Патент на изобретение №2184112
|
||||||||||||||||||||||||||
(54) ПРОИЗВОДНЫЕ ПИПЕРИДИНА И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ НА ИХ ОСНОВЕ
(57) Реферат: Изобретение относится к новым производным 4-гидроксипиперидина формулы I 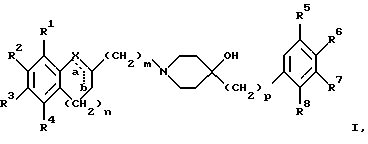 где Х обозначает -О-, -NH-, -СН2-, -СН=, -СНОН- или -СО-; R1-R4 независимо друг от друга обозначают водород, гидроксигруппу, (низший) алкилсульфониламиногруппу или ацетамидогруппу; R5-R8 независимо друг от друга обозначают водород, гидроксигруппу, (низший)алкил, галоген, (низший)алкоксигруппу, трифторметил или трифторметилоксигруппу; a и b могут обозначать двойную связь при условии, что когда a обозначает двойную связь, b не может обозначать двойную связь; n = 0-2; m = 1-3; p = 0 или 1, и их фармацевтически приемлемым аддитивным солям. Соединения по настоящему изобретению являются избирательными блокаторами подтипа NMDA-(N-метил-D-аспартат)-рецептора, который может принимать участие в медиирующих процессах, лежащих в основе развития ЦНС, включая формирование и функционирование способности к обучению и памяти. 2 с. и 9 з.п. ф-лы, 3 табл. Настоящее изобретение относится к производным 4-гидроксипиперидина общей формулы 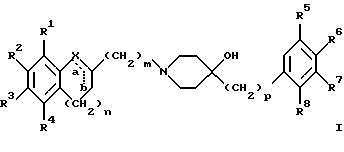 где Х обозначает -О-, -NH-, -СН2-, -СН=, -СНОН-, -СО-, -S-, -SO- или -SO2-; R1-R4 независимо друг от друга обозначают водород, гидроксигруппу, (низший)алкилсульфониламиногруппу, 1- или 2-имидазолил или ацетамидогруппу; R5-R8 независимо друг от друга обозначают водород, гидроксигруппу, (низший)алкил, галоген, (низший)алкоксигруппу, трифторметил или трифторметилоксигруппу; а и b могут обозначать двойную связь при условии, что когда “а” обозначает двойную связь, “b” не может обозначать двойную связь; n равно 0-2; m равно 1-3; р равно 0 или 1, и к их фармацевтически приемлемым аддитивным солям. Соединения формулы I и их соли обладают терапевтическими свойствами. Соединения по настоящему изобретению являются избирательными блокаторами подтипа NMDA-рецептора (N-метил-D-аспартат-рецептора), который обладает ключевой функцией в модуляции нейронной активности и пластичности, что делает их ключевыми участниками медиирующих процессов, лежащих в основе развития ЦНС, в том числе в формировании и функционировании способности к обучению и памяти. В патологических условиях острой и хронической форм нейродегенерации избыточная активность NMDA-рецепторов является ключевым фактором, инициирующим гибель нервных клеток. NMDA-рецепторы состоят из представителей семейств двух субъединиц, называемых NR-1 (8 различных сплайсинг-вариантов) и NR-2 (от А до D), являющихся производными разных генов. Для представителей семейств двух субъединиц характерно определенное распределение в различных участках головного мозга. Гетеромерные комбинации представителей NR-1 с различными субъединицами NR-2 обусловливают проявление NMDA-рецепторами различных фармацевтических свойств. Возможные терапевтические показания по применению специфичных блокаторов подтипа NMDA-рецептора включают острые формы нейродегенерации, вызванные, например, ударом и травмой головного мозга, и хронические формы нейродегенерации, такие как болезнь Альцгеймера, болезнь Паркинсона, болезнь Гентингтона, боковой амиотрофический склероз (БАС), и нейродегенерацию, связанную с бактериальными или вирусными инфекциями. Изобретение относится к соединениям формулы I и их фармацевтически приемлемым аддитивным солям, рацемическим смесям и соответствующим энантиомерам, к получению вышеуказанных соединений, к содержащим их лекарствам и способам их изготовления, а также к применению вышеуказанных соединений для лечения или предупреждения заболеваний, прежде всего заболеваний и расстройств указанных выше типов, или при изготовлении соответствующих лекарств. Следующие определения общих понятий, используемых в настоящем описании, применимы независимо от того, рассматриваются ли эти понятия отдельно или в комбинации. В контексте данного описания понятие “(низший)алкил” обозначает алкильную группу с прямой или разветвленной цепью, содержащую от 1 до 4 атомов углерода, например метил, этил, пропил, изопропил, н-бутил, изобутил, 2-бутил и трет-бутил. Понятие “галоген” включает хлор, йод, фтор и бром. Понятие “(низший)алкокси” обозначает группу, где алкильный радикал имеет значения, указанные выше. Понятие “уходящая группа” имеет общепринятое значение и относится, например, к галогену, алкилсульфонилокси-, арилсульфонилоксигруппе и т.п. Наиболее предпочтительной уходящей группой в данном случае является галоген. Понятие “фармацевтически приемлемые аддитивные соли” включает соли с неорганическими и органическими кислотами, хорошо известными специалисту в данной области техники, такими как соляная кислота, азотная кислота, серная кислота, фосфорная кислота, лимонная кислота, муравьиная кислота, фумаровая кислота, малеиновая кислота, уксусная кислота, янтарная кислота, винная кислота, метансульфоновая кислота, пара-толуолсульфоновая кислота и т.п. Предпочтительными являются соединения формулы I, в которых Х обозначает -О-, -NH-, – СНОН- или -СН2-. Примерами предпочтительных соединений, в которых Х обозначает -О-, являются: (RS)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)-4-(4-метилбензил)пиперидин-4-ол, (RS)-4-бензил-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)пиперидин -4-ол, (RS)-4-(4-фторбензил)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ол, (RS)-4-(4-этилбензил)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил) пиперидин-4-ол, (S)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)-4-(4-метилбензил)пиперидин-4-ол, (S)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)-4-(4-хлорбензил)пиперидин-4-ол, (RS)-N-[2-{ 4-хлор-4-(4-метилбензил)пиперидин-1-илметил]-2,3-дигидробензофуран-5-ил]метансульфонамид и (RS)-N-[2-{4-гидрокси-4-(4-метилбен зил)пиперидин-1-илметил}- 2,3-дигидробензофуран-5-ил] метансульфонамид. Примерами предпочтительных соединений, в которых Х обозначает -СНОН-, являются: (1RS, 2RS)- и (1RS, 2SR)-2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-илметил] индан-1,5-диол, (1RS, 2RS)-1-(1,6-дигидрокси-1,2,3,4-тетрагидронафталин-2-илметил)-4-(4-метилбензил)пиперидин-4-ол и (1RS, 2RS)-2-(4-бензил-4-гидроксипиперидин-1-илметил)-6-гидрокси-1,2,3,4-тетрагидронафталин-1-ол. Другими примерами предпочтительных соединений, в которых Х обозначает -СН2-, являются: (RS)-1-(5-гидроксииндан-2-илметил)-4- (4-метилбензил) пиперидин-4-ол и (RS)-1-(6-гидрокси-1,2,3,4-тетрагидронафталин-2-илметил)-4-(4-метилбензил) пиперидин-4-ол. Кроме того, другим предпочтительным соединением, в котором Х обозначает -NH-, является (RS)-2-14-гидрокси-4-(4-метилбензил)пиперидин-1-илметил] -2,3-дигидро-1Н-индол-5-ол. Соединения формулы I по настоящему изобретению и их фармацевтически приемлемые соли могут быть получены методами, хорошо известными в данной области техники, например описанными ниже способами, включающими: а) взаимодействие соединения формулы 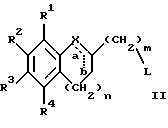 где R1-R4, X, a, b, n и m имеют указанные выше значения и L обозначает ОН или уходящую группу, например галоген или -О-тозил, с соединением формулы 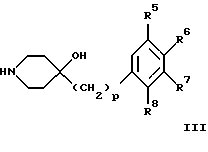 где R5-R8 и р имеют указанные выше значения, или б) взаимодействие соединения формулы 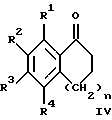 где заместители имеют указанные выше значения, с соединением формулы 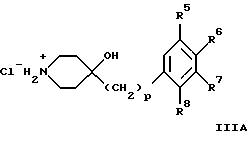 в присутствии параформальдегида с получением соединения формулы 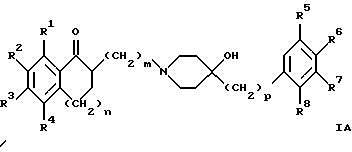 где m равно 1 и другие заместители имеют указанные выше значения, или в) дегидратацию соединения формулы 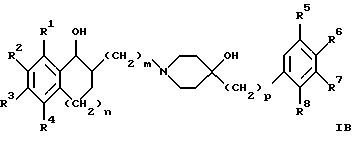 с получением соединения формулы 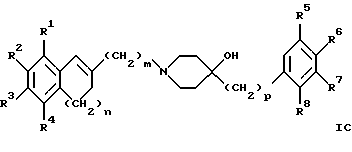 где m равно 1 и другие заместители имеют указанные выше значения, г) восстановление соединения формулы IA с получением соединения формулы IB, или д) дебензилирование соединения формулы 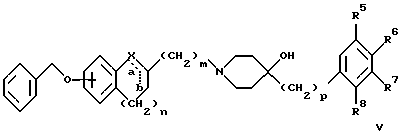 е) взаимодействие соединения формулы I, где один из радикалов R1-R4 обозначает аминогруппу, с (низший)алкилсульфонилгалогенидом с получением соединения формулы I, где один из радикалов R1-R4 обозначает (низший)алкилсульфониламиногруппу, или ж) гидрирование изолированной двойной связи в соединении формулы I, или з) удаление гидрокси- или аминозащитных(ной) групп(ы), присутствующих(ей) в виде заместителей(я) R1-R4 или в виде группы X’, означающей -N(защитная группа)-, или и) окисление соединения формулы I, в котором Х обозначает -S- или -SO-, с получением соответствующего сульфонильного (-SO2-) соединения и к) при необходимости превращение полученного соединения формулы I в фармацевтически приемлемую аддитивную соль. В соответствии с вариантом а) указанного выше способа смесь соединения формулы II и соединения формулы III, где уходящая группа L в соединении формулы II представляет собой, например, бром, растворяют в пригодном растворителе, например, в ДМФ, и нагревают приблизительно до 80-90oС. Эту реакцию проводят в присутствии основания, предпочтительно триэтиламина. Затем соединение формулы I выделяют общепринятым способом. Когда один из радикалов R1-R4 в формуле II представляет собой гидроксигруппу, эти группы защищают с помощью обычно применяемых для этой цели защитных групп. Примеры таких групп описаны у Green Т., Protective Groups in Organic Synthesis, глава 7, John Wiley and Sons, Inc. (1981), стр. 218-287. Наиболее предпочтительны бензилокси- и алкоксигруппы. Эта реакция может быть осуществлена известными способами, например, гидрированием в присутствии Pd/C (10%) или раствора трибромид бора-дихлорметан. В варианте б) способа описан процесс получения соединений формулы I, в которых Х обозначает группу -СО-. Соединение формулы IV нагревают в пригодном растворителе, например в ДМФ, вместе с соединением формулы IIIA в присутствии параформальдегида. Эту реакцию проводят общепринятым способом при температуре приблизительно 80oС. В соответствии с вариантом в) указанного выше способа соединение формулы IB может быть дегидратировано общепринятым способом в присутствии этанольного раствора НС1. При этом получают соединения формулы I, в которых “а” обозначает двойную связь. В варианте г) описан способ восстановления соединений формулы IA с получением соединений формулы IB. Эту реакцию проводят общепринятым способом, предпочтительно в присутствии LiA1H4 в ТГФ и при температуре приблизительно 5-10oС. В соответствии с вариантом д) указанного выше способа получают соединение формулы I, в котором один из радикалов 1-R4 обозначает гидроксигруппу. Этот процесс осуществляют с помощью дебензилирования соединения формулы V при условии, что ни один из радикалов R5-R8 не обозначает галоген. Дебензилирование проводят общепринятым способом. Например, соединение формулы V растворяют в пригодном растворителе или в смеси растворителей, таких как этанол и этилацетат, и гидрируют в присутствии палладия на угле при комнатной температуре и атмосферном давлении. В соответствии с вариантом е) указанного выше способа может быть получено соединение формулы I, в котором один из радикалов R1-R4 обозначает (низший)алкилсульфониламиногруппу. Эту реакцию проводят при комнатной температуре путем обработки соединения формулы I, в котором один из радикалов R1-R4 обозначает аминогруппу, (низший)алкилсульфонилгалогенидом, таким как метансульфонилхлорид, в пригодном растворителе, таком как метиленхлорид, в присутствии пиридина. Гидрирование соединения формулы I, в котором один из символов “а” или “b” обозначает двойную связь, в соответствии с вариантом ж) указанного выше способа проводят общепринятым способом, например, в присутствии Pd/C в этилацетате в атмосфере водорода в течение приблизительно 24 часов при комнатной температуре. Защитные группы, например гидроксизащитная группа, могут быть удалены описанными выше методами. Пригодные защитные группы и способы их удаления должны быть известны специалисту в данной области техники, хотя, как очевидно, следует применять только те защитные группы, которые могут быть удалены в условиях, которые не оказывают воздействия на другие структурные элементы в соединениях. Окисление соединений формулы I, в которых Х обозначает -S- или -SO-, проводят общепринятым способом. В соответствии с вариантом и) указанного выше способа соединения формулы I, в котором Х обозначает -S- или -SO-, окисляют с получением соответствующего сульфонильного (SO2) соединения. Окисление может быть осуществлено в присутствии Oxone  (тройная соль моноперсульфата калия) при комнатной температуре или в присутствии метахлорпербензойной кислоты.
