Патент на изобретение №2184125
|
||||||||||||||||||||||||||
(54) ВОДНАЯ ГЕТЕРОПОЛИМЕРНАЯ ДИСПЕРСИЯ ДЛЯ ИЗГОТОВЛЕНИЯ ПОКРЫТИЙ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ
(57) Реферат: Изобретение относится к дисперсиям для изготовления покрытий, в частности прозрачных, ударопрочных покрытий на стеклянных изделиях, например светотехнических изделиях, экранах дисплеев, стеклянных волокнах и т.д. Частицы водной гетерополимерной дисперсии для изготовления покрытий содержат сополимеры, включающие звенья акриловых мономеров, и состоят из ядра, промежуточной оболочки и внешней оболочки. Сополимер ядра является сополимером (мет)акрилового эфира или смеси (мет)акриловых эфиров, этиленненасыщенного мономера, содержащего две или три связи С=С, и, возможно, этиленненасыщенного мономера, содержащего карбоксильные группы, и имеет температуру стеклования менее oС. Сополимер промежуточной оболочки является сополимером метакрилового эфира или смеси метакриловых эфиров, этиленненасыщенного мономера, содержащего две или три связи С=С, и, возможно, этиленненасыщенного мономера, содержащего карбоксильные группы, и имеет температуру стеклования 50-85oС. Сополимер внешней оболочки является сополимером метакрилового эфира или смеси метакриловых эфиров и этиленненасыщенного мономера, содержащего карбоксильные группы, и имеет температуру стеклования 25-70oС. Массовое соотношение сополимеров ядра, промежуточной и внешней оболочек находится в пределах (60-75): (7,5-20): (10-22,5), а средний диаметр частиц дисперсии составляет 50-220 нм. Водную гетерополимерную дисперсию для изготовления покрытий получают путем многостадийной эмульсионной сополимеризации, включающей последовательные стадии эмульсионной сополимеризации мономеров ядра и мономеров оболочки при температуре менее 100oС в присутствии водорастворимого свободнорадикального инициатора и анионного поверхностно-активного вещества (ПАВ). Стадию получения сополимера ядра проводят в два этапа, на первом из которых проводят эмульсионную сополимеризацию в присутствии инициатора и анионного ПАВ при единовременной загрузке 10-30% от общего количества мономерной смеси ядра, а на втором этапе в образовавшийся затравочный латекс дозируют оставшуюся часть мономеров ядра или эмульсию этой части мономеров ядра, стабилизированную анионным ПАВ. Стадию получения сополимера промежуточной оболочки проводят путем непрерывного дозирования мономерной смеси, образующей промежуточную оболочку, в дисперсию сополимера ядра. Перед началом этой стадии в дисперсию сополимера ядра вводят водный раствор инициатора. Получение сополимера промежуточной оболочки можно производить в два этапа, причем составы мономерной смеси на указанных этапах различаются. Стадию получения сополимера внешней оболочки проводят путем эмульсионной сополимеризации мономеров, образующих внешнюю оболочку, в присутствии дисперсии, полученной на предыдущей стадии. Полученные по упрощенной технологии гетерополимерные водные дисперсии обеспечивают прозрачность, твердость не липкость и ударопрочность покрытий. 2 с. и 5 з. ф-лы, 1 ил., 2 табл. Изобретение относится к водным гетерополимерным дисперсиям, используемым для изготовления покрытий, в частности прозрачных, ударопрочных покрытий на стеклянных изделиях. Такие дисперсии могут найти применение при получении безосколочных стекол, защитных покрытий на стеклянных светотехнических изделиях, на экранах дисплеев, на стеклянных волокнах и т.д. Водная основа полимерных дисперсий создает благоприятные технологические и экологические условия при формировании полимерных покрытий на стеклянных изделиях распылением, маканием и другими методами. Известен ряд водных полимерных дисперсий или композиций на их основе, пригодных для получения покрытий на стекле. Известна водная композиция на основе эластомерного латекса, пластификатора и небольших количеств других ингредиентов (патент США 4263362, В 32 В 7/00, 1981). В качестве эластомерного латекса композиция содержит латекс полибутадиена или карбоксилированного стирол-бутадиенового сополимера. Покрытия, полученные на основе этих полимеров, являются липкими и недостаточно твердыми вследствие того, что входящие в их состав полимеры, особенно в присутствии пластификаторов, имеют низкую температуру стеклования. Известна композиция, включающая латекс на основе сополимера бутадиена, ненасыщенной карбоновой кислоты и винилового соединения (патент США 3919440, В 32 В 17/10, 1975). Покрытия на стеклянных изделиях, полученные на основе этой композиции, имеют недостаточную тепло- и светостойкость, а также низкую прочность, так как они частично разрушаются даже при толщине 145-250 мкм. Известны водные полимерные композиции, также включающие латексы полибутадиена или сополимеров на основе стирола, бутадиена и винилпиридина (патенты США 4060658, В 32 В 9/00, D 02 G 3/00, 1977; 3853605, В 32 В 17/06, В 32 В 17/10, С 03 С 25/02, 1974). В состав этих композиций также входят резорцинформальдегидные смолы, что позволяет рассчитывать на образование более прочных и твердых покрытий. Однако высокое содержание в полимерах и сополимерах бутадиена углерод-углеродных двойных связей снижает химическую стойкость и атмосферостойкость получаемых покрытий. Полимерные композиции на основе акриловых мономеров более соответствуют требованиям к покрытиям на изделиях из стекла. Так, например, известны водные дисперсии термореактивных композиций, включающих акриловые сополимеры и эпоксидные смолы (патент США 3970628, C 08 L 63/10, 1976), которые могли бы быть использованы для покрытий на стекле, однако сведения об их прочности и прозрачности отсутствуют. Известны композиции, включающие акриловый латекс, в частности латекс сополимера акрилонитрила и этилакрилата, сшивающий агент и загуститель (патенты США 3944100, B 65 D 23/00, C 08 L 61/20, 1976, и 4098934, В 32 В 17/10, 1978). Эти композиции используются для покрытия стеклянных изделий, но для их отверждения требуются высокие температуры так же, как и для композиций по патенту США 3970628. Известны также водные акриловые полимерные дисперсии, используемые для изготовления покрытий на стекловолокнах (патент США 5827612, D 02 G 3/00, 1998), свободные от указанного недостатка. Процесс получения полимерных покрытий на основе этих дисперсий состоит из следующих стадий: 1. Нанесение первой акриловой полимерной дисперсии с использованием известных методов на поверхность стеклянных волокон. 