Патент на изобретение №2179977
|
||||||||||||||||||||||||||
(54) ГИПОЛИПИДЕМИЧЕСКИЕ 1,4-БЕНЗОТИАЗЕПИН-1,1-ДИОКСИДЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ
(57) Реферат: Изобретение относится к новым 1,4-бензотиазепин-1,1-диоксидам формулы (I), где R1 представляет неразветвленную C1-6алкильную группу, R2 представляет неразветвленную C1-6алкильную группу, R3 представляет водород, R4 представляет фенил, R5 R6 и R8 выбирают из водорода, R7 представляет группу формул (Iа) и (Iв), где гидроксигруппы могут быть замещены ацетилом, R16 представляет -СООН, -CH2-OH, -СН2-O-ацетил, -СООМе, R9 и R10 одинаковы или различны и каждый представляет водород или C1-6алкильную группу, Х представляет -О-, или к его солям, сольватам и физиологически приемлемым производным. Раскрыто также соединение формулы (V) и способы получения этих соединений. Соединения по изобретению проявляют активность ингибирования поглощения желчных кислот и могут найти применение в качестве активного ингредиента лекарственного средства для профилактики или лечения гиперлипидемических состояний. 6 с. и 3 з.п. ф-лы, 1 табл. 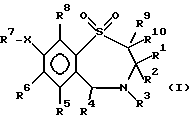 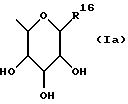 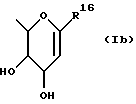 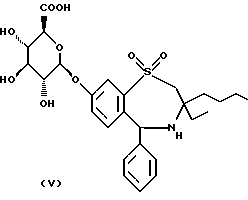
Настоящее изобретение относится к новым гиполипидемическим соединениям, к способам их получения и к новым промежуточным соединениям для их получения, к содержащим их фармацевтическим композициям и к их использованию в медицине, особенно для профилактики и лечения гиперлипидемических состояний, таких, как атеросклероз. Гиперлипидемические состояния часто связаны с повышенными концентрациями в плазме низкой плотности липопротеин (ЛНП) холестерина и очень низкой плотности липопротеин (ЛОНП) холестерина. Такие концентрации можно снизить, уменьшив абсорбцию желчных кислот в кишечнике. Одним из способов достижения этой цели является ингибирование системы активного поглощения желчных кислот в конечной подвздошной кишке. Такое ингибирование стимулирует превращение холестерина в желчные кислоты печенью, что приводит к повышению потребности в холестерине, и, в свою очередь, приводит к соответствующему повышению скорости выведения ЛНП и ЛОНП холестерина из плазмы или сыворотки крови. Был обнаружен новый класс гетероциклических соединений, которые снижают концентрации ЛНП и ЛОНП холестерина в плазме или сыворотке, и следовательно, особенно полезны в качестве гиполипидемических агентов. Уменьшая концентрации холестерина и сложного эфира холестерина в плазме, соединения настоящего изобретения замедляют образование атеросклеротических бляшек, и снижают вероятность проявлений, связанных с коронарной болезнью сердца. Такие проявления определяют, как сердечные заболевания, связанные с повышенными концентрациями холестерина и сложного эфира холестерина в плазме или сыворотке. Для целей настоящего описания гиперлипидемическое состояние определяют как любое состояние, при котором полная концентрация холестерина (ЛНП+ЛОНП) в плазме или сыворотке превышает 240 мг/дл (6/21 ммоль/л) (J/Amer.Med.Assn./ 256, 20, 2849-2858 (1986)). В международной патентной заявке W96/05188 раскрыты соединения формулы (0): 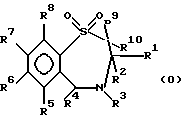 Авторами была обнаружена группа соединений, которые обладают более высокой гиполипидемической активностью ин виво, нежели соединения, раскрытые в международной патентной заявке WO 96/05188. Эти соединения отличаются определением группы R7. Соответственно, в настоящем изобретении предложены соединения формулы (I): 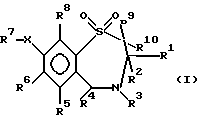 где R1 представляет неразветвленную C1-6алкильную группу; R2 представляет неразветвленную C1-6алкильную группу; R3 представляет водород или группу OR11, где R11 представляет водород, необязательно замещенный C1-6алкил или C1-6алкилкарбонильную группу; R4 представляет пиридин или необязательно замещенный фенил; R5, R6, и R8 одинаковы или различны, и каждый выбирают из водорода, галогена, циано, R15-ацетилида, OR15, необязательно замещенного C1-6алкила, COR15, CH(OH)R15, S(О)nR15, Р(О)(ОR15)2, OCOR15, ОСF3, OCN, SCN, NHCN, CH2OR15, CHO, (CH2)pCN, CONR12R13, (CH2)pCО2R15, (CH2)pNR12R13, CО2R15, NHCOCF3, NHSО2R15, OCH2OR15, OCH=CHR15, О(CH2CH2О)nR15, О(СН2)рSО3R15, О(CH2)pNR12R13 и O(CH2)pN+R12R13R14, где р представляет целое число от 1 до 4; n представляет целое число от 0 до 3; R12, R13, R14 и R15 независимо выбирают из водорода и необязательно замещенного C1-6алкила; R7 представляет группу формулы: 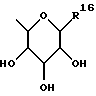 или 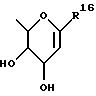 где гидроксильные группы могут быть замещены ацетилом, бензилом или -(C1-С6)-алкил-R17 где алкильная группа может быть замещена одной или более из гидроксильных групп; R16 представляет -СООН, -CH2-OH, -СН2-О-ацетил, СООМе, -СООЕт; R17 представляет Н, -ОН, -NH2, -СООН или COOR18; R18 представляет (C1-C4)-алкил или -NH-(C1-C4)-алкил; Х представляет -NH- или -О-; и R9 и R10 одинаковы или различны, и каждый представляет водород или С1-6алкил; и их соли, сольваты и физиологически функциональные производные. Если R4 представляет замещенную фенильную группу, могут быть от одного до пяти, предпочтительно один или два заместителя, которые одинаковы или различны, и каждый выбирают из галогена, гидрокси, нитро, фенил-С1-6алкокси, C1-6алкокси, необязательно замещенного C1-6алкила, S(О)nR15, CO2R15, О(CH2CH2О)nR15, О(СН2)рSО3R15, O(CH2)pNR12R13 и О(СН2)pN+R12R13R14, где R12-R15, n и р имеют указанные ранее