Патент на изобретение №2179972
|
||||||||||||||||||||||||||
(54) БИС(АМИДЫ) АЗОТСОДЕРЖАЩИХ ТРИЦИКЛИЧЕСКИХ КАРБОНОВЫХ КИСЛОТ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ
(57) Реферат: Изобретение относится к новым бис(амидам)азотсодержащих трициклических карбоновых кислот ф-лы (I), где каждый из X, которые могут быть одинаковыми или различными в данной молекуле, представляет -СН= или -N=, каждый из R1-R4, которые могут быть одинаковыми или различными, представляет Н, С1-С4-алкил, NH2, С1-С4-алкокси, фенил, фенокси, NHR, N(R)2, СF3 или галоген, R – С1-С4-алкил, каждый из R5 и R6 – Н или С1-С4-алкил, Z представляет (CH2)nN(R7)(CH2)n, (CH2)nN(R7)(CH2)N(R7)(CH2)n, (CH2)nNH(CH2CH2)NH(CH2), R7 – Н или С1-С4-алкил, n и m = 1-4, или их фармацевтически приемлемым солям, за исключением соединений, где каждый Х – N, каждый из R1 – R6 – Н, карбоксамидная часть присоединена в положении I каждого кольца феназина и Z представляет (СН2)2NН(CН2)2, (СН2)3NН(CН2)3, (CН2)3NН(CН2CН2)2NН(CН2)3, (СН2)2NН(CН2)2NН(CН2)2 или (CН2)3NН(CН2)2NН(CН2)3. Соединения ф-лы (I) обладают противоопухолевой активностью и могут найти применение в медицине. 3 с. и 4 з.п. ф-лы, 1 ил., 2 табл. 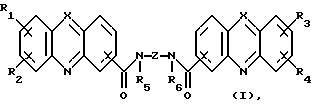
Изобретение относится к соединениям, полезным в качестве противоопухолевых агентов, к их получению и к содержащим их фармацевтическим композициям. Известно, что многие соединения, которые обратимо связываются с ДНК посредством интеркаляции, обладают клеточными антипролиферативными свойствами и in vivo противоопухолевым действием, медиированным, главным образом, посредством ингибирования ими ферментов топоизомераз. Например, производные акридина амсакрин [Issel, Cancer Treat. Rev. 1980, 7, 73], азулакрин [Harvey et al., Eur. J. Cancer, 1991, 27, 1617] и акридинкарбоксамид [Finlay et al, Eur. J. Cancer 1996, 32A, 708] являются клиническими противораковыми лекарственными средствами или проявляют такие свойства в клиническом испытании, и было описано, что родственные им 9-азаакридины (феназины) обладают in vivo противоопухолевыми свойствами на моделях животных [Rewcastle et al., J. Med. Chem., 1987, 30, 843]. В настоящее время обнаружено, что ряд производных бис(акридинкарбоксамида) и бис(9-феназинкарбоксамида) обладают противоопухолевыми свойствами. Данное изобретение, следовательно, относится к соединению, которое представляет собой производное бис(акридинкарбоксамида) и бис(феназинкарбоксамида) формулы (I): 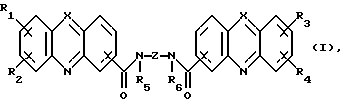 где каждый X, которые могут быть одинаковыми или различными в данной молекуле, представляет -СН= или -N=; каждый из от R1 до R4, которые могут быть одинаковыми или различными, представляет Н, С1-С4-алкил, ОН, SH, NH2, С1-С4-алкокси, арил, арилокси, NHR, N(R)2, SR или SO2R, где R представляет С1-С4-алкил, СF3, NO2 или галоген, или R1 и R2 вместе с атомами углерода, к которым они присоединены, образуют метилендиоксигруппу; каждый из R5 и R6, которые могут быть одинаковыми или различными, представляет Н или С1-С4-алкил; Z представляет (СН2)n, (СН2)nO(СН2)n, (CH2)nN(R7)(СН2)n, (СН2)nN(R7)(СН2)mN(R7)(СН2)n или (CH2)nN(CH2CH2)2N(CH2)n, (CH2)nCONH(CH2)m или (СН2)nCONH(CH2)mNHCO(CH2)n, где R7 представляет Н или С1-С4-алкил и n и m, которые могут быть одинаковыми или различными, каждый равен целому числу от 1 до 4; или его фармацевтически приемлемой кислотно-аддитивной соли или N-оксиду, за исключением соединений, где каждый Х представляет N, каждый из от R1 до R6 представляет Н, карбоксамидная часть присоединяется в положении 1 каждого кольца феназина и Z представляет (СН2)2NН(СН2)2, (СН2)3NН(СН2)3, (СН2)3N(СН2СН2)2N(CH2)3, (СН2)2NН(СН2)2NH(CH2)2 или (СН2)3NН(СН2)2NH(CH2)3. В формуле (I) нумерация в кольцевых атомах в каждом трициклическом хромофоре различается в зависимости от того, представляет ли Х -СН= (производные акридина) или -N= (производные феназина). Нумерация, используемая в каждом случае со ссылкой на одну трициклическую часть, является следующей: 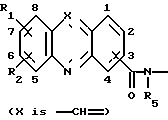 (Х представляет -СН=) 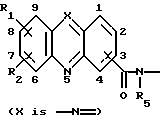 (X представляем -N=) В формуле (I) заместители R1 и R2 и R3 и R4 могут занимать любое одно из доступных положений кольца в их соответствующих трициклических хромофорах. Таким образом, когда Х в трициклической части представляет -СН=, то R1 и R2 или R3 и R4, если возможно, каждый может быть связан с любым одним из доступных положений кольца от 1 до 8, незанятым карбоксамидной частью -С(О)-N(R5)-. Карбоксамидная часть, в свою очередь, может быть связана с любым одним из положений кольца 1, 2, 3 и 4, предпочтительно 1 или 4. Типично, когда Х в данном трициклическом хромофоре представляет собой -СН=, один из R1 и R2 или R3 и R4, если возможно, представляет водород, и другой представляет водород или представляет заместитель, определенный выше для формулы (I), связанный любым одним из положений кольца от 1 до 8, и карбоксамидная часть присоединена в положении 1 или 4. Когда Х в трициклической части представляет -N=, то R1 и R2 или R3 и R4, если возможно, каждый, может быть связан с любым одним из доступных положений кольца от 1 до 4 и от 6 до 9, которые не заняты карбоксамидной частью, особенно одним из положений от 6 до 9. Карбоксамидная часть, в свою очередь, может быть связана с любым из положений кольца 1 и 2, предпочтительно 1. Типично, когда Х в данном трициклическом хромофоре представляет -N=, один из R1 и R2 или R3 и R4, если возможно, представляет водород и другой представляет водород или представляет заместитель, определенный выше для формулы (I) в любом одном из положений кольца от 1 до 4 или от 6 до 9, особенно 6, 7, 8 или 9, и карбоксамидная часть присоединена в положении 1 или 4. Когда в трициклических хромофорах левая и правая стороны в формуле (I) идентичны, соединения являются структурно симметричными. Когда два хромофора различаются в определении одного или нескольких заместителей, соединения структурно асимметричны. В предпочтительном варианте изобретения соединения являются структурно симметричными. С1-С4-Алкильная группа может быть линейной или разветвленной группой, например метилом, этилом, н-пропилом, изопропилом, н-бутилом, втор-бутилом или трет-бутилом. С1-С4-Алкоксигруппа аналогично может быть линейной или разветвленной группой, например, метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси или трет-бутокси. Галоген представляет, например, фтор, хлор, бром или иод. В одном варианте изобретения соединение представляет собой производное бис(акридинкарбоксамида) формулы (1а): 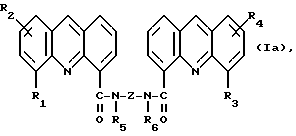 где каждый из R1 и R3, которые одинаковы или различны, представляет С1-С4-алкокси, С1-С4-алкил или галоген, каждый из R2 и R4, которые одинаковы или различны, представляет водород, С1-С4-алкокси, С1-С4-алкил или галоген и каждый из R5 и R6 представляет Н, или его фармацевтически приемлемую соль или N-оксид. В другом варианте изобретения соединение представляет собой производное бис(феназинкарбоксамида) формулы (Ib): 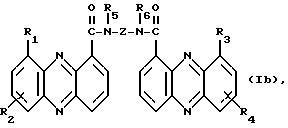 где каждый из R1 и R2, которые одинаковы или различны, представляет С1-С4-алкокси, С1-С4-алкил или галоген, каждый из R2 и R4, которые одинаковы или различны, представляет водород, С1-С4-алкокси, С1-С4-алкил или галоген и каждый из R5 и R6 представляет Н или его фармацевтически приемлемую соль или N-оксид. Примеры конкретных соединений изобретения приведены в следующей таблице 1 при помощи указания их заместителей. Все соединения симметричны, другими словами, левая сторона трициклического хромофора идентична его правой стороне. Таким образом, в формуле (I) R1=R3 и R2=R4. Соединения формулы (I) и их фармацевтически приемлемые кислотно-аддитивные соли и N-оксиды получают способом, который включает реакцию двух молей производного акридинкарбоновой кислоты или 9-азаакридинкарбоновой кислоты (феназинкарбоновой кислоты) формулы (II): 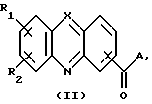 где R1, R2 и Х такие, как определено выше, и А представляет ОН, Сl или N-имидазолил, с одним молем бис(амина) формулы (III): NHR5-Z-NHR6 (III), где R5, R6 и Z такие, как определено выше, и, если необходимо, превращение полученного соединения в его фармацевтически приемлемую кислотно-аддитивную соль или N-оксид. Эту реакцию присоединения обычно проводят в негидроксильном растворителе, предпочтительно, ТГФ. Реакцию удобно проводить при температурах от -10oС до 50oС. Производные формулы (I) можно превратить в фармацевтически приемлемые кислотно-аддитивные соли и соли можно превратить в свободное соединение общепринятыми способами. Например, кислотно-аддитивные соли можно получить контактированием свободного основания с подходящим количеством желаемой кислоты общепринятым способом. Подходящие соли включают соли как с органическими, так и с неорганическими кислотами. Примерами подходящих кислот являются хлористоводородная, серная, фосфорная, уксусная, лимонная, щавелевая, малоновая, салициловая, яблочная, фумаровая, янтарная, аскорбиновая, метансульфоновая и подобные. В зависимости от структуры и от условий соединения могут образовать поликатионные формы. Необязательное превращение соединения формулы (I) в другое соединение формулы (I) можно проводить общепринятыми способами. Например, фторгруппу в соединении формулы можно заменить на амино или тиольную группу, с получением амина или простого тиоэфира, соответственно; фенольную или тиольную группу в соединении формулы (I) можно алкилировать с получением простого эфира или тиоэфира, соответственно; аминогруппу можно ацилировать с получением N-ацетата; и нитрогруппу можно восстановить с получением амина. Все эти превращения являются обычными в органической химии. Соединения формулы (I) можно превратить в соответствующий N-оксид общепринятыми методиками, например, растворением их в СНгСl2 и обработкой 2-(фенилсульфонил)-3-фенилоксаридином в течение от 15 мин до 24 ч, предпочтительно в течение 1 ч, при 0 – 60oС, предпочтительно 20oС. Формы свободных оснований можно регенерировать обработкой формы соли основанием. Формы свободных оснований отличаются от их соответствующих форм солей по некоторым физическим свойствам, но в остальном эквивалентны соответствующим солевым формам для целей данного изобретения. Бис(амины) общей формулы (III) являются известными соединениями, которые коммерчески доступны или которые можно получить способами, описанными в литературе. Конкретные примеры таких соединений включают NH2(CH2)6NH2, NH2(СН2)3О(СН2)3NH2, NH2(CH2)3NH(СН2)3NH2, NH2(СН2)3NMe(CH2)3NH2, NH2(CH2)2NH(CH2)2NH(CH2)2NH2 и NH2(СН2)3N(СН2СН2)2N(СН2)3NH2. Замещенные акридинкислоты формулы II, где Х представляет СН и А представляет ОН, можно получить способом, описанным в Atwell et al., J. Med. Chem., 1984, 27, 1481. Он включает восстановление при помощи Аl/Нg соответствующих акридон-4-карбоновых кислот формулы IV с последующим повторным окислением FеСl3 или другим подходящим мягким окислителем, как указано на приведенной ниже схеме 1 (см. в конце описания). Акридон-4-карбоновые кислоты формулы V можно, в свою очередь, получить различными известными способами, включая сочетание пo Jourdan-Ullman замещенных антраниловых кислот и галогенкислот, как описано в Denny et al., J. Med. Chem. , 1987, 30, 658, сочетание по Jourdan-Ullman иодизофталевой кислоты с замещенными анилинами, как описано в Rewcastle and Denny, Synthesis, 1985, 217, или реакцию замещенных карбоксилатов дифенилиодония с замещенными анилинами, как описано в Rewcastle and Denny, Synthesis, 1985, 220, с последующей циклизацией образованных таким образом дифениламиндикислот и сложных дифениламинмоноэфиров при помощи РРА или РРЕ с получением акридонов. Реакция акридин-4-карбоновых кислот формулы II, где А представляет ОН, с 1,1 эквивалентом 1,1′-карбонилдиимидазола в ДМФ при 10-40oС в течение от 10 мин до 24 ч дает соответствующие производные имидазолида соединений формулы II. Их можно выделить фильтрованием и подвергнуть реакции с 0,5 эквивалентом бисамина формулы III с получением целевых соединений формулы I. Некоторые из замещенных акридин-4-карбоновых кислот формулы (II) и их производные, которые можно использовать в качестве промежуточных продуктов для получения соединений общей формулы (I), являются новыми соединениями. В соответствии с этим, изобретение далее относится к соединению формулы (IIа): 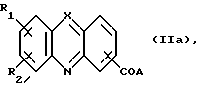 где X, R1 и R2 такие, как определено для формулы (I), и А представляет ОН, Сl, N-имидазолил или OR, где R представляет разветвленный или неразветвленный, насыщенный или ненасыщенный C1-C6-алкил или арил, за исключением соединений, где: (i) Х представляет СН, А представляет ОН, один из R1 и R2 представляет Н и другой представляет Н, Сl, ОМе или Me и часть -СОА находится в 4-положении трициклического хромофора, и (ii) X представляет N, А представляет ОН, один из R1 и R2 представляет Н и другой представляет Н, Сl, ОМе или Me и часть -СОА находится в 1-положении трициклического хромофора. Эти новые соединения формулы (IIа) можно получить способом, который описан полностью в приведенной ниже схеме 2. В соответствии с этим, изобретение далее относится к способу получения соединения формулы (IIа), который включает циклизацию соединения формулы (X): 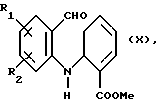 где R1 и R2 такие, как определено выше. Циклизацию обычно проводят в кислотных условиях, например, с использованием неразбавленной трифторуксусной кислоты в течение периода 12 час при температуре от 20 до 60oС. Соединения формулы (X) получают, как указано в приведенной ниже схеме 2 (см. в конце описания). В схеме 2 R1 и R2 такие, как определено выше для общей формулы (I). Замещенные дифениламинокислоты (VII) получают сочетанием замещенных антраниловых кислот (V) с метил-2-галогенбензоатами (VI), обычно иодбензоатом, медным катализатором в полярном растворителе, обычно бутан-2,3-диоле. Этот способ описан в Rewcastle and Denny, Synth. Comm., 1987, 17, 309. Реакция замещенных дифениламинокислот с 1,1′-карбонилдиимидазолом (CDI) в полярном растворителе, обычно ТГФ, дает имидазолиды формулы (VIII), которые восстанавливают in situ избытком восстанавливающего агента на основе металла, приемлемо, борогидридом натрия, в спирты формулы (IX). Окисление спиртов твердым МnO2 в полярном растворителе, обычно этилацетате или ацетоне, дает соответствующие альдегиды формулы (X), которые циклизуют, как указано выше, с образованием сложных замещенных акридинэфиров, которые представляют собой соединения формулы (II), где А представляет собой ОМе. Эти эфиры можно гидролизовать в кислотных или основных условиях в соответствующие кислоты, которые представляют собой соединения формулы (II), где А представляет собой ОН. Их можно, в свою очередь, активировать реакцией с 1,1′-карбонилдиимидазолом в полярном растворителе (предпочтительно, сухом ДМФ) с получением соответствующих производных имидазолида, которые представляют собой соединения формулы (IIа), где А представляет N-имидазолил, или обработкой тионилхлоридом в инертном растворителе, обычно дихлорэтане, с получением соответствующих хлорангидридов, которые представляют собой соединения формулы (II), где А представляет собой Сl. Эти последние промежуточные продукты можно сочетать с бис(аминами) формулы (III), как описано выше, с получением целевых соединений общей формулы (I). Альтернативно, сложные замещенные акридинэфиры формулы (II), где А=ОМе, можно подвергнуть реакции непосредственно с бис(аминами) общей формулы III в полярном растворителе, предпочтительно, этаноле или изопропаноле, чтобы получить соединения общей формулы (I). Феназинкислоты, а именно, соединения формулы II, где Х представляет собой N и А представляет собой ОН, можно получить способами, списанными в литературе. Примеры включают J. Med. Chem. 1987, 30, 843; Synth. Comm. 1987, 17, 1171; Европейский патент ЕР-А-172744 and Chem. Abstr. 1986, 105, 97496р. Их можно затем превратить в соединения общей формулы I способами, указанными выше. Другие соединения общей формулы (I) можно также получить общим способом, указанным на схеме 3, которая приведена в конце описания. В этом способе реакция соединений формулы (II) (где А=имидазолид) с монозащищенным амином формулы (XI) дает промежуточный продукт формулы (XII). Удаление защитной группы R9 дает амины формулы (XIII), которые можно сочетать с дополнительным эквивалентом соединения формулы (II) (где А=имидазолид), с получением соединения формулы (I). На схеме 3 R1-R5, Z и R6 такие, как определено для формулы (I), за исключением того, что R7 может также быть БОК, тритилом, СО2СF3 или другой подходящей аминозащитной группой и R8 определяют как БОК, тритил, СО2СF3 или другая подходящая аминозащитная группа. В биологических испытаниях было обнаружено, что соединения формулы (I) и их соли и N-оксиды обладают противоопухолевой активностью. Результаты показаны в примере 78, который приведен ниже. Соединения обладают незначительной токсичностью, и их можно, следовательно, использовать в качестве противоопухолевых средств. Пациента-человека или животного, имеющего опухоль, можно, таким образом, лечить способом, который включает введение ему соединения формулы (I) или его соли или N-оксида. Состояние пациента, страдающего раком, можно тем самым улучшить. Примеры опухолей, для лечения которых можно использовать соединения формулы (I), включают опухоли легких и ободочной кишки, меланому и опухоли центральной нервной системы (ЦНС). Данные соединения можно вводить в различных дозированных формах, например, для перорального введения, таких как формы в виде таблеток, капсул, таблеток, покрытых сахаром или пленкой, жидких растворов или суспензий, или парентерального введения, например, внутримышечно, внутривенно или подкожно. Данные соединения можно, следовательно, вводить инъекцией или инфузией. Доза зависит от различных факторов, включая возраст, массу и состояние пациента и пути введения. Обычно, однако, доза, выбранная для каждого пути введения, когда соединение изобретения вводят как таковое взрослым людям, составляет от 50 мг/м2 до 5 г/м2. Такую дозу можно давать, например, от 1 до 5 раз в день, инфузией болюса, например 3 раза в день, инфузией на протяжении нескольких часов, например 3 ч, и/или многократным введением. Данные соединения формулы (I) и их соли и N-оксиды изготовляют для использования в виде фармацевтической или ветеринарной композиции. Данное изобретение, следовательно, далее предлагает фармацевтическую или ветеринарную композицию, которая включает фармацевтически или ветеринарно приемлемый носитель или разбавитель и в качестве активного ингредиента соединение формулы (I) или его соль или N-оксид. Композиции обычно готовят следующими общепринятыми способами и вводят в фармацевтически или ветеринарно пригодной форме. Например, твердые пероральные формы могут содержать вместе с активным соединением разбавители, такие как лактоза, декстроза, сахароза, целлюлоза, кукурузный крахмал или картофельной крахмал; смазывающие вещества, такие как диоксид кремния, тальк, стеариновая кислота, стеарат магния или кальция и/или полиэтиленгликоли; связывающие агенты, такие как крахмалы, аравийские камеди, желатин, метилцеллюлоза, карбоксиметилцеллюлоза или поливинилпирролидон, разрыхляющие агенты, такие как крахмал, альгиновая кислота, альгинаты или натриевая соль гликолята крахмала; смеси, вызывающие вскипание, красители; подслащивающие средства, смачивающие агенты, такие как лецитин, полисорбаты, лаурилсульфаты. Такие препараты можно изготовить известным способом, например при помощи смешивания, гранулирования, таблетирования, покрытия сахаром или покрытия пленкой. Жидкими дисперсиями для перорального введения могут быть сиропы, эмульсии и суспензии. Сиропы могут содержать в качестве носителя, например, сахарозу или сахарозу с глицерином и/или маннитом, и/или сорбитом. В частности, сироп может содержать в качестве носителя, например, сахарозу или сахарозу с глицерином и/или маннитом, и/или сорбитом. В частности, сироп для диабетических пациентов может содержать в качестве носителей только продукты, например сорбит, которые не метаболизируют в глюкозу или которые метаболизируют только в небольшом количестве в глюкозу. Суспензии и эмульсии могут содержать в качестве носителя, например, природную камедь, агар, альгинат натрия, пектин, метилцеллюлозу, карбоксиметилцеллюлозу или поливиниловый спирт. Суспензии или растворы для внутримышечных инъекций могут содержать, вместе с активным соединением, фармацевтически приемлемый носитель, такой как стерильная вода, оливковое масло, этилолеат, гликоли, такие как пропиленгликоль, и, если необходимо, подходящее количество гидрохлорида лидокаина. Обычно соединения формулы (I) изготовляют в виде водных растворов гидрохлоридных или других фармацевтически приемлемых солей. Растворы для внутривенной инъекции или инфузии могут содержать носитель, например, стерильную воду, которая обычно представляет собой воду для инъекций. Изобретение далее описано в следующих примерах со ссылкой на сопровождающий чертеж, на котором представлен график зависимости объема опухоли в мм3 (ось y) от времени в днях (ось х) для демонстрации замедления роста клеток опухоли 38 ободочной кишки у мышей в испытании in vivo, как описано в примере 80, который представлен ниже. Пример 1: Получение соединения 22 таблицы 1 способом по схеме 1 Смесь 2-иодизофталевой кислоты (2,92 г, 10 ммоль), 4-этиланилина (1,82 г, 15 ммоль), CuCl (1 г), 2,3-бутандиола (12 мл) и бензола (10 мл) нагревали и перемешивали, давая бензолу отгоняться. Когда внутренняя температура реакционной смеси достигала 100oС, добавляли N-этилморфолин (6 мл), и реакционную смесь перемешивали в течение дополнительных 4 ч при 120oС. Реакционную смесь затем разбавляли 0,5 М NH4OН (50 мл), обрабатывали древесным углем и фильтровали через целит. Подкисление 2 М НСl вызывало образование осадка, который экстрагировали в EtOAc (2  100 мл), фильтровали через целит и далее экстрагировали 0,5 М NH3 (100 мл). Подкисление концентрированной НСl и перекристаллизация выделенного продукта давали 2-[(4-этил) фениламино]изофталевую кислоту (2,36 г, 83%), т. пл. (EtOAc/петролейный эфир) 194-195,4oС. 1Н ЯМР [(CD3)2SO] 100 мл), фильтровали через целит и далее экстрагировали 0,5 М NH3 (100 мл). Подкисление концентрированной НСl и перекристаллизация выделенного продукта давали 2-[(4-этил) фениламино]изофталевую кислоту (2,36 г, 83%), т. пл. (EtOAc/петролейный эфир) 194-195,4oС. 1Н ЯМР [(CD3)2SO]  1,15 (т, J=7,6, 3 Н, СН3), 2,51 (к, J=7,6 Гц, 2 Н, СН2), 6,84 (д, J=8,4 Гц, 2 Н, Н-2′ и Н-6′ или Н-3′ и Н-5′), 6,97 (т, J=7,7 Гц, 1 Н, Н-5), 7,04 (д, J=8,4 Гц, 2 Н, Н-3′ и Н-5′ или Н-2′ и Н-6′), 7,92 (д, J= 7,6 Гц, 2 Н, Н-4 и Н-6), 9,65 (шир. с, 1 Н, NH) и 12,90 (шир. с, 2 Н, 2СООН). Элементный анализ (C16H14NO4) С, Н, N.
