особенно этилена и стирола, с использованием этих катализаторов. Технический результат – новый катализатор полимеризации олефинов, использование которого приводит к высокоэффективному образованию высокомолекулярных полиолефинов в широком интервале условий полимеризации. 3 с. и 4 з.п.ф-лы, 1 табл.
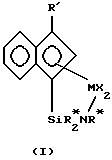
Изобретение относится к комплексам металлов 4 группы и катализаторам полимеризации, образованным из них, которые, в частности, пригодны для применения в процессе полимеризации для получения гомополимеров и сополимеров -олефинов, в особенности сополимеров, содержащих моновинилароматический мономер и этилен. Комплексы металлов со стесненной геометрией и способы их получения описываются в заявке на патент США серийный номер 545403, поданной 3 июля 1990 (ЕР-А-416815). В этой публикации также описывается получение некоторых новых сополимеров этилена и затрудненного винилового мономера, включая моновинилароматические мономеры, с псевдостатистическим включением в них затрудненного мономера. Описание катализаторов со стесненной геометрией можно найти также в заявке на патент США серийный номер 547718, поданной 3 июля 1990 (ЕР-А-468651); заявке на патент США серийный номер 702475, поданной 20 мая 1991 (ЕР-А-514828); заявке на патент США серийный номер 876268, поданной 1 мая 1992 (ЕР-А-520732) и в заявке на патент США серийный номер 8003, поданной 21 января 1993 (WO 93/19104), а также в US-A-5055438, US-A-5057475, US-A-5096867, US-A-5064802, US-A-5132380, US-A-5470993, WO 95-00526 и в предварительной заявке на патент США 60-005913. Комплексы металлов, содержащие по-разному замещенный инденил, описаны в заявке США, серийный номер 592756, поданной 26 января 1996, а также в WO 95/14024.
Соответственно, настоящее изобретение относится к комплексам металлов, соответствующим формуле (I)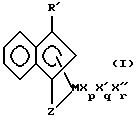 где М – титан, цирконий или гафний в состоянии формального окисления +2, +3 или +4; R’ – арильный лиганд или его галоген-, силил-, алкил-, циклоалкил-, дигидрокарбиламино-, гидрокарбилокси- или гидрокарбиленаминозамещенное производное, причем указанный R’ содержит от 6 до 40 неводородных атомов; Z является двухвалентной группой или группой, содержащей одну 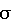 -связь и нейтральную электронную пару, способную образовать координационно-ковалентную связь с М, причем указанный Z содержит бор или член 14 группы Периодической таблицы элементов, а также содержащей азот, фосфор, серу или кислород; -связь и нейтральную электронную пару, способную образовать координационно-ковалентную связь с М, причем указанный Z содержит бор или член 14 группы Периодической таблицы элементов, а также содержащей азот, фосфор, серу или кислород;Х представляет одновалентную анионную лигандную группу, содержащую до 60 атомов, исключительно из класса лигандов, которые представляют циклические делокализованные  -связанные лигандные группы; -связанные лигандные группы;X’ представляет, каждый раз независимо, лигирующее соединение – нейтральное основание Льюиса, содержащее до 20 атомов; X” представляет двухвалентную анионную лигандную группу, содержащую до 60 атомов; р = 0, 1, 2 или 3; q = 0, 1 или 2; r = 0 или 1. Вышеописанные комплексы могут существовать в виде выделенных кристаллических веществ необязательно в чистой форме или в форме смеси с другими комплексами, в форме сольватированного аддукта необязательно в растворителе, особенно в органической жидкости, а также в форме димера или его хелатированного производного, где хелатообразователь представляет органическое вещество, такое как этилендиаминтетрауксусная кислота (ЭДТК). Также настоящее изобретение относится к катализатору для полимеризации олефинов, содержащему A) 1) комплекс металла формулы (I) и 2) активирующий сокатализатор, причем молярное соотношение 1) и 2) составляет от 1:10000 до 100:1, или B) продукт реакции, образовавшийся при превращении комплекса металла формулы (I) в активный катализатор с помощью методов активирования. Настоящее изобретение также относится к способу полимеризации олефинов, включающему контактирование в условиях полимеризации одного или нескольких С2-20– -олефинов с катализатором, содержащимA) 1) комплекс металла формулы (I) и 2) активирующий сокатализатор, причем молярное соотношение 1) и 2) составляет от 1:10000 до 100:1, или B) продукт реакции, образовавшийся при превращении комплекса металла формулы (I) в активный катализатор с помощью методов активирования. Наконец, настоящее изобретение относится к способу получения сополимеров моновинилароматического мономера и этилена, включающему приведение в контакт, в условиях полимеризации, смеси, содержащей один или несколько моновинилароматических мономеров и этилен, с катализатором, содержащим A) 1) комплекс металла формулы (I) и 2) активирующий сокатализатор, причем молярное соотношение 1) и 2) составляет от 1:10000 до 100:1 или B) продукт реакции, образовавшийся при превращении комплекса металла формулы (I) в активный катализатор с помощью методов активирования. Применение катализаторов и способов по настоящему изобретению приводит, в результате, к высокоэффективному образованию высокомолекулярных полиолефинов в широком интервале условий полимеризации, а особенно при повышенных температурах. Они особенно пригодны для образования сополимеров этилена и стирола (ЕS-полимеров) и сополимеров этилена со стиролом и диеном (ESDM-полимеров), где диен представляет этилиденнорборнен, 1,4-гексадиен или подобный неконъюгированный диен. Применение новых катализаторов полимеризации по настоящему изобретению позволяет включить большие количества сомономера, особенно – винилароматического сомономера, в сополимер на единицу катализатора из-за сочетания высокой каталитической эффективности с высоким содержанием сомономера. Катализаторы данного изобретения можно также наносить на материал-носитель и использовать в процессах полимеризации олефинов в суспензии или в газовой фазе. Катализатор можно предварительно полимеризовать с одним или несколькими олефиновыми мономерами in situ в реакторе для полимеризации или в отдельном процессе, с извлечением промежуточного соединения из форполимеризованного катализатора перед процессом первичной полимеризации. Подробное описание Все ссылки на Периодическую таблицу элементов здесь будут соотноситься с Периодической таблицей элементов, опубликованной и обеспеченной авторскими правами CRC Press, Inc., 1995. Также любая ссылка на группу или группы будет относиться к группе или группам, как они отражены в Периодической таблице элементов, с использованием системы ИЮПАК для нумерации групп. Олефины, используемые здесь, представляют C2-100000 алифатические или ароматические соединения, содержащие виниловую ненасыщенность, а также циклические соединения, такие как циклобутен, циклопентен и норборнен, в том числе норборнен, замещенный в положении 5 и 6 C1-20-углеводородными группами. Включаются также смеси таких олефинов, а также смеси таких олефинов с С4-40-диолефиновыми соединениями. Примерами последних соединений являются этилиденнорборнен, 1,4-гексадиен и норборнадиен. В процессе полимеризации могут образоваться длинноцепочечные мономеры с концевыми виниловыми группами, например, за счет явления  -гидридного исключения протона из растущей полимерной цепи. Этот процесс приводит в результате к включению в получающийся в результате полимер исключительно длинных цепей, т.е. к ответвлениям с длинными цепями. Катализаторы и способы, описанные здесь, особенно подходят для применения при получении сополимеров этилена и 1-бутена, этилена и 1-гексена, этилена и стирола и этилена и 1-октена, а также терполимеров этилена, пропилена и неконъюгированного диена, называемых EPDM-полимерами, этилена, стирола и неконъюгированного диена, называемых ESDM-полимерами, или этилена, пропилена и стирола, называемых EPS-полимерами. Наиболее предпочтительные описанные здесь катализаторы и способы применяют для получения сополимеров этилена и стирола.
Моновинилароматические мономеры для применения в настоящем изобретении представляют С8-20-арилзамещенные соединения этилена формулы -гидридного исключения протона из растущей полимерной цепи. Этот процесс приводит в результате к включению в получающийся в результате полимер исключительно длинных цепей, т.е. к ответвлениям с длинными цепями. Катализаторы и способы, описанные здесь, особенно подходят для применения при получении сополимеров этилена и 1-бутена, этилена и 1-гексена, этилена и стирола и этилена и 1-октена, а также терполимеров этилена, пропилена и неконъюгированного диена, называемых EPDM-полимерами, этилена, стирола и неконъюгированного диена, называемых ESDM-полимерами, или этилена, пропилена и стирола, называемых EPS-полимерами. Наиболее предпочтительные описанные здесь катализаторы и способы применяют для получения сополимеров этилена и стирола.