Аддитивные соли соединений формулы I наиболее пригодны для фармацевтического применения.
Исходные продукты для получения соединений формулы I являются известными или могут быть получены известными методами, например, в соответствии с приведенными ниже реакционными схемами. Эти реакции более детально описаны в примерах 33-75.
Как указано ранее, соединения формулы I и их фармацевтически приемлемые аддитивные соли обладают полезными фармакодинамическими свойствами. Они являются избирательными блокаторами подтипа NMDA-рецептора, который обладает ключевой функцией в модуляции нейронной активности и пластичности, что делает их основными участниками медиирующих процессов, лежащих в основе развития ЦНС, а также в формировании способности к обучению и памяти.
Соединения исследовали в соответствии с приведенными ниже тестами.
Метод 1 (тройная соль моноперсульфата калия) при комнатной температуре или в присутствии метахлорпербензойной кислоты.
Аддитивные соли соединений формулы I наиболее пригодны для фармацевтического применения.
Исходные продукты для получения соединений формулы I являются известными или могут быть получены известными методами, например, в соответствии с приведенными ниже реакционными схемами. Эти реакции более детально описаны в примерах 33-75.
Как указано ранее, соединения формулы I и их фармацевтически приемлемые аддитивные соли обладают полезными фармакодинамическими свойствами. Они являются избирательными блокаторами подтипа NMDA-рецептора, который обладает ключевой функцией в модуляции нейронной активности и пластичности, что делает их основными участниками медиирующих процессов, лежащих в основе развития ЦНС, а также в формировании способности к обучению и памяти.
Соединения исследовали в соответствии с приведенными ниже тестами.
Метод 1Связывание 3Н-Ro 25-6981 (Ro 25-6981 обозначает [R-(R*,S*)]-a-(4-гидроксифенил)-b-метил-4-(фенилметил)-1-пиперидинпропанол) Использовали самцов крыс-альбиносов линии Fullinsdorf весом 150-200 г. Мембраны получали гомогенизацией всего головного мозга без мозжечка и продолговатого мозга с помощью центрифуги типа Polytron (10000 об/мин, 30 с) в 25 объемах холодного буфера, содержащего 50 мМ Трис-HCl, 10 мМ ЭДТК, рН 7,1. Гомогенат центрифугировали при 48000xg в течение 10 минут при 4oС. Дебрис ресуспендировали с помощью Polytron в таком же объеме буфера и гомогенат инкубировали при 37oС в течение 10 минут. После центрифугирования дебрис гомогенизировали в этом же буфере и выдерживали при -80oС в течение по крайней мере 16 часов, но не более 10 дней. Для анализа связывания гомогенат оттаивали при 37oС, центрифугировали и дебрис промывали трижды, как указано выше, в 5 мМ Трис-НСl холодного буфера, рН 7,4. Образовавшийся в результате дебрис ресуспендировали в этом же буфере и использовали в конечной концентрации 200 мкг протеина/мл. Эксперименты по связыванию 3Н-Ro 25-6981 проводили, используя 50 мМ буфер Трис-HCl, рН 7,4. Для экспериментов по замещению применяли 5 нМ 3H-Ro 25-6981 и неспецифическое связывание оценивали с использованием 10 мкМ тетрагидроизохинолина, и обычно его содержание составляло до 10% от общего количества. Время инкубации при 4oС составляло 2 часа, и опыт прекращали с помощью фильтрации через стекловолоконные фильтры типа Whatmann GF/B (Unifilter-96, фирма Packard, Цюрих, Швейцария). Фильтры промывали 5 раз холодным буфером. Радиоактивность на фильтре подсчитывали с помощью микропланшетного сцинтилляционного счетчика типа Packard Top-count после добавления 40 мл микросцинтилляционной жидкости 40 (фирма Canberra Packard, Цюрих, Швейцария). Воздействия соединений оценивали, используя минимум 8 концентраций и повторяя опыт по крайней мере еще один раз. Суммированные нормализованные значения анализировали, используя программу для расчета по методу нелинейной регрессии, позволяющую получить значения IС50 с 95%-ными верхними и нижними доверительными пределами (RS1, BBN, США). Метод 2 Связывание 3Н-празозина Использовали самцов крыс-альбиносов линии Fullinsdorf весом 150-200 г. Мембраны получали гомогенизацией всего головного мозга без мозжечка и продолговатого мозга с помощью центрифуги типа Polytron (10000 об/мин, 30 с) в 25 объемах холодного буфера, содержащего 50 мМ Трис-HCl, 10 мМ ЭДТК, рН 7,1. Гомогенат центрифугировали при 48000xg в течение 10 минут при 4oС. Дебрис ресуспендировали с помощью Polytron в таком же объеме буфера и гомогенат инкубировали при 37oС в течение 10 минут. После центрифугирования дебрис гомогенировали в этом же буфере и выдерживали при -80oС в течение по крайней мере 16 часов, но не более 10 дней. Для анализа связывания гомогенат оттаивали при 37oС, центрифугировали и дебрис промывали трижды, как указано выше, в 5 мМ холодном буфере Трис-HCl, рН 7,4. Образовавшийся в результате дебрис ресуспендировали в этом же буфере и использовали в конечной концентрации 200 мкг протеина/мл. Эксперименты по связыванию 3Н-празозина проводили, используя 50 мМ буфер Трис-HCl, рН 7,4. Для экспериментов по замещению применяли 0,2 нМ 3Н-празозина и неспецифическое связывание оценивали с использованием 100 мкМ хлорпромазина. Время инкубации при комнатной температуре составляло 30 минут, и опыт прекращали с помощью фильтрации через стекловолоконные фильтры типа Whatmann GF/B (Unifilter-96, фирма Packard, Цюрих, Швейцария). Фильтры промывали 5 раз холодным буфером. Радиоактивность на фильтре подсчитывали с помощью микропланшетного сцинтилляционного счетчика типа Packard Top-count после добавления 40 мл микросцинтилляционной жидкости 40 (фирма Canberra Packard, Цюрих, Швейцария). Воздействия соединений оценивали, используя минимум 8 концентраций и повторяя опыт по крайней мере еще один раз. Суммированные нормализованные значения анализировали, используя программу для расчета по методу нелинейной регрессии, позволяющую получить значения IС50 с 95%-ными верхними и нижними доверительными пределами (RS1, BBN, США). Метод 3 Электрофизиологическое исследование на рекомбинантных NMDA-рецепторах Клоны кДНК, кодирующие субъединицы NR1C и NR2A NMDA-рецептора (данные о номенклатуре субъединиц NMDA-рецептора см. у Hollmann и Heinemann, 1994, Annu. Rev. Neurosci. 17: 31), выделяли из библиотеки кДНК  gt11 головного мозга крысы, как описано у Sigel и др. (1994, J. Biol. Chem. 269: 8204). Клон для субъединицы NR2B NMDA-рецептора головного мозга крысы получали от S. Nakanishi (Киото, Япония). кДНК, кодирующие субъединицы NR1C, NR2A и NR2B крысы, субклонировали в векторе экспрессии pBC/CMV (Bertocci и др., 1991, Proc. Natl. Acad. Sci. USA 88:1416), осуществляя транскрипцию кДНК под контролем промотора из цитомегаловируса человека. Очищенные с помощью CsCl плазмиды экспрессии смешивали в соотношении 1:3 с NR1С:NR2A или NR1C:NR2B в буфере для инъекции (88 мМ NaCl, 1 мМ КС1, 15 мМ HEPES, рН 7,0). Для экспрессии либо комбинации субъединиц NR1C и NR2A, либо субъединиц NR1C и NR2B использовали ооциты южноафриканской шпорцевой лягушки (Xenopus laevis). От 12 до 120 пг смеси 1:3 (NR1C:NR2B) соответствующих видов кДНК инъецировали в ядро каждого ооцита. В течение двух следующих дней поток ионов через каналы NMDA-рецептора измеряли в экспериментах с фиксацией напряжения (описания методов экспрессии кДНК в ооцитах и фиксации напряжения см., например, у Bertrand и др., 1991, Methods in Neurosciences 4: 174). Мембранный потенциал фиксировали на уровне -80 мВ и рецепторы активировали, добавляя модифицированный раствор Рингера, содержащий такие агонисты NMDA-рецептора, как L-глутамат (Glu) и глицин (Gly). Для каждой комбинации субъединиц выбирали различные концентрации агониста с целью определить различные уровни чувствительности к агонистам двух типов рецепторов (2,7 мкМ Glu плюс 0,9 мкМ Gly для NR1C+NR2A и 1,3 мкМ Glu плюс 0,07 мкМ Gly для NR1C+NR2B). Агонисты вносили с интервалом 15 с каждые 2,5 мин путем быстрой суперфузии ооцита раствором, содержащим агонист, и амплитуду вызванного агонистом потока измеряли непосредственно перед концом каждой обработки. После серии начальных контрольных обработок тестируемый антагонист добавляли как к основному раствору Рингера, так и к раствору, содержащему агонист. Концентрация антагониста, которым обрабатывали ооциты, экспрессирующие субъединицу NR2A, составляла 10 мкмоль/л, а ооциты, экспрессирующие NR2B, обрабатывали концентрацией 0,1 мкмоль/л. Для каждого соединения и подтипа NMDA-рецептора тестировали от 4 до 6 ооцитов. Ооциты обрабатывали соединениями в течение 5-30 мин в зависимости от времени, необходимого для достижения равновесного ингибирования потока ионов через NMDA-рецептор. Для каждого ооцита уменьшение амплитуды потока выражали в виде процента от амплитуды контрольного потока, измеренной перед обработкой соединением. Данные в таблицах представляют собой средние арифметические значения этих процентов.
Определенная таким образом активность некоторых соединений по изобретению представлена в табл.1 (см. в конце описания).
Путем скрининга среди соединений формулы I могут быть выявлены избирательные блокаторы подтипа NMDA-рецептора, и для некоторых соединений с помощью электрофизиологического метода с использованием экспрессируемых ооцитами клонированных подтипов NMDA-рецептора может быть продемонстрировано предпочтительное действие по отношению к NMDAR-2В-субъединицам.
Соединения формулы I и их соли по настоящему изобретению могут быть включены в стандартные фармацевтические дозируемые формы, предназначенные, например, для орального или парентерального введения, вместе с обычными фармацевтическими адъювантами, например органическими или неорганическими инертными носителями, такими как вода, желатин, лактоза, крахмал, стеарат магния, тальк, растительные масла, смолы, полиалкенгликоли и т.п. Фармацевтические препараты могут применяться в твердой форме, например в виде таблеток, суппозиториев, капсул, или в жидкой форме, например в виде растворов, суспензий или эмульсий. Можно добавлять фармацевтические адъюванты, которые могут включать консерванты, стабилизаторы, смачивающие или эмульгирующие агенты, соли для изменения осмотического давления или действующие в качестве буферов. Фармацевтические препараты также могут содержать другие терапевтически активные соединения.
Суточная доза вводимого соединения формулы I изменяется в зависимости от конкретного используемого соединения, выбранного пути введения и реципиента. Характерными способами применения соединений формулы формулы I являются оральный или парентеральный пути введения. Композицию для орального применения соединения формулы I предпочтительно назначают взрослым в дозе от 150 мг до 1,5 г в день. Композицию для парентерального применения соединения формулы I предпочтительно назначают взрослым в дозе от 5 до 500 мг в день.
Ниже изобретение более подробно проиллюстрировано на примерах. Все температуры указаны в градусах Цельсия.
