Патент на изобретение №2183631
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ 8-МЕТИЛ-8-АЗАБИЦИКЛО[3,2,1]ОКТ-3-ИЛОВОГО ЭФИРА ИНДОЛ-3-КАРБОНОВОЙ КИСЛОТЫ
(57) Реферат: Изобретение относится к усовершенствованному способу получения 8-метил-8-азабицикло[3,2,1]окт-3-илового эфира индол-3-карбоновой кислоты, гидрохлорид которого является субстанцией трописетрона и применяется как противорвотное средство, эффективное при рвоте, обусловленной химиотерапией противоопухолевыми препаратами. Описывается способ получения 8-метил-8-азабицикло[3,2,1] окт-3-илового эфира индол-3-карбоновой кислоты путем взаимодействия N-метил-8-азабицикло[3,2,1]октан-3-ола с производным индола, при этом в качестве производного индола используют 3-трихлорацетилиндол, реакционную смесь кипятят в среде кислородсодержащих растворителей в присутствии следов едких щелочей и целевой продукт выделяют известным способом. Технический результат – предложенный способ исключает применение пожаровзрывоопасного бутиллития, не требуется трудоемкой очистки конечного продукта и используемое исходное соединение является доступным. Изобретение относится к области органической химии и синтеза медицинских препаратов, конкретно к способу получения 8-метил-8-азабицикло [3,2,1] окт-3-илового эфира индол-3-карбоновой кислоты формулы I 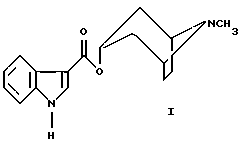 гидрохлорид которого является субстанцией трописетрона и применяется как противорвотное средство, эффективное при рвоте, обусловленной химиотерапией противоопухолевыми препаратами (Машковский М.Д. Лекарственные препараты. – М.: Медицина, 1993г.-T.I.-235с.) Известен способ получения 8 -метил-8-азабицикло [3,2,1] окт-3-илового эфира индол-3-карбоновой кислоты взаимодействием раствора хлорангидрида индол-3-карбоновой кислоты в тетрагидрофуране (ТГФ) с литиевым алкоголятом N-метил-8-азабицикло [3,2,1] октан-3-ола, приготовленным из N-метил-8-азабицикло [3,2,1] октан-3-ола (тропина) и бутиллития в гексане в среде абсолютного ТГФ при 20oС. После разложения реакционной смеси раствором соды продукт реакции экстрагируют дихлорметаном. Сырой продукт очищают колоночной хроматографией и кристаллизуют из смеси дихлорметана-этилацетата. Выход не приведен. Температура плавления 201-202oС (Температура плавления гидрохлорида 8-метил-8-азабицикло [3,2,1] окт-3-илового эфира индол-3-карбоновой кислоты 283-285oС (с разложением)). (Патент 4789673 США, МКИ4 А 61 К 31/46. Heterocyclic corboxylic acid amides and esters. / Peter Donatsch (Switzerland). Gunter Engel (Germany), Bruno Hugl (Switzerland). – N 119360; Заяв. 10.11.1987, Опубл. 6.12.1988; НКИ 546/112.-10с.) Недостатками известного способа являются: а) использование пожаровзрывоопасного бутиллития; б) применение дорогостоящей индол-3-карбоновой кислоты; в) большой расход силикагеля для очистки целевого соединения (250 г на 6,35 г и 4,8 г исходных N-метил-8-азабицикло [3,2,1] октан-3-ола (тропина) и хлорангидрида индол-3-карбоновой кислоты соответственно). Задачей настоящего изобретения является создание способа получения 8-метил-8-азабицикло [3,2,1] окт-3-илового эфира индол-3-карбоновой кислоты, лишенного указанных выше недостатков. Поставленная задача может быть решена с помощью предложенного нового способа получения 8-метил-8-азабицикло [3,2,1] окт-3-илового эфира индол-3-карбоновой кислоты. Способ заключается