Патент на изобретение №2180342
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ МЕТИЛОВОГО ЭФИРА ФЕОФОРБИДА (А)
(57) Реферат: Изобретение относится к химии природных соединений, а именно к получению метилового эфира (МЭ) феофорбида (а), променяемого в отраслях медицины, парфюмерии, косметики и др. Способ включает извлечение производных хлорофиллов (а) и (б) из сырья растительного происхождения, их метилирование кипячением в 5%-ном растворе концентрированной серной кислоты в метаноле, хроматографическое разделение МЭ феофорбидов (а) и (б). При этом в качестве сырья используют липофильную фракцию экстрактивных веществ серпухи венценосной (Serratula coronata L.). Извлечение производных хлорофиллов (а) и (б) проводят путем обработки фракции хлороформом с последующим переосаждением производных хлорофиллов (а) и (б) из хлороформенного раствора в гептан. После метилирования осуществляют выделение МЭ феофорбидов (а) и (б) кристаллизацией их из смешанного растворителя: хлороформ – метанол (1 : 1, по объему), а после хроматографического разделения МЭ феофорбидов (а) и (б) проводят очистку МЭ феофорбида (а) кристаллизацией из смешанного растворителя: хлороформ – метанол (1 : 1,5, по объему). Способ позволяет выделить МЭ феофорбида (а) с чистотой 95-97% из липофильной фракции экстрактивных веществ серпухи венценосной (Serratula coronata L.) – отходов производства концентрата экдистероидов. 2 табл. Изобретение относится к химии природных соединений, а именно к получению метилового эфира феофорбида (а) из растительного сырья для медицины, парфюмерии, косметики и др. Производные хлорофилла, в частности феофорбид (а) и его метиловый эфир (М. Э. ), используются во всем мире в составе лекарственных препаратов, пищевых и косметических композициях, в качестве биодобавок, пищевых красителей и др. Интерес к получению индивидуальных хлорофиллов и их производных был поначалу связан с важной ролью этих пигментов в природе. Для изучения процесса фотосинтеза было важно установить структуру этих пигментов. Кроме того, производные хлорофилла используются для получения модельных соединений, при помощи которых изучают протекание фотосинтеза. До относительно недавнего времени других применений производные хлорофилла как индивидуальные соединения не находили. Однако оказалось, что производные хлорофилла в силу своих спектральных свойств перспективны как фотосенсибилизаторы (ФС) для фотодинамической терапии (ФДТ), в частности терапии злокачественных новообразований. Эти же вещества могут использоваться и как диагностические препараты в онкологии. Так, натриевая соль феофорбида (а) показала хорошую фотосенсибилизирующую активность. Аргинил-хлорин e6 и лизил-хлорин р6 Несмотря на то, что все эти вещества находятся еще на различных стадиях испытания, считается весьма вероятным, что препараты, полученные на основе производных феофорбида (а) и хлоринов e6 и р6, найдут применение в клинической практике наряду с такими препаратами как “Фотофрин” и другие. Таким образом, поиск новых доступных источников природного сырья и получение из них индивидуальных производных хлорофилла в больших количествах является актуальной задачей. 4:ацетон 96:4) и тонкослойной хроматографией (ТСХ) на силикагеле (элюент – ССl4: ацетон 9:1). Недостатком этого способа является необходимость предварительного получения производных хлорофиллов – феофитинов с минимальным количеством примесей непосредственно из растительного сырья. Задачей настоящего изобретения является разработка способа выделения М. Э. феофорбида (а) из липофильной фракции – отхода переработки растения серпухи венценосной (Serratula coronata L.) после извлечения экдистероидов. Способ получения концентрата экдистероидов из серпухи венценосной (Serratula coronata L.) состоит в следующем: наземная часть растения экстрагируется 70%-ным водным этанолом, полученный экстракт упаривается на роторном испарителе при температуре 40-55oС до 20% от исходного объема. Технический результат заключается в том, что предлагаемое изобретение позволяет выделить М. Э. феофорбида (а) с чистотой 95-97% из липофильной фракции экстрактивных веществ серпухи венценосной (Serratula coronata L.) – отхода производства концентрата экдистероидов. Общим с выбранным прототипом является то, что способ получения метилового эфира феофорбида (а) включает извлечение производных хлорофиллов (а) и (б) из сырья растительного происхождения, их метилирование кипячением в 5%-ном растворе концентрированной серной кислоты в метаноле, хроматографическое разделение М.Э. феофорбидов (а) и (б). Отличительным является то, что в качестве сырья используют липофильную фракцию экстрактивных веществ серпухи венценосной (Serratula coronata L.), извлечение производных хлорофиллов (а) и (б) проводят путем обработки фракции хлороформом с последующим переосаждением производных хлорофиллов (а) и (б) из хлороформенного раствора в гептан, после метилирования осуществляют выделение М.Э. феофорбидов (а) и (б) кристаллизацией их из смешанного растворителя: хлороформ – метанол (1:1, по объему), а после хроматографического разделения М. Э. феофорбидов (а) и (б) проводят очистку М.