2. Высушивание полученного первого полимерного слоя при комнатной или повышенной температуре. 3. Нанесение второго полимерного слоя, например, маканием изделия во вторую акриловую полимерную дисперсию, отличающуюся по составу от первой. 4. Высушивание второго полимерного слоя. Недостатком данных композиций является многостадийность получения покрытий на их основе. Предпочтительнее было бы использовать водные полимерные композиции, которые могут обеспечить получение полимерного покрытия за один цикл “нанесение покрытия – сушка”. Известна водная полимерная композиция, которая позволяет получать безосколочные покрытия на стекле с улучшенными механическими характеристиками, на основе акриловых полимерных дисперсий, например Нycar 2679, в которые вводили 1%-ный раствор полиакриламида Polyhall 295, вероятно, для загущения дисперсии, что необходимо для регулирования толщины покрытия (заявка Германии 2424329, В 65 D 23/08, С 03 с 17/32, 1974). Композиция образует покрытие на стеклянных изделиях, имеющее улучшенные механические свойства и обеспечивающее отсутствие осколков при разрушении изделия; такие покрытия и изделия, в которых они использованы, здесь и далее именуются безосколочными. Покрытия на основе этой композиции получали маканием стеклянных изделий в полимерную дисперсию, последующим подсушиванием при комнатной температуре и окончательным отверждением покрытия при 232oС в течение 4 мин. Ударопрочность покрытия оценивали путем разбивания в определенных условиях с определением числа осколков, на которое распадается изделие при разрушении. В зависимости от типа стеклянное изделие без полимерного покрытия разрушалось с образованием 120-155 осколков (таблица 1 из описания изобретения к заявке Германии 2424329). При нанесении покрытия на стеклянное изделие число осколков при разрушении уменьшалось в зависимости от толщины покрытия до 18-43 (примеры F, G, Н в указанной табл.1). Таким образом, стеклянные изделия с указанным акриловым полимерным покрытием нельзя считать безосколочными. Ударопрочность улучшается и изделие становится безосколочным, если перед нанесением полимерного акрилового покрытия поверхность стекла сначала обрабатывается смачивателем – тетраизопропилтитанатом, а затем полиэтиленовой эмульсией SC-100 (пример К, табл.1). Недостаток описанной композиции состоит в многостадийности процесса получения покрытия на ее основе. Для получения безосколочных стеклянных изделий с использованием этой композиции оказывается необходимым проведение по крайней мере трех последовательных операций “нанесение-сушка”. Покрытия на основе различных латексных систем, описанных выше, являются, как правило, однофазными полимерными системами, которые могут содержать твердые включения отвержденных термореактивных смол. Для получения ударопрочных покрытий более предпочтительным оказывается использование микрогетерогенных полимерных систем. Наиболее близкой к заявляемой по своей технической сущности является известная водная гетерополимерная дисперсия для изготовления покрытий, частицы которой содержат сополимеры, включающие звенья акриловых мономеров, и состоят из ядра и одной наружной оболочки (патент США 4009317, В 32 В 17/02, С 08 L 23/02, 1977). Водную полимерную композицию, приготовленную с использованием указанной дисперсии, применяли для однократной обработки стекловолокон. Использование мономеров акрилового типа на стадиях получения полимеров ядра и оболочки позволяет предполагать, что покрытия на базе этих дисперсий могут быть оптически прозрачными, а при наличии в составе большого количества полимера оболочки – также и ударопрочными. Однако в патенте США 4009317 отсутствуют какие-либо показатели, характеризующие прозрачность и ударопрочность получаемых покрытий. Основной недостаток покрытий, изготавливаемых на основе водных полимерных композиций, получаемых согласно этому патенту, состоит в низкой температуре стеклования полимера оболочки и наличии в композиции различных добавок (парафин, воск, катионная смазка и др.). Оба указанных фактора, повышая липкость покрытия, препятствуют получению покрытий с достаточной поверхностной твердостью. Известны способы получения водных гетерополимерных дисперсий для изготовления покрытий, частицы которых содержат сополимеры, включающие звенья акриловых мономеров, путем эмульсионной сополимеризации указанных мономеров в несколько стадий в присутствии анионного поверхностно-активного вещества (ПАВ) и водорастворимого свободнорадикального инициатора (патенты США 4469825, С 08 F 265/10, 1984; 5157084, С 08 F 265/02, 1992). Наиболее близким к предложенному по технической сущности является известный способ получения водной гетерополимерной дисперсии для изготовления покрытий, применяемых для обработки стекловолокон, частицы которой содержат сополимеры, включающие звенья акриловых мономеров, и состоят из ядра и одной наружной оболочки, путем двухстадийной эмульсионной сополимеризации, включающей последовательные стадии эмульсионной сополимеризации мономеров ядра и мономеров внешней оболочки при температуре менее 100oС в присутствии водорастворимого свободнорадикального инициатора и анионного ПАВ (патент США 4009317, В 32 В 17/02, С 08 L 23/02, 1977). Ядро частиц этой дисперсии получали эмульсионной полимеризацией алкилметакрилатов, обычно метилметакрилата. Затем в присутствии полученной дисперсии осуществляли затравочную эмульсионную полимеризацию мономеров, обычно алкилакрилатов, с образованием полимерной оболочки частиц, имеющей температуру стеклования 10oС или ниже. Полученная таким образом полимерная дисперсия не используется непосредственно для получения покрытий, а смешивается с предварительно приготовленной эмульсией некоторых добавок (парафин, воск и др.), так что процесс получения конечной композиции для покрытий включает по крайней мере три процесса, а именно: получение гетерополимерной дисперсии, получение эмульсии и смешение двух компонентов. Технический результат, достижение которого обеспечивает настоящее