значения. Предпочтительные варианты соединений формулы (I) включают соединения формул (III), (IV) или (IVa): 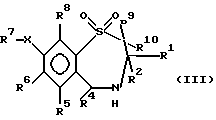 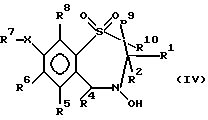 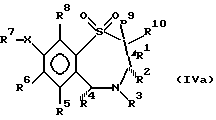 где R1-R10 и X имеют указанные ранее значения. Если один или более из R3-R6, R8 или R11-R14 замещен C1-6алкильной группой, или включает C1-6алкильную группу, заместители могут быть одинаковы или различны, и каждый выбирают из гидрокси, галогена, C1-6алкила, C1-6алкокси, COR20, нитрила, CO2R20, SО3R20, NR21R22; N+R21R22R23, где R20-R23 одинаковы или различны, и каждый выбирают из водорода или C1-6алкила. Удобно, чтобы R1 представлял метил, этил или н-пропил, и предпочтительно, чтобы R1 представлял этил. Удобно, чтобы R2 представлял метил, этил, н-пропил, н-бутил или н-пентил. Предпочтительно, чтобы R2 представлял н-бутил. Предпочтительно, чтобы R5 представлял водород. Удобно R7 выбирать из: 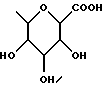 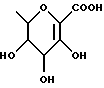 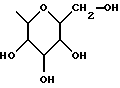 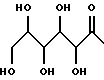 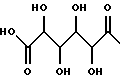 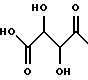 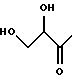 Удобно, чтобы Х представлял -О-. Удобно, чтобы R9 и R10 представляли водород, метил или этил, водород. Предпочтительно, чтобы R9 и R10 оба представляли водород. Удобно, чтобы R4 представлял пиридил или фенил, необязательно замещенный предпочтительно в 4- и/или 3-положении, галогеном, метилом, этилом, метокси, этокси, трифторметилом, гидрокси, карбокси, или О(СН2)3SО3Н. Предпочтительно, чтобы R4 представлял незамещенный фенил. В соединениях формулы (III): удобно, чтобы по крайней мере один, и предпочтительно, чтобы все из R5, R6 и R8 представляли водород. Если R5, R6 и R8 отличны от водорода, тогда удобно, чтобы они представляли С1-4алкил, необязательно замещенный фтором, С1-4алкокси, галогеном или гидрокси, наиболее удобно – метил, метокси, гидрокси, трифторметил или хлором и предпочтительно – метокси. В соединениях формулы (IV): удобно, чтобы два или три из R5, R6 и R8 представляли водород, а другие представляли бы С1-4алкил, необязательно замещенный фтором, С1-4алкокси, галоген или гидрокси, наиболее удобно – метил, метокси, гидрокси, трифторметил или хлор и предпочтительно – метокси. В соединениях формулы (IV): удобно, чтобы по крайней мере один, и предпочтительно все из R5, R6 и R8 представляли водород. Если R5, R6 и R8 отличны от водорода, тогда удобно, чтобы они представляли С1-4алкил, необязательно замещенный фтором, С1-4алкокси, галоген или гидрокси, наиболее удобно, чтобы они представляли метил, метокси, гидрокси, трифторметил или хлор, и предпочтительно метокси. Наиболее предпочтительно, чтобы R1 представлял н-бутил, R2 представлял этил, R3, R5, R6, R8, R9 и R10 представляли водород, R4 представлял фенил, а R7 представлял: 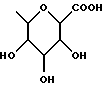 Фармацевтически приемлемые соли наиболее подходят для медицинских целей, благодаря их более высокой водорастворимости по сравнению с исходными, т.е. основными соединениями. Такие соли, очевидно, должны иметь фармацевтически приемлемый анион или катион. Подходящие фармацевтически приемлемые соли присоединения кислот соединений настоящего изобретения включают соли, полученные из неорганических кислот, таких, как соляная, бромистоводородная, фосфорная, метафосфорная, азотная, сульфоновая и серная кислоты, и таких органических кислот, как уксусная, бензолсульфоновая, бензойная, лимонная, этансульфоновая, фумаровая, глюконовая, гликолевая, изотионовая, молочная, лактобионовая, малеиновая, малоновая, метансульфоновая, янтарная, пара-толуолсульфоновая, винная и трифторуксусная кислота. Для медицинских целей наиболее подходят хлориды. Подходящие фармацевтически приемлемые основные соли включают аммонийные соли, соли щелочных металлов, такие, как соли натрия и калия, соли щелочноземельных металлов, такие, как соли магния и кальция. Соли, содержащие фармацевтически неприемлемый анион, попадают в объем настоящего изобретения, и пригодны в качестве удобных промежуточных соединений для получения или очистки фармацевтически приемлемых солей и/или для использования в не терапевтических применениях, например в применениях ин витро. Термин “физиологически функциональное производное” в том смысле, как здесь использован, относится к любому физиологически приемлемому производному соединения настоящего изобретения, например к сложному эфиру, который, после введения такому млекопитающему, как человек, способен обеспечить (прямо или косвенно) такое соединение или его активный метаболит. Другим аспектом настоящего изобретения являются пролекарственные формы соединений настоящего изобретения. Такие пролекарства в результате метаболизма могут ин виво давать соединения настоящего изобретения. Такие пролекарства могут быть активны или неактивны сами по себе. Соединения настоящего изобретения могут существовать в различных полиморфных формах, например в аморфной и кристаллической полиморфных формах. Все полиморфные формы соединений настоящего изобретения попадают в объем настоящего изобретения и составляют его дополнительный аспект. Термин “алкил” в том смысле, как здесь использован, если нет других указаний, относится к одновалентному разветвленному или неразветвленному цепочечному радикалу. Аналогично, термин “алкокси” относится к одновалентному разветвленному или неразветвленному цепочечному радикалу, присоединенному к основному молекулярному фрагменту через