Реакцию вышеуказанной дикислоты (1,43 г, 5 ммоль) с полифосфорной кислотой (38 г) проводили нагреванием смеси при 120oС в течение 2 час. Охлажденную смесь выливали в кипящую воду (250 мл), что приводило к образованию суспензии желтого осадка. Осадок удаляли фильтрованием и растворяли в смеси МеОН (100 мл) и 1 М NaOH (100 мл), которую нагревали и затем фильтровали горячей. Подкисление этой смеси с использованием ледяной уксусной кислоты давало желтый твердый продукт, который перекристаллизовывали, получая 7-этил-9-оксоакридан-4-карбоновую кислоту (1,14 г, 89%), т. пл. (Н2О) 301oС (разложение). 1Н ЯМР [(СD3)2SO] 1,26 (т, J=7,6 Гц, 3 Н, СН3), 2,74 (к, J= 7,6 Гц, 2 Н, СН2), 7,33 (т, J=7,7 Гц, 1 Н, Н-2), 7,64 (дд, J=8,5, 2,1 Гц, 1 Н, Н-6), 7,71 (д, J=8,5 Гц, 1 Н, Н-5), 8,04 (шир. с, 1 Н, Н-8), 8,42 (дд, J= 7,5, 1,7 Гц, 1Н, Н-3), 8,52 (дд, J=8,0, 1,7 Гц, 1 Н, Н-1), 11,98 (с, 1 Н, NH) и 13,85 (шир. с, 1 Н, СООН). Элементный анализ (С16Н13NО3) С, Н, N.
Суспензию вышеуказанной акридонкислоты (1,00 г, 3,75 ммоль) в 50% водном EtOH перемешивали и нагревали, затем добавляли достаточное количество триэтиламина с получением прозрачного желтого раствора. Порции алюминиевой фольги (0,83 г) амальгамировали в растворе HgCl2 (3 г) в EtOH, затем добавляли к вышеуказанному энергично кипящему раствору в течение 30 мин. После того, как ТСХ показала, что реакция завершалась, реакционную смесь фильтровали, и собранное твердое вещество промывали раствором КОН в водном EtOH. Фильтрат затем сильно подкислили концентрированной НСl и обрабатывали FeCl3 при кипячении с обратным холодильником в течение 45 мин. Реакционную смесь концентрировали при пониженном давлении и добавляли достаточное количество ацетата калия до тех пор, пока значение рН не было нейтральным. Кристаллизация продукта, происходящая при охлаждении, и фильтрование и перекристаллизация получаемого коричневого твердого продукта давали 7-этилакридин-4-карбоновую кислоту (0,77 г, 82%), т. пл. (ацетон) 210-211,5oС. 1H ЯМР [(СD3)2SO] 1,35 (т, J=7,5 Гц, 3 Н, СН3), 2,91 (к, J=7,5 Гц, 2 Н, СН2), 7,83 (дд, J=8,3, 7,2 Гц, 1 Н, Н-2), 7,97 (дд, J=9,0, 1,9 Гц, 1 Н, Н-6), 8,09 (шир. с, 1 Н, Н-8), 8,26 (д, J=9,0 Гц, 1 Н, Н-5), 8,54 (дд, J=8,4, 1,2 Гц, 1 Н, Н-1), 8,71 (шир. д, J= 7,0 Гц, 1 Н, Н-3), 9,43 (с, 1 Н, Н-9), 17,09 (шир. с, 1 Н, СООН). Элементный анализ (C16H13NO2) С, Н, N.
Вышеуказанной акридинкислоте (0,30 г, 1,2 ммоль) давали реагировать с CDI (карбонилдиимидазол) (0,38 г, 2,39 ммоль) в сухом ДМФ (50 мл) в течение 4 ч при 50oС. ДМФ удаляли при пониженном давлении, и полученное масло растворяли в смеси петролейного эфира и СН2Сl2 (20 мл, 3:1). При охлаждении имидазолид кристаллизовался, и этот сырой материал использовали в следующей реакции сочетания. Сочетание проводили растворением 3,3′-диамино-N-метилдипропиламина (0,09 г) в ТГФ (50 мл), охлаждением до 0oС (лед/вода) и добавлением по частям сырого имидазолида. Реакционную смесь оставляли при перемешивании в течение ночи при комнатной температуре, ТГФ удаляли, и остаток разбавляли CH2Cl2 (100 мл), который затем промывали 1 М Na2СО3 (50 мл). Слой СН2Сl2 сушили Na2SO4, растворитель удаляли при пониженном давлении, и полученное коричневое масло очищали хроматографией на оксиде алюминия (0,5% МеОН в СН2Сl2 в качестве элюента), получая бис[(7-этилакридин-4-карбоксамидо)пропил] метиламин (22) (0,17 г, 47%) в виде желтого масла. 1H ЯМР (СDСl3) 1,28 (т, J=7,5, 6 Н, 2СH3), 2,03-2,11 (м, 4 Н, 2CH2CH2CН2), 2,40 (с, 3 Н, NCН3), 2,70 (к, J= 7,6 Гц, 4 Н, 2СН2СН3), 2,79 (т, J=7,4 Гц, 4 Н, 2СН2NСН3), 3,74 (к, J=6,2 Гц, 4 Н, 2CH2NH), 7,46 (шир. с, 2 Н, Н-8), 7,48 (дд, J=8,9, 1,8 Гц, 2 Н, Н-6), 7,56 (дд, J=8,3, 7,2 Гц, 2 Н, Н-2), 7,89 (д, J=8,8 Гц, 2 Н, Н-5), 7,98 (дд, J=8,3, 1,5 Гц, 2 Н, Н-1), 8,51 (с, 2 Н, Н-9), 8,86 (дд, J= 7,1, 1,5 Гц, 2 Н, Н-3) и 11,80 (шир. т, J=5,0 Гц, 2 H, CONH). HRMS (масс-спектр высокого разрешения) (FAB+) (бомбардировка быстрыми атомами) m/z вычислено для С39Н42N5O2 612,3339 (МН+), найдено 612,3333.
Пример 2: Получение соединения 1 таблицы 1 способом по схеме 1.
Активирование и сочетание известной [Atwell et al., J. Med. Chem. 1987, 30, 664] 1-метилакридин-4-карбоновой кислоты давало бис[(1-метилакридин-4-карбоксамидо)пропил]метиламин (1) в виде желтого масла (91%). 1H ЯМР (CDCl3) 2,02-2,09 (м, 4 H, 2CH2CH2CH2), 2,38 (с, 3 H, NСН3), 2,75 (т, J=7,5 Гц, 4 H, 2СН2NСН3), 2,80 (д, J= 0,7 Гц, 6 H, 2СН3), 3,73 (к, J=6,1 Гц, 4 H, 2CH2NH), 7,36 (ддд, J=8,2, 6,7, 0,9 Гц, 2 H, H-6 или Н-7), 7,43 (дд, J=7,6, 0,8 Гц, 2 H, H-2), 7,66 (ддд, J=8,7, 6,7, 1,4 Гц, 2 Н, Н-7 или H-6), 7,80 (д, J=8,0 Гц, 2 H, Н-5 или Н-8), 7,99 (дд, J=8,7, 0,8 Гц, 2 H, H-8 или Н-5), 8,78 (с, 2 H, H-9), 8,80 (д, J=7,3 Гц, 2 H, Н-3) и 11,77 (шир. т, J=5,1 Гц, 2 H, 2NH). HRMS (FAB+) m/z вычислено для C37H38N5O2 584,3026 (МН+), найдено 584,3041. Элементный анализ (С37Н37N5О2 0,5 Н2O) С, H, N.
Пример 3: Получение соединения 2 таблицы 1 способом по схеме 1 1,15 (т, J=7,6, 3 Н, СН3), 2,51 (к, J=7,6 Гц, 2 Н, СН2), 6,84 (д, J=8,4 Гц, 2 Н, Н-2′ и Н-6′ или Н-3′ и Н-5′), 6,97 (т, J=7,7 Гц, 1 Н, Н-5), 7,04 (д, J=8,4 Гц, 2 Н, Н-3′ и Н-5′ или Н-2′ и Н-6′), 7,92 (д, J= 7,6 Гц, 2 Н, Н-4 и Н-6), 9,65 (шир. с, 1 Н, NH) и 12,90 (шир. с, 2 Н, 2СООН). Элементный анализ (C16H14NO4) С, Н, N.
Реакцию вышеуказанной дикислоты (1,43 г, 5 ммоль) с полифосфорной кислотой (38 г) проводили нагреванием смеси при 120oС в течение 2 час. Охлажденную смесь выливали в кипящую воду (250 мл), что приводило к образованию суспензии желтого осадка. Осадок удаляли фильтрованием и растворяли в смеси МеОН (100 мл) и 1 М NaOH (100 мл), которую нагревали и затем фильтровали горячей. Подкисление этой смеси с использованием ледяной уксусной кислоты давало желтый твердый продукт, который перекристаллизовывали, получая 7-этил-9-оксоакридан-4-карбоновую кислоту (1,14 г, 89%), т. пл. (Н2О) 301oС (разложение). 1Н ЯМР [(СD3)2SO] 1,26 (т, J=7,6 Гц, 3 Н, СН3), 2,74 (к, J= 7,6 Гц, 2 Н, СН2), 7,33 (т, J=7,7 Гц, 1 Н, Н-2), 7,64 (дд, J=8,5, 2,1 Гц, 1 Н, Н-6), 7,71 (д, J=8,5 Гц, 1 Н, Н-5), 8,04 (шир. с, 1 Н, Н-8), 8,42 (дд, J= 7,5, 1,7 Гц, 1Н, Н-3), 8,52 (дд, J=8,0, 1,7 Гц, 1 Н, Н-1), 11,98 (с, 1 Н, NH) и 13,85 (шир. с, 1 Н, СООН). Элементный анализ (С16Н13NО3) С, Н, N.
Суспензию вышеуказанной акридонкислоты (1,00 г, 3,75 ммоль) в 50% водном EtOH перемешивали и нагревали, затем добавляли достаточное количество триэтиламина с получением прозрачного желтого раствора. Порции алюминиевой фольги (0,83 г) амальгамировали в растворе HgCl2 (3 г) в EtOH, затем добавляли к вышеуказанному энергично кипящему раствору в течение 30 мин. После того, как ТСХ показала, что реакция завершалась, реакционную смесь фильтровали, и собранное твердое вещество промывали раствором КОН в водном EtOH. Фильтрат затем сильно подкислили концентрированной НСl и обрабатывали FeCl3 при кипячении с обратным холодильником в течение 45 мин. Реакционную смесь концентрировали при пониженном давлении и добавляли достаточное количество ацетата калия до тех пор, пока значение рН не было нейтральным. Кристаллизация продукта, происходящая при охлаждении, и фильтрование и перекристаллизация получаемого коричневого твердого продукта давали 7-этилакридин-4-карбоновую кислоту (0,77 г, 82%), т. пл. (ацетон) 210-211,5oС. 1H ЯМР [(СD3)2SO] 1,35 (т, J=7,5 Гц, 3 Н, СН3), 2,91 (к, J=7,5 Гц, 2 Н, СН2), 7,83 (дд, J=8,3, 7,2 Гц, 1 Н, Н-2), 7,97 (дд, J=9,0, 1,9 Гц, 1 Н, Н-6), 8,09 (шир. с, 1 Н, Н-8), 8,26 (д, J=9,0 Гц, 1 Н, Н-5), 8,54 (дд, J=8,4, 1,2 Гц, 1 Н, Н-1), 8,71 (шир. д, J= 7,0 Гц, 1 Н, Н-3), 9,43 (с, 1 Н, Н-9), 17,09 (шир. с, 1 Н, СООН). Элементный анализ (C16H13NO2) С, Н, N.
Вышеуказанной акридинкислоте (0,30 г, 1,2 ммоль) давали реагировать с CDI (карбонилдиимидазол) (0,38 г, 2,39 ммоль) в сухом ДМФ (50 мл) в течение 4 ч при 50oС. ДМФ удаляли при пониженном давлении, и полученное масло растворяли в смеси петролейного эфира и СН2Сl2 (20 мл, 3:1). При охлаждении имидазолид кристаллизовался, и этот сырой материал использовали в следующей реакции сочетания. Сочетание проводили растворением 3,3′-диамино-N-метилдипропиламина (0,09 г) в ТГФ (50 мл), охлаждением до 0oС (лед/вода) и добавлением по частям сырого имидазолида. Реакционную смесь оставляли при перемешивании в течение ночи при комнатной температуре, ТГФ удаляли, и остаток разбавляли CH2Cl2 (100 мл), который затем промывали 1 М Na2СО3 (50 мл). Слой СН2Сl2 сушили Na2SO4, растворитель удаляли при пониженном давлении, и полученное коричневое масло очищали хроматографией на оксиде алюминия (0,5% МеОН в СН2Сl2 в качестве элюента), получая бис[(7-этилакридин-4-карбоксамидо)пропил] метиламин (22) (0,17 г, 47%) в виде желтого масла. 1H ЯМР (СDСl3) 1,28 (т, J=7,5, 6 Н, 2СH3), 2,03-2,11 (м, 4 Н, 2CH2CH2CН2), 2,40 (с, 3 Н, NCН3), 2,70 (к, J= 7,6 Гц, 4 Н, 2СН2СН3), 2,79 (т, J=7,4 Гц, 4 Н, 2СН2NСН3), 3,74 (к, J=6,2 Гц, 4 Н, 2CH2NH), 7,46 (шир. с, 2 Н, Н-8), 7,48 (дд, J=8,9, 1,8 Гц, 2 Н, Н-6), 7,56 (дд, J=8,3, 7,2 Гц, 2 Н, Н-2), 7,89 (д, J=8,8 Гц, 2 Н, Н-5), 7,98 (дд, J=8,3, 1,5 Гц, 2 Н, Н-1), 8,51 (с, 2 Н, Н-9), 8,86 (дд, J= 7,1, 1,5 Гц, 2 Н, Н-3) и 11,80 (шир. т, J=5,0 Гц, 2 H, CONH). HRMS (масс-спектр высокого разрешения) (FAB+) (бомбардировка быстрыми атомами) m/z вычислено для С39Н42N5O2 612,3339 (МН+), найдено 612,3333.
Пример 2: Получение соединения 1 таблицы 1 способом по схеме 1.
Активирование и сочетание известной [Atwell et al., J. Med. Chem. 1987, 30, 664] 1-метилакридин-4-карбоновой кислоты давало бис[(1-метилакридин-4-карбоксамидо)пропил]метиламин (1) в виде желтого масла (91%). 1H ЯМР (CDCl3) 2,02-2,09 (м, 4 H, 2CH2CH2CH2), 2,38 (с, 3 H, NСН3), 2,75 (т, J=7,5 Гц, 4 H, 2СН2NСН3), 2,80 (д, J= 0,7 Гц, 6 H, 2СН3), 3,73 (к, J=6,1 Гц, 4 H, 2CH2NH), 7,36 (ддд, J=8,2, 6,7, 0,9 Гц, 2 H, H-6 или Н-7), 7,43 (дд, J=7,6, 0,8 Гц, 2 H, H-2), 7,66 (ддд, J=8,7, 6,7, 1,4 Гц, 2 Н, Н-7 или H-6), 7,80 (д, J=8,0 Гц, 2 H, Н-5 или Н-8), 7,99 (дд, J=8,7, 0,8 Гц, 2 H, H-8 или Н-5), 8,78 (с, 2 H, H-9), 8,80 (д, J=7,3 Гц, 2 H, Н-3) и 11,77 (шир. т, J=5,1 Гц, 2 H, 2NH). HRMS (FAB+) m/z вычислено для C37H38N5O2 584,3026 (МН+), найдено 584,3041. Элементный анализ (С37Н37N5О2 0,5 Н2O) С, H, N.
Пример 3: Получение соединения 2 таблицы 1 способом по схеме 1Активирование и сочетание известной [Atwell et al., J. Med. Chem, 1987, 30, 664] 1-хлоракридин-4-карбоновой кислоты давало бис[(1-хлоракридин-4-карбоксамидо)пропил]метиламин (2), (83%), т. пл. (СН2Сl2,/н-гексан) 94-96oС. 1Н ЯМР (CDCl3) 2,02-2,09 (м, 4 H, 2CH2CH2CH2), 2,39 (с, 3 H, NСН3), 2,79 (т, J=7,4 Гц, 4 Н, 2СН2NCН3), 3,72 (к, J=6,1 Гц, 4 Н, 2CH2NH), 7,31 (ддд, J= 8,2, 6,7, 0,8 Гц, 2 Н, Н-6 или Н-7), 7,63 (ддд, J=8,7, 6,7, 1,3 Гц, 2 Н, Н-7 или Н-6), 7,67 (д, J=7,9 Гц, 2 Н, Н-2), 7,74 (шир. д, J=8,0 Гц, 2 Н, Н-5 или Н-8), 7,91 (шир. д, J=8,7 Гц, 2 Н, Н-8 или Н-5), 8,75 (д, J=8,0 Гц, 2 Н, Н-3), 8,98 (с, 2 Н, Н-9), 11,45 (шир. т, J=4,8 Гц, 2 Н, 2CONH). HRMS (FAB+) m/z вычислено для С35Н32 35Cl2N5O2 624,1933 (МН+), найдено 624,1935. Элементный анализ (С35Н31Сl2N5О2) С, Н, N.
Пример 4: Получение соединения 3 таблицы 1 способом по схеме 1.
Активирование и сочетание известной [Atwell et al., J. Med. Chem. 1987, 30, 664] 2-метилакридин-4-карбоновой кислоты давало бис[(2-метилакридин-4-карбоксамидо)пропил]метиламин (3) в виде желтого масла (90%). 1Н ЯМР (СDСl3) 2,01-2,08 (м, 4 Н, 2СН2СН2СН2), 2,38 (с, 3 Н, NСН3), 2,60 (с, 3 Н, 2СН3), 2,73 (т, J= 7,4 Гц, 4 Н, 2СН2NСН3), 3,70-3,77 (м, 4 Н, 2 CH2NH), 7,38 (шир. т, J=7,8 Гц, 2 Н, Н-6 или Н-7), 7,64 (ддд, J=8,6, 6,9, 1,5 Гц, 2 Н, Н-7 или Н-6), 7,76 (шир, с, 2 Н, Н-1), 7,79 (д, J=8,3 Гц, 2Н, Н-5 или Н-8), 8,00 (д, J=8,2 Гц, 2 Н, Н-8 или Н-5), 8,55 (с, 2 Н, Н-9), 8,76 (д, J= 2,2 Гц, 2 Н, Н-3) и 11,79 (шир. т, J=5,0 Гц, 2 Н, 2CONH). HRMS (FAB+) m/z вычислено для C37H38N5O2 584,3026 (МН+), найдено 584,3031. Элементный анализ (C37H37N5O2) С, Н, N.
Пример 5: Получение соединения 4 таблицы 1 способом по схеме 1.
Активирование и сочетание известной [Atwell et al., J. Med. Chem. 1987, 30, 664] 2-хлоракридин-4-карбоновой кислоты давало бис[(2-хлоракридин-4-карбоксамидо) пропил] метиламин (4) в виде желтого твердого продукта (54%), т. пл, (СН2Сl2/н-гексан) 175,5-176,5oС. 1H ЯМР (СDСl3) 2,00-2,07 (м, 4 Н, 2CH2CH2CH2), 2,38 (с, 3 Н, NСН3), 2,75 (т, J=7,4 Гц, 4 Н, 2CH2NCH3), 3,71 (к, J= 6,2 Гц, 4 Н, 2CH2NH), 7,42 (ддд, J=8,3, 6,6, 0,9 Гц, 2 Н, Н-6 или Н-7), 7,68 (ддд, J=8,7, 6,6, 1,3 Гц, 2 Н, Н-7 или Н-6), 7,74 (шир. д, J=7,9 Гц, 2 Н, Н-5 или Н-8), 7,93-7,96 (м, 4Н, Н-1 и Н-8 или Н-5), 8,49 (с, 2 Н, Н-9), 8,74 (д, J=2,5 Гц, 2 Н, Н-3) и 11,52 (шир. т, J=5,0 Гц, 2NH). HRMS (FAB+) m/z вычислено для С35Н32 35Cl2N5O2 624,1933 (МН+), найдено 624,1900. Элементный анализ С35Н31Сl2N5O2) С, Н, N.
Пример 6: Получение соединения 5 таблицы 1 способом по схеме 1.
Активирование и сочетание известной [Atwell et al., J. Med. Chem. 1987, 30, 664] 3-метилакридин-4-карбоновой кислоты с последующей очисткой на оксиде алюминия и затем хроматографией на силикагеле давали бис[(3-метилакридин-4-карбоксамидо) пропил]метиламин (5) в виде бледно-желтой смолы (15%). 1Н ЯМР (СDСl3) 1,78-1,84 (м, 4 Н, 2CН2CH2CH2), 2,18 (с, 3 Н, NСН3), 2,48 (с, 6 Н, 2СН3), 2,67 (т, J=6,5 Гц, 4 Н, 2CH2NCH3), 3,56 (к, J=6,1 Гц, 4 Н, 2CH2NH), 7,19 (д, J=8,6 Гц, 2 Н, Н-1 или Н-2), 7,33 (ддд, J=8,4, 6,6, 1,0 Гц, 2 Н, Н-6 или Н-7), 7,39 (шир. т, J=5,3 Гц, 2Н, 2NH), 7,58 (ддд, J=8,8, 6,6, 1,4 Гц, 2 Н, Н-7 или Н-6), 7,66 (д, J=8,7 Гц, Н-2 или Н-1), 7,70 (шир. д, J=7,9 Гц, 2 Н, Н-5 или Н-8), 7,94 (д, J=8,6 Гц, 2 Н, Н-8 или Н-5) и 8,37 (с, 2 Н, Н-9). HRMS (FAB+) m/z вычислено для C37H38N5O2 584,3026 (МН+), найдено 584,3016.
Пример 7: Получение соединения 6 таблицы 1 способом по схеме I.