изобретение, заключается в упрощении технологии получения ударопрочных полимерных покрытий на стекле благодаря использованию водной полимерной дисперсии определенного состава, получаемой заявленным способом. Изобретение обеспечивает получение ударопрочного безосколочного покрытия за один цикл нанесение-сушка. Указанный технический результат достигается за счет того, что для получения покрытий, в частности прозрачных ударопрочных покрытий на стекле, предложена водная гетерополимерная дисперсия для изготовления покрытий, частицы которой содержат сополимеры, включающие звенья акриловых мономеров, и состоят из ядра и по крайней мере одной наружной оболочки, отличающаяся тем, что ее частицы состоят из ядра, промежуточной оболочки и внешней оболочки, сополимер ядра является сополимером (мет)акрилового эфира или смеси (мет)акриловых эфиров, этиленненасыщенного мономера, содержащего две или три связи С= С, и, возможно, этиленненасыщенного мономера, содержащего карбоксильные группы, и имеет температуру стеклования менее 7oС, сополимер промежуточной оболочки является сополимером метакрилового эфира или смеси метакриловых эфиров, этиленненасыщенного мономера, содержащего две или три связи С= С, и, возможно, этиленненасыщенного мономера, содержащего карбоксильные группы, и имеет температуру стеклования 50-85oС, а сополимер внешней оболочки является сополимером метакрилового эфира или смеси метакриловых эфиров и этиленненасыщенного мономера, содержащего карбоксильные группы, и имеет температуру стеклования 25-70oС, причем массовое соотношение сополимеров ядра, промежуточной и внешней оболочек находится в пределах (60-75):(7,5-20): (10-22,5), а диаметр частиц дисперсии составляет 50-220 нм. При получении гетерополимерной дисперсии предпочтительно используют – в качестве (мет)акрилового эфира – соединение общей формулы CH2= C(R)-C(О)OR1, где R= Н или СН3, R1-C1-C18 алкил, например, выбранное из группы, включающей метил-, этил-, н.-бутил-, изобутил-, н.-октил-, 2-этилгексил-, додецилакрилат, метил-, этилметакрилат; – в качестве этиленненасыщенного мономера, содержащего две или три связи С=С, -ди(мет)акрилат этилен-, диэтилен- или триэтиленгликоля, аллил(мет)акрилат, триметилолпропантриметакрилат, триаллилцианурат, дивинилбензол; – в качестве этиленненасыщенного мономера, содержащего карбоксильные группы, – акриловую кислоту или ее  -(C1-C5 алкил)-замещенные, малеиновую, фумаровую, итаконовую кислоты или их моно-(С1-С4 алкиловые) эфиры.
Предпочтительно сополимеры, входящие в состав частиц дисперсии, могут характеризоваться следующим образом: сополимер ядра является сополимером (мет)акрилового эфира или смеси (мет)акриловых эфиров, ди(мет)акрилового эфира и, возможно, (мет)акриловой кислоты, содержит 0,1-0,5 мас.% звеньев ди(метакрилового) эфира и 0-5,0 мас.% звеньев (мет)акриловой кислоты, сополимер промежуточной оболочки является сополимером метакрилового эфира или смеси метакриловых эфиров, ди(мет)акрилового эфира и, возможно, (мет)акриловой кислоты, содержит 0,5-2,0 мас.% звеньев ди(мет)акрилового эфира и 0-7,5 мас. % звеньев (мет)акриловой кислоты, а сополимер внешней оболочки является сополимером метакрилового эфира или смеси метакриловых эфиров и (мет)акриловой кислоты и содержит 7,5-10,0 мас.% звеньев (мет)акриловой кислоты.
Для получения указанной дисперсии предложен способ получения водной гетерополимерной дисперсии для изготовления покрытий, частицы которой содержат сополимеры, включающие звенья акриловых мономеров, и состоят из ядра и по крайней мере одной наружной оболочки, путем многостадийной эмульсионной сополимеризации, включающей последовательные стадии эмульсионной сополимеризации мономеров ядра и мономеров оболочек при температуре менее 100oС в присутствии водорастворимого свободнорадикального инициатора и анионного поверхностно-активного вещества (ПАВ), отличающийся тем, что стадию получения сополимера ядра проводят в два этапа, на первом из которых проводят эмульсионную сополимеризацию в присутствии инициатора и анионного ПАВ при единовременной загрузке 10-30% от общего количества мономерной смеси ядра, а на втором этапе в образовавшийся затравочный латекс дозируют оставшуюся часть мономеров ядра или эмульсию этой части мономеров ядра, стабилизированную анионным ПАВ, стадию получения сополимера промежуточной оболочки проводят путем непрерывного дозирования мономерной смеси, образующей промежуточную оболочку, в дисперсию сополимера ядра, причем перед началом данной стадии в дисперсию сополимера ядра вводят водный раствор инициатора, стадию получения сополимера внешней оболочки проводят путем эмульсионной сополимеризации мономеров, образующих внешнюю оболочку, в присутствии дисперсии, полученной на предыдущих стадиях, причем загрузку мономеров внешней оболочки производят либо двумя равными порциями, вводя при этом в дисперсию водные растворы инициатора и анионного ПАВ перед загрузкой мономеров внешней оболочки, либо путем непрерывного дозирования в виде водной эмульсии мономеров, стабилизированной анионным ПАВ, одновременно дозируя водный раствор инициатора, который может содержать растворенное анионное ПАВ, и после завершения процесса в полученную дисперсию вводят водный раствор аммиака, летучего алифатического амина, либо водорастворимого гидроксида или соли двухвалентного металла в количестве 0,1-1,0 моль на 1 моль звеньев мономера, содержащего карбоксильные группы.
Согласно предложенному способу стадию получения сополимера ядра проводят в два приема. Вначале в водную фазу, содержащую растворенный инициатор и анионное ПАВ, загружают 10-30% мономерной смеси от общего количества мономерной смеси ядра, проводят процесс полимеризации и получают затравочный латекс. На втором этапе в затравочный латекс начинают дозировать оставшуюся часть мономерной смеси. Указанная часть мономерной смеси может дозироваться в виде заранее приготовленной эмульсии, стабилизированной анионным ПАВ. Для получения сополимера ядра предпочтительно использовать алкилакрилат или смесь алкилакрилата и алкилметакрилата, причем массовое соотношение последних выбирают таким образом, чтобы температура стеклования сополимера ядра была не выше 7o, предпочтительно от -25 до -54oС.