атом кислорода. Термин “фенилалкокси” относится к одновалентной фенильной группе, присоединенной к двухвалентной C1-6 алкиленовой группе, которая сама присоединена к основному молекулярному фрагменту через атом кислорода. Соединения формулы (I) существуют в формах, где углеродные центры -C(R1)(R2)- и -CHR4– являются хиральными. Настоящее изобретение включает в свой объем каждый из возможных оптических изомеров практически в чистой, т. е. содержащей менее 5% любого другого оптического изомера (изомеров), форме, и смеси одного или более из оптических изомеров в любой пропорции, включая рацемические смеси. Для целей настоящего описания абсолютные хиральности вышеуказанных углеродных центров даны в порядке -C(R1)(R2), затем -CHR4-. В тех случаях, когда абсолютная стереохимия у -C(R1)- и CHR4– не определена, соединения настоящего изобретения определяют в терминах относительных положений R1/R2 и H/R4 заместителей, и таким образом, те соединения, в которых наиболее массивный из R1 и R2 заместителей, т.е., заместитель с большей массой, и заместитель R4 оба расположены с одной стороны тиазепинового кольца, в данном контексте называют “цис”, а те соединения, в которых более массивный из заместителей R1 и R2 расположены на противоположных сторонах кольца, называют “транс”, и они являются предпочтительными. Специалистам должно быть очевидно, что как “цис”, так и “транс” соединения настоящего изобретения могут существовать в двух энантиомерных формах, которые по отдельности обозначают как “(+)” – или “(-)” – в соответствии с направлением вращения плоскости поляризации света при прохождении его через образец соединений. Цис- и транс-соединения настоящего изобретения, в которых отдельные энантиомеры не были разделены, здесь обозначают, используя приставку “(+)-“. В соответствии со следующими аспектами настоящего изобретения предложены также: (a) соединения формулы (I) и их фармацевтически приемлемые соли, сольваты и физиологически функциональные производные для использования в качестве терапевтических агентов, особенно для профилактики и лечения клинических состояний, при которых показан ингибитор поглощения желчных кислот, например, при таких гиперлипидемических состояниях, как атеросклероз. (b) фармацевтические композиции, включающие соединение формулы (I), или одно из фармацевтически приемлемых солей, сольватов или физиологически функциональных производных по крайней мере один из фармацевтически приемлемых носителей и, необязательно, один или более из физиологически активных агентов; (c) использование соединения формулы (I) или его фармацевтически приемлемой соли, сольвата или физиологически функционального производного при изготовлении лекарственного средства для профилактики или лечения клинических состояний, при которых показан ингибитор поглощения желчных кислот, например, таких гиперлипидемических состояний, как атеросклероз; (d) способ ингибирования абсорбции желчных кислот из кишечника млекопитающего, например человека, который включает введение эффективного ингибирующего абсорбцию желчных кислот количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата или физиологически функционального производного, млекопитающему; (e) способ снижения концентраций в плазме крови или сыворотке ЛНП и ЛОНП холестерина у млекопитающих, например у человека, который включает введение эффективного снижающего холестерин количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата или физиологически функционального производного млекопитающему; (f) способ снижения концентраций холестерина и сложного эфира холестерина в плазме или сыворотке крови такого млекопитающего, как человек, который включает введение млекопитающему эффективного снижающего содержание холестерина или сложного эфира холестерина количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата или физиологически функционального производного; (g) способ повышения фекальной экскреции желчных кислот у такого млекопитающего, как человек, который включает введение эффективно повышающего фекальную экскрецию желчных кислот количества соединения формулы (I) или его фармацевтически приемлемой соли, сольвата или физиологически функционального производного, млекопитающему; (h) способ профилактики или лечения клинических состояний у таких млекопитающих, как человек, для которого показан ингибитор поглощения желчных кислот, например, гиперлипидемических состояний, таких, как атеросклероз, который включает введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли сольвата или физиологически функционального производного млекопитающему; (i) способ снижения числа случаев заболеваний, связанных с коронарной болезнью сердца у таких млекопитающих, как человек, который включает введение эффективного количества, снижающего число случаев заболеваний, связанных с коронарной болезнью сердца, соединения формулы (I) или его фармацевтически приемлемой соли, сольвата или физиологически функционального производного; (j) способ снижения концентрации холестерина в плазме крови или сыворотке у млекопитающего, такого, как человек, который включает введение эффективного количества, снижающего холестерин, соединения формулы (I); (k) способы получения соединений формулы (I) (включая его соли, сольваты и физиологически функциональные производные, как здесь определено); и (1) новые химические производные для получения соединений формулы (I); (m) соединения примеров получения 1-5, как здесь раскрыто. Здесь и далее все ссылки на “соединение(соединения) формулы (I)” относятся к