в том, что 8-метил-8-азабицикло [3,2,1] окт-3-иловый эфир индол-3-карбоновой кислоты получают взаимодействием N-метил-8-азабицикло [3,2,1] октан-3-ола (тропина) ф.III и 3-трихлорацетилиндола ф.II в присутствии следов едких щелочей. Реакция осуществляется при нагревании исходных реагентов в среде кислородсодержащих растворителей (ТГФ, диоксан, диметиловый эфир диэтиленгликоля, диметиловый эфир этиленгликоля и другие циклические и нециклические эфиры). Процесс осуществляется по следующей схеме: 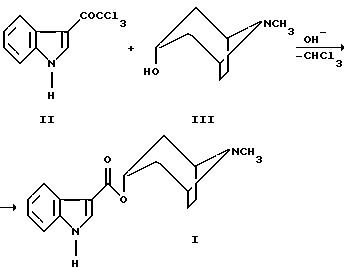 Механизм реакции заключается в галоформенном разложении 3-трихлорацетилиндола при взаимодействии с алкоголятом N-метил-8-азабицикло [3,2,1] октан-3-ола. Такая реакция хорошо протекает при синтезе эфиров индол-3-карбоновой кислоты и различных первичных спиртов (Келарев В.И. и др. Синтез функционально-замещенных производных индол-3- карбоновых кислот. – ЖОрХ – 1992. – Т.28, в.12 – с.2561-2568). Нами найдены условия, при которых эта реакция успешно протекает со вторичными спиртами, представителем которых является N-метил-8-азабицикло [3,2,1] октан-3-ол (тропин). Пример 1. В 250 мл 1,4-диоксана растворяют 26,6 г (0,1 моля) 3-трихлорацетилиндола и 21,1 г (0,15 моля) безводного N-метил-8-азабицикло [3,2,1] октан-3-ола, затем добавляют 2 капли концентрированного раствора едкого натрия и нагревают реакционную смесь до кипения. В течение 1 часа отгоняют 25-35 мл растворителя, после чего продолжают нагревать до кипения в течение 24 часов (контроль за ходом и окончанием реакции определяют путем тонкослойной хроматографии на пластинках Silyfol, элюент-хлороформ). Растворитель отгоняют при пониженном давлении (25-30 мм рт. ст.). К охлажденному остатку приливают 50 мл серного эфира, перемешивают, фильтруют N-метил-8-азабицикло [3,2,1] октан-3-иловый эфир индол-3-карбоновой кислоты. Получают 19,9 г (70%) целевого продукта (I). Температура плавления 203oС (из ацетона). Аналогичные результаты получены при использовании в качестве растворителя диметилового эфира диэтиленгликоля и производных 1,3-диоксана. Пример 2. 2,8 г (0,01 моля) 8-метил-8-азабицикло [3,2,1] окт-3-илового эфира индол-3-карбоновой кислоты растворяют в 20 мл ацетона и приливают насыщенный раствор хлористого водорода до слабой кислой реакции. Полученную смесь выливают в 70 мл серного эфира, перемешивают. Спустя 1 час выделившейся осадок отфильтровывают, промывают 20 мл серного эфира. Получают 2,88 г (90%) гидрохлорида 8-метил-8-азабицикло [3,2,1] окт-3-илового эфира индол-3-карбоновой кислоты с температурой плавления 282-284oС (с разложением). Заявленный способ получения 8 -метил-8-азабицикло [3,2,1] окт-3-илового эфира индол-3-карбоновой кислоты имеет следующие преимущества перед известным: а) не требуется применение пожаровзрывоопасного бутиллития; б) не требуется трудоемкой очистки конечного продукта; в) исходный 3-трихлорацетилиндол является доступным химическим соединением, получаемым в одну стадию из индола и хлорангидрида трихлоруксусной кислоты с выходом 71% ( Bergmann I. The Reaction of N-Acylpyridinium Salts with Indole. – J. Heterosycl. Chem. – 1970. – Vol. 7, N 5. – P. 1071-1076). Формула изобретения
MM4A Досрочное прекращение действия патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 21.09.2004
Извещение опубликовано: 20.02.2006 БИ: 05/2006
|
||||||||||||||||||||||||||