Стадию получения сополимера промежуточной оболочки можно проводить при непрерывном дозировании мономерной смеси в дисперсию сополимера ядра. Перед началом дозирования мономерной смеси, образующей сополимер промежуточной оболочки, в дисперсию сополимера ядра вводят водный раствор инициатора. На данной стадии в качестве алкилметакрилата можно использовать смесь двух алкилметакрилатов с различными заместителями, но их массовое соотношение выбирают таким образом, чтобы сополимер промежуточной оболочки имел температуру стеклования в пределах 50-85oС, предпочтительно 50-70oС.
Получение сополимера промежуточной оболочки можно также проводить в два этапа, на каждом из которых состав мономерной смеси различается, при этом на втором этапе одновременно дозируют мономерную смесь и водный раствор инициатора и анионного ПАВ.
Стадию получения сополимера внешней оболочки проводят путем эмульсионной сополимеризации мономеров внешней оболочки в присутствии дисперсии, полученной на предыдущей стадии и содержащей частицы, состоящие из ядра и промежуточной оболочки. Загрузка мономеров может производиться двумя равными порциями. В этом случае перед загрузкой мономеров, образующих сополимер внешней оболочки, вводят водные растворы инициатора и анионного ПАВ. Предпочтительнее мономерную смесь внешней оболочки дозировать непрерывно в виде водной эмульсии, стабилизированной анионным ПАВ, при этом одновременно может дозироваться водный раствор, содержащий растворенные инициатор и анионное ПАВ. В качестве алкилметакрилата на данной стадии может быть использован один мономер или смесь двух алкилметакрилатов с различными алкильными заместителями, но их массовое соотношение выбирают таким образом, чтобы сополимер внешней оболочки имел температуру стеклования в пределах 25-70oС, предпочтительнее 30-60oС.
После завершения полимеризационного процесса в полученную водную гетерополимерную дисперсию вводят водный раствор аммиака, летучего алифатического амина, такого как триметиламин, диэтиламин, триэтиламин, диэтилэтаноламин, либо водорастворимого гидроксида или соли двухвалентного металла, например, бария, цинка, предпочтительно гидроксид бария, ацетат цинка или его аммиачный комплекс, в количестве 0,1-1,0 моль на 1 моль звеньев использованного в процессе этиленненасыщенного мономера, содержащего карбоксильные группы. Введение этих агентов приводит по крайней мере к частичной нейтрализации карбоксильных групп упомянутого ненасыщенного мономера. Кроме того, соединения двухвалентных металлов не только нейтрализуют карбоксильные группы, но действуют также как сшивающие агенты, повышая поверхностную твердость покрытия. При использовании соединений двухвалентных металлов перед их загрузкой вводят неионное ПАВ (например, оксиэтилированный алифатический спирт) в количестве 0,5-1,0% от массы дисперсии в расчете на содержание в ней сухого вещества.
Сополимеризацию на каждой стадии процесса проводят практически до полного превращения мономеров в сополимер, т.е. состав образующихся сополимеров соответствует составу мономерной смеси. Это достигается выбранными концентрациями инициатора и анионного ПАВ, продолжительностью процесса и дополнительной выдержкой полимерной дисперсии после каждой стадии при температуре реакции после окончания загрузки компонентов в течение 30 мин. Предпочтительно использовать водорастворимый инициатор и анионное ПАВ, соответственно, в количестве 0,3-0,4% и 0,2-2,0% от массы мономеров при получении сополимера ядра, 0,3-0,9% и 0-0,5% от массы мономеров при получении сополимера промежуточной оболочки, 0,3-1,2% и 0,3-3,0% от массы мономеров при получении сополимера внешней оболочки.
В качестве инициаторов эмульсионной сополимеризации используют известные водорастворимые соединения, например персульфаты аммония, натрия, калия.
В качестве анионных ПАВ предпочтительно использовать C12-C18-алкилсульфонаты натрия, например, Elfan OS-46 (смесь алкилов C14-C16 – фирма Akzo Nobel), либо C10-C18-алкиларилсульфонаты или С12-С18-алкилсульфаты натрия.
При получении гетерополимерной водной дисперсии очень важным является соблюдение предложенных соотношений количеств мономеров, загружаемых на различных стадиях процесса. При отклонении от вышеуказанных соотношений покрытия, получаемые с использованием гетерополимерной водной дисперсии, являются либо липкими, либо не обладают необходимой ударопрочностью.
Полученные по настоящему изобретению водные гетерополимерные дисперсии характеризуются следующими свойствами: -(C1-C5 алкил)-замещенные, малеиновую, фумаровую, итаконовую кислоты или их моно-(С1-С4 алкиловые) эфиры.
Предпочтительно сополимеры, входящие в состав частиц дисперсии, могут характеризоваться следующим образом: сополимер ядра является сополимером (мет)акрилового эфира или смеси (мет)акриловых эфиров, ди(мет)акрилового эфира и, возможно, (мет)акриловой кислоты, содержит 0,1-0,5 мас.% звеньев ди(метакрилового) эфира и 0-5,0 мас.% звеньев (мет)акриловой кислоты, сополимер промежуточной оболочки является сополимером метакрилового эфира или смеси метакриловых эфиров, ди(мет)акрилового эфира и, возможно, (мет)акриловой кислоты, содержит 0,5-2,0 мас.% звеньев ди(мет)акрилового эфира и 0-7,5 мас. % звеньев (мет)акриловой кислоты, а сополимер внешней оболочки является сополимером метакрилового эфира или смеси метакриловых эфиров и (мет)акриловой кислоты и содержит 7,5-10,0 мас.% звеньев (мет)акриловой кислоты.