соединению(соединениям) формулы (I), как описано выше, наряду с их солями, сольватами и физиологически функциональными производными, как здесь определено. Количество соединения формулы (I), которое необходимо для достижения необходимого биологического эффекта, будет естественно зависеть от ряда факторов, например от конкретно выбранного соединения, предполагаемого использования, способа введения и клинического состояния реципиента. Обычно дневная доза находится в интервале от 0,3 мг до 100 мг (обычно от 3 мг до 50 мг) в день на килограмм веса тела, например 3-10 мг/кг/день. Доза для внутривенного введения может быть, например, в интервале от 0,3 мг до 1,0 мг/кг, которую можно обычно вводить в виде вливаний со скоростью от 10 нг до 100 нг/кг в минуту. Жидкости для внутривенных вливаний для этих целей могут содержать, например, от 0,1 нг до 10 мг, и обычно от 1 нг до 10 мг на миллилитр. Единичная доза может содержать, например, от 1 мг до 10 г активного соединения. Так, ампулы для инъекций могут содержать, например, от 1 мг до 100 мг, а единичные дозы для перорального приема в таком виде, как таблетки или капсулы, могут содержать, например, от 1,0 до 1000 мг, обычно от 10 до 600 мг. В случае фармацевтически приемлемых солей указанный вес относится к весу бензотиазепинового иона, полученного из соли. Для профилактики или лечения состояний, указанных выше, соединения формулы (I) можно использовать как сами по себе, но предпочтительно представлять их с приемлемым носителем в форме фармацевтической композиции. Носитель естественно должен быть приемлемым в смысле совместимости с другими ингредиентами композиции и не должен оказывать вредного воздействия на реципиента. Носитель может быть как твердым, так и жидким, или обоими сразу, и его предпочтительно приготавливают вместе с соединением в виде композиции в единичной дозе, например в форме таблетки, которая может содержать от 0,05% до 95% по весу активного соединения. Могут присутствовать и другие фармакологически активные вещества, включая другие соединения формулы (I). Фармацевтические композиции настоящего изобретения можно получить любым из хорошо известных методов фармацевтики, которые состоят, по существу, из смешивания компонентов. Фармацевтические композиции настоящего изобретения включают композиции для перорального, ректального, поверхностного, буккального (например, под язык) и парентерального (например, подкожного, внутримышечного, через кожу или внутривенного) введения, хотя наиболее удобный способ в любом из указанных случаев будет зависеть от характера и тяжести подлежащего лечению заболевания, и от природы конкретного соединения формулы (I), которое предполагают использовать. В объем настоящего изобретения включены также композиции с противожелудочным покрытием и с противожелудочным покрытием, обеспечивающим контролируемое выделение. Предпочтительны композиции, устойчивые к кислотам и желудочному соку. Подходящие противожелудочные покрытия включают ацетатфталат целлюлозы, поливинилацетатфталат, фталат гидроксипропилметилцеллюлозы и анионные полимеры метакриловой кислоты и метилового эфира метакриловой кислоты. Фармацевтические композиции для перорального введения могут присутствовать в такой дискретной единичной форме, как капсулы, пастилки или таблетки, каждая из которых содержит заранее определенное количество соединения формулы (I) в виде порошка или гранул; в виде растворов или суспензий в водных или неводных жидкостях; или в виде эмульсий типа вода в масле или масло в воде. Как указано, такие композиции можно получить любым известным в фармацевтике способом, который включает стадию смешивания активного соединения и носителя (который может содержать один или более из вспомогательных ингредиентов). Обычно, такие композиции получают в результате равномерного и тщательного перемешивания активного соединения с жидким или тонко измельченным носителем, или обоими сразу, а затем, при необходимости, придавая продукту нужную форму. Так, например, таблетки можно получить, прессуя или плавя порошок или гранулы соединения, необязательно с одним или более из вспомогательных ингредиентов. Прессованные таблетки можно получить, прессуя в подходящем устройстве, соединение в свободно пересыпающейся форме, например, в виде порошка или гранул, необязательно в смеси со связующим, смазкой, инертным разбавителем и/или поверхностно активным/диспергирующим агентом (агентами). Плавленые таблетки можно получить, расплавляя в соответствующем устройстве, порошкообразное соединение, увлажненное инертным жидким разбавителем. Фармацевтические композиции, пригодные для буккального (под язык) введения, включают соединение формулы (I) во вкусовой основе, обычно в сахарозе, смоле акации или трагаканте, и пастилки, содержащие соединение в такой инертной основе, как желатин и глицерин или сахароза и акация. Фармацевтические композиции для парентерального введения обычно включают стерильные водные препараты соединения формулы (I) предпочтительно изотоничные с кровью предполагаемого реципиента. Такие препараты предпочтительно вводят внутривенно, хотя такое введение можно осуществить с помощью подкожных, внутримышечных или через кожу инъекций. Такие препараты обычно приготавливают, смешивая соединение с водой, стерилизуя полученный раствор и придавая ему изотоничность с кровью. Инъектируемые композиции настоящего изобретения обычно содержат от 0,1 до 5% вес/вес активного соединения. Фармацевтические композиции, пригодные для ректального введения предпочтительно представляют