Активирование и сочетание известной [Atwell et al., J. Med. Chem. 1987, 30, 664] 5-метилакридин-4-карбоновой кислоты давало бис[(5-метилакридин-4-карбоксамидо)пропил]метиламин (6) (53%) в виде желтого масла. 1Н ЯМР (CDCl3) 1,61 (с, 6 Н, 2СН3), 1,97-2,00 (м, 4 Н, 2СН2СН2СН2), 2,30 (с, 3 Н, NСН3), 2,58 (т, J=7,5 Гц, 4 Н, 2СН2NСН3), 3,70 (к, J=6,5 Гц, 4 Н, 2CH2NH), 7,37 (дд, J=8,5, 6,8 Гц, 2 Н, Н-7), 7,57 (шир. д, J=6,7 Гц, 2 Н, Н-8), 7,61 (дд, J=8,3, 7,2 Гц, 2 Н, Н-2), 7,74 (шир. д, J=8,6 Гц, 2 Н, Н-6), 8,03 (дд, J= 8,4, 1,5 Гц, 2 Н, Н-1), 8,69 (с, 2 Н, Н-9), 8,94 (дд, J=7,1, 1,5 Гц, 2 Н, Н-3), 11,79 (шир. т, J=5,1 Гц, 2 Н, 2NH). HRMS (FAB+) m/z вычислено для C37H38N5O2 584,3026 (МН+), найдено 584,3044.
Пример 8: Получение соединения 7 таблицы 1 способом по схеме 1.
Аналогичное восстановление известной [Rewcastle and Denny, Synthesis, 1985, 217] 5-этил-9-оксоакридин-4-карбоновой кислоты, как указано выше, давало 5-этилакридин-4-карбоновую кислоту (79%), т. пл. (ацетон) 239-240,5oС. 1Н ЯМР [(СD3)2SO) 1,43 (т, J=7,5 Гц, 3 Н, СН3), 3,27-3,38 (м, 2 Н, СН2 – затемняется Н2О), 7,73 (шир. т, J=7,2 Гц, 1 Н, Н-2), 7,87 (шир. т, J= 7,8 Гц, 1Н, Н-7), 7,93 (шир, д, J=6,6 Гц, 1 Н, Н-1), 8,19 (шир. д, J=8,4 Гц, 1 Н, Н-6), 8,57 (шир. д, J=8,2 Гц, 1 Н, Н-8), 8,76 (шир. д, J=6,9 Гц, 1 Н, Н-3), 9,54 (с, 1 Н, Н-9) и 17,44 (шир. с, 1, COOH) Элементный анализ (C16H13NO2) С, Н, N.
Активирование и сочетание предыдущего соединения, как указано выше, давало бис[(5-этилакридин-4-карбоксамидо)пропил] метиламин (7) (57%) в виде желтого масла. 1Н ЯМР (CDCl3) 1,42 (т, J=7,5 Гц, 6 Н, 2СН3), 1,97-2,04 (м, 4 Н, 2CH2CH2CH2), 2,32 (с, 3 Н, NСН3), 2,61 (т, J=7,4 Гц, 4 Н, 2СН2NСН3), 3,24 (к, J= 7,5 Гц, 4 Н, 2СН2СН3), 3,69 (к, J=6,2 Гц, 4 Н, 2CH2NH), 7,40 (дд, J=8,4, 6,8 Гц, 2 Н, Н-7), 7,53 (дд, J=6,7 Гц, 1.0, 2 Н, Н-6), 7,60 (дд, J=8,3, 7,1 Гц, 2 Н, Н-2), 7,74 (дд, J=8,2, 1,0 Гц, 2 Н, Н-8), 8,02 (дд, J=8,4, 1,5 Гц, 2 Н, Н-1), 8,69 (с, 2 Н, Н-9), 8,93 (дд, J=7,1, 1,6 Гц, 2 Н, Н-3) и 11,77 (шир. т, J=5,5 Гц, 2CONH). HRMS (FAB+) m/z вычислено для C39H42N5O2 612,3339 (МН+), найдено 612,3343.
Пример 9: Получение соединения 8 таблицы 1 способом по схеме 1.
Аналогичная реакция 2-иодизофталевой кислоты и 2-изопропиланилина давала 2-[(2-изопропил)фениламино]изофталевую кислоту (38%), т. пл. (EtOAc/петролейный эфир) 217-219oС. 1Н ЯМР [(СD3)2SO] 1,25 (д, J=6,8 Гц, 6 Н, 2СН3), 3,22-3,29 (м, 1 Н, СН), 6,81 (дд, J=7,4, 1,8 Гц, 1 Н, Н-3′ или Н-6′), 6,93 (т, J=7,7 Гц, 1 Н, Н-2), 6,29-7,02 (м, 2 Н, Н-4′ и Н-5′), 7,26 (дд, J=7,1, 2,2 Гц, 1 Н, Н-6′ или Н-3′), 7,90 (д, J=7,7 Гц, 2 Н, Н-4 и Н-6), 9,69 (с, 1 Н, NH), 12,93 (шир. с, 2 Н, 2СООН). Анализ (C17H17NO4) С, Н, N.
Циклизация предыдущего соединения, как указано выше, давала 5-изопропил-9-оксоакридан-4-карбоновую кислоту (91%), т. пл. (Н2O) 304oС (разложение). 1Н ЯМР [(СD3)2SO) 1,42 (д, J=6,8 Гц, 6 Н, 2СН3), 3,29-3,41 (м, 1 Н, СН), 7,31-7,40 (м, 2 Н, Н-2 и Н-7), 7,74 (дд, J=7,4, 1,2 Гц, 1 Н, Н-6), 8,15 (дд, J= 8,1, 1,2 Гц, 1 Н, Н-8), 8,47 (дд, J=7,6, 1,6 Гц, 1 Н, Н-3), 8,53 (дд, J= 8,0, 1,6 Гц, 1 Н, Н-1), 12,48 (с, 1 Н, NH), 14,07 (шир. с, 1 Н, СООН). Элементный анализ (C17H15NO3 0,25 H2O) С, Н, N.
Восстановление предыдущего соединения, как указано выше, давало 5-изопропилакридин-4-карбоновую кислоту (70%), т. пл. (ацетон) 238oС (разложение). 1Н ЯМР [(СD3)2SO) 1,45 (д, J=6,8 Гц, 6 Н, 2СН3), 3,94-4,05 (м, 1 Н, СН), 7,75 (дд, J= 8,4, 7,1 Гц, 1 Н, Н-2 или Н-7), 7,86 (дд, J=8,4, 7,1 Гц, 1 Н, Н-7 или Н-2), 7,95 (шир. д, J=6,9 Гц, 1 Н, Н-6), 8,18 (дд, J=8,4, 1,0 Гц, 1 Н, Н-8), 8,55 (дд, J=8,4, 1,4 Гц, 1 Н, Н-1), 8,75 (дд, J=7,1, 1,4 Гц, 1 Н, Н-3), 9,52 (с, 1 Н, Н-9), 17,39 (шир. с, 1 Н, СООН). Элементный анализ (C17H15NO2) С, Н, N.
Активирование и циклизация предыдущего соединения, как указано выше, давало бис[(5-изопропилакридин-4-карбоксамидо) пропил]метиламин (8) (70%) в виде пены. 1Н ЯМР (CDCl3) 1,47 (д, J=7,0 Гц, 12 Н, 4СН3), 1,96-2,03 (м, 4 Н, 2СН2СН2СН2), 2,32 (с, 3 Н, NСН3), 2,59 (т, J=7,4 Гц, 4 Н, 2СН2NСН3), 3,70 (к, J= 7,1 Гц, 6,3 Гц, 4 Н, 2CH2NH), 4,15-4,18 (м, 2 Н, 2СН), 7,53 (дд, J= 8,4, 6,9 Гц, 2 Н, Н-7), 7,63 (дд, J=8,3, 7,2 Гц, 2 Н, Н-2), 7,67 (шир. д, J=6,6 Гц, 2 Н, Н-6), 7,83 (дд, J=8,4, 1,1 Гц, 2 Н, Н-8), 8,07 (дд, J= 8,3, 1,5 Гц, 2 Н, Н-1), 8,80 (с, 2 Н, Н-9), 8,95 (дд, J=7,1, 1,6 Гц, 2 Н, Н-3), 11,80 (шир. т, J=5,6 Гц, 2NH). HRMS (FAB+) m/z вычислено для C41H46N5O2 640,3652 (MH+), найдено 640,3657. Элементный анализ (C41H45N5O2) С, Н, N.
Пример 10: Получение соединения 9 таблицы 1 способом по схеме 1.
Активирование и сочетание известной [Atwell et al., J. Med. Chem. 1987, 30, 664] 5-фенилакридин-4-карбоновой кислоты давало бис[(5-фенилакридин-4-карбоксамидо)пропил]метиламин (9) (64%) в виде желтого масла. 1H ЯМР (CDCl3) 1,24-1,26 (м, 4 Н, 2СН2СН2СН2), 2,04 (с, 3 Н, NСН3), 2,06-2,10 (шир. т, J= 7,7 Гц, 4 Н, 2СН2NСН3), 3,13 (к, J=7,0, 6,7 Гц, 4 Н, 2СН2NH), 7,43-7,45 (м, 2 Н, Н-4′), 7,49-7,53 (м, 4 Н, Н-3′ и Н-5′), 7,59-7,67 (м, 8 Н, Н-2, Н-2′, Н6′, Н-7), 7,75 (дд, J=6,7, 1,4 Гц, 2 Н, Н-6), 7,99 (дд, J=8,5, 1,3 Гц, 2Н, Н-8), 8,09 (дд, J=8,4, 1,5 Гц, 2 Н, Н-1), 8,88 (с, 2 Н, Н-9), 8,94 (дд, J= 7,2, 1,5 Гц, 2 Н, Н-3), 11,06 (шир. т, J=6,0 Гц, 2NH). HRMS (FAB+) m/z вычислено для C47H42N5O2 708,3339 (МН+), найдено 708,3345.
Пример 11: Получение соединения 10 таблицы 1 способом по схеме 1.
Активирование и сочетание известной [Atwell et al., J. Med. Chem. 1987, 30, 664] 5-метоксиакридин-4-карбоновой кислоты давало бис[(5-метоксиакридин-4-карбоксамидо)пропил] метиламин (10) (71%) в виде желтого масла. 1H ЯМР (СDСl3) 1,99-2,06 (м, 4 Н, 2СН2СН2CH2), 2,38 (с, 3 Н, МСН3), 2,72 (т, J= 7,6 Гц, 4 Н, 2СН2NСН3), 3,65 (к, J=7,0, 5,2 Гц, 4 Н, 2СН2NH), 3,87 (с, 6 Н, 2ОСН3), 6,67 (дд, J=6,5, 2,0 Гц, 2 Н, Н-6), 7,10-7,16 (м, 4 Н, Н-7 и Н-8), 7,54 (дд, J=8,2, 7,2 Гц, 2 Н, Н-2), 7,86 (дд, J=8,4, 1,3 Гц, 2 Н, Н-1), 8,36 (с, 2 Н, Н-9), 8,82 (дд, J=7,1, 1,5 Гц, 2 Н, Н-3), 12,04 (шир. т, J= 4,6 Гц, 2 Н, 2CONH). HRMS (FAB+) m/z вычислено для С37Н38N5O4 616,2924 (МН+), найдено 616,2943.
Пример 12: Получение соединения 11 таблицы 1 способом по схеме 1.
Восстановление известной [Rewcastle and Denny, Synthesis, 1985, 217] 5-фтор-9-оксосиакридан-4-карбоновой кислоты, как описано выше, давало 5-фторакридин-4-карбоновую кислоту (90%), т.пл. (MeOH/H2O) 295-298oС (разложение) 1Н ЯМР [(СD3)2SO) 7,74-7,80 (м, 1 Н АrН), 7,90-7,96 (м, 2 Н, ArH), 8,19 (д, J=8,6 Гц, 1 Н, ArH), 8,61 (дд, J=8, 6, 1,2 Гц, 1Н, ArH), 8,81 (дд, J= 7,0, 1,0 Гц, 1Н, ArH), 9,65 (с, 1 Н, Н-9), Элементный анализ (C14H8FNO2) С, Н, N, F.
Активирование и сочетание предыдущего соединения, как описано выше, давало бис[(5-фторакридин-4-карбоксамидо) пропил] метиламин (11) (96%), т. пл. (соль НСl) 188oС (разложение). 1Н ЯМР (СDСl3) 2,06 (квинтет, J=7,2 Гц, 4 Н, 2СН2СН2СН2), 2,38 (с, 3 Н, СН3), 2,73 (т, J=7,6 Гц, 4 Н, 2СН2NСН3), 3,72 (к, J= 6,3 Гц, 4 Н, 2CH2NH), 7,26 (м, 4 Н, ArH), 7,56 (м, 2 Н, АrН), 7,59 (дд, J=8,4, 7,2 Гц, 2 Н, Н-2), 7,99 (дд, J=8,4, 1,4 Гц, 2 Н, Н-1), 8,64 (д, J= 0,6 Гц, 2 Н, Н-9), 8,93 (дд, J=7,1, 1,5 Гц, 2 Н, Н-3), 11,61 (т, J= 4,57 Гц, 2 Н CONH).
Пример 13: Получение соединения 12 таблицы 1 способом по схеме 1.
Активирование и сочетание известной [Atwell et al., J. Med, Chem. 1987, 30, 664] 5-хлоракридин-4-карбоновой кислоты давало бис[(5-хлоракридин-4-карбоксамидо)пропил] метиламин (12) (62%) в виде желтого масла. 1H ЯМР (CDCl3) 2,01-2,05 (м, 4 Н, 2СН2CH2СН2), 2,33 (с, 3 Н, NСН3), 2,64 (т, J= 7,5 Гц, 4 Н, 2СН2NСН3), 3,72 (к, J=6,4 Гц, 4 Н, 2СН2NH), 7,33 (дд, J=8,4, 7,3 Гц, 2 Н, Н-7), 7,58 (дд, J=8,2, 7,2 Гц, 2 Н, Н-2), 7,74-7,77 (м, 4 Н, Н-6 и Н-8), 7,96 (дд, J=8,4, 1,5 Гц, 2 Н, Н-1), 8,64 (с, 2 Н, Н-9), 8,91 (дд, J= 7,1, 1,5 Гц, 2 Н, Н-3), 11,74 (шир. т, J=5,3 Гц, 2 Н, 2NH). HRMS (FAB+) m/z вычислено для С35Н32 35Cl2N5O2 624,1933 (МН+), найдено 624,1940. Пример 14: Получение соединения 13 таблицы 1 способом по схеме 1.
Восстановление известной [Rewcastle and Denny, Syntnesis, 1985, 217] 5-бром-9-оксосиакридан-4-карбоновой кислоты, как описано выше, давало 5-бромакридин-4-карбоновую кислоту (70%), т. пл. (МеОН/Н2O) 327oС (разложение). 1H ЯМР [(СD3)2SO) 7,71 (дд, J=8,3, 7,4 Гц, 1 Н, Н-2), 7,94 (дд, J=8,4, 7,1 Гц, 1 Н, Н-7), 8,40 (дд, J=8,7, 0,8 Гц, 1 Н, ArH), 8,50 (дд, J= 7,3, 1,0 Гц, 1Н, ArH), 8,64 (дд, J=8,3, 1,3 Гц, 1 H, ArH), 8,85 (дд, J= 7,1, 1,3 Гц, 1 Н, ArH), 9,66 (с, 1 Н, Н-9), 16,77 (шир. с, 1 Н, СООН). Элементный анализ (C14H8BrNO2) С, Н, N, Вr.
Активирование и сочетание предыдущего соединения, как описано выше, давало бис[(5-бромакридин-4-карбоксамидо) пропил] метиламин (13) (80%), т. пл. (соль НСl) 212-214oС. 1H ЯМР (СDСl3) 2,05 (квинтет, J=7,3 Гц, 4 Н, 2СН2СН2СН2), 2,33 (с, 3 Н, СН3), 2,63 (т, J=7,6 Гц, 4 Н, 2СН2NСН3), 3,73 (к, J= 6,7 Гц, 4 Н, 2СН2NH), 7,28 (дд, J=8,2, 7,2 Гц, 2 Н, Н-2), 7,57 (дд, J= 8,4, 7,2 Гц, 2 H, Н-7), 7,81 (дд, J=8,5, 0,9 Гц, 2 Н, Н-1), 7,91 (м, 4 H, ArH), 8,64 (с, 2 Н, Н-9), 8,90 (дд, J=7,1, 1,5 Гц, 2 Н, Н-3), 11,72 (т, J= 5,6 Гц, 2 Н).
Пример 15: Получение соединения 15 таблицы 1 способом по схеме 1.
Активирование и сочетание известной [Atwell et al., J. Med. Chem. 1987, 30, 664] 6-метоксиакридин-4-карбоновой кислоты давало бис[(6-метоксиакридин-4-карбоксамидо) пропил]метиламин (15) (24%), т. пл. (соль НСl) 204-206oС (разложение). 1H ЯМР [(СD3)2SO] 2,20 (м, 4 Н, 2СН2CH2СН2), 2,85 (д, J= 4,65 Гц, 3 Н, СН3), 3,59-3,69 (м, 8 Н, 4СН2), 3,95 (с, 6 Н, 2ОСН3), 7,17 (д, J= 8,9 Гц, 1Н, ArH), 7,57 (шир. с, 1 Н, ArH), 7,64 (т, J=7,7 Гц, 1 Н, Н-2), 7,70 (д, J=0,8 Гц, 1 Н, Н-5), 7,97 (д, J=8,35 Гц, 1 Н, ArH), 8,25 (д, J= 8,35 Гц, 1 Н), 8,63 (д, J=6,55 Гц, 1 Н, ArH), 9,11 (с, 1 Н, Н-9), 10,74 (шир. с, 1 Н, NH), 11,21 (шир. с, 2 Н, 2CONH), 14,43 (шир. с, 1 Н, NH).
Пример 16: Получение соединения 16 таблицы 1 способом по схеме 1.
Восстановление 6-фтор-9-оксосиакридан-4-карбоновой кислоты, как описано выше, давало 6-фторакридин-4-карбоновую кислоту (91%), т. пл. (MeOH/H2O) 268-270oС. 1H ЯМР [(СD3)2SO] 7,76 (тд, J=8,9, 2,5 Гц, 1 Н, Н-7), 7,86 (тд, J= 8,9, 2,5 Гц, 1 Н, Н-2), 8,21 (дд, J=10,6, 2,4 Гц, 1 Н, Н-6), 8,45 (дд, J= 9,3, 6,4 Гц, 1 Н, Н-1), 8,58 (дд, J=8,4, 1,3 Гц, 1 Н, ArH), 8,77 (дд, J=7,1, 1,5 Гц, 1 H, ArH), 9,60 (с, 1 Н, Н-9), 16,67 (шир. с, 1 Н, СООН).
Активирование и сочетание предыдущего соединения, как описано выше, давало бис[(6-фторакридин-4-карбоксамидо) пропил] метиламин (16) (57%), т. пл. (соль НСl) 165,5oС (разложение). 1H ЯМР (CDCl3) 2,04 (квинтет, J=7,1 Гц, 4 Н, 2СН2СН2СН2), 2,39 (с, 3 Н, СН3), 2,72 (т, J=7,4 Гц, 6 Н, 2СН2NСН3), 3,45 (к, J=6,4 Гц, 4 H, 2СН2NH), 7,28 (ддд, J=9,2, 8,0, 2,4 Гц, 2 Н, Н-2), 7,62 (дд, J=8,35, 7,2 Гц, 2 Н, Н-7), 7,69 (дд, J=7,6, 2,4 Гц, 2 Н, Н-3), 7,89 (дд, J=9,2, 6,1 Гц, 2 Н, Н-8), 8,05 (дд, J=8,3, 1,5 Гц, 2 Н, Н-1), 8,74 (с, 2 Н, Н-9), 8,95 (дд, J=7,2, 1,5 Гц, 2 H, Н-5), 11,57 (т, J= 4,85 Гц, 2 Н, 2CONH). Элементный анализ (С35Н31F2N5O2 НСl 4H2O) С, Н, N.
Пример 17: Получение соединения 17 таблицы 1 способом по схеме 1.
Активирование и сочетание известной [Atwell et al., J. Med. Chem. 1987, 30, 664] 6-хлоракридин-4-карбоновой кислоты давало бис[(6-хлоракридин-4-карбоксамидо)пропил] метиламин (17) (76%), т. пл., (соль НСl) 216-218oС. 1H ЯМР [(СD3)2SO] 2,17 (квинтет, J=6,9 Гц, 4 Н, 2СН2CH2CH2), 2,87 (д, J=4,7 Гц, 3 Н, СН3), 3,30 (м, 4 Н, 2СН2), 3,63 (м, 4 Н, 2СН2), 7,47 (дд, J=8,9, 2,0 Гц, 1 Н, АrН), 7,69 (т, J=7,7 Гц, 1 Н, АrН), 8,06 (д, J=9,0 Гц, 1 Н, Н-8), 8,28 (дд, J=8,3, 1,3 Гц, 1 Н, АrН), 8,38 (д, J=1,7 Гц, 1 Н, Н-5), 8,64 (дд, J=7,1, 1,3 Гц, 1 Н, АrН), 9,16 (с, 1 Н, Н-9), 10,17 (шир. с, 1 Н, NH), 11,09 (т, J=5,7 Гц, 2 Н, 2NH), 11,44 (шир. с, 2 Н, 2CONH).
Пример 18: Получение соединения 21 таблицы 1 способом по схеме 1.
Активирование и сочетание известной [Atwell et al., J. Med. Chem. 1987, 30, 664] 7-метилакридин-4-карбоновой кислоты давало бис[(7-метилакридин-4-карбоксамидо)пропил] метиламин (21) (73%) в виде желтого масла. 1H ЯМР (CDCl3) 2,03-2,10 (м, 4 Н, 2СН2СН2СН2), 2,34 (с, 6 Н, 2СН3), 2,39 (с, 3 Н, NCH3), 2,80 (т, J= 7,6 Гц, 4 Н, 2СН2NCH3), 3,73 (к, J=6,1 Гц, 4 Н, 2СН2NH), 7,30 (шир. с, 2 Н, Н-8), 7,35 (дд, J=8,8, 1,9 Гц, 2 Н, Н-6), 7,55 (дд, J=8,4, 7,1 Гц, 2 Н, Н-2),7,77 (д, J=8,9 Гц, 2 Н, Н-5), 7,92 (дд, J=8,4, 1,5 Гц, 2 Н, Н-1), 8,36 (с, 2 Н, Н-9), 8,84 (дд, J=7,1, 1,5 Гц, 2 Н, Н-3), 11,74 (шир. т, J=5,0 Гц, 2 Н, 2 CONH). HRМS (FAB+) m/z вычислено для С37Н38N5O2 584,3026 (МН+), найдено 584,3043.
Пример 19: Получение соединения 23 таблицы 1 способом по схеме 1.
Аналогичная реакция 2-иодизофталевой кислоты и 4-изопропиланилина давала 2-[(4-изопропил)фениламино] изофталевую кислому (62%), т. пл. (EtOAc/петролейный эфир) 208oС (разложение). 1H ЯМР [(СD3)2SО] 1,16 (д, J= 6,9 Гц, 6 Н, 2СН3), 2,78-2,82 (м, 1 Н, СН), 6,83 (д, J=8,4 Гц, 2 Н, Н-2′ и Н-6′ или Н-3′ и Н-5′), 6,97 (т, J=7,7 Гц, 1Н, Н-5), 7,07 (д, J=8,5 Гц, 2 Н, Н-3′ и Н-5′ или Н-2′ и Н-6′), 7,92 (д, J=7,7 Гц, 2 Н, Н-4 и Н-6), 9,66 (шир. с, 1 Н, NH), 12,89 (шир. с, 2 Н, 2COOH). Элементный анализ (C17H17NO4) С, Н, N.