Пример 1 gt11 головного мозга крысы, как описано у Sigel и др. (1994, J. Biol. Chem. 269: 8204). Клон для субъединицы NR2B NMDA-рецептора головного мозга крысы получали от S. Nakanishi (Киото, Япония). кДНК, кодирующие субъединицы NR1C, NR2A и NR2B крысы, субклонировали в векторе экспрессии pBC/CMV (Bertocci и др., 1991, Proc. Natl. Acad. Sci. USA 88:1416), осуществляя транскрипцию кДНК под контролем промотора из цитомегаловируса человека. Очищенные с помощью CsCl плазмиды экспрессии смешивали в соотношении 1:3 с NR1С:NR2A или NR1C:NR2B в буфере для инъекции (88 мМ NaCl, 1 мМ КС1, 15 мМ HEPES, рН 7,0). Для экспрессии либо комбинации субъединиц NR1C и NR2A, либо субъединиц NR1C и NR2B использовали ооциты южноафриканской шпорцевой лягушки (Xenopus laevis). От 12 до 120 пг смеси 1:3 (NR1C:NR2B) соответствующих видов кДНК инъецировали в ядро каждого ооцита. В течение двух следующих дней поток ионов через каналы NMDA-рецептора измеряли в экспериментах с фиксацией напряжения (описания методов экспрессии кДНК в ооцитах и фиксации напряжения см., например, у Bertrand и др., 1991, Methods in Neurosciences 4: 174). Мембранный потенциал фиксировали на уровне -80 мВ и рецепторы активировали, добавляя модифицированный раствор Рингера, содержащий такие агонисты NMDA-рецептора, как L-глутамат (Glu) и глицин (Gly). Для каждой комбинации субъединиц выбирали различные концентрации агониста с целью определить различные уровни чувствительности к агонистам двух типов рецепторов (2,7 мкМ Glu плюс 0,9 мкМ Gly для NR1C+NR2A и 1,3 мкМ Glu плюс 0,07 мкМ Gly для NR1C+NR2B). Агонисты вносили с интервалом 15 с каждые 2,5 мин путем быстрой суперфузии ооцита раствором, содержащим агонист, и амплитуду вызванного агонистом потока измеряли непосредственно перед концом каждой обработки. После серии начальных контрольных обработок тестируемый антагонист добавляли как к основному раствору Рингера, так и к раствору, содержащему агонист. Концентрация антагониста, которым обрабатывали ооциты, экспрессирующие субъединицу NR2A, составляла 10 мкмоль/л, а ооциты, экспрессирующие NR2B, обрабатывали концентрацией 0,1 мкмоль/л. Для каждого соединения и подтипа NMDA-рецептора тестировали от 4 до 6 ооцитов. Ооциты обрабатывали соединениями в течение 5-30 мин в зависимости от времени, необходимого для достижения равновесного ингибирования потока ионов через NMDA-рецептор. Для каждого ооцита уменьшение амплитуды потока выражали в виде процента от амплитуды контрольного потока, измеренной перед обработкой соединением. Данные в таблицах представляют собой средние арифметические значения этих процентов.
Определенная таким образом активность некоторых соединений по изобретению представлена в табл.1 (см. в конце описания).
Путем скрининга среди соединений формулы I могут быть выявлены избирательные блокаторы подтипа NMDA-рецептора, и для некоторых соединений с помощью электрофизиологического метода с использованием экспрессируемых ооцитами клонированных подтипов NMDA-рецептора может быть продемонстрировано предпочтительное действие по отношению к NMDAR-2В-субъединицам.
Соединения формулы I и их соли по настоящему изобретению могут быть включены в стандартные фармацевтические дозируемые формы, предназначенные, например, для орального или парентерального введения, вместе с обычными фармацевтическими адъювантами, например органическими или неорганическими инертными носителями, такими как вода, желатин, лактоза, крахмал, стеарат магния, тальк, растительные масла, смолы, полиалкенгликоли и т.п. Фармацевтические препараты могут применяться в твердой форме, например в виде таблеток, суппозиториев, капсул, или в жидкой форме, например в виде растворов, суспензий или эмульсий. Можно добавлять фармацевтические адъюванты, которые могут включать консерванты, стабилизаторы, смачивающие или эмульгирующие агенты, соли для изменения осмотического давления или действующие в качестве буферов. Фармацевтические препараты также могут содержать другие терапевтически активные соединения.
Суточная доза вводимого соединения формулы I изменяется в зависимости от конкретного используемого соединения, выбранного пути введения и реципиента. Характерными способами применения соединений формулы формулы I являются оральный или парентеральный пути введения. Композицию для орального применения соединения формулы I предпочтительно назначают взрослым в дозе от 150 мг до 1,5 г в день. Композицию для парентерального применения соединения формулы I предпочтительно назначают взрослым в дозе от 5 до 500 мг в день.
Ниже изобретение более подробно проиллюстрировано на примерах. Все температуры указаны в градусах Цельсия.
Пример 11-(6-гидрокси-3,4-дигидронафталин-2-илметил)-4-(4-метилбензил)пиперидин-4-ол (1RS, 2RS)(1-1,6-дигидрокси-1,2,3,4-тетрагидронафталин-2-илметил)-4(4-метилбензил)пиперидин-4-ол (286 мг, 0,75 ммоль) растворяли в EtOAc (25 мл) и обрабатывали 6,4 н. НС1 в EtOH при комнатной температуре. Затем смесь в течение 1 ч кипятили с обратным холодильником. После охлаждения добавляли Н2О (25 мл) и смесь нейтрализовали 10%-ным раствором NаНСО3. Затем реакционную смесь экстрагировали EtOAc (2  50 мл), промывали насыщенным раствором NaCl (25 мл), сушили над Na2SО4, фильтровали и упаривали, получая указанное в заголовке соединение в виде твердого вещества розового цвета (238,8 мг, 0,657 ммоль, 87%); МС: m/e=363,2 (M+H+).
Пример 2 50 мл), промывали насыщенным раствором NaCl (25 мл), сушили над Na2SО4, фильтровали и упаривали, получая указанное в заголовке соединение в виде твердого вещества розового цвета (238,8 мг, 0,657 ммоль, 87%); МС: m/e=363,2 (M+H+).
Пример 2Гидрохлорид (RS)-1-(5-гидрокси-2,3-дигидробензофуран-2- илметил)-4-(4-метилбензил) пиперидин-4-ола (RS)-1-(5-бензилокси-2,3-дигидробензофуран-2-илметил)-4-(4-метилбензил)пиперидин-4-ол (1,0 г, 2,2 ммоль) в МеОН (100 мл) гидрировали в присутствии Pd/C (10%) (200 мг) в течение 17 ч при температуре окружающей среды. После удаления катализатора и упаривания получали пену желтого цвета (720 мг, 2,0 ммоль, 92%). Затем этот продукт (618 мг, 1,75 ммоль) растворяли в EtOH (20 мл) и обрабатывали 1,45 н. НС1/ЕtOН (1,1 экв.) при 0-5oС, получая гидрохлорид (RS)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)-4-(4-метилбензил)пиперидин-4-ола в виде белой/бежевой пены, смесь E/Z-изомеров (679 мг, количественный выход), МС: т/е=354,2 (M+H+). По способу, описанному в примере 2, получали соединения из примеров 3-12. Пример 3 Гидрохлорид (RS)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)-4-(4-метоксибензил) пиперидин-4-ола Указанное в заголовке соединение [МС: т/е=370,2 (М+Н+)] получали из (RS)-1-(5-бензилокси-2,3-дигидробензофуран-2-илметил)-4-(4-метоксибензил)пиперидин-4-ола. Пример 4 Гидрохлорид (RS)-4-бензил-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил) пиперидин-4-ола Указанное в заголовке соединение [МС: m/e=340,2 (M+H+)] получали из (RS)-4-бензил-1-(5-бензилокси-2,3-дигидробензофуран-2-илметил) пиперидин-4-ола. Пример 5 Гидрохлорид (RS)-4-(4-фторбензил)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ола Указанное в заголовке соединение [МС: m/e=358,2 (M+H+)] получали из (RS)-4-(4-фторбензил)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ола. Пример 6 Гидрохлорид (RS)-4-(3,4-диметилбензил)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил) пиперидин-4-ола Указанное в заголовке соединение [МС: m/e=368,2 (M+H+)] получали из (RS)-4-(3,4-диметилбензил)-1-(5-бензилокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ола. Пример 7 Гидрохлорид (RS)-4-(4-этилбензил)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ола Указанное в заголовке соединение [МС: т/е=368,2 (M+H+)] получали из (RS)-4-(4-этилбензил)-1-(5-бензилокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ола. Пример 8 Гидрохлорид (RS)-4-(4-изопропилбензил)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил) пиперидин-4-ола Указанное в заголовке соединение [МС: m/e=382,2 (М+Н+)] получали из (RS)-4-(4-изопропилбензил)-1-(5-бензилокси-2,3-дигидробензофуран-2-илметил) пиперидин-4-ола. Пример 9 Гидрохлорид (RS)-4-(2-метилбензил)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ола Указанное в заголовке соединение [МС: m/e=354,2 (М+Н+)] получали из (RS)-4-(2-метилбензил)-1-(5-бензилокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ола. Пример 10 Гидрохлорид (RS)-4-(2,4-дифторбензил)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ола Указанное в заголовке соединение [МС: m/e=376,2 (М+Н+)] получали из (RS)-4-(2,4-дифторбензил)-1-(5-бензилокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ола. Пример 11 Гидрохлорид (RS)-4-(4-трифторметоксибензил)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ола Указанное в заголовке соединение [МС: m/e=424,2 (М+Н+)] получали из (RS)-4-(4-трифторметоксибензил)-1-(5-бензилокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ола. Пример 12 Гидрохлорид (RS)-4-(3-трифторметилбензил)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ола Указанное в заголовке соединение [МС: m/e=408,2 (М+Н+)] получали из (RS)-4-(3-трифторметилбензил)-1-(5-бензилокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ола. Пример 13 Гидрохлорид (S)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)-4-(4-метилбензил) пиперидин-4-ола (S)-1-(5-метокси-2,3-дигидробензофуран-2-илметил)-4-(4-метилбензил) пиперидин-4-ол (2,10 г, 5,71 ммоль), растворенный в 50 мл СН2Сl2, охлаждали до -78oС и по каплям в атмосфере аргона добавляли 1М раствор BBr3-CH2Cl2 (12,5 мл, 2,2 экв.). Суспензии бежевого цвета давали нагреться до температуры окружающей среды в течение 30 мин, а затем перемешивали в течение еще 1 ч, в течение которого образовывался клейкий твердый осадок желтого цвета. Далее для прекращения реакции добавляли МеОН (10 мл), а затем дистиллированную Н2О (100 мл) и насыщенный раствор NаНСО3 (25 мл); после этого смесь интенсивно перемешивали в течение 15 мин. Органическую фазу отделяли, затем к водной фазе добавляли насыщенный раствор NaCl (100 мл) и экстрагировали СН2Сl2 (2 50 мл). Объединенные органические экстракты сушили над Na2SО4, а затем фильтровали и упаривали. Образовавшуюся желтую пену хроматографировали на SiO2 (Merck, 230-400 меш), элюируя CH2Cl2, a затем МеОН-СН2Сl2 (3:97), далее МеОН-СН2Сl2 (7:93), получая (S)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)-4-(метилбензил)пиперидин-4-ол (1,7 г, 4,81 ммоль, выход 84%). Это соединение (1,62 г, 4,58 ммоль) суспендировали в EtOH и обрабатывали 1,1 экв. этанольного раствора НС1 при 0-5oС, получая гидрохлорид (S)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)-4-(4-метилбензил)пиперидин-4-ола, смесь E/Z-изомеров, в виде пены белого цвета (1,74 г, 4,46 ммоль, 97%), МС: m/e=354,2 (M+H+) [ ]20D =+57,8o (с=1,0, EtOH).
По описанному в примере 13 способу получали соединения из примеров 14 и 15.
Пример 14 ]20D =+57,8o (с=1,0, EtOH).
По описанному в примере 13 способу получали соединения из примеров 14 и 15.
Пример 14Гидрохлорид (S)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)-4-(4-хлор6ензил)пиперидин-4-ола Указанное в заголовке соединение [MC: m/e=374,2 (M+H+) [ ]20D =+32,4o (с= 1,0, ДМФ)] , выход >99% е.е. получали с помощью ЖХВР с хиральной фазой из (S)-1-(5-метокси-2,3-дигидробензофуран-2-илметил)-4-(4-хлорбензил)пиперидин-4-ола.
Пример 15Гидрохлорид (R)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил) -4-(метилбензил) пиперидин-4-ола Указанное в заголовке соединение, представляющее собой смесь E/Z-изомеров, в виде пены белого цвета (1,64 г, 4,20 ммоль, 100%) [МС: m/e=354,2 (M+H+) [ ]20D =-58,0o (с= 1,0, ЕtOН)] получали из (R)-1-(5-метокси-2,3-дигидробензофуран-2-илметил)-4-(метилбензил)пиперидин -4-ола.