в виде однодозовых суппозиториев. Их можно получить, смешивая соединение формулы (I) с одним или более из обычных твердых носителей, например с маслом какао, а затем придавая форму полученной смеси. Фармацевтические композиции, пригодные для поверхностного нанесения на кожу предпочтительно имеют форму мазей, кремов, лосьонов, паст, готовых спреев, аэрозолей или масел. Носители, которые можно использовать, включают вазелин, ланолин, полиэтиленгликоли, спирты и сочетания двух или более из них. Активное соединение обычно присутствует в концентрациях от 0,1 до 15% (вес/вес) композиции, например от 0,5 до 2%. Возможно также трансдермальное введение. Фармацевтические композиции, пригодные для трансдермального введения, могут иметь вид дискретных пластырей, приспособленных для того, чтобы оставаться в тесном контакте с эпидермисом реципиента в течение продолжительного промежутка времени. Такие пластыри могут содержать активное соединение и необязательно буферированный, водный раствор, растворенные и/или диспергированные в адгезиве, или диспергированные в полимере. Подходящие концентрации активного соединения составляют величину от около 1% до 35%, предпочтительно от около 3% до 15%. В частности, активное соединение возможно доставлять из пластыря с помощью электротранспорта или ионтофореза, например, способом, описанным в Pharmaceutical Research, 2(6), 318 (1986). Соединения настоящего изобретения можно получить обычными способами, известными специалистам или способами, аналогичными опубликованным. Так, например, соединения формулы (I) можно получить способом, который включает: а) ацилирование соединения формулы (II): 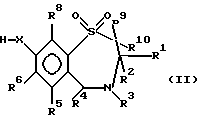 стандартным способом (например, с помощью N,N-карбонилдиимидазола) по -Х-Н группе, или а) алкилирование соединения формулы (II) стандартными способами по -Х-Н группе, или a) гликозилирование или глюкуронидирование соединения формулы (II) по -Х-Н группе, особенно, используя имидатный способ, и b) удаление защитных групп, особенно при гидрокси- и амино-функциональных группах, например ацетила с помощью гидролиза, а бензила – с помощью гидрогенолиза. Соединения формулы (II) можно получить способом получения, раскрытым в WO 96/05188. Соединения формулы (I), практически не содержащие других оптических изомеров, можно получить либо с помощью хирального синтеза, например, используя соответствующий исходный хиральный материал (материалы), такой, как азиридин, или разделяя продукты, полученные при ахиральном синтезе, например, с помощью хиральной ВЭЖХ, или с помощью классического разделения хиральными кислотами. Необязательное превращение соединения формулы (I), или соединения формулы (I), содержащего основной заместитель, в соответствующую соль присоединения кислоты можно осуществить в результате реакции с раствором соответствующей кислоты, например, одной из указанных ранее. Необязательное превращение соединения формулы (I), содержащего кислотный заместитель, в соответствующую основную соль можно осуществить при взаимодействии с раствором соответствующего основания, например гидроксида натрия. Необязательное превращение в такое функциональное производное, как сложный эфир, можно осуществить способами, известными специалистам или доступными из химической литературы. Кроме того, соединения формулы (I) можно превратить в другие соединения формулы (I) стандартными способами, известными специалистам, или доступными из литературы, например с помощью алкилирования гидроксильной группы. Сравнение гиполипидемической активности соединений настоящего изобретения с соединением 11 WO 96/05188. Для доказательства более высокой гиполипидемической активности соединений настоящего изобретения проводились тесты с тремя генетически модифицированными клеточными линиями. Они представляли производные общеизвестной клеточной линии “Chinese hamster ovary” (СНЩ) (клетки яичников китайского хомяка), которая за счет встроенных экспрессионных плазмид дополнительно продуцирует натрий-зависимые транспортеры желчных кислот. Первая клеточная линия (CHO/pPIBAT8) в данном случае представляет транспортер подвздошной кишки кролика (RIBAT), вторая (CHO/pHIBAT8) транспортер подвздошной кишки человека (HIBAT), а третья (СНО/рНLВАТ5) печеночный транспортер человека. Все плазмиды основаны на стандартной плазмиде pCDNAlneo, которая, как важные элементы, содержит цитомегаловирусный промотор для постоянной экспрессии гетерологичных генов, и ген для выработки устойчивости клеток к веществу G418. Исходным материалом для получения плазмид для RIBAT-продуцирующей клеточной линии (pRIBAT8) была полная РНК терминальной подвздошной кишки кролика. Из нее с помощью RT-PCR процедуры (реакция обратной транскриптазы, с последующей полимеразной цепной реакцией) с помощью олигонуклеотидов 5′-gtcagaccagaagcttgggcttctgcagac-3′ и 5′-atcttaataatattctagacagtttttctttg-3′, синтезируют кДНК, которая содержит полный протеин-кодирующий участок RIBAT, и также 41 пару оснований на прилегающем к 5′, и 31 пару оснований на прилегающем к 3′-нетранслируемом участке. Этот участок фланкируют, отщепляя сайты для рестрикционных ферментов Hind3 (с 5′- конца) и Xbal (с 3′-конца). Полученные кДНК и ДНК плазмиды pcDNAlneo обрабатывают, используя два рестрикционных фермента, и полученные фрагменты объединяют с помощью лигазы до получения экспрессионной плазмиды pRIBAT8. Плазмиду для HIBAT-продуцирующей клеточной линии (pHIBAT8) получают аналогично pRIBAT8. В этом случае полная РНК концевой подвздошной кишки человека и олигонуклеотиды 