Для получения указанной дисперсии предложен способ получения водной гетерополимерной дисперсии для изготовления покрытий, частицы которой содержат сополимеры, включающие звенья акриловых мономеров, и состоят из ядра и по крайней мере одной наружной оболочки, путем многостадийной эмульсионной сополимеризации, включающей последовательные стадии эмульсионной сополимеризации мономеров ядра и мономеров оболочек при температуре менее 100oС в присутствии водорастворимого свободнорадикального инициатора и анионного поверхностно-активного вещества (ПАВ), отличающийся тем, что стадию получения сополимера ядра проводят в два этапа, на первом из которых проводят эмульсионную сополимеризацию в присутствии инициатора и анионного ПАВ при единовременной загрузке 10-30% от общего количества мономерной смеси ядра, а на втором этапе в образовавшийся затравочный латекс дозируют оставшуюся часть мономеров ядра или эмульсию этой части мономеров ядра, стабилизированную анионным ПАВ, стадию получения сополимера промежуточной оболочки проводят путем непрерывного дозирования мономерной смеси, образующей промежуточную оболочку, в дисперсию сополимера ядра, причем перед началом данной стадии в дисперсию сополимера ядра вводят водный раствор инициатора, стадию получения сополимера внешней оболочки проводят путем эмульсионной сополимеризации мономеров, образующих внешнюю оболочку, в присутствии дисперсии, полученной на предыдущих стадиях, причем загрузку мономеров внешней оболочки производят либо двумя равными порциями, вводя при этом в дисперсию водные растворы инициатора и анионного ПАВ перед загрузкой мономеров внешней оболочки, либо путем непрерывного дозирования в виде водной эмульсии мономеров, стабилизированной анионным ПАВ, одновременно дозируя водный раствор инициатора, который может содержать растворенное анионное ПАВ, и после завершения процесса в полученную дисперсию вводят водный раствор аммиака, летучего алифатического амина, либо водорастворимого гидроксида или соли двухвалентного металла в количестве 0,1-1,0 моль на 1 моль звеньев мономера, содержащего карбоксильные группы.
Согласно предложенному способу стадию получения сополимера ядра проводят в два приема. Вначале в водную фазу, содержащую растворенный инициатор и анионное ПАВ, загружают 10-30% мономерной смеси от общего количества мономерной смеси ядра, проводят процесс полимеризации и получают затравочный латекс. На втором этапе в затравочный латекс начинают дозировать оставшуюся часть мономерной смеси. Указанная часть мономерной смеси может дозироваться в виде заранее приготовленной эмульсии, стабилизированной анионным ПАВ. Для получения сополимера ядра предпочтительно использовать алкилакрилат или смесь алкилакрилата и алкилметакрилата, причем массовое соотношение последних выбирают таким образом, чтобы температура стеклования сополимера ядра была не выше 7o, предпочтительно от -25 до -54oС.
Стадию получения сополимера промежуточной оболочки можно проводить при непрерывном дозировании мономерной смеси в дисперсию сополимера ядра. Перед началом дозирования мономерной смеси, образующей сополимер промежуточной оболочки, в дисперсию сополимера ядра вводят водный раствор инициатора. На данной стадии в качестве алкилметакрилата можно использовать смесь двух алкилметакрилатов с различными заместителями, но их массовое соотношение выбирают таким образом, чтобы сополимер промежуточной оболочки имел температуру стеклования в пределах 50-85oС, предпочтительно 50-70oС.
Получение сополимера промежуточной оболочки можно также проводить в два этапа, на каждом из которых состав мономерной смеси различается, при этом на втором этапе одновременно дозируют мономерную смесь и водный раствор инициатора и анионного ПАВ.
Стадию получения сополимера внешней оболочки проводят путем эмульсионной сополимеризации мономеров внешней оболочки в присутствии дисперсии, полученной на предыдущей стадии и содержащей частицы, состоящие из ядра и промежуточной оболочки. Загрузка мономеров может производиться двумя равными порциями. В этом случае перед загрузкой мономеров, образующих сополимер внешней оболочки, вводят водные растворы инициатора и анионного ПАВ. Предпочтительнее мономерную смесь внешней оболочки дозировать непрерывно в виде водной эмульсии, стабилизированной анионным ПАВ, при этом одновременно может дозироваться водный раствор, содержащий растворенные инициатор и анионное ПАВ. В качестве алкилметакрилата на данной стадии может быть использован один мономер или смесь двух алкилметакрилатов с различными алкильными заместителями, но их массовое соотношение выбирают таким образом, чтобы сополимер внешней оболочки имел температуру стеклования в пределах 25-70oС, предпочтительнее 30-60oС.
После завершения полимеризационного процесса в полученную водную гетерополимерную дисперсию вводят водный раствор аммиака, летучего алифатического амина, такого как триметиламин, диэтиламин, триэтиламин, диэтилэтаноламин, либо водорастворимого гидроксида или соли двухвалентного металла, например, бария, цинка, предпочтительно гидроксид бария, ацетат цинка или его аммиачный комплекс, в количестве 0,1-1,0 моль на 1 моль звеньев использованного в процессе этиленненасыщенного мономера, содержащего карбоксильные группы. Введение этих агентов приводит по крайней мере к частичной нейтрализации карбоксильных групп упомянутого ненасыщенного мономера. Кроме того, соединения двухвалентных металлов не только нейтрализуют карбоксильные группы, но действуют также как сшивающие агенты, повышая поверхностную твердость покрытия. При использовании соединений двухвалентных металлов перед их загрузкой вводят неионное ПАВ (например, оксиэтилированный алифатический спирт) в количестве 0,5-1,0% от массы дисперсии в расчете на содержание в ней сухого вещества.
Сополимеризацию на каждой стадии процесса проводят практически до полного превращения мономеров в сополимер, т.е. состав образующихся сополимеров соответствует составу мономерной смеси. Это достигается выбранными концентрациями инициатора и анионного ПАВ, продолжительностью процесса и дополнительной выдержкой полимерной дисперсии после каждой стадии при температуре реакции после окончания загрузки компонентов в течение 30 мин. Предпочтительно использовать водорастворимый инициатор и анионное ПАВ, соответственно, в количестве 0,3-0,4% и 0,2-2,0% от массы мономеров при получении сополимера ядра, 0,3-0,9% и 0-0,5% от массы мономеров при получении сополимера промежуточной оболочки, 0,3-1,2% и 0,3-3,0% от массы мономеров при получении сополимера внешней оболочки.
В качестве инициаторов эмульсионной сополимеризации используют известные водорастворимые соединения, например персульфаты аммония, натрия, калия.
В качестве анионных ПАВ предпочтительно использовать C12-C18-алкилсульфонаты натрия, например, Elfan OS-46 (смесь алкилов C14-C16 – фирма Akzo Nobel), либо C10-C18-алкиларилсульфонаты или С12-С18-алкилсульфаты натрия.