Циклизация предыдущего соединения, как указано выше, давала 7-изопропил-9-оксоакридан-4-карбоновую кислоту (95%), т. пл. (Н2О/МеОН/ТЭА/АсОН) 289-291oС. 1H ЯМР [(СD3)2SO] 1,28 (д, J=6,9 Гц, 6 Н, 2СН3), 3,03-3,07 (м, 1 Н, СН), 7,34 (т, J=7,7 Гц, 1 Н, Н-2), 7,70 (дд, J=8,6, 1,6 Гц, 1 Н, Н-6), 7,74 (д, J=8,5 Гц, 1 Н, Н-5), 8,07 (д, J=1, 6 Гц, Н-8), 8,43 (дд, J=7,5, 1,6 Гц, 1 Н, Н-3), 8,54 (дд, J=7,9, 1,6 Гц, 1 Н, Н-1), 11,93 (с, 1 Н, NH), 13,80 (шир. с, 1 Н, COOH). Элементный анализ (С17Н15NО3 0,5 Н2O) С, Н, N.
Восстановление предыдущего соединения, как указано выше, давало 7-изопропилакридин-4-карбоновую кислоту (51%), т. пл. (ацетон) 186-187oС. 1H ЯМР [(СD3)2SO] 1,37 (д, J=6,9 Гц, 6 Н, 2СН3), 3,15-3,25 (м, 1 Н, СН), 7,84 (дд, J= 8,3, 7,2 Гц, 1 Н, Н-2), 8,03 (дд, J=9,0, 1,8 Гц, 1 Н, Н-6), 8,11 (шир. с, 1 Н, Н-8), 8,27 (д, J=9,0 Гц 1 Н, Н-5), 8,54 (дд, J=8,5, 1,0 Гц, 1 Н, Н-1), 8,73 (дд, J=7,0, 1,2 Гц, 1 Н, Н-3), 9,45 (с, 1 Н, Н-9), 17,10 (шир. с, 1 Н, СООН). Элементный анализ (C17H15NO2) С, Н, N.
Активация и сочетание предыдущего соединения, как указано выше, давало бис[(7-изопропилакридин-4-карбоксамидо) пропил] метиламин (23) (73%) в виде желтого масла. 1H ЯМР (СDСl3) 1,33 (д, J=6,9 Гц, 12 Н, 4СН3), 2,04-2,08 (м, 4 Н, 2СН2CH2СН2), 2,38 (с, 3 Н, NСН3), 2,74 (т, J=7,4 Гц, 4 Н, 2СН2NСН3), 3,03-3,06 (м, 2 Н, 2СН), 3,74 (к, J=6,2 Гц, 4 Н, 2СН2NH), 7,58 (дд, J=8,3, 7,2 Гц, 2 Н, Н-2), 7,60-7,66 (м, 4 Н, Н-6 и Н-8), 8,01 (д, J=9,5 Гц, 2 Н, Н-5), 8,03 (дд, J=8,3, 1,5 Гц, 2 Н, Н-1), 8,66 (с, 2 Н, Н-9), 8,88 (дд, J= 7,2, 1,5 Гц, 2 Н, Н-3), 11,85 (шир. т, J=5,1 Гц, 2 Н, 2NH). HRMS (FAB+) m/z вычислено для С41Н46N5O2 640,3652 (МН+), найдено 640,3657.
Пример 20: Получение соединения 24 таблицы 1 способом по схеме 1.
Аналогичная реакция 2-иодизофталевой кислоты и 4-трет-бутиланилина давала 2-[(4-трет-бутил)фениламино]изофталевую кислоту (93%), т. пл. (EtOAc/петролейный эфир) 221-222oС. 1H ЯМР [(СD3)2SO] 1,24 (с, 9 Н, 3СН3), 6,84 (д, J=8,7 Гц, 2 Н, Н-2′ и Н-6′ или Н-3′ и Н-5′), 6,99 (т, J=7,7 Гц, 1 Н, Н-5), 7,21 (д, J=8,6 Гц, 2 Н, Н-3′ и Н-5′ или Н-2′ и Н-6′), 7,93 (д, J=7,8 Гц, 2 Н, Н-4 и Н-6), 9,65 (шир. с, 1 Н, NH) и 12,99 (шир. с, 2 Н, 2СООН). Элементный анализ (C18H13NO2) C, H, N.
Циклизация предыдущего соединения, как указано выше, давала 7-трет-бутил-9-оксоакридан-4-карбоновую кислоту (79%), т. пл. (H2O/MeOH) 326-327,5oС. 1H ЯМР [(СD3)2SO] 1,37 (с, 9 Н, 3СН3), 7,34 (т, J=7,8 Гц, 1 Н, Н-2), 7,74 (д, J=8,8 Гц, 1 Н, Н-5), 7,88 (дд, J=8,8, 2,3 Гц, 1 Н, Н-6), 8,19 (д, J=2,4 Гц, 1Н, Н-8), 8,43 (дд, J=7,6, 1,6 Гц, 1 Н, Н-3), 8,53 (дд, J=8,0, 1,6 Гц, 1 Н, Н-1), 11,96 (с, 1Н, NH) и 13,85 (шир. с, 1 Н, СООН). Элементный анализ (C18H17NO3) С, Н, N.
Восстановление предыдущего соединения, как указано выше, давало 7-трет-бутилакридин-4-карбоновую кислоту (62%), т. пл. (ацетон) 253-253,5oС. 1H ЯМР [(СD3)2SO] 1,46 (с, 9 Н, 3СН3), 7,83 (дд, J=8,4, 7,1 Гц, 1 Н, Н-2), 8,18 (д, J=1,7 Гц, 1 Н, Н-8), 8,22 (дд, J=9,2, 2,0 Гц, 1 Н, Н-6), 8,27 (д, J= 9,2 Гц, 1 Н, Н-5), 8,52 (дд, J=8,4, 1,2 Гц, 1 Н, Н-1), 8,72 (дд, J= 7,1, 1,2 Гц, 1 Н, Н-3), 9,46 (с, 1 Н, Н-9) и 17,11 (шир. с, 1Н, СООН). Элементный анализ (C18H17NO2) С, Н, N.
Активирование и циклизация предыдущего соединения, как указано выше, давало бис[(7-трет-бутилакридин-4-карбоксамидо) пропил]метиламин (24) (82%) а виде желтого масла. 1H ЯМР (CDCl3) 1,43 (с, 18 Н, 6СН3), 2,04-2,07 (м, 4 Н, 2СН2СН2СН2), 2,38 (с, 3 Н, NCH3), 2,72 (т, J=7,4 Гц, 4 Н, 2СН2NCH3), 3,74 (к, J= 6,8 Гц, 5,6 Гц, 4 Н, 2 СН2NH), 7,59 (дд, J=8,3, 7,2 Гц, 2 Н, Н-2), 7,81 (д, J=2,1 Гц, 2 Н, Н-8), 7,88 (дд, J=9,2, 2,1 Гц, 2 Н, Н-6), 8,05 (дд, J= 8,3, 1,4 Гц, 2 Н, Н-1), 8,07 (д, J=9,3 Гц, 2 Н, Н-5), 8,73 (с, 2 Н, Н-9), 8,89 (дд, J= 7,2, 1,5 Гц, 2 Н, Н-3) и 11,87 (шир. т, J=5,1 Гц, 2 Н, 2(CONH)). HRMS (FAB+) m/z вычислено для С43Н50N5O2 668,3965 (МН+), найдено 668,3963.
Пример 21: Получение соединения 25 таблицы 1 способом по схеме 1.
Восстановление известной [Rewcastle et al., J. Med. Chem., 1987, 30, 658] 7-фенил-9-оксоакридан-4-карбоновой кислоты, как описано выше, давало 7-фенилакридин-4-карбоновую кислоту (69%), т. пл. (ацетон) 239-241oС. 1H ЯМР [(СD3)2SO] 7,49 (т, J=7,3 Гц, 1 Н, Н-4′), 7,60 (т, J=7,3 Гц, 2 Н, Н-3′ и Н-5′), 7,86 (дд, J=8,4, 7,1 Гц, 1 Н, Н-2), 7,96 (д, J=7,3 Гц, 2 Н, Н-2′ и Н-6′), 8,43 (шир. с, 2 Н, Н-6 и Н-8), 8,58 (дд, J=8,5, 1,2 Гц, 1 Н, Н-1), 8,64 (шир. с, 1 Н, Н-5), 8,74 (шир. д, J=7,1 Гц, 1 Н, Н-3), 9,56 (с, 1 Н, Н-9), 16,93 (шир. с, 1 Н, СООН). Элементный анализ (C20H13NO2) С, Н, N.
Активирование и сочетание предыдущего соединения, как указано выше, давало бис[(7-фенилакридин-4-карбоксамидо) пропил] метиламин (25) (90%), т. пл. (СН2Cl2/MeOH) 162-163oС. 1H ЯМР (СDСl3) 2,07-2,14 (м, 4 Н, 2CH2CH2СН2), 2,42 (с, 3 Н, NCH3), 2,82 (т, J=7,4 Гц, 4 Н, 2СH2NСН3), 3,77 (к, J= 6,5, 5,6 Гц, 4 Н, 2СН2NH), 7,40-7,52 (м, 4 Н, Н-2 и Н-4′ или Н-3′ и Н-5′), 7,48-7,52 (м, 4Н, Н-3′ и Н-5′ или Н-2 и Н-4′), 7,63-7,65 (м, 4 Н, Н-2′ и Н-6′), 7,82-7,84 (м, 4 Н, Н-6 и Н-8), 7,87 (дд, J=8,4, 1,4 Гц, 2 Н, Н-1), 7,97 (д, J=9,5 Гц, 2 Н, Н-5), 8,54 (с, 2 Н, Н-9), 8,80 (дд, J=7,1, 1,5 Гц, 2 Н, Н-3), 11,69 (шир. т, J=5,2 Гц, 2 Н, 2NH). HRMS (FAB+) m/z вычислено для С47Н42N5O2 708,3339 (МН+), найдено 708,3351.
Пример 22: Получение соединения 26 таблицы 1 способом по схеме 1.
Активирование и сочетание известной [Atwell et al., J. Med. Chem., 1987, 30, 664] 7-метоксиакридин-4-карбоновой кислоты давало бис[(7-метоксиакридин-4-карбоксамидо) пропил]метиламин (26) (93%) в виде желтого масла. 1H ЯМР (СDСl3) 2,02-2,06 (м, 4 Н, 2СН2СН2СН2), 2,37 (с, 3 Н, NСН3), 2,74 (т, J= 7,4 Гц, 4 Н, 2СН2NСН3), 3,72 (к, J=6,7, 5,6 Гц, 4 Н, 2СН2NH), 3,88 (с, 6 Н, ОСН3), 6,89 (д, J=2,7 Гц, 2 Н, Н-8), 7,32 (дд, J=9,3, 2,7 Гц, 2 Н, Н-6), 7,54 (дд, J=8,2, 7,2 Гц, 2 Н, Н-2), 7,85 (д, J=9,3 Гц, 2 Н, Н-5), 7,94 (дд, J=8,4, 1,5 Гц, 2 Н, Н-1), 8,44 (с, 2 Н, Н-9)), 8,82 (дд, J=7,1, 1,5 Гц, 2 Н, Н-3), 11,69 (шир. т, J= 5,1 Гц, 2CONH). HRMS (FAB+) m/z вычислено для С37Н38N5O4 616,2924 (МН+), найдено 616,2927.
Пример 23: Получение соединения 27 таблицы 1 способом по схеме 1.
Восстановление известной [Atwell et al., J. Med. Chem. 1987, 30, 658] 7-фтор-9-оксосиакридан-4-карбоновой кислоты, как описано выше, давало 7-фторакридин-4-карбоновую кислоту (95%), т. пл. (MeOH/H2O) 267-268oС. 1H ЯМР [(СD3)2SO] 7,87 (дд, J=8,4, 7,0 Гц, 1 Н, Н-2), 8,01 (ддд, J=9,5, 8,5, 2,3 Гц, 1 Н, Н-6), 8,13 (дд, J=9,3, 2,8 Гц, 1 HJ, H-8), 8,45 (дд, J=9,6, 5,3 Гц, 1 Н, Н-5), 8,54 (дд, J=8,5, 1,3 Гц, 1 Н, Н-1), 8,73 (дд, J=6,9, 1,4 Гц, 1 Н, Н-3), 9,47 (c, 1 H, Н-9), 16,53 (шир. с, 1 Н, СООН). Элементный анализ (C14H8FNO2) С, H, N, F.
Активирование и сочетание предыдущего соединения, как указано выше, давало бис[(7-фторакридин-4-карбоксамидо]пропил]метиламин (27) (57%), т. пл. (соль НСl) 173,5oС (разложение). 1H ЯМР (СDСl3) 2,04 (квинтет, J=7,07 Гц, 4 Н, 2СН2СН2СН2), 2,73 (т, J=7,4 Гц, 4 Н, 2СН2NСН3), 3,73 (к, J=6,3 Гц, 4 Н, 2СН3NН), 7,32 (дд, J=8,6, 2,7 Гц, 2 Н, Н-8), 7,42 (ддд, J=9,4, 8,1, 2,7 Гц, 4 Н, Н-6), 7,63 (дд, J=7,7, 7,2 Гц, 2 Н, Н-2), 7,96 (дд, J=9,1, 4,9 Гц, 2 Н, Н-5), 7,99 (дд, J=7,7, 1,5 Гц, 2 Н, Н-1), 8,56 (с, 2 Н, Н-9), 8,89 (дд, J=7,0, 1,5 Гц, 2 Н, Н-3), 11,50 (т, J=4,95 Гц, 2 Н, 2CONH).
Пример 24: Получение соединения 28 таблицы 1 способом по схеме 1.
Активирование и сочетание известной [Atwell et al., J. Med. Chem. 1987, 30, 664] 7-хлоракридин-4-карбоновой кислоты давало бис[(7-хлоракридин-4-карбоксамидо)пропил] метиламин (28) (75%), в виде желтого масла. 1H ЯМР (СDСl3) 2,03-2,07 (м, 4 Н, 2СН2СН2СН2), 2,38 (с, 3 Н, NСН3), 2,75 (т, J= 7,4 Гц, 4 Н, 2СН2NCH3), 3,73 (к, J=6,1 Гц, 4 Н, 2CH2NH), 7,45 (дд, J=9,2, 2,3 Гц, 2 Н, Н-6), 7,63-7,67 (м, 4 Н, Н-2 и Н-8), 7,84 (д, J=9,2 Гц, 2 Н, Н-5), 7,99 (дд, J=8,4, 1,5 Гц, 2 Н, Н-1), 8,48 (с, 2 Н, Н-9), 8,93 (дд, J= 1,2, 1,5 Гц, 2 Н, Н-3), 11,42 (шир. т, J=5,0 Гц, 2CONH). HRMS (FAB+) m/z вычислено для С35Н32 35Cl2N5O2 624,1933 (МН+), найдено 624,1923.
Пример 25: Получение соединения 29 таблицы 1 способом по схеме 1.
Восстановление 7-бром-9-оксосиакридан-4-карбоновой кислоты, как описано выше, давало 7-бромакридин-4-карбоновую кислоту (59%), т. пл. (MeOH/H2O) 304oС (разложение). 1H ЯМР [(СD3)2SO] 7,87 (дд, J=8,4, 7,2 Гц, 1 Н, Н-2), 8,13 (дд, J=9,2, 2,2 Гц, 1 Н, Н-6), 8,32 (д, J-9,2 Гц, 1 Н, Н-5), 8,56 (дд, J=8,5, 1,3 Гц, 1 Н, Н-1), 8,66 (д, J=2,1 Гц, 1 Н, Н-8), 8,74 (дд, J=7,1, 1,4 Гц, 1 Н, Н-3), 9,48 (с, 1 Н, Н-9), 16,49 (с, 1 Н, СООН). Элементный анализ (С14Н8ВrNO2) С, Н, N, Вr.
Активирование и сочетание предыдущего соединения, как описано выше, давало бис[(7-бромакридин-4-карбоксамидо)пропил]метиламин (29) (25%), т, пл. (соль НСl) 138-142oС. 1H ЯМР (СDСl3) 2,04 (квинтет, J=7,0 Гц, 4 Н, 2СН2СН2СН2), 2,38 (с, 3 Н, СН3), 2,75 (т, J=7,4 Гц, 4 Н, 2СН2NСН3), 3,73 (к, J= 6,3 Гц, 4 Н, 2CH2NH), 7,55 (дд, J=9,1, 2,1 Гц, 2 Н, Н-6), 7,66 (дд, J=8,3, 7,2 Гц, 2 Н, Н-2), 7,76 (д, J=9,2 Гц, 2 Н, Н-5), 7,83 (д, J=2,1 Гц, 2 Н, Н-8), 7,99 (дд, J=8,2, 1,4 Гц, 2 Н, Н-1), 8,46 (с, 2 Н, Н-9), 8,94 (дд, 7,0, 1,5 Гц, 2 Н, Н-3), 11,41 (т, J=4,9 Гц, 2 Н, 2CONH).
Пример 26: Получение соединения 30 таблицы 1 способом по схеме 1.
Активирование и сочетание известной [Atwell et al., J. Med. Chem., 1987, 30, 664] 8-метил-9-оксоакридан-4-карбоновой кислоты давало бис[(8-метилакридин-4-карбоксамидо) пропил] метиламин (30) в виде желтого масла (61%). 1H ЯМР (CDCl3) 2,03-2,10 (м, 4 Н, 2СН2СН2СН2), 2,39 (с, 3 Н, NСН3), 2,66 (с, 6 Н, 2СН3), 2,79 (т, J=7,5 Гц, 4 Н, 2СН2NСН3), 3,74 (к, J=6,2 Гц, 4 Н, 2CH2NH), 7,09 (д, J=6,9 Гц, 2 Н, Н-5 или Н-7), 7,49 (дд, J=8,8, 6,8 Гц, 2 Н, Н-6), 7,62 (дд, J=8,4, 7,1 Гц, 2 Н, Н-2), 7,83 (д, J=8,7 Гц, 2 Н, Н-7 или Н-5), 8,04 (дд, J=8,3, 1,5 Гц, 2 Н, Н-1), 8,74 (с, 2 Н, Н-9), 8,91 (дд, J= 7,1, 1,5 Гц, 2 Н, Н-3) и 11,78 (шир. т, J=4,8 Гц, 2 Н, 2CONH); HRMS (FAB+) m/z вычислено для C37H38N5O2 584,3026 (MН+), найдено 584,3033. Элементный анализ (С37Н37N5O2 0,5 Н2O) С, Н, N.
Пример 27: Получение соединения 31 таблицы 1 способом по схеме 1.
Активирование и сочетание известной хлоракридин-4-карбоновой кислоты давало бис[(8-хлоракридин-4-карбоксамидо)пропил] метиламин (31) в виде желтого масла (88%). 1H ЯМР (СDСl3) 2,04-2,10 (м, 4 Н, 2СН2СН2СН2), 2,40 (с, 3 Н, NСН3), 2,84 (т, J=7,4 Гц, 4 Н, 2СH2NСН3), 3,74 (к, J=6,1 Гц, 4 Н, 2СН2NH), 7,10 (дд, J=7,3, 0,8 Гц, 2 Н, Н-5 или Н-7), 7,33 (дд, J=8,8, 7,3 Гц, 2 Н, Н-2 или Н-6), 7,65 (дд, J=8,3, 7,2 Гц, 2 Н, Н-6 или Н-2), 7,73 (д, J= 8,7 Гц, 2 Н, Н-7 или Н-5), 8,06 (дд, J=8,8, 1,5 Гц, 2 Н, Н-1), 8,86 (с, 2 Н, Н-9), 8,90 (дд, J=7,2, 1,5 Гц, 2 Н, Н-3) и 11,36 (шир. т, J=5,0 Гц, 2 Н, 2CONH); HRMS (FAB+) m/z вычислено для С35Н32 35Cl2N5O2 624/1933 (МN+), найдено 624,1939. Элементный анализ (C37H31Cl2N5O2) С, Н, N.
Пример 28: Получение соединения 37 таблицы 1 способом по схеме 1.
Активирование и сочетание акридин-2-карбоновой кислоты давало бис[(акридин-2-карбоксамидо)пропил]метиламин (37) (60%), т. пл. (СН2Сl2/петролейный эфир) 199-200oС. 1H ЯМР (СDСl3) 1,85-1,91 (м, 4 Н, 2СН2СH2СН2), 2,36 (с, 3 Н, NСН3), 2,59 (т, J=6,2 Гц, 4 Н, 2СH2NСН3), 3,65 (дд, J= 6,3, 5,9 Гц, 4 Н, 2СН2NH), 7,41 (ддд, J=8,4, 6,6, 1,0 Гц, 2 Н, Н-6 или Н-7), 7,64 (шир. т, J=5,3 Гц, 2 Н, 2NH), 7,70 (ддд, J=8,9, 6,6, 1,4 Гц, 2 Н, Н-7 или Н-6), 7,75 (д, J=8,1 Гц, 2 Н, Н-5 или Н-8), 8,06 (дд, J=9,1, 1,9 Гц, 2 Н, Н-3), 8,13 (дд, J=8,9, 0,8 Гц, 2 Н, Н-8 или Н-5), 8,20 (д, J= 9,1 Гц, 2 Н, Н-4), 8,36 (д, J=1,9 Гц, 2 Н, Н-1) и 8,52 (с, 2 Н, Н-9). HRMS (FAB+) m/z вычислено для С35Н34N5O2 556,2713 (МН+), найдено 556,2694. Элементный анализ (С35Н33N5O2) С, Н, N.
Пример 29: Получение соединения 33 таблицы 1 способом по схеме 2Раствор метил-2-[N-(2-карбоксифенил)амино]бензоата [Rewcastle and Denny, Synthesis, 1987, 37, 309] (10 г, 36,9 ммоль) в сухом ТГФ (200 мл) обрабатывали CDI (8,97 г, 55,4 ммоль). Реакционную смесь оставляли для перемешивания при комнатной температуре в течение 1 часа, затем медленно добавляли к суспензии NaBH4 (7,00 г) в Н2О (200 мл) без выделения промежуточного имидазолида. Когда определяли, что реакция завершена (ТСХ, приблизительно 30 мин), смесь гасили концентрированной НСl и распределяли между СН2Сl2 (200 мл) и NаНСО3 (200 мл) и органический слой сушили Na2SO4. Удаление растворителя и фильтрование остатка через слой силикагеля флэш-сорта в смеси петролейный эфир/EtOAc (4:1) давало метил-2-[N-(2-гидроксиметил)фениламино]бензоат (7,85 г, 83%), т. пл. (СН2Сl2/петролейный эфир) 69,0-71,0oС. 1H ЯМР (CDCl3) 1,93 (шир. с, 1 Н, ОН), 3,91 (с, 3 Н, СООСН3), 4,72 (с, 2 Н, СН2OН), 6,74 (ддд, J= 8,0, 7,0, 1,1 Гц, 1 Н, Н-5), 7,08-7,44 (м, 6 Н, Н-3,3′, 4,4′, 5′, 6′), 7,97 (дд, J=8,0, 1,6 Гц, 1 Н, Н-6), 9,59 (шир. с, 1 Н, NH). Элементный анализ (C15Н14NО3) С, Н, N.
