Патент на изобретение №2184116
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ ПИПЕРИДИНИЛАМИНОМЕТИЛ-ТРИФТОРМЕТИЛОВЫХ ЦИКЛИЧЕСКИХ ЭФИРОВ
(57) Реферат: Изобретение относится к новому способу получения диастереомерной смеси пиперидиниламинометил-трифторметиловых циклических эфиров формул Iа и Ib и их фармацевтически приемлемых солей, где R1 является C1-С6 алкилом, R2 является C1-С6 алкилом, галоген C1-С6 алкилом или фенилом или замещенным фенилом, R3 является водородом или галогеном; m = 0, 1 или 2, в котором указанная смесь является высокообогащенной соединением формулы Iа. Описываемый способ заключается во взаимодействии смеси соединений Iа и Ib с кислотой формулы HX, которая выбрана из группы, состоящей из (S)-(+)-миндальной кислоты, D-(-)-винной кислоты, ди-п-толуоил-D-винной кислоты и других кислот, кристаллизации HX соли диастереомерной смеси продуктов из ее раствора, обработки образовавшейся смеси соединений Iа и Ib, которая обогащена соединением формулы Iа. Использование определенных, указанных выше кислот для разделения диастереомерной смеси позволяет достичь высокую степень диастереомерной чистоты (более 95%). Изобретение относится также к смеси соединений Iа и Ib, обогащенной Iа, и к промежуточным соединениям, используемым при получении соединений формул Iа и Ib. 9 с. и 13 з.п. ф-лы. 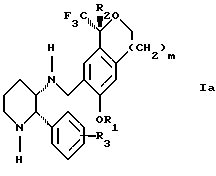 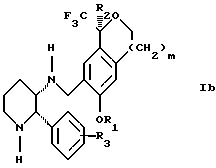
Изобретение относится к новому способу получения диастереомерной смеси пиперидиниламинометилтрифторметиловых циклических эфиров формул Iа и Ib: 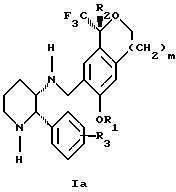 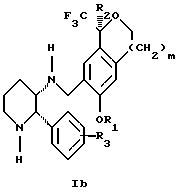 и их фармацевтически приемлемых солей, где R1 является C1-6 алкилом; R2 является C1-6 алкилом, галоген C1-6 алкилом или фенилом или замещенным фенилом; R3 является водородом или галогеном; m означает нуль, один или два. Кроме того, настоящее изобретение также относится к способу получения диастереомерной смеси соединений формул Iа и Ib и их фармацевтически приемлемых солей, высокообогащенной соединением формулы Ia. Способ по настоящему изобретению позволяет с помощью селективной кристаллизации выделить диастереомерные смеси соединений формул Iа и Ib, в которых соотношение соединений формулы Iа к Ib превышает 90:10. Кроме того, настоящее изобретение относится к новым способам получения соединения формулы II: 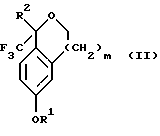 промежуточного соединения, полезного для получения соединений формул Iа и Ib. Кроме того, настоящее изобретение направлено также на другие новые промежуточные соединения, полезные в способе получения смеси соединений формул Iа и Ib. Настоящее изобретение также относится к новому способу очистки некоторых промежуточных соединений для использования в способах по изобретению. Соединения формулы Iа и Ib, особенно соединения формулы Iа, и их фармацевтически приемлемые соли являются полезными в качестве антагонистов вещества Р, природного ундекапептида, принадлежащего к тахикининовому семейству пептидов, которое широко вовлечено в патофизиологию многочисленных заболеваний, включая нарушения центральной нервной системы, такие как депрессия, чувство тревоги и шизофрения, респираторные и воспалительные заболевания, такие как астма и ревматоидный артрит, желудочно-кишечные нарушения и заболевания желудочно-кишечного тракта, такие как язвенный колит и болезнь Крона, и в передачу боли, включая мигрень. Диастереомерная смесь соединений формул Iа и Ib и способ получения диастереомерной смеси описываются в международной патентной публикации WO 99/25714, опубликованной 27 мая 1999 года. Этот источник касается способов получения диастереомерной смеси с использованием способов, иных, чем способы по настоящему изобретению, и он включен в качестве источника во всей его полноте. Настоящее изобретение обеспечивает более практичный, более прямой и дающий больший выход способ получения смеси диастереомеров соединений формул Iа и Ib, высокообогащенной соединением формулы Iа, с помощью новых путей синтеза. Краткое описание изобретения Настоящее изобретение относится к способу получения смеси соединений формул Iа и Ib: 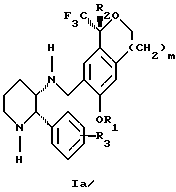 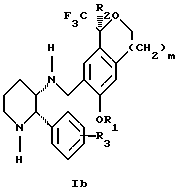 высокообогащенной присутствием соединения формулы Iа, и их фармацевтически приемлемых солей, где R1 является C1-6 алкилом; R2 является C1-6 алкилом, галоген C1-6 алкилом или фенилом или замещенным фенилом; R3 является водородом или галогеном; m означает нуль, один или два; включающему стадии (а1) взаимодействия смеси соединений формул Iа и Ib: 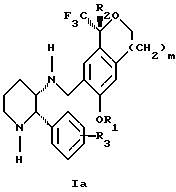 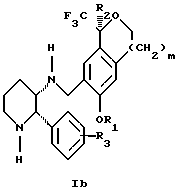 с кислотой формулы НХ, где НХ выбирается из группы, состоящей из (S)-(+)-миндальной кислоты, D-(-)-винной кислоты, ди-п-толуоил-D-винной кислоты, ((1R)-эндо, анти)-(+)-3-бромкамфора-8-сульфоновой кислоты, хинной кислоты, уксусной кислоты и бромистоводородной кислоты с образованием смеси диастереомерных соединений формул Va и Vb соответственно, обогащенной присутствием соединения формулы Va: 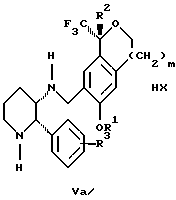 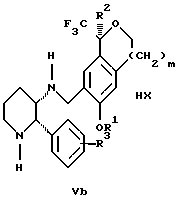 (b1) предоставления возможности НХ соли смеси диастереомерных продуктов стадии (а1) кристаллизоваться из ее раствора в соответствующем растворителе; и (с1) обработки образовавшейся смеси соединений, полученной со стадии (b1), основанием. Наиболее предпочтительным воплощением изобретения является случай, когда кислота НХ стадии (а1) является (3)-(+)-миндальной кислотой. Более предпочтительным воплощением изобретения является такое, когда соответствующий растворитель стадии (а1) выбирается из группы, состоящей из метанола, этанола, изопропанола, тетрагидрофурана, этилацетата, изопропилацетата, метил-трет-бутилового эфира, диизопропилового эфира, толуола, ацетонитрила, ацетона, воды и смеси любых из вышеуказанных растворителей. В наиболее предпочтительном воплощении соответствующим растворителем стадии (а1) является этанол. Более предпочтительным воплощением изобретения является такое, когда основание стадии (с1) выбирается из группы, состоящей из гидроокиси натрия, гидроокиси калия, карбоната натрия, бикарбоната натрия, карбоната калия и бикарбоната калия. Настоящее изобретение также относится к получению фармацевтически приемлемых солей смеси соединений формул Iа и Ib, высокообогащенной соединением формулы Iа, которое включает обработку смеси соединений Iа и Ib, которая обогащена одним из диастереомерных соединений формулы Iа, протонной кислотой H+Y–, где анион Y– выбран из группы, состоящей из гидрохлорида, гидробромида, сульфата, бисульфата, фосфата, кислого фосфата, ацетата, лактата, цитрата, кислого цитрата, тартрата, битартрата, сукцината, малеата, фумарата, глюконата, сахарата, бензоата, метансульфоната, этансульфоната, бензолсульфоната, п-толуолсульфоната и памоата (т.е. 1,1′-метилен-бис-(2-гидрокси-3-нафтоата)) с образованием смеси соединений VIa и VIb, высокообогащенной кислотно-аддитивной солью диастереомерного соединения формулы VIa: 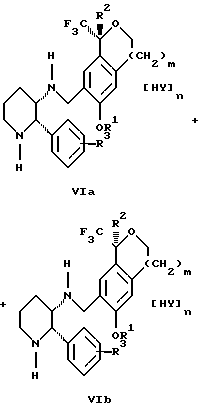 где n определяется присущими характеристиками формы соединений Iа и Ib, когда они закомплексованы с определенной кислотой НY, и n является целым числом от одного до двух. Способ по изобретению также относится к получению гидратов соединений формул VIa и VIb, в которых между нулем и тремя молекулами воды может быть связано с каждой молекулой соединений формул VIa и VIb, причем указанные гидраты образуются на стадии, на которой соединения формул Iа и Ib обрабатываются протонной кислотой. Более предпочтительным воплощением изобретения является такое, при котором используемой протонной кислотой является соляная кислота, и n равно 2. Предпочтительным воплощением изобретения является случай, когда полученное соотношение соединений VIa и VIb составляет 90:10 или выше. Более предпочтительным воплощением изобретения является такое, когда полученное соотношение соединений VIa и VIb составляет 98:2 или выше. Настоящее изобретение также относится к способу получения соединений формул Iа и Ib, высокообогащенных присутствием соединения формулы Iа, дополнительно включающему стадию взаимодействия соединения формулы III: 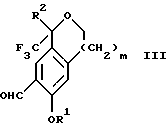 с соединением формулы IV: 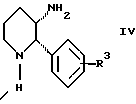 в присутствии восстанавливающего агента с получением смеси соединений формул Iа и Ib. В предпочтительном воплощении изобретения восстанавливающий агент выбирается из группы, состоящей из триацетоксиборгидрида натрия, цианоборгидрида натрия и боргидрида натрия. В более предпочтительном воплощении изобретения восстанавливающим агентом является триацетоксиборгидрид натрия. Настоящее изобретение относится также к способу получения соединений формул Iа и Ib, высокообогащенных присутствием соединения формулы Iа, дополнительно включающему стадию формилирования соединения формулы II: 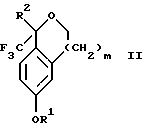 где R1, R2 и R3 имеют значения, определенные выше; m означает 0, 1 или 2, гексаметилентетрамином в присутствии кислоты с образованием соединения формулы III. В предпочтительном воплощении изобретения кислота в реакции формилирования является трифторуксусной кислотой, глицероборной кислотой, уксусной кислотой или соляной кислотой. Наиболее предпочтительной кислотой является трифторуксусная кислота. Настоящее изобретение также относится к способу получения соединений формул Iа и Ib, высокообогащенных присутствием соединения формулы Iа, в котором соединение формулы II: 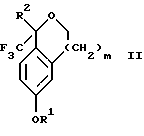 в которой R1, R2 и R3 имеют значения, определенные выше; m означает 0, 1 или 2, получают способом, включающим стадии: (а2) взаимодействия соединения формулы VII: 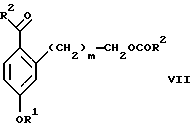 с соединением формулы CF3SiR4 3, где R4 является (C1-С6) алкилом или фенилом, в присутствии фторидного источника с образованием соединения формулы VIII: 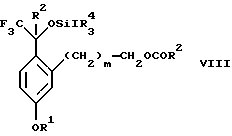 (b2) удаления силильной защитной группы из продукта стадии (а2) с помощью обработки основанием или фторидным источником с образованием соединения формулы IX: 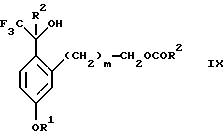 (с2) гидролиза сложно-эфирной группы продукта стадии (b2) в присутствии основания с образованием соединения формулы Х: 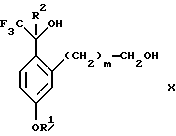 и (d2) проведения реакции циклизации кольца продукта стадии (с2) в присутствии основания и активирующего агента, выбранного из группы, состоящей из метансульфонилхлорида, метансульфонового ангидрида, п-толуолсульфонилхлорида, п-толуолсульфонового ангидрида и трифторметансульфонового