Моновинилароматические мономеры для применения в настоящем изобретении представляют С8-20-арилзамещенные соединения этилена формулы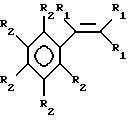 где каждый из R1 – независимо водород или C1-4-алкил, и каждый из R2 – независимо R1 или галоген. В комплексах металлов предпочтительными группами X’ являются монооксид углерода; фосфины, особенно триметилфосфин, триэтилфосфин, трифенилфосфин и бис(1, 2-диметилфосфино)этан; Р(ОR)3, где R представляет C1-20-гидрокарбил (углеводородный радикал); простые эфиры, в особенности тетрагидрофуран; амины, особенно пиридин, бипиридин, тетраметилэтилендиамин (TMEDA) и триэтиламин; олефины и конъюгированные диены, содержащие от 4 до 40 атомов углерода. К комплексам, содержащим диеновые Х’ -группы, относятся комплексы, в которых металл находится в формальной степени окисления +2. Что также касается комплексов металлов, это то, что Х выбирают предпочтительно из группы, состоящей из галогена, гидрокарбила и N,N-диалкиламинозамещенного гидрокарбила. Число групп Х зависит от степени окисления М, от того, является ли Х анионной, дианионной или нейтральной группой, является ли Z двухвалентным или нет и присутствуют ли какие-либо двухвалентные группы X”. Специалист в этой области техники поймет, что количество различных заместителей и особенности Z выбирают для обеспечения равновесия заряда, причем посредством этого получают в результате нейтральный комплекс металла. Например, когда Z двухвалентный, а г = 0, р на два меньше формальной степени окисления М. Когда Z содержит один нейтральный двухэлектронный центр координационно-ковалентного связывания и М находится в состоянии формального окисления +3, р может равняться нулю, а г = 1, или р может равняться 2, а г = 0. Наконец, например, если М имеет формальную степень окисления +2, Z может представлять двухвалентную лигандную группу, р и г могут оба равняться нулю и может присутствовать одна нейтральная лигандная группа. Предпочтительными координационными комплексами, применяемыми по настоящему изобретению, являются комплексы, соответствующие формуле (II) 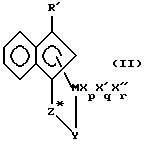 где R’ – фенил, бифенил или нафтил; М – титан; Y – -О-, -S-, -NR*-, -PR*-; -NR2* или -PR2*; Z* – SiR*2, CR*2, SiR*2SiR*2, CR*2CR*2, CR*=CR*, CR*2SiR*2 или GeR*2; каждый из R* – независимо водород или его выбирают среди гидрокарбила, гидрокарбилокси, силила, галогенированного алкила, галогенированного арила и их сочетаний, причем указанный R* содержит до 20 неводородных атомов и необязательно две R* -группы из Z (когда R* не является водородом) или R*-группa из Z и R*-группа из Y образуют циклическую систему; X, X’ и X” имеют установленные ранее значения; р = 0, 1 или 2; q = 0 или 1; г = 0 или 1; при условии, что когда р = 2, q и г = 0, М находится в состоянии формального окисления +4 (или М находится в состоянии формального окисления +3, если Y представляет -NR*2 или -PR*2), и Х представляет анионный лиганд, выбранный из группы, состоящей из галогенидной, гидрокарбильной, гидрокарбилокси-, ди(гидрокарбил)амидо-, ди(гидрокарбил)фосфидо-, гидрокарбилсульфидо- и силильной групп, а также их галоген-, ди(гидрокарбил)амино-, гидрокарбилокси- и ди(гидрокарбил )фосфинозамещенных производных, причем указанная группа Х содержит до 30 неводородных атомов, когда г = 1, р и q = 0, М находится в состоянии формального окисления +4, и X” представляет дианионный лиганд, выбранный из группы, состоящей из гидрокарбадиильных, оксигидрокарбильных и гидрокарбилендиоксигрупп, причем указанная группа Х содержит до 30 неводородных атомов, когда р = 1, q и г = 0, М находится в состоянии формального окисления +3 и Х представляет стабилизирующую анионную лигандную группу, выбранную из группы, состоящей из аллила, 2-(N,N-диметиламино)фенила, 2-(N,N-диметиламинометил)фенила и 2-(N,N-диметиламино)бензила, и когда р и г = 0, q = 1, М находится в состоянии формального окисления +2 и X’ представляет нейтральный диен с сопряженными или несопряженными двойными связями, необязательно замещенный одной или несколькими углеводородными группами, причем указанный X’ содержит до 40 атомов углерода и образует -комплекс с М.
Наиболее предпочтительными комплексами металлов являются комплексы, соответствующие приведенным выше формулам (II) или (III), где М, X, X’, X”, R’, Z*, Y, p, q и г имеют установленные ранее значения, при условии, чтокогда р = 2, q и г = 0, М находится в состоянии формального окисления +4, и каждый Х представляет независимо метил, бензил или галогенид; когда р и q = 0, г = 1 и М находится в состоянии формального окисления +4, X” представляет 1,4-бутадиенильную группу, которая образует с М металлоциклопентеновое кольцо, когда р = 1, q и г = 0, М находится в состоянии формального окисления +3 и Х представляет 2-(N,N-диметиламино)бензил и когда р и г = 0, q = 1 и М находится в состоянии формального окисления +2, X’ представляет 1,4-дифенил-1,3-бутадиен или 1,3-пентадиен. Особенно предпочтительным координационным комплексом является комплекс, соответствующий формуле 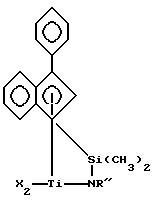 где R” представляет трет-бутил и Х представляет хлор, метил или бензил. Примерами комплексов металлов, которые можно использовать при практическом применении настоящего изобретения, являются 3-фенилинденидьные комплексы: (трет-бутиламидо)диметил(  5-3-фенилинденил)силантитан(II)-1,4-дифенил-1,3-бутадиен, 5-3-фенилинденил)силантитан(II)-1,4-дифенил-1,3-бутадиен,(трет-бутиламидо) диметил( 5-3-фенилинденил)силантитан(II)-1,3-пентадиен,(трет-бутиламидо)диметил( 5-3-фенилинденил)силантитан(III)-2- (N, N-диметиламино) бензил,(трет-бутиламидо)диметил( 5-3-фенилинденил)силантитан(IV)диметил,(трет-бутиламидо)диметил( 5-3-фенилинденил)силантитан(IV)дибензил,(н-бутиламидо)диметил( 5-3-фенилинденил)силантитан(II) -1,4-дифенил-1,3-бутадиен,(н-бутиламидо)диметил( 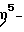 3-фeнилиндeнил)силантитан (II) -1,3-пентадиен, 3-фeнилиндeнил)силантитан (II) -1,3-пентадиен,(н-бутиламидо)диметил( 5-3-фeнилиндeнил)силантитан (III)-2-(N,N-диметиламино)бензил,(н-бутиламидо)диметил( 5-3-фенилинденил)силантитан(IV)диметил,(н-бутиламидо)диметил( 5-3-фенилинденил)силантитан(IV)дибензил,(циклододециламидо)диметил( 5-3-фенилинденил)силантитан(II)-1, 4-дифенил-1,3-бутадиен,(циклододециламидо)диметил( 5-3-фенилинденил) силантитан(II)-1,3-пентадиен,(циклододециламидо)диметил( 5-3-фенилинденил)силантитан(III)-2-(N,N-диметиламино)бензил,(циклододециламидо)диметил( 5-3-фeнилиндeнил)силантитан(IV)диметил,(циклододециламидо)диметил( 5-3-фенилинденил)силантитан(IV)дибензил,(2, 4, 6-триметиланилидо)диметил( 5-3-фенилинденил)силантитан(II)-1,4-дифенил-1,3-бутадиен,(2, 4, 6-триметиланилидо)диметил( 5-3-фенилинденил)силантитан(II)-1,3-пентадиен,(2,4, 6-триметиланилидо)диметил( 5-3-фeнилиндeнил)силантитан(III)-2-(N, N-диметиламино)бензил,(2, 4, 6-триметиланилидо)диметил( 5-3-фенилинденил)силантитан(IV)диметил,(2,4, 6-триметиланилидо)диметил( 5-3-фенилинденил)силантитан(IV)дибензил,(трет-бутиламидо)диметокси( 5-3-фенилинденил)силантитан(II)-1,4-дифенил-1,3-бутадиен,(трет-бутиламидо)диметокси( 5-3-фенилинденил)силантитан(II)-1,3-пентадиен,(трет-бутиламидо)диметокси( 5-3-фенилинденил)силантитан(III)-2-(N, N-диметиламино)бензил,(трет-бутиламидо)диметокси( 5-3-фенилинденил)силантитан(IV)диметил,(трет-бутиламидо)диметокси( 5-3-фенилинденил)силантитан(IV)дибензил;3-нафтилинденильные комплексы: (трет-бутиламидо)диметил( 5-3-нафтилинденил)силантитан(II)-1,4-дифенил-1,3-бутадиен,(трет-бутиламидо)диметил( 5-3-нaфтилиндeнил)силантитан(II)-1,3-пентадиен,(трет-бутиламидо)диметил( 5-3-нафтилинденил)силантитан(III)-2-(N,N-димeтилaминo)бензил,(трет-бутиламидо)диметил( 5-3-нaфтилиндeнил)силантитан(IV)-диметил,(трет-бутиламидо)диметил ( 5-3-нафтилинденил)силантитан(IV)-дибензил;3-бифенилинденильные комплексы: (трет-бутиламидо)диметил( 5-3-бифенилинденил)силантитан(II)-1,4-дифенил-1,3-бутадиен,(трет-бутиламидо)диметил( 5-3-бифeнилиндeнил)силантитан(II)-1,3-пентадиен,(трет-бутиламидо)диметил( 5-3-бифенилинденил)силантитан(III)-2-(N, N-диметиламино)бензил,(трет-бутиламидо)диметил( 5-3-бифeнилиндeнил)силантитан(IV)диметил и(трет-бутиламидо)диметил( 5-3-бифенилинденил)cилантитан(IV)дибензил.