Пример 16Фумарат (1RS, 2RS)- и (1RS,2SR)-2-[4-гидрокси-4-(4-метилбензил) пиперидин-1-илметил] индан-1,5-диола (1RS,2RS)- и (1RS,2SR)-2-[4-бензилокси-4-(4-метилбензил) пиперидин-1-илметил] индан-1,5-диол (674 мг, 1,84 ммоль) растворяли в ЕtOН (20 мл), добавляли фумаровую кислоту (106 мг, 0,92 ммоль), смесь перемешивали в течение 2 ч при комнатной температуре и после выпаривания растворителя получали фумарат (1RS, 2RS)- и (1RS,2SR)-2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-илметил] индан-1,5-диола в виде пены белого цвета (0,78 г, количественный выход), МС: m/e=368,2 (M+H+). Пример 17 (1RS,2RS)- и (1RS,2SR)-2-[4-гидрокси-4-(4-метилбензил) пиперидин-1-илметил] индан-1,5-диол Раствор, содержащий (1RS,2RS)- и (1RS,2SR)-2-14-бензилокси-4-(4-метилбензил) пиперидин-1-илметил]индан-1,5-диол (0,83 г, 1,82 ммоль) в МеОН (100 мл) и 10%-ный Pd/C (100 мг), интенсивно перемешивали в атмосфере водорода в течение 1 ч при температуре окружающей среды. После удаления катализатора и выпаривания растворителя получали указанное в заголовке соединение: (1RS, 2RS)- и (1RS, 2SR)-2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-илметил] индан-1,5-диол (664 мг, 1,81 ммоль, 99%) в виде аморфной пены белого цвета, смесь (1:1) диастереоизомеров, МС: m/е=368,2 (M+H+). Пример 18 Гидрохлорид (RS)-1-(5-гидроксииндан-2-илметил)-4-(4-метилбензил)пиперидин-4-ола (RS)-1-(5-гидроксииндан-2-илметил)-4-(4-метилбензил)пиперидин-4-ол (177 мг, 0,503 ммоль) растворяли в EtOH, добавляли этанольный раствор НС1 (1,7 экв. ) и через 15 мин продукт осаждали, добавляя при охлаждении до 4oС диэтиловый эфир. Таким путем получали продукт, гидрохлорид (RS)-1-(5-гидроксииндан-2-илметил)-4-(4-метилбензил)пиперидин-4-ола (95,7 мг, 0,246 ммоль, 49%), в виде вспененного твердого вещества белого цвета, tпл 88-90oС. МС: m/е=352,2 (М+Н+). Пример 19 (RS)-1-(5-гидроксииндан-2-илметил)-4-(4-метилбензил)пиперидин-4-ол 1-(6-гидрокси-1Н-инден-2-илметил)-4-(4-метилбензил)пиперидин-4-ол (256 мг, 0,732 ммоль) и 10%-ный Pd/C (50 мг) в EtOAc (15 мл) интенсивно перемешивали в атмосфере водорода в течение 24 ч при комнатной температуре. После удаления катализатора и выпаривания растворителя получали (RS)-1-(5-гидроксииндан-2-илметил)-4-(4-метилбензил)пиперидин-4-ол в виде бесцветного масла (235 мг, 0,667 ммоль, 91%), МС: m/е= 352,2 (М+Н+). Пример 20 1-(6-гидрокси-1Н-инден-2-илметил)-4-(4-метил6ензил)пиперидин-4-ол (1RS, 2RS)- и (1RS,2SR)-2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-илметил] индан-1,5-диол (310 мг, 0,84 ммоль) и этанольный раствор НС1 (5 экв.) выдерживали в EtOAc (30 мл) при 65oС в течение 1,5 ч. Добавляли дистиллированную Н2О (30 мл) и 10%-ный раствор NаНСО3 (30 мл) и смесь встряхивали, а затем водную фазу экстрагировали EtOAc (2 20 мл) и объединенные органические экстракты промывали насыщенным раствором NaCl (30 мл), сушили (Na2SO4) и фильтровали. После выпаривания растворителя получали 1-(6-гидрокси-1Н-инден-2-илметил)-4-(4-метилбензил)пиперидин-4-ол в виде твердого вещества бежевого цвета (308 мг, 0,84 ммоль, 100%), tпл 154-157oС, МС: m/e=350,2 (M+H+).
Пример 21(RS)-2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-илметил] -2,3-дигидро-1Н-индол-5-ол К раствору (RS)-1-(5-метокси-2,3-дигидро-1Н-индол-2-илметил)-4-(4-метилбензил)пиперидин-4-ола (131 мг, 0,357 ммоль) в CH2Cl2 (10 мл) добавляли при -78oС в течение 5 мин 1М ВВr3-СН2Сl2 (2,14 мл, 2,74 ммоль, 6 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 48 ч, затем добавляли МеОН (20 мл), а затем 10%-ный раствор NаНСО3 (20 мл), после чего водную фазу экстрагировали СН2Сl2 (2 50 мл) и объединенные экстракты промывали насыщенным раствором NaCl (50 мл), сушили (Na2SO4), фильтровали и упаривали. После очистки неочищенного продукта на SiO2 (Merck, 230-400 меш), элюируя СН2Сl2-МеОН (9: 1), получали (RS)-2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-илметил] -2,3-дигидро-1Н-индол-5-ол в виде твердого вещества коричневого цвета (64,6 мг, 0,183 ммоль, 51%), tпл 90-94oС, МС: m/е=353,3 (М+Н+).
Пример 22Гидрохлорид (RS)-N-{ 2-[4-(4-хлорбензил)-4-гидроксипиперидин-1-илметил] -2,3-дигидробензофуран-5-ил} метансульфонамида (RS)-N-(2-бромметил-2,3-дигидробензофуран-5-ил)метансульфонамид (600 мг, 1,96 ммоль), 4-(4-хлорбензил)пиперидин-4-ол (514 мг, 1,96 ммоль), Еt3N (400 мг, 3,96 ммоль) растворяли в ДМФ (50 мл) и выдерживали при 60oС в течение 90 ч. Затем ДМФ выпаривали и остаток растворяли в CH2Cl2 и промывали Н2О. Органическую фазу сушили над Na2SО4 и концентрировали. Остаток хроматографировали на SiO2 (Merck, 230-400 меш), элюируя CH2Cl2-MeOH-NH4ОH (65:10:1), с получением пены бежевого цвета, которую растворяли в ТГФ (50 мл) и обрабатывали 1,2 н. НС1 (1 мл), получая гидрохлорид (RS)-N-{2-[4-(4-хлорбензил)-4-гидроксипиперидин-1-илметил] -2,3-дигидробензофуран-5-ил} метансульфонамида, смесь E/Z-изомеров, в виде пены белого цвета. МС: m/е=451,3 (М+Н+). По способу, описанному в примере 22, получали соединение из примера 23. Пример 23 Гидрохлорид (RS)-N-(2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-илметил] -2,3-дигидробензофуран-5-ил] метансульфонамида Указанное в заголовке соединение [МС: m/e=431,5 (M+H+)] получали из (RS)-N-(2-бромметил-2,3-дигидробензофуран-5-ил)метансульфонамида и 4-(4-метилбензил)пиперидин-4-ола. Пример 24 Гидрохлорид (RS)-1-(6-гидоокси-2,3-дигидробензофуран-2-илметил)-4-(4-метилбензил)пиперидин-4-ола (RS)-1-(6-метокси-2,3-дигидробензофуран-2-илметил)-4-(4-метилбензил)пиперидин-4-ол (1,24 г, 3,37 ммоль), растворенный в 35 мл СН2Сl2, охлаждали до -8oС и по каплям в атмосфере аргона добавляли 1М раствор BBr3-CH2Cl2 (6,8 мл, 2 экв.). Суспензии фиолетового цвета давали нагреться до комнатной температуры и затем перемешивали в течение 30 мин. Затем реакционную смесь охлаждали до 0oС и для прекращения реакции добавляли МеОН (9 мл), а затем насыщенный раствор NаНСО3 (50 мл). Органическую фазу отделяли и водную фазу экстрагировали CH2Cl2 (2 50 мл). Объединенные органические экстракты сушили над Na2SO4, затем фильтровали и упаривали. Образовавшуюся пену желтого цвета хроматографировали на SiO2 (Merck, 230-400 меш), элюируя MeOH-CH2Cl2 (1: 19), а затем МеОН-СН2Сl2 (1:9), с получением пены желтого цвета, которую растворяли в МеОН (5 мл) и обрабатывали 1 н. НС1 (3,4 мл), получая гидрохлорид (RS)-1-(6-гидрокси-2,3-дигидробензофуран-2-илметил)-4-(4-метилбензил)пиперидин-4-ола (0,92 г, 70%), смесь E/Z-изомеров, в виде твердого вещества белого цвета, tпл 203-205oС, МС: m/е=354,3 (М+Н+).
По описанному в примере 24 способу получали соединение из примера 25.
Пример 25Гидрохлорид 1-(6-гидроксибензофуран-2-илметил)-4-(4-метилбензил)пиперидин-4-ола Указанное в заголовке соединение [tпл 214oС, МС: m/е=352,2 (М+Н+)] получали из 1-(6-метоксибензофуран-2-илметил)-4-(4-метилбензил)пиперидин-4-ола. Пример 26 Гидрохлорид (RS)-4-бензил-1-(6-гидроксихроман-2-илметил) пиперидин-4-ола (RS)-4-бензил-1-(6-бензилоксихроман-2-илметил) пиперидин-4-ол (0,58 г, 1,31 ммоль) растворяли в ЕtOАс (30 мл) и добавляли 10%-ный Pd/C (135 мг), смесь помещали в атмосферу водорода и интенсивно перемешивали в течение 17 ч при температуре окружающей среды. После удаления катализатора и хроматографирования на SiO2 (Merck, 230-400 меш), элюируя CH2Cl2-MeOH-NH4ОH (100:5: 0,25), получали пену белого цвета (0,37 г, 1,04 ммоль, 80%), которую растворяли в EtOH (10 мл) и при 0-5oС добавляли HCl/EtOH (1,1 экв.). После удаления растворителя получали гидрохлорид (RS)-4-бензил-1-(6-гидроксихроман-2-илметил) пиперидин-4-ола в виде пены белого цвета (0,38 г, 0,98 ммоль, 95%), МС: m/е=354,4 (М+Н+). Пример 27 Фумарат (1RS, 2RS)-1-(1,6-дигидрокси-1,2,3,4-тетрагидронафталин-2-илметил)-4-(4-метилбензил)пиперидин-4-ола (1RS, 2RS)-1-(1,6-дигидрокси-1,2,3,4-тетрагидронафталин-2-илметил)-4-(4-метилбензил)пиперидин-4-ол (0,581 г, 1,53 ммоль) в EtOH (20 мл) перемешивали с фумаровой кислотой (88 мг, 0,765 ммоль, 0,5 экв.) в течение 2 ч при комнатной температуре. Затем смесь полностью упаривали и сушили в глубоком вакууме, получая указанное в заголовке соединение в виде аморфной пены белого цвета (0,66 г, количественный выход), МС: m/е=382,3 (М+Н+). Пример 28 (1RS, 2RS)-1-(1,6-дигидрокси-1,2,3,4-тетрагидронафталин-2-илметил)-4-(4-метилбензил)пиперидин-4-ол 1-(6-бензилокси-1-гидрокси-1,2,3,4-тетрагидронафталин-2-илметил)-4-(4-метилбензил)пиперидин-4-ол (0,22 г, 0,46 ммоль) в ЕtOАс (60 мл) обрабатывали 10%-ным Pd/C (50 мг) и перемешивали в течение 6 ч при температуре окружающей среды в атмосфере водорода. После удаления катализатора и выпаривания растворителя получали указанное в заголовке соединение в виде пены белого цвета (175 мг, количественный выход), МС: m/e=382,3 (M+H+). По описанному в примере 28 способу получали соединение из примера 29. Пример 29 (1RS, 2RS)-2-(4-бензил-4-гидроксипиперидин-1-илметил)-6-гидоокси-1,2,3,4-тетрагидронафталин-1-ол Указанное в заголовке соединение в виде твердого вещества белого цвета tпл 94-98oС, МС: m/e= 368,4 (M+H+)] получали из (1RS,2RS)-2-(4-бензил-4-гидроксипиперидин-1-илметил)-6-бензилокси-1,2,3,4-тетрагидро-нафталин-1-ола. Пример 30 Гидрохлорид (RS)-1-(6-гидрокси-1,2,3,4-тетрагидронафталин-2-илметил)-4-(4-метилбензил)пиперидин-4-ола К (RS)-1-(6-гидрокси-1,2,3,4-тетрагидронафталин-2-илметил)-4-(4-метилбензил) пиперидин-4-олу (500 мг, 1,36 ммоль) в ЕtOН (4 мл) при 4oС добавляли этанольный раствор НС1 (1,1 экв.). После выпаривания ЕtOН получали указанное в заголовке соединение в виде пены белого цвета (530 мг, 1,32 ммоль, 97%), МС: m/е=366,2 (М+Н+). Пример 31 (RS)-1-(6-гидрокси-1,2,3,4-тетрагидронафталин-2-илметил)-4-(4-метилбензил) пиперидин-4-ол К (RS)-1-(6-метокси-1,2,3,4-тетрагидронафталин-2-илметил)-4-(4-метилбензил)пиперидин-4-олу (843 мг, 2,22 ммоль) в CH2Cl2 (20 мл) добавляли в течение 15 мин при -78oС 1М ВВr3/СН2Сl2 (4,88 мл, 4,88 ммоль, 2,2 экв.), а затем смеси давали нагреться до комнатной температуры в течение 40 мин. Добавляли МеОН (3 мл), Н2О (20 мл) и NаНСО3 (20 мл) и смесь экстрагировали CH2Cl2 (4 50 мл). Экстракты промывали насыщенным раствором NaCl (30 мл), сушили (Na2SO4), фильтровали и упаривали, получая неочищенный продукт в виде пены желтого цвета. После хроматографирования на SiO2 (Merck, 230-400 меш), элюируя CH2Cl2-MeOH (97: 3), получали (RS)-1-(6-гидрокси-1,2,3,4-тетрагидронафталин-2-илметил)-4-(4-метилбензил)пиперидин-4-ол в виде пены белого цвета (500 мг, 13,68 ммоль, 61%), МС: m/e=366,2 (M+H+).