Э. феофорбида (а) кристаллизацией из смешанного растворителя: хлороформ – метанол (1:1.5, по объему). Получение концентрата экдистероидов серпухи венценосной состоит в следующем. 6 кг наземной части сухого растения экстрагируют 75 л 70% этанола. Полученный экстракт упаривают до 15% от исходного объема. При этом в растворе содержатся экдистероиды, а в осадок выпадает липофильная фракция, содержащая производные хлорофиллов (а) и (б), липиды и бесцветные примеси. Эта неиспользуемая ранее липофильная фракция экстрактивных веществ серпухи венценосной (Serratula coronata L.) после отделения экдистероидов и служит сырьем для получения производных хлорофиллов. Способ получения М.Э. феофорбида (а) осуществляют следующим образом. Из липофильной фракции экстрактивных веществ серпухи венценосной (Serratula coronata L.), содержащей производные хлорофилла и примеси, извлекают производные хлорофилла, очищая от примесей. Для этого липофильную фракцию смешивают с хлороформом при расходе хлороформа 800-1000 мл на 100 г фракции, нерастворимые в хлороформе примеси отделяют фильтрованием, а полученный хлороформенный раствор упаривают до объема 50-80 мл, затем приливают гептан (200-300 мл). При этом в осадок выпадают производные хлорофиллов, а в растворе остаются липиды. Выпавший осадок отфильтровывают и промывают на фильтре гептаном (50-70 мл). Полученный осадок, содержащий производные хлорофиллов: феофорбиды (а) и (б) и их этиловые эфиры метилируют 5%-ным раствором концентрированной H2SO4 в метаноле кипячением с обратным холодильником в течение 0,5-1 ч. Затем выделяют смесь метиловых эфиров. Для этого реакционную смесь охлаждают, разбавляют хлороформом и промывают водой до рН 5-7. Хлороформенный раствор смешивают с метанолом при объемном соотношении 1:1 соответственно, затем смесь упаривают и получают кристаллический порошок, представляющий собой по данным ТСХ и ВЭЖХ смесь М.Э. феофорбидов (а) и (б). Полученную смесь М.Э. феофорбидов (а) и (б) разделяют колоночной хроматографией на оксиде алюминия L40/250 (элюент бензол-хлороформ 100:5). Выделяют хроматографическую фракцию, состоящую по данным высокоэффективной жидкостной хроматографии (ВЭЖХ) из двух компонентов – М.Э. феофорбида (а) и М.Э. феофорбида (а’) в соотношении 4:1. Выделение М. Э. феофорбида (а) из хроматографической фракции, состоящей из двух компонентов – М.Э. феофорбидов (а) и (а’), осуществляют кристаллизацией из смешанного растворителя хлороформ-метанол при объемном соотношении 1: 1.5. Для этого полученные соединения растворяют в хлороформе, к раствору прибавляют метанол, при этом в осадок выпадает М.Э. феофорбида (а) с чистотой 95-97% (данные ВЭЖХ). Пример 1 450 г липофильной фракции смешивают с 4,5 л хлороформа, нерастворимые в хлороформе примеси отделяют фильтрованием, полученный хлороформенный раствор упаривают до объема 80 мл, затем приливают 250 мл гептана. Выпавший осадок отфильтровывают и промывают на фильтре 60 мл гептана. Полученный осадок (около 15 г), содержащий производные хлорофиллов: феофорбиды (а) и (б) и их этиловые эфиры, метилируют следующим образом: осадок растворяют в смеси 50 мл метанола и 2,5 мл концентрированной H2SO4, раствор кипятят с обратным холодильником в течение 1 ч. Реакционную смесь охлаждают, разбавляют 100 мл хлороформа и промывают водой до рН 6. 100 мл хлороформенного раствора смешивают со 100 мл метанола, затем смесь упаривают до 40 мл, получают 3,5 г кристаллического порошка, представляющего собой по данным ТСХ и ВЭЖХ смесь М.Э. феофорбидов (а) и (б). Полученную смесь М. Э. феофорбидов (а) и (б) разделяют колоночной хроматографией на оксиде алюминия L40/250 (элюент бензол-хлороформ 100:5). Выделяют хроматографическую фракцию (около 850 мг), состоящую по данным ВЭЖХ из двух компонентов – М.Э. феофорбида (а) и М.Э. феофорбида (а’) в соотношении 4:1. Хроматографическую фракцию растворяют в 20 мл хлороформа, к раствору прибавляют 30 мл метанола, при этом в осадок выпадает около 500 мг М.Э. феофорбида (а) с чистотой 95-97% (данные ВЭЖХ). Пример 2 Выделение М.Э. феофорбида (а) проводят так же, как в примере 1, за исключением хроматографического разделения. Полученную смесь М.Э. феофорбидов (а) и (б) разделяют колоночной хроматографией на силикагеле L40/100 (элюент CCL4 – ацетон 10:1). Выход около 500 мг М.Э. феофорбида (а) с чистотой 95-97% (данные ВЭЖХ). 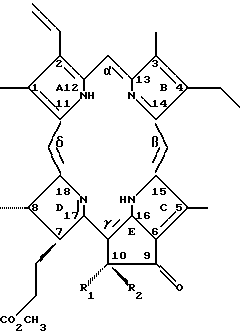 R1=СООСН3, R2=H метиловый эфир феофорбида (а’) R1=H, R2=COOCH3 метиловый эфир феофорбида (а’) Структурная формула метиловых эфиров феофорбида Анализ метилового эфира феофорбида (а). ВЭЖХ: Милихром Al, Silosorb 600; бензол-пропанол 100:7;  =360 нм.
ЯМР 1H и 13С: =360 нм.
ЯМР 1H и 13С:Bruker AM 300, 300 МГц, CDCl3. Величины химических сдвигов в спектрах ЯМР 1Н и 13С полученного М.Э. феофорбида (а) соответствуют литературным данным: – Electrophylic reactions of chlorin derivatives and a comprehensive collection of 13 Химические сдвиги атомов С7 и С10 М. Э. феофорбидов (а) и (а+а’) в спектрах ЯМР 13С приведены в табл. 1. Спектр ПМР М.Э. феофорбида (а) представлен в табл. 2. Формула изобретения
РИСУНКИ
MM4A Досрочное прекращение действия патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 25.03.2003
Номер и год публикации бюллетеня: 19-2004
Извещение опубликовано: 10.07.2004
|
||||||||||||||||||||||||||