Патент на изобретение №2184733
|
||||||||||||||||||||||||||
(54) ЗАМЕЩЕННЫЕ ПИПЕРИДИН-2,6-ДИОНЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ
(57) Реферат: Изобретение относится к новым замещенным пиперидин-2,6-дионам формулы (I) 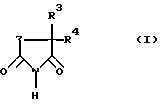 где Z – -C(R1R2)-CH2 или -C(R1)=CH-; R1 – фталимид, когда Z – -C(R1R2)-CH2-, или одно- либо двукратно замещенный гидрокси-, метокси- или аминогруппами фталимидный радикал, когда Z обозначает -C(R1)=CH-, R2 – водород или C1-6 алкильная группа; R3 – водород, С1-6 алкильная группа или ароматическая кольцевая система; R4 – C1-6 алкил или ароматическое кольцо. Соединения формулы (I) обладают ингибирующим действием на высвобождение TNF- где Z – -C(R1R2)-CH2 или -C(R1)=CH-; R1 – фталимид, когда Z – -C(R1R2)-CH2-, или одно- либо двукратно замещенный гидрокси-, метокси- или аминогруппами фталимидный радикал, когда Z обозначает -C(R1)=CH-, R2 – водород или C1-6 алкильная группа; R3 – водород, С1-6 алкильная группа или ароматическая кольцевая система; R4 – C1-6 алкил или ароматическое кольцо. Соединения формулы (I) обладают ингибирующим действием на высвобождение TNF- , а также на индуцированный антигеном синтез интерлейкина 2. Соединения формулы (I) могут быть получены конденсацией фталевого ангидрида с замещенной глутаминовой кислотой с последующей циклизацией полученного продукта и переводом его в имид. Другой способ получения соединений формулы (I) заключается в конденсации фталевого ангидрида с замещенным 3-аминоглутаконимидом или 5-замещенным 3-аминоглутаримидом. 3 с. и 2 з.п. ф-лы, 1 табл. , а также на индуцированный антигеном синтез интерлейкина 2. Соединения формулы (I) могут быть получены конденсацией фталевого ангидрида с замещенной глутаминовой кислотой с последующей циклизацией полученного продукта и переводом его в имид. Другой способ получения соединений формулы (I) заключается в конденсации фталевого ангидрида с замещенным 3-аминоглутаконимидом или 5-замещенным 3-аминоглутаримидом. 3 с. и 2 з.п. ф-лы, 1 табл.
Изобретение относится к аналогам талидомида из класса пиперидин-2,6-дионов, способу их получения и их применению в лекарственных средствах. При патогенезе синдрома реакции “трансплантат против хозяина”, рассеянного склероза, отторжения трансплантата, афтозного стоматита, эритемы нодозной лепроматозной, болезни Бека, ревматоидного артрита и целого ряда других заболеваний, сопровождаемых воспалительными явлениями, чрезмерное образование являющегося цитокином фактора- некроза опухоли (TNF-, Tumor-Necrosis-Factor-) играет главную роль. Один из подходов в терапии этих болезней состоит в целенаправленном подавлении высвобождения   F- введением в качестве активных веществ иммунодепрессантов или модуляторов, например дексаметазона, пентоксифиллина или талидомида.
Однако среди показаний к применению следует проводить различия между такими, которые требуют общей иммуносупрессии, и такими, при которых требуется в полной мере учитывать все достоинства и недостатки иммуносупрессии. Было установлено, что при лечении афтозного стоматита талидомид превосходит классические иммунодепрессанты. Другими примерами заболеваний, в которых талидомид показал хорошую эффективность, не приводя к общей иммуносупрессии, являются кожная красная волчанка, (H+G 69, 816-822 (1994)), гангренозная пиодермия и орогенитальные язвы при болезни Бехчета (The Lancet, 20.05.89, 1093-1095). Патогенными факторами этих ограниченных кожей и слизистыми оболочками поражений являются эндогенные медиаторы с действием на эндотелий и циркулирующие лейкоциты. Под влиянием F- и других цитокинов адгезивность эндотелия по отношению к лейкоцитам фокально возрастает, что решающим образом способствует образованию васкулитидов. При системных картинах болезни действие самого талидомида на кожу и слизистые оболочки ограничено, что (дополнительно) требует иммуносупрессии. Примерами этого является системная красная волчанка, которая наряду с кожными явлениями вызывает также опасные для жизни изменения внутренних сосудов, в частности почек, лепрареакция типа II с поражением глаз и/или суставов, а также болезнь Бехчета с поражением глаз и/или суставов.