ангидрида. В более предпочтительном воплощении настоящего изобретения источник фтора на стадии (а2) выбирается из группы, состоящей из фторида цезия, фторида калия и фторида алкиламмония. Наиболее предпочтительным фторидом алкиламмония является фторид тетрабутиламмония. В наиболее предпочтительном воплощении изобретения источником фтора на стадии (а2) является фторид цезия. Предпочтительными растворителями для стадии (а2) являются диметилформамид, диметилацетамид, толуол, дихлорметан, дихлорэтан и тетрагидрофуран. Наиболее предпочтительным растворителем для стадии (а2) является диметилформамид. На стадии (b2) предпочтительными основаниями являются гидроокись натрия или гидроокись калия, и предпочтительными фторидными источниками являются фторид тетрабутиламмония, фторид цезия, комплекс фтористоводородной кислоты-пиридина и фтористоводородная кислота. Наиболее предпочтительным фторидным источником является фторид тетрабутиламмония. Предпочтительными растворителями для стадии (b2) являются тетрагидрофуран, диизопропиловый эфир, ацетонитрил, метил-трет-бутиловый эфир, дихлорметан и толуол. Наиболее предпочтительным растворителем для стадии (b2) является тетрагидрофуран. Предпочтительными основаниями на стадии (с2) являются гидроокись натрия, гидроокись калия, карбонат натрия, бикарбонат натрия, карбонат калия и бикарбонат калия. Предпочтительным основанием на стадии (с2) является гидроокись натрия. Предпочтительными растворителями для стадии (с2) являются вода, тетрагидрофуран, метанол, этанол, изопропанол, 1,4-диоксан и сочетание любых из этих растворителей. Наиболее предпочтительным растворителем для стадии (с2) является смесь воды и тетрагидрофурана. На стадии (d2) наиболее предпочтительным активирующим агентом является метансульфонилхлорид. Предпочтительными основаниями для стадии (d2) являются триэтиламин, диизопропилэтиламин, 2,6-лутидин, пиридин, гидроокись натрия, гидроокись калия, карбонат цезия и карбонат калия. Наиболее предпочтительным основанием для стадии (d2) является триэтиламин. Предпочтительными растворителями для стадии (d2) являются дихлорметан, тетрагидрофуран, толуол, диизопропиловый эфир и метил-трет-бутиловый эфир. Наиболее предпочтительным растворителем для стадии (d2) является дихлорметан. Настоящее изобретение относится также к способу получения соединений формул Iа и Ib, высокообогащенных присутствием соединения формулы Iа, в котором соединение формулы II: 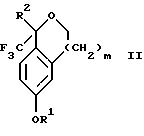 в которой R1, R2 и R3 имеют значения, определенные выше; m означает 0, 1 или 2; получают с помощью способа, включающего стадии: (а3) взаимодействия соединения формулы XI: 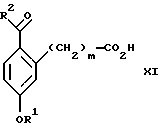 со спиртом формулы R1OH в присутствии кислоты, где R1 имеет значения, определенные выше, с образованием соединения формулы XII: 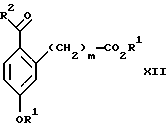 (b3) взаимодействия продукта стадии (а3) с соединением формулы CF3SiR4 3, где R4 является (C1-C6) алкилом или фенилом, с образованием соединения формулы XIII: 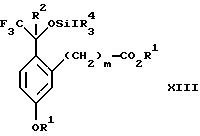 (с3) взаимодействия продукта стадии (b3) с фторидным источником с получением лактона формулы XIV: 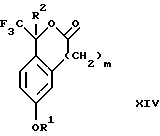 (d3) взаимодействия лактонового продукта стадии (с3) с восстанавливающим агентом необязательно в присутствии кислоты Льюиса с получением соединения формулы XV: 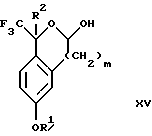 и (е3) взаимодействия продукта стадии (d3) с восстанавливающим агентом необязательно в присутствии кислоты Льюиса с получением соединения формулы II. Согласно еще одному предпочтительному воплощению изобретения кислота стадии (а3) выбирается из группы, состоящей из серной кислоты, соляной кислоты, бромистоводородной кислоты, трифторуксусной кислоты и метансульфоновой кислоты. Наиболее предпочтительной кислотой для стадии (а3) является серная кислота. На стадии (b3) предпочтительными фторидными источниками являются фторид цезия, фторид калия и фторид алкиламмония, такой как фторид тетрабутиламмония. Наиболее предпочтительным фторидным источником является фторид цезия. Предпочтительными растворителями для стадии (b3) являются диметилформамид, диметилацетамид, дихлорметан и тетрагидрофуран. Наиболее предпочтительным растворителем для стадии (b3) является диметилформамид. Предпочтительными фторидными источниками для стадии (с3) являются фторид тетрабутиламмония, фторид цезия, комплекс фтористоводородной кислоты и пиридина и фтористоводородная кислота. Наиболее предпочтительным фторидным источником для стадии (с3) является фторид тетрабутиламмония. Предпочтительными растворителями для стадии (с3) являются тетрагидрофуран, диизопропиловый эфир, ацетонитрил, метил-трет-бутиловый эфир, дихлорметан и толуол. Наиболее предпочтительным растворителем для стадии (с3) является тетрагидрофуран. Предпочтительными восстанавливающими агентами для стадии (d3) являются боргидрид натрия, комплекс борана и тетрагидрофурана, комплекс борана и диметилсульфида, диборан, боргидрид лития, боргидрид кальция, алюмогидрид лития, гидрид диизобутилалюминия, L-селектрид и К-селектрид. Наиболее предпочтительным восстанавливающим агентом является боргидрид натрия. Предпочтительной кислотой Льюиса для стадии (d3) является комплекс трифторида бора и диэтилового эфира. Предпочтительными растворителями для стадии (d3) являются тетрагидрофуран, диизопропиловый эфир, метил-трет-бутиловый эфир и диметоксиэтан. Наиболее предпочтительным растворителем для стадии (d3) является тетрагидрофуран. Предпочтительными восстанавливающими агентами для стадии (е3) являются триэтилсилан или трифенилсилан в присутствии кислоты Льюиса, такой как эфират трифторида бора или трифторуксусная кислота, предпочтительно трифторуксусная кислота. Предпочтительными растворителями для стадии (е3) являются дихлорметан, дихлорэтан и хлороформ. Наиболее предпочтительным растворителем для стадии (е3) является дихлорметан. В другом предпочтительном воплощении на стадии (е3) соединение формулы XIV обрабатывают катализатором, таким как платина, окись платины или гидроокись палладия, предпочтительно платина, в растворителе, таком как метанол, этанол или изопропанол, предпочтительно этанол, в атмосфере водорода, необязательно под давлением выше, чем атмосферное давление. Настоящее изобретение относится также к способу получения соединений формул Iа и Ib, высокообогащенных присутствием соединения формулы Iа, в котором соединение формулы III: 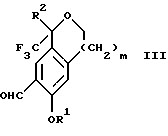 очищают способом, включающим стадии (а4) образования гидразона с помощью реакции соединения формулы III с гидразоном формулы XVI: 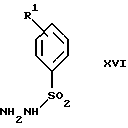 в присутствии кислоты, дающим соединение формулы XVII: 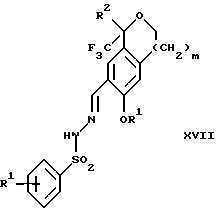 и (b4) гидролиза продукта стадии (а4) с помощью обработки реагентом, выбранным из группы, состоящей из хлорида меди (II), йодида меди (II), ацетата меди (II), сульфата меди, серной кислоты, уксусной кислоты и соляной кислоты. Предпочтительные кислоты для стадии (а4) включают уксусную кислоту, серную кислоту, соляную кислоту, метансульфоновую кислоту и п-толуолсульфоновую кислоту. Наиболее предпочтительной кислотой для стадии (а4) является уксусная кислота. Предпочтительными растворителями для стадии (а4) являются метанол, этанол, изопропанол, тетрагидрофуран, вода и смесь любых из вышеуказанных растворителей. Наиболее предпочтительным растворителем для стадии (а4) является смесь метанола и воды. Наиболее предпочтительным реагентом для стадии (b4) является хлорид меди (II). Предпочтительными растворителями для стадии (b4) являются трет-бутиловый спирт, метанол, этанол, изопропанол, тетрагидрофуран, вода и смесь любых из вышеуказанных растворителей. Наиболее предпочтительным растворителем для стадии (b4) является смесь трет-бутилового спирта и воды. Кроме того, настоящее изобретение охватывает способы получения фармацевтических композиций смесей соединений формул Iа или Ib или их фармацевтически приемлемых солей. Способ получения такой фармацевтической композиции включает добавление смеси соединений формулы Iа и Ib или их фармацевтически приемлемых солей к фармацевтически приемлемому носителю или разбавителю. Настоящее изобретение относится также к новым промежуточным соединениям, используемым в способе изобретения, включая, но не ограничиваясь ими, соединения формул VII, IX, XIII, XIV, XV и XVII и их соли. Термин “алкил”, используемый здесь, если не указано иначе, включает насыщенные одновалентные углеводородные радикалы, имеющие нормальные, разветвленные или циклические группы и их сочетания. Термин “замещенный фенил”, используемый здесь, если не указано иначе, означает фенил, замещенный одним или более, предпочтительно одним или двумя заместителем(ями), такими как галоген, гидрокси, (С1-С6) алкил или (C1-С6) алкокси. Термин “гало” или “галоген”, используемый здесь, если не указано иначе, означает фтор, хлор, бром или йод. Термин “подходящий растворитель” или “соответствующий растворитель”, используемый здесь, если не указано иначе, означает среду, которая служит для растворения конкретного указанного вещества(в), соединения(ний) или реагента(тов) для образования однородно диспергированной смеси этого вещества или соединения на молекулярном или ионном уровне. Термин “протонная кислота”, используемая для получения кислотно-аддитивных солей соединений способа данного изобретения, представляет такие кислоты, которые образуют нетоксичные кислотно-аддитивные соли, т.е. соли, содержащие фармакологически приемлемые анионы, такие соли, как гидрохлорид, гидробромид, сульфат, бисульфат, фосфат, кислый фосфат, ацетат, лактат, цитрат, кислый цитрат, тартрат, битартрат, сукцинат, малеат, фумарат, глюконат, сахарат, бензоат, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат и памоат (т.