При получении гетерополимерной водной дисперсии очень важным является соблюдение предложенных соотношений количеств мономеров, загружаемых на различных стадиях процесса. При отклонении от вышеуказанных соотношений покрытия, получаемые с использованием гетерополимерной водной дисперсии, являются либо липкими, либо не обладают необходимой ударопрочностью.
Полученные по настоящему изобретению водные гетерополимерные дисперсии характеризуются следующими свойствами:– сухой остаток 36-50 мас.%, – диаметр частиц 50-220 нм, – рН 7-8,4 при использовании аммиака и 2,2-2,3 при использовании ацетата цинка, – вязкость по вискозиметру ВЗ-4 30-53 с. Покрытия, изготовленные с использованием полученных дисперсий, характеризуются высокой прозрачностью (интегральное светопропускание 91-92%), твердостью и ударопрочностью. Стеклянные изделия с покрытием являются истинно безосколочными, поскольку при их разрушении все фрагменты стекла остаются в полимерной оболочке. Свойства полученных водных гетерополимерных дисперсий и покрытий на их основе определяли по следующим методикам: 1. рН дисперсий определяли на универсальном иономере ЭВ-74. 2. Размер частиц дисперсии определяли широко известным методом рассеяния света разбавленной дисперсией, основанным на логарифмической зависимости оптической плотности от длины волны (В.И. Кленин и др. Характеристические функции светорассеяния дисперсных систем. Саратов: Изд-во Саратовского университета, 1977, 177 с.). 3. Содержание сухого остатка в дисперсии определяли гравиметрическим методом по ГОСТ 25709-83. 4. Адгезию сополимеров определяли методом отслаивания латексной пленки от стеклянной пластины под углом 180o. 5. Твердость покрытий определяли методом затухания колебаний маятника по ГОСТ 5233-89 (вес маятника 120  1 г, длина маятника 5001 мм, диаметр шарика из нержавеющей стали 7,938 мм). Относительную твердость Н рассчитывали по уравнению 1 г, длина маятника 5001 мм, диаметр шарика из нержавеющей стали 7,938 мм). Относительную твердость Н рассчитывали по уравнениюH=t/t1, где t – время затухания от отклонения в 5o до отклонения в 2o на покрытии, нанесенном на стеклянную основу, t1 – время затухания от отклонения в 5o до отклонения в 2o на стеклянной основе без покрытия. 6. Условную вязкость дисперсий определяли вискозиметрически с использованием вискозиметра ВЗ-4 по ГОСТ 8420-74 (объем воронки 120 1 см3, диаметр отверстия 4 мм).
7. Коэффициент светопропускания определяли с помощью фотометра в соответствии с ГОСТ 15875-80.
8. Липкость покрытия определяли по качественному тесту. На покрытие толщиной не менее 100 мкм, нанесенное на ровную стеклянную поверхность, накладывали лист стандартной писчей бумаги и прижимали ее к покрытию с усилием 0,005 МПа. Покрытие считается не липким, если не наблюдается следов бумаги, прилипшей к покрытию.
9. Температуру стеклования Тст. сополимеров рассчитывали по уравнению Фокса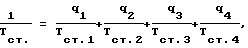 где qn – массовая доля мономера n, Тст..n – температура стеклования полимера n. 10. Минимальную температуру пленкообразования (МТП) определяли как границу перехода между непрерывной и дискретной частью пленки при формировании дисперсного покрытия на горизонтальной поверхности прибора с линейным градиентом температуры. 11. Ударное растяжение пленок определяли по специально разработанной методике. Для испытаний ударопрочности пленок использовали ударный маятник в соответствии с DIN 53448. Испытывали образцы пленок толщиной 0,3-0,8 мм. Образец, изображенный на чертеже, толщиной 0,3-0,8 мм, фиксировали на основании измеряющего ударопрочность прибора двумя зажимами, один из которых жестко закреплен на станине, а другой подвижен. Подвижный зажим имеет выступ в поперечном направлении, по которому ударяет П-образный молоток. Подвижный зажим перемещается под действием ударного импульса от молотка, и образец разрушается. Зажимы для фиксации образцов имеют большую важность. В случае, если образец слабо закреплен зажимами, он начинает перемещаться, и происходит потеря энергии на трение. Другой источник потерь представляет собой работу, совершаемую при извлечении подвижного зажима с остатками образца. Эта энергия определяется холостыми опытами. Прочность при ударном растяжении Qt определяли в кДж/м2 по формуле 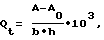 где А – энергия, затрачиваемая на разрушение образца (Дж), А0 – энергия, затрачиваемая на извлечение подвижного зажима (Дж), b – ширина образца (мм), h – толщина образца (мм). 12. Ударопрочность стеклянных изделий с покрытием на основе предложенной дисперсии определяли методом разрушения толстостенных стеклянных трубок (длина 300 мм, диаметр 36 мм, толщина стенки 1,5 мм) с нанесенным покрытием толщиной не менее 100 мкм при их падении с высоты 1,5 м на массивное бетонное основание. Покрытие считалось выдержавшим испытание, если не наблюдалось разрывов и отслоений полимерной оболочки при разрушении стекла, а также деформации стеклянного изделия. Все осколки стекла оставались в полимерной оболочке. Покрытия на стеклянных трубках готовили путем их макания в дисперсию с последующим высушиванием при 90oС в течение 2 часов. Изобретение иллюстрируется приведенными ниже примерами 1-13, которые иллюстрируют состав предложенной дисперсии и способ ее получения. Во всех примерах описано проведение процесса в три стадии с получением дисперсий, частицы которых содержат ядро, промежуточную и внешнюю оболочки. Пример 1 Стадия 1 В реактор объемом 0,5 л загружают 45 г деионизованной воды, в которой растворено 0,9 г анионного ПАВ Elfan OS-46 (С14-С16-алкилсульфонат натрия; продукт фирмы Akzo Nobel), продувают азотом, нагревают до 80oС и загружают 22,17 г н.