Вещества, которые аналогично талидомиду подавляют это изменение эндотелия, но одновременно полностью или частично блокируют реакции специфической клеточной иммунной защиты, могут способствовать значительному прогрессу в терапии названных системных картин болезни. Ключевым информационным веществом клеточного иммунного ответа является интерлейкин 2, от которого зависит пролиферация антигенспецифичных лимфоцитов.
Поэтому при разработке новых лекарственных средств ставится цель в полной мере использовать противовоспалительные свойства талидомида совместно с иммуносупрессивными активными компонентами, которыми талидомид при его индивидуальном клиническом применении не обладает.
В основу изобретения была положена задача создать аналоги талидомида из класса пиперидин-2,6-дионов, подавляющих обусловленное воспалением высвобождение F-, а также индуцированный антигеном синтез интерлейкина 2.
Было установлено, что указанным требованиям удовлетворяют предлагаемые согласно изобретению соединения.
Таким образом, объектом настоящего изобретения являются замещенные в положении 3 и 5 пиперидин-2,6-дионы общей формулы (I) F- введением в качестве активных веществ иммунодепрессантов или модуляторов, например дексаметазона, пентоксифиллина или талидомида.
Однако среди показаний к применению следует проводить различия между такими, которые требуют общей иммуносупрессии, и такими, при которых требуется в полной мере учитывать все достоинства и недостатки иммуносупрессии. Было установлено, что при лечении афтозного стоматита талидомид превосходит классические иммунодепрессанты. Другими примерами заболеваний, в которых талидомид показал хорошую эффективность, не приводя к общей иммуносупрессии, являются кожная красная волчанка, (H+G 69, 816-822 (1994)), гангренозная пиодермия и орогенитальные язвы при болезни Бехчета (The Lancet, 20.05.89, 1093-1095). Патогенными факторами этих ограниченных кожей и слизистыми оболочками поражений являются эндогенные медиаторы с действием на эндотелий и циркулирующие лейкоциты. Под влиянием F- и других цитокинов адгезивность эндотелия по отношению к лейкоцитам фокально возрастает, что решающим образом способствует образованию васкулитидов. При системных картинах болезни действие самого талидомида на кожу и слизистые оболочки ограничено, что (дополнительно) требует иммуносупрессии. Примерами этого является системная красная волчанка, которая наряду с кожными явлениями вызывает также опасные для жизни изменения внутренних сосудов, в частности почек, лепрареакция типа II с поражением глаз и/или суставов, а также болезнь Бехчета с поражением глаз и/или суставов.
Вещества, которые аналогично талидомиду подавляют это изменение эндотелия, но одновременно полностью или частично блокируют реакции специфической клеточной иммунной защиты, могут способствовать значительному прогрессу в терапии названных системных картин болезни. Ключевым информационным веществом клеточного иммунного ответа является интерлейкин 2, от которого зависит пролиферация антигенспецифичных лимфоцитов.
Поэтому при разработке новых лекарственных средств ставится цель в полной мере использовать противовоспалительные свойства талидомида совместно с иммуносупрессивными активными компонентами, которыми талидомид при его индивидуальном клиническом применении не обладает.
В основу изобретения была положена задача создать аналоги талидомида из класса пиперидин-2,6-дионов, подавляющих обусловленное воспалением высвобождение F-, а также индуцированный антигеном синтез интерлейкина 2.
