Патент на изобретение №2184113
|
||||||||||||||||||||||||||
(54) НОВЫЕ СОЕДИНЕНИЯ
(57) Реферат: Предложены новые соединения общей формулы (I), где R1 и R2 являются низшим алкилом, R3, который находится в положении 3, 4 или 5 фенильного кольца, является Н, галогеном, низшим алкилом; R4 – Н, низшим алкилом, X, который соединен с гетероциклом в положении 4 или 7, является NH или О, или их фармацевтически приемлемые соли, представляющие собой гидрохлорид или соль метансульфоновой кислоты; три разных способа их получения, фармацевтический препарат, обладающий активностью ингибитора секреции желудочной кислоты; способ ингибирования секреции желудочной кислоты, способ лечения желудочно-кишечных воспалительных заболеваний или состояний, при которых имеет место инфекция Helicobacter pylori слизистой оболочки млекопитающего. 9 с. и 19 з.п. ф-лы, 1 табл. Изобретение относится к новым соединениям и их терапевтически приемлемым солям, которые ингибируют экзогенно или эндогенно стимулированную секрецию желудочной кислоты и, таким образом, могут быть применимы для предотвращении или лечения желудочно-кишечных воспалительных заболеваний. В дополнительных аспектектах, изобретение относится к соединениям по изобретению для применения в терапии; к способам получения таких новых соединений; к фармацевтическим композициям, содержащим по меньшей мере одно соединение по изобретению или его терапевтически приемлемую соль в качестве активного ингредиента; и к применению активных соединений в производстве лекарственных средств для медицинского применения, описанного выше. Производные бензимидазола следующей формулы, активные в качестве противоязвенных агентов, описаны в Европейском патенте ЕР-В-0266326 и американском патенте US 5106862: 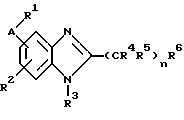 где R1 среди прочих является арильной группой формулы 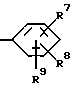 в которой каждый из R7, R8 и R9 независимо представляет собой среди прочих Н или C1-6алкил или галоген; R2 является среди прочих Н; R3 является среди прочих Н или C1-6алкилом; N является целым числом 0-6; R4, R5 и R6 являются Н или C1-6-алкилом; А является алкиленом, содержащим до 6 атомов углерода, возможно прерванных гетероатомом, таким как О или N. Для обзора фармакологии желудочной кислой помпы (Н+, К+-АТФаза) смотри Sachs et al. (1995) Annu. Rev. Pharmacol. Toxicol. 35: 277-305. Неожиданно было найдено, что соединения Формулы I, которые являются замещенными производными бензимидазола, в которых фенильная группировка замещена низшим алкилом в 2- и 6-положении, являются особенно эффективными как ингибиторы желудочно-кишечной Н+ К+-АТФазы и таким образом как ингибиторы секреции желудочной кислоты. В одном аспекте, изобретение, таким образом, относится к соединениям общей формулы I: 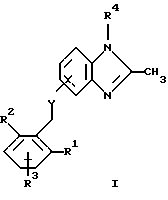 или их фармацевтически приемлемым солям, где R1 является низшим алкилом; R2 является низшим алкилом; R3, который находится в положении 3, 4 или 5 фенильного кольца, является (а) Н, (б) галогеном, или (в) низшим алкилом; R4 является (а) Н, или (б) низшим алкилом; X, который соединен с гетероциклом в положении 4 или 7, является (a) NH, или (6) O. Применяемый здесь термин “низший алкил” обозначает прямую или разветвленную алкильную группу, имеющую от 1 до 6 атомов углерода. Примеры указанного низшего алкила включают в себя метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, и пентил, и гексил с прямой и разветвленной цепью. Термин “галоген” включает в себя фтор, хлор, бром и йод. Оба чистых энантиомера, рацемические смеси и неравные смеси двух энантиомеров входят в объем изобретения. Следует понимать, что все возможные диастереомерные формы (чистые энантиомеры, рацемические смеси и неравные смеси двух энантиомеров) входят в объем изобретения. Также в изобретение включены производные соединений формулы I, которые имеют биологическую функцию соединений формулы I. В зависимости от условий способа конечные продукты формулы I получают или в нейтральной или в солевой форме. Как свободное основание, так и соли этих конечных продуктов входят в объем изобретения. Соли новых соединений, полученные присоединением кислоты способом, по существу известным, могут быть превращены в свободное основание, применяя основные агенты, такие как щелочь, или путем йонообмена. Полученное свободное основание также может образовывать соли с органическими или неорганическими кислотами. При получении солей, полученных присоединением кислоты, предпочтительно применяют такие кислоты, которые образуют подходящие терапевтически приемлемые соли. Примерами таких кислот являются галогеноводородные кислоты, такие как соляная кислота, серная кислота, фосфорная кислота, азотная кислота, алифатические, алициклические, ароматические или гетероциклические карбоновые или сульфоновые кислоты, такие как муравьиная кислота, уксусная кислота, пропионовая кислота, янтарная кислота, гликолевая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, аскорбиновая кислота, малеиновая кислота, гидроксималеиновая кислота, пировиноградная кислота, пара-гидроксибензойная кислота, эмбоновая кислота, метансульфоновая кислота, этансульфоновая кислота, гидроксиэтансульфоновая кислота, галогенбензолсульфоновая кислота, толуолсульфоновая кислота или нафталинсульфоновая кислота. Предпочтительными соединениями согласно изобретению являются такие соединения формулы I, где R1 является СН3 или СН2СН3; R2 является СН3 или СН2СН3; R3 является Н, 4-F или 4-Cl; R4 является Н или СН3 и Х является NH или О, связанным с гетероциклом в 4-м положении. Особенно предпочтительными соединениями по изобретению являются: 4-(2,6-диметилбензиламино)-2-метилбензимидазол; 4-(2,6-диметилбензилокси)-2-метилбензимидазол; 