-бутилакрилата (БА) и 0,07 г диметакрилата этиленгликоля (ДМЭГ), перемешивают 5 мин, добавляют раствор 0,13 г персульфата аммония (ПА), растворенного в 10 г воды, и проводят процесс затравочной сополимеризации в течение 30 мин. Отдельно приготавливают эмульсию мономеров: БА 66,5 г, ДМЭГ 0,20 г, Elfan OS-46 (37% раствор) 2,71 г, вода 28 г. В полученную затравочную дисперсию добавляют раствор 0,13 г ПА в 5 г воды, перемешивают 5 мин и начинают дозировать эмульсию мономеров со скоростью 0,812 г/мин. Общее время дозирования 120 мин. После окончания дозирования выдерживают полученную дисперсию при 80oС в течение 30 мин. Стадия 2 В полученную на стадии 1 дисперсию загружают раствор 0,16 г ПА в 3 г воды, перемешивают 5 мин и начинают дозировать смесь мономеров, состоящую из 5,32 г метилметакрилата (ММА), 11,44 г н.-бутилметакрилата (БМА), 0,88 г метакриловой кислоты (МАК) и 0,09 г ДМЭГ, со скоростью 0,296 г/мин. Общее время дозирования 60 мин. После окончания дозирования выдерживают полученную дисперсию при 80oС в течение 30 мин. Стадия 3 Отдельно приготавливают эмульсию мономеров: ММА 1,33 г, БМА 23,34 г, МАК 2,0 г, Elfan OS-46 (37% раствор) 1,3 г, вода 12 г. В полученную на стадии 2 дисперсию загружают раствор 0,05 г ПА в 9 г воды, перемешивают 5 мин и начинают дозировать эмульсию мономеров со скоростью 0,222 г/мин. Общее время дозирования 180 мин. Через 90 мин после начала дозирования в дисперсию загружают раствор 0,05 г ПА в 9 г воды. После окончания дозирования эмульсии мономеров выдерживают дисперсию при 80oС в течение 30 мин. Содержание остаточных мономеров в дисперсии: БА, МАК, ДМЭГ, ММА – отсутствие; БМА – 0,01 мас.%. Дисперсию охлаждают и нейтрализуют 1,61 г 24,5% водного раствора аммиака. Общее время процесса 500 мин. Составы загрузок по этому и последующим примерам приведены в таблице 1, составы и свойства дисперсий и покрытий на их основе – в таблице 2. Пример 2 Стадия 1 Проводят, как в примере 1. Стадия 2 В дисперсию, полученную на стадии 1, используемую как затравочную, загружают раствор 0,09 г ПА в 9,9 г воды, перемешивают 5 мин и начинают дозировать смесь мономеров, состоящую из 5,60 г ММА, 3,61 г БМА, 0,76 г МАК и 0,20 г ДМЭГ со скоростью 0,1695 г/мин. Общее время дозирования 60 мин. После окончания дозирования мономерной смеси выдерживают дисперсию при 80oС в течение 30 мин. Стадия 3 Отдельно приготавливают эмульсию мономеров и водную фазу. Состав эмульсии: ММА 14,26 г, БМА 13,33 г, МАК 2,24 г, Elfan OS-46 (37% раствор) 1,21 г, вода 10 г. Состав водной фазы: ПА 0,09 г, Elfan OS-46 (37% раствор) 0,20 г, вода 12,2 г. В дисперсию, полученную на стадии 2, используемую как затравочную, одновременно начинают дозировать эмульсию мономеров со скоростью 0,1710 г/мин и водную фазу со скоростью 0,0520 г/мин. Общее время дозирования 240 мин. После окончания дозирования мономеров и водной фазы дисперсию выдерживают при 80oС в течение 30 мин. После этого дисперсию охлаждают и нейтрализуют 1,81 г 24,5% водного раствора аммиака. Общее время процесса 555 мин. Пример 3 Отличается от примера 1 рецептурой загрузки компонентов по стадиям процесса. Температура процесса 90oС. Общее время процесса 470 мин. Пример 4 Отличается от примера 1 рецептурой загрузки компонентов по стадиям процесса и тем, что вместо раствора аммиака используют ацетат цинка в виде 0,05 М водного раствора. Для усиления стабилизации дисперсии вводят неионогенное ПАВ ОС-20 (оксиэтилированный алифатический спирт С16-C18) в виде 5% водного раствора. Общая продолжительность процесса 500 мин. Пример 5 Отличается от примера 2 рецептурой загрузки компонентов по стадиям процесса и тем, что вместо раствора аммиака используют ацетат цинка в виде 0,05 М водного раствора. Для усиления стабилизации дисперсии вводят неионогенное ПАВ ОС-20 в виде 5% водного раствора. Общая продолжительность процесса 535 мин. Пример 6 Отличается от примера 1 рецептурой загрузки компонентов по стадиям процесса. Общая продолжительность процесса 500 мин. Пример 7 Отличается от примера 2 рецептурой загрузки компонентов по стадиям процесса и тем, что вместо ДМЭГ используют диакриловый эфир этиленгликоля (ДАЭГ). Общая продолжительность процесса 540 мин. Пример 8 Отличается от примера 2 рецептурой загрузки компонентов по стадиям процесса и тем, что вместо метакриловой используют акриловую кислоту (АК). Общая продолжительность процесса 500 мин. Пример 9 Стадия 1 Проводят, как в примере 1. Стадия 2 В полученную на стадии 1 дисперсию загружают раствор 0,16 г ПА в 5 г воды, перемешивают 5 мин и начинают дозировать смесь мономеров, состоящую из 5,32 г ММА, 11,44 г БМА, 0,88 г МАК и 0,09 г ДМЭГ со скоростью 0,296 г/мин. Общее время дозирования 60 мин. Отдельно приготавливают водную фазу: ПА 0,08 г, Elfan OS-46 (37% р-р) 0,12 г, вода – 3 г. В дисперсию одновременно начинают дозировать водную фазу со скоростью 0,053 г/мин и смесь мономеров, состоящую из 4,47 г ММА, 3,98 г БМА, 0,45 г МАК и 0,04 г ДМЭГ, со скоростью 0,149 г/мин. Общее время дозирования 60 мин. После окончания дозирования дисперсию выдерживают при 80oС 30 мин. Стадия 3 Проводят, как в примере 2. Общая продолжительность процесса 620 мин. Пример 10 Отличается от примера 1 рецептурой загрузки компонентов по стадиям процесса и тем, что вместо БА используют метилакрилат (МА). Общая продолжительность процесса 500 мин. Пример 11 Отличается от примера 1 рецептурой загрузки