Комплексы можно получить с применением хорошо известных методов синтеза. Можно необязательно использовать восстановитель, чтобы получить комплексы с меньшей степенью окисления. Такой способ описывается в заявке на патент США серийный номер 08/241523, поданной 13 мая 1994, опубликованной как WO 95-00526. Реакции проводят в подходящем, не влияющем на взаимодействие растворителе при температуре от -100 до 300oС, предпочтительно от -78 до 100oС, наиболее предпочтительно от 0 до 50oС. Термином “восстановитель” здесь обозначается металл или соединение металла, которое в условиях восстановления вызывает восстановление металла М от состояния с большей степенью окисления до состояния с меньшей степенью окисления. Примерами подходящих восстановителей-металлов являются щелочные металлы, щелочноземельные металлы, алюминий и цинк, сплавы щелочных металлов или щелочноземельных металлов, такие как амальгама натрия и сплава натрия и калия. Примерами подходящих соединений-восстановителей являются нафталинид натрия, калийграфит, литийалкилы, алкадиенилы лития или калия и реактивы Гриньяра. Наиболее предпочтительными восстановителями являются щелочные металлы или щелочноземельные металлы, в особенности металлы литий и магний.
Подходящей реакционной средой для образования комплексов являются алифатические и ароматические углеводороды, галоидоуглероды, галогенированные углеводороды, простые эфиры и простые циклические эфиры, в частности разветвленные углеводороды, такие как изобутан, бутан, пентан, гексан, гептан, октан и их смеси; циклические и алициклические углеводороды, такие как циклогексан, циклогептан, метилциклогексан, метилциклогептан, и их смеси; ароматические и гидрокарбилзамещенные ароматические соединения, такие как бензол, толуол и ксилол, простые C1-4-диалкилэфиры, C1-4-диалкилэфирные производные (поли)алкиленгликолей и тетрагидрофуран. Подходящими являются также смеси указанных выше растворителей.
Комплексы приводят в каталитически активное состояние посредством сочетания с активирующим сокатализатором или путем применения методов активирования. Подходящими активирующими катализаторами в данном случае являются полимерные или олигомерные алюмоксаны, в особенности метилалюмоксан, модифицированный триизобутилалюминием метилалюмоксан или изобутилалюмоксан; нейтральные кислоты Льюиса, такие как C1-30-гидрокарбилзамещенные соединения элементов 13 группы, в особенности соединения три(гидрокарбил)алюминия или три(гидрокарбил)бора, и их галогенированные (в том числе, пергалогенированные) производные, содержащие от 1 до 10 атомов углерода в каждой гидрокарбильной или галогенированной гидрокарбильной группе, в особенности перфторированные соединения три(арил)бора, и наиболее предпочтителен трис(пентафторфенил)боран; а подходящими методами активирования для применения в данном случае являются применение неполимерных совместимых, не образующих координационных связей ионообразующих соединений (включая применение таких соединений в условиях окисления), в особенности применение аммониевых, фосфониевых, оксониевых, карбониевых, силилиевых или сульфониевых солей с совместимыми, не образующими координационных связей анионами, или ферроцениевых солей с совместимыми, не образующими координационных связей анионами; объемный электролиз (подробнее поясняется ниже) и комбинации указанных выше активирующих сокатализаторов и методов активации. Указанные выше активирующие сокатализаторы и методы активирования описаны ранее для комплексов различных металлов в таких работах, как ЕР-А-277003, US-A-5153157, US-A-5064802, ЕР-А-468651, ЕР-А-520732 и ЕР-А-529732.
Особенно подходящими активирующими сокатализаторами являются комбинации нейтральных кислот Льюиса, в особенности сочетание триалкилалюминия с 1-4 атомами углерода в каждой алкильной группе и галогенированного три(гидрокарбил)бора с 1-20 атомами углерода в каждой углеводородной группе, в особенности трис(пентафторфенил)борана, также сочетания таких смесей нейтральных кислот Льюиса с полимерным или олигомерным алюмоксаном и сочетания одной нейтральной кислоты Льюиса, в особенности трис(пентафторфенил)борана, с полимерным или олигомерным алюмоксаном. Предпочтительные молярные соотношения комплекса металла 4 группы, трис(пентафторфенил)борана и алюмоксана составляют от 1:1:1 до 1:5:20, предпочтительнее от 1:1:1,5 до 1:5:10.
Подходящие ионообразующие соединения, полезные в качестве сокатализаторов, в одном из вариантов воплощения настоящего изобретения содержат катион, представляющий кислоту Бренстеда, способную отдавать протон, и совместимый, не образующий координационных связей анион А–. Используемый здесь термин “не образующий координационных связей” относится к аниону или веществу, которые либо не образуют координационной связи с предшественником комплекса, содержащего металл 4 группы, и полученным из него каталитическим производным, либо образуют только слабую координационную связь с такими комплексами, в результате чего остаются достаточно лабильными, чтобы замещаться нейтральным основанием Льюиса. Не образующий координационных связей анион относится, конкретно, к аниону, который, когда он функционирует как зарядово-уравновешивающий анион в катионсодержащем комплексе металла, не переносит анионный заместитель или его фрагмент к указанному катиону, посредством чего образуются нейтральные комплексы. “Совместимые анионы” представляют анионы, которые не разрушаются до нейтрального состояния, когда разлагается первоначально образовавшийся комплекс, и на них не влияет последующая требуемая полимеризация или иные процессы, в которых применяют комплекс.
Предпочтительными анионами являются анионы, содержащие один координационный комплекс, содержащий несущий заряд центральный атом металла или металлоида, и способные уравновесить заряд активной группы катализатора (металлокатион), который может образоваться при соединении двух компонентов. Кроме того, указанный катион должен быть достаточно лабильным, чтобы замещаться олефиновыми, диолефиновыми и ацетиленовыми ненасыщенными соединениями или другими нейтральными основаниями Льюиса, такими как простые эфиры или нитрилы. Подходящими металлами являются, но не ограничиваются перечисленным, алюминий, золото и платина. Подходящими металлоидами являются, но не ограничиваются перечисленным, бор, фосфор и кремний. Соединения, содержащие анионы, включающие координационные комплексы, содержащие один атом металла или металлоида, конечно, хорошо известны, и многие такие соединения, в частности, содержащие в анионной части один атом бора, доступны коммерчески.