4-(2,6-диметил-4-фторбензиламино)-2-метилбензимидазол; 4-(2,6-диметил-4-фторбензилокси)-2-метилбензимидазол; 4-(2,6-диметилбензиламино)-1,2-диметилбензимидазол; 4-(2-этил-6-метилбензиламино)-2-метилбензимидазол; 4-(2,6-диэтилбензиламино)-2-метилбензимидазол; 4-(2,6-диметил-4-фторбензиламино)-1,2-диметилбензимидазол; 4-(2,6-диметил-4-фторбензилокси)-1,2-диметилбензимидазол. Получение В настоящем изобретении также предлагаются следующие способы от А до В для производства соединений общей формулы I: Способ А Способ А для производства соединений общей формулы I включает в себя следующие стадии: а) Соединения общей формулы II 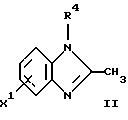 где X1 является NH2 или ОН-группой, связанной с гетероциклом в положении 4 или 7, и R4 такой, как определен для формулы I, могут быть подвергнуты взаимодействию с соединениями общей формулы III 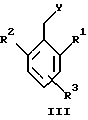 где R1, R2 и R3 те, что определены для формулы I, и Y является уходящей группой, такой как галогенид, тозилокси или мезилокси, с получением соединений формулы I. Удобно проводить эту реакцию в инертном растворителе, например ацетоне, ацетонитриле, диметоксиэтане, метаноле, этаноле или диметилформамиде с основанием или без. Основанием является, например, гидроксид щелочного металла, такой как гидроксид натрия или гидроксид калия; алкоголят натрия, такой как метоксид натрия или этоксид натрия; гидрид щелочного металла, такой как гидрид натрия и гидрид калия; карбонат щелочного металла, такой как карбонат калия и карбонат натрия; или огранический амин, такой как триэтиламин. Способ Б Способ Б для производства соединений общей формулы 1 включает в себя следующие стадии: а) Соединение общей формулы IV 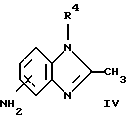 где R4 тот, что определен для формулы I, и NН2 группа связана с гетероциклом в положении 4 или 7, может быть подвергнуто взаимодействию с соединениями общей формулы V 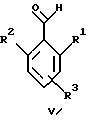 где R1, R2 и R3 те, что определены для формулы I, в присутствии кислоты Льюиса, например хлорида цинка с получением соединений формулы VI 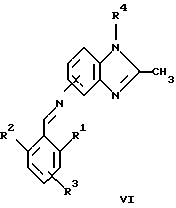 где R4 тот, что определен для формулы 1 и имин азота связан с гетероциклом в положении 4 или 7, таким образом, соединения общей формулы VI восстанавливают, например, применяя боргидрид натрия или боргидрид натрийциано, получая соединения общей формулы I. Реакции можно проводить при стандартных условиях в инертном растворителе, например, метаноле или этаноле. Способ В Соединения общей формулы I, где R4 является “низшим алкилом” могут быть приготовлены путем реакции алкилирования соединений общей формулы I, где R4 является Н в инертном растворителе, таком как ацетонитрил или ацетон, с основанием или без него, с соединениями общей формулы VII R4X2 (VII) где R4 тот, что определен для формулы I и X2 является уходящей группой, например галидом, мезилатом или тозилатом. Основанием является, например, гидроксид щелочного металла, такой как гидроксид натрия или гидроксид калия; алкоголят натрия, такой как метоксид натрия или этоксид натрия; гидрид щелочного металла, такой как гидрид натрия или гидрид калия; карбонат щелочного металла, такой как карбонат калия и карбонат натрия; или огранический амин, такой как триэтиламин. Медицинское применение В дополнительном аспекте, изобретение относится к соединениям формулы I для применения в терапии, в частности для применения против желудочно-кишечных воспалительных заболеваний. В изобретении также предлагается применение соединения формулы I в производстве лекарственного средства для ингибирования секреции желудочной кислоты или для лечения желудочно-кишечных воспалительных заболеваний. Соединения по изобретению, таким образом, могут быть использованы для предотвращения и лечения у млекопитающих, включая человека, желудочно-кишечных воспалительных заболеваний и заболеваний, связанных с желудочной кислотой, таких как гастрит, язва желудка, язва двенадцатиперстной кишки, рефлюкс-эзофагит и синдром Золлингера-Эллисона. Более того, соединения можно применять для лечения других желудочно-кишечных расстройств, где желателен желудочный противосекреторный эффект, например, у пациентов с гастриномами и у пациентов с острым верхним желудочно-кишечным кровотечением. Они также могут быть применимы у пациентов в ситуациях с интенсивным уходом и до- и постоперативно для предотвращения аспирации кислоты и стрессорного образования язвы. Фармацевтические препараты В еще одном дополнительном аспекте, изобретение относится к фармацевтическим композициям, содержащим по меньшей мере одно соединение по изобретению или его терапевтически приемлемую соль в качестве активного ингредиента. Соединения по изобретению также могут быть применимы в препаратах вместе с другими активными ингредиентами, например антибиотиками, такими как амоксициллин. Для клинического применения, соединения по изобретению приготовляют в виде фармацевтических препаратов для перорального, ректального, парентерального или другого способа введения. Фармацевтический препарат содержит соединение по изобретению в комбинации с одним или более чем одним фармацевтически примелемыми ингредиентами. Носитель может быть в форме твердого вещества, полутвердого вещества или жидкого разбавителя или капсулы. Эти фармацевтические препараты являются дополнительным объектом по изобретению. Обычно количество активных соединений находится между 0,1-95 мас.% препарата, предпочтительно между 0,1-20 мас.% в препаратах для парентерального применения и предпочтительно между 0,1 и 50 мас.