Было установлено, что указанным требованиям удовлетворяют предлагаемые согласно изобретению соединения.
Таким образом, объектом настоящего изобретения являются замещенные в положении 3 и 5 пиперидин-2,6-дионы общей формулы (I)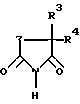 где Z обозначает одну из групп: 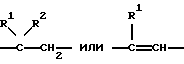 причем атом углерода с заместителем R1 связан по карбонильной группе, и где R1 обозначает фталимидный радикал (когда Z обозначает -C(R1R2)-CH2-) или одно- либо двукратно замещенный гидрокси-, метокси- или аминогруппами фталимидный радикал (когда Z обозначает -C(R1)=СН-), R2 обозначает водород или С1-6 алкил (линейный или разветвленный), R3 обозначает водород, С1-6 алкильную группу (линейную или разветвленную) или ароматическую либо гетероароматическую кольцевую систему и R4 обозначает С1-6 алкильную группу (линейную или разветвленную) или ароматическую либо гетероароматическую кольцевую систему. Из пиперидин-2,6-дионов формулы (I), в которой Z обозначает -C(R1R2)-CH2-, R1 обозначает фталимид и R2 и R3 обозначают водород, наиболее предпочтительно соединение, в котором R4 является фенилом. Из пиперидин-2,6-дионов формулы (I), в которой Z обозначает -C(R1)=CH-, R3 обозначает этил и R4 обозначает фенил, наиболее предпочтительно соединение, в котором R1 является 3,4-диметоксифталимидом. Изобретение далее относится к способу получения аналогов талидомида из класса пиперидин-2,6-дионов общей формулы (I). Соединения общей формулы (I), в которой Z обозначает -C(R1R2)-СН2-, могут быть получены конденсацией фталевого ангидрида с помощью замещенной глутаминовой кислоты, например 4-фенилглутаминовой кислоты или 4-метилглутаминовой кислоты, в органическом растворителе, предпочтительно пиридине, циклизацией продукта в ацетангидриде и последующим переводом в имид. Превращение ангидрида в имид при этом осуществляется плавлением в присутствии мочевины. Эти целевые соединения формулы (I) могут быть получены также взаимодействием фталевого ангидрида с 5-замещенным 3-аминоглутаримидом, предпочтительно путем нагревания в уксусной кислоте. Соединения общей формулы (I), в которой Z обозначает -C(R1)=СН-, могут быть получены конденсацией замещенного фталевого ангидрида, например 3,4-диметоксифталевого ангидрида, с помощью 5-замещенных 3-амино-3,4-дегидропиперидин-2,6-дионов, таких как, например 3-амино-5-этил-5-фенилглутаконимид, в органическом растворителе, например, в уксусной кислоте. Пример 1. 2-(5-Метил-2,6-диоксопиперидин-3-ил)-1,3-дигидро-2Н-изоиндол-1,3-дион (1) 2,00 г (11 ммоль) 4-Метилглутаминовой кислоты и 1,95 г (13 ммоль) фталевого ангидрида нагревают с обратным холодильником в 15 мл сухого пиридина в течение 6 ч. После отгонки растворителя остаток нагревают в течение 1 ч в 10 мл ацетангидрида до кипения. Выпавшее в осадок при охлаждении твердое вещество подвергают вакуум-фильтрации и фильтрат концентрируют. После смешения фильтрата с простым эфиром образовавшийся осадок подвергают вакуум-фильтрации и объединенные осадки перекристаллизовывают из абсолютного толуола. 