е. 1,1′-метилен-бис-(2-гидрокси-3-нафтоат)). Термин “обогащенный”, используемый здесь, если не указано иначе, означает преобладание в соотношении выше, чем 1:1, одного конкретного вещества или изомера над другим или другими компонентами в смеси. Термин “высокообогащенный”, используемый здесь, если не указано иначе, означает преобладание в соотношении по меньшей мере 90:10 одного конкретного соединения или изомера над другим или другими компонентами в смеси. Если не указано иначе, данное изобретение относится ко всем оптическим изомерам, таутомерам и стереоизомерам любого из соединений, описанных здесь. Термин “фармацевтически приемлемая соль”, используемый здесь, если не указано иначе, относится к кислотно-аддитивной соли протонной кислоты, определенной здесь, или гидрату кислотно-аддитивной соли. Подробное описание изобретения Диастереомерную смесь пиперидиниламинометилтрифторметиловых циклических эфиров формул Iа и Ib, высокообогащенную соединением формулы Iа, можно получить в соответствии с новым способом, представленным на реакционной схеме 1 (см. в конце описания). Новые способы для получения критического промежуточного соединения при получении пиперидиниламинометилтрифторметиловых циклических эфиров соединения формулы II могут осуществляться в соответствии со схемами 2 и 3 (см. в конце описания). Новый способ очистки ключевого промежуточного соединения в способе по схеме 1 представлен на схеме 4 (см. в конце описания). Если не указано иначе, переменные R1, R2, R3, R4, m и n имеют значения, описанные выше. Стадия 1 на схеме 1 является формилированием. Соединение формулы II обрабатывают гексаметилентетрамином в присутствии кислоты, такой как трифторуксусная кислота, глицероборная кислота, уксусная кислота или соляная кислота, предпочтительно трифторуксусная кислота, необязательно в растворителе, таком как дихлорметан, дихлорэтан, гептан или нитрометан, предпочтительно без растворителя, при температуре между 0 и 100oС, предпочтительно при 70oС, в течение периода времени между 10 мин и 24 ч, предпочтительно 3 ч, с последующим добавлением воды для получения соединения формулы III. В этот момент соединение формулы III может очищаться по способу изобретения, как показано ниже на схеме 4, перед проведением стадии 2. Стадия 2 на схеме 1 является восстановительным сочетанием. Альдегид формулы III обрабатывают амином формулы IV или его солью в присутствии восстанавливающего агента, такого как триацетоксиборгидрид натрия, цианоборгидрид натрия или боргидрид натрия, предпочтительно триацетоксиборгидрид натрия, в растворителе, таком как дихлорметан, дихлорэтан, тетрагидрофуран, толуол, уксусная кислота, диизопропиловый эфир или метил-трет-бутиловый эфир, предпочтительно дихлорметан, при температуре между -20 и 60oС, предпочтительно 0oС, в течение периода времени между 30 мин и 24 ч, предпочтительно 3 ч, для получения смеси соединений формул Iа и Ib. Стадия 3 на схеме 1 представляет образование соли. Смесь соединений Iа и Ib обрабатывают кислотой формулы НХ, такой как (S)-(+)-миндальная кислота, D-(-)-винная кислота, ди-п-толуоил-D-винная кислота, ((1R)-эндо,анти)-(+)-3-бромкамфора-8-сульфоновая кислота, хинная кислота, уксусная кислота, бромистоводородная кислота, предпочтительно (S)-(+)-миндальная кислота, в растворителе, таком как метанол, этанол, изопропанол, тетрагидрофуран, этилацетат, изопропилацетат, метил-трет-бутиловый эфир, диизопропиловый эфир, толуол, ацетонитрил, ацетон, вода или смесь вышеуказанных растворителей, предпочтительно этанол, при температуре между -20 и 70oС, предпочтительно при комнатной температуре, в течение периода времени между 30 мин и 48 ч, предпочтительно 18 ч, с получением смеси соединений формул Va и Vb, которая обогащена соединением формулы Va. Стадия 3 позволяет выделить смеси соединений формул Va и Vb, в которых соотношение соединений формулы Va к Vb составляют выше, чем 70:30, и в основном 80:20 или выше. Стадия 4 на схеме 1 представляет образование кислотно-аддитивной соли. Смесь соединений формул Va и Vb, высокообогащенной соединением Va, обрабатывают основанием, таким как гидрооокись натрия, гидроокись калия, карбонат натрия, бикарбонат натрия, карбонат калия или бикарбонат калия, в воде в присутствии сорастворителя, такого как толуол, диизопропиловый эфир, метил-трет-бутиловый эфир, этилацетат или дихлорметан, предпочтительно диизопропиловый эфир, при температуре между 0 и 40oС, предпочтительно при комнатной температуре, в течение периода времени между 10 мин и 48 ч, предпочтительно 18 ч, с получением смеси соединений формул Iа и Ib, которая обогащена соединением формулы Iа. Соотношение соединения Iа и Ib, полученное на данном этапе стадии 4, составляет 70:30 или выше, но в основном 80:20 или выше. Данную смесь обрабатывают протонной кислотой HY, определенной выше, предпочтительно соляной кислотой, в растворителе, таком как метанол, этанол, изопропанол, тетрагидрофуран, диизопропиловый эфир, вода и смесь вышеуказанных растворителей, предпочтительно смесь метанола и воды, при температуре между 0 и 60oС, предпочтительно при комнатной температуре, в течение периода времени между 1 и 48 ч, предпочтительно 18 ч, с получением смеси соединений формул VIa и VIb, которая высокообогащена соединением формулы VIa, и где n имеет значения, как определено выше. Стадия 4 позволяет выделить смеси соединений формул VIa и VIb, в которых соотношение соединения формулы VIa к VIb выше, чем 90:10, и может приблизиться к 98:2 или выше. Стадию 4, если нужно, можно повторить для получения более высоких соотношений. Стадия 1 на схеме 2 является ацилированием арена, которое протекает с защитой спирта по способу, аналогичному известному способу (Sternberg E.D.; Vollhardt К. Р. С. , J. Org. Chem. 1984, 49, 1574-1583). Арен формулы XVIII обрабатывают ацилирующим агентом формулы R2(C=O)-X’, где R2 имеет значения, определенные выше, и X’ является галогеном, R2(C=O)-O- или другой подходящей группой в ацилирующем агенте, как это известно специалистам в этой области, в присутствии кислоты, такой как трибромид алюминия, трихлорид алюминия, тетрахлорид олова, тетрахлорид титана или полифосфорная кислота, предпочтительно трибромид алюминия, в растворителе, таком как дихлорметан, дихлорэтан, нитрометан, нитробензол, дисульфид углерода или хлорбензол, предпочтительно дихлорметан, при температуре между -20 и 125oС, предпочтительно между 0 и 20oС, в течение периода времени между 10 мин и 10 ч, предпочтительно примерно 1 ч, с получением соединения формулы VII. Стадия 2 на схеме 2 представляет присоединение трифторметильной группы к кетону с использованием модификации известного способа (Prakash G.K.S.; Krishnamurti R.; Olah G.A., J. Am. Chem. 1989, 111, 393-395). Кетон формулы VII обрабатывают соединением формулы CF3SiR4 3, где R4 имеет значения, определенные выше, в присутствии фторидного источника, такого как фторид цезия, фторид калия или фторид алкиламмония, такой как фторид тетрабутиламмония, предпочтительно фторид цезия, в присутствии растворителя, такого как диметилформамид, диметилацетамид, толуол, дихлорметан, дихлорэтан или тетрагидрофуран, предпочтительно диметилформамид, при температуре между -78 и 50oС, предпочтительно при комнатной температуре в течение периода времени между 10 мин и 18 ч, предпочтительно 45 мин, с получением соединения формулы VIII. Стадия 3 на схеме 2 является снятием защиты со спирта. Соединение формулы VIII обрабатывают реагентом, таким как гидроокись натрия, гидроокись калия или фторидный источник, такой как фторид тетрабутиламмония, фторид цезия, комплекс фтористоводородной кислоты-пиридина или фтористоводородная кислота, предпочтительно фторид тетрабутиламмония, в растворителе, таком как тетрагидрофуран, диизопропиловый эфир, ацетонитрил, метил-трет-бутиловый эфир, дихлорметан или толуол, предпочтительно тетрагидрофуран, при температуре между -40 и 60oС, предпочтительно при комнатной температуре, в течение периода времени между 5 мин и 5 ч, предпочтительно 1 ч, с получением соединения формулы IX. Стадия 4 на схеме 2 является гидролизом сложного эфира. Соединение формулы IX обрабатывают реагентом, таким как гидроокись натрия, гидроокись калия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия, предпочтительно гидроокись натрия, в растворителе, таком как вода, тетрагидрофуран, метанол, этанол, изопропанол, 1,4-диоксан или сочетание вышеуказанных растворителей, предпочтительно смесь воды и тетрагидрофурана, при температуре между 0 и 75oС, предпочтительно при комнатной температуре, в течение периода времени между 1 и 48 ч, предпочтительно 12 ч, с получением соединения формулы X. Стадия 5 на схеме 2 является циклизацией. Соединение формулы Х обрабатывают активирующим агентом, таким как метансульфонилхлорид, метансульфоновый ангидрид, п-толуолсульфонилхлорид, п-толуолсульфоновый ангидрид или трифторметансульфоновый ангидрид, предпочтительно метансульфонилхлорид, и основанием, таким как триэтиламин, диизопропилэтиламин, 2,6-лутидин, пиридин, гидроокись натрия, гидроокись калия, карбонат цезия или карбонат калия, предпочтительно триэтиламин, в растворителе, таком как дихлорметан, тетрагидрофуран, толуол, диизопропиловый эфир или метил-трет-бутиловый эфир, предпочтительно дихлорметан, при температуре между -40 и 75oС, предпочтительно между 0oС и комнатной температурой, в течение периода времени между 1 и 48 ч, предпочтительно 12 ч, с получением соединения формулы II. Стадия 1 на схеме 3 является ацилированием арена. Арен формулы XIX обрабатывают ацилирующим агентом формулы R2(С=О)-X’, где R2 имеет значения, определенные выше, и X’ является галогеном, R2 (С=О)-О- или другой подходящей группой в ацилирующем агенте, известной для специалистов в данной области, в присутствии кислоты, такой как трибромид алюминия, трихлорид алюминия, тетрахлорид олова, тетрахлорид титана или полифосфорная кислота, предпочтительно трибромид алюминия, в растворителе, таком как дихлорметан, дихлорэтан, нитрометан, нитробензол, дисульфид углерода или хлорбензол, предпочтительно дихлорметан, при температуре между -20 и 125oС, предпочтительно между 0 и 20oС, в течение периода времени 10 мин и 10 ч, предпочтительно примерно 1 ч, с получением соединения формулы XI. Стадия 2 на схеме 3 является этерификацией. Карбоновую кислоту формулы XI обрабатывают спиртом формулы R1OH, где R1 имеет значения, определенные выше, в присутствии кислоты, такой как серная кислота, соляная кислота, бромистоводородная кислота, трифторуксусная кислота или метансульфоновая кислота, предпочтительно серная кислота, при температуре между 0 и 100oС, предпочтительно при комнатной температуре, в течение периода времени между 10 мин и 48 ч, предпочтительно 16 ч, с получением соединения формулы XII. Стадия 3 на схеме 3 представляет присоединение трифторметильной группы к кетону с использованием модификации известного способа (Prakash G.K.S.; Krishnamurti R.; Olah G.A., J. Am. Chem. Soc. 1989, 111, 393-395). Кетон формулы XII обрабатывают соединением формулы CF3SiR4 3, где R4 имеет значения, определенные выше, в присутствии фторидного источника, такого как фторид цезия, фторид калия или фторид алкиламмония, такой как фторид тетрабутиламмония, предпочтительно фторид цезия, в присутствии растворителя, такого как диметилформамид, диметилацетамид, дихлорметан или тетрагидрофуран, предпочтительно диметилформамид, при температуре