% в препаратах для перорального введения. При получении фармацевтических препаратов, содержащих соединение по настоящему изобретению в форме стандартных доз для перорального введения, выбранное соединение может быть смешано с твердыми, порошкообразными ингредиентами, такими как лактоза, сахароза, сорбит, маннит, крахмал, амилопектин, производные целлюлозы, желатин или другой подходящий ингредиент, а также с дезинтегрирующими агентами и смазывающими агентами, такими как стеарат магния, стеарат кальция, натрий стеарил фумарат и полиэтиленгликолевые воски. Затем смесь перерабатывают в гранулы или прессуют в таблетки. Мягкие желатиновые капсулы могут быть получены при использовании капсул, содержащих смесь активного соединения или соединений по изобретению, растительного масла, жира или другого подходящего наполнителя для мягких желатиновых капсул. Твердые желатиновые капсулы могут содержать гранулы активного соединения. Твердые желатиновые капсулы могут также содержать активное соединение в комбинации с твердыми порошкообразными ингредиентами, такими как лактоза, сахароза, сорбит, маннит, картофельный крахмал, кукурузный крахмал, амилопектин, производные целлюлозы или желатин. Формы стандартных доз для ректального введения могут быть приготовлены (i) в форме суппозиториев, которые содержат активное вещество, смешанное с нейтральным жирным основанием; (ii) в форме желатиновой ректальной капсулы, которая содержит активное вещество в смеси с растительным маслом, парафиновым маслом или другим подходящим наполнителем желатиновых ректальных капсул; (iii) в форме готовой микроклизмы; или (iv) в форме препарата сухой микроклизмы, разводимого в подходящем растворителе непосредственно перед введением. Жидкие препараты для перорального введения могут быть получены в форме сиропов или суспензий, например растворов или суспензий, содержащих от 0,1% до 20 мас.% активного ингредиента и остальной части, состоящей из сахара или сахарных спиртов и смеси этанола, воды, глицерина, пропиленгликоля и полиэтиленгликоля. При необходимости такие жидкие препараты могут содержать красящие агенты, вкусовые агенты, сахарин и карбоксиметилцеллюлозу или другие загустители. Жидкие препараты для перорального введения могут быть также получены в форме сухого порошка для разведения в подходящем растворителе перед применением. Растворы для парентерального введения могут быть получены в виде раствора соединения по изобретению в фармацевтически приемлемом растворителе, предпочтительно в концентрации от 0,1% до 10 мас.%. Эти растворы могут также содержать стабилизирующие ингредиенты и/или буферные ингредиенты и быть разделены на стандартные дозы в форме ампул или флаконов. Растворы для парентерального введения могут быть также приготовлены в виде сухого препарата и произвольным образом растворены подходящим растворителем перед применением. Типичная дневная доза активного вещества варьирует в широком диапазоне и будет зависеть от различных факторов, таких как, например, индивидуальная потребность каждого пациента, путь введения и заболевание. В общем, пероральные и парентеральные дозировки будут находиться в диапазоне от 5 до 1000 мг в день активного вещества. Соединения в соответствии с изобретением могут быть также применимы в лекарственных препаратах вместе с другими активными ингредиентами, например, для лечения или профилактики состояний, при которых имеет место инфекция Helicobacter pylori слизистой оболочки желудка человека. Такие дополнительные активные ингредиенты могут быть антимикробными агентами, в частности: – бета-лактамные антибиотики, такие как амоксициллин, ампициллин, цефалотин, цефаклор или цефиксим; – макролиды, такие как эритромицин или кларитромицин; – тетрациклины, такие как тетрациклин или доксициклин; – аминогликозиды, такие как гентамицин, канамицин или амикацин; – хинолоны, такие как норфлоксацин, ципрофлоксацин или эноксацин; – другие, такие как метронидазол, нитрофурантоин или хлорамфеникол; или – препараты, содержащие соли висмута, такие как субцитрат висмута, субсалицилат висмута, субкарбонат висмута, субнитрат висмута или субгаллат висмута. ПРИМЕРЫ 1. ПОЛУЧЕНИЕ СОЕДИНЕНИЙ ПО ИЗОБРЕТЕНИЮ Пример 1.1 Синтез 4-(2,6-диметилбензиламино)-2-метилбензимидазола 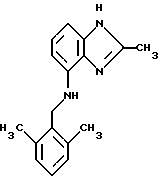 4-амино-2-метилбензимидазол (6,0 г, 0,041 моль) растворяют в ацетонитриле (120 мл). К раствору добавляют 2,6-диметилбензилхлорид (6,3 г, 0,041 моль), карбонат натрия (16 г, 0,15 моль) и каталитическое количество йодида натрия, и реакционную смесь кипятят с обратным холодильником в течение 3 часов. Карбонат натрия удаляют путем фильтрации и промывают метиленхлоридом. Вакуумное выпаривание растворителя дает масляный остаток, который подвергают флэш-хроматографии на силикагеле, метиленхлорид: метанол (10:1). Остаток кристаллизуют путем обработки этилацетатом, получая 3,3 г (30%) соединения, указанного в заголовке. (1Н ЯМР, 300 МГц, CDCl3): 2,35 (s, 6Н), 2,50 (s, 3Н), 4,35 (s, 2Н), 4,45 (bs, 1H), 6,50 (d, 1H), 6,75 (d, 1H), 6,95-7,15 (m, 4H). Пример 1.2 Синтез 4-(2,6-диметилбензилокси)-2-метилбензимидазола 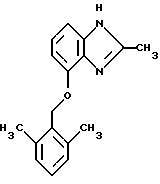 4-Гидрокси-2-метилбензимидазол (0,59 г, 4 ммоль) растворяют в ацетонитриле (15 мл). К раствору добавляют 2,6-диметилбензилхлорид (0,52 г, 4 ммоль) и гидроксид натрия (0,16 г, 4 ммоль) (растворенный в 1 мл воды) и реакционную смесь кипятят с обратным холодильником в течение 2 часов. Растворитель выпаривают при пониженном давлении, остаток растворяют в метиленхлориде и промывают 2М NaOH. Органический слой отделяют, сушат над сульфатом натрия и выпаривают при пониженном давлении. Масляный остаток очищают путем колоночной хроматографии на силикагеле. Продукт элюируют метиленхлоридом, содержащим 5% метанол. Кристаллизация из ацетонитрила дает 0,18 г (18%) соединения, указанного в заголовке. (1Н ЯМР, 300 МГц, ДМСО-d6): 2,3 (s, 6Н), 2,40 (s, 3Н), 5,25 (s, 2H), 6,9 (d, 1H), 7,05-7,15 (m, 4H), 