Предпочтительно такие сокатализаторы можно отобразить общей формулой(L*-H)d +(A)d-, где L* – нейтральное основание Льюиса; (L*-H)+ представляет кислоту Бренстеда; Аd- – не образующий координационных связей совместимый анион с зарядом d– и d =1-3, целое число. Предпочтительнее Аd- соответствует формуле [M’Q4] , где М’ – бор или алюминий с формальной степенью окисления +3, и каждый Q выбирают независимо среди гидридных, диалкиламидных, галогенидных, углеводородных, гидрокарбилоксидных, галогензамещенных углеводородных, галогензамещенных гидрокарбилоксидных и галогензамещенных силилгидрокарбильных радикалов (включая пергалогенированные углеводородные, пергалогенированные гидрокарбилоксидные и пергалогенированные силилгидрокарбильные радикалы), причем указанный Q содержит до 20 атомов углерода, при условии, что не более чем один Q представляет галогенид. Примеры подходящих гидроксикарбилоксидных групп Q приводятся в патенте США 5296433. В более предпочтительном варианте воплощения изобретения d равен 1, т.е. , контрион имеет единичный отрицательный заряд и представляет А–. Активирующие сокатализаторы, содержащие бор, которые особенно полезны при получении катализаторов данного изобретения, можно представить общей формулой (L*-H)+(BQ4)–, где L* имеет установленные ранее значения; В – бор в состоянии формального окисления 3; Q представляет углеводородную, гидрокарбилокси-, фторированную углеводородную, фторированную гидрокарбилокси- или фторированную силилгидрокарбильную группу с числом неводородных атомов до 20 при условии, что не более чем в одном случае Q представляет углеводородный радикал. Предпочтительными солями оснований Льюиса являются аммониевые соли, предпочтительнее соли триалкиламмония, содержащие одну или несколько С12-40-алкильных групп. Наиболее предпочтительно, когда каждый из Q представляет фторированную арильную группу, особенно пентафторфенильную группу. Пояснительными, но не ограничивающими, примерами соединений бора, которые могут применяться в качестве активирующих сокатализаторов при получении улучшенных катализаторов данного изобретения, являются соли тризамещенного аммония, такие как триметиламмоний-тетракис(пентафторфенил)борат, триэтиламмоний-тетракис(пентафторфенил)борат, трипропиламмоний-тетракис(пентафторфенил)борат, три(н-бутил)аммоний-тетракис(пентафторфенил)борат, три(втор-бутил)аммоний-тетракис(пентафторфенил)борат, N,N-диметиланилиний-тетракис(пентафторфенил)борат, N,N-диметиланилиний-н-бутилтрис(пентафторфенил)борат, N,N-диметиланилиний-бензилтрис(пентафторфенил)борат, N, N-диметиланилиний-тетракис(трет-бутилдиметилсилил)-2, 3, 5, 6-тетрафторфенил)борат, N, N-диметиланилиний-тетракис(4-триизопропилсилил)-2, 3, 5, 6-тетрафторфенил)борат, N,N-диметиланилиний-пентафторфенокситрис(пентафторфенил)борат, N,N-диметиланилиний-тетракис(пентафторфенил)борат, N,N-диметил-2, 4, 6-триметиланилиний-тетракис(пентафторфенил)борат, диметилоктадециламмоний-тетракис(пентафторфенил)-борат, метилдиоктадециламмоний-тетракис(пентафторфенил)-борат, диалкиламмониевые соли, такие как ди(изопропил)аммоний-тетракис(пентафторфенил)борат, метилоктадециламмоний-тетракис(пентафторфенил)борат, метилоктадодециламмоний-тетракис(пентафторфенил)борат и диоктадециламмоний-тетракис(пентафторфенил)борат; соли тризамещенного фосфония, такие как трифенилфосфоний-тетракис(пентафторфенил)борат, метилдиоктадецилфосфоний-тетракис(пентафторфенил)-борат и три(2, 6-диметилфенил)фосфоний-тетракис(пентафторфенил) борат; соли дизамещенного оксония, такие как дифенилоксоний-тетракис(пентафторфенил)борат, ди(о-толил)оксоний-тетракис(пентафторфенил)борат и ди(октадецил)оксоний-тетракис(пентафторфенил)борат; соли дизамещенного сульфония, такие как ди(о-толил)сульфоний-тетракис(пентафторфенил)борат и метилоктадецилсульфоний-тетракис(пентафторфенил)борат. Предпочтительными (L*-H)+– катионами являются метилдиоктадециламмоний и диметилоктадециламмоний. Другие подходящие ионообразующие активирующие сокатализаторы содержат соль катионного окисляющего агента и не образующего координационной связи совместимого аниона, изображаемую формулой (Охe+)d(Аd-)e, где Охe+ – катионный окисляющий агент с зарядом е+; е = 1-3, целое число; Аd- и d имеют установленные ранее значения. Примерами катионных окисляющих агентов являются ферроцений, гидрокарбилзамещенный ферроцений, Аq+ или Рb+2. Предпочтительными вариантами Аd- являются анионы, определение которым дается в связи с активирующими сокатализаторами, содержащими кислоту Бренстеда, в особенности тетракис(пентафторфенил)борат. Еще один подходящий ионообразующий активирующий сокатализатор содержит соединение, представляющее соль карбений-иона и не образующего координационной связи совместимого аниона, изображаемую формулой 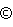 +A–, +A–,где + – C1-20-карбений-ион;А– имеет установленные ранее значения. Предпочтительным карбений-ионом является тритил-катион, т.е. трифенилметилий. Другой подходящий ионообразующий активирующий сокатализатор содержит соединение, представляющее соль силилий-иона и не образующего координационной связи совместимого аниона, изображаемую формулой R3Si(Х’)q+А–, где R – C1-10-гидрокарбил, а X’, q и А– имеют установленные ранее значения. Предпочтительными активирующими сокатализаторами – солями силилия являются триметилсилилий-тетракиспентафторфенилборат, триэтилсилилий-тетракиспентафторфенилборат и их эфирозамещенные аддукты. Соли силилия в общих чертах описаны ранее в J. Chem. Soc. Chem. Comm., 1993, 383-384, а также в Lambert, J. B. et al., Organometallics, 1994, 13, 2430-2443. Применение вышеуказанных солей силилия в качестве активирующих сокатализаторов для добавления к катализаторам полимеризации описывается в заявке на патент США, регистрационный номер 304314, зарегистрированной 12 сентября 1994, опубликованной в равноценной форме как WO 96/08519 21 марта 1996. Некоторые комплексы спиртов, меркаптанов, силанолов и оксимов с трис(пентафторфенил)бораном также являются эффективными активаторами катализаторов и могут применяться по настоящему изобретению. Такие сокатализаторы описываются в патенте США 5296433. Метод объемного электролиза подразумевает электрохимическое окисление комплекса металла в условиях электролиза в присутствии фонового электролита, содержащего не образующий координационных связей инертный анион. При этом методе используются такие растворители, фоновые электролиты и электролитические потенциалы для электролиза, что побочные продукты электролиза, которые могут сделать комплекс металла каталитически неактивным, во время реакции, по существу, не образуются. Точнее, подходящими растворителями и материалами являются жидкости, способные в условиях электролиза (обычно, при температурах от 0 до 100oС) растворять фоновый электролит и являющиеся инертными. “Инертными растворителями” являются растворители, которые не восстанавливаются или не окисляются в условиях реакции, используемой для электролиза. Обычно, с учетом нужного взаимодействия при электролизе, можно подобрать растворитель и фоновый электролит, которые не изменяются под действием электрического потенциала, используемого в случае нужного процесса электролиза. Предпочтительными растворителями являются дифторбензол (все изомеры), диметоксиэтан (DME) и их смеси. Электролиз можно осуществлять в стандартном электролизере, содержащем анод и катод (называемые также рабочим электродом и противоэлектродом соответственно). Подходящими конструкционными материалами для электролизера являются стекло, пластик, керамика и металл со стеклянным покрытием. Электроды изготавливают из инертных проводящих материалов, под которыми подразумеваются проводящие материалы, не изменяющиеся под воздействием реакционной смеси или условий реакции. Предпочтительными инертными проводящими материалами являются платина или палладий. Обычно проницаемая для ионов мембрана, например тонкая стеклообразная фритта, разделяет электролизер на отдельные камеры – камеру рабочего электрода и камеру противоэлектрода. Рабочий электрод погружают в реакционную среду, содержащую комплекс металла, который активируют, растворитель, фоновый электролит и любые другие материалы, нужные для умеренного электролиза или стабилизации получающегося в результате комплекса. Противоэлектрод погружают в смесь растворителя и фонового электролита. Нужное напряжение можно определить расчетным путем или экспериментально путем промывки электролизера с использованием опорного электрода, например, серебряного электрода, погруженного в электролит электролизера. Определяют также фоновый ток ячейки – ток, протекающий в отсутствие нужного электролиза. Электролиз завершают, когда ток падает с нужного уровня до уровня