Пример 32Гидрохлорид (RS)-N-[6-(4-бензил-4-гидроксипиперидин-1-илметил)-5-оксо-5,6,7,8-тетрагидронафталин-2-ил]ацетамида N-(5,6,7,8-тетрагидро-5-оксо-2-нафтил)ацетамид (1,0 г, 4,92 ммоль), гидрохлорид 4-(бензил)пиперидин-4-ола (1,12 г, 4,92 ммоль) и параформальдегид (148 мг, 4,92 ммоль) вместе выдерживали в ДМФ (50 мл) при 80oС в течение 4 ч. Затем ДМФ выпаривали, остаток растворяли в СН2Сl2 (50 мл) и промывали 10%-ным раствором NаНСО3 (25 мл), а затем водную фазу экстрагировали СН2Сl2 (50 мл) и объединенные СН2Сl2-экстракты сушили (Na2SО4), фильтровали и упаривали. Неочищенный продукт хроматографировали на SiO2 (Merck, 230-400 меш), элюируя CH2Cl2-MeOH-NH4OH (110: 10:1) и получая 0,79 г пены желтого цвета. Этот продукт растворяли в EtOH, охлажденном до 0-4oС, и добавляли этанольный раствор НС1 (1,1 экв.), осадок в виде белого твердого вещества собирали и сушили, получая гидрохлорид (RS)-N-[6-(4-бензил-4-гидроксипиперидин-1-илметил)-5-оксо-5,6,7,8-тетрагидронафталин-2-ил] ацетамида (639 мг, 1,44 ммоль, 29%), tпл 123-126oС, МС: m/е=407,5 (М+Н+). N-(5,6,7,8-тетрагидро-5-оксо-2-нафтил)ацетамид получали в соответствии с методом, изложенным в литературе: Biggs D.F. и др., J. Med. Chem., 19, 1976, 472-475; Allinger N.L., Jones E.S., J. Org. Chem., 27, 1962, 70-76. Получение промежуточных продуктов Общий способ получения бензилпиперидиновых промежуточных продуктов Пример 33 (RS)-1-(5-бензилокси-2,3-дигидробензофуран-2-илметил)-4-(4-метилбензил) пиперидин-4-ол (RS)-5-бензилокси-2-бромметил-2,3-дигидробензофуран (840 мг, 2,63 ммоль) и 4-(4-метилбензил)пиперидин-4-ол (1,08 г, 5,26 ммоль) суспендировали в толуоле (20 мл) и нагревали до 110oС в течение 17 ч. Смесь фильтровали и упаривали, получая масло оранжевого цвета, которое хроматографировали на SiO2 (Merck, 230-400 меш), элюируя MeOH-CH2Cl2 (3:97) и получая (RS)-1-(5-бензилокси-2,3-дигидробензофуран-2-илметил)-4-(4-метилбензил)пиперидин-4-ол в виде масла желтого цвета (1,03 г, 2,32 ммоль, 88%), МС: m/e=445,5 (M+H+). Получение промежуточных продуктов для примеров 2-12 По описанному в примере 33 способу получали соединения из примеров 34-43. Пример 34 1-(5-бензилокси-2,3-дигидробензофуран-2-илметил)-4-(4-метоксибензил) пиперидин-4-ол Указанное в заголовке соединение [МС: m/е=460,2 (М+Н+)] получали из 4-(4-метоксибензил)пиперидин-4-ола и 5-бензилокси-2-(RS)-бромметил-2,3-дигидробензофурана. Пример 35 (RS)-4-бензил-1-(5-бензилокси-2,3-дигидробензофуран-2-илметил) пиперидин-4-ол Указанное в заголовке соединение [MC: m/e=430,6 (M+H+)] получали из 4-(бензил)пиперидин-4-ола и 5-бензилокси-2-(RS)-бромметил-2,3-дигидробензофурана. Пример 36 (RS)-4-(4-фторбензил)-1-(5-бензилокси-2,3-дигидробензофуран-2-илметил) пиперидин-4-ол Указанное в заголовке соединение [МС: m/е=448,6 (M+H+)] получали из 4-(4-фторбензил)пиперидин-4-ола и 5-бензилокси-2-(RS)-бромметил-2,3-дигидробензофурана. Пример 37 (RS)-4-(3,4-диметилбензил)-1-(5-бензилокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ол Указанное в заголовке соединение [MC: m/e=458,6 (М+Н+)] получали из 4-(3,4-диметилбензил)пиперидин-4-ола и 5-бензилокси-2-(RS)-бромметил-2,3-дигидробензофурана. Пример 38 (RS)-4-(4-этилбензил)-1-(5-бензилокси-2,3-дигидробензофуран-2-илметил) пиперидин-4-ол Указанное в заголовке соединение [MC: m/е=458,6 (M+H+)] получали из 4-(4-этилбензил)пиперидин-4-ола и 5-бензилокси-2-(RS)-бромметил-2, 3-дигидробензофурана. Пример 39 (RS)-4-(4-изопропилбензил)-1-(5-бензилокси-2,3-дигидробензофуран-2-илметил) пиперидин-4-ол Указанное в заголовке соединение [МС: m/е=472,6 (М+Н+)] получали из 4-(4-изопропилбензил)пиперидин-4-ола и 5-бензилокси-2-(RS)-бромметил-2,3-дигидробензофурана. Пример 40 (RS)-4-(2-метилбензил)-1-(5-6ензилокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ол Указанное в заголовке соединение [МС: m/e=444,6 (M+H+)] получали из 4-(2-метилбензил)пиперидин-4-ола и 5-бензилокси-2-(RS)-бромметил-2,3-дигидробензофурана. Пример 41 (RS)-4-(2,4-дифторбензил)-1-(5-бензилокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ол Указанное в заголовке соединение [МС: m/е=466,6 (M+H+)] получали из 4-(2,4-дифторбензил)пиперидин-4-ола и 5-бензилокси-2-(RS)-бромметил-2,3-дигидробензофурана. Пример 42 (RS)-4-(4-трифторметоксибензил)-1-(5-бензилокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ол Указанное в заголовке соединение [МС: m/e=514,6 (M+H+)] получали из 4-(4-трифторметоксибензил)пиперидин-4-ола и 5-бензилокси-2-(RS)-бромметил-2,3-дигидробензофурана. Пример 43 (RS)-4-(3-трифторметилбензил)-1-(5-бензилокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ол Указанное в заголовке соединение [MC: m/е=498,6 (M+H+)] получали из 4-(3-трифторметилбензил)пиперидин-4-ола и 5-бензилокси-2-(RS)-бромметил-2,3-дигидробензофурана. Пример 44 (RS)-5-бензилокси-2-(бромметил)-2,3-дигидробензофуран 4-бензилокси-2-(2,3-дибромпропил)фениловый эфир (RS)-уксусной кислоты (5,05 г, 11,3 ммоль) суспендировали в ЕtOН (50 мл), добавляли метоксид натрия (620 мг, 11,3 ммоль) и смесь перемешивали в течение 2 ч при температуре окружающей среды. Затем добавляли дистиллированную Н2О (100 мл) и CH2Cl2 (100 мл) и отделяли органическую фазу. Водную фазу экстрагировали CH2Cl2 (100 мл) и объединенные органические экстракты промывали насыщенным раствором NaCl (100 мл). Масло желтого цвета, полученное после сушки над Na2SO4, фильтрации и упаривания хроматографировали на SiO2 (Merck, 230-400 меш), элюируя CH2Cl2 и получая (RS)-5-бензилокси-2- (бромметил)-2,3-дигидробензофуран в виде масла желтого цвета (3,0 г, 9,39 ммоль, 83%), МС: m/е= 318,0 (М+). Пример 45 4-бензилокси-2-(2,3-дибромпропил)фениловый эфир (RS)-уксусной кислоты К раствору 2-аллил-4-бензилоксифенилового эфира уксусной кислоты (3,30 г, 11,7 ммоль) в ССl4 (30 мл) при 0-5oС в течение 10 мин добавляли бром (0,6 мл, 11,7 ммоль) и образовавшуюся смесь перемешивали в течение 1 ч при 5-10oС. Для прекращения реакции добавляли раствор Nа2СО3 (10%-ный, 4 мл) и дистиллированную Н2О (10 мл). Органическую фазу отделяли, сушили над Na2SO4 и затем упаривали, получая бесцветное масло, из которого после перекристаллизации при стоянии получали 4-бензилокси-2-(2,3-дибромпропил)фениловый эфир (RS)-уксусной кислоты (5,05 г, 11,4 ммоль, 97%), tпл 72-74oС, МС: m/е= 440,0 (М+). Пример 46 2-аллил-4-бензилоксифениловый эфир уксусной кислоты 2-аллил-4-бензилоксифенол (2,87 г, 12 ммоль) растворяли в уксусном ангидриде (40 мл), добавляли NaOAc (150 мг, 1,8 ммоль) и смесь выдерживали при 80oС в течение 18 ч. После охлаждения реакционную смесь упаривали, получая масло, которое распределяли между EtOAc и Н2О (100 мл), водную фазу экстрагировали EtOAc (100 мл) и объединенные органические фазы сушили над MgSO4, фильтровали и упаривали, получая 2-аллил-4-бензилоксифениловый эфир уксусной кислоты в виде светло-желтого масла (3,30 г, 11,7 ммоль, 97%), МС: m/е=282,1 (М+). Пример 47 2-аллил-4-бензилоксифенол 1-бензилокси-4-аллилоксибензол (20,4 г, 84,9 ммоль), растворенный в мезитилене (150 мл), выдерживали при 165oС в течение 48 ч в атмосфере аргона. После охлаждения до температуры окружающей среды образовавшееся масло коричневого цвета хроматографировали на SiO2 (Merck, 230-400 меш), элюируя EtOAc-н-гексаном (1: 9) и получая 2-аллил-4-бензилоксифенол в виде светло-желтого масла (17,45 г, 72,6 ммоль, 85%), МС: m/e=240,l (M+). Пример 48 1-бензилокси-4-аллилоксибензол 4-бензилоксифенол (20 г, 100 ммоль), К2СО3 (20,8 г, 150 ммоль) и аллилбромид (12,7 мл, 150 ммоль) кипятили с обратным холодильником в ацетоне (200 мл) в течение 18 ч. После фильтрации и выпаривания растворителя получали 1-бензилокси-4-аллилоксибензол в виде твердого вещества бежевого цвета (23,8 г, 99 ммоль, 99%), tпл 56-57oС, МС: m/е=240,1 (M+). Получение промежуточного продукта для примеров 13 и 14 Пример 49 (S)-1-(5-метокси-2,3-дигидробензофуран-2-илметил)-4-(4-метилбензил) пиперидин-4-ол 5-метокси-2,3-дигидробензофуран-2-илметиловый эфир (S)-толуолсульфоновой кислоты (2,30 г, 6,88 ммоль), 4-(4-метилбензил)пиперидин-4-ол (1,62 г, 7,9 ммоль) и Na2CO3 (1,10 г, 10,3 ммоль) суспендировали в ДМФ и выдерживали при 110oС в течение 1 ч. После охлаждения до температуры окружающей среды добавляли дистиллированную Н2О и EtOAc (по 100 мл), полученную смесь встряхивали, органическую фазу отделяли и водную фазу экстрагировали EtOAc (10 мл). Затем объединенные органические экстракты промывали насыщенным раствором NaCl (100 мл) и органическую фазу сушили над Na2SО4, фильтровали и упаривали, получая масло желтого цвета. Полученный продукт хроматографировали на SiO2 (Merck, 230-400 меш), элюируя CH2Cl2, а затем CH2Cl2-MeOH (97:3) и получая (S)-1-(5-метокси-2,3-дигидробензофуран-2-илметил)-4-(4-метилбензил)пиперидин-4-ол в виде масла желтого цвета (2,20 г, 6,0 ммоль, выход 87%), МС: m/е=368,2 (М+Н+); [ ]20D =+45,8o (с=1,0, СНС13).
По описанному в примере 49 способу получали соединение из примера 50.
Пример 50(S)-1-(5-метокси-2,3-дигидробензофуран-2-илметил)-4-(4-хлорбензил)пиперидин-4-ол Указанное в заголовке соединение [МС: m/e=387,9 (М+Н+)] получали из 4-(4-хлорбензил)пиперидин-4-ола и 5-метокси-2,3-дигидробензофуран-2-илметилового эфира (S)-толуолсульфоновой кислоты. Пример 51 5-метокси-2,3-дигидробензофуран-2-илметиловый эфир (S)-толуолсульфоновой кислоты Раствор (S)-5-метокси-2,3-дигидробензофуран-2-карбоновой кислоты (2,4 г, 12,4 ммоль) в ТГФ (30 мл) добавляли по каплям в течение 15 мин при охлаждении к суспензии LiA1H4 (0,71 г, 18,5 ммоль) в ТГФ (20 мл). Затем смесь кипятили с обратным холодильником в течение 1 ч, после охлаждения добавляли дистиллированную Н2О (0,7 мл), а затем 4 н. NaOH (1,4 мл) и дистиллированную Н2О (2,1 мл). Всю смесь сушили над Na2SO4, фильтровали и упаривали, получая масло светло-желтого цвета (2,20 г, 12,2 ммоль, 100%). Это масло растворяли в пиридине (22 мл), добавляли пара-толуолсульфонилхлорид (3,48 г, 18,3 ммоль) и смесь перемешивали при температуре окружающей среды в течение 1 ч. Затем к неочищенной смеси добавляли Н2О и интенсивно перемешивали в течение 10 мин, экстрагировали EtOAc (2 100 мл) и промывали органическую фазу 2 н. НС1 (150 мл), а затем насыщенным раствором NaCl (100 мл), сушили над Na2SO4 и упаривали, получая масло желтого цвета. Это масло хроматографировали на SiO2 (Merck, 230-400 меш), элюируя циклогексаном-EtOAc (4:1) и получая 5-метокси-2,3-дигидробензофуран-2-илметиловый эфир (S)-толуолсульфоновой кислоты в виде твердого вещества белого цвета (2,4 г, 7,2 ммоль, 57%), tпл 82-84oС, МС: m/e=334,1 (М+); []20D =+75,1o (с=1,0, СНС13).