7,2 (t, 1H). Пример 1.3 Синтез 4-(2,6-диметил-4-фторбензиламино)-2-метилбензимидазола 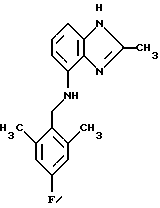 4-Амино-2-метилбензимидазол (0,6 г, 4,1 ммоль) растворяют в ацетонитриле (10 мл). К раствору добавляют 2,6-диметил-4-фторбензилбензилбромид (0,89 г, 4,1 ммоль) и карбонат калия (0,68 г, 4,9 ммоль), и смесь нагревают при 70-80oС в течение 3 часов. Реакционную смесь охлаждают до комнатной температуры и добавляют метиленхлорид (25 мл) и воду (25 мл). Органический слой отделяют, сушат над сульфатом натрия и выпаривают при пониженном давлении. Остаток дважды кристаллизуют из этилацетата и очищают путем колоночной хроматографии на силикагеле, применяя в качестве элюента метиленхлорид, содержащий 10% метанола. Получают 0,26 г (22%) соединения, указанного в заголовке. (1Н ЯМР, 300 МГц, СDСl3): 2,3 (s, 6Н), 2,45 (s, 3Н), 4,25 (s, 2H), 4,35 (bs, 1H), 6,45 (d, 1H), 6,65 (d, 2H), 6,7 (d, 1H), 7,1 (t, 1H). Пример 1.4 Синтез 4-(2,6-диметил-4-фторбензилокси)-2-метилбензимидазола 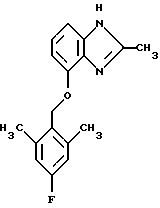 4-Гидрокси-2-метилбензимидазол (0,57 г, 3,8 ммоль) растворяют в ацетонитриле (4 мл). К раствору добавляют гидроксид натрия (0,17 г, 43 ммоль) (растворенный в 1 мл воды) и 2,6-диметил-4-фторбензилбромид (0,82 г, 3,8 ммоль), растворенный в ацетонитриле (6 мл), реакционную смесь кипятят с обратным холодильником в течение 80 минут и перемешивают при температуре окружающей среды в течение 20 часов. К реакционной смеси добавляют метиленхлорид (25 мл) и воду (25 мл). Органический слой отделяют, три раза промывают 2М NaOH, сушат над сульфатом натрия и выпаривают при пониженном давлении. Остаток кристаллизуют из этилацетата и затем дважды очищают путем колоночной хроматографии на силикагеле а) метиленхлорид:метанол (10:1) б) этилацетат: метанол: уксусная кислота:вода (96:6:6:4). Получают 0,066 г (6%) соединения, указанного в заголовке. (1Н ЯМР, 300 МГц, ДМСО-d6): 2,3 (s, 6Н), 2,5 (s, 3Н), 5,05 (s, 2H), 6,7 (d, 2H), 6,8 (d, 1Н), 7,15 (t, 1H), 7,25 (d, 1H). Пример 1.5 Синтез 4-(2,6-диметилбензиламино)-1,2-диметилбензимидазола 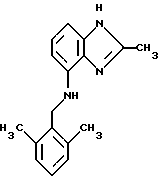 4-(2,6-Диметилбензиламино)-2-метилбензимидазол (0,2 г, 0,75 ммоль) растворяют в ацетонитриле (15 мл). К раствору добавляют метилйодид (0,05 мл 0,82 ммоль) и карбонат калия (0,2 г 1,4 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 20 часов. Твердые вещества удаляют путем фильтрации и промывают метиленхлоридом. Вакуумное выпаривание растворителя дает масляный остаток, который подвергают флэш-хроматографии на силикагеле, метиленхлорид: метанол (10:1). Кристаллизация из ацетонитрила дает 0,018 г (9%) соединения, указанного в заголовке. (1Н ЯМР, 500 МГц, СDСl3): 2,4 (s, 6H), 2,55 (s, 3Н), 3,65 (s, 3Н), 4,4 (d, 2H), 4,55 (bs, 1Н), 6,55 (d, 1H), 6,7 (d, 1Н), 7,05 (d, 2H), 7,1 (t, 1Н), 7,2 (t, 1H). Пример 1.6 Синтез 4-(2-этил-6-метилбензиламино)-2-метилбензимидазола 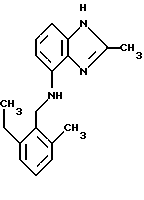 4-Амино-2-метилбензимидазол (0,4 г, 2,7 ммоль) и 2-этил-6-метилбензилхлорид (0,46 г, 2,7 ммоль) растворяют в 10 мл диметоксиэтана. К раствору добавляют карбонат натрия (0,5 г, 4,7 ммоль) и (0,2 г, 4.7 ммоль) йодид калия. Реакционную смесь кипятят с обратным холодильником в течение 2 часов. Неорганические соли удаляют путем фильтрации и промывают диметоксиэтаном. Фильтрат выпаривают до сухости. Остаток очищают путем колоночной хроматографии на силикагеле. Продукт элюируют смесью 50:50 метиленхлорида и этилацетата. Получают 0,21 г соединения, указанного в заголовке. (1Н ЯМР, 400 МГц, СDСl3): 1,1 (t, 3Н), 2,35 (s, 3Н), 2,45 (s, 3Н), 2,70 (q, 2H), 4,35 (s, 2H), 4,45 (bs, 1H), 6,55 (d, 1H), 6,75 (d, 1H), 6,95-7,20 (m, 4H). Пример 1.7 Синтез 4-(2,6-диэтилбензиламино)-2-метилбензимидазола 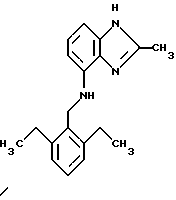 4-Амино-2-метилбензимидазол (0,79 г, 5,4 ммоль) и 2,6-диэтилбензальдегид (1,1 г, 6,6 ммоль) растворяют в метаноле (30 мл). Добавляют ZnCl2 (0,9 г, 6,6 ммоль) и последовательно маленькими порциями NaBH3CN (0,42 г, 6,6 ммоль), смесь кипятят с обратным холодильником в атмосфере аргона в течение 3 часов и перемешивают при комнатной температуре в течение 16 часов. Смесь выливают на водный раствор 1М NaOH (50 мл). Полученную в результате желтую суспензию экстрагируют ДХМ, органический раствор промывают рассолом, сушат над Na2SO4 и затем выпаривают при пониженном давлении. Масляный остаток (1,8 г) очищают путем кристаллизации из смеси этилацетата и ацетонитрила, получая 0,65 г (41%) соединения, указанного в заголовке. (1Н ЯМР, 300 МГц, СDСl3): 1,2 (t, 6Н), 2,5 (s, 3Н), 2,27 (q, 4H), 4,35 (d, 2H), 4,55 (bs, 1H), 6,05 (d, 1H), 6,75 (d, 1H), 7,0-7,25 (m, 4H), 9,0 (br, 1H) Пример 1.8 Синтез 4-(2,6-диметил-4-фторбензиламино)-1,2-диметилбензимидазола 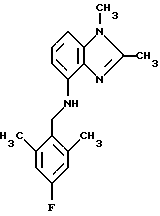 4-(2,6-Диметил-4-фторбензиламино)-2-метилбензимидазол (0,1 г, 0,35 ммоль) растворяют в 1,2-диметоксиэтане (3 мл). Добавляют твердый гидроксид натрия (25 мг, 0,63 ммоль) и тетрабутиламмоний бромид (5 мг, 0,016 ммоль). Смесь перемешивают в течение 15 минут при температуре окружающей среды. Добавляют метилйодид (60 мг 0,42 ммоль) и реакционную смесь перемешивают в течение 2,5 часов при температуре окружающей среды. Растворитель выпаривают при пониженном давлении. Остаток очищают путем колоночной хроматографии на силикагеле, применяя в качестве элюента метиленхлорид:этилацетат (50:50). Получают 80 мг (76%) соединения, указанного в заголовке. (1Н ЯМР, 300 МГц, CDCl3): 2,38 (s, 6Н), 2,53 (s, 3H), 3,67 (s, 3Н), 4,32 (d, 2H), 4,44 (bs, 1Н), 6,51 (d, 1Н), 6,68 (d, 1Н), 6,75 (d, 2H), 7,18 (t, 1H). Пример 1.9 Синтез 4-(2,6-диметил-4-фторбензилокси)-1,2-диметилбензимидазола. 