между -78 и 50oС, предпочтительно при 0oС, в течение периода времени между 10 мин и 18 ч, предпочтительно 7 ч, для получения соединения формулы XIII. Стадия 4 на схеме 3 является лактонизацией. Соединение формулы XIII обрабатывают фторидным источником, таким как фторид тетрабутиламмония, фторид цезия, комплекс фтористоводородной кислоты-пиридина или фтористоводородная кислота, предпочтительно фторид тетрабутиламмония, в растворителе, таком как тетрагидрофуран, диизопропиловый эфир, ацетонитрил, метил-трет-бутиловый эфир, дихлорметан или толуол, предпочтительно тетрагидрофуран, при температуре между -40 и 60oС, предпочтительно при комнатной температуре, в течение периода времени между 5 мин и 5 ч, предпочтительно 1 ч, с получением соединения формулы XIV. Стадия 5 на схеме 3 является восстановлением лактона. Соединение формулы XIV обрабатывают восстанавливающим агентом, таким как боргидрид натрия, комплекс борана-тетрагидрофурана, комплекс борана-диметилсульфида, диборан, боргидрид лития, боргидрид кальция, алюмогидрид лития, гидрид диизобутилалюминия, L-селектрид или К-селектрид, необязательно в присутствии кислоты Льюиса, такой как комплекс трифторида бора-диэтилового эфира, предпочтительно боргидрид натрия, в присутствии комплекса трифторида бора-диэтилового эфира, в растворителе, таком как тетрагидрофуран, диизопропиловый эфир, метил-трет-бутиловый эфир или диметоксиэтан, предпочтительно тетрагидрофуран, при температуре между -78 и 60oС, предпочтительно между 0oС и комнатной температурой, в течение периода времени между 30 мин и 48 ч, предпочтительно 16 ч, с получением соединения формулы XV. Стадия 6 на схеме 3 является восстановлением. Соединение формулы XV обрабатывают восстанавливающим агентом, таким как триэтилсилан или трифенилсилан, в присутствии кислоты Льюиса, такой как эфират трифторида бора или трифторуксусная кислота, предпочтительно трифторуксусная кислота, в растворителе, таком как дихлорметан, дихлорэтан или хлороформ, предпочтительно дихлорметан, при температуре между -78 и 60oС, предпочтительно при комнатной температуре, в течение периода времени между 5 мин и 5 ч, предпочтительно 2 ч, с получением соединения формулы II. Альтернативно соединение формулы XV обрабатывают восстанавливающим агентом, то есть катализатором, таким как платина, окись платины или гидроокись палладия, предпочтительно платина, в растворителе, таком как метанол, этанол или изопропанол, предпочтительно этанол, в атмосфере водорода, необязательно под давлением, при температуре между комнатной температурой и 100oС, предпочтительно при комнатной температуре, в течение периода времени между 1 и 48 ч, предпочтительно 5 ч, с получением соединения формулы II. Альтернативно соединение III можно очистить дериватизацией. Стадия 1 на схеме 4 является образованием гидразона. Соединение формулы III обрабатывают гидразоном формулы XVI с кислотой, такой как уксусная кислота, серная кислота, соляная кислота, метансульфоновая кислота или п-толуолсульфоновая кислота, предпочтительно уксусная кислота, в растворителе, таком как метанол, этанол, изопропанол, тетрагидрофуран, вода или смесь любых из вышеуказанных растворителей, предпочтительно смесь метанола и воды, при температуре между 0 и 110oС, предпочтительно при кипячении с обратным холодильником, в течение периода времени между 30 мин и 10 ч, предпочтительно 90 мин, с получением соединения формулы XVII. Стадия 2 на схеме 4 является гидролизом гидразона. Соединение формулы XVII обрабатывают реагентом, таким как хлорид меди (II), йодид меди (II), ацетат меди (II), сульфат меди, серная кислота, уксусная кислота или соляная кислота, предпочтительно хлорид меди (II), в растворителе, таком как трет-бутиловый спирт, метанол, этанол, изопропанол, тетрагидрофуран, вода или смесь любых из вышеуказанных растворителей, предпочтительно смесь трет-бутилового спирта и воды, при температуре между 0 и 110oС, предпочтительно при 70oС, в течение периода времени между 30 мин и 10 ч, предпочтительно 2,5 ч, с получением соединения формулы III. Получение других соединений по настоящему изобретению, конкретно не описанных в вышеупомянутом экспериментальном разделе, можно проводить, используя сочетания реакций, описанных выше, которые знакомы специалистам в данной области. В каждой из реакций, обсуждаемых или изображенных на схемах 1-4 выше, давление не является решающим фактором, если не указано иначе. Обычно предпочитается давление примерно от 0,9 атмосфер до примерно 2 атмосфер и давление окружающей среды, т.е. примерно 1 атмосфера, что является вопросом удобства. Промежуточные соединения изобретения, на которые ссылаются выше, могут содержать хиральные центры и, следовательно, могут существовать в различных энантиомерных и диастереомерных формах; данное изобретение относится ко всем таким оптическим и стереоизомерам указанных промежуточных соединений, а также к их смесям. Данное изобретение также относится к меченым изотопами соединениям, идентичным тем, которые были перечислены для формул Iа и Ib или их фармацевтически приемлемым солям, но с учетом того, что один или более атомов замещены в них атомом, имеющим атомную массу или массовое число, отличные от атомной массы или массового числа, обычно обнаруживаемых в природе. Примеры изотопов, которые могут быть включены в соединения данного изобретения, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2Н, 3Н, 13С, 14С, 15N, 18О, 17О, 31P, 32Р, 35S, 18F и 36Cl соответственно. Соединения настоящего изобретения, их пролекарства и фармацевтически приемлемые соли указанных соединений или указанных пролекарств, которые содержат вышеуказанные изотопы и/или другие изотопы других атомов, охватываются объемом данного изобретения. Некоторые меченые изотопами соединения настоящего изобретения, например, такие, в которые включены радиоактивные изотопы, такие как 3H и 14С, являются полезными, например, для анализа распределения лекарственного препарата и/или субстратной ткани. Особенно предпочтительными являются тритиированные, т.е. 3H и углерод-14, т.е. 14С, изотопы вследствие простоты их получения и способности к обнаружению. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2H, может предоставить некоторые терапевтические преимущества в результате более высокой метаболической стабильности, например, повышенного периода полураспада in vivo или требований пониженной дозировки, и, следовательно, при некоторых обстоятельствах оно может быть предпочтительным. Меченые изотопами соединения формул Iа и Ib данного изобретения и их пролекарства обычно могут получаться с помощью процедур, приведенных здесь путем замены немеченых изотопами реагентов легко доступными мечеными изотопами реагентами. Активность, способы испытания активности, дозировки, лекарственные формы, способы введения и информация о предпосылках, касающаяся соединений формул Iа и Ib, представлены в международной патентной публикации WO 99/25714, опубликованной 27 мая 1999 г. Пиперидиниламинометилтрифторметиловые циклические эфиры, полученные с помощью способов настоящего изобретения, проявляют значительную активность в отношении связывания с рецепторами вещества Р и являются ценными для лечения широкого разнообразия клинических состояний, характеризующихся наличием избытка активности указанного вещества Р. Такие состояния включают сердечно-сосудистые заболевания, аллергические нарушения, ангиогенез, желудочно-кишечные нарушения, расстройства центральной нервной системы, воспалительные заболевания, рвоту, недержание мочи, боль, мигрень, солнечный ожог и заболевания, расстройства и состояния, вызываемые Helicobacter pylori, у млекопитающих, особенно у людей. Для лечения рвоты эти соединения могут предпочтительно использоваться в сочетании с антагонистом 5-НТ3. Активные пиперидиниламинометилтрифторметиловые циклические эфиры формул Iа и Ib можно вводить млекопитающим пероральным, парентеральным (например, внутривенно, внутримышечно или подкожно) или местным путем. Эти соединения можно вводить по одному или в сочетании с фармацевтически приемлемыми носителями или разбавителями одним из путей, указанных выше, и можно вводить в виде одной или многократных доз. Соединения, полученные способами изобретения, можно вводить в широком разнообразии различных лекарственных доз, например, в сочетании с различными фармацевтически приемлемыми инертными носителями в форме таблеток, капсул, лепешек, пастилок, твердых конфет, порошков, спреев, кремов, бальзамов, суппозиториев, желе, гелей, паст, лосьонов, мазей, водных суспензий, инъецируемых растворов, эликсиров, сиропов и т.д. ПРИМЕРЫ Настоящее изобретение иллюстрируется следующими примерами. Однако понятно, что изобретение не ограничивается специфическими деталями этих примеров. Пример 1 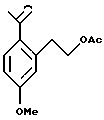 2-(2-Ацетил-5-метоксифенил)-этиловый эфир уксусной кислоты Данное соединение получали с помощью модификации известной процедуры (Sternberg E. D. , Vollhardt К.Р.С., J. Org. Chem. 1984, 49, 1574-1583). К раствору трибромида алюминия (43,8 г, 164 ммоль) в дихлорметане (70 мл) при 0oС медленно добавляли ацетилбромид (14,6 мл, 197 ммоль). Реакционную смесь нагревали до 15oС и в течение 45 мин добавляли 2-(3-метоксифенил)-этанол (10,0 г, 65,7 ммоль) в дихлорметане (20,0 мл). Реакционную смесь перемешивали в течение одного часа и затем выливали на лед (100 мл). К смеси добавляли 1Н водный раствор соляной кислоты (100 мл). Органический слой отделяли и водный слой экстрагировали дихлорметаном (100 мл). Объединенные органические экстракты промывали 1Н водным раствором гидроокиси натрия (100 мл), сушили над сульфатом магния, фильтровали через целит, сконцентрировали с получением 2-(2-ацетил-5-метоксифенил)-этилового эфира уксусной кислоты в виде масла (14,8 г, 95%). 1H ЯМР (300 МГц, СDСl3)  2,05 (с, 3), 2,59 (с, 3), 3,29 (т, 2, J=6,9), 3,89 (с, 3), 4,33 (т, 2, J=6,9), 6,81 (д, 1, J=2,5), 8,85 (дд, 1, J= 8,6, 2,6), 7,82 (д, 1, J=8,6). 13С ЯМР (75 МГц, СDСl3) 22,28, 30,37, 35,36, 56,63, 66,11, 101,21, 112,63, 119,17, 131,00, 134,23, 142,90, 163,24, 172,32. ИК 1737, 1674, 1604, 1567, 1358, 1239, 1037 см-1. Анализ. Вычислено для C13H16O4: С 66,09; Н 6,83. Найдено: С 65,71; Н 7,21.
Пример 2 2,05 (с, 3), 2,59 (с, 3), 3,29 (т, 2, J=6,9), 3,89 (с, 3), 4,33 (т, 2, J=6,9), 6,81 (д, 1, J=2,5), 8,85 (дд, 1, J= 8,6, 2,6), 7,82 (д, 1, J=8,6). 13С ЯМР (75 МГц, СDСl3) 22,28, 30,37, 35,36, 56,63, 66,11, 101,21, 112,63, 119,17, 131,00, 134,23, 142,90, 163,24, 172,32. ИК 1737, 1674, 1604, 1567, 1358, 1239, 1037 см-1. Анализ. Вычислено для C13H16O4: С 66,09; Н 6,83. Найдено: С 65,71; Н 7,21.