5′-taaaagttggatccggtagaagtaaacg-3′ и 5′-tctgttttgtcctctagatgtctacttttc-3′ служат в качестве исходного материала. Помимо полного протеин-кодирующего участка HIBAT, полученная кДНК содержит также 97 пар оснований на 5′-прилегающем и 5 пар оснований на 3′-прилегающем нетранслируемом участке. Этот участок фланкируют, отщепляя сайты для рестрикционных ферментов BamHl (с 5′-конца) и Xbal (с 3′-конца). Полученные кДНК и ДНК плазмиды pcDNAlneo обрабатывают, используя указанные два рестрикционных фермента и полученные фрагменты объединяют с помощью лигазы до получения экспрессионной плазмиды pRIBAT8. Коммерчески доступный кДНК генный банк, полученный из печени человека, служит исходным материалом для плазмиды для получения HLВАТ-продуцирующей клеточной линии (pHLBAT5). Из нее с помощью PCR-процедуры (полимеразной цепной реакции) с помощью олигонуклеотидов 5′-ggagtggtcttccactggatcccaggaggatggagg-3′ и 5′-ccagaatccaggccacctctagaagggctaggctgt-3′, синтезируют кДНК, которая содержит полный протеин-кодирующий участок Н ВАТ, и также 7 пар оснований, прилегающих к 5′- и 6 пар оснований, прилегающих к 3′-нетранслируемому участку. Этот участок фланкируют, отщепляя сайты для рестрикционных ферментов ВамН1 (с 5′-конца) и Xbal (с 3′-конца). Полученные кДНК и ДНК плазмиды pcDNAlneo обрабатывают, используя два рестрикционных фермента, и полученные фрагменты соединяют с помощью лигазы до получения экспрессионной плазмиды pHLBAT5. Для получения генетически модифицированных клеточных линий, СНО клетки трансфектируют с помощью ДНК из pRIBAT8, pHIBAT8 или pHLBAT5, и клетки, которые вырабатывают устойчивость к выбранному веществу G418, селективно дополнительно культивируют, добавляя это вещество к клеточной среде. Клетки CHO/pRIBAT8, CHO/pHIBAT8 и CHO/pHLBAT5 далее выделяют из количества G418-устойчивых клеток, и из них культивируют чистые клональные линии. Инструментом, который используют для последующего выделения в этом случае является флуоресцентное производное желчных кислот (3  -NBD-NCT; N-[7-(4-нитробензо-2-окса-1, 3-диазол)] -3-амино-7а, 12а-дигидрокси -NBD-NCT; N-[7-(4-нитробензо-2-окса-1, 3-диазол)] -3-амино-7а, 12а-дигидрокси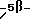 холан-24-оил)-2′-аминоэтаносульфонат. Клетки с интактными транспортерами желчных кислот быстро абсорбируют это вещество из клеточной среды, и в результате становятся флуоресцентными. Поэтому их становится легко дифференцировать от клеток без интактных транспортеров желчных кислот с помощью флуоресцентного микроскопа.
Все три клеточные линии эффективно транспортируют радиоактивномеченую таурохолевую кислоту из внеклеточной среды внутрь клетки. Этот процесс является натрий-зависимым. В противоположность этому СНО клетки без интактных транспортеров желчных кислот только абсорбируют лишь очень небольшие количества таурохолевой кислоты. На основании этого, характеризацию тестовых соединений по способу настоящего изобретения осуществляют следующим образом: клетки типов CHO/pRIBAT8, CHO/pHIBAT8 или CHO/pHLBAT5 одновременно экспонируют в культуральных чашках радиоактивномеченой таурохолевой кислоте, и измеряют тестовое вещество и абсорбцию клетками радиоактивного материала. Концентрации тестового вещества изменяют систематически от чашки к чашке, сохраняя постоянными все остальные параметры. Для подготовки их для эксперимента клетки обычным образом культивируют в среде (минимальная незаменимая среда (MEM); 1% MEM раствор заменимых аминокислот; 10% фетальная телячья сыворотка; 400 г/мл G418) в культуральных колбах; при необходимости удаляют из их окружения с помощью трипсина, инокулируют в разбавленной форме в культуральные чашки (диаметр 3,5 см) и дополнительно культивируют в среде. Незадолго до достижения конфлюэнтности клеток среду удаляют, а содержимое в каждой чашке дважды промывают 1,5 мл PBS (Дюльбеко фосфатом буферированный физиологический раствор). После удаления промывочного раствора в каждую чашку добавляют по 1 мл определенной концентрации тестового соединения в PBS, и инкубируют при 21oС в течение 30 минут. Этот предварительно инкубированный раствор заменяют затем тестовым раствором, содержащим |24-14C|-таурохолевую кислоту в концентрации 4,3 М и удельной радиоактивностью 7400Bq/мл, но во всем остальном имеющим тот же объем и состав, что и преинкубационный раствор. Клетки экспонируют тестовому раствору при 21oС в течение 30 минут, а затем промывают 5 раз в 1,5 мл PBS на чашку. Для осуществления лизиса клеток, 1 мл водного раствора, содержащего 0,1 моль/л NaOH и 0,1% (вес/объем) SDS добавляют в каждую чашку, которую инкубируют в течение 30 минут при 21oС и тщательно растирают.
И, наконец, содержимое каждой чашки смешивают с 10 мл коммерчески доступного сцинтилляционного раствора, и определяют радиоактивность, удержанную клетками с помощью прибора, измеряющего сцинтилляцию.
Для оценки результатов, относящихся к транспорту, величины радиоактивности не откладывают на графике непосредственно, но берут их процентное отношение к контрольному значению в тех случаях, если измерения были осуществлены без ингибирования тестового вещества. Половину максимального ингибирующего значения (IC50) получают либо из графика, либо арифметически: холан-24-оил)-2′-аминоэтаносульфонат. Клетки с интактными транспортерами желчных кислот быстро абсорбируют это вещество из клеточной среды, и в результате становятся флуоресцентными. Поэтому их становится легко дифференцировать от клеток без интактных транспортеров желчных кислот с помощью флуоресцентного микроскопа.