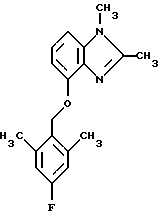 4-(2,6-Диметил-4-фторбензилокси)-2-метилбензимидазол (180 мг, 0,63 ммоль) растворяют в 1,2-диметоксиэтане (6 мл). К раствору добавляют гидроксид натрия (26 мг, 0,65 ммоль) и метилйодид (0,47 мл, 0,76 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 3,5 часов. После выпаривания растворителя остаток очищают путем хроматографии на силикагеле, метиленхлорид: метанол 10:1. Хроматография одной из полученных фракций на силикагеле с применением в качестве элюента этилацетата дает 40 мг (21%) соединения, указанного в заголовке. (1Н ЯМР, 300 МГц, СDОCl3): 2,40 (s, 6H), 2,58 (s, 3Н), 3,70 (s, 3Н), 5,24 (s, 2H), 6,75 (d, 2H), 6,82 (d, 1H), 6,93 (d, 1H), 7,19 (t, 1H). Пример 1.10 Получение гидрохлоридной соли 4-(2,6-диметилбензиламино)-2-метилбензимидазола 4-(2,6-Диметилбензиламино)-2-метилбензимидазол (0,5 г, 1,9 ммоль) растворяют в этилацетате (20 мл) и метаноле (6 мл). К раствору добавляют соляную кислоту (12М) (0,16 мл, 1,9 моль) и смесь перемешивают при температуре окружающей среды в течение 5 минут. Осажденную соль отфильтровывают и сушат, получая желаемый продукт в виде белого кристаллического твердого вещества (0,45 г, 79%). (1Н ЯМР, 500 МГц, MeOD): 2,4 (s, 6H), 2,8 (s, 3H), 4,45 (s, 2H), 6,9 (d, 1H), 7,0 (d, 1H), 7,1 (d,2H), 7,15 (t, 1H), 7,4 (t, 1H). Пример 1.11 Получение соли метансульфоновой кислоты 4-(2,6-диметилбензиламино)-2-метилбензимидазола 4-(2,6-Диметилбензиламино)-2-метилбензимидазол (0,5 г, 1,9 ммоль) растворяют в этилацетате (20 мл) и метаноле (7 мл). К раствору добавляют метансульфоновую кислоту (0,18 г, 1,9 ммоль) и смесь перемешивают при температуре окружающей среды в течение 5 минут. Осажденную соль отфильтровывают и сушат, получая желаемый продукт в виде белого кристаллического твердого вещества (0,58 г, 85%). (1Н ЯМР, 500 МГц, MeOD): 2,4 (s, 6H), 2,65 (s, 3H), 2,75 (s, 3H), 4,45 (s, 2H), 6,9 (d, 1H), 7,0 (d, 1H), 7,1 (d, 2H), 7,15 (t, 1H), 7,4 (t, 1H). 2. ПОЛУЧЕНИЕ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ Пример 2.1 Синтез 2,6-диметил-4-фторбензилбромида Смесь 3,5-диметилфторбензола (5 г, 0,04 моль), параформальдегида (15 г), бромистоводородной кислоты (70 мл) (30% в уксусной кислоте) и уксусной кислоты (25 мл) перемешивают при температуре окружающей среды в течение 4,5 часов. К смеси добавляют воду и петролейный эфир, органический слой отделяют, сушат над безводным сульфатом натрия и осторожно выпаривают при пониженном давлении. Остаток очищают путем колоночной хроматографии на силикагеле с петролейным эфиром в качестве элюента, получая продукт, указанный в заголовке (3,7 г, 43%). (1Н ЯМР, 300 МГц, CDCl3): 2,5 (s, 6H), 4,55 (s, 2H), 6,75 (d, 2H) Пример 2.2 Синтез 2-этил-6-метилбензилхлорида 2-Этил-6-метилбензиловый спирт (1,0 г, 6,67 ммоль) растворяют в 10 мл метиленхлорида. Добавляют тионилхлорид (1,0 г, 8,5 ммоль). Смесь перемешивают в течение ночи при температуре окружающей среды. Реакционную смесь выпаривают. Остаток растворяют в метиленхлориде и фильтруют через 5 г силикагеля. Фильтрат выпаривают. Получают 1,0 г (89%) соединения, указанного в заголовке (масло). (1Н ЯМР, 300 МГц, CDCl3): 1,29 (t, 3Н), 2,46 (s, 3H), 2,76 (q, 2H), 4,71 (s, 2H), 7,0-7,2 (m, 3H). 3. БИОЛОГИЧЕСКИЕ ТЕСТЫ 3.1 Эксперименты in vitro Ингибирование секреции кислоты в изолированных желудочных железах кролика Ингибирующее действие на секрецию кислоты in vitro в изолированных желудочных железах кролика измеряют как описано Berglindh et al. (1976) Acta Physiol. Scand. 97, 401-414. Соединения по Примерам 1.1-1.7 показывают значения ИК50 менее, чем 1,6 мкмоль. Для сравнения соединение (А) (4-бензиламино-2-метилбензимидазол) является соединением как заявлено в п.1 формулы изобретения Европейского патента ЕР-В-0266326, где: А является CH2NH; R2, R3, R4, R5 и R6 представляют собой Н; n = 1; и R1 является группой формулы II’, где R7, R8 и R9 представляют собой Н. Для сравнения соединение (А) показывает значение ИК50 12,9 мкмоль. 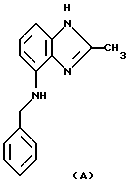 Для сравнения соединение (Б) (4-бензилокси-2-метилбензимидазол) является соединением как заявлено в п. 1 формулы изобретения Европейского патента ЕР-В-0266326, где: А является СН2O; R2, R3, R4, R5 и R6 представляют собой Н; n = 1; и R1 является группой формулы II’, где R7, R8 и R9 представляют собой Н. Для сравнения соединение (Б) показывает значение ИK50 7,8 мкмоль. 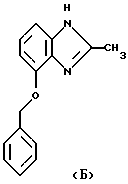 Следовательно, соединения по изобретению проявляют значительно лучшее ингибирующее действие на секрецию кислоты in vitro по сравнению с соединениями (А) и (Б), которые несут незамещенную фенильную группу. Заключают, что улучшенный эффект наблюдается благодаря фенильной группе в соединениях по изобретению, которая замещена низшим алкилом во 2- и 6-м положении. 3.2 Эксперименты in vivo Ингибирующее действие на секрецию кислоты у самок крыс Используют самок крыс линии Sprague-Dawly. В просвет желудка и верхнюю часть двенадцатиперстной кишки крысам вводят канюлированные фистулы для сбора желудочных секретов и введения тестируемых веществ, соответственно. После хирургического вмешательства до начала тестирования в течение 14 дней проводят восстановительный период. Перед секреторным тестом животных в течение 20 часов лишают пищи, но не воды. Желудок через желудочную канюлю несколько раз промывают водопроводной водой (37oС) и подкожно вводят 6 мл раствора Рингера-глюкозы. Секрецию кислоты стимулируют инфузией в течение 2,5-4 часов (1,2 мл/час, подкожно) пентагастрина и карбахола (20 и 110 нмоль/кг  час, соответственно), во время чего собирают 30-минутные фракции желудочных секретов. Тестируемые вещества или наполнители дают или через 60 минут после начала стимуляции (внутривенное и интрадуоденальное дозирование, 1 мл/кг) или за 2 часа до начала стимуляции (пероральное дозирование, 5 мл/кг, желудочная канюля закрыта). Для изучения длительности действия временной интервал между дозированном и стимуляцией может быть увеличен. Образцы желудочного сока титруют до рН 7,0 при помощи NaOH, 0,1 М, и выход кислоты рассчитывают из объема титранта и концентрации.