фона. Таким образом можно легко обнаружить полную конверсию исходного комплекса металла. Подходящими фоновыми электролитами являются соли, содержащие катион и совместимый, не образующий координационных связей анион А–. Предпочтительными фоновыми электролитами являются соли, соответствующие формуле G+A–, где G+ – катион, нереакционноспособный в отношении исходного и получающегося в результате комплекса, и А– имеет установленные ранее значения. Примерами катионов G+ являются катионы – тетрагидрокарбилзамещенный аммоний или фосфоний, содержащие до 40 неводородных атомов. Предпочтительными катионами являются тетра(н-бутиламмоний) и тетраэтиламмоний. В процессе активации комплексов настоящего изобретения путем объемного электролиза катион фонового электролита направляется к противоэлектроду, а А– мигрирует к рабочему электроду и становится анионом получающегося в результате окисленного продукта. Или растворитель, или катион фонового электролита восстанавливается у противоэлектрода в количестве, эквимолярном количеству окисленного комплекса металла, образовавшегося на рабочем электроде. Предпочтительными фоновыми электролитами являются соли тетрагидрокарбиламмоний-тетракис(перфторарил)бораты с 1-10 атомами углерода в каждой углеводородной или перфторарильной группе, в особенности тетра(н-бутиламмоний)-тетракис(пентафторфенил)борат. Другим недавно описанным электрохимическим методом получения активирующих сокатализаторов является электролиз дисиланового соединения в присутствии источника не образующего координационных связей совместимого аниона. Этот метод полнее описывается и заявляется в упомянутой ранее заявке на патент США за регистрационным номером 304314. Вышеуказанные метод электрохимического активирования и активирующие сокатализаторы также можно использовать в сочетании. Особенно предпочтительной комбинацией является смесь соединения три(гидрокарбил)алюминия и три(гидрокарбил)борана с 1-4 атомами углерода в каждой углеводородной группе с олигомерным или полимерным алюмоксаном. Молярное соотношение используемых катализатора и сокатализатора находится предпочтительно в интервале от 1:10000 до 100:1, предпочтительнее от 1: 5000 до 10: 1, наиболее предпочтительно от 1:1000 до 1:1. Алюмоксан, когда его самого используют в качестве сокатализатора, используется в большем количестве, обычно по меньшей мере в 100 раз больше, в молярном соотношении, количества комплекса металла. Когда в качестве активирующего сокатализатора используют трис(пентафторфенил)боран, его используют в молярном соотношении с комплексом металла от 0,5:1 до 10:1, предпочтительнее от 1:1 до 6:1, наиболее предпочтительно от 1:1 до 5:1. Другие активирующие сокатализаторы обычно используют приблизительно в эквимолярном количестве с комплексом металла. Катализаторы, нанесены ли они на носители или нет в каком-либо из вышеописанных способов, могут применяться для полимеризации этилен- или ацетиленненасыщенных мономеров, содержащих от 2 до 100000 атомов углерода, или отдельных или в сочетании. Предпочтительными мономерами являются С2-20– -олефины, особенно этилен, пропилен, изобутилен, 1-бутен, 1-пентен, 1-гексен, 3-метил-1-пентен, 4-метил-1-пентен, 1-октен, 1-децен, длинноцепочечные макромолекулярные -олефины и их смеси. Другими предпочтительными мономерами являются стирол, C1-4-алкилзамещенный стирол, тетрафторэтилен, винилбензоциклобутан, этилиденнорборнен, 1,4-гексадиен, 1,7-октадиен, винилциклогексан, 4-винилциклогексан, дивинилбензол и их смеси с этиленом. Длинноцепочечные макромолекулярные -олефины представляют остатки полимеров с концевыми винильными группами, образовавшиеся in situ во время реакций при непрерывной полимеризации в растворе. При подходящих условиях обработки такие длинноцепочечные макромолекулярные звенья легко полимеризуются до полимера вместе с этиленом и другими олефиновыми мономерами с короткими цепями, и дают небольшое количество длинноцепочечных разветвлений в получающемся в результате полимере.
Предпочтительными мономерами являются сочетания этилена и одного или нескольких сомономеров, выбранных среди моновинилароматических мономеров, 4-винилциклогексена, винилциклогексана, норборнадиена, этилиденнорборнена, С3-10-алифатических -олефинов (особенно, пропилена, изобутилена, 1-бутена, 1-гексена, 3-метил-1-пентена, 4-метил-1-пентена и 1-октена) и С4-40-диенов. Наиболее предпочтительными мономерами являются смеси этилена со стиролом; смеси этилена, пропилена и стирола; смеси этилена, стирола и неконъюгированного диена, в особенности этилиденнорборнена.
Вообще, полимеризацию можно осуществлять при условиях, хорошо известных на известном уровне техники для реакций полимеризации типа полимеризации с катализаторами Циглера-Натта или Каминского-Синна, т.е. при температурах 0-250oС, предпочтительно при 30-200oС, и давлении от атмосферного до 10000 атмосфер. При желании можно использовать бисерную полимеризацию, полимеризацию в растворе, суспензионную, газофазную полимеризацию, твердофазную порошковую полимеризацию или другие условия процесса. Можно использовать носитель, в особенности диоксид кремния, оксид алюминия или полимер (особенно, поли(тетрафторэтилен) или полиолефин), и его желательно использовать, когда катализаторы применяют при процессе газофазной полимеризации. Носитель используют предпочтительно в количестве, обеспечивающем массовое соотношение катализатора (в расчете на металл) и носителя от 1:100000 до 1:10, предпочтительнее от 1:50000 до 1:20, и наиболее предпочтительно от 1:10000 до 1:30.
При большинстве реакций полимеризации молярное соотношение используемых катализатора и полимеризуемых соединений составляет от 10-12:1 до 10-1:1, предпочтительнее от 10-9:1 до 10-5:1.
Подходящими растворителями для применения при полимеризации в растворе являются инертные жидкости. Примеры включают линейные или разветвленные углеводороды, такие как изобутан, бутан, пентан, гексан, гептан, октан и их смеси; циклические и алициклические углеводороды, такие как циклогексан, циклогептан, метилциклогексан, метилциклогептан и их смеси; перфторированные углеводороды, такие как перфторированные С4-10-алканы, и алкилзамещенные ароматические соединения, такие как бензол, толуол, ксилол и этилбензол. Подходящими растворителями также являются жидкие олефины, которые могут действовать как мономеры или сомономеры.
Катализаторы можно использовать в сочетании по меньшей мере с одним дополнительным катализатором гомогенной или гетерогенной полимеризации в отдельных реакторах, соединенных в ряд или параллельно, и получить полимерные смеси с нужными свойствами. Пример такого способа описывается в WO 94/00500, соответствующей заявке на патент США с серийным номером 07/904770, а также в заявке на патент США с серийным номером 08/10958, поданной 29 января 1993.
С использованием катализаторов настоящего изобретения легко получить, высокоэффективным способом, гомополимеры и сополимеры -олефинов с плотностью от 0,85 до 0,96 г/см3 и скоростями течения расплава от 0,001 до 10,0 дг/мин.
Катализаторы настоящего изобретения имеют особые преимущества при получении гомополимеров этилена и сополимеров этилена и -олефинов с высоким содержанием длинноцепочечных разветвлений. Применение катализаторов настоящего изобретения в непрерывных способах полимеризации, особенно в непрерывных способах полимеризации в растворе, допускает повышенные температуры в реакторе, что благоприятно для образования полимерных цепей с концевыми винильными группами, которые могут включаться в растущий полимер, причем посредством этого образуется длинноцепочечное ответвление. Применение системы катализаторов настоящего изобретения выгодно для экономичного получения этилен--олефиновых сополимеров, обладающих технологичностью, подобной технологичности полиэтилена пониженной плотности, полученного свободнорадикальной полимеризацией при высоком давлении.
Систему катализаторов по настоящему изобретению можно выгодно использовать для получения полиолефинов с улучшенными технологическими свойствами посредством полимеризации одного этилена или смесей этилена с -олефинами с низкой степенью “Н”-ответвлений, включая диен, такой как норборнадиен, 1,7-октадиен или 1,9-декадиен. Уникальное сочетание повышенных температур в реакторе, высокой молекулярной массы (или низких индексов расплава) при высоких температурах в реакторе и высокой реакционной способности сомономеров выгодно для экономичного получения полимеров с превосходными физическими свойствами и способностью к переработке. Предпочтительно такие полимеры содержат этилен, С3-20–-олефин и “Н” -разветвляющий сомономер. Предпочтительно такие полимеры получают в растворе, наиболее предпочтительно-непрерывным способом в растворе.