Все три клеточные линии эффективно транспортируют радиоактивномеченую таурохолевую кислоту из внеклеточной среды внутрь клетки. Этот процесс является натрий-зависимым. В противоположность этому СНО клетки без интактных транспортеров желчных кислот только абсорбируют лишь очень небольшие количества таурохолевой кислоты. На основании этого, характеризацию тестовых соединений по способу настоящего изобретения осуществляют следующим образом: клетки типов CHO/pRIBAT8, CHO/pHIBAT8 или CHO/pHLBAT5 одновременно экспонируют в культуральных чашках радиоактивномеченой таурохолевой кислоте, и измеряют тестовое вещество и абсорбцию клетками радиоактивного материала. Концентрации тестового вещества изменяют систематически от чашки к чашке, сохраняя постоянными все остальные параметры. Для подготовки их для эксперимента клетки обычным образом культивируют в среде (минимальная незаменимая среда (MEM); 1% MEM раствор заменимых аминокислот; 10% фетальная телячья сыворотка; 400 г/мл G418) в культуральных колбах; при необходимости удаляют из их окружения с помощью трипсина, инокулируют в разбавленной форме в культуральные чашки (диаметр 3,5 см) и дополнительно культивируют в среде. Незадолго до достижения конфлюэнтности клеток среду удаляют, а содержимое в каждой чашке дважды промывают 1,5 мл PBS (Дюльбеко фосфатом буферированный физиологический раствор). После удаления промывочного раствора в каждую чашку добавляют по 1 мл определенной концентрации тестового соединения в PBS, и инкубируют при 21oС в течение 30 минут. Этот предварительно инкубированный раствор заменяют затем тестовым раствором, содержащим |24-14C|-таурохолевую кислоту в концентрации 4,3 М и удельной радиоактивностью 7400Bq/мл, но во всем остальном имеющим тот же объем и состав, что и преинкубационный раствор. Клетки экспонируют тестовому раствору при 21oС в течение 30 минут, а затем промывают 5 раз в 1,5 мл PBS на чашку. Для осуществления лизиса клеток, 1 мл водного раствора, содержащего 0,1 моль/л NaOH и 0,1% (вес/объем) SDS добавляют в каждую чашку, которую инкубируют в течение 30 минут при 21oС и тщательно растирают.
И, наконец, содержимое каждой чашки смешивают с 10 мл коммерчески доступного сцинтилляционного раствора, и определяют радиоактивность, удержанную клетками с помощью прибора, измеряющего сцинтилляцию.
Для оценки результатов, относящихся к транспорту, величины радиоактивности не откладывают на графике непосредственно, но берут их процентное отношение к контрольному значению в тех случаях, если измерения были осуществлены без ингибирования тестового вещества. Половину максимального ингибирующего значения (IC50) получают либо из графика, либо арифметически:Пример 3 IC50(RIBAT): 70 нМ = 0,07 мкМ Пример 11 WO 96/05188 IC50(RIBAT): 4 мкМ Аналогичное исследование влияния тех же веществ на транспорт клеточной линии CHO/pHIBAT8 показывает, что в этом случае соответствующие IС50 значения меняются примерно в том же порядке величин. В противоположность этому IС50 величина, определенная для клеточной линии CHO/pHLBAT5 была на несколько порядков выше. Это показывает, что соединения настоящего изобретения могут оказывать сравнительное действие на ортологичные натрий-зависимые транспортеры желчных кислот различных видов, и, в противоположность этому, действие на паралогичные транспортеры других органов оказывается гораздо меньше. Для лучшего понимания изобретения приводится следующий пример для иллюстрации (например, с N,N-карбонилдиимидазолом), и его не следует рассматривать как в какой-либо степени ограничивающий объем изобретения. Пример 1 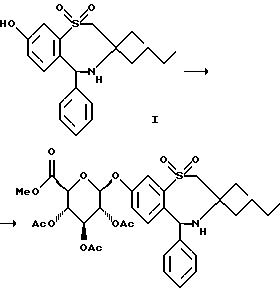 К раствору 2,9 г метил-2,3,4-три-О-ацетилглюкуроната в 100 мл сухого дихлорметана при комнатной температуре в атмосфере аргона добавляют 4,6 мл трихлорацетонитрила, и полученный раствор перемешивают в течение 10 минут. Затем добавляют 730 мг карбоната калия. Спустя 30 минут перемешивания при комнатной температуре смесь фильтруют через небольшой слой двуокиси кремния, элюируя эфиром. Полученный фильтрат концентрируют в вакууме до получения неочищенного продукта в виде твердого вещества бледно-желтого цвета (3,7 г). 1,0 г этого продукта растворяют в 15 мл сухого дихлорметана, и добавляют к раствору Фенола 1 (транс-рацемат) в 30 мл сухого дихлорметана. После охлаждения до -10oС добавляют 0,32 мл ВF3  ОЕt2 и спустя 30 минут при -10oС, полученную смесь перемешивают в течение 20 часов при комнатной температуре. Затем реакционную смесь разбавляют дихлорметаном и промывают водным раствором бикарбоната натрия и рассолом. Объединенные органические фазы сушат над сульфатом натрия и выпаривают в вакууме. Сырой продукт очищают хроматографически на силикагеле (н-гептан/этилацетат 2:1) до получения 625 мг соединения примера 1 Rf=0,17 (н-гептан/этилацетат 1:1).
C34H43NO12S (689): MS (бомбардировка быстрыми атомами, 3-NBA):690(M+H+).