2,00 г (7 ммоль) кристаллизата и 0,23 г (3,8 ммоль) мочевины тщательно перемешивают и расплавляют в масляной бане при приблизительно 200oС в течение 30 мин. Застывший расплав быстро нагревают до кипения последовательно с 4 мл ацетангидрида и 6 мл этанола. Выпавшее в осадок твердое вещество подвергают вакуум-фильтрации и перекристаллизовывают из смеси ДМФ/вода. Таким путем получают 1,35 г (67% от теории) 2-(5-метил-2,6-диоксопиперидин-3-ил)-1,3-дигидро-2Н-изоиндол-1,3-диона (1) с температурой плавления 270-272oС. Пример 2. 2-(5-Фенил-2,6-диоксопиперидин-3-ил)-1,3-дигидро-2Н-изоиндол-1,3-дион (2) 3,00 г (12 ммоль) 4-Фенилглутаминовой кислоты и 2,12 г (14 ммоль) фталевого ангидрида нагревают с обратным холодильником в 40 мл сухого пиридина в течение 6 ч. После отгонки растворителя остаток растворяют в 50 мл 5%-ной НС1 и экстрагируют этилацетатом. Органическую фазу промывают водой, обесцвечивают активированным углем и сушат над сульфатом натрия. После отгонки растворителя остаток нагревают с обратным холодильником в течение 1 ч в 40 мл ацетангидрида. Затем раствор концентрируют и смешивают с простым эфиром. Выпавший осадок подвергают вакуум-фильтрации и перекристаллизовывают из сухого толуола. 2,00 г (6 ммоль) кристаллизата и 0,19 г (3 ммоль) мочевины расплавляют в масляной бане при приблизительно 200oС в течение 30 мин. Застывший расплав быстро нагревают до кипения последовательно с 4 мл ацетангидрида и 8 мл этанола. Выпавшее в осадок твердое вещество перекристаллизовывают из смеси ДМФ/вода. Таким путем получают 0,80 г (40% от теории) 2-(5-фенил-2,6-диоксопиперидин-3-ил)-1,3-дигидро-2Н-изоиндол-1,3-диона (2) с температурой плавления 228-231oС. Пример 3. 2-(5-Этил-5-фенил-2,6-диоксопиперидин-3-ил)-1,3-дигидро-2Н-изоиндол-1,3-дион (3) 1,00 г (4 ммоль) 3-Амино-5-этил-5-фенилглутаконимида растворяют в 40 мл безводного этанола, раствор смешивают с 0,1 г палладия на угле (10% Pd/C) и перемешивают в атмосфере водорода в течение 8,5 ч. Затем катализатор отфильтровывают и фильтрат концентрируют досуха. Остаток нагревают с обратным холодильником с 0,70 г (5 ммоль) фталевого ангидрида в 40 мл ледяного уксуса в течение 4 ч. После отгонки растворителя остаток перекристаллизовывают из этанола. Таким путем получают 0,99 г (63% от теории) 2-(5-этил-5-фенил-2,6-диоксопиперидин-3-ил)-1,3-дигидро-2Н-изоиндол-1,3-диона (3) с точкой плавления 174-177oС. Пример 4. 2-(5-Этил-5-фенил-2,6-диоксо-1,2,5,6-тетрагидропиридин-3-ил)-4,5-диметокси-1,3-дигидро-2Н-изоиндол-1,3-дион (4) 0,45 г (2 ммоль) 3-амино-5-этил-5-фенилглутаконимида и 0,45 г (2 ммоль) 4,5-диметоксифталевого ангидрида нагревают с обратным холодильником в 15 мл ледяного уксуса в течение 5 ч. Затем концентрируют досуха и остаток перекристаллизовывают из этанола. Таким путем получают 0,55 г (67% от теории) 2-(5-этил-5-фенил-2,6-диоксо-1,2,5,6-тетрагидропиперидин-3-ил)-4,5-диметокси-1,3-дигидро-2Н-изоиндол-1,3-диона (4) с температурой плавления 203-205oС. Соединения по изобретению безопасны с токсикологической точки зрения и поэтому пригодны для использования в качестве фармацевтически активных веществ. Соответственно этому изобретение также относится к применению аналогов талидомида из класса пиперидин-2,6-дионов формулы (I) в качестве активных веществ в лекарственных средствах, предпочтительно в качестве супрессантов для обусловленного воспалением высвобождения F-, a также индуцированного антигеном синтеза интерлейкина 2.