Пример 52(S)-5-метокси-2,3-дигидробензофуран-2-карбоновая кислота К раствору R(+)-1-(1-нафтил)этиламина (3,80 г, 22,2 ммоль) в ацетоне (40 мл) добавляли (RS)-5-метокси-2,3-дигидробензофуран-2-карбоновую кислоту (4,1 г, 21,1 ммоль) в ацетоне (80 мл), и после выдерживания при перемешивании в течение 2-3 мин при температуре окружающей среды осаждалось твердое вещество бежевого цвета, после чего смесь охлаждали до 0-5oС и перемешивали еще в течение 30 мин. Твердый продукт фильтровали и промывали холодным (4oС) ацетоном (3 20 мл), получая 6 г кристаллов белого цвета, tпл 183-190oС. Этот твердый продукт дважды перекристаллизовывали из горячего EtOH (60 мл), получая 4,0 г кристаллов белого цвета, tпл 190-194oС. Этот продукт суспендировали в EtOAc (100 мл) и промывали 1 н. НС1 (50 мл), кислую водную фазу экстрагировали EtOAc (50 мл), затем объединенные органические экстракты промывали дистиллированной Н2О (50 мл), после чего сушили над Na2SO4 и упаривали, получая (S)-5-метокси-2,3-дигидробензофуран-2-карбоновую кислоту в виде светло-желтого кристаллического твердого вещества (2,3 г, 11,8 ммоль, 77%), tпл 85-87oС, МС: m/е=194,2 (М+); []20D =-9,2o (с=1,0, EtOH).
Пример 53(RS)-5-метокси-2,3-дигидробензофуран-2-карбоновая кислота К суспензии 5-метоксибензофуран-2-карбоновой кислоты (14 г, 72,8 ммоль) в МеОН (600 мл) добавляли магниевую стружку (10,6 г, 437 ммоль) и смесь интенсивно перемешивали механической мешалкой в течение 2 ч, поддерживая температуру ниже 30oС. Через 2 ч дополнительно добавляли магний (10,6 г, 437 ммоль) и смесь перемешивали еще в течение 4 ч, вновь поддерживая температуру ниже 30oС. Через 6 ч смесь концентрировали до ~100 мл, добавляли дистиллированную Н2О (600 мл) и значение рН доводили до 1-2 с помощью 1 н. серной кислоты. Неочищенный продукт экстрагировали EtOAc (2 300 мл) и объединенные органические экстракты промывали Н2О (200 мл). Объединенную органическую фазу сушили над Na2SО4, фильтровали и упаривали, получая желтоватое твердое вещество, которое перекристаллизовывали из горячего толуола (100 мл), получая кристаллы белого цвета (9,2 г, 47,4 ммоль, 65%), tпл 98-100oС, МС: m/e=194,1 (M+).
По описанному в примере 49 способу получали соединение из примера 54.
Пример 54(R)-1-(5-метокси-2,3-дигидробензофуран-2-илметил)-4-(4-метилбензил)пиперидин-4-ол Указанное в заголовке соединение (2,40 г, 6,53 ммоль, выход 76%) в виде масла желтого цвета [МС: m/e=368,2 (М+Н+); [ ]20D =-44,0o (c=1,0, CHCl3)] получали из 5-метокси-2,3-дигидробензофуран-2-илметилового эфира (R)-толуолсульфоновой кислоты.
По описанному в примере 51 способу получали соединение из примера 55.
Пример 555-метокси-2,3-дигидробензофуран-2-илметиловый эфир (R)-толуолсульфоновой кислоты Указанное в заголовке соединение (3,0 г, 9,0 ммоль, 67%) в виде твердого вещества белого цвета [tпл 82-84oС, МС: m/e=334,l (M+); [ ]20D =-74,9o (c=l, O, СНС13)]>99% е.е. получали из (R)-5-метокси-2,3-дигидробензофуран-2-карбоновой кислоты с помощью ЖХВР с хиральной фазой.
Пример 56(R)-5-метокси-2,3-дигидробензофуран-2-карбоновая кислота К раствору S(-)-1-(1-нафтил)этиламина (7,21 г, 41,6 ммоль) в ацетоне (80 мл) добавляли (RS)-5-метокси-2,3-дигидробензофуран-2-карбоновую кислоту (7,70 г, 39,6 ммоль) в ацетоне (150 мл), и после выдерживания при перемешивании в течение 2-3 мин при температуре окружающей среды осаждалось твердое вещество бежевого цвета, после чего смесь охлаждали до 0-5oС и перемешивали еще в течение 30 мин. Твердый продукт фильтровали и промывали холодным (4oС) ацетоном (3 20 мл), получая 9,90 г кристаллов белого цвета, tпл 145-160oС. Этот твердый продукт дважды перекристаллизовывали из горячего EtOH (125 мл), получая 4,75 г кристаллов белого цвета, tпл 185-194oС. Этот продукт суспендировали в EtOAc (100 мл) и промывали 1 н. НС1 (50 мл), кислую водную фазу экстрагировали EtOAc (50 мл), затем объединенные органические экстракты промывали дистиллированной Н2О (50 мл), после чего сушили над Na2SO4 и упаривали, получая (R)-5-метокси-2,3-дигидробензофуран-2-карбоновую кислоту в виде светло-желтого кристаллического твердого вещества (2,5 г, 12,9 ммоль, 65%), tпл 85-87oС, МС: m/е=194,1 (М+); []20D =+9,8o (с=1,0, EtOH).
Пример 57Смесь (1RS, 2RS)- и (1RS,2SR)-2-[4-бензилокси-4-(4-метилбензил) пиперидин-1-илметил] индан-1,5-диола 5-бензилоксииндан-1-он (2,38 г, 10 ммоль), гидрохлорид 4-(4-метилбензил) пиперидин-4-ола (2,41 г, 10 ммоль) и параформальдегид (0,3 г, 10 ммоль) в ДМФ (20 мл) выдерживали при 70oС в течение 20 ч. После охлаждения добавляли EtOAc (150 мл), дистиллированную Н2О (200 мл) и 25%-ный NH4OH (4 мл) и смесь встряхивали. Затем водную фазу экстрагировали EtOAc (150 мл), объединенные органические экстракты промывали насыщенным раствором NaCl (2 100 мл) и сушили над Nа2SO4, получая после упаривания масло желтого цвета. Это масло растворяли в ТГФ (40 мл) и добавляли в течение 30 мин к суспензии LiAlH4 (1,87 г, 50 ммоль, 5 экв.) в ТГФ (50 мл) при охлаждении на льду (5-15oС). Затем смеси давали перемешиваться в течение еще 2 ч при комнатной температуре, реакцию прекращали, добавляя дистиллированную Н2О (2 мл), 4 н. NH4ОH (4 мл), а затем Н2О (4 мл), и интенсивно перемешивали в течение 15 мин. Затем всю смесь сушили над Na2SO4, фильтровали, промывали ТГФ и упаривали, получая вязкое масло. Неочищенный продукт хроматографировали на SiO2 (Merck, 230-400 меш), элюируя СН2Сl2-МеОН (97:3), а затем CH2Cl2-MeOH (9:1) и получая пену белого цвета, которая может быть перекристаллизована из EtOAc-Et2O с получением (1RS, 2RS)- и (1RS,2SR)-2-[4-бензилокси-4-(4-метилбензил) пиперидин-1-илметил] индан-1,5-диола в виде кристаллов белого цвета (1,89 г, 4,13 ммоль, 41%), tпл 131-132oC, MC: m/e=458,4 (М+Н+).
Пример 585-бензилоксииндан-1-он Смесь 5-гидроксииндан-1-она (5,3 г, 35,7 ммоль), KI (0,6 г, 3,6 ммоль), К2СО3 (6,17 г, 44,6 ммоль) и бензилбромида (4,66 мл, 39,3 ммоль) в ДМФ (50 мл) выдерживали при 100oС в течение 1 ч. После добавления Н2О (150 мл) и экстракции EtOAc (3 50 мл), промывки органической фазы насыщенным раствором NaCl (100 мл), сушки над MgSO4 и упаривания получали твердое вещество коричневого цвета. Этот продукт дважды перекристаллизовывали из EtOH, получая 5-бензилоксииндан-1-он в виде кристаллов желтого цвета (6 г, 25,17 ммоль. 70%), tпл 105-106oC, MC: m/e= 238,1 (M+).
Пример 595-гидроксииндан-1-он 5-метоксииндан-1-он (8 г, 49,3 ммоль) и натриевую соль 4-трет-бутил-2-метилбензолтиола (11,92 г, 59,2 ммоль) выдерживали при 142oС в течение 1 ч в атмосфере аргона. После охлаждения до комнатной температуры добавляли воду (80 мл), а затем 1 н. НС1 (80 мл) и EtOAc (120 мл). Смесь встряхивали и водную фазу экстрагировали EtOAc (100 мл), объединенные органические экстракты промывали насыщенным раствором NaCl (100 мл) и сушили над Na2SО4, фильтровали и упаривали. Остаток хроматографировали на SiO2 (Merck, 230-400 меш), элюируя ЕtOАс-н-гексаном (2:3) и получая 5-гидроксииндан-1-он в виде кристаллов оранжевого цвета (3,55 г, 22,6 ммоль, 45,9%), tпл 185-187oC, MC: m/e= 148,2 (М+). Пример 60 (RS)-1-(5-метокси-2,3-дигидро-1Н-индол-2-илметил)-4-(4-метилбензил) пиперидин-4-ол К раствору (RS)-1-[5-метокси-1-(толуол-4-сульфонил)-2,3-дигидро-1Н-индол-2-илметил1-4-(4-метилбензил) пиперидин-4-ола (300 мг, 0,576 ммоль) в толуоле (15 мл) добавляли бис(2-метоксиэтокси)алюмогидрид натрия (3,5 М в ТГФ) (0,66 мл, 2,31 ммоль) и смесь кипятили с обратным холодильником в течение 18 ч. После охлаждения добавляли 2 н. NaOH до рН 14, смесь экстрагировали EtOAc (3 25 мл) и объединенные экстракты промывали насыщенным раствором NaCl (25 мл), сушили (Na2SO4), затем фильтровали и упаривали. Остаток хроматографировали на SiO2 (Merck, 230-400 меш), элюируя смесью EtOAc-циклогексан-Et3N (9: 10: 1) и получая (RS)-1-(5-метокси-2,3-дигидро-1Н-индол-2-илметил)-4-(4-метилбензил)пиперидин-4-ол в виде вязкого масла желтого цвета (158,4 мг, 0,433 ммоль, 75%), МС; m/е=367,3 (М+Н+).
Пример 61(RS)-1-[5-метокси-1-(толуол-4-сульфонил)-2,3-дигидро-1Н-индол-2-илметил] -4-(4-метилбензил)пиперидин-4-ол 5-метокси-1-(толуол-4-сульфонил)-2,3-дигидро-1Н-индол-2-илметиловый эфир (RS)-толуол-4-сульфоновой кислоты (200 мг, 0,41 ммоль) и 4-(4-метилбензил)пиперидин-4-ол (337 мг, 1,64 ммоль) выдерживали в мезитилене (10 мл) в течение 20 ч при 140oС. После охлаждения добавляли 4 н. НС1 до рН 1 и смесь экстрагировали EtOAc (3 25 мл), экстракты сушили (Nа2SO4), фильтровали и упаривали. После очистки неочищенного продукта на SiO2 (Merck, 230-400 меш), элюируя смесью EtOAc-циклогексан-Et3N (9:10:1), получали указанное в заголовке соединение в виде масла желтого цвета (155,9 мг, 0,3 ммоль, 73%), МС: m/е=521,4 (М+Н+).
Пример 625-метокси-1-(толуол-4-сульфонил)-2,3-дигидро-1Н-индол-2-илметиловый эфир (RS)-толуол-4-сульфоновой кислоты (RS)-(5-метокси-2,3-дигидро-1-Н-индол-2-ил)метанол (100 мг, 0,46 ммоль), пара-толуолсульфонилхлорид (177 мг, 0,93 ммоль), триэтиламин (0,21 мл, 1,48 ммоль) и N,N-диметиламинопиридин (2 мг) перемешивали в течение 18 ч при комнатной температуре в СН2Сl2 (5 мл). Добавляли 1 н. НС1 (10 мл), смесь экстрагировали СН2Сl2 (2 25 мл), экстракты промывали насыщенным раствором NaCl (20 мл) и сушили (Na2SO4). Неочищенный продукт хроматографировали на SiO2 (Merck, 230-400 меш), элюируя смесью EtOAc-циклогексан-Еt3N (9:10:1) и получая указанное в заголовке соединение в виде масла желтого цвета (172,8 мг, 0,35 ммоль, 76%), МС: m/е=488,0 (М+Н+).
Пример 63(RS)-(5-метокси-2,3-дигидро-1-Н-индол-2-ил)метанол Метиловый эфир (RS)-5-метокси-2,3-дигидро-1Н-индол-2-карбоновой кислоты (2,5 г, 12,06 ммоль) в ТГФ (130 мл) добавляли по каплям к суспензии LiAlH4 в ТГФ (80 мл) при 4oС и смеси давали перемешиваться при комнатной температуре в течение ночи, а затем выдерживали при 65oС в течение 4 ч. После охлаждения осторожно добавляли Н2О (20 мл) и смесь перемешивали в течение 20 мин, затем фильтровали и фильтрат экстрагировали EtOAc (3 50 мл), экстракты промывали насыщенным раствором NaCl (50 мл), сушили (Na2SО4), фильтровали и упаривали. Неочищенное масло хроматографировали на SiO2 (Merck, 230-400 меш), элюируя CH2Cl2-MeOH (9: 1) и получая (RS)-(5-метокси-2,3-дигидро-1-Н-индол-2-ил)метанол в виде масла желтого цвета (1,41 г, 7,86 ммоль, 65%), МС: m/e= 179,l (M+).