Пример 2 ОЕt2 и спустя 30 минут при -10oС, полученную смесь перемешивают в течение 20 часов при комнатной температуре. Затем реакционную смесь разбавляют дихлорметаном и промывают водным раствором бикарбоната натрия и рассолом. Объединенные органические фазы сушат над сульфатом натрия и выпаривают в вакууме. Сырой продукт очищают хроматографически на силикагеле (н-гептан/этилацетат 2:1) до получения 625 мг соединения примера 1 Rf=0,17 (н-гептан/этилацетат 1:1).
C34H43NO12S (689): MS (бомбардировка быстрыми атомами, 3-NBA):690(M+H+).
Пример 2Пример 3 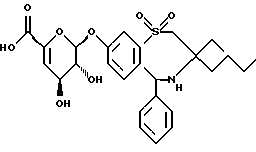 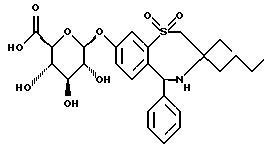 К раствору 900 мг соединения примера 1 в 45 мл метанола добавляют 15 мл 1н. NaOH. Через 4 часа при комнатной температуре добавляют 150 мл Н2O, и органический растворитель выпаривают в вакууме. Водный раствор доводят до рН 3 с помощью 2н. НСl и выпаривают досуха. В результате хроматографической обработки на силикагеле (CH2Cl2/MeOH/ 33% водн. NH3=30:10:3) получают 2 фракции. 1 фракция: пример 2, Rf= 0,85 (СН2Сl2/МеОН/33% водн. NН3=30:10:3) (ионизация распылением электронов) (С27Н35NO8S (531): MS(ESI):532 (M+H+) 2 фракция: пример 3, Rf= 0,52 (СН2Сl2/МеОН/33% водн. NH3, 30:10:3) (C27H35NO9S (549):MS, (бомбардировка быстрыми атомами, 3-NBA) 550(M+H+) Пример 4 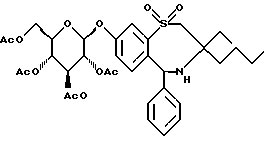 Соединение примера 4 получают по способу примера 1 Rf=0,20 (н-гептан/этилацетат 1:1) C35H45NO12S (703) Масс-спектр (ионизация распылением электронов): 704(M+H+) Пример 5 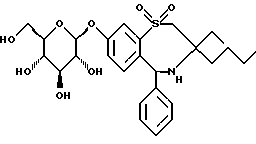 Соединение примера 5 получают по способу примера 2 Rf=0,20 (СН2С12/МеОН/33% водн. NН3, 60:10:3) C27H37NO8S (535): Масс-спектр (бомбардировка быстрыми атомами 3-NBA): 536 (М+Н+)д Формула изобретения
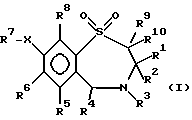 где R1 представляет неразветвленную C1-6 алкильную группу; R2 представляет неразветвленную C1-6 алкильную группу; R3 представляет водород; R4 представляет фенил; R5, R6 и R8 выбирают из водорода; R7 представляет группу формулы 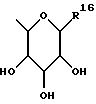 или 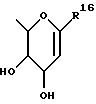 где гидроксигруппы могут быть замещены ацетилом; R16 представляет -СООН, -СН2-ОН, -СН2-О-ацетил, -СООМе; R9 и R10 одинаковы или различны и каждый представляет водород или C1-6 алкильную группу; Х представляет -О-; или его соли, сольваты и физиологически функциональные производные. 2. Соединения по п. 1 формулы (III) 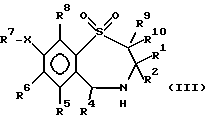 где R1-R10 и Х имеют указанные в п. 1 значения. 3. Соединения по п. 1, которые имеют формулу (IVa) 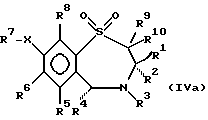 где R1-R10 и Х имеют указанные в п. 1 значения. 4. Соединение по любому из пп. 1-3, где R7 выбирают из соединений формул 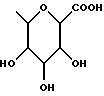 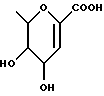 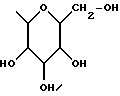 5. 1,4-бензотиазепин-1,1-диоксид формулы (V) 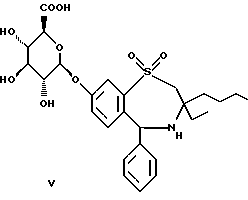 6. Соединение по любому из пп. 1-5 или его фармацевтически приемлемая соль, сольват или физиологически функциональное производное в качестве активного ингредиента лекарственного средства для профилактики или лечения клинического состояния, при котором показан ингибитор поглощения желчных кислот. 7. Способ получения соединения по любому из пп. 1-5 и его солей, сольватов и физиологически функциональных производных, отличающийся тем, что включает a) ацилирование соединения формулы II 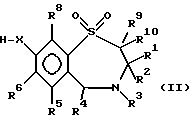 стандартными способами по -Х-Н группе и b) удаление защитных групп, особенно, при гидроксильных и аминофункциональных группах. 8. Способ получения соединения по любому из пп. 1-5 и его солей, сольватов и физиологически функциональных производных, отличающийся тем, что включает a) алкилирование соединения формулы II стандартными способами по -Х-Н группе и b) удаление защитных групп, особенно, при гидроксильных и амино-функциональных группах. 9. Способ получения соединения по любому из пп. 1-5 и его солей, сольватов и физиологически функциональных производных, отличающийся тем, что включает a) гликозилирование или глюкуронидирование соединения формулы II по -Х-Н группе и b) удаление защитных групп, особенно, при гидроксильных и амино-функциональных группах. РИСУНКИ
MM4A Досрочное прекращение действия патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 13.03.2004
Извещение опубликовано: 10.05.2005 БИ: 13/2005
|
||||||||||||||||||||||||||