Лекарственные средства по изобретению наряду по меньшей мере с одним соединением общей формулы (I) содержат носители, наполнители, растворители, разбавители, красители и/или связующие. Выбор вспомогательных веществ, а также применяемые количества зависят от метода введения, а именно, должно ли лекарство применяться орально, внутривенно, внутрибрюшинно, внутрикожно, внутримышечно, назально, буккально или локально. Для орального применения пригодны препараты в форме таблеток, жевательных таблеток, драже, капсул, гранул, капель, микстур или сиропов, для парентерального, местного и ингаляционного применения пригодны растворы, суспензии, легко реконституируемые сухие препараты, а также аэрозоли. Примерами пригодных форм для чрескожного применения являются предлагаемые соединения в депо в растворенной форме, в пленке-носителе или в пластыре, при необходимости с добавкой средств, способствующих проникновению через кожу. Высвобождение предлагаемых соединений в организме из препаративных форм, применяемых орально или чрескожно, может происходить пролонгированно.
Назначаемая пациенту доза активных веществ зависит от веса пациента, метода введения, показания и степени тяжести заболевания. Обычно применяемая доза составляет от 1 до 150 мг/кг по меньшей мере одного аналога талидомида формулы (I).
Фармакологические исследованияВысвобождение F- может быть исследовано in vitro на мононуклеарных клетках периферической крови человека (Т-лимфоциты, В-лимфоциты и моноциты) после стимуляции липополисахаридом (ЛПС). ЛПС является составной частью стенки бактериальной клетки и стимулирует моноциты и макрофаги.
Наряду со стимуляцией с помощью ЛПС высвобождение F- может также провоцироваться стимуляцией мононуклеарных клеток периферической крови человека специфическими в отношении Т-лимфоцитов моноклональными антителами к активирующим антигенам (антиСD2-антиСD28) или бактериальным суперантигеном токсином-1, вырабатываемым при токсическом шоке (Toxic Shock Syndrome Toxin-1 или TSST-1). Помимо высвобождения F- эти стимуляторы приводят, в частности, к образованию интерлейкина-2 (IL-2).
Стимуляция ЛПС мононуклеарных клеток: действие на F-Высвобождение F- может быть исследовано in vitro на мононуклеарных клетках периферической крови человека, т.е. Т-лимфоцитах, В-лимфоцитах и моноцитах, после стимуляции липополисахаридом (ЛПС). ЛПС является составной частью стенки бактериальной клетки и стимулирует моноциты и макрофаги.
Мононуклеарные клетки получали из гепаринизированной крови по крайней мере трех добровольных доноров. С этой целью по 20 мл крови фракционировали по известной методике в градиентах плотности с использованием фиколл-пак (Ficoll-Paque). Затем клетки собирали и трижды промывали клеточной культуральный средой. Используемая клеточная культуральная среда состояла из среды RPMI 1640, дополненной 2 мМ глутамином (фирма Life Technologies, Эггенштейн), 10%-ной фетальной телячьей сывороткой (фирма Life Technologies), 50 мкг/мл стрептомицина (фирма Sigma, Дейзенхофен), 50 МЕ/мл пенициллина (фирма Sigma) и 100 мкМ  -меркаптоэтанолом (фирма Merck, Дармштадт). Затем мононуклеарные клетки вносили в 15 мл клеточной культуральной среды и полученную смесь распределяли порциями по 1 мл по стерильным инкубационным 24-луночным планшетам (фирма Sigma). К 1-миллилитровым порциям смеси добавляли соответственно по 1 мкл диметилсульфоксида (ДМСО, фирма Merck) или по 1 мкл раствора тестируемого вещества (в ДМСО; конечные концентрации в тесте: 0,5; 5; 12,5 и 50 мкг/мл) и смеси инкубировали в течение одного часа в инкубаторном шкафу в атмосфере СО2 (5% СО2, 90%-ная влажность воздуха). Затем добавляли, за исключением контроля, соответственно по 2,5 мкг ЛПС (Е. coli 0127: В8; фирма Sigma, Дейзенхофен). Инкубацию культур продолжали еще в течение 20 ч. Концентрацию F- в над осадочной жидкости культуры клеток определяли после окончания инкубации с помощью коммерческого набора для твердофазного иммуноферментного анализа (ELISA-анализ; фирма Boehringer-Mannheim). На основе данных, полученных для не обработанных активным веществом контрольных смесей и инкубированных тестируемыми соединениями смесей, рассчитывали степень ингибирования F-. С помощью прямой регрессии определяли концентрации, обеспечивающие 50%-ное ингибирование высвобождения F- (значения IC50).