Пример 64Метиловый эфир (RS)-5-метокси-2,3-дигидро-1Н-индол-2-карбоновой кислоты К этиловому эфиру 5-метокси-1Н-индол-2-карбоновой кислоты (5 г, 22,8 ммоль) в МеОН (150 мл) добавляли магниевую стружку (2,2 г, 91,6 ммоль, 4 экв. ) и смесь интенсивно перемешивали механической мешалкой (10-30oС) в течение 2 ч. Добавляли 4 н. НС1 (до рН 1-2), а затем 25%-ный NH4OH (до рН 7) и смесь экстрагировали EtOAc (4 100 мл). Затем объединенные экстракты промывали насыщенным раствором NaCl (50 мл), сушили (Na2SО4), фильтровали и упаривали. Неочищенное масло зеленого цвета хроматографировали на SiO2 (Merck, 230-400 меш), элюируя EtOAc-н-гексаном (1:3), с получением указанного в заголовке соединения в виде твердого вещества желтого цвета (3,21 г, 15,5 ммоль, 68%). пл 59-61oС, МС: m/e=207,l (M+).
Пример 65Этиловый эфир 5-метокси-1Н-индол-2-карбоновой кислоты 5-метокси-1Н-индол-2-карбоновую кислоту (10 г, 52,3 ммоль) в ЕtOН (400 мл), содержащем 36% H2SO4 (7 мл), кипятили с обратным холодильником в течение 18 ч. После охлаждения смесь нейтрализовали 2 н. NaOH (до рН 7) и экстрагировали EtOAc (3 150 мл), объединенные экстракты промывали 10%-ным NaHCO3 (225 мл), сушили (MgSO4), фильтровали и упаривали, получая этиловый эфир 5-метокси-1Н-индол-2-карбоновой кислоты в виде твердого вещества коричневого цвета (9,52 г, 43,3 ммоль, 82%), tпл 154-155oС, МС: m/е=219,1 (М+).
Получение промежуточных продуктов для примеров 22 и 23Пример 66 (RS)-N-(2-бромметил-2,3-дигидробензофуран-5-ил)метансульфонамид (RS)-2-бромметил-2,3-дигидробензофуран-5-иламин (410 мг, 1,80 ммоль), метансульфонилхлорид (206 мг, 1,8 ммоль), Et3N (182 мг, 1,80 ммоль) растворяли в CH2Cl2 (50 мл) и перемешивали при комнатной температуре в течение ночи. Затем растворитель выпаривали и остаток хроматографировали на SiO2 (Merck, 230-400 меш), элюируя СН2Сl2-МеОН (95:5) и получая (RS)-N-(2-бромметил-2,3-дигидробензофуран-5-ил)метансульфонамид (516 мг, 94%) в виде масла, МС: m/е=305,9 (М-Н+). (RS)-2-бромметил-2,3-дигидробензофуран-5-иламин получали по способу, описанному в примере 44. Получение промежуточных продуктов для примеров 24 и 25 Пример 67 (RS)-1-(6-метокси-2,3-дигидробензофуран-2-илметил)-4-(4-метилбензил)пиперидин-4-ол 6-метокси-2,3-дигидробензофуран-2-илметиловый эфир (RS)-толуол-4-сульфоновой кислоты (110 мг, 0,33 ммоль) и 4-(4-метилбензил)пиперидин-4-ол (148 мг, 0,72 ммоль) растворяли в ДМФ (3 мл) и выдерживали при 130oС в течение 1 ч. Затем ДМФ выпаривали, остаток растворяли в CH2Cl2 (5 мл) и промывали Н2О (5 мл). Органическую фазу сушили над Na2SO4 и концентрировали. Остаток хроматографировали на SiO2 (Merck, 230-400 меш), элюируя CH2Cl2-MeOH (19:1) и получая (RS)-1-(6-метокси-2,3-дигидробензо-фуран-2-илметил)-4-(4-метилбензил)пиперидин-4-ол в виде масла желтого цвета (80 мг, 66%), МС: m/e=368,4 (M+H+). Пример 68 6-метокси-2,3-дигидробензофуран-2-илметиловый эфир (RS)-толуол-4-сульфоновой кислоты К раствору (RS)-(6-метокси-2,3-дигидробензофуран-2-ил)метанола (0,09 г, 0,5 ммоль), Et3N (0,1 мл, 0,75 ммоль), 4-диметиламинопиридина (ДМАП) (1 мг, 0,01 ммоль) в CH2Cl2 (1,5 мл) при 0oС по каплям добавляли раствор пара-толуолсульфонилхлорида (115 мг, 0,6 ммоль). Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 4 ч перед добавлением Н2О (2 мл). Водную фазу экстрагировали CH2Cl2 (2 5 мл). Объединенные органические фазы сушили над Na2SO4 и концентрировали. Остаток хроматографировали на SiO2 (Merck, 230-400 меш), элюируя н-гексаном-EtOAc (4:1) и получая 6-метокси-2,3-дигидробензофуран-2-илметиловый эфир (RS)-тoлyoл-4-cульфoнoвoй кислоты в виде бесцветного масла (120 мг, 72%). МС: m/e=334 (M+).
Пример 691-(6-метоксибензофуран-2-илметил)-4-(4-метилбензил)пиперидин-4-ол К раствору 6-метокси-2-бензофуранметанола (89 мг, 0,5 ммоль) в диоксане (3 мл) при комнатной температуре добавляли по каплям SOCl2 (0,11 мл, 1,5 ммоль). Через 1,5 ч реакционную смесь концентрировали при комнатной температуре в глубоком вакууме. Остаток растворяли в диоксане (3 мл) и обрабатывали 4-(4-метилбензил)пиперидин-4-олом (225 мг, 1,1 ммоль). После перемешивания в течение 17 ч при комнатной температуре растворитель выпаривали. Остаток растворяли в Н2О (4 мл) и экстрагировали CH2Cl2 (6 4 мл). Объединенные органические фазы сушили над Nа2SO4 и концентрировали. Остаток хроматографировали на SiO2 (Merck, 230-400 меш), элюируя CH2Cl2-МеОН (19:1) и получая 1-(6-метоксибензофуран-2-илметил)-4-(4-метилбензил)пиперидин-4-ол в виде масла желтого цвета (140 мг, 77%), МС: m/e=366,3 (M+H+).
Пример 70(RS)-(6-метокси-2,3-дигидробензофуран-2-ил)метанол Раствор (RS)-6-метокси-2,3-дигидробензофуран-2-карбоновой кислоты (158 мг, 0,813 ммоль) в ТГФ (2 мл) добавляли по каплям при 0oС в суспензию LiAlH4 (31 мг, 0,813 ммоль) в ТГФ (2 мл). После перемешивания в течение 30 мин при 0oС реакционную смесь кипятили с обратным холодильником в течение 30 мин. Затем реакционную смесь охлаждали до 0oС и последовательно обрабатывали Н2О (0,05 мл), 5 н. NaOH (0,05 мл), Н2О (0,15 мл). После перемешивания в течение 20 мин при комнатной температуре добавляли EtOAc, а затем Na2SO4. Полученный таким образом твердый продукт фильтровали и фильтрат упаривали. Остаток хроматографировали на SiO2 (Merck, 230-400 меш), элюируя н-гексаном-ЕtOАс (4: 1) и получая (RS)-(6-метокси-2,3-дигидробензофуран-2-ил)метанол в виде бесцветного масла (102 мг, 70%), МС: m/е=180 (M+). По способу из примера 70 получали соединение из примера 71. Пример 71 6-метокси-2-бензофуранметанол Указанное в заголовке соединение [МС: m/e=178 (M+)] получали из 6-метокси-2-бензофуранкарбоновой кислоты. По способу из примера 53 получали соединение из примера 72. Пример 72 (RS)-6-метокси-2,3-дигидробензофуран-2-карбоновая кислота Указанное в заголовке соединение [tпл 108-110oС, МС: m/e=194 (M+)] получали из 6-метокси-2-бензофуранкарбоновой кислоты. 6-метокси-2-бензофуранкарбоновую кислоту получали в соответствии с методом, описанным в литературе: S. Tanaka, J. Am. Chem. Soc. 73, 1951, 872. Пример 73 (RS)-4-бензил-1-(6-бензилоксихроман-2-илметил)пиперидин-4-ол (RS)-6-бензилокси-2-бромметилхроман (0,52 г, 1,56 ммоль) и 4-(бензил) пиперидин-4-ол (0,66 г, 3,43 ммоль) в толуоле (20 мл) кипятили с обратным холодильником в течение 18 ч в атмосфере аргона. После удаления растворителя неочищенную смесь хроматографировали на SiO2 (Merck, 230-400 меш), элюируя CH2Cl2-MeOH-NH4ОH (100: 5:0,25) и получая указанное в заголовке соединение, (RS)-4-бензил-1-(6-бензилоксихроман-2-илметил) пиперидин-4-ол в виде масла желтого цвета (0,6 г, 1,35 ммоль, 86%), МС: m/е= 445,5 (М+Н+). Пример 74 (RS)-6-бензилокси-2-бромметилхроман (RS)-6-бензилокси(хроман-2-ил)метанол (0,97 г, 3,58 ммоль) и 1,1′-карбонилдиимидазол (0,58 г, 3,58 ммоль) и аллилбромид (19,9 ммоль) выдерживали в ацетонитриле при 80oС в течение 4 ч. Затем неочищенную смесь упаривали досуха и хроматографировали на SiO2 (Merck, 230-400 меш), элюируя CH2Cl2. Таким путем получали (RS)-6-бензилокси-2-бромметилхроман в виде твердого вещества белого цвета (0,28 г, 0,84 ммоль, 23%), tпл 61-63oС, МС: m/е=332,0 (М+). Пример 75 (RS)-6-бензилокси (хроман-2-ил) метанол К суспензии LiAlH4 (0,352 г, 9,26 ммоль) в ТГФ в течение 15 мин при перемешивании при 10oС добавляли раствор (RS)-6-гидроксихроман-2-карбоновой кислоты (1,2 г, 6,18 ммоль) в ТГФ (25 мл). Смеси давали нагреться до температуры окружающей среды, добавляли 4 н. НС1 (25 мл) и EtOAc (100 мл) и смесь встряхивали. Водную фазу экстрагировали EtOAc (100 мл) и объединенные органические экстракты промывали насыщенным раствором NaCl (50 мл), сушили над Na2SО4 и упаривали. Образовавшийся твердый продукт растворяли в ацетоне (50 мл), затем добавляли К2СО3 (0,94 г, 6,8 ммоль) и бензилбромид (0,81 мл, 6,8 ммоль) и смесь кипятили с обратным холодильником в течение 18 ч. Реакционную смесь фильтровали, упаривали досуха и хроматографировали на SiO2 (Merck, 230-400 меш), элюируя EtOAc-циклогексаном (3:7) и получая (RS)-6-бензилокси(хроман-2-ил)метанол в виде твердого вещества белого цвета (1,0 г, 3,7 ммоль, 60%), tпл 80-82oС, МС: m/е=270,1 (М+). Пример 76 (RS)-6-гидроксихроман-2-карбоновая кислота (RS)-6-метоксихроман-2-карбоновую кислоту (1,12 г, 5,37 ммоль) и пиридинийгидрохлорид (11,2 г, 96,6 ммоль) выдерживали вместе при перемешивании при 80oС в течение 2 ч в атмосфере аргона. После охлаждения до температуры окружающей среды добавляли Н2О (100 мл) и EtOAc (50 мл) и встряхивали. Водную фазу экстрагировали EtOAc (50 мл) и объединенные органические экстракты промывали насыщенным раствором NaCl (50 мл), а затем сушили над Na2SО4 и упаривали, получая (RS)-6-гидроксихроман-2-карбоновую кислоту в виде кристаллов белого цвета (0,94 г, 4,84 ммоль, 90%), tпл 175-177oС, МС: m/е=194,1 (М+). Пример 77 (RS)-6-метоксихроман-2-карбоновая кислота Смесь 6-метоксихроменон-2-карбоновой кислоты (2,20 г, 10 ммоль) и Pd/C (10%) (300 мг) в уксусной кислоте (200 мл) при интенсивном перемешивании выдерживали при 60oС в течение 5 ч в атмосфере водорода. Катализатор удаляли и раствор упаривали, получая (RS)-6-метоксихроман-2-карбоновую кислоту в виде кристаллов желтоватого цвета (2,04 г, 9,8 ммоль, 98%), tпл 131-135oС, МС: m/е=208,1 (М+). Литература: N. Cohen и др., J. Med. Chem., 1989, 32, 1842-1860; и D.T. Witaik и др., J. Med. Chem., 1971, 14, 758-766. Пример 78 (1RS, 2RS)-1-(6-бензилокси-1-гидрокси-1,2,3,4-тетрагидронафталин-2-илметил)-4-(4-метилбензил)пиперидин-4-ол (RS)-6-бензилокси-2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-илметил] -3,4-дигидро-2Н-нафталин-1-он (1,30 г, 2,77 ммоль) в ТГФ (15 мл) добавляли по каплям в течение 15 мин (5-10oС) к суспензии LiAlH4 (0,6 г, 15,5 ммоль) в ТГФ (20 мл) и затем перемешивали при комнатной температуре в течение 2,5 ч. Для прекращения реакции добавляли дистиллированную Н2О (1 мл) и 4 н. NaOH (2 мл), а затем еще раз Н2О (2 мл) и интенсивно перемешивали в течение 15 мин, после чего смесь сушили над