Каталитическую композицию можно получить в виде однородного катализатора путем добавления необходимых компонентов в растворитель, в котором будет осуществляться полимеризация методами полимеризации в растворе. Каталитическую композицию можно также получить и использовать в виде гетерогенного катализатора путем адсорбирования необходимых компонентов на инертном неорганическом или органическом твердом веществе в виде частиц. Примерами таких твердых веществ являются диоксид кремния, силикагель, оксид алюминия, соединения триалкилалюминия и органические или неорганические полимерные материалы, в особенности полиолефины. Впредпочтительном варианте воплощения изобретения гетерогенный катализатор получают путем соосаждения комплекса металла, инертного неорганического соединения и активатора, в особенности соли аммонийгидроксиарил(триспентафторфенил)-бората, такой как аммоний(4-гидрокси-3,5-ди-трет-бутилфенил)(триспентафторфенил)борат. Предпочтительным инертным неорганическим соединением для применения в таком варианте является три(C1-4-алкил)алюминий. Когда катализатор получают в гетерогенной форме или на носителе, каталитическую композицию используют при суспензионной или газофазной полимеризации. Практическим ограничением является то, что суспензионная полимеризация происходит в жидких разбавителях, в которых полимерный продукт, по существу, не растворяется. Предпочтительно разбавителем для суспензионной полимеризации является один или несколько углеводородов с числом атомов углерода менее 5. Если желательно, как весь разбавитель или его часть можно использовать насыщенные углеводороды, такие как этан, пропан или бутан, как весь разбавитель или его часть. Подобным образом, в качестве всего разбавителя или его части можно использовать мономер -олефин или смесь разных -олефинов. Наиболее предпочтительно, когда по меньшей мере основную часть разбавителя составляет -олефиновый мономер или мономеры, которые полимеризуют.
На всем протяжении процесса отдельные ингредиенты, а также извлекаемые компоненты катализатора должны быть защищены от воздействия кислорода и влаги. Следовательно, компоненты катализаторов и катализаторы должны приготовляться и извлекаться в атмосфере, свободной от кислорода и влаги. Поэтому предпочтительно осуществлять реакции в присутствии сухого инертного газа, такого как, например, азот.
Полимеризацию можно осуществлять как полимеризацию периодическим или непрерывным способом. Предпочтителен непрерывный способ, при котором нужный катализатор, этилен, сомономер и необязательно растворитель непрерывно подаются в реакционную зону, а полимер как продукт непрерывно удаляется из нее.
Без какого-либо ограничения объема изобретения способ осуществления такого процесса полимеризации следующий. В реактор с мешалкой непрерывно загружают мономеры, которые полимеризуют вместе с растворителем и необязательным регулятором степени полимеризации. Реактор содержит жидкую фазу, состоящую, по существу, из мономеров с каким-либо растворителем или добавленным разбавителем и растворенного полимера. Если желательно, можно также добавить небольшое количество вызывающего “Н”-ветвление диена, такого как норборнадиен, 1,7-октадиен или 1,9-декадиен. В жидкую фазу реактора непрерывно вводят катализатор и сокатализатор. Температуру реактора и давление можно регулировать, подбирая соотношение растворителя и мономера, скорость добавления катализатора, а также путем охлаждения или нагревания змеевиков, кожухов или тех и других. Скорость полимеризации регулируется скоростью добавления катализатора. Содержание этилена в полимере определяется соотношением этилена и сомономера в реакторе, которое регулируют путем манипуляций с соответствующими скоростями загрузки этих компонентов в реактор. Молекулярную массу образующегося полимера регулируют необязательно путем регулирования других переменных полимеризации, таких как температура, концентрация мономера, или с помощью ранее упомянутого регулятора степени полимеризации, такого как поток водорода, вводимого в реактор, что хорошо известно в технике. Вытекающий из реактора поток приводят в контакт с отравляющим катализатор агентом, таким как вода. Полимерный раствор нагревают необязательно и извлекают полимер посредством отгона газообразных мономеров, а также оставшегося растворителя или разбавителя при пониженном давлении и, при необходимости, проведением дальнейшего удаления летучих в таком оборудовании, как экструдер для дегазации. При непрерывном способе среднее время пребывания катализатора в реакторе обычно составляет от 5 минут до 8 часов, а предпочтительно от 10 минут до 6 часов. При применении катализатора, который включает большое количество затрудненного моновинилового мономера, существенно снижается количество затрудненного гомополимера винилового мономера, образовавшегося из оставшегося мономера.
Способ настоящего изобретения можно использовать для выгодного осуществления газофазной сополимеризации олефинов. Газофазные способы полимеризации олефинов, в особенности – гомополимеризации и сополимеризации этилена и пропилена и сополимеризации этилена с более высокими -олефинами, такими как, например, 1-бутен, 1-гексен, 4-метил-1-пентен, хорошо известны в технике. При таких процессах охлаждение реактора можно обеспечить путем использования циркулирующего газа, который подают в слой в виде летучей жидкости, обеспечивающей при испарении эффект охлаждения. Летучая жидкость, используемая в таком случае, может представлять, например, летучую инертную жидкость, например, насыщенный углеводород с 3-8, предпочтительно 4-6, атомами углерода. В случае, когда сам мономер или сомономер представляет летучую жидкость (или может конденсироваться с образованием такой жидкости), он может быть подходящим для загрузки в слой для обеспечения эффекта охлаждения при испарении. Примерами олефиновых мономеров, которые можно использовать таким образом, являются олефины, содержащие от 3 до 8, предпочтительно от 3 до 6, атомов углерода. Летучая жидкость испаряется в горячем псевдоожиженном слое с образованием газа, который смешивается с флюидизирующим газом. Если летучая жидкость представляет мономер или сомономер, она будет претерпевать в слое полимеризацию. Испарившаяся жидкость затем выходит из реактора как часть горячего циркулирующего газа и входит в узел сжатия и теплообмена контура циркуляции. Циркулирующий газ охлаждается в теплообменнике, и, если температура, до которой охлаждается газ, ниже точки росы, из газа выпадает жидкость. Эту жидкость желательно непрерывно возвращать в псевдоожиженный слой. Возможно возвращать выпавшую жидкость в слой в виде капель жидкости, уносимых потоком циркулирующего газа. Способ такого типа описывается, например, в ЕР 89691; патенте США 4543399; WO 94/25495 и в патенте США 5352749. Особенно предпочтительный способ возвращения жидкости в слой состоит в выделении жидкости из потока циркулирующего газа и повторном впрыскивании этой жидкости непосредственно в слой предпочтительно с использованием способа, при котором в слое образуются мелкие капельки жидкости. Этот тип процесса описывается в WO 94/28032, ВР Chemicals.
Реакция полимеризации, происходящая в газовом псевдоожиженном слое, катализируется непрерывным или полунепрерывным добавлением катализатора. Такой катализатор может быть нанесен на неорганический или органический носитель, как описано выше.
Полимер получают непосредственно в псевдоожиженном слое путем катализированной сополимеризации мономера и одного или нескольких сомономеров на псевдоожиженных частицах катализатора, катализатора на носителе или форполимера в слое. Начала реакции полимеризации добиваются с использованием слоя предварительно сформированных полимерных частиц, которые предпочтительно подобны целевому полиолефину, и приведением слоя в нужное состояние методами, хорошо известными в технике. Такие способы применяют коммерчески в крупном масштабе для производства полиэтилена высокой плотности (ПЭВП), полиэтилена средней плотности (ПЭСП), линейного полиэтилена низкой плотности (ЛПЭНП) и полипропилена.
Используемый газофазный способ может быть, например, типа, при котором в качестве реакционной зоны полимеризации используют механически перемешиваемый слой или псевдоожиженный газом слой. Предпочтительным является способ, при котором реакция полимеризации осуществляется в вертикальном цилиндрическом реакторе полимеризации, содержащем псевдоожиженный слой полимерных частиц, поддерживаемый выше сетчатой тарелки – флюидизирующей решетки – потоком флюидизирующего газа.
Газ, используемый для флюидизации слоя, содержит мономер или мономеры, которые полимеризуют, а также служит в качестве теплообменной среды для удаления тепла реакции из слоя. Горячие газы выходят из верхней части реактора, обычно через спокойную зону, также известную как зона снижения скорости, имеющую большую площадь поперечного сечения, чем псевдоожиженный слой, где мелкие частицы, находящиеся в газовом потоке, имеют удобный случай, чтобы притянуться назад к слою. Также может быть выгодным использовать циклон для удаления ультрамелких частиц из потока горячего газа. Затем газ снова возвращается к слою с помощью вентилятора или компрессора и одного или нескольких теплообменников, которые освобождают газ от тепла, выделившегося при полимеризации. Образовавшийся полимер выгружается из псевдоожиженного слоя непрерывно или периодически, как желательно.