Дальнейшие расчеты основаны на групповом среднем значении ответов 4-6 крыс. В случае введения во время стимуляции; выход кислоты во время указанных периодов после введения тестируемого вещества или наполнителя выражают как фракционные ответы, устанавливая выход кислоты в 30-минутный период, предшествующий введению за 1,0. Процент ингибирования рассчитывают из фракционных ответов, вызываемых тестируемым соединением и наполнителем. В случае введения до стимуляции; процент ингибирования рассчитывают непосредственно из выхода кислоты, регистрируемого после тестируемого соединения и наполнителя.
Биологическая доступность у крыс час, соответственно), во время чего собирают 30-минутные фракции желудочных секретов. Тестируемые вещества или наполнители дают или через 60 минут после начала стимуляции (внутривенное и интрадуоденальное дозирование, 1 мл/кг) или за 2 часа до начала стимуляции (пероральное дозирование, 5 мл/кг, желудочная канюля закрыта). Для изучения длительности действия временной интервал между дозированном и стимуляцией может быть увеличен. Образцы желудочного сока титруют до рН 7,0 при помощи NaOH, 0,1 М, и выход кислоты рассчитывают из объема титранта и концентрации.
Дальнейшие расчеты основаны на групповом среднем значении ответов 4-6 крыс. В случае введения во время стимуляции; выход кислоты во время указанных периодов после введения тестируемого вещества или наполнителя выражают как фракционные ответы, устанавливая выход кислоты в 30-минутный период, предшествующий введению за 1,0. Процент ингибирования рассчитывают из фракционных ответов, вызываемых тестируемым соединением и наполнителем. В случае введения до стимуляции; процент ингибирования рассчитывают непосредственно из выхода кислоты, регистрируемого после тестируемого соединения и наполнителя.
Биологическая доступность у крысИспользуют взрослых крыс линии Sprague-Dawley. За 1-3 дня до экспериментов всем крысам под анестезией в левую сонную артерию вводят канюлю. Крысам, используемым для внутривенных экспериментов, также вводят канюлю в яремную вену (Popovic (1960) J. Appl. Physiol. 15, 727-728). Канюли выводят на заднюю часть шеи. Образцы крови (0,1-0,4 г) неоднократно отбирают из сонной артерии с интервалами до 5,5 часов после введенной дозы. Образцы замораживают до анализа тестируемого соединения. Биологическую доступность оценивают, рассчитывая соотношение между площадью под кривой концентрации кровь/плазма (ППК) после (i) интрадуоденального (i.d.) или перорального (р.о.) введения и (ii) внутривенного (i.v.) введения крысе или собаке, соответственно. Площадь под кривой концентрации крови от времени (ППК) определяют с помощью правила log/линейной трапеции и экстраполируют до бесконечности, деля последнюю определенную концентрацию крови на константу скорости элиминации в терминальной фазе. Системную биологическую доступность (F%) после интрадуоденального или перорального введения раcсчитывают как F(%)=(ППК (р.о. или i.d.)/ППК (i.v.)(100. Ингибирование секреции желудочной кислоты и биологической доступности у находящейся в сознании собаки Используют лабрадорских ретриверов или гончих любого пола. Им вводят дуоденальную фистулу для введения тестируемых соединений или наполнителя и канюлированную желудочную фистулу или мешок Heidenhaim для сбора желудочного секрета. Перед секреторными тестами животных в течение 18 часов лишают еды, но вода свободно доступна. Секрецию желудочной кислоты стимулируют в течение до 6,5 часов инфузии дигидрохлорида гистамина (12 мл/час) при дозе, вызывающей около 80% индивидуального максимального секреторного ответа, и собирают желудочный сок последовательными 30-минутными фракциями. Тестируемое вещество или наполнитель дают перорально, i.d. или i.v., через 1 или 1,5 часа после начала инфузии гистамина, в объеме 0,5 мл/кг веса тела. В случае перорального введения следует указать, что тестируемое соединение вводят в главный секретирующий кислоту желудок мешка Heidenham собаки. Кислотность образцов желудочного сока определяют путем титрования до рН 7,0 и раcсчитывают выход кислоты. Выход кислоты в указанные периоды сбора после введения тестируемого вещества или наполнителя выражают как фракционные ответы, устанавливая выход кислоты во фракцию, предшествующей введению за 1.0. Процент ингибирования расcчитывают из фракционных ответов, вызванных тестируемым соединением и наполнителем. Образцы крови для анализа концентрации тестируемого соединения в плазме берут с интервалами до 4 часов после дозирования. Плазму отделяют и замораживают в течение 30 минут после сбора и после анализируют. Системную биологическую доступность (F%) после перорального или i.d. введения рассчитывают как описано выше в крысиной модели. Заявитель также представляет данные по биологической активности In vitro и in vivo для заявленных соединений, которые были получены в примерах 1.1-1.9, представленных в описании изобретения (см. приложения). Данные тестов in vitro иллюстрируют активность ингибитора Н+, К+-АТФазы (ИК50, мкмоль) и ингибирующее действие на секрецию кислоты in vitro в изолированных желудочных железах кролика (ИК50, мкмоль), как указано на стр.18 описания изобретения (раздел 3.1). Активность Н+, К+-АТФазы определяли по следующей методике: желудочные мембранные пузырьки, содержащие Н+, К+-АТФазу, получали из желудка собаки, как описано ранее Saccomani et al в Biochem. Biophys. Acta, 1977, 465, pp. 311-330. Мембранную фракцию разводили в 1 мМ буфере PIPES/Tris, pH 7,4, с получением 