Пример 2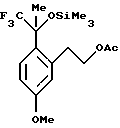 2-[5-Метокси-2-(2,2,2-трифтор-1-метил-1-триметилсиланилоксиэтил)-фенил] -этиловый эфир уксусной кислоты К раствору 2-(2-ацетил-5-метоксифенил)-этилового эфира уксусной кислоты (12,5 г, 52,9 ммоль) и фторида цезия (0,964 г, 6,35 ммоль) в диметилформамиде (75 мл) при 0oС медленно добавляли трифторметилтриметилсилан (10,2 мл, 69,0 ммоль). Реакционную смесь перемешивали в течение 45 мин, после чего анализ ГХ/МС и ВЭЖХ показал отсутствие исходного вещества. Для целей характеристики реакционную смесь выливали в воду и экстрагировали метил-трет-бутиловым эфиром (100 мл). Органический слой промывали водой (2 х 75 мл) и рассолом (50 мл), сушили над сульфатом магния, фильтровали и концентрировали, получая 2-[5-метокси-2-(2,2,2-трифтор-1-метил-1-триметилсиланилоксиэтил)-фенил] -этиловый эфир уксусной кислоты в виде сырого масла. 1Н ЯМР (300 МГц, СDС13) 0,19 (с, 9), 1,93 (с, 3), 2,10 (с, 3), 3,23-3,33 (м, 1), 3,42-3,52 (м, 1), 3,83 (с, 3), 4,26-4,32 (м, 2), 6,77 (дд, 1, J=8,9, 2,8), 6,86 (д, 1, J= 2,9), 7,32 (д, 1, J=8,9). 13С ЯМР (100 МГц, СDС13) 2,03, 21,03, 24,64, 32,86, 55,11, 65,54, 78,90 (кв, J=30,3), 111,26, 117,44, 125,70 (кв, J=287), 129,56, 129,79, 139,77, 159,17, 171,09. ИК 2961, 1741, 1610, 1383, 1286, 1255, 1165, 1140, 1039, 864, 846 см-1. Анализ. Вычислено для C17H25F3О4Si: С 53,95; Н 6,66. Найдено: С 53,72; Н 6,53.
Пример 3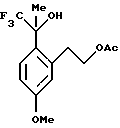 2-[5-Метокси-2-(2,2,2-трифтор-1-гидрокси-1-метилэтил)-фенил] -этиловый эфир уксусной кислоты К сырой реакционной смеси, описанной в примере 2, содержащей раствор 2-[5-метокси-2-(2,2,2-трифтор-1-метил-1-триметилсиланилоксиэтил)-фенил] -этилового эфира уксусной кислоты добавлялся фторид тетрабутиламмония (52,9 мл 1,0 М раствора в тетрагидрофуране, 52,9 ммоль). Реакционная смесь перемешивалась в течение одного часа, после чего анализ ГХ/МС и ВЭЖХ показал отсутствие исходного вещества. Для целей характеристики реакционная смесь выливалась в воду и экстрагировалась метил-трет-бутиловым эфиром (75 мл). Органический слой промывался водой (75 мл) и рассолом (50 мл), сушился над сульфатом магния, фильтровался и концентрировался, давая сырое масло. 1H ЯМР (400 МГц, СDСl3) 1,82 (с, 3), 2,01 (с, 3), 2,98-3,06 (м, 2), 3,55 (дт, 1, J= 13,7, 6,8), 3,79 (с, 3), 4,27-4,32 (м, 2), 6,73-6,77 (м, 2), 7,28 (д, 1, J= 8,5). 13С ЯМР (100 МГц, CDCl3) 20,92, 25,50, 34,16, 55,10, 66,49, 76,67 (кв, J= 30,3), 111,55, 118,25, 126,02 (кв, J=286), 128,67, 129,56, 139,70, 159,20, 171,32. ИК 3453, 1720, 1610, 1249, 1161, 1134, 1038 см-1. Анализ. Вычислено для C14H17F3O4: С 54,90; Н 5,59. Найдено: С 55,03; Н 5,85.
Пример 4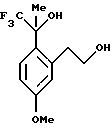 1,1,1-Трифтор-2-[2-(2-гидроксиэтил)-4-метоксифенил]-пропан-2-ол К сырой реакционной смеси, описанной в примере 3, содержащей 2-[5-метокси-2-(2,2,2-трифтор-1-гидрокси-1-метилэтил)-фенил]-этиловый эфир уксусной кислоты, добавлялся 1Н водный раствор гидроокиси натрия (75,0 мл, 75 ммоль). Реакционную смесь оставляли подогреться до комнатной температуры и перемешивали в течение 12 часов. Реакционную смесь выливали в воду (75 мл) и экстрагировали метил-трет-бутиловым эфиром(150 мл). Органический слой промывали водой (75 мл) и рассолом (75 мл), сушили над сульфатом магния и концентрировали до масла. К сырому маслу добавляли гексан (20 мл) и метил-трет-бутиловый эфир (4 мл) и в осадок выпадало твердое вещество. Смесь перемешивалась течение 30 мин и фильтровалась, давая 1,1,1-трифтор-2-[2-(2-гидроксиэтил)-4-метоксифенил] -пропан-2-ол (7,3 г, 52% общий выход из 2-(2-ацетил-5-метокси-фенил)-этилового эфира уксусной кислоты). Т. пл. 110-111oС. 1H ЯМР (300 МГц, СDС13) 1,83 (с, 3), 2,91 (дт, 1, J=13,7, 3,9), 3,76 (ддд, 1, J= 13,7, 9,3, 4,4), 3,85 (с, 3), 3,85-3,93 (м, 1), 4,08 (дт, 1, J=9,3, 3,7), 6,80-6,83 (м, 2), 7,38 (д, 1, J=8,4). 13С ЯМР (100 МГц, СDС13)  26,01, 36,12, 55,19, 64,13, 76,52 (кв, J=28,9), 111,47, 117,43, 125,99 (кв, J=287), 129,69, 129,94, 140,86, 159,55. ИК 3395, 3162, 1610, 1513, 1467, 1248, 1157, 1087, 1046 см-1. Анализ. Вычислено для С12H15F3О3: С 54,54; Н 5,72. Найдено: С 54,65; Н 5,70.
Пример 5 26,01, 36,12, 55,19, 64,13, 76,52 (кв, J=28,9), 111,47, 117,43, 125,99 (кв, J=287), 129,69, 129,94, 140,86, 159,55. ИК 3395, 3162, 1610, 1513, 1467, 1248, 1157, 1087, 1046 см-1. Анализ. Вычислено для С12H15F3О3: С 54,54; Н 5,72. Найдено: С 54,65; Н 5,70.
Пример 5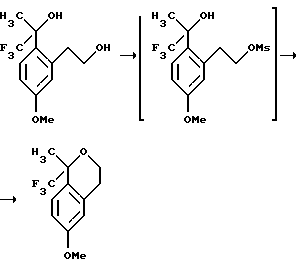 6-Метокси-1-метил-1-трифторметилизохроман К раствору 1,1,1-трифтор-2-[2-(2-гидроксиэтил)-4-метоксифенил]-пропан-2-ола (5,00 г, 18,9 ммоль) в дихлорметане (30 мл) добавляли триэтиламин (9,20 мл, 66,3 ммоль). Раствор охлаждали до 0oС и добавляли по каплям метансульфонилхлорид (1,61 мл, 20,8 ммоль). Реакционной смеси давали подогреться до комнатной температуры и перемешивали в течение 12 часов. Образование 2-[5-метокси-2-(2,2,2-трифтор-1-гидрокси-1-метилэтил)-фенил] -этилового эфира метансульфоновой кислоты было быстрым и его исчезновение контролировали ВЭЖХ (время удерживания 4,5 мин, колонка Zorbax Rx-C6 4,6Х150 мм, 40oC, 50% СН3СN/50% (0,2% Еt3N, 0,1% водный раствор Н3РO4, рН 3,2, буфер), 1 мл/мин). В конце реакции смесь выливали в 1Н водный раствор соляной кислоты (30 мл) и экстрагировали дихлорметаном (20 мл). Органические экстракты сушились над сульфатом магния, фильтровались и концентрировались, давая 6-метокси-1-метил-1-трифторметилизохроман в виде масла (3,40 г, 73%). 1H ЯМР (300 МГц, СDС13) 1,69 (с, 3), 2,85-2,90 (м, 2), 3,85 (с, 3), 3,90-3,98 (м, 1), 4,14-4,21 (м, 1), 6,72 (д, 1, J=2,6), 6,85 (дд, 1, J=8,7, 2,6), 7,31 (д, 1, J=8,7). 13С ЯМР (100 МГц, СDС13) 23,25, 29,42, 55,19, 61,37, 76,10 (кв, J= 27,4), 112,84, 113,43, 124,85, 125,96 (кв, J=289), 127,86, 136,49, 158,98. ИК 2946, 2839, 1738, 1611, 1505, 1162, 1137, 1101 см-1. Анализ. Вычислено для С12Н13F3О2: С 58,54; Н 5,32. Найдено: С 58,27; Н 5,35.
Пример 6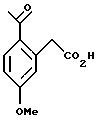 (2-Ацетил-5-метоксифенил)-уксусная кислота К раствору трибромида алюминия (57,6 г, 216 ммоль) в дихлорметане (90 мл) при 0oС медленно добавляли ацетилхлорид (11,5 мл, 162 ммоль). К реакционной смеси добавляли (3-метоксифенил)-уксусную кислоту (17,9 г, 108 ммоль) в дихлорметане (20,0 мл). Реакционную смесь перемешивали в течение одного часа и затем выливали на лед (100 мл). Органический слой отделяли и добавляли 1Н водный раствор гидроокиси натрия (100 мл). Двухфазную смесь интенсивно перемешивали в течение 90 мин и слои разделяли. Органический слой отбрасывали и к водному слою добавляли концентрированную соляную кислоту до тех пор, пока не достигалось значение рН 1. В осадок выпадало твердое вещество, оно отфильтровывалось и сушилось на воздухе, давая (2-ацетил-5-метоксифенил)-уксусную кислоту (16,8 г, 75%). Т. пл. 153-155oС. 1H ЯМР (300 МГц, СDС13) 2,68 (с, 3), 3,91 (с, 3), 3,92 (с, 2), 6,92-6,95 (м, 2), 7,88 (д, 1, J= 9,5). 13С ЯМР (100 МГц, СDС13) 28,33, 41,43, 55,46, 112,54, 118,26, 129,17, 133,08, 136,94, 162,65, 174,80, 200,96. ИК 3435, 1704, 1663, 1609, 1568, 1258 см-1. Анализ. Вычислено для C11H12O4: С 63,45; Н 5,81. Найдено: С 63,35; Н 5,46.