Na2SО4, фильтровали и упаривали. Образовавшуюся неочищенную жидкость хроматографировали на SiO2 (Merck, 230-400 меш), элюируя CH2Cl2-MeOH-NH4OH (100:5:0,25) и получая указанное в заголовке соединение в виде пены белого цвета (0,75 г, 1,6 ммоль, 57%), МС: m/e=472,4 (M+H+). По способу из примера 78 получали соединение из примера 79. Пример 79 (1RS, 2RS)-1-(6-бензилокси-1-гидрокси-1,2,3,4-тетрагидронафталин-2-илметил)-4-бензилпиперидин-4-ол Указанное в заголовке соединение в виде бесцветного масла [МС: m/e=458,6 (М+Н+)] получали из (RS)-2-(4-бензил-4-гидроксипиперидин-1-илметил)-(6-бензилокси-3,4-дигидро-2Н-нафталин-1-она. Пример 80 (RS)-6-бензилокси-2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-илметил] -3,4-дигидро-2Н-нафталин-1-он 6-бензилокси-1-тетралон (1,26 г, 5 ммоль), гидрохлорид 4-(4-метилбензил) пиперидин-4-ола (1,20 г, 5 ммоль) и параформальдегид (150 мг, 5 ммоль) в ДМФ (10 мл) выдерживали при 70oС в течение 17 ч. Неочищенную смесь распределяли между дистиллированной Н2О (150 мл), 25%-ным NH4ОH (2 мл) и EtOAc (100 мл). Затем водную фазу экстрагировали EtOAc (100 мл) и объединенные органические экстракты дважды промывали насыщенным раствором NaCl (100 мл), сушили над Na2SО4, фильтровали и упаривали, получая масло оранжевого цвета. После хроматографии на SiO2 (Merck, 230-400 меш), элюируя ЕtOАс-Еt3N (19:1), получали (RS)-6-бензилокси-2-[4-гидрокси-4-(4-метилбензил) пиперидин-1-илметил]-3,4-дигидро-2Н-нафталин-1-он в виде масла янтарного цвета (1,34 г, 2,85 мл, 57%), МС: m/е=470,3 (M+H+). По способу из примера 80 получали соединение из примера 81. Пример 81 (RS)-2-(4-бензил-4-гидроксипиперидин-1-илметил)-6-бензилокси-3,4-дигидро-2Н-нафталин-1-он Указанное в заголовке соединение в виде масла желтого цвета [МС: m/е= 456,6 (М+Н+)] получали из 6-бензилокси-1-тетралона, гидрохлорида 4-бензил-4-гидроксипиперидина и параформальдегида. Пример 82 6-бензилокси-1-тетралон 6-гидрокси-1-тетралон (10 г, 61,65 ммоль), бензилбромид (8,1 мл, 67,82 ммоль) и К2СО3 (21,3 г, 154 ммоль) в ацетоне (40 мл) кипятили с обратным холодильником в течение 2,5 ч. После фильтрации и выпаривания растворителя неочищенный продукт подкисляли с помощью 1 н. НС1 до рН 1 и распределяли между Н2О (150 мл) и EtOAc (150 мл). Затем водную фазу экстрагировали EtOAc (100 мл) и экстракты сушили (Na2SO4), фильтровали и упаривали, получая 6-бензилокси-1-тетралон в виде твердого вещества оранжевого цвета (12,37 г, 49,3 ммоль, 80%), tпл 97-100oС, МС: m/е=252,1 (М+). Пример 83 6-гидрокси-1-тетралон 6-метокси-1-тетралон (80 г, 453 ммоль) растворяли в 48%-ной НВr (270 мл) при 4oС, а затем перемешивали при температуре окружающей среды в течение 4 ч. При охлаждении до 4oС добавляли дистиллированную Н2О (1 л) и осажденное твердое вещество фильтровали, примывали Н2О и дважды перекристаллизовывали из ЕtOН-Н2О (4: 1), получая 6-гидрокси-1-тетралон в виде твердого вещества бежевого цвета (59,7 г, 368 ммоль, 81%), tпл 153-155oС. МС: m/е=162,1 (М+). Пример 84 (RS)-1-(6-метокси-1,2,3,4-тетрагидронафталин-2-илметил)-4-(4-метилбензил) пиперидин-4-ол 6-метокси-1,2,3,4-тетрагидронафталин-2-илметиловый эфир (RS)-толуол-4-сульфоновой кислоты (1,32 г, 3,81 ммоль) и 4-(метилбензил)пиперидин-4-ол (3,13 г, 15,24 ммоль) в мезитилене (100 мл) выдерживали при 140oС в течение 20 ч. После выпаривания растворителя неочищенный продукт распределяли между CH2Cl2 (50 мл) и 5%-ным NаНСО3 (30 мл) и водную фазу экстрагировали CH2Cl2 (2 100 мл), промывали насыщенным раствором NaCl (50 мл), сушили (Na2SО4), фильтровали и упаривали. Неочищенный продукт хроматографировали на SiO2 (Merck, 230-400 меш), элюируя смесью EtOAc-н-гексана (1:3), а затем (1:1) и получая (RS)-1-(6-метокси-1,2,3,4-тетрагидронафталин-2-илметил)-4-(4-метилбензил)пиперидин-4-ол в виде твердого вещества белого цвета (972 мг, 2,56 ммоль, 67,3%), tпл 91-94oС, МС: m/e=380,3 (M+H+).
Пример 856-метокси-1,2,3,4-тетрагидронафталин-2-илметиловый эфир (RS)-толуол-4-сульфоновой кислоты (RS)-(6-метокси-1,2,3,4-тетрагидронафталин-2-ил)метанол (939 мг, 4,88 ммоль), пара-толуолсульфонилхлорид (934 мг, 4,9 ммоль), триэтиламин (1,12 мл, 7,88 ммоль) и N,N-диметиламинопиридин (10 мг) в CH2Cl2 (10 мл) перемешивали вместе при температуре окружающей среды в течение 24 ч. После подкисления 1 н. НС1 (рН 1-2), экстракции водной фазы CH2Cl2 (2 20 мл), сушки (Nа2SO4), фильтрации и упаривания получали неочищенный продукт, который хроматографировали на SiO2 (Merck, 230-400 меш), элюируя EtOAc-н-гексаном (1:3) и получая указанное в заголовке соединение в виде бесцветного масла (1,37 г, 3,95 ммоль, 81%), МС: m/e=346,l (M+).
Пример 86(RS)-(6-метокси-1,2,3,4-тетрагидронафталин-2-ил) метанол Этиловый эфир (RS)-6-метокси-1,2,3,4-тетрагидронафталин-2-карбоновой кислоты (1,39 мг, 5,93 ммоль) в ТГФ (20 мл) добавляли по каплям при 0-10oС в течение 15 мин к суспензии LiAlH4 (473 мг, 12,46 ммоль). Реакционную смесь перемешивали в течение 1 ч при комнатной температуре, а затем реакцию прекращали добавлением 4 н. NaOH (15 мл) и воды (20 мл). После интенсивного перемешивания в течение 15 мин смесь экстрагировали EtOAc (25 мл), затем водную фазу экстрагировали EtOAc (2 25 мл), объединенные органические экстракты промывали насыщенным раствором NaCl (30 мл), сушили (Na2SO4), фильтровали и упаривали, получая неочищенный продукт в виде прозразного масла. После хроматографической очистки на SiO2 (Merck, 230-400 меш), элюируя EtOAc-н-гексаном (1: 3), получали (RS)-(6-метокси-1,2,3,4-тетрагидронафталин-2-ил) метанол в виде бесцветного масла (989 мг, 5,14 ммоль, 86%). МС: m/е=192,1 (M+).
Пример 87Этиловый эфир (RS)-6-метокси-1,2,3,4-тетрагидронафталин-2-карбоновой кислоты Этиловый эфир (RS)-6-метокси-1-оксо-1,2,3,4-тетрагидронафталин-2-карбоновой кислоты (2,0 г, 8,05 ммоль) в МеОН (20 мл) и Pd/C (10%) (200 мг) интенсивно перемешивали в атмосфере водорода в течение 3 ч при температуре окружающей среды. Катализатор удаляли и растворитель выпаривали с получением масла, которое очищали хроматографией на SiO2 (Merck, 230-400 меш), элюируя EtOAc-н-гексаном (1: 9) и получая этиловый эфир (RS)-6-метокси-1,2,3,4-тетрагидронафталин-2-карбоновой кислоты в виде бесцветного масла (1,88 г, 6,14 ммоль, 76%), МС: m/е=234,1 (М+). Пример 88 Этиловый эфир (RS)-6-метокси-1-оксо-1,2,3,4-тетрагидронафталин-2-карбоновой кислоты 6-метокситетралон (10 г, 56,75 ммоль), диэтилкарбонат (20,6 мл, 170, 2 ммоль) и гидрид натрия (5,06 г, 210,8 ммоль) выдерживали вместе при 65oС в ТГФ (400 мл) в течение 18 ч. После охлаждения осторожно добавляли ледяную уксусную кислоту (15 мл, 250 ммоль), а затем толуол (2 200 мл), который совместно выпаривали для удаления избытка уксусной кислоты. Остаток растворяли в EtOAc (400 мл), Н2О (400 мл) и встряхивали, водную фазу экстрагировали EtOAc (2100 мл) и объединенные органические экстракты промывали насыщенным раствором NaCl (100 мл), сушили (Na2SO4), фильтровали и промывали. Неочищенный продукт кристаллизовали из EtOAc-н-гексана, получая указанное в заголовке соединение в виде твердого вещества желтого цвета (8,3 г, 33,4 ммоль, 58%), МС: m/е=248,1 (М+).
В табл. 2 и 3 приведены примеры фармацевтических препаратов.
Формула изобретения
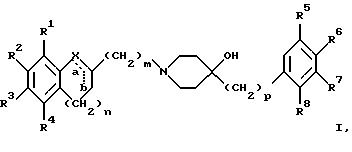 где Х обозначает -О-, -NH-, -СН2-, -СН= , -СНОН- или -СО-; R1 – R4 независимо друг от друга обозначают водород, гидроксигруппу, (низший) алкилсульфониламиногруппу или ацетамидогруппу; R5 – R8 независимо друг от друга обозначают водород, гидроксигруппу, (низший)алкил, галоген, (низший)алкоксигруппу, трифторметил или трифторметилоксигруппу; a и b могут обозначать двойную связь, при условии, что когда а обозначает двойную связь, b не может обозначать двойную связь; n = 0-2; m = 1-3; р равно 0 или 1, и их фармацевтически приемлемые аддитивные соли. 2. Соединение по п. 1, где Х обозначает -О-. 3. Соединение по п. 2, выбранное из группы, включающей: (RS)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)-4-(4-метилбензил)пиперидин-4-ол, (RS)-4-бензил-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ол, (RS)-4-(4-фторбензил)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ол, (RS)-4-(4-этилбензил)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)пиперидин-4-ол, (S)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)-4-(4-метилбензил)пиперидин-4-ол, (S)-1-(5-гидрокси-2,3-дигидробензофуран-2-илметил)-4-(4-хлорбензил)пиперидин-4-ол, (RS)-N-[2-{ 4-хлор-4-(4-метилбензил)пиперидин-1-илметил} -2,3-дигидробензофуран-5-ил] метансульфонамид и (RS)-N-[2-{ 4-гидрокси-4-(4-метилбензил)пиперидин-1-илметил} -2,3-дигидробензофуран-5-ил] метансульфонамид. 4. Соединение по п. 1, где Х обозначает -СНOН-. 5. Соединение по п. 4, выбранное из группы, включающей: (1RS, 2RS)- и (1RS, 2SR)-2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-илметил] индан-1,5-диол, (1RS, 2RS)-1-(1,6-дигидрокси-1,2,3,4-тетрагидронафталин-2-илметил)-4-(4-метилбензил)пиперидин-4-ол и (1RS, 2RS)-2-(4-бензил-4-гидроксипиперидин-1-илметил)-6-гидрокси-1,2,3,4-тетрагидронафталин-1-ол. 6. Соединение по п. 1, где Х обозначает -СН2– или -СН= . 7. Соединение по п. 6, выбранное из группы, включающей: (RS)-1-(5-гидроксииндан-2-илметил)-4-(4-метилбензил)пиперидин-4-ол и (RS)-1-(6-гидрокси-1,2,3,4-тетрагидронафталин-2-илметил)-4-(4-метилбензил)пиперидин-4-ол. 8. Соединение по п. 1, где Х обозначает -NH-. 9. Соединение по п. 8, представляющее собой (RS)-2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-илметил] -2,3-дигидро-1Н-индол-5-ол. 10. Соединение по п. 1, где Х обозначает -СО-. 11. Фармацевтический препарат, обладающий ингибирующей NMDA-рецепторы активностью, содержащий одно или несколько соединений формулы I по пп. 1-10 или их фармацевтически приемлемую соль. РИСУНКИ
MM4A Досрочное прекращение действия патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 04.12.2003
Извещение опубликовано: 20.10.2005 БИ: 29/2005
|
||||||||||||||||||||||||||