1% концентрации сахарозы, гомогенизировали и центрифугировали при 100000 g в течение 2 часов. Полученный в результате осадок суспендировали в воде и лиофилизировали дважды. Проницаемые мембранные пузырьки (2,5-5 мг) инкубировали в течение 15 минут при 37oС в 18 мМ буфере PIPES/Tris, pH=7,4, содержащем 2 мМ MgCl2, 10 мМ KCl и 2 мМ АТФ. Активность АТФазы оценивали по высвобождению неорганического фосфата из АТФ, как описано LeBel et al в Anal. Biochem, 1978, 85, рр.86-89. В отношении представленных данных по активности заявленных соединений заявитель сообщает следующее. Данные, представленные в таблице, приведены в сравнении с активностью соединений, являющихся наиболее близкими аналогами заявленных соединений, представляющими собой производные бензимидазола, в которых бензильная группировка является незамещенной, которые описаны в патенте ЕР 266326. Авторами изобретения неожиданно обнаружено, что введение низших алкильных заместителей в ортоположение бензильной группы увеличивает активность заявленных соединений в 4-25 раз (в тестах in vitro). В таблице также приведены данные тестов in vivo, проведенные на крысах и собаках по методикам, изложенным выше. Как видно из представленных данных, соединения по настоящему изобретению проявляют повышенную эффективность в ингибировании секреции желудочной кислоты в дозировках, которые в 2-6,7 раз ниже дозировок соединений из ЕР 266326. Заявитель также сообщает, что проведенные эксперименты in vivo позволили установить, что заявленные соединения не токсичны в использованных дозировках. В отношении подтверждения заявленных средств и способов для лечения желудочно-кишечных воспалительных заболеваний заявитель, в соответствии с указаниями экспертизы, приводит следующие дополнительные доводы. Как следует из материалов заявки, предложенные соединения являются ингибиторами Н+, К+-АТФазы и, следовательно, ингибиторами секреции желудочной кислоты, что подтверждено данными биологических испытаний. Гиперсекреция желудочной кислоты является известным решающим фактором, влияющим на патогенез целого ряда широко распространенных гастроэнтерологических заболеваний, поэтому основной принцип их лечения заключается в ингибировании гиперсекреции желудочной кислоты. То есть, путем ингибирования продукции желудочной кислоты можно эффективно лечить кислотозависимые воспалительные заболевания желудочно-кишечного тракта. Представленные в описании опыты in vivo на крысах и собаках являются адекватной моделью воспалительных желудочно-кишечных заболеваний, в которой секрецию кислоты стимулируют путем введения испытуемому животному пентагастрина и карбахола (в случае опытов на крысах) либо дигидрохлорида гистамина (в случае опытов на собаках). Доказанная ингибирующая активность заявленных соединений в этой модели позволяет утверждать об эффективности заявленных соединений для лечения воспалительных желудочно-кишечных заболеваний. К ингибиторам протонной помпы относятся производные бензимидазола, в частности омепразол, пантопразол, лансопразол и рабепразол. Механизм действия ингибиторов протонной помпы установлен и заключается в связывании SH-групп протонной помпы, расположенных на поверхности апикальной мембраны, которая обращена в просвет желудочных желез. “Выключение из работы” протонной помпы обеспечивает тем самым подавление кислотной продукции (Харкевич Д.А. Фармакология: Учебник. – 6-ое изд., перераб. и доп. – М.: ГЭОТАР МЕДИЦИНА, 1999. – с.328-333). Известно, что ингибиторы протонного насоса (Н+, К+ В отношении возможности использования заявленных соединений для лечения заболеваний, при которых имеет место инфекция Helicobacter pylori, заявитель дополнительно сообщает, что в настоящее время доказан факт определенной этиологической и патогенетической роли бактерий Helicobacter pylori в развитии распространенных желудочно-кишечных заболеваний – острого и хронического гастрита, язвенной болезни желудка и двенадцатиперстной кишки, образовании некоторых видов опухолей желудка, диспепсии неязвенной этиологии (Noach L, Tytgat G. Helicobacter pylori infection. Aspects of pathogenesis and therapy. – Amsterdam, 1994, 165 p.). Общепринято. что при заболеваниях, ассоциированных с инфекцией Helicobacter pylori, необходимо проводить антихеликобактерную терапию. В 1996-1997 гг. были предложены единые стандартные схемы лечения заболеваний, ассоциированных с инфекцией Helicobacter pylori (Европейский Маастрихтский консенсус, Американская Интернациональная конференция фонда здоровья, Азиатский консенсус), при которых используются 1 антисекреторный препарат, наиболее предпочтительно ингибитор протонной помпы, и 2 или 3 антимикробных препарата (Consensus conference Helicobacter pylori in peptic ulcer disease, 1994; The European Maastricht Consensus, 1997; Lam S. K. , Talley N.J., 1998). Таким образом, специалисту в данной области очевидно, что применение соединения, обладающего активностью ингибитора секреции кислоты, в комбинации с противомикробным агентом, будет эффективно в лечении заболеваний, ассоциированных с Helicobacter pylori. Данные по биологической активности заявленных соединений даны в таблице. Формула изобретения