Пример 7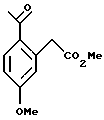 Метиловый эфир (2-ацетил-5-метоксифенил)-уксусной кислоты К раствору (2-ацетил-5-метоксифенил)-уксусной кислоты (5,00 г, 24,0 ммоль) в метаноле (50 мл) добавляли концентрированную серную кислоту (1,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, после чего ее концентрировали до небольшого объема. Добавляли дихлорметан (50 мл) и раствор промывали 1Н раствором гидроокиси натрия (50 мл). Слои разделяли и органический слой сушился над сульфатом магния, фильтровался и концентрировался до масла, которое затвердело при стоянии, давая метиловый эфир (2-ацетил-5-метоксифенил)-уксусной кислоты (4,70 г, 88%). Т. пл. 74-76oС. 1H ЯМР (300 МГц, СDС13) 2,58 (с, 3), 3,74 (с, 3), 3,89 (с, 3), 3,95 (с, 2), 6,78 (д, 1, J=2,6), 6,89 (дд, 1, J=8,7, 2,6), 7,89 (д, 1, J= 8,6). 13С ЯМР (75 МГц, CDCl3) 29,65, 42,35, 53,11, 56,69, 113,17, 120,00, 130,52, 134,39, 138,90, 163,54, 173,23, 200,35. ИК 1739, 1665, 1605, 1568, 1321, 1247, 1165 см-1. Анализ. Вычислено для C12H14О4: С 65,85; Н 6,35. Найдено: С 64,87; Н 6,44.
Пример 8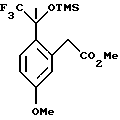 Метиловый эфир [5-метокси-2-(2,2,2-трифтор-1-метил-1-триметилсиланилоксиэтил)-фенил]-уксусной кислоты К раствору метилового эфира (2-ацетил-5-метоксифенил)-уксусной кислоты (2,00 г, 9,00 ммоль) и фторида цезия (96,0 мг, 0,632 ммоль) в диметилформамиде (12 мл) при 0oС медленно добавляли трифторметилтриметилсилан (1,73 мл, 11,7 ммоль). Реакционную смесь перемешивали при 0oС в течение 7 часов. В целях характеристики реакционную смесь выливали в воду и экстрагировали метил-трет-бутиловым эфиром (50 мл). Органический слой промывался водой (2 х 75 мл) и рассолом (50 мл), сушился над сульфатом магния, фильтровался и концентрировался, давая метиловый эфир [5-метокси-2-(2,2,2-трифтор-1-метил-1-триметилсиланилоксиэтил)-фенил] -уксусной кислоты в виде масла. 1H ЯМР (400 МГц, СDС13) 0,11 (с, 9), 1,89 (с, 2), 3,68 (с, 3), 3,77 (с, 3), 3,98 (д, 1, J=17,2), 4,28 (д, 1, J=17,0), 6,74-6,77 (м, 2), 7,29 (д, 1, J=9,1). 13С ЯМР (100 МГц, СDС13)  1,87, 24,25, 39,32, 51,75, 55,12, 78,67 (кв, J= 30,3), 111,97, 118,30, 125,70 (кв, J=286), 129,50, 129,57, 136,10, 159,17, 172,81. ИК 2956, 1745, 1611, 1577, 1467, 1436, 1290, 1256, 1166, 1092, 989, 863, 847 см-1. Анализ. Вычислено для С16Н23F3O4Si: С 52,73; Н 6,36. Найдено: С 52,84; Н 6,36.
Пример 9 1,87, 24,25, 39,32, 51,75, 55,12, 78,67 (кв, J= 30,3), 111,97, 118,30, 125,70 (кв, J=286), 129,50, 129,57, 136,10, 159,17, 172,81. ИК 2956, 1745, 1611, 1577, 1467, 1436, 1290, 1256, 1166, 1092, 989, 863, 847 см-1. Анализ. Вычислено для С16Н23F3O4Si: С 52,73; Н 6,36. Найдено: С 52,84; Н 6,36.
Пример 9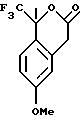 6-Метокси-1-метил-1-трифторметилизохроман-3-он К сырой реакционной смеси, описанной в примере 8, содержащей раствор метилового эфира [5-метокси-2-(2,2,2-трифтор-1-метил-1-триметилсиланилоксиэтил)-фенил]-уксусной кислоты, добавляли фторид тетрабутиламмония (9,00 мл 1,0 М раствора в тетрагидрофуране, 9,00 ммоль). Реакционную смесь перемешивали в течение 1 часа, после чего ее выливали в воду (50 мл) и экстрагировали метил-трет-бутиловым эфиром (50 мл). Органический слой промывался водой (50 мл) и рассолом (30 мл), сушился над сульфатом магния, фильтровался и концентрировался, давая 6-метокси-1-метил-1-трифторметилизохроман-3-он в виде масла (1,26 г, 54%). 1H ЯМР (400 МГц, CDCl3) 1,89 (с, 3), 3,71 (д, 1, J= 20,6), 3,79 (с, 3), 3,89 (д, 20,8), 6,65 (д, 1, J=1,5), 6,85-6,89 (м, 1), 7,29 (д, 1, J=8,7). 13С ЯМР (100 МГц, CDCl3) 21,45, 34,32, 55,33, 83,01 (кв, J=30,3), 112,21, 113,88, 120,57, 124,68 (кв, J=285,7), 127,73, 132,18, 160,75, 167,45. ИК 1765, 1614, 1509, 1322, 1301, 1274, 1259, 1183, 1101, 997, 813 см-1. Анализ. Вычислено для С12Н11F3О3: С 55,39; Н 4,26. Найдено: С 55,03; Н 4,54.
Пример 10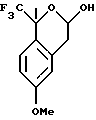 6-Метокси-1-метил-1-трифторметилизохроман-3-ол К раствору 6-метокси-1-метил-1-трифторметилизохроман-3-она (1,50 г, 5,76 ммоль) в тетрагидрофуране (30 мл) при 0oС добавляли боргидрид натрия (0,240 г, 6,34 ммоль), затем комплекс трифторида бора-диэтилового эфира (0,992 г, 8,07 ммоль). Реакционную смесь подогревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь добавляли к воде (75 мл) и экстрагировали метил-трет-бутиловым эфиром (75 мл). Слои разделялись и органический слой промывался 1Н водным раствором соляной кислоты (50 мл), сушился над сульфатом магния, фильтровался и концентрировался, давая 6-метокси-1-метил-1-трифторметилизохроман-3-ол в виде масла и смесь а и b аномеров (1,19 г, 79%). Приведенные данные для основного диастереоизомера. 1H ЯМР (400 МГц, СDС13) 1,74 (с, 3), 2,85 (дд, 1, J=15,7, 4,3), 2,88-2,99 (м, 1), 3,11 (дд, 1, J=15,7, 3,2), 3,80 (с, 3), 5,63 (т, 1, J=3,7), 6,69 (д, 1, J= 2,7), 6,82 (дд, 1, J=8,7, 2,7), 7,22-7,27 (м, 1). 13С ЯМР (100 МГц, СDС13), (приведенные данные для идентифицируемых сигналов основного диастереоизомера) 24,52, 35,46, 55,16, 90,71, 113,11, 113,98, 125,22, 127,57, 132,98, 159,59. ИК 3439, 2949, 1735, 1613, 1506, 1166, 1141, 1070 см-1.
Пример 11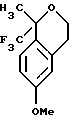 6-Метокси-1-метил-1-трифторметилизохроман К раствору 6-метокси-1-метил-1-трифторметилизохроман-3-ола (8,36 г, 31,9 ммоль) в дихлорметане (84 мл) добавляли триэтилсилан (15,3 мл, 95,8 ммоль), затем трифторуксусную кислоту (14,7 мл, 191 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и выливали в 1Н водный раствор гидроокиси натрия (250 мл). Органический слой отделяли и промывали 1Н водным раствором гидроокиси натрия (100 мл). Органический слой сушился над сульфатом магния, фильтровался и концентрировался, давая 6-метокси-1-метил-1-трифторметилизохроман в виде масла (6,88 г, 88%). 1H ЯМР (300 МГц, CDCl3) 1,69 (с, 3), 2,85-2,90 (м, 2), 3,85 (с, 3), 3,90-3,98 (м, 1), 4,14-4,21 (м, 1), 6,72 (д, 1, J=2,6), 6,85 (дд, 1, J=8,7, 2,6), 7,31 (д, 1, J=8,7). 13С ЯМР (100 МГц, СDС13) 23,25, 29,42, 55,19, 61,37, 76,10 (кв, J= 27,4), 112,84, 113,43, 124,85, 125,96 (кв, J=289), 127,86, 136,49, 158,98. ИК 2946, 2839, 1738, 1611, 1505, 1162, 1137, 1101 см-1. Анализ. Вычислено для С12Н13F3О2: С 58,54; Н 5,32. Найдено: С 58,27; Н 5,35.
Пример 12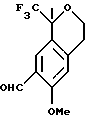 6-Метокси-1-метил-1-трифторметилизохроман-7-карбальдегид К гексаметилентетрамину (31,3 г, 223 ммоль) добавляли трифторуксусную кислоту (400 мл) и смесь нагревали до 70oС в течение 90 мин. Затем к реакционной смеси в течение 40 мин добавляли раствор 6-метокси-1-метил-1-трифторметилизохромана (50,0 г, 203 ммоль) в трифторуксусной кислоте (100 мл). Раствор перемешивали в течение 3 часов и добавляли воду (450 мл). Реакционную смесь перемешивали в течение 16 часов, охлаждали до комнатной температуры и выливали в метил-трет-бутиловый эфир (500 мл). Органический слой отделяли и промывали водой (3 х 300 мл). Органический слой выливали в круглодонную колбу и охлаждали до 0oС. Порциями добавляли 6Н раствор гидроокиси натрия до тех пор, пока рН не поднялся до 10 (  500 мл). Органический слой отделялся, промывался водой (200 мл), сушился над сульфатом магния, фильтровался и концентрировался, давая 6-метокси-1-метил-1-трифторметилизохроман-7-карбальдегид в виде масла (54,2 г смеси 12:1 региоизомеров, 97%). 1H ЯМР (400 МГц, СDС13) 1,71 (с, 3), 2,95 (дт, 2, J=2,6, 5,3), 3,90-3,97 (м, 1), 3,97 (с, 3), 4,19 (дт, 1, J=11,2, 5,6), 6,81 (д, 1, J=1,2), 10,4 (с, 1). 13С ЯМР (75 МГц, СDС13) 500 мл). Органический слой отделялся, промывался водой (200 мл), сушился над сульфатом магния, фильтровался и концентрировался, давая 6-метокси-1-метил-1-трифторметилизохроман-7-карбальдегид в виде масла (54,2 г смеси 12:1 региоизомеров, 97%). 1H ЯМР (400 МГц, СDС13) 1,71 (с, 3), 2,95 (дт, 2, J=2,6, 5,3), 3,90-3,97 (м, 1), 3,97 (с, 3), 4,19 (дт, 1, J=11,2, 5,6), 6,81 (д, 1, J=1,2), 10,4 (с, 1). 13С ЯМР (75 МГц, СDС13) 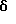 23,07, 29,98, 55,73, 60,83, 76,03 (кв, J=27,4), 111,81, 112,50, 123,65, 125,32, 125,64 (кв, J=287), 127,06, 160,89, 188,92. ИК 1683, 1616, 1498, 1296, 1271, 1163, 1149, 1120, 1096, 874 см-1. Анализ. Вычислено для С13H13F3О3: С 57,13; Н 5,05. Найдено: С 56,94; Н 4,78.
Пример 13 23,07, 29,98, 55,73, 60,83, 76,03 (кв, J=27,4), 111,81, 112,50, 123,65, 125,32, 125,64 (кв, J=287), 127,06, 160,89, 188,92. ИК 1683, 1616, 1498, 1296, 1271, 1163, 1149, 1120, 1096, 874 см-1. Анализ. Вычислено для С13H13F3О3: С 57,13; Н 5,05. Найдено: С 56,94; Н 4,78.