В таблице представлено ингибирующее влияние соединений согласно изобретению на индуцированное липополисахаридом высвобождение F-. -меркаптоэтанолом (фирма Merck, Дармштадт). Затем мононуклеарные клетки вносили в 15 мл клеточной культуральной среды и полученную смесь распределяли порциями по 1 мл по стерильным инкубационным 24-луночным планшетам (фирма Sigma). К 1-миллилитровым порциям смеси добавляли соответственно по 1 мкл диметилсульфоксида (ДМСО, фирма Merck) или по 1 мкл раствора тестируемого вещества (в ДМСО; конечные концентрации в тесте: 0,5; 5; 12,5 и 50 мкг/мл) и смеси инкубировали в течение одного часа в инкубаторном шкафу в атмосфере СО2 (5% СО2, 90%-ная влажность воздуха). Затем добавляли, за исключением контроля, соответственно по 2,5 мкг ЛПС (Е. coli 0127: В8; фирма Sigma, Дейзенхофен). Инкубацию культур продолжали еще в течение 20 ч. Концентрацию F- в над осадочной жидкости культуры клеток определяли после окончания инкубации с помощью коммерческого набора для твердофазного иммуноферментного анализа (ELISA-анализ; фирма Boehringer-Mannheim). На основе данных, полученных для не обработанных активным веществом контрольных смесей и инкубированных тестируемыми соединениями смесей, рассчитывали степень ингибирования F-. С помощью прямой регрессии определяли концентрации, обеспечивающие 50%-ное ингибирование высвобождения F- (значения IC50).
В таблице представлено ингибирующее влияние соединений согласно изобретению на индуцированное липополисахаридом высвобождение F-.Стимуляция Т-лимфоцитов: ингибирование IL-2 Высвобождение интерлейкина-2 может быть исследовано путем стимуляции in vitro мононуклеарных клеток периферической крови человека, содержащей наряду с Т-лимфоцитами также В-лимфоциты и моноциты. Благодаря стимуляции поликлонов через постоянные эпитопы рецептора Т-лимфоцитов или так называемые добавочные сигнальные молекулы клеточной поверхности получают более показательные данные измерений по сравнению со стимуляцией антигенами отдельных малых популяций Т-лимфоцитов. Применяют комбинацию двух таких добавочных сигналов, а именно тех, воздействия которых опосредованы поверхностными молекулами CD2 и CD28. Мононуклеарные клетки получали из гепаринизированной крови по крайней мере трех добровольных доноров. С этой целью по 20 мл крови фракционировали по известной методике в градиентах плотности с использованием фиколл-пак (Ficoll-Paque). Затем клетки собирали и трижды промывали клеточной культуральный средой. Используемая клеточная культуральная среда состояла из среды RPMI 1640, дополненной 2 мМ глутамином (фирма Life Technologies, Эггенштейн), 10%-ной фетальной телячьей сывороткой (фирма Life Technologies), 50 мкг/мл стрептомицина (фирма Sigma, Дейзенхофен), 50 МЕ/мл пенициллина (фирма Sigma) и 100 мкМ -меркаптоэтанолом (фирма Merck, Дармштадт). Затем мононуклеарные клетки вносили в 15 мл клеточной культуральной среды и полученную смесь распределяли порциями по 1 мл по стерильным инкубационным 24-луночным планшетам (фирма Sigma). К 1-миллилитровым порциям смеси добавляли соответственно по 1 мкл диметилсульфоксида (ДМСО, фирма Merck) или по 1 мкл раствора тестируемого вещества (в ДМСО; конечные концентрации в тесте: 0,5; 5; 12,5 и 50 мкг/мл) и смеси инкубировали в течение одного часа в инкубаторном шкафу в атмосфере СО2 (5% СО2, 90%-ная влажность воздуха). Затем добавляли, за исключением контроля, соответственно по 0,1 мкг/мл моноклинальных антител (Клон AICD2.M1, предоставлен проф. д-ром Мойером; анти-СD28, получен от CLB, Амстердам). Инкубацию культур продолжали еще в течение 20 ч. Концентрацию IL-2 в надосадочной жидкости культуры клеток определяли после окончания инкубации с помощью набора для ELISA-анализа (фирма Boehringer-Mannheim). На основе данных, полученных для не обработанных активным веществом контрольных смесей и инкубированных тестируемыми соединениями смесей, рассчитывали степень ингибирования IL-2.