Перемешиваемый раствор вышеуказанного спирта (7,74 г, 30 ммоль) в ацетоне (200 мл) обрабатывали суспензией МnО2 (10 г) при комнатной температуре в течение 3 дней до тех пор, пока не был израсходован весь исходный материал (ТСХ). МnО2 удаляли фильтрованием (целит), и ацетон удаляли при пониженном давлении, получая метил-2-[N-(2-формил)фениламино]бензоат в виде ярко-желтого твердого продукта (7,70 г, 100%). Образец, кристаллизованный из смеси (EtOAc/петролейный эфир), имел т. пл. 110,0-112,0oС. 1H ЯМР (СDСl3) 3,95 (с, 3 Н, СООСН3), 6,95-7,03 (м, 2 Н, Н-4′,5), 7,41-7,45 (м, 2 Н, Н-5′,6), 7,50 (шир. д, J= 8,5 Гц, 1 Н, Н-3 или Н-6′), 7,61 (шир. д, J=8,2 Гц, 1 Н, Н-6′ или Н-3), 7,65 (дд, J=7,7, 1,7 Гц, 1 Н, Н-3′), 8,01 (дд, J=7,9, 1,7 Гц, 1 Н, Н-6), 10,00 (с, 1 Н, СНО), 11,26 (шир. с, 1 Н, NH). Элементный анализ (С15Н13NО3) С, Н, N.
Вышеуказанный альдегид (210 мг, 0,8 ммоль) помещали в двухгорлую колбу, которую затем продували струей N2, добавляли трифторуксусную кислоту (10 мл), и раствор перемешивали в течение 15 часов при комнатной температуре в атмосфере N2. Трифторуксусную кислоту затем удаляли при пониженном давлении, и колбу, содержащую полученный сырой метилакридин-4-карбоксилат, продували струей азота#. Затем добавляли дегазированный 2 М раствор NaOH в водном EtOH (1: 1) (35 мл), и смесь перемешивали в течение 3 часов при 50oС в атмосфере N2 до получения прозрачного раствора, затем нейтрализовали ледяной АсОН и экстрагировали EtOAc (350 мл). Выпаривание органического слоя и хроматография остатка на силикагеле с элюированием смесью EtOAc/петролейный эфир (1: 4) давало акридин-4-карбоновую кислоту (160 мг, 87%), т. пл. (Ме2СО) 196-197oС [Atwell et al., J. Med. Chem. 1987, 30, 664, зарегистрирована т. пл. 202-204oС].
# Разбавление остатка в этой точке СН2Cl2 и осторожная нейтрализация раствора Et3N с последующим удалением растворителя при пониженном давлении и фильтрованием остатка через короткую колонку флэш-силикагеля в смеси EtOAc/петролейный эфир (1: 3) давали чистый метилакридин-4-карбоксилат в виде оранжевого масла. 1H ЯМР (CDCl3) 4,12 (с, 3 Н, СООСН3), 7,53-7,58 (м, 2 Н, Н-2 и Н-6 или Н-7), 7,79 (ддд, J=8,8, 6,6, 1,4 Гц, 1 Н, Н-7 или Н-6), 8,00 (дд, J= 8,0, 1,0 Гц, 1 Н, Н-1), 8,12-8,14 (м, 2 Н, Н-5,8), 8,30 (дд, J=8,7, 0,9 Гц, 1 Н, Н-3), 8,80 (с, 1 Н, Н-9).
Суспензию акридин-4-карбоновой кислоты (4,00 г, 17,9 ммоль) в ДМФ (25 мл) обрабатывали CDI (3,49 г, 21,5 ммоль) и перемешивали при 30oС в течение 2 часов. После охлаждения смесь разбавляли СН2Сl2 (25 мл), затем петролейным эфиром (75 мл) для полного осаждения продукта, который собирали, промывали смесью петролейный эфир/СН2Сl2 (4:1) и сушили, получая чувствительный к влаге имидазолид (3,81 г, 78%). Этот продукт подвергали реакции с N,N-бис- (3-аминопропил) метиламином по методике, описанной выше. Продукт очищали хроматографией на оксиде алюминия-90 с элюированием смесью СН2Cl2/MeOH (20: 1), получая бис[(акридин-4-карбоксамидо) пропил] метиламин (33) (83%) в виде пены. 1H ЯМР [(CD3)2SO] 9,06 (с, 2 Н, Н-9), 8,65 (д, J=7,1 Гц, 2 Н, Н-3), 8,24 (д, J=8,5 Гц, 2 Н, АrН), 8,00 (т, J=9,5 Гц, 4 Н, АrН), 7,8-7,6 (м, 4 Н, АrН), 7,45 (т, J=7,5 Гц, 2 Н, АrН), 3,58 (к, J=6,2 Гц, 4 Н, 2NHСН2), 2,65 (т, J=7,0 Гц, 4 Н, 2СН2NСН3), 2,29 (с, 3 Н, СН3), 1,91 (квинтет, J=6,9 Гц, 4 Н, 2СН2СН2СН2). Кристаллизация из MeOH/EtOAc/HCl давала тригидрохлоридную соль, т. пл. 168-170oС. Элементный анализ (C35H33N5O2 3HCl) С, Н, N, Сl.
Пример 30: Получение соединения 34 таблицы 1 способом по схеме 2.
Реакция имидазолида акридин-4-карбоновой кислоты с N,N-бис-(2-аминоэтил)амином, как указано выше, с последующей кристаллизацией сырого продукта из МеОН/Н2O давала бис [2-(акридин-4-карбоксамидо)этил]амин (34) (84%). 1H ЯМР [(CD3)2SO] 11,57 (т, J=5,0 Гц, 2 Н, 2CONH), 8,80 (с, 2 Н, Н-9), 8,46 (д, J=7,1 Гц, 2 Н, Н-3), 8,08 (д, J=8,4 Гц, 2 H, ArH), 7,92 (д, J=8,7 Гц, 2 H, ArH), 7,79 (д, J=8,1 Гц, 2 H, ArH), 7,55 (т, J=7,7 Гц, 2 H, ArH), 7,41 (т, J=7,6 Гц, 2 H, ArH), 7,27 (т, J=7,4 Гц, 2 H, ArH), 3,73 (к, J=5,5 Гц, 4 H, 2NHСН2), 3,11 (т, J= 5, 6 Гц, 4 H, СН2NHCH). Тригидрохлоридная соль кристаллизовалась из MeOH/EtOAc/HCl, т. пл. 182-184oС. Элементный анализ (С32Н27N5O2 3НСl) С, H, N, Cl.
Пример 31: Получение соединения 35 таблицы 1 способом по схеме 2.
Подобная реакция имидазолида акридин-4-карбоновой кислоты с N,N-бис-(3-аминопропил)амином, как указано выше, с последующей очисткой продукта хроматографией на оксиде алюминий-90 с элюированием СН2Cl2/MeOH (20:1) давала бис[3-(акридин-4-карбоксамидо)пропил] амин (35) (80%) в виде масла, 1H ЯМР [(СD3)2SО] 11,40 (т, J=5,4 Гц, 2 Н, 2CONH), 9,19 (с, 2 Н, Н-9), 8,71 (д, J= 7,1 Гц, 2 Н, Н-3), 8,32 (д, J=8,5 Гц, 2 Н, ArH), 8,18-8,00 (м, 4 Н, ArH), 7,83-7,62 (м, 4 Н, ArH), 7,51 (т, J=7,5 Гц, 2 Н, ArH), 3,64 (к, J=6,0 Гц, 4 Н, 2CONHСН2), 2,86 (т, J=6,7 Гц, 4 Н, СН2NHСН2), 1,92 (квинтет, J=6,5 Гц, 4 Н, 2CH2СН2СН2). Кристаллизация из MeOH/EtOAc/HCl давала тригидрохлоридную соль, т. пл. 171-173oС. Элементный анализ (С34Н31N5O2 2НСl 2Н2O) С, Н, N, Cl.
Пример 32: Получение соединения 36 таблицы 1 способом по схеме 2.
Аналогичная реакция имидазолида акридин-4-карбоновой кислоты с 1,4-бис-(3-аминопропил)пиперазином, как указано выше, и кристаллизация сырого продукта из СН2Cl2/EtOAc/изоo-Pr2O давала N’,N-4-бис[акридин-4-карбоксамидо)пропил] пиперазин (36) (91%). 1H ЯМР [(СD3)2SO] 11,39 (т, J=5,2 Гц, 2 Н, 2CONH), 9,33 (с, 2 Н, Н-9), 8,73 (д, J=7,0 Гц, 2 Н, Н-3), 8,38 (д, J=8,5 Гц, 2 Н, АrН), 8,32-8,20 (м, 4 Н, АrН), 7,97 (т, J=7,8 Гц, 2 Н, АrН), 7,82-7,63 (м, 4 Н, АrН), 3,57 (к, J=6,0 Гц, 4 Н, 2NHСН2), 2,6-2,3 (м, 12 Н, Н-пиперазин, 2СН2СН2СН2N), 1,85 (квинтет, J=6,7 Гц, 4 Н, СН2CH2CH2). Кристаллизация из MeOH/EtOAc/HCl давала тетрагидрохлоридную соль, т. пл. 248-253oС. Элементный анализ (C38H38N5O2 4HCl) С, Н, N, Cl.
Пример 33: Получение соединения 18 таблицы 1 способом по схеме 2.
Реакция метил-2-иодбензоата и 4-бромантраниловой кислоты по описанному способу [Rewcastle and Denny, Synth. Cornm, 1987, 17, 309] давала 4-бром-2-[(2-метоксикарбонилфенил) амино] бензойную кислоту (70%), т. пл. (МеОН/Н2О) 218-219,5oС. 1H ЯМР [(СD3)2SО] 3,85 (с, 3 Н, СООСН3), 7,08-7,12 (м, 2 Н, 2АrН), 7,50 (д, J= 1,9 Гц, 1Н, Н-3), 7,57 (д, J=3,8 Гц, 3 Н, АrН), 7,84 (д, J=8,4 Гц, 1 Н, АrН), 7,93 (д, J=7,7 Гц, 1 Н, АrН), 10,80 (с, 1 Н, NH), 13,33 (шир. с, 1 Н, СООН). Элементный анализ (C15H12BrNO4) С, Н, N.
Образование имидазолида и восстановление его, как указано выше, давали сырой метил-2-[N-(5-бром-2′-гидроксиметил) фениламино] бензоат (81%). 1H ЯМР [(СD3)2SО] 3,91 (с, 3 Н, СООСН3), 4,68 (д, J=4,8 Гц, 2 Н, СН2), 6,79-6,84 (м, 1 Н, АrН), 7,17-7,21 (м, 2 Н, 2АrН), 7,25 (д, J=8,5 Гц, 1 H, ArH), 7,40-7,43 (м, 2 H, ArH), 7,55 (д, J=1,8 Гц, 1 H, H-6′), 9,66 (с, 1 H, NH). Окисление его, как указано выше, давало метил-2-[N-(5′-бром-2-формил)фениламино] бензоат (67% по двум стадиям), т. пл. (МеОН/Н2O) 122-123oС. 1H ЯМР [(СD3)2SО] 3,95 (с, 3 H, СО2СН3), 7,05-7,11 (м, 2 H, ArH), 7,41-7,52 (м, 2 H, 2ArH), 7,58-7,62 (м, 2 H, 2ArH), 8,03 (дд, J=7,9, 1,6 Гц, 1 H, ArH), 9,93 (c, 1 H, CH), 11,33 (шир. с, 1 H, NH). Элементный анализ (С15Н12ВrNО3) С, H, N.
Циклизация соединения, полученного выше, давала сырой метил-6-бромакридин-4-карбоксилат, который сразу гидролизовали, как описано выше, получая 6-бромакридин-4-карбоновую кислоту (100% за две стадии), т. пл. (MeOH/H2O) 283-285oС. 1H ЯМР [(СD3)2SО] 7,87 (дд, J=8,3, 7,15 Гц, 1 H, H-2), 7,99 (дд, J= 9,0, 1,9 Гц, 1 H, H-7), 8,23 (д, J=9,1 Гц, 1 H, H-8), 8,56 (дд, J= 8,4, 1,4 Гц, 1 H, H-1), 8,70 (c, 1 H, H-5), 8,73 (дд, J=7,06, 1,4 Гц, Н-4), 9,57 (c, 1 H, H-9), 16,44 (шир. с, 1 H, COOH). Элементный анализ (C14H8BrNO2) C, H, N.
Активирование и сочетание соединения, полученного выше, давало бис-[(6-бромакридин-4-карбоксамидо)пропил] метиламин (18) (91%), т. пл. (соль НСl) 218-221oС. 1H ЯМР [(CD3)2SO] 2,07 (квинтет., J=7,0 Гц, 4 H, 2СН2CH2СН2), 2,41 (с, 3 H, СН3), 2,76 (т, J=7,4 Гц, 4 H, 2СН2NCН3), 3,75 (к, J=6,4 Гц, 4 H, 2СН2NH), 7,42 (дд, J=8,95, 1,8 Гц, 2 H, ArH), 7,65 (м, 4 H, ArH), 8,03 (дд, J=8,4, 1,5 Гц, 2 H, ArH), 8,25 (д, J=0,9 Гц, H-5), 8,67 (c, 2 H, H-9), 8,95 (дд, J=7,15, 1,5 Гц, 2 H, ArH), 11,45 (т, J=5,0 Гц, 2 H, CONH).
Пример 34: Получение соединения 14 таблицы 1 способом по схеме 2.
Подобная реакция 2-амино-3-трифторметилбензойной кислоты и метил-2-иодбензоата описанным способом [Rewcastle and Denny, Synth. Соmm, 1987, 17, 309] давала 3-трифторметил-2-[(2-метоксикарбонил) фенил)амино] бензойную кислоту (51%), т. пл. (MeOH/H2O) 113-115oС. 1H ЯМР [СD3)2SО] 3,89 (с, 3 H, СО2СН3), 6,35 (д, J=8,5 Гц, 1 H, ArH), 6,78 (т, J=7,5 Гц, 1 H, ArH), 7,30 (ддд, J=7,8, 7,8, 1,6 Гц, 1 H, ArH), 7,59 (т, J=7,8 Гц, 1 H, ArH), 7,88 (дд, J= 8,0, 1,5 Гц, 1 H, ArH), 8,03 (д, J=7,4 Гц, 1 H, ArH), 8,07 (д, J=8,07 Гц, 1 H, ArH), 9,49 (c, 1 H, NH), 13,15 (шир. с, 1 H, COOH). Элементный анализ (C16H12F3NO2) C, H, N, F.
Образование соответствующего имидазолида и немедленное восстановление его, как указано выше, давало сырой метил-2-[N-(2-гидроксиметил-6-трифторметил)фениламино]бензоат (100%) в виде масла. 1H ЯМР [(СD3)2SО] 3,94 (с, 3 H, СO2СН3), 4,50 (д, J=14,0 Гц, 1 H, CH), 4,72 (д, J=14,0 Гц, 1 H, CH), 6,18 (дд, J= 8,6, 0,7 Гц, 1 H, ArH), 6,72 (ддд, J=7,7, 7,5, 1,0 Гц, 1 H, ArH), 7,23 (ддд, J=8,5, 7,1, 1,5 Гц, 1 H, ArH), 7,46 (т, J=7,8 Гц, 1 H, ArH), 7,70 (д, J=7,1 Гц, 1 H, ArH), 7,83 (д, J=7,7 Гц, 1 H, ArH), 7,98 (дд, J=8,0, 1,6 Гц, 1 H, ArH), 9,25 (c, 1 H, NH).
Раствор вышеуказанного сырого эфира окисляли, как описано выше, получая метил-2-[N-(6-трифторметил-2-формил) фениламино] бензоат (100%), т. пл. (МеОН/Н2О) 122-123oС. 1H ЯМР (CDCl3) 3,96 (с, 3 Н, СO2СН3), 6,49 (дд, J= 8,3, 0,8 Гц, 1 Н, ArH), 6,79 (тд, J=7,5, 1,0 Гц, 1 Н, ArH), 7,25 (ддд, J= 8,3, 6,5, 1,6 Гц, 1 Н, ArH), 7,50 (т, J=7,8 Гц, 1 H, ArH), 7,9-8,01 (м, 2 Н, 2ArH), 8,14 (дд, J=7,8, 1,4 Гц, 1 H, ArH), 9,71 (шир. с, 1 Н, СНО), 10,09 (с, 1 Н, NH). Элементный анализ (С16Н12F3NО3) С, Н, N.
Циклизация этого соединения с последующим гидролизом, как описано выше, давала 5-трифторметилакридин-4-карбоновую кислоту (76%), т. пл. (МеОН/Н2O) 287-288,5oС. 1H ЯМР 7,89-7,98 (м, 2 Н, 2ArH), 8,55 (д, J=7,0 Гц, 1 Н, ArH), 8,65 (тд, J=8,7, 1,3 Гц, 2 Н, 2ArH), 8,86 (дд, J=6,9, 1,4 Гц, 1 H, ArH), 9,74 (с, 1 Н, Н-9), 16,13 (шир. с, 1 Н, СООН). Элементный анализ (C18H8F3NO4) С, Н, N.
Активация и сочетание соединения, полученного выше, давало бис[(5-трифторметилакридин-4-карбоксамидо)пропил] метиламин (14) (52%), т. пл. (EtOAc/MeOH) 231-233oС. 1H ЯМР [СD3)2SО] 1,81 (квинтет, J=7,1 Гц, 4 Н, СН2СН2CH2), 2,42 (с, 3 Н, NСН3), 2,44 (т, J=7,1 Гц, 4 Н, СН2NH3), 3,51 (к, J= 6,8 Гц, 4 H, NHСН2СН2) 7,73 (к, J=7,4 Гц, 4 Н, 4ArH), 8,24-8,29 (м, 4 Н, 4ArH), 8,42 (д, J=8,1 Гц, 2 H, ArH), 8,78 (дд, J=7,1, 1,5 Гц, 2 H, ArH), 9,30 (с, 2 Н, Н-9), 10,97 (т, J=5,8 Гц, 2 H, CONH). Элементный анализ (С37Н31F6N5O2 3НСl 2H2O) С, Н, N.
Пример 35: Получение соединения 19 таблицы 1 способом по схеме 2.
Реакция 4-трифторметилантраниловой кислоты и метил-2-иодбензоата описанным способом [Rewcastle and Denny, Synth. Comm, 1987, 17, 309] давала 4-трифторметил-2-(2-метоксикарбонилфениламино) бензойную кислоту (43%), т. пл. (МеОН/Н2О) 206-207oС. 1H ЯМР [(СD3)2SО] 3,87 (с, 3 Н, СО2СН3), 7,12 (ддд, J= 8,0, 6,1, 2,1 Гц, 1 Н, Н-5‘), 7,23 (дд, J=8,3, 1,0 Гц, 1 Н, АrН), 7,55-7,62 (м, 3 Н, 3ArH), 7,95 (дд, J=8,0, 1,3 Гц, 1 Н, ArH), 8,12 (д, J= 8,2 Гц, 1 Н, ArH). Образование соответствующего имидазолида и немедленное восстановление его, как указано выше, давало метил-2-[N-(5′-трифторметил-2′-гидроксиметил) фениламино]бензоат (86%), т. пл. (гексан) 86-87oС. 1H ЯМР (СDСl3) 2,00 (т, J=5,6 Гц, 1 Н, ОН), 3,92 (с, 3 Н, СО2Н3), 4,78 (д, J=5,3 Гц, 2 Н, СН2), 6,84 (тд, J=7,6, 1,1 Гц, 1 Н, ArH), 7,15 (дд, J=8,6, 0,8 Гц, 1 H, ArH), 7,31-7,39 [м, 2 Н, 2ArH), 7,52 (д, J=7,7 Гц, 1 Н, ArH), 7,70 (с, 1 Н, Н-6′), 8,76 (дд, J= 8,0, 1,6 Гц, 1 H, ArH), 9,72 (с, 1 H, NH).
Окисление предыдущего соединения, как указано выше, давало метил-2-[N-(5′-трифторметил-2′-формил)фениламино] бензоат (85%), т, пл. (MeOH/H2O) 79,5-80,5oC. 1H ЯМР [(СD3)2SО] 3,86 (с, 3 Н, СО2Н3), 7,20 (ддд, J=8,0, 6,2, 2,0 Гц, 1 Н, ArH), 7,34 (дд, J=7,5, 0,8 Гц, 1 H, ArH), 7,60-7,66 (м, 3 Н, 3ArH), 7,98 (дд, J=8,0, 1,4 Гц, 1 H, ArH), 8,09 (д, J=8,0 Гц, 1 H, ArH), 10,09 (c, 1H, NH), 11,16 (c, 1 Н, СНО).
Циклизация предыдущего соединения, как указано выше, с последующим немедленным гидролизом сырого метил-6-трифторметилакридин-4-карбоксилата давала 6-трифторметилакридин-4-карбоновую кислоту (81%), т. пл. (MeOH/H2O) 244-246oC. 1H ЯМР [(СD3)2SО] 7,93 (т, J=7,9 Гц, 1 Н, Н-3), 7,98 (дд, J= 8,9, 1,5 Гц, 1 Н, АrН), 8,56 (д, J=8,8 Гц, 1 Н, АrН), 8,60 (д, J=8,5 Гц, 1 Н, АгН), 8,79 (дд, J=7,0, 1,1 Гц, 1 Н, АrН), 8,86 (с, 1 Н, Н-5), 9,66 (с, 1 Н, Н-9).
Активирование и сочетание предыдущего соединения, как указано выше, давало бис[(6-трифторметилакридин -4-карбоксамидо) пропил] метиламин (19) (60%), т. пл. (гексан) 169-171oС. 1H ЯМР [(СD3)2SО] 1,89 (квинтет, J=6,6 Гц, 4 Н, СН2СН2СН2), 2,28 (с, 3 Н, NСН3), 2,66 (т, J=6,8 Гц, 4 Н, СН2СН3), 3,56 (к, J=6,1 Гц, 4 Н, NHСН2СН2), 7,60 (дд, J=8,8, 1,5 Гц, 2 Н, Н-7), 7,68 (дд, J= 8,3, 7,2 Гц, 2 Н, Н-2), 8,14 (д, J=8,8 Гц, 2 Н, Н-8), 8,23 (дд, J= 8,4, 1,4 Гц, 2 H, ArH), 8,38 (с, 2 Н, Н-5), 8,55 (дд, J=7,2, 1,5 Гц, 2 Н, АrН), 9,13 (с, 2 Н, Н-9), 10,78 (т, J=5,5 Гц, 2 H, CONH). Элементный анализ (С37Н31F6N5O2 0,5 Н2О) С, Н, N.
Пример 36: Получение соединения 32 таблицы 1 способом по схеме 2.
Реакция 2-амино-3,5-диметилбензойной кислоты и метилиодбензоата, как описано (Rewcastle and Denny, Synth. Comm., 1987, 17, 309), и очистка продукта на силикагеле с элюированием смесью EtOAc/петролейный эфир (1:4) давали 3,5-диметил-2-[(2-метоксикарбонил)фениламинобензойную кислоту (73%), т. пл. (EtOAc/петролейный эфир) 210-211,5oС. 1H ЯМР (СDСl3) 2,07 (с, 3 Н, СН3), 2,40 (с, 3 Н, СН3), 3,97 (с, 3 Н, СООСН3), 6,40 (дд, J=8,3, 0,9 Гц, 1 Н, Н-6′), 6,90-6,94 (м, 1 Н, Н-4′), 7,28-7,32 (м, 2 Н, Н-5′ и Н-4 или Н-6), 8,00 (д, J=1,8 Гц, 1 Н, Н-6 или Н-4), 8,03 (дд, J=8,0, 1,6 Гц, 1 Н, Н-3′), 9,45 (шир. с, 1 Н, NH).