Пример 13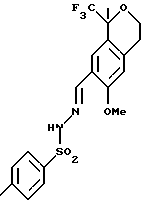 N’-1-[(E)-1-(6-метокси-1,1-диметил-3,4-дигидро-1Н-изохромен-7-ил)метилиден]-4-метил-1-бензолсульфонгидразид К раствору сырого 6-метокси-1-метил-1-трифторметилизохроман-7-карбальдегида (54,2 г, 198 ммоль), полученного в примере 12, в метаноле (542 мл) добавляли п-толуолсульфонгидразид (36,9 г, 198 ммоль), затем 2% водный раствор уксусной кислоты (81,3 мл). Реакционную смесь нагревали до кипячения с обратным холодильником в течение 90 мин и охлаждали до комнатной температуры. В осадок выпадало твердое вещество, которое фильтровалось, давая N’-1-[(Е)-1-(6-метокси-1,1-диметил-3,4-дигидро-1Н-изохромен-7-ил) метилиден]-4-метил-1-бензолсульфонгидразида (45,46 г, 52%). Т. пл. 181-183oС. 1H ЯМР (300 МГц, СDС13) 1,71 (д, 3, J=0,7), 2,44 (с, 3), 2,85-2,89 (м, 2), 3,84 (с, 3), 3,93 (дт, 1, J=11,2, 5,6), 4,16 (дт, 1, J=11,2, 5,6), 6,65 (с, 1), 7,33 (д, 2, J=8,1), 7,79 (д, 1, J=1,2), 7,89 (д, 2, J=8,4), 8,13 (с, 1). 13С ЯМР (75 МГц, CDCl3) 21,48, 23,07, 29,50, 55,47, 60,99, 76,02 (кв, J=27,4), 110,91, 120,45, 124,74, 125,04, 125,72 (кв, J=287), 127,95, 129,45, 134,97, 138,97, 143,34, 144,22, 157,04. ИК 3223, 1623, 1505, 1417, 1325, 1289, 1275, 1172, 1157, 1123, 1098, 918, 658 см-1. Анализ. Вычислено для С20Н21F3N2О4S: С 54,29; Н 4,78; N 6,33. Найдено: С 54,34; Н 4,73; N 6,37.
Пример 14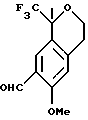 6-Метокси-1-метил-1-трифторметилизохроман-7-карбальдегид Смесь хлорида меди (II) (52,7 г, 309 ммоль) и N’-1-[(Е)-1-(6-метокси-1,1-диметил-3,4-дигидро-1Н-изохромен-7-ил)метилиден] -4-метил-1-бензолсульфонгидразида (45,5 г, 103 ммоль) в трет-бутиловом спирте (910 мл) и воде (228 мл) нагревали до 70oС в течение 2,5 часов. Реакционную смесь охлаждали до комнатной температуры, концентрировали примерно до 300 мл и выливали в метил-трет-бутиловый эфир (500 мл) и воду (500 мл). Смесь перемешивали в течение 15 мин и фильтровали. Фильтрат выливали в метил-трет-бутиловый эфир (200 мл) и слои разделяли. Органический слой промывался водой (4 х 250 мл), сушился над сульфатом магния, фильтровался и концентрировался, давая 6-метокси-1-метил-1-трифторметилизохроман-7-карбальдегид в виде масла, которое затвердевало при стоянии (26,8 г, 95%). Т. пл. 82-93oС. 1H ЯМР (400 МГц, СDС13) 1,71 (с, 3), 2,95 (дт, 2, J=2,6, 5,3), 3,90-3,97 (м, 1), 3,97 (с, 3), 4,19 (дт, 1, J=11,2, 5,6), 6,81 (д, 1, J=1,2), 10,4 (с, 1). 13С ЯМР (75 МГц, СDС13) 23,07, 29,98, 55,73, 60,83, 76,03 (кв, J=27,4), 111,84, 112,50, 123,65, 125,32, 125,64 (кв, J=287), 127,06, 160,89, 188,92. ИК 1683, 1616, 1498, 1296, 1271, 1163, 1149, 1120, 1096, 874 см-1. Анализ. Вычислено для С13Н13F3O3: С 57,13; Н 5,05. Найдено С 56,94; Н 4,78.
Пример 15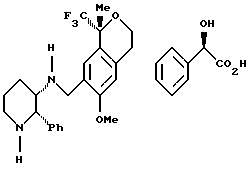 (S)-(+)-Манделат (2S, 3S)-[(1R)-6-метокси-1-метил-1-трифторметилизохроман-7-илметил]-(2-фенилпиперидин-3-ил)-амина Триацетоксиборгидрид натрия (11,61 г, 54,8 ммоль) добавляли одной порцией к охлажденной на водяной бане суспензии 6-метокси-1-метил-1-трифторметилизохроман-7-карбальдегида (7,51 г, 27,4 ммоль) и диманделата (2S-3S)-2-фенилпиперидин-3-иламина (13,8 г, 28,7 ммоль) в дихлорметане (150 мл). В течение 15 мин большая часть исходного вещества растворялась, и вскоре после этого начиналось медленное осаждение продукта. Реакционную смесь перемешивали в течение 2,5 ч при комнатной температуре, охлаждали до 0oС и медленно добавляли 1Н водный раствор гидроокиси натрия (150 мл). Слои разделяли, водный слой (рН 9) экстрагировали дихлорметаном (50 мл). Объединенные органические экстракты перемешивались в течение одного часа с 1Н водным раствором гидроокиси натрия (100 мл), слои разделялись и органический слой промывался водой (50 мл), рассолом (50 мл), сушился над Na2SО4 и фильтровался. Растворитель выпаривался и образовавшаяся не совсем белая пена сушилась в вакууме, давая 11,08 г (93%) сырого продукта. S-(+)миндальную кислоту (7,55 г, 49,6 ммоль), растворенную в этаноле (100 мл), добавляли к раствору смеси диастереомеров (6-метокси-1-метил-1-трифторметилизохроман-7-илметил)-(2-фенилпиперидин-3-ил)-амина (10,78 г, 24,8 ммоль) в этаноле (300 мл) при комнатной температуре. Смесь перемешивали, и начиналась кристаллизация. После перемешивания в течение ночи смесь фильтровалась, давая 4,66 г (32%) (S)-(+)-манделата (6-метокси-1-метил-1-трифторметилизохроман-7-илметил)-(2-фенилпиперидин-3-ил)-амина в виде смеси диастереомеров (соотношение 81:19 по результатам анализа ВЭЖХ). 1H ЯМР (400 МГц, СDС13), (представленные данные для основного диастереоизомера) 1,42-1,64 (м, 2), 1,53 (с, 3), 1,72-1,79 (м, 1), 1,94-1,98 (м, 1), 2,46-2,89 (м, 4), 3,15-3,28 (м, 3), 3,45 (с, 3), 3,47-3,78 (м, 1), 3,92-3,97 (м, 2), 4,27 (шир.с., 1), 4,52 (с, 1), 6,66 (с, 1), 7,04-7,19 (м, 4), 7,27-7,36 (м, 7). 13С ЯМР (100 МГц, СDС13) 16,99, 22,53, 25,96, 28,58, 45,05, 45,46, 53,52, 53,95, 55,08, 60,61, 61,86, 73,25, 75,54 (кв, J=28,2), 110,36, 126,02, 126,27, 126,32, 126,42, 126,55, 127,01, 127,43, 127,57, 128,27, 135,04, 137,83, 143,16, 156,51, 174,59. ИК 3441, 1576, 1358, 1160, 1136, 1098, 1038, 775, 756, 698 см-1. Анализ. Вычислено для С32Н37F3N2О5: С 65,52; Н 6,36; N 4,78. Найдено: С 65,55; Н 6,03; N 4,84.
Пример 16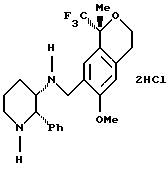 Дигидрохлорид (2S,3S)-[(1R)-6-метокси-1-метил-1-трифторметилизохроман-7-илметил]-(2-фенилпиперидин-3-ил)-амина (S)-(+)-Манделат (6-метокси-1-метил-1-трифторметилизохроман-7-илметил)-(2-фенилпиперидин-3-ил)-амина (2,25 г смеси диастереоизомеров 81:19, 3,84 ммоль) перемешивали в течение ночи в диизопропиловом эфире (23 мл) и 1Н водном растворе гидроокиси натрия (23 мл). Слои разделяли и органический слой промывали водой (20 мл) и рассолом (20 мл). Органический слой концентрировался до сырого воскообразного твердого вещества и добавлялся метанол (15 мл). Раствор перемешивался при комнатной температуре и добавлялся по каплям 1,5Н водный раствор соляной кислоты (5,0 мл). Сразу же выпадал в осадок дигидрохлорид, а белая суспензия перемешивалась в течение ночи при комнатной температуре, фильтровалась и сушилась в вакууме, давая дигидрохлорид (6-метокси-1-метил-1-трифторметилизохроман-7-илметил)-(2-фенилпиперидин-3-ил)-амина (1,282 г, 66%) в виде смеси диастереоизомеров 96:4. Соотношение диастереоизомеров можно было дополнительно увеличить кристаллизацией из метанола/воды (75/25). 1H ЯМР (400 МГц, D2O), (представленные данные для основного диастереоизомера) 1,52 (с, 3), 1,80-1,92 (м, 2), 1,95-2,50 (м, 1), 2,21-2,26 (м, 1), 2,63-2,71 (м, 2), 3,04-3,11 (м, 1), 3,36 (с, 3), 3,45-3,49 (м, 1), 3,65-3,81 (м, 3), 3,90-3,96 (м, 1), 4,09 (д, 1, J=13,5), 6,46 (с, 1), 6,98-7,07 (м, 3), 7,23-7,25 (м, 2), 7,30 (т, 1, J=7,5). ИК 2958, 1457, 1377, 1143, 749, 692 см-1. Анализ. Вычислено для C24H31Cl2F3N2О2: С 56,81; Н 6,16; Cl 13,97; N 5,52. Найдено: С 56,69; Н 6,31; Cl 14,13; N 5,55.5
Формула изобретения