Газофазные процессы, подходящие для практического осуществления данного изобретения, являются предпочтительно непрерывными процессами, предусматривающими непрерывную загрузку реагентов в реакционную зону реактора и удаление продуктов реакции из реакционной зоны реактора, посредством чего обеспечивается стационарная макросреда в реакционной зоне реактора.
Типично, с псевдоожиженным слоем при газофазном способе работают при температурах, превышающих 50oС, предпочтительно от 60 до 110oС, предпочтительнее от 70 до 110oС.
Молярное соотношение сомономера и мономера, используемое при полимеризации, зависит от нужной плотности получаемой композиции и обычно составляет 0,5 или менее. При получении материалов с плотностью в интервале от 0,91 до 0,93 соотношение сомономера и мономера составляет, желательно, менее 0,2, предпочтительно менее 0,05, и еще предпочтительнее менее 0,02, и даже может составлять величину менее 0,01. Как правило, соотношение водорода и мономера меньше 0,5, предпочтительно менее 0,2, предпочтительнее менее 0,05 и еще предпочтительнее менее 0,02, и даже может составлять величину менее 0,01.
Указанные выше интервалы технологических параметров подходят для газофазного способа данного изобретения и могут оказаться подходящими для других способов, пригодных для практического осуществления данного изобретения.
В ряде патентов и заявок на патенты описываются газофазные процессы, пригодные для применения в способе данного изобретения, в частности в патентах США 4588790; 4543399; 5352749; 5436304; 5405922; 5462999; 5461123; 5453471; 5032562; 5028670; 5473028; 5106804; 5541270, в заявках на европейский патент 659773; 692500 и в заявках РСТ WO 94/29032, WO 94/25497, WO 94/25495, WO 94/28032; WO 95/13305; WO 94/26793 и WO 95/07942.
ПримерыСпециалисту в этой области техники будет понятно, что изобретение, описываемое здесь, можно осуществить на практике в отсутствие какого-либо компонента, который не описан конкретно. Приведенные далее примеры представляют дополнительное пояснение изобретения и не рассматриваются как ограничивающие изобретение. Если нет иных указаний, все части и проценты являются массовыми. Спектры 1H и 13С ЯМР регистрируют на спектрометре Varian XL (300 МГц). Тетрагидрофуран (ТГФ), диэтиловый эфир, толуол и гексан используют после пропускания через сдвоенные колонки, набитые активированным оксидом алюминия и смешанным (медь/марганец) металлооксидным катализатором (доступным от Engelhard Corp. ). Соединения н-BuLi, все реактивы Гриньяра, КН, инден, 1-инданон и Ni(dppp)Cl2 закупают у Aldrich. 2-Броминден, 2-метил-4-фенилинден и 2,4,6-триметилинданон синтезируют так, как описано в литературе. Все синтезы осуществляют в атмосфере сухого азота с использованием сочетания скафандра и высоковакуумной технологии. Пример 1 Синтез дихлор[N-(трет-бутил)-1,1-диметил-[3-фенилинден-1-ил]силанамин-(2-)-N] титана (называемого также диметилсилил(3-фенилинденил)(трет-бутиламидо)ТiСl2) 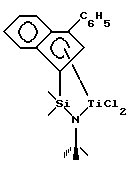 Получение 1-фенилиндена 1-Инданон (13,30 г, 0,1006 моль) перемешивают в диэтиловом эфире (300 мл) при -78oС и в то же время добавляют PhMgBr (0,150 моль, 50,00 мл 3,0 М раствора в диэтиловом эфире). Смесь затем постепенно нагревают до 20-25oС и затем перемешивают в течение 16 часов. По завершении периода реакции смесь выливают на лед и затем экстрагируют водными растворами 1 М НСl (1  100 мл), 1 М NаНСО3 (1100 мл) и затем Н2О (1100 мл). Органический слой затем сушат над МgSO4. В результате фильтрации с последующим удалением летучих веществ выделяют желтое масло, в котором, как показывает ЯМР, еще содержится некоторое количество 1-фенил-1-инданола. Это масло затем перегоняют в вакууме и получают нужный продукт в виде светло-желтого масла (18,56 г, 95,9%).
Получение литий-1-фенилинденида 100 мл), 1 М NаНСО3 (1100 мл) и затем Н2О (1100 мл). Органический слой затем сушат над МgSO4. В результате фильтрации с последующим удалением летучих веществ выделяют желтое масло, в котором, как показывает ЯМР, еще содержится некоторое количество 1-фенил-1-инданола. Это масло затем перегоняют в вакууме и получают нужный продукт в виде светло-желтого масла (18,56 г, 95,9%).
Получение литий-1-фенилинденидаПеремешивают в гексане (300 мл) 1-фенилинден (6,00 г, 0,0312 моль) и в то же время добавляют по каплям 12,5 мл 2,5 М раствора н-BuLi (0,0312 моль) в гексане. Затем смесь перемешивают в течение 48 часов при 20-25oС и в это время в осадок выпадает твердое вещество. По завершении периода реакции твердое вещество собирают путем фильтрации под вакуумом и используют без дополнительной очистки или анализа (5,53 г, 89,4%). Получение диметилсилил(3-фенилинденил)(трет-бутил-амина) К раствору диметилсилил(трет-бутиламино)хлорида (4,62 г, 0,0279 моль) в ТГФ (100 мл) добавляют по каплям раствор литий-1-фенилинденида (5,53 г, 0,0279 моль) в ТГФ (50 мл). Эту смесь перемешивают в течение 16 часов. По завершении периода реакции летучие вещества удаляют, а остаток экстрагируют и фильтруют с использованием гексана. После удаления гексана выделяют в результате нужный продукт в виде масла (8,63 г, 96,2%). Получение диметилсилил(3-фенилинденид)(трет-бутил-амидо)дилитида Диметилсилил(3-фенилинденил)(трет-бутиламин) (8,63 г, 0,0268 моль) перемешивают в гексане (200 мл) и в то же время постепенно добавляют н-BuLi (0,0537 моль, 21,5 мл 2,5 М раствора в гексане). Затем эту смесь перемешивают в течение 16 часов и в это время образуется липкий осадок. Затем удаляют летучие вещества, а получающийся в результате желтый остаток промывают гексаном. По завершении периода реакции твердое вещество сушат и выделяют в виде желтого порошка, который используют без дополнительной очистки или анализа (8,13 г, 91,0%). Получение диметилсилил(3-фенилинденил)(трет-бутиламидо)ТiСl2 К суспензии ТiСl3(ТГФ)3 (9,03 г, 0,0244 моль) в ТГФ (30 мл) добавляют диметилсилил(3-фенилинденил)(трет-бутиламидо)Li2 (8,13 г, 0,0243 моль) в виде твердого вещества. Эту смесь перемешивают в течение 30 минут. Затем добавляют в виде твердого вещества PbCl2 (3,38 г, 0,0122 моль) и смесь перемешивают еще в течение 45 минут. По завершении периода реакции летучие вещества удаляют, а остаток экстрагируют и фильтруют с использованием толуола. В результате удаления толуола выделяют темный остаток, который затем экстрагируют гексаном, и концентрируют экстракт до выпадения твердых веществ. Осадок затем собирают посредством фильтрации и промывают холодным гексаном, выделяют в результате красно-коричневое твердое кристаллическое вещество (7,39 г, 69,1%). 1H ЯМР (CDCl3):  0,39 (с, 3Н), 0,59 (с, 3Н), 1,01 (с, 9Н), 6,64 (с, 1Н), 7,0-7,25 (м, 4 Н), 7,65 (м, 4Н), 7,78 (д, 1Н).
Пример 2 0,39 (с, 3Н), 0,59 (с, 3Н), 1,01 (с, 9Н), 6,64 (с, 1Н), 7,0-7,25 (м, 4 Н), 7,65 (м, 4Н), 7,78 (д, 1Н).