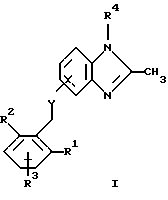 или его фармацевтически приемлемая соль, где R1 является низшим алкилом; R2 – низшим алкилом; R3, который находится в положении 3, 4 или 5 фенильного кольца, является (а) Н, (б) галогеном или (в) низшим алкилом; R4 является (а) Н или (б) низшим алкилом; Х, который соединен с гетероциклом в положении 4 или 7, является (а) NH или (б) О. 2. Соединение по п. 1 или его фармацевтически приемлемая соль, где Х соединен с гетероциклом в положении 4. 3. Соединение по п. 2 или его фармацевтически приемлемая соль, где R1 – СН3 или СН2СН3; R2 – СН3 или СН2СН3; R3 – Н, 4-F или 4-Сl и R4 – Н или СН3. 4. Соединение по п. 3, которое представляет собой соединение 4-(2,6-диметилбензиламино)-2-метилбензимидазол или его фармацевтически приемлемую соль. 5. Соединение по п. 3, которое представляет собой соединение 4-(2,6-диметилбензилокси)-2-метилбензимидазол или его фармацевтически приемлемую соль. 6. Соединение по п. 3, которое представляет собой соединение 4-(2,6-диметил-4-фторбензиламино)-2-метилбензимидазол или его фармацевтически приемлемую соль. 7. Соединение по п. 3, которое представляет собой соединение 4-(2,6-диметил-4-фторбензилокси)-2-метилбензимидазол или его фармацевтически приемлемую соль. 8. Соединение по п. 3, которое представляет собой соединение 4-(2,6-диметилбензиламино)-1,2-диметилбензимидазол или его фармацевтически приемлемую соль. 9. Соединение по п. 3, которое представляет собой соединение 4-(2-этил-6-метилбензиламино)-2-метилбензимидазол или его фармацевтически приемлемую соль. 10. Соединение по п. 3, которое представляет собой соединение 4-(2,6-диэтилбензиламино)-2-метилбензимидазол или его фармацевтически приемлемую соль. 11. Соединение по п. 3, которое представляет собой соединение 4-(2,6-диметил-4-фторбензиламино)-1,2-диметилбензимидазол или его фармацевтически приемлемую соль. 12. Соединение по п. 3, которое представляет собой соединение 4-(2,6-диметил-4-фторбензилокси)-1,2-диметилбензимидазол или его фармацевтически приемлемую соль. 13. Соединение, которое является гидрохлоридной солью соединения по любому из пп. 1-12. 14. Соединение, которое является солью метансульфоновой кислоты соединения по любому из пп. 1-12. 15. Соединение по любому из пп. 1-14 для применения в терапии. 16. Способ получения соединения по любому из пп. 1-14, при котором соединение общей формулы II 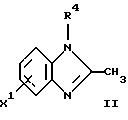 где Х1 является NH2 или ОН-группой, связанной с гетероциклом в положении 4 или 7, и R4 такой, как определено для формулы I, подвергают взаимодействию с соединением общей формулы III 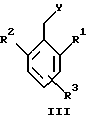 где R1, R2 и R3 такие, как определены для формулы I; Y является уходящей группой, в инертном растворителе с основанием или без него с получением соединения формулы I. 17. Способ получения соединения по любому из пп. 1-14, при котором а) соединение общей формулы IV 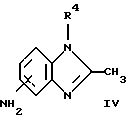 где R4 такой, как определено для формулы I; NH2-группа связана с гетероциклом в положении 4 или 7, подвергают взаимодействию с соединением формулы V 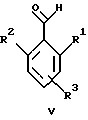 где R1, R2 и R3 такие, как определены для формулы I, в присутствии кислоты Льюиса в инертном растворителе с получением соединения формулы VI 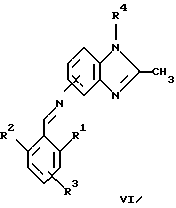 где R4 такой, как определен для формулы I; имин азота связан с гетероциклом в положении 4 или 7; (б) соединение формулы VI в инертном растворителе при стандартных условиях подвергают восстановлению до соединения общей формулы I. 18. Способ получения соединения по любому из пп. 1-3 или 8, где R4 является “низшим алкилом”, при котором соединение формулы I, где R4 – Н, в инертном растворителе с основанием или без него подвергают алкилированию с соединением формулы VII R4X2, (VII) где R4 такой, как определено для формулы I; Х2 является уходящей группой, с получением соединения формулы I, где R4 является “низшим алкилом”. 19. Фармацевтический препарат, обладающий активностью ингибитора секреции желудочной кислоты, содержащий соединение по любому из пп. 1-14 и дополнительно содержащий фармацевтически приемлемый носитель. 20. Соединение по любому из пп. 1-14 для применения при производстве лекарственного средства для ингибирования секреции желудочной кислоты. 21. Соединение по любому из пп. 1-14 для применения при производстве лекарственного средства для лечения желудочно-кишечных воспалительных заболеваний. 22. Соединение по любому из пп. 1-14 для применения при производстве лекарственного средства для лечения или профилактики состояний, при которых имеет место инфекция Helicobacter pylori слизистой оболочки желудка человека, где указанная соль адаптирована для введения в комбинации с по меньшей мере одним противомикробным агентом. 23. Способ ингибирования секреции желудочной кислоты, при котором млекопитающему, включая человека, нуждающемуся в таком ингибировании, вводят эффективное количество соединения по любому из пп. 1-14. 24. Способ лечения желудочно-кишечных воспалительных заболеваний, при котором млекопитающему, включая человека, нуждающемуся в таком лечении, вводят эффективное количество соединения по любому из пп. 1-14. 25. Способ лечения или профилактики состояний, при которых имеет место инфекция Helicobacter pylori слизистой оболочки желудка человека, при котором млекопитающему, включая человека, нуждающемуся в таком лечении, вводят эффективное количество соединения по любому из пп. 1-14, где указанную соль вводят в комбинации с по меньшей мере одним противомикробным агентом. 26. Фармацевтический препарат, обладающий способностью ингибировать секрецию желудочной кислоты, где активным ингредиентом является соединение по любому из пп. 1-14. 27. Фармацевтический препарат для применения в лечении желудочно-кишечных воспалительных заболеваний, где активным ингредиентом является соединение по любому из пп. 1-14. 28. Фармацевтический препарат для применения в лечении или профилактике состояний, при которых имеет место инфекция He1icobacter pylori слизистой оболочки желудка человека, где активным ингредиентом является соединение по любому из пп. 1-14 в комбинации с по меньшей мере одним противомикробным агентом. РИСУНКИ
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 06.06.2006
Извещение опубликовано: 20.12.2007 БИ: 35/2007
|
||||||||||||||||||||||||||