Восстановление предыдущего соединения, как указано выше, через образование имидазолида давало метил-2-[N-(4,6-диметил-2-гидроксиметил)фениламино] бензоат (86%), т. пл. (EtOAc/петролейный эфир) 105-106oС. 1H ЯМР (СDСl3) 1,83 (шир. с, 1 Н, ОН), 2,13 (с, 3 Н, СН3), 2,36 (с, 3 Н, СН3), 3,92 (с, 3 Н, СООСН3), 4,51 (д, J= 12,8 Гц, 1 Н, СН2OH), 4,63 (д, J=12,8 Гц, 1 Н, СН2OН), 6,22 (д, J= 8,3 Гц, 1 Н, Н-3), 6,65 (шир. т, J=7,6 Гц, 1 Н, Н-5), 7,07 (шир. с, 1 Н, Н-3′ или Н-5′), 7,16 (шир. с, 1 Н, Н-5′ или Н-3′), 7,16-7,22 (м, 1 Н, Н-4), 7,95 (дд, J=8,0, 1,4 Гц, 1 Н, Н-6), 9,01 (шир. с, 1 Н, NH).
Окисление предыдущего соединения, как указано выше, давало метил-2-[N-(4,6-диметил-2-формил)фениламино] бензоат (95%), т. пл. (EtOAc/петролейный эфир) 103-104oС. 1H ЯМР (СDСl3) 2,19 (с, 3 Н, СН3), 2,40 (с, 3 Н, СН3), 3,95 (с, 3 Н, СООСН3), 6,31 (дд, J=8,3, 0,8 Гц, 1 Н, Н-3), 6,69-6,73 (м, 1 Н, Н-5), 7,20-7,24 (м, 1 Н, Н-4), 7,37 (д, J=1,6 Гц, 1 Н, Н-3′ или Н-5′), 7,60 (д, J=1,7 Гц, 1 Н, Н-5′ или Н-3′), 7,97 (дд, J=8,0, 1,6 Гц, 1 Н, Н-6), 9,42 (шир. с, 1 Н, NH), 10,14 (с, 1 Н, СНО).
Циклизация предыдущего соединения, как указано выше, давала сырой метил-5,7-диметилакридин-4-карбоксилат (99%). 1H ЯМР (СDСl3) 2,53 (с, 3 Н, СН3), 2,88 (с, 3 Н, СН3), 4,12 (с, 3 Н, СООСН3), 7,49 (шир, с, 1 Н, Н-6 или Н-8), 7,52 (дд, J=8,5, 7,0 Гц, 1 Н, Н-2), 7,57 (шир. с, 1 Н, Н-8 или Н-6), 8,03 (дд, J= 6,8, 1,4 Гц, 1 Н, Н-1 или Н-3), 8,05 (дд, J=8,5, 1,4 Гц, 1 Н, Н-3 или Н-1), 8,61 (с, 1 Н, Н-9). Гидролиз этого соединения, как указано выше, давал 5,7-диметилакридин-4-карбоновую кислоту (73%), т. пл. (МеОН/ТЭА/АсОН) 312-315oC. 1H ЯМР [(СD3)2SO/NaOD] 2,49 (с, частично затемненный ДМСО, 3 Н, СН3), 7,39-7,45 (м, 2 Н, Н-1 и Н-2), 7,49 (шир. с, 1 Н, Н-6 или Н-8), 7,67 (шир. с, 1 Н, Н-8 или Н-6), 7,85 (дд, J=7,7, 2,1 Гц, 1 Н, Н-3) и 8,76 (с, 1 Н, Н-9). Элементный анализ (C16H13NO2) С, Н, N.
Активирование и сочетание предыдущего соединения, как указано выше, давало бис[(5,7-диметилакридин-4-карбоксамидо) пропил] метиламин (32) в виде оранжевого масла (56%). 1H ЯМР (СDСl3) 1,94-2,05 (м, 4 Н, 2СН2СН2СН2), 2,31 (с, 3 Н, NСН3), 2,45 (с, 6 Н, 2СН3), 2,58 (т, J=7,4 Гц, 4 Н, 2СН2NСН3), 2,70 (с, 6 Н, 2СН3), 3,68 (дд, J=7,2, 5,7 Гц, 4 Н, 2СН2NH), 7,32 (шир. с, 2 Н, Н-6 или Н-8), 7,41 (шир. с, 2 Н, Н-8 или Н-6), 7,57 (дд, J= 8,3, 7,2 Гц, 2 Н, Н-2), 7,96 (дд, J=8,4, 1,4 Гц, 2 Н, Н-1), 8,49 (с, 2 Н, Н-9), 8,89 (дд, J= 7,2, 1,5 Гц, 2 Н, Н-3) и 11,75 (шир. т, J=5,3 Гц, 2 Н, 2CONH). HRMS (FAB+) m/z вычислено для C39H42N5O2 612,3339 (МН+), найдено 612,3330. Элементный анализ (C39H41N5O2 0,5 H2O) С, Н, N.
Пример 37: Получение соединения 20 таблицы 1.
Тригидрохлорид бис(6-фтор)соединения (16) (0,52 г, 0,7 ммоль) нагревали с 40% водным диметиламином (10 мл) в МеОН (10 мл) в автоклаве при 100oС в течение одной недели. Растворитель и избыток реагента выпаривали при пониженном давлении, добавляли аммиак и смесь экстрагировали СН2Cl2. Выпаривание и хроматография остатка на оксиде алюминия с элюированием градиентом МеОН в СН2Сl2 давали бис[(6-(диметиламино) акридин-4-карбоксамидо) пропил]метиламин (20) (84%), т. пл. (соль НСl из MeOH/EtOAc) 100oС (разложение), 1H ЯМР (свободное основание в CDCl3) 2,03 (квинтет, J=7,0 Гц, 4 Н, 2СН2СН2СН2), 2,37 (с, 3 Н, NСН3), 2,82 (т, J=7,6 Гц, 4 Н, 2СН2NСН3), 2,85 (с, 12 Н, 2N(СН3)2), 3,73 (к, J=6,1 Гц, 4 Н, 2СН2NH), 6,54 (д, J=2,2 Гц, 2 Н, Н-5), 6,67 (дд, J=9,2, 2,4 Гц, 2 Н, Н-7), 7,31 (д, J=9,2 Гц, 2 Н, Н-8), 7,40 (т, J=7,6 Гц, 2 Н, Н-2), 7,86 (дд, J=8,2, 1,6 Гц, 2 Н, Н-1), 8,21 (с, 2 Н, Н-9), 8,81 (дд, J=7,2, 1,6 Гц, 2 Н, Н-3), 12,15 (т, J=5,0 Гц, 2 Н, 2CONH).
Пример 38: Получение соединения 38 таблицы 1.
Суспензию феназин-1-карбоновой кислоты [Rewcastle and Denny, Synth. Comm, 1987, 17, 1171] (1,30 г, 5,8 ммоль) в ДМФ (8 мл) обрабатывал и 1,1′-карбонилдиимидазолом (1,13 г, 7,0 ммоль), и смесь перемешивали при 45oС в течение 30 мин. После охлаждения смесь разбавляли смесью СНСl2/петролейный эфир (1: 1) для полного осаждения сырого имидазолида, который собирали, промывали петролейным эфиром и сушили. К охлажденному льдом раствору N,N-бис-(3-аминопропил)-метиламина (0,35 г, 2,41 ммоль) в ТГФ (15 мл) добавляли сырой имидазолид (1,33 г, 4,85 ммоль), и смесь перемешивали при 20oС в течение 4 ч. Летучую часть удаляли при пониженном давлении, и остаток распределяли между СН2Cl2 и водным Nа2СО3. Органический слой промывали водой, сушили и выпаривали, и остаток хроматографировали на оксиде алюминия-90. Элюирование СН2Cl2/MeOH (20:1) и последующая кристаллизация из EtOAc/изо-Рr2О давали бис[(феназин-1-карбоксамидо)пропил]метиламин (38) (1,02 г, 63% от кислоты), т. пл. 153-154oС. 1H ЯМР [(СD3)2SO] 1,87 (квинтет, J=6,5 Гц, 4 Н, 2СН2СН2СН2), 2,27 (с, 3 Н, СН3), 2,62 (т, J=6,7 Гц, 4 Н, 2СH2NСН3), 3,53 (к, J= 5,7 Гц, 4 Н, 2СН2NH), 7,5-7,8 (м, 4 Н, АrН), 7,8-8,1 (м, 6 Н, АrН), 8,16 (д, J=8,6 Гц, 2 Н, АrН), 8,47 (д, J=6,9 Гц, 2 Н, АrН), 10,14 (т, J= 5,0 Гц, 2 Н, 2NH). Обработка смесью MeOH/EtOAc/HCl (1 эквив.) давала моногидрохлоридную соль, т. пл. (MeOH/EtOAc) 233-235oC. Элементный анализ (С33Н31N7O2 НCl 0,5 Н2О) С, Н, N, Cl.
Пример 39: Получение соединения 39 таблицы 1.
Активирование и сочетание феназин-2-карбоновой кислоты, как указано выше, давало бис[(феназин-2-карбоксамидо)пропил]метиламин (39) в виде желтого твердого продукта (88%), т. пл. 196-197,5oС. 1H ЯМР (СDСl3) 1,90-1,96 (м, 4 Н, 2СН2СН2СН2), 2,34 (с, 3 Н, NСН3), 2,64 (т, J=6,2 Гц, 4 Н, 2СН2NСН3), 3,71 (к, J=6,0 Гц, 2 Н, 2СН2NH), 7,66 (ддд, J=8,6, 6,6, 1,5 Гц, 2 Н, Н-7 или Н-8), 7,72 (ддд, J=8,7, 6,6, 1,5 Гц, 2 Н, Н-8 или Н-7), 7,99 (дд, J=8,7, 1,3 Гц, 2 Н, Н-6 или Н-9), 8,12 (дд, J=8,4, 1,3 Гц, 2 Н, Н-9 или Н-6), 8,16 (м, 4 Н, Н-4 и NH), 8,21 (дд, J=9,1, 1,9 Гц, 2 Н, Н-3) и 8,44 (д, J=1,6 Гц, 2 Н, Н-1). Элементный анализ (С33Н31N7О2) С, Н, N. HRMS (FAB+) m/z вычислено для С33Н32N7О2 558,2617 (МН+), найдено 558,2599.
Пример 40: Получение соединения 40 таблицы 1.
Активирование и сочетание известной [Rewcastle et al., J. Med. Chem., 1987, 30, 843] 6-метилфеназин-1-карбоновой кислоты, как указано выше, давало бис[(6-метилфеназин-1-карбоксамидо) пропил] метиламин (40) (47%), т. пл. (соль НСl) 228-230oC (MeOH/EtOAc). 1H ЯМР (СDСl3) 2,06 (квинтет, J=6,9 Гц, 4 Н, 2СН2СН2СН2), 2,39 (с, 3 Н, NСН3), 2,79 (с, 6 Н, 2АrСН3), 2,81 (к, J= 7,0 Гц, 4 Н, 2СН2NСН3), 3,75 (к, J=6,1 Гц, 4 Н, 2СН2NH), 7,42 (т, J=7,8 Гц, 2Н, Н-8), 7,61 (д, J=8,8 Гц, 2 Н, 2АrН), 7,87 (дд, J=8,5, 7,1 Гц, 4 Н, Н-3, 2АrН), 8,27 (дд, J=8,7, 1,5 Гц, 2 Н, Н-4), 8,88 (дд, J=7,0, 1,5 Гц, 2 Н, Н-2), 10,93 (шир. с, 2 Н, 2CONH).
Пример 41: Получение соединения 41 таблицы 1.
Активирование и сочетание известной [Rewcastle et al., J. Med. Chem., 1987, 30, 843] 6-хлорфеназин-1-карбоновой кислоты, как указано выше, давало бис[(6-хлорфеназин-1-карбоксамидо)пропил] метиламин (41) (56%), т. пл. (СН2Cl2/MeOH) 198-200oС. 1H ЯМР (СDСl3) 2,01-2,08 (м, 4Н, 2СН2СН2СН2), 2,37 (с, 3 Н, NСН3), 2,73 (т, J=7,2 Гц, 4 Н, 2СН2NСН3), 3,72 (к, J=6,2 Гц, 2 Н, 2СН2NH), 7,62 (дд, J=8,7, 7,2 Гц, 2 Н, Н-8), 7,74 (дд, J=7,2, 1,2 Гц, 2 Н, Н-7 или Н-9), 7,91 (дд, J=8,8, 1,2 Гц, 2 Н, Н-9 или Н-7), 7,93 (дд, J= 8,7, 7,1 Гц, 2 Н, Н-3), 8,39 (дд, J=8,7, 1,6 Гц, 2 Н, Н-4), 8,88 (дд, J=7,1, 1,6 Гц, 2 Н, Н-2), 10,59 (шир, т, J=5,1 Гц, 2 Н, 2CONH). HRMS (FAB+) m/z вычислено для С33Н29Сl2N7O4 626,1838 (МН+), найдено 618,1840. Элементный анализ (С33Н29Сl2N7O4) С, Н, N, Cl.
Пример 42: Получение соединения 42 таблицы 1.
Активирование и сочетание известной [Rewcastle et al., J. Med. Chem., 1987, 30, 843] 7-метилфеназин-1-карбоновой кислоты, как указано выше, давало бис[(7-метилфеназин-1-карбоксамидо)пропил]метиламин (42) (63%), т. пл. (соль НСl) 213-215oС (MeOH/EtOAc). 1H ЯМР (СDСl3) 2,06 (квинтет, J=6,9 Гц, 4 Н, 2СН2СН2СН2), 2,38 (с, 3 Н, NСН3), 2,44 (с, 6 Н, 2АrСН3), 2,79 (т, J=7,4 Гц, 4 Н, 2Сl2NСН3), 3,75 (к, J=6,2 Гц, 4 Н, 2СН2NH), 7,40 (дд, J=8,9, 1,6 Гц, 2 Н, Н-8), 7,62 (шир. с, 2 Н, Н-6), 7,77 (д, J=8,9 Гц, 2 Н, Н-9), 7,86 (дд, J=8,5, 7,1 Гц, 2 Н, Н-3), 8,22 (дд, J=8,6, 1,5 Гц, 2 Н, Н-4), 8,86 (дд, J= 7,2, 1,5 Гц, 2 Н, Н-2), 10,85 (т, J=4,9 Гц, 2 Н, 2CONH). Элементный анализ (С35Н35N7O2 HCl) С, Н, N, Cl.
Пример 43: Получение соединения 43 таблицы 1.
Активирование и сочетание известной [Rewcastle et al., J. Med. Chem., 1987, 30, 843] 7-метоксифеназин-1-карбоновой кислоты, как указано выше, давало бис[(7-метоксифеназин-1-карбоксамидо) пропил]метиламин (43) (60%), т. пл. (соль HCl) 225-229oС (MeOH/EtOAc). 1H ЯМР (СDСl3) 2,03 (квинтет, J=6,9 Гц, 4 Н, 2СН2СН2СН2), 2,37 (с, 3 Н, NСН3), 2,74 (т, J=7,2 Гц, 4 Н, 2СН2NСН3), 3,73 (к, J=6,3 Гц, 4 Н, 2СН2NH), 3,93 (с, 6 Н, 2АrОСН3), 7,10 (д, J= 2,7 Гц, 2 Н, Н-6), 7,28 (дд, J=9,1, 3,1 Гц, 2 Н, 2АrН), 7,78 (д, J= 9,5 Гц, 2 Н, Н-9), 7,83 (дд, J=8,7, 1,5 Гц, 2 Н, Н-8), 8,17 (дд, J=8,6, 1,5 Гц, 2 Н, 2ArH), 8,81 (J=7,2, 1,5 Гц, 2 Н, Н-2), 10,77 (т, J=4,6 Гц, 2 Н, 2CONH). Элементный анализ (С35Н35N7O4 2НСl 3Н2O) С, Н, N.
Пример 44: Получение соединения 44 таблицы 1.
Активирование и сочетание известной [Rewcastle et al., J. Med. Chem., 1987, 30, 843] 7-хлорфеназин-1-карбоновой кислоты, как указано выше, давало бис[(7-хлорфеназин-1 карбоксамидо)пропил] метиламин (44) (71%), т. пл. (СН2Cl2/MeOH) 173-175oС. 1H ЯМР (СDСl3) 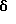 1,99-2,06 (м, 4 Н, СН2СН2СН2), 2,37 (с, 3 Н, NСН3), 2,73 (т, J=7,2 Гц, 4 Н, СН2NСН3), 3,73 (к, J=6,5, 5,8 Гц, 2 Н, СН2NH), 7,54 (дд, J=9,3, 2,4 Гц, 2 Н, Н-8), 7,84 (д, J=9,3 Гц, 2 Н, Н-9), 7,90 (д, J=2,5 Гц, 2 Н, Н-6), 7,92 (дд, J=8,7, 7,1 Гц, 2 Н, Н-3), 8,20 (дд, J= 8,7, 1,6 Гц, 2 Н, Н-4), 8,88 (дд, J=7,1, 1,5 Гц, 2 Н, Н-2), 10,54 (шир. т, J=5,1 Гц, 2 Н, 2CONH). HRMS (FAB+) m/z вычислено для С33Н29Сl2Н7O4 626,1838 (МН+), найдено 618,1844. Элементный анализ (C33H29C12N7O4) С, Н, N, Сl.
Пример 45: Получение соединения 45 таблицы 1.
Активирование и сочетание известной [Rewcastle and Denny, Synth. Comm., 1987, 17, 1171] 8-метилфеназин-1-карбоновой кислоты, как указано выше, давало бис[(8-метилфеназин-1-карбоксамидо) пропил]метиламин (45) (76%), т. пл. (соль НСl) 215oС (разложение). (MeOH/EtOAc). 1H ЯМР (СDСl3) 2,16 (квинтет, J= 6,6 Гц, 4 Н, 2СН2СН2СН2), 2,52 (с, 9 Н, NСН3, 2АrСН3), 2,93 (м, 4 Н, 2СН2NСН3), 3,76 (к, J=6,3 Гц, 4 Н, 2СН2NH), 7,41 (д, J=8,6 Гц, 2 Н, Н-6), 7,77 (шир. с, 2 Н, Н-9), 7,86 (дд, J=8,5, 7,1 Гц, 4 Н, Н-3,7), 8,26 (дд, J= 8,6, 1,5 Гц, 2 Н, Н-4), 8,87 (дд, J=7,7, 1,5 Гц, 2 Н, Н-2), 11,0 (шир. с, 2 Н, 2CONH).
Пример 46: Получение соединения 46 таблицы 1.
Активирование и сочетание известной [Rewcastle and Denny, Synth. Comm., 1987, 17, 1171] 8-метоксифеназин-1-карбоновой кислоты, как указано выше, давало бис[(8-метоксифеназин-1-карбоксамидо)пропил] метиламин (46) (99%), т. пл. 182-186oС (разложение). (MeOH/EtOAc). 1H ЯМР (СDСl3) 1,92 (м, 4 Н, 2СН2), 2,30 (с, 3 Н, NСН3), 2,71 (м, 4 Н, 2СН2), 3,60 (к, J=6,1 Гц, 4 Н, 2СН2NН), 3,85 (с, 6 Н, 2АrОСН3), 7,06 (с, 2 Н, Н-9), 7,19 (дд, J=9,4, 2,4 Гц, 2 Н, Н-7), 7,69 (д, J=9,4 Гц, 2 Н, Н-6), 7,80 (дд, J=8,6, 7,2 Гц, 2 Н, Н-3), 8,11 (дд, J=8,5, 1,4 Гц, 2 Н, Н-4), 8,48 (J=7,1, 1,5 Гц, 2 Н, Н-2), 10,39 (т, J=5,4 Гц, 2 Н, 2CONH). Элементный анализ (С35Н35N7O4) С, Н, N.
Пример 47: Получение соединения 47 таблицы 1.
Активирование и сочетание известной [Rewcastle et al., J. Med. Chem., 1987, 30, 843] 9-метилфеназин-1-карбоновой кислоты, как указано выше, давало бис[(9-метилфеназин-1-карбоксамидо) пропил] метиламин (47) (82%), т. пл. (соль НСl) 262-264oС (MeOH/EtOAc). 1H ЯМР (СDСl3) 1,99 (квинтет, J=7,3 Гц, 4 Н, 2СН2СН2СН2), 2,32 (с, 3 Н, NСН3), 2,60 (т, J=7,4 Гц, 4 Н, 2 СН2NСН3), 2,79 (с, 6 Н, 2АrСН3), 3,75 (к, J=6,7 Гц, 4 Н, 2СН2NH), 7,56 (д, J= 6,73 Гц, 2 Н, Н-8), 7,65 (дд, J=8,7, 7,2 Гц, 2 Н, Н-7), 7,89 (дд, J=8,7, 7,2 Гц, 2 Н, Н-3), 7,97 (д, J=8,6 Гц, 2 Н, Н-6), 8,27 (дд, J=8,7, 1,5 Гц, 2 Н, Н-4), 8,93 (дд, J=7,2, 1,5 Гц, 2 Н, Н-2), 10,94 (шир. с, 2 Н, 2CONH). Элементный анализ (С35Н35N7O2 НСl) С, Н, N, Cl.
Пример 48: Получение соединения 48 таблицы 1.
Активирование и сочетание 9-метилфеназин-1-карбоновой кислоты, как указывается выше, и последующее сочетание с 1,4-бис(аминопропил)пиперазином давало сырой продукт, который растворяли в МеОН/АсОН, обрабатывали системой уголь/целит и фильтровали, затем делали основным при помощи Et3N, получая бис[(9-метилфеназин-1-карбоксамидо)пропил] -1,4-пиперазин (48) (45%) в виде свободного основания, т. пл. (МеОН) 252-253oС. 1H ЯМР (гидрохлоридная соль в D2O) 2,07 (квинтет, J=7,5 Гц, 4 Н, 2СН2СН2СН2), 2,89 (с, 6 Н, 2СН3), 3,10 (т, J= 7,0 Гц, 6 Н, 3СН2), 3,29 (шир. с, 6 Н, 3СН2), 3,64 (т, J=6,7 Гц, 6 Н, 3СН2), 7,92-7,98 (м, 4 Н, 4ArH), 8,11 (дд, J=9,6, 7,2 Гц, 2 Н, 2ArH), 8,15 (д, J= 8,4 Гц, 2 Н, 2ArH), 8,45 (дд, J=8,7, 1,3 Гц, 2 Н, 2ArH), 8,69 (дд, J=7,1, 1,3 Гц, 2 Н, Н-2). Элементный анализ (С38Н40N8O2 0,5 H2O) С, Н, N.
Пример 49: Получение соединения 49 таблицы 1.
Активирование и сочетание 9-метилфеназин-1-карбоновой кислоты, как указывается выше, и последующее сочетание с этилентриамином давало сырой продукт, который растворяли в МеОН/АсОН, обрабатывали системой уголь/целит и фильтровали, затем делали основным при помощи EtsN, получая бис[(9-метилфеназин-1-карбоксамидо)этил] -1,4-этилендиамин (49) (33%), т. пл. (соль НСl из MeOH/EtOAc) 281oC (разложение). 1H ЯМР (гидрохлоридная соль в D2O) 2,89 (с, 6 Н, 2СН3), 3,38 (м, 8 Н, 4СН2), 3,90 (к, J=6,9 Гц, 4 Н, 2СН2), 7,90 (м, 4 Н, 4АrН), 8,07 (дд, J=8,6, 7,2 Гц, 2 Н, Н-3), 8,13 (д, J=8,2 Гц, 2 Н, 2АrН), 8,44 (дд, J=8,7, 1,4 Гц, 2 Н, 2АrН), 8,71 (дд, J=7,1, 1,4 Гц, 2 Н, Н-2). HRMS (FAB+) m/z вычислено для C34H34N8O2 586,61 (МН+), найдено 587,29.