компонентов по стадиям процесса, условиями проведения стадии 1. После проведения затравочной сополимеризации в полученную затравочную дисперсию вместо эмульсии мономеров дозируют мономерную смесь со скоростью 0,556 г/мин и тем, что вместо БА используют 2-этилгексилакрилат (ЭГА), а вместо ДМЭГ – аллилметакрилат (АМА). Пример 12 Отличается от примера 1 тем, что вместо МАК используют монобутилмалеинат, а в качестве ПАВ на всех стадиях – додецилбензолсульфонат натрия. Пример 13 Стадия 1 В реактор объемом 0,5 л загружают 45 г деионизованной воды, в которой растворено 0,12 г Elfan OS-46, продувают азотом, нагревают до 80oС и загружают 8,87 г БА и 0,03 г ДМЭГ, перемешивают 5 мин, добавляют раствор 0,13 г ПА, растворенного в 10 г воды, и проводят процесс затравочной сополимеризации в течение 30 мин. Отдельно приготавливают эмульсию мономеров: БА 79,8 г, ДМЭГ 0,24 г, Elfan OS-46 0,70 г, вода 28 г. В полученную затравочную дисперсию добавляют раствор 0,17 г ПА в 5 г воды, перемешивают 5 мин и начинают дозировать эмульсию мономеров со скоростью 0,812 г/мин. Общее время дозирования 135 мин. После окончания дозирования выдерживают полученную дисперсию при 80oС в течение 30 мин. Остальные стадии процесса проводят, как в примере 1. Общее время процесса 515 мин. Как видно из приведенных в примерах данных, гетерополимерные водные дисперсии, получаемые, в отличие от прототипа, по упрощенной технологии, обеспечивают получение прозрачных, твердых, не липких и ударопрочных покрытий. Формула изобретения
-(С1-С5 алкил)-замещенные, малеиновую, фумаровую, итаконовую кислоты или их моно-(С1-С4 алкиловые) эфиры.
3. Водная гетерополимерная дисперсия по п. 2, отличающаяся тем, что сополимер ядра является сополимером (мет)акрилового эфира или смеси (мет)акриловых эфиров, ди(мет)акрилового эфира и, возможно, (мет)акриловой кислоты, содержит 0,1-0,5 мас. % звеньев ди(мет)акрилового эфира и 0-5,0 мас. % звеньев (мет)акриловой кислоты, сополимер промежуточной оболочки является сополимером метакрилового эфира или смеси метакриловых эфиров, ди(мет)акрилового эфира и, возможно, (мет)акриловой кислоты, содержит 0,5-2,0 мас. % звеньев ди(мет)акрилового эфира и 0-7,5 мас. % звеньев (мет)акриловой кислоты, а сополимер внешней оболочки является сополимером метакрилового эфира или смеси метакриловых эфиров и (мет)акриловой кислоты и содержит 7,5-10,0 мас. % звеньев (мет)акриловой кислоты.
4. Способ получения водной гетерополимерной дисперсии для изготовления покрытий, частицы которой содержат сополимеры, включающие звенья акриловых мономеров, и состоят из ядра и, по крайней мере, одной наружной оболочки, путем многостадийной эмульсионной сополимеризации, включающий последовательные стадии эмульсионной сополимеризации мономеров ядра и мономеров оболочки при температуре менее 100oС в присутствии водорастворимого свободнорадикального инициатора и анионного поверхностно-активного вещества (ПАВ), отличающийся тем, что стадию получения сополимера ядра проводят в два этапа, на первом из которых проводят эмульсионную сополимеризацию в присутствии инициатора и анионного ПАВ при единовременной загрузке 10-30% от общего количества мономерной смеси ядра, а на втором этапе в образовавшийся затравочный латекс дозируют оставшуюся часть мономеров ядра или эмульсию этой части мономеров ядра, стабилизированную анионным ПАВ, стадию получения сополимера промежуточной оболочки проводят путем непрерывного дозирования мономерной смеси, образующей промежуточную оболочку, в дисперсию сополимера ядра, причем перед началом данной стадии в дисперсию сополимера ядра вводят водный раствор инициатора, стадию получения сополимера внешней оболочки проводят путем эмульсионной сополимеризации мономеров, образующих внешнюю оболочку, в присутствии дисперсии, полученной на предыдущей стадии, причем загрузку мономеров внешней оболочки проводят либо двумя равными порциями, вводя при этом в дисперсию водные растворы инициатора и анионного ПАВ перед загрузкой мономеров внешней оболочки, либо путем непрерывного дозирования в виде водной эмульсии мономеров, стабилизированной анионным ПАВ, одновременно дозируя водный раствор инициатора, который может содержать растворенное анионное ПАВ, причем смесь мономеров, по крайней мере, на одной из стадий включает этиленненасыщенный мономер, содержащий карбоксильные группы, и после завершения процесса в полученную дисперсию вводят водный раствор аммиака, летучего алифатического амина, либо водорастворимого гидроксида или соли двухвалентного металла в количестве 0,1-1,0 моль на 1 моль звеньев этиленненасыщенного мономера, содержащего карбоксильные группы.
5. Способ получения водной гетерополимерной дисперсии по п. 4, отличающийся тем, что перед введением гидроксида или соли двухвалентного металла в дисперсию добавляют неионное ПАВ в количестве 0,5-1,0% от массы сополимера.
6. Способ получения водной гетерополимерной дисперсии по пп. 4 и 5, отличающийся тем, что получение сополимера промежуточной оболочки производят в два этапа, причем составы мономерной смеси на указанных этапах различаются.
7. Способ получения водной гетерополимерной дисперсии по пп. 4-6, отличающийся тем, что водорастворимый инициатор и анионное ПАВ вводят в реакционную смесь, соответственно, в количестве 0,3-0,4 и 0,2-2,0% от массы мономеров при получении сополимера ядра, 0,3-0,9 и 0-0,5% от массы мономеров при получении сополимера промежуточной оболочки, 0,3-1,2 и 0,3-3,0% от массы мономеров при получении сополимера внешней оболочки.
РИСУНКИ
|
||||||||||||||||||||||||||