Вещество из примера 4 в этих условиях при концентрации 50 мкг/мл ингибирует стимулированный CD2/CD28 синтез IL-2 на 86 6%. При применении стафилоккового суперантигена (Е. coli 0127: В8; Sigma, Дайзенгофен) TSST-1 (0,1 мкг/мл) в качестве стимулятора Т-лимфоцитов синтез IL-2 подавляется на 7720%.
Вышеприведенные исследования показывают, что аналоги талидомида из класса пиперидин-2,6-дионов формулы (I) ингибируют как обусловленное воспалением высвобождение F-, так и индуцированный антигенами синтез интерлейкина-2. 6%. При применении стафилоккового суперантигена (Е. coli 0127: В8; Sigma, Дайзенгофен) TSST-1 (0,1 мкг/мл) в качестве стимулятора Т-лимфоцитов синтез IL-2 подавляется на 7720%.
Вышеприведенные исследования показывают, что аналоги талидомида из класса пиперидин-2,6-дионов формулы (I) ингибируют как обусловленное воспалением высвобождение F-, так и индуцированный антигенами синтез интерлейкина-2.
Формула изобретения
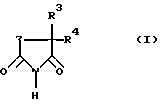 в которой Z – -C(R1R2)-CH2– или -C(R1)= CH-; R1 – фталимид, когда Z – -С(R1R2)-CH2-, или одно- либо двукратно замещенный гидрокси-, метокси- или аминогруппами фталимидный радикал, когда Z – -C(R1)= CH-; R2 – водород или С1-6 алкильная группа; R3 – водород, C1-6 алкильная группа или ароматическая кольцевая система; R4 – C1-6 алкил или ароматическое кольцо. 2. Замещенные пиперидин-2,6-дионы формулы (I) по п. 1, отличающиеся тем, что Z – -С(R1R2)-CH2-; R1 – фталимид; R2 – водород; R3 – водород или этил и R4 – метил или фенил. 3. Замещенные пиперидин-2,6-дионы формулы (I) по п. 1, отличающиеся тем, что Z – -С(R1)= СН2-; R1 – 3,4-диметоксифталимид; R3 – этил и R4 – фенил. 4. Способ получения замещенных пиперидин-2,6-дионов формулы (I) по пп. 1 и 2, отличающийся тем, что фталевый ангидрид конденсируют с замещенной глутаминовой кислотой, продукт циклизуют до ангидрида и последний переводят в имид. 5. Способ получения замещенных пиперидин-2,6-дионов формулы (I) по пп. 1-3, отличающийся тем, что фталевый ангидрид конденсируют с замещенным 3-аминоглутаконимидом или 5-замещенным 3-аминоглутаримидом. РИСУНКИ
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 31.01.2006
Извещение опубликовано: 20.12.2007 БИ: 35/2007
|
||||||||||||||||||||||||||