Пример 50: Получение соединения 50 таблицы 1.
Активирование и сочетание известной [Rewcastle et al., J. Med. Chеmm., 1987, 30, 843] 9-метоксифеназин-1-карбоновой кислоты, как указано выше, давало бис[(9-метоксифеназин-1-карбоксамидо) пропил] метиламин (50) (86%), т. пл. (СН2Cl2/MeOH) 220-222oС. 1H ЯМР (CDCl3) 1,99-2,05 (м, 4 Н, 2CH2СН2CH2), 2,39 (с, 3 Н, NСН3), 2,73 (с, J=7,6 Гц, 4 Н, 2CH2NCH3), 3,66 (к, J=6,0 Гц, 2 Н, 2СН2NH), 3,90 (с, 6 Н, ОСН3), 6,60 (дд, J=6,7, 1,9 Гц, 2 Н, Н-6 или Н-8), 7,32-7,38 (м, 4 Н, Н-7 и Н-8 или Н-6), 7,84 (дд, J=8,7, 7,2 Гц, 2 Н, Н-3), 8,11 (дд, J=8,7, 1,5 Гц, 2 Н, Н-4), 8,83 (дд, J=7,1, 1,5 Гц, 2 Н, Н-2), 11,12 (шир. т, J=4,7 Гц, 2 Н, NH). HRMS (FAB+) m/z вычислено для С35Н35N7O4 618,2829 (МН+), найдено 618,2847. Элементный анализ (С35Н35N7O4) С, Н, N.
Пример 51: Получение соединения 51 таблицы 1.
Активирование и сочетание известной [Atwell et al., Eur. Pat. Appl. EP 172,744, Feb 1986; Chem. Abstr. 1986, 105, 97496р] 9-феноксифеназин-1-карбоновой кислоты, как указано выше, давало бис[(9-феноксифеназин-1-карбоксамидо)пропил]метиламин (51) в виде оранжевого масла (51%). 1H ЯМР (CDCl3) 1,69-1,73 (м, 4 Н, 2СН2СН2СН2), 1,97 (с, 3 Н, NСН3), 2,31 (т, J=7,3 Гц, 4 Н, 2СН2NСН3), 3,43 (к, J=6,4 Гц, 2 Н, 2СН2NH), 7,11-7,14 (м, 6 Н, Н-2′, Н-6′ и Н-6 или Н-8), 7,18 (т, J=7,5 Гц, 2 Н, Н-4′), 7,39 (т, J=7,5 Гц, 4 Н, Н-3′ и Н-5′), 7,69 (дд, J=8,7, 7,6 Гц, 2 Н, Н-7), 7,89 (дд, J=8,7, 1,0 Гц, 2 Н, Н-3, Н-8 или Н-6), 8,26 (дд, J=8,7, 1,5 Гц, 2 Н, Н-4), 8,90 (дд, J=7,1, 1,5 Гц, 2 Н, Н-2) и 10,98 (шир. т, J=5,2 Гц, 2 Н, 2CONH). HRMS (FAB+) m/z вычислено для C45H40N7O4 742,3142 (МН+), найдено 742,3147.
Пример 52: Получение соединения 52 таблицы 1.
Активирование и сочетание известной [Rewcastle et al., Synth. Соmm., 1987, 17, 1171] 9-фторфеназин-1-карбоновой кислоты, как указано выше, давало бис[(9-фторфеназин-1-карбоксамидо)пропил] метиламин (52) (87%), т. пл. (СН2Cl2/MeOH) 186-187oС. 1H ЯМР (СDСl3) 2,00-2,04 (м, 4 Н, 2СН2СН2СН2), 2,36 (с, 3 Н, NСН3), 2,72 (т, J=7,4 Гц, 4 Н, 2СН2NСН3), 3,73 (к, J=6,2 Гц, 2 Н, 2СН2NH), 7,30-7,35 (м, 2 Н, Н-7 или Н-8), 7,54-7,60 (м, 2 Н, Н-8 или Н-7), 7,84 (д, J=9,0 Гц, 2 Н, Н-6), 7,94 (дд, J=8,7, 7,0 Гц, 2 Н, Н-3), 8,25 (дд, J= 8,7, 1,5 Гц, 2 Н, Н-4), 8,95 (дд, J=7,0, 1,5 Гц, 2 Н, Н-2), 10,94 (шир. т, J= 5,0 Гц, 2 Н, 2CONH). HRMS (FAB+) вычислено для С33Н29F2N7O4 594,2429 (МН+), найдено 594,2403. Элементный анализ (С33Н29N7O4 0,5 H2O) С, Н, N.
Пример 53: Получение соединения 53 таблицы 1.
Активирование и сочетание известной [Rewcastle et al., J. Med. Chem., 1987, 30, 843] 9-хлорфеназин-1-карбоновой 1,99-2,06 (м, 4 Н, СН2СН2СН2), 2,37 (с, 3 Н, NСН3), 2,73 (т, J=7,2 Гц, 4 Н, СН2NСН3), 3,73 (к, J=6,5, 5,8 Гц, 2 Н, СН2NH), 7,54 (дд, J=9,3, 2,4 Гц, 2 Н, Н-8), 7,84 (д, J=9,3 Гц, 2 Н, Н-9), 7,90 (д, J=2,5 Гц, 2 Н, Н-6), 7,92 (дд, J=8,7, 7,1 Гц, 2 Н, Н-3), 8,20 (дд, J= 8,7, 1,6 Гц, 2 Н, Н-4), 8,88 (дд, J=7,1, 1,5 Гц, 2 Н, Н-2), 10,54 (шир. т, J=5,1 Гц, 2 Н, 2CONH). HRMS (FAB+) m/z вычислено для С33Н29Сl2Н7O4 626,1838 (МН+), найдено 618,1844. Элементный анализ (C33H29C12N7O4) С, Н, N, Сl.
Пример 45: Получение соединения 45 таблицы 1.
Активирование и сочетание известной [Rewcastle and Denny, Synth. Comm., 1987, 17, 1171] 8-метилфеназин-1-карбоновой кислоты, как указано выше, давало бис[(8-метилфеназин-1-карбоксамидо) пропил]метиламин (45) (76%), т. пл. (соль НСl) 215oС (разложение). (MeOH/EtOAc). 1H ЯМР (СDСl3) 2,16 (квинтет, J= 6,6 Гц, 4 Н, 2СН2СН2СН2), 2,52 (с, 9 Н, NСН3, 2АrСН3), 2,93 (м, 4 Н, 2СН2NСН3), 3,76 (к, J=6,3 Гц, 4 Н, 2СН2NH), 7,41 (д, J=8,6 Гц, 2 Н, Н-6), 7,77 (шир. с, 2 Н, Н-9), 7,86 (дд, J=8,5, 7,1 Гц, 4 Н, Н-3,7), 8,26 (дд, J= 8,6, 1,5 Гц, 2 Н, Н-4), 8,87 (дд, J=7,7, 1,5 Гц, 2 Н, Н-2), 11,0 (шир. с, 2 Н, 2CONH).
Пример 46: Получение соединения 46 таблицы 1.
Активирование и сочетание известной [Rewcastle and Denny, Synth. Comm., 1987, 17, 1171] 8-метоксифеназин-1-карбоновой кислоты, как указано выше, давало бис[(8-метоксифеназин-1-карбоксамидо)пропил] метиламин (46) (99%), т. пл. 182-186oС (разложение). (MeOH/EtOAc). 1H ЯМР (СDСl3) 1,92 (м, 4 Н, 2СН2), 2,30 (с, 3 Н, NСН3), 2,71 (м, 4 Н, 2СН2), 3,60 (к, J=6,1 Гц, 4 Н, 2СН2NН), 3,85 (с, 6 Н, 2АrОСН3), 7,06 (с, 2 Н, Н-9), 7,19 (дд, J=9,4, 2,4 Гц, 2 Н, Н-7), 7,69 (д, J=9,4 Гц, 2 Н, Н-6), 7,80 (дд, J=8,6, 7,2 Гц, 2 Н, Н-3), 8,11 (дд, J=8,5, 1,4 Гц, 2 Н, Н-4), 8,48 (J=7,1, 1,5 Гц, 2 Н, Н-2), 10,39 (т, J=5,4 Гц, 2 Н, 2CONH). Элементный анализ (С35Н35N7O4) С, Н, N.
Пример 47: Получение соединения 47 таблицы 1.
Активирование и сочетание известной [Rewcastle et al., J. Med. Chem., 1987, 30, 843] 9-метилфеназин-1-карбоновой кислоты, как указано выше, давало бис[(9-метилфеназин-1-карбоксамидо) пропил] метиламин (47) (82%), т. пл. (соль НСl) 262-264oС (MeOH/EtOAc). 1H ЯМР (СDСl3) 1,99 (квинтет, J=7,3 Гц, 4 Н, 2СН2СН2СН2), 2,32 (с, 3 Н, NСН3), 2,60 (т, J=7,4 Гц, 4 Н, 2 СН2NСН3), 2,79 (с, 6 Н, 2АrСН3), 3,75 (к, J=6,7 Гц, 4 Н, 2СН2NH), 7,56 (д, J= 6,73 Гц, 2 Н, Н-8), 7,65 (дд, J=8,7, 7,2 Гц, 2 Н, Н-7), 7,89 (дд, J=8,7, 7,2 Гц, 2 Н, Н-3), 7,97 (д, J=8,6 Гц, 2 Н, Н-6), 8,27 (дд, J=8,7, 1,5 Гц, 2 Н, Н-4), 8,93 (дд, J=7,2, 1,5 Гц, 2 Н, Н-2), 10,94 (шир. с, 2 Н, 2CONH). Элементный анализ (С35Н35N7O2 НСl) С, Н, N, Cl.
Пример 48: Получение соединения 48 таблицы 1.
Активирование и сочетание 9-метилфеназин-1-карбоновой кислоты, как указывается выше, и последующее сочетание с 1,4-бис(аминопропил)пиперазином давало сырой продукт, который растворяли в МеОН/АсОН, обрабатывали системой уголь/целит и фильтровали, затем делали основным при помощи Et3N, получая бис[(9-метилфеназин-1-карбоксамидо)пропил] -1,4-пиперазин (48) (45%) в виде свободного основания, т. пл. (МеОН) 252-253oС. 1H ЯМР (гидрохлоридная соль в D2O) 2,07 (квинтет, J=7,5 Гц, 4 Н, 2СН2СН2СН2), 2,89 (с, 6 Н, 2СН3), 3,10 (т, J= 7,0 Гц, 6 Н, 3СН2), 3,29 (шир. с, 6 Н, 3СН2), 3,64 (т, J=6,7 Гц, 6 Н, 3СН2), 7,92-7,98 (м, 4 Н, 4ArH), 8,11 (дд, J=9,6, 7,2 Гц, 2 Н, 2ArH), 8,15 (д, J= 8,4 Гц, 2 Н, 2ArH), 8,45 (дд, J=8,7, 1,3 Гц, 2 Н, 2ArH), 8,69 (дд, J=7,1, 1,3 Гц, 2 Н, Н-2). Элементный анализ (С38Н40N8O2 0,5 H2O) С, Н, N.
Пример 49: Получение соединения 49 таблицы 1.
Активирование и сочетание 9-метилфеназин-1-карбоновой кислоты, как указывается выше, и последующее сочетание с этилентриамином давало сырой продукт, который растворяли в МеОН/АсОН, обрабатывали системой уголь/целит и фильтровали, затем делали основным при помощи EtsN, получая бис[(9-метилфеназин-1-карбоксамидо)этил] -1,4-этилендиамин (49) (33%), т. пл. (соль НСl из MeOH/EtOAc) 281oC (разложение). 1H ЯМР (гидрохлоридная соль в D2O) 2,89 (с, 6 Н, 2СН3), 3,38 (м, 8 Н, 4СН2), 3,90 (к, J=6,9 Гц, 4 Н, 2СН2), 7,90 (м, 4 Н, 4АrН), 8,07 (дд, J=8,6, 7,2 Гц, 2 Н, Н-3), 8,13 (д, J=8,2 Гц, 2 Н, 2АrН), 8,44 (дд, J=8,7, 1,4 Гц, 2 Н, 2АrН), 8,71 (дд, J=7,1, 1,4 Гц, 2 Н, Н-2). HRMS (FAB+) m/z вычислено для C34H34N8O2 586,61 (МН+), найдено 587,29.
Пример 50: Получение соединения 50 таблицы 1.
Активирование и сочетание известной [Rewcastle et al., J. Med. Chеmm., 1987, 30, 843] 9-метоксифеназин-1-карбоновой кислоты, как указано выше, давало бис[(9-метоксифеназин-1-карбоксамидо) пропил] метиламин (50) (86%), т. пл. (СН2Cl2/MeOH) 220-222oС. 1H ЯМР (CDCl3) 1,99-2,05 (м, 4 Н, 2CH2СН2CH2), 2,39 (с, 3 Н, NСН3), 2,73 (с, J=7,6 Гц, 4 Н, 2CH2NCH3), 3,66 (к, J=6,0 Гц, 2 Н, 2СН2NH), 3,90 (с, 6 Н, ОСН3), 6,60 (дд, J=6,7, 1,9 Гц, 2 Н, Н-6 или Н-8), 7,32-7,38 (м, 4 Н, Н-7 и Н-8 или Н-6), 7,84 (дд, J=8,7, 7,2 Гц, 2 Н, Н-3), 8,11 (дд, J=8,7, 1,5 Гц, 2 Н, Н-4), 8,83 (дд, J=7,1, 1,5 Гц, 2 Н, Н-2), 11,12 (шир. т, J=4,7 Гц, 2 Н, NH). HRMS (FAB+) m/z вычислено для С35Н35N7O4 618,2829 (МН+), найдено 618,2847. Элементный анализ (С35Н35N7O4) С, Н, N.
Пример 51: Получение соединения 51 таблицы 1.
Активирование и сочетание известной [Atwell et al., Eur. Pat. Appl. EP 172,744, Feb 1986; Chem. Abstr. 1986, 105, 97496р] 9-феноксифеназин-1-карбоновой кислоты, как указано выше, давало бис[(9-феноксифеназин-1-карбоксамидо)пропил]метиламин (51) в виде оранжевого масла (51%). 1H ЯМР (CDCl3) 1,69-1,73 (м, 4 Н, 2СН2СН2СН2), 1,97 (с, 3 Н, NСН3), 2,31 (т, J=7,3 Гц, 4 Н, 2СН2NСН3), 3,43 (к, J=6,4 Гц, 2 Н, 2СН2NH), 7,11-7,14 (м, 6 Н, Н-2′, Н-6′ и Н-6 или Н-8), 7,18 (т, J=7,5 Гц, 2 Н, Н-4′), 7,39 (т, J=7,5 Гц, 4 Н, Н-3′ и Н-5′), 7,69 (дд, J=8,7, 7,6 Гц, 2 Н, Н-7), 7,89 (дд, J=8,7, 1,0 Гц, 2 Н, Н-3, Н-8 или Н-6), 8,26 (дд, J=8,7, 1,5 Гц, 2 Н, Н-4), 8,90 (дд, J=7,1, 1,5 Гц, 2 Н, Н-2) и 10,98 (шир. т, J=5,2 Гц, 2 Н, 2CONH). HRMS (FAB+) m/z вычислено для C45H40N7O4 742,3142 (МН+), найдено 742,3147.
Пример 52: Получение соединения 52 таблицы 1.
Активирование и сочетание известной [Rewcastle et al., Synth. Соmm., 1987, 17, 1171] 9-фторфеназин-1-карбоновой кислоты, как указано выше, давало бис[(9-фторфеназин-1-карбоксамидо)пропил] метиламин (52) (87%), т. пл. (СН2Cl2/MeOH) 186-187oС. 1H ЯМР (СDСl3) 2,00-2,04 (м, 4 Н, 2СН2СН2СН2), 2,36 (с, 3 Н, NСН3), 2,72 (т, J=7,4 Гц, 4 Н, 2СН2NСН3), 3,73 (к, J=6,2 Гц, 2 Н, 2СН2NH), 7,30-7,35 (м, 2 Н, Н-7 или Н-8), 7,54-7,60 (м, 2 Н, Н-8 или Н-7), 7,84 (д, J=9,0 Гц, 2 Н, Н-6), 7,94 (дд, J=8,7, 7,0 Гц, 2 Н, Н-3), 8,25 (дд, J= 8,7, 1,5 Гц, 2 Н, Н-4), 8,95 (дд, J=7,0, 1,5 Гц, 2 Н, Н-2), 10,94 (шир. т, J= 5,0 Гц, 2 Н, 2CONH). HRMS (FAB+) вычислено для С33Н29F2N7O4 594,2429 (МН+), найдено 594,2403. Элементный анализ (С33Н29N7O4 0,5 H2O) С, Н, N.
Пример 53: Получение соединения 53 таблицы 1.
Активирование и сочетание известной [Rewcastle et al., J. Med. Chem., 1987, 30, 843] 9-хлорфеназин-1-карбоновойкислоты, как указано выше, давало бис[(9-хлорфеназин-1-карбоксамидо) пропил]метиламин (53) (86%), т.пл. (СН2Cl2/MeOH) 169-171,5oС. 1H ЯМР (CDCl3) 1,99-2,03 (м, 4 Н, 2СН2СН2CH2), 2,32 (с, 3 Н, NСН3), 2,62 (т, J=7,4 Гц, 4 Н, 2СН2NСН3), 3,70 (к, J=6,2 Гц, 2 Н, 2СН2NH), 7,64 (дд, J=8,8, 7,4 Гц, 2 Н, Н-7), 7,80 (дд, J=7,2, 1,0 Гц, 2 Н, Н-6 или Н-8), 7,95 (дд, J=8,7, 7,2 Гц, 2 Н, Н-3), 8,01 (дд, J=8,7, 1,0 Гц, 2 Н, Н-8 или Н-6), 8,27 (дд, J=8,7, 1,5 Гц, 2 Н, Н-4), 8,99 (дд, J=7,2, 1,5 Гц, 2 Н, Н-2), 10,94 (шир. т, J=5,0 Гц, 2 Н, 2CONH). HRMS (FAB+) вычислено для С33Н29Сl2N7O4 626,1838 (МН+), найдено 618,1848. Элементный анализ С33Н29Сl2N7O4 С, Н, N.
Пример 54: Получение соединения 54 таблицы 1.
Раствор 9-фторфеназин-1-карбоновой кислоты [Rewcastle et al., J. Synth Comm, 1987, 1730, 1171] (200 мг, 0/8 ммоль) в Me2NH (40% водный, 20 мл) нагревали при 100oС в автоклаве в течение 3 ч. Получаемый интенсивно темно-красный раствор разбавляли водой и затем нейтрализовали АсОН. Водный раствор затем экстрагировали СНСl3 (350 мл) до тех пор, пока не была удалена вся окраска раствора. Органический слой далее промывали водой (1150 мл), затем сушили над Na2SO4, и растворитель удаляли при пониженном давлении. Получаемый темно-красный твердый продукт растворяли в минимальном количестве СН2Cl2, и петролейный эфир добавляли до тех пор, пока не происходила кристаллизация, получая 9-(диметиламино)феназин-1-карбоновую кислоту в виде темно-красных игл (210 г, 95%), т. пл. 186-187,5oС. 1H ЯМР (СDСl3) 3,16 [с, 6 Н, N(СН3)2], 7,26 (дд, J=6,8, 1,8 Гц, 1 Н, Н-6 или Н 8), 7,81-7,88 (м, 2 Н, Н-7 и Н-8 или Н-6), 8,01 (дд, J=8,7, 7,0 Гц, 1 Н, Н-3), 8,48 (дд, J= 8,7, 1,2 Гц, 1 Н, Н-4) и 8,91 (дд, J=7,0, 1,3 Гц, 1 Н, Н-2). Элементный анализ (С15Н13N3О3) С, Н, N.
Активирование и сочетание предыдущего соединения, как указано выше, давало бис[((9-диметиламино) феназин-1-карбоксамидо) пропил]метиламин (54) в виде темно-красного масла (78%). 1H ЯМР (СDСl3) 1,91-2,00 (м, 4 Н, 2CH2СН2СН2), 2,29 (с, 3 Н, NСН3), 2,57 (т, J=7,3 Гц, 4 Н, СН2NСН3), 3,05 (с, 12 Н, 2N(СН3)2, 3,68 (к, J=6,5 Гц, 2 Н, СН2NH), 7,07 (дд, J=7,2, 1,3 Гц, 2 Н, Н-6 или Н-8), 7,65 (дд, J=8,7, 7,3 Гц, 2 Н, Н-7), 7,70 (дд, J=8,7, 1,3 Гц, 2 Н, Н-8 или Н-6), 7,90 (дд, J=8,6, 7,1 Гц, 2 Н, Н-3), 8,27 (дд, J= 8,6, 1,4 Гц, 2 Н, Н-4), 8,87 (дд, J=7,1, 1,4 Гц, 2 Н, Н-2) и 10,99 (шир. т, J= 5,1 Гц, 2 Н, 2CONH); HRMS (FAB+) m/z вычислено для С37Н42O2 644,3461 (МН+), найдено 644,3485.
Пример 55: Получение соединения 55 таблицы 1.
Бис(5-фтор)аналог (11) нагревали при 100oС в избытке смеси 40% водный диметиламин/МеОН в течение 8 недель в аппарате высокого давления, растворитель затем удаляли выпариванием и остаток хроматографировали на оксиде алюминия, получая бис-[3-(5-(диметиламино)-акридин-4-карбоксамидо)пропил] метиламин (55) (60%) в виде пены. 1H ЯМР(СDСl3) 1,97 (квинтет, J=7,3 Гц, 4 Н, 2СН2СН2CH2), 2,30 (с, 3 Н, NСН3), 2,59 (т, J=7,3 Гц, 4 Н, СН2N(СН3)СН2), 3,01 (с, 12 Н, 2N(СН3)2), 3,68 (к, J=6,7 Гц, 4 Н, 2СН2NH), 7,12 (дд, J= 7,2, 0,9 Гц, 2 Н, Н-6), 7,39 (дд, J=8,4, 7,3 Гц, 2 Н, Н-7), 7,51 (дд, J=8,2, 0,8 Гц, 2 Н, Н-8), 7,62 (дд, J=8,3, 7,2 Гц, 2 Н, Н-2), 8,04 (дд, J=8,4, 1,4 Гц, 2 Н, Н-1), 8,70 (с, 2 Н, Н-9), 8,91 (дд, J=7,1, 1,5 Гц, 2 Н, Н-3), 11,94 (шир. с, 2 Н, 2CONH), Элементный анализ (С39Н43N7O2 Н2О) С, Н.
Пример 56: Получение соединения 56 таблицы 1.