Пример 2Синтез диметил[N-(трет-бутил)-1,1-диметил-[3-фенилинден-1-ил] силанамин-(2-)-N] титана (называемого также диметилсилил(3-фенилинденил)(трет-бутиламидо)TiMe2) Перемешивают диметилсилил(3-фенилинденил)(трет-бутиламидо) TiCl2 (0,330 г, 0,000753 моль) в диэтиловом эфире (30 мл) и в то же время добавляют по каплям 0,525 мл 3 М раствора МеМgI (0,00158 моль) в диэтиловом эфире. Эту смесь затем перемешивают в течение 40 минут. По завершении периода реакции летучие вещества удаляют, а остаток экстрагируют и фильтруют с использованием гексана. В результате удаления гексана выделяют нужный продукт (0,210 г, 70,2%). 1H ЯМР (CDCl3): 0,02 (с, 3Н), 0,43 (с, 3Н), 0,61 (с, 3Н), 0,69 (с, 3Н), 1,45 (с, 9Н), 6,39 (с, 1Н), 6,90 (т, 1Н), 7,15 (т, 2Н), 7,25 (т, 2Н), 7,52 (д, 1Н), 7,65 (д, 2Н), 7,80 (д, 1Н).
Пример 3Синтез дихлор[N-(циклогексил)-1,1-диметил-[3-фенилинден-1-ил] силанамин-(2-)-N]титана (также называемого диметилсилил (3-фенилинденил)(циклогексиламидо)ТiСl2) 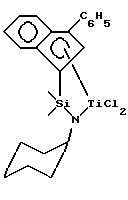 Получение диметилсилил(3-фенилинденил)хлорида К раствору Me2SiCl2 (51,3673 моль) в ТГФ (150 мл) добавляют по каплям Li-1-фенилинденид (21,8736 г, 0,1104 моль) в ТГФ (75 мл). Эту смесь затем перемешивают в течение 16 часов. По завершении периода реакции летучие вещества удаляют, а остаток экстрагируют и фильтруют с использованием гексана. В результате удаления гексана выделяют нужный продукт в виде желто-красного масла (28,6183 г, 91,0%). Получение диметилсилил(3-фенилинденил)(циклогексиламина) Me2Si(3-Фенилинденил)Сl (4,7327 г, 0,01661 моль) перемешивают в гексане (100 мл) и в то же время добавляют Net3 (5,1786 г, 0,05118 моль) и циклогексиламин (1,6960 г, 0,01710 моль). Эту смесь затем перемешивают в течение 16 часов. По завершении периода реакции смесь фильтруют и удаляют летучие вещества, причем в результате выделяют нужный продукт в виде желтого масла (5,2800 г, 91,7%). Получение диметилсилил(3-фенилинденил)(циклогексиламидо)Li2 Me2Si(3-Фенилинденил)(циклогексиламин) (5,2800 г, 0,001524 моль) перемешивают в гексане (150 мл) и в то же время постепенно добавляют н-BuLi (0,034 моль, 17,00 мл 2,5 М раствора в гексане). Эту смесь перемешивают в течение 16 часов и за это время смесь становится густым однородным маслом. По завершении периода реакции летучие вещества удаляют, причем в результате выделяют светло-желтое твердое вещество. Это твердое вещество затем промывают гексаном, а затем сушат и в результате выделяют светло-желтый порошок, который используют без дополнительной очистки или анализа (4,4410 г, 81,35%). Получение диметилсилил(3-фенилинденил)(циклогексиламидо)TiCl2 Me2Si(3-Фенилинденил)(циклогексиламидо)Li2 (3,7346 г, 0,01042 моль) в ТГФ (50 мл) добавляют по каплям к суспензии ТiСl3(ТГФ)3 (3,8613 г, 0,01042 моль) в ТГФ (100 мл). Эту смесь перемешивают в течение 2 часов. Затем добавляют PbCl2 (1,4513 г, 0,005219 моль) в виде твердого вещества и смесь перемешивают еще в течение 45 минут. По завершении периода реакции летучие вещества удаляют, а остаток экстрагируют и фильтруют с использованием толуола. В результате удаления толуола выделяют темный остаток, который затем суспендируют в гексане и охлаждают до 0oС. Затем посредством фильтрации собирают нужный продукт в виде красно-коричневого твердого кристаллического вещества (2,7475 г, 56,8%). 1H ЯМР (CDCl3): 0,69 (с, 3Н), 0,91 (с, 3Н), 0,8-2,1 (м, 10Н), 4,6-4,8 (м, 1Н), 6,73 (с, 1Н), 7,1-7,8 (м, 8Н), 8,03 (д, 3Jнн=8,7 Гц, 1Н).
Пример 4Синтез диметил[N-(циклогексил)-1,1-диметил-[3-фенилинден-1-ил] силанамин-(2-)-N]титана (называемого также диметилсилил(3-фенилинденил)(циклогексиламидо)TiMe2) Me2Si(3-Фенилинденил)(циклогексиламидо)ТiСl2 (0,3936 г, 0,0008476 моль) перемешивают в диэтиловом эфире (75 мл) и в то же время добавляют по каплям 0,57 мл 3,0 М раствора МеМgI (0,0017 моль) в диэтиловом эфире. Эту смесь затем перемешивают в течение 3 часов. По завершении периода реакции летучие вещества удаляют, а остаток экстрагируют и фильтруют с использованием гексана. В результате удаления гексана выделяют нужный продукт в виде желто-красного остатка (0,1404 г, 39,1%). 1H ЯМР (C6D6): 0,044 (с, 3Н), 0,48 (с, 3Н), 0,64 (с, 3Н), 0,68 (с, 3Н), 0,9-2,2 (м, 10Н), 4,6-4,8 (м, 1Н), 6,49 (с, 1Н), 6,99 (т, 3Jнн=7,4 Гц, 1Н), 7,14-7,25 (м, 2Н), 7,34 (т, 3Jнн=7,5 Гц, 2Н), 7,59 (д, 3Jнн=8,7 Гц, 1Н), 7,75 (д, 3Jнн=7,7 Гц, 2Н), 8,08 (д, 3Jнн=8,5 Гц, 1Н).
ПолимеризацияПолимеризацию проводят в следующих условиях. В 2-литровый реактор Парра загружают приблизительно 360 г смеси алканов – растворителя isopar-ETM (доступного от Exxon Chemicals Inc.) и 460 г стирольного сомономера. В качестве регулятора молекулярной массы добавляют водород, путем расширения при различном давлении, из 75-миллилитровой дополнительной емкости при давлении 25 ф/д2 (2070 кПа). Реактор нагревают до 90oС и насыщают этиленом при 200 ф/д2 (1,4 МПа). Предварительно смешивают в сухой камере соответствующее количество катализатора и сокатализатора (триспентафторфенилборан) в виде 0,005 М растворов в толуоле для получения молярного соотношения катализатора и сокатализатора 1:1. После нужного для предварительного смешивания периода времени раствор переносят в дополнительную емкость для катализатора и инъецируют в реактор. Условия полимеризации поддерживают в течение 30 минут по расходу этилена. Периодически добавляют дополнительное количество предварительной смеси катализатора. Получающийся в результате раствор удаляют из реактора, гасят изопропиловым спиртом и к получающемуся в результате раствору добавляют антиоксидант – фенол с пониженной реакционной способностью (IrganoxTM 1010 от Ciba Geigy Corporation). Образовавшиеся полимеры сушат в вакуумной печи при 135oС в течение 20 часов. Результаты, полученные с использованием катализаторов изобретения и катализатора для сравнения (инденил)диметил(трет-бутиламидо)силантитандиметила, приводятся в таблице. Формула изобретения
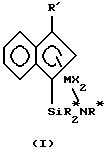 где М – титан, цирконий или гафний в формальной степени окисления +2 +3 или +4; R’ – бифенил или нафтил; R* – водород или гидрокарбил; Х – галоген или метил. 2. Комплекс металла по п. 1, соответствующий формуле (I), где R* – трет-бутил или циклогексил. 3. Комплекс металла по п. 2, который представляет (3-фенилинденил)диметил(трет-бутиламидо)силантитандихлорид, (3-фенилинденил)диметил(трет-бутиламидо)силантитандиметил, (3-фенилинденил)диметил(циклогексиламидо)силантитандихлорид или (3-фенилинденил)диметил(циклогексиламидо)силантитандиметил. 4. Катализатор полимеризации олефинов, содержащий 1) комплекс металла по п. 1 и 2) активирующий сокатализатор, содержащий триспентафторфенилборан. 5. Способ полимеризации олефинов, включающий контактирование в условиях полимеризации одного или нескольких С2-100000 – -олефинов с катализатором, отличающийся тем, что катализатором является катализатор по п. 4.
6. Способ по п. 5, включающий сополимеризацию этилена и стирола.
7. Способ по п. 6, осуществляемый в условиях полимеризации в растворе.
РИСУНКИ
MM4A Досрочное прекращение действия патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 18.10.2003
Извещение опубликовано: 10.05.2005 БИ: 13/2005
|
||||||||||||||||||||||||||