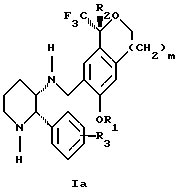 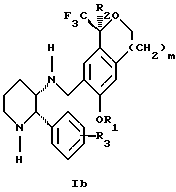 обогащенной соединением формулы Iа, и их фармацевтически приемлемых солей, где R1 является C1-6 алкилом, R2 является C1-6 алкилом, галоген C1-6 алкилом или фенилом или замещенным фенилом; R3 является водородом или галогеном; m = 0, 1 или 2; включающий стадии (а1) взаимодействия смеси соединений формул Iа и Ib 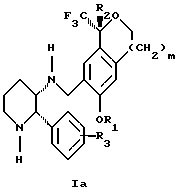 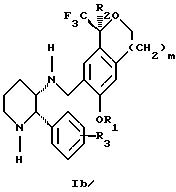 с кислотой формулы НХ, где НХ выбрана из группы, состоящей из (S)-(+)-миндальной кислоты, D-(-)-винной кислоты, ди-п-толуоил-D-винной кислоты, ((1R)-эндо, анти)-(+)-3-бромкамфора-8-сульфоновой кислоты, хинной кислоты, уксусной кислоты и бромистоводородной кислоты с образованием диастереомерной смеси соединений формул Va и Vb соответственно 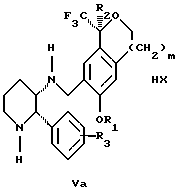 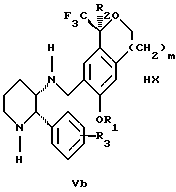 (b1) кристаллизации НХ соли диастереомерной смеси продуктов стадии (а1) из ее раствора в соответствующем растворителе, (с1) обработки образовавшейся смеси соединений, полученной на стадии (b1), основанием с получением смеси соединений Iа и Ib, которая обогащена соединением формулы Iа. 2. Способ по п. 1, дополнительно включающий обработку смеси соединений Iа и Ib, которая обогащена соединением формулы Ia 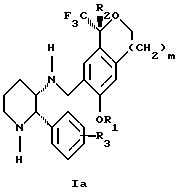 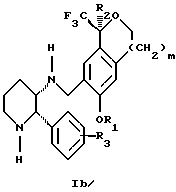 протонной кислотой H+Y–, где анион Y– выбран из группы, состоящей из хлорида, бромида, сульфата, бисульфата, фосфата, кислого фосфата, ацетата, лактата, цитрата, кислого цитрата, тартрата, битартрата, сукцината, малеата, фумарата, глюконата, сахарата, бензоата, метансульфоната, этансульфоната, бензолсульфоната, п-толуолсульфоната и 1,1′-метилен-бис-(2-гидрокси-3-нафтоата), с образованием смеси соединений VIa и VIb, высокообогащенной кислотноаддитивной солью диастереомерного соединения формулы VIa 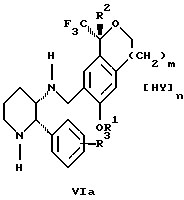 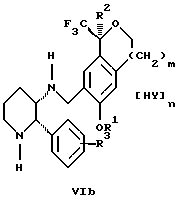 где n является целым числом от 1 до 2. 3. Способ по п. 2, в котором протонной кислотой является соляная кислота, и n = 2. 4. Способ по п. 1, дополнительно включающий стадию взаимодействия соединения формулы III 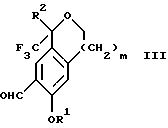 в которой R1 является C1-6 алкилом; R2 является C1-6 алкилом, галоген C1-6 алкилом или фенилом или замещенным фенилом; m = 0, 1 или 2; с соединением формулы IV 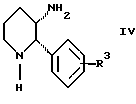 в которой R3 является водородом или галогеном; в присутствии восстанавливающего агента с образованием диастереомерной смеси соединений формул Iа и Ib 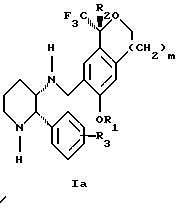 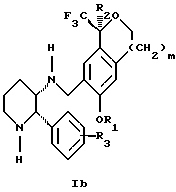 5. Способ по п. 4, в котором восстанавливающий агент выбирают из группы, состоящей из триацетоксиборгидрида натрия, цианоборгидрида натрия и боргидрида натрия. 6. Способ по п. 1, в котором кислота НХ на стадии (а1) является (S)-(+)-миндальной кислотой. 7. Смесь соединений формул Iа и Ib 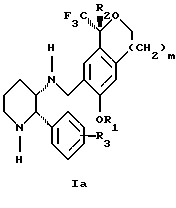 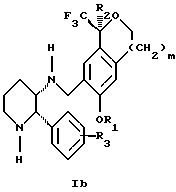 или их фармацевтически приемлемых солей, в которых R1 является C1-C6 алкилом; R2 является C1-C6 алкилом, галоген C1-C6 алкилом или фенилом или замещенным фенилом; R3 является водородом или галогеном; m = 0, 1 или 2, и соотношение соединения формулы Iа и Ib составляет 90: 10 или выше. 8. Смесь по п. 7, в которой соотношение составляет 98: 2 или выше. 9. Смесь соединений формул Va и Vb 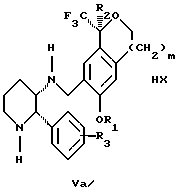 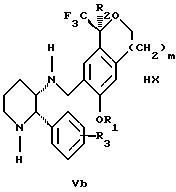 высокообогащенная соединением формулы Va, в которых R1 является C1-C6 алкилом; R2 является C1-C6 алкилом, галоген C1-C6 алкилом или фенилом или замещенным фенилом; R3 является водородом или галогеном; m = 0, 1 или 2; НХ выбрана из группы, состоящей из (S)-(+)-миндальной кислоты, D-(-)-винной кислоты, ди-п-толуол-D-винной кислоты, ((1R)-эндо, анти)-(+)-3-бромкамфора-8-сульфоновой кислоты, хинной кислоты, уксусной кислоты и бромистоводородной кислоты. 10. Смесь по п. 9, в которой НХ является (S)-(+)-миндальной кислотой. 11. Способ по п. 1, дополнительно включающий стадию формилирования соединения формулы II 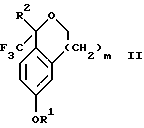 в которой R1 является C1-C6 алкилом; R2 является C1-C6 алкилом, галоген C1-C6 алкилом или фенилом или замещенным фенилом; R3 является водородом или галогеном; m = 0, 1 или 2, по реакции с гексаметилентетрамином в присутствии кислоты с образованием соединения формулы III 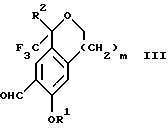 12. Способ по п. 11, в котором кислоту выбирают из группы, состоящей из трифторуксусной кислоты, глицероборной кислоты, уксусной кислоты и соляной кислоты. 13. Способ по п. 1, в котором соединение формулы II 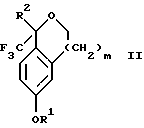 в которой R1 является C1-C6 алкилом; R2 является C1-C6 алкилом, галоген C1-C6 алкилом или фенилом или замещенным фенилом; R3 является водородом или галогеном; m = 0, 1 или 2; получают по способу, включающему стадии (а2) взаимодействия соединения формулы VII 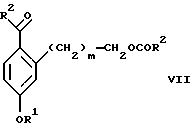 с соединением формулы CF3SiR3 4, в которой R4 является алкилом или фенилом, в присутствии фторидного источника с образованием соединения формулы VIII 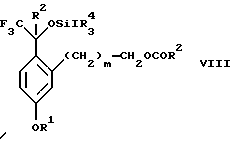 (b2) удаления силильной защитной группы из продукта стадии (а2) с помощью обработки основанием или фторидным источником с образованием соединения формулы IX 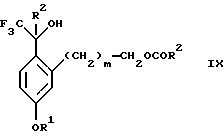 (с2) гидролиза эфирной группы продукта стадии (b2) в присутствии основания с образованием соединения формулы Х 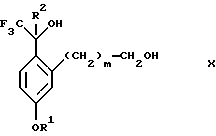 и (d2) проведения реакции циклизации кольца у продукта стадии (с2) в присутствии основания и активирующего агента, выбранного из группы, состоящей из метансульфонилхлорида метансульфонового ангидрида п-толуолсульфонилхлорида, п-толуолсульфонового ангидрида и трифторметансульфонового ангидрида. 14. Способ по п. 13, в котором фторидный источник на стадии (а2) выбирают из группы, состоящей из фторида цезия, фторида калия и фторида алкиламмония. 15. Способ по п. 1, в котором соединение формулы II 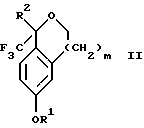 в которой R1 является C1-С6 алкилом; R2 является C1-С6 алкилом, галоген C1-С6 алкилом или фенилом или замещенным фенилом; R3 является водородом или галогеном; m = 0, 1 или 2, получают по способу, включающему стадии (а3) взаимодействия соединения формулы XI 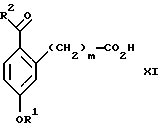 со спиртом формулы R1ОН в присутствии кислоты, где R1 имеет значения, определенные выше, с образованием соединения формулы XII 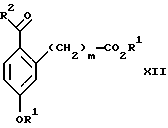 (b3) взаимодействия продукта стадии (а3) в присутствии фторидного источника с соединением формулы CF3SiR3 4, в которой R4 является C1-C6 алкилом или фенилом, с образованием соединения формулы XIII 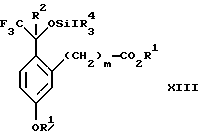 (с3) взаимодействия продукта стадии (b3) с фторидным источником с получением лактона формулы XIV 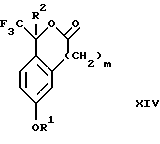 (d3) взаимодействия лактонового продукта стадии (с3) с восстанавливающим агентом необязательно в присутствии кислоты Льюиса с получением соединения формулы XV 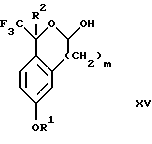 и (е3) взаимодействия продукта стадии (d3) с восстанавливающим агентом необязательно в присутствии кислоты Льюиса. 16. Способ по п. 1, дополнительно включающий стадию очистки соединения формулы III 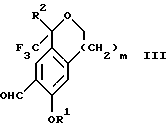 в которой R1 является C1-C6 алкилом; R2 является C1-C6 алкилом, галоген C1-C6 алкилом или фенилом или замещенным фенилом; m = 0, 1 или 2, включающий стадии (а4) образования гидразона по реакции соединения формулы III с гидразоном формулы XVI 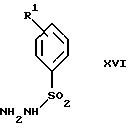 в которой R1 имеет значения, определенные выше, в присутствии кислоты с получением соединения формулы XVII 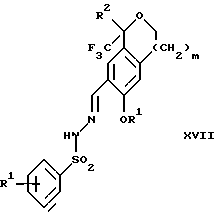 и (b4) гидролиза продукта стадии (а4) с помощью обработки реагентом, выбранным из группы, состоящей из хлорида меди (II), йодида меди (II), ацетата меди (II), сульфата меди, серной кислоты, уксусной кислоты и соляной кислоты. 17. Соединение формулы VIII 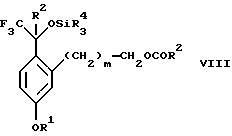 в которой R1 является C1-С6 алкилом; R2 является C1-С6 алкилом; R4 является C1-С6 алкилом; m = 0, 1 или 2. 18. Соединение формулы IX 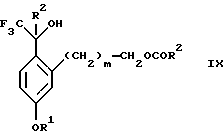 в которой R1 является C1-C6 алкилом; R2 является C1-C6 алкилом; m = 0, 1 или 2. 19. Соединение формулы XIII 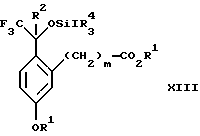 в которой R1 является C1-C6 алкилом; R2 является C1-C6 алкилом; R4 является C1-C6 алкилом; m = 0, 1 или 2. 20. Соединение формулы XIV 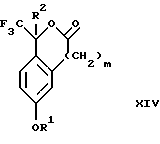 в которой R1 является C1-C6 алкилом; R2 является C1-C6 алкилом; m = 0, 1 или 2. 21. Соединение формулы XV 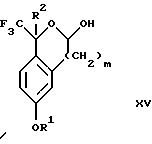 в которой R1 является C1-C6 алкилом; R2 является C1-C6 алкилом; m = 0, 1 или 2. 22. Соединение формулы XVII 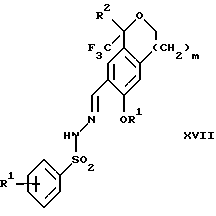 в которой R1 является C1-С6 алкилом; R2 является C1-С6 алкилом; m = 0, 1 или 2. РИСУНКИ
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 18.10.2008
Извещение опубликовано: 20.07.2010 БИ: 20/2010
|
||||||||||||||||||||||||||