Бис(7-фтор)аналог (27) нагревали при 100oС в избытке смеси 40% водный диметиламин/МеОН в течение 6 недель в аппарате высокого давления, растворитель затем удаляли выпариванием, и остаток хроматографировали на оксиде алюминия, получая бис-[3-(7-(диметиламино)-акридин-4-карбоксамидо) пропил] метиламин (56) (89%) в виде пены. 1H ЯМР (СDСl3) 2,08 (квинтет, J=7,0 Гц, 4 Н, 2СH2CH2СН2), 2,40 (с, 3 Н, NСН3), 2,86 (т, J=7,6 Гц, 4 Н, CH2N(СН3)СН2), 2,99 (с, 12 Н, 2N(СН3)2), 3,75 (к, J=6,1 Гц, 4 Н, 2СН2NH), 6,30 (д, J=2,8 Гц, 2 Н, Н-8), 7,18 (дд, J=9,5, 2,8 Гц, 2 Н, Н-6), 7,44 (дд, J=8,2, 7,2 Гц, 2 Н, Н-2), 7,67 (д, J=9,5 Гц, 2 Н, Н-5), 7,82 (дд, J=8,5, 1,4 Гц, 2 Н, Н-1), 8,13 (с, 2 Н, Н-9), 8,69 (дд, J=7,1, 1,5 Гц, 2 Н, Н-3), 11,84 (т, J=5,0 Гц, 2 Н, 2CONH). Элементный анализ (C39H43N7O2) С, Н, N.
Пример 57: Получение соединения 57 таблицы 1 способом по схеме 1.
Реакция 2,5-диметиланилина с 2-иодизофталевой кислотой в условиях, описанных в примере 1, давала сырую N-(2,5-диметилфенил)изофталевую кислоту. Ее циклизовали непосредственно при помощи РРА, получая 5,8-диметилакридон-4-карбоновую кислоту (общий выход 46%); т. пл. (МеОН/Н2О) 343-346oС; 1H ЯМР [(СD3)2SO] 2,87 (с, 6 Н, 2СН3), 6,98 (д, J=7,3 Гц, 1 Н, Н-6), 7,33 (т, J= 7,7 Гц, 1 Н, Н-2), 7,51 (д, J=7,5 Гц, 1 Н, Н-7), 8,41 (дд, J=7,6, 1,6 Гц, 1 Н, Н-1), 8,46 (дд, J=7,9, 1,6 Гц, 1 Н, Н-3), 12,00 (шир. с, 1 Н, NH), 13,93 (шир. с, 1 Н, СООН). Элементный анализ (С16Н31NО3) С, Н, N.
Восстановление 5,8-диметилакридон-4-карбоновой кислоты, как указано выше, давало 5,8-диметилакридин-4-карбоновую кислоту (82%); т. пл. (МеОН/Н2O) 239-241oС; 1H ЯМР [(CD3)2SO] 2,78 (с, 3 Н, СН3), 2,83 (с, 3 Н, СН3), 7,50 (д, J=6,7 Гц, 1 Н, Н-6), 7,81 (д, J=7,0 Гц, 1 Н, Н-7), 7,88 (дд, J= 8,3, 7,2 Гц, 1 Н, Н-2), 8,62 (дд, J=8,4, 1,4 Гц, 1 Н, Н-1), 8,76 (дд, J= 7,0, 1.4 Гц, 1Н, Н-3), 8,61 (с, 1 Н, Н-9), 17,48 (с, 1 Н, СООН): Элементный анализ (C12H13NO2) С, Н, N.
Активирование и сочетание 5,8-диметилакридин-4-карбоновой кислоты, как описано выше, давало бис-[3-(5,8-диметилакридин-4-карбоксамидо)пропил]метиламин (57%) (79%), т. пл. (СН2Сl2/гексан) 119-124oС. 1H ЯМР (СDСl3) 2,00 (квинтет, J= 7,3 Гц, 4 Н, 2СН2СH2СН2), 2,31 (с, 3 Н, NСН3), 2,60 (т, J=7,4 Гц, 4 Н, СН2N(СН3)СН2), 2,70 (с, 6 Н, 2СН3), 2,73 (с, 6 Н, 2СН3), 3,70 (к, J= 6,7 Гц, 4 Н, 2СН2NH), 7,16 (д, J=7,1 Гц, 2 Н, Н-6), 7,40 (д, J=6,8 Гц, 2 Н, Н-7), 7,61 (дд, J=8,1, 7,3 Гц, 2 Н, Н-2), 8,06 (дд, J=8,3, 1,4 Гц, 2 Н, Н-1), 8,81 (с, 2 Н, Н-9), 8,93 (дд, J=7,1, 1,5 Гц, 2 Н, Н-3), 11,81 (шир. с, 2 Н, 2CONH). Элементный анализ (С39Н41N5O2) С, Н, N.
Пример 58: Получение соединения 58 таблицы 1 способом по схеме 1.
Реакция 3-метилантраниловой кислоты и 2-бром-4-метилбензойной кислоты в условиях, описанных в примере 1, давала сырую N-(2-метил-6-карбоксифенил)-4- метилантраниловую кислоту. Ее циклизовали в РРА, как указано выше, получая 1,5-диметилакридон 4-карбоновую кислоту (общий выход 49%), т. пл. (МеОН) 317-318oС; 1H ЯМР [(СD3)2SO] 2,51 (с, 3 Н, СН3), 2,91 (с, 3 Н, СН3), 7,07 (д, J=8,1 Гц, 1 Н, Н-2), 7,20 (т, J=7,0 Гц, 1 Н, Н-7), 7,51 (д, J=7,0 Гц, 1 Н, Н-6), 8,05 (д, J=7,7 Гц, 1 Н, Н-3), 8,26 (д, J=7,8 Гц, 1 Н, Н-8), 12,45 (шир. с, 1 Н, СO2Н). Элементный анализ (С16Н13NО3) С, Н, N.
Восстановление 1,5-диметилакридон-4-карбоновой кислоты, как описано выше, давало 1,5-диметилакридин-4-карбоновую кислоту (98%), т. пл. (МеОН) 267oС (разложение); 1H ЯМР [(СD3)2SO] (с, 3 Н, СН3, 2,93 (с, 3 Н, СН3), 7,70 (дд, J= 8,1, 7,2 Гц, 2 Н, Н-2, 7), 7,95 (д, J=6,7 Гц, 1 Н, Н-6), 8,25 (д, J= 8,4 Гц, 1 Н, Н-8), 8,67 (д, J=7,3 Гц, 1 Н, Н-3), 9,63 (с, 1 Н, Н-9), 17,55 (с, 1 Н, СO2Н). Элементный анализ (C16H13NO3) С, Н, N.
Активирование и сочетание 1,5-диметилакридин 4-карбоновой кислоты, как указано выше, давало бис[3-(1,5-диметилакридин-4-карбоксамидо)пропил]метиламин (58) (82%), т. пл. (СН2Cl2/гексан) 110-116oC (разложение); 1H ЯМР (СDСl3) 2,01 (квинтет, J=6,9 Гц, 4 Н, 2СН2СН2СН2), 2,34 (с, 3 Н, NСН3), 2,64 (шир. т, 4 Н, СН2N(СН3)NСН2), 2,77 (с, 12 Н, 4СН3), 3,69 (к, J=6,7 Гц, 4 Н, 2CH3NH), 7,36 (дд, J=8,4, 6,9 Гц, 2 Н, Н-7), 7,42 (дд, J=7,2, 0,8 Гц, 2 Н, Н-6), 7,52 (д, J=6,8 Гц, 2 Н, Н-8), 7,75 (д, J=8,4 Гц, 2 Н, Н-2), 8,80 (с, 2 Н, Н-9), 8,8 (д, J=8,6 Гц, 2 Н, Н-3), 11,80 (шир. с, 2 Н, 2CONH). Элементный анализ (C39H41N5O2 2Н2O) С, Н, N.
Пример 59: Получение соединения 59 таблицы 1 способом по cxeме 1.
Реакция 2-метил-5-хлорантраниловой кислоты и 2-иодизофталевой кислоты в условиях, описанных в примере 1, давала сырую N-(2-метил-5-хлорфенил)изофталевую кислоту. Ее циклизовали непосредственно при помощи РРА, получая 8-хлор-5-метилакридон-4-карбоновую кислоту (общий выход 51%), т. пл. (МеОН) 325-330oС; 1H ЯМР [(СD3)2SО] 2,50 (с, 3 Н, СН3; перекрывается пиком ДМСО), 7,81 (д, J=7,2 Гц, 1 Н, Н-6), 7,38 (т, J=7,8 Гц, 1 Н, Н-2), 7,61 (д, J= 7,7 Гц, 1 Н, Н-7), 8,43-8,48 (м, 2 Н, Н-1, 3), 12,18 (шир. с, 1 Н, NH), 14,10 (с, 1 Н, СO2Н). Элементный анализ (C15H10ClNO3) С, Н, N.
Восстановление 8-хлор-5-метилакридон-4-карбоновой кислоты, как описано выше, давало 8-хлор-5-метилакридин-4-карбоновую кислоту (84%), т. пл. (МеОН) 259-260oС; 1H ЯМР [(СD3)2SO] 2,81 (с, 3 Н, СН3), 7,86-7,95 (м, 3 Н, Н-1,2,3), 8,74 (д, J=8,4 Гц, 1 Н, Н-6), 8,80 (д, J=7,0 Гц, 1 Н, Н-7), 9,70 (с, 1 Н, Н-9), 16,83 (с, 1 Н, СO2Н). Элементный анализ (C15H10ClNO2) С, Н, N.
Активирование и сочетание 8-хлор-5-метилакридин-4 карбоновой кислоты, как указано выше, давало бис-[3-(8-хлор- 5- метилакридин-4-карбоксамидо) пропил] метиламин (59) (81%), т. пл. (СН2Сl2/гексан) 212-215oС; 1H ЯМР (СDСl3) 1,98 (квинтет, J=7,3 Гц, 4 Н, 2СН2СН2СН2), 2,02 (с, 3 Н, NСН3), 2,60 (т, J=7,4 Гц, 4 H, СН2N(CH3)СН2), 2,67 (с, 6 Н, 2СН3), 3,70 (к, 4 Н, 2СН2NH), 7,28 (дд, J=7,7, 0,9 Гц, 2 Н, Н-7), 7,32 (д, J=7,4 Гц, 2 Н, Н-6), 7,65 (дд, J=8,3, 7,2 Гц, 2 Н, Н-2), 8,07 (дд, J=8,6, 1,5 Гц, 2 Н, Н-1), 8,96 (дд, J=7,2, 1,5 Гц, 2 Н, Н-3), 9,01 (с, 2 Н, Н-9), 11,41 (т, J=5,3 Гц, 2 H, 2CONH). Элементный анализ (С37Н35Сl2Н2O5 0,5 Н2О) С, H, N, Cl.
Пример 60: Получение соединения 60 таблицы 1 способом по схеме 2.
Смесь 3-метилантраниловой кислоты (7,6 г, 50 ммоль), метил-4-хлор-2-иодбензоата (19,2 г, 65 ммоль), Сu и CuI (каталитическое количество) в 2,3-бутаноле (20 мл) нагревали с бензолом (30 мл) на масляной бане. После того, как бензол был отогнан, добавляли N-этилморфолин (50 мл) и перемешиваемую смесь нагревали при 110oС в течение 18 ч, затем разбавляли разбавленной НСl, экстрагировали в EtOAc и фильтровали для удаления солей Сu. Органический слой отделяли и экстрагировали в разбавленный NH4OH, затем осаждали аммониевую соль продукта. Ее собирали и перемешивали в разбавленной НСl, и смесь фильтровали и промывали водой, получая 2-[[(5-хлор-2-метоксикарбонил)фенил] амино] -3-метилбензойную кислоту (6,6 г, 41%); т. пл. (МеОН) 187-188,5oС; 1H ЯМР [(СD3)2SO] 2,10 (с, 3 Н, СН3), 3,87 (с, 3 Н, ОСН3), 6,12 (д, J=2,0 Гц, 1 Н, Н-6′), 6,79 (дд, J=8,6, 2,0 Гц, 1 Н, Н-4′), 7,29 (т, J= 7,6 Гц, 1 Н, Н-5), 7,52 (д, J=7,4 Гц, 1 Н, Н-4), 7,74 (д, J=7,4 Гц, 1 Н, Н-6), 7,89 (д, J= 8,5 Гц, 1 Н, Н-3′), 9,90 (шир. с, 1 Н, NH). Элементный анализ (C16H14ClNO4) С, Н, N, Cl.
Раствор 2-[[(5-хлор-2-метоксикарбонил)фенил] амино] -3-метилбензойной кислоты (6,0 г, 18,8 ммоль) в сухом ТГФ (100 мл) обрабатывали CDl (6,0 г, 37,6 ммоль) при 20oС в течение 18 ч, и раствор затем добавляли по каплям к суспензии NaBH4 (0,69 г, 5 эквив.) в Н2О (50 мл). Когда реакция была завершена (30 мин, как установлено ТСХ), смесь гасили разбавленной НСl и экстрагировали СН2Cl2. Отфильтрованный слой СН2Сl2 сушили, получая сырой продукт, который хроматографировали на силикагеле с элюированием градиентом 1% МеОН в СН2Cl2, получая метил-4-хлор-2-[N-(2-гидроксиметил-6-метил) фениламино] бензоат (1,0 г, 17%), т. пл. (СН2Сl2/гексан) 114-115oС. 1H ЯМР (СDСl3) 1,78 (шир. с, 1 Н, ОН), 2,18 (с, 3 Н, СН3), 3,92 (с, 3 Н, СО2СН3), 4,54 (дд, J=12,8, 4,3 Гц, 1 Н, СНОН), 4,67 (дд, J=12,8, 4,3 Гц, 1 Н, СНОН), 6,01 (д, J=2,0 Гц, 1 Н, Н-3), 6,63 (дд, J=8,3, 2,0 Гц, 1 Н, Н-5), 7,24-7,29 (м, 2 Н, 2 АrН), 7,35-7,39 (м, 1 Н, АrН), 7,89 (д, J=8,6 Гц, 1 Н, Н-6), 9,22 (с, 1 Н, NH). Элементный анализ (C16H16ClNO3) С, Н, N.
Раствор метил-4-хлор-2-[N-(2-гидроксиметил-6-метил) фениламино]бензоата (0,72 г, 2,35 ммоль) в EtOAc (100 мл) кипятили с обратным холодильником в течение 7 ч с МnO2 (1 г). Смесь фильтровали через целит для удаления остатков Мn, и растворитель выпаривали и остаток фильтровали через колонку силикагеля в СН2Cl2, получая метил-4-хлор-2-[N-(2-формил-6-метил) -фениламино] бензоат (0,7 г, 98%), т пл. (МеОН/Н2O) 81-82oС. 1H ЯМР (СDСl3) 2,23 (с, 3 Н, СН3), 3,95 (с, 1 Н, СO2СН3), 6,27 (д, J=2,0 Гц, 1 Н, Н-3), 6,70 (дд, J= 8,7, 2,0 Гц, 1 Н, Н-5), 7,37 (т, J=7,6 Гц, 1 Н, Н-4′), 7,58 (д, J=7,9 Гц, 1 Н, Н-5′), 7,81 (дд, J=7,7, 1,3 Гц, 1 Н, Н-3′), 7,92 (д, J=8,6 Гц, 1 Н, Н-6), 9,68 (шир. с, 1 Н, NH), 10,15 (с, 1 Н, СНО). Анализ для метил-4-хлор-2-[N-(2-формил-6-метил) -фениламино]бензоата. Элементный анализ (С16Н14СlNО3) С, Н, N.
Раствор 4-хлор-2-[N-(2-формил-6-метил)фениламино] бензоата (0,65 г, 2,1 ммоль) в трифторуксусной кислоте (8 мл) перемешивали при 40oС в течение 4 ч в атмосфере азота. Избыток реагента удаляли при пониженном давлении при 40oС, и остаток суспендировали в 2 н. NaOH (25 мл) и EtOH (18 мл) и нагревали в течение 1 ч до тех пор, пока не получали прозрачный раствор. Охлажденную реакционную смесь нейтрализовали АсОН, и полученный осадок собирали, промывали водой и сушили, получая 1-хлор-5-метилакридин-4-карбоновую кислоту (0,56 г, 96%), т. пл. (MeOH/H2O) 260oC (разложение); 1H ЯМР (СDСl3) 2,93 (с, 3 Н, СН3), 7,64 (дд, J=8,4, 7,9 Гц, 1 Н, Н-7), 7,83-7,86 (м, 2 Н, Н-2 и Н-6 или Н-8), 8,08 (д, J=8,6 Гц, 1 Н, Н-8 или Н-6), 8,84 (д, J=7,8 Гц, 1 Н, Н-3), 9,4 (с, 1 Н, Н-9), 17,26 (с, 1 Н, СО2Н). Элементный анализ (C15H10ClNO2) С, Н, N.
Активирование и сочетание 1-хлор-5-метилакридин-4-карбоновой кислоты, как указано выше, давало бис[3-(1-хлор-5- метилакридин-4-карбоксамидо) пропил] метиламин (60) (84%), т. пл. (СН2Сl2/гексан) 156-158,5oС. 1H ЯМР (CDCl3) 1,85 (квинтет, J=7,2 Гц, 4 Н, 2CH2CH2СН2), 2,32 (с, 3 Н, NСН3), 2,60 (т, 4 Н, СН2N(СН3)СН2), 2,74 (с, 6 Н, 2СН3), 3,68 (к, J=6,7 Гц, 4 Н, 2СН2NH), 7,38 (дд, J=8,3, 6,8 Гц, 2 Н, Н-7), 7,52 (д, J=6,8 Гц, 2 Н, Н-6), 7,65 (д, J=8,0 Гц, 2 Н, Н-2), 7,76 (д, J=8,5 Гц, 2 Н, Н-8), 8,80 (д, J=8,0 Гц, 2 Н, Н-3), 9,04 (с, 2 Н, Н-9), 11,50 (шир. с, 2 Н, 2CONH). Анализ для бис[3-(1-хлор-5-метилакридин-4-карбоксамидо) пропил] метиламина (60). Элементный анализ (С37Н35Сl2N5O2) С, Н, N, Cl.
Пример 61: Получение соединения 61 таблицы 1.
Активирование и сочетание известной [Rewcastle et al., J. Med. Chem. 1987, 30, 843] 3-метил-феназин-1-карбоновой кислоты давало бис[(2-(3-метилфеназин-1-карбоксамидо)пропил] метиламин (61) в виде желтого твердого продукта (84%), т. пл. 75-78oС (СН2Сl2/н-гексан). 1H ЯМР (CDCl3) 2,03 (квинтет, J= 7,0 Гц, 4 Н, 2СН2СН2СН2), 2,37 (с, 3 Н, NСН3), 2,67 (д, J=0,9 Гц, 6 Н, 2СН3), 2,73 (т, J=7,2 Гц, 4 H, СН2N(СН3)СН2), 3,73 (к, J=6,3 Гц, 2 Н, 2СН2NH), 7,62-7,70 (м, 4 Н, Н-7 и Н-8), 7,98-8,03 (м, 6 Н, Н-6, Н-9 и Н-2 или Н-4), 8,73 (д, J=2,1 Гц, 2 Н, Н-2) и 10,88 (шир. т, J=5,2 Гц, 2 Н, 2CONH), HRMS (FAB+) m/z вычислено для С35Н36N7O2 586,2930 (МН+), найдено 586,2931. Элементный анализ (C35H35N7O2) С, Н, N.
Пример 62: Получение соединения 62 таблицы 1.
Активирование и сочетание известной [Rewcastle et al., J. Med. Chem. 1987, 30, 843] 3-хлорфеназин-1-карбоновой кислоты давало бис[(2-(3-хлорфеназин-1-карбоксамидо) пропил] метиламин (62) в виде желтого твердого продукта (76%), т. пл. 169-170oС (СН2Cl2/гексан). 1H ЯМР (СDСl3) 2,02 (квинтет, J= 6,9 Гц, 4 Н, 2СН2СН2СН2), 2,36 (с, 3 Н, NСН3), 2,72 (т, J=7,3 Гц, 4 Н, 2СН2NСН3), 3,71 (к, J=6,3 Гц, 2 Н, 2СН2NH), 7,69-7,6 (м, 4 Н, Н-7 И Н-8), 7,94-8,00 (м, 4 Н, Н-6 и Н-9), 8,20 (д, J=2,5 Гц, 2 Н, Н-4), 8,74 (д, J=2,5 Гц, 2 Н, Н-2) и 10,65 (шир. т, J=5,2 Гц, 2 Н, 2CONH), HRMS (FAB+) m/z вычислено для C33H29 35Cl2N7O2 626,1838 (МН+), найдено 626,1824. Элементный анализ (С33Н29Сl2N7O2 Н2О) С, Н, N.
Пример 63: Получение соединения 63 таблицы 1.
Активирование и сочетание известной [Rewcastle et al., J. Med. Chem. 1987, 30, 843] 2-хлорфеназин-1-карбоновой кислоты давало бис[(3-(2-хлорфеназин-1-карбоксамидо) пропил] метиламин (63) в виде желтого твердого продукта (45%), т. пл. 206-207oС (СН2Сl2/гексан). 1H ЯМР (СDСl3) 1,83 (квинтет, J= 6,0 Гц, 4 Н, 2СН2СН2СН2), 2,17 (с, 3 Н, NСН3), 2,72 (т, J=6,1 Гц, 4 Н, СН2N(СН3)СН2), 3,67 (к, J=6,0 Гц, 2 Н, 2СН2NH), 7,03 (шир. т, J= 5,9 Гц, 2 Н, 2CONH), 7,47 (д, J=9,4 Гц, 2 Н, Н-3 или Н-4), 7,60-7,68 (м, 4 Н, Н-7 и Н-8), 7,89 (д, J=9,3 Гц, 2 Н, Н-4 или Н-3), 7,91-7,97 (м, 4 Н, Н-6 и Н-9); HRMS (FAB+) m/z вычислено для С33Н29 35Cl2N7O2 626,1838 (МН+), найдено 626,1854. Элементный анализ (С33Н29Сl2N7O2) С, Н, N.
Пример 64: Получение соединения 64 таблицы 1.
Активирование и сочетание известной [Rewcastle et al., J. Med. Chem. 1987, 30, 843] 8-хлорфеназин-1-карбоновой кислоты давало бис[(3-(8-хлорфеназин-1-карбоксамидо)пропил] метиламин (64) в виде бледно-желтого твердого продукта (85%), т. пл. 210-212oС (СН2Сl2/н-гексан). 1H ЯМР (CDCl3) 2,04 (квинтет, J= 7,0 Гц, 4 Н, 2СН2СН2СН2), 2,39 (с, 3 Н, NСН3), 2,73 (т, J=7,2 Гц, 4 Н, СН2N(СН3)СН2), 3,74 (к, J=6,3 Гц, 4 Н, 2CH2NH), 7,56 (дд, J=9,2, 2,4 Гц, 2 Н, Н-7), 7,92 (дд, J=8,7, 7,2 Гц, 2 Н, Н-3), 7,98 (д, J=9,2, 2 Н, Н-6), 8,03 (д, J=2,2 Гц, 2 Н, Н-9), 8,26 (дд, J=8,7, 1,5 Гц, 2 Н, Н-4), 8,92 (дд, J=7,2, 1,5 Гц, 2 Н, Н-2) и 10,64 (шир. т, J=5,2 Гц, 2 Н, 2CONH); HRMS (FAB+) m/z вычислено для С33Н30 35Cl2N7O2 626,1838 (МН+), найдено 626,1860. Элементный aнализ (С33H30Cl2N7O2) С, Н, N, Cl.
Пример 65: Получение соединения 65 таблицы 1.
Реакция 2-бром 3-нитробензойной кислоты и 2,5-ксилидина давала 2-(2,5-диметил-фениламино)-3-нитробензойную кислоту (65%); т. пл. (бензол/ацетон) 215-217oС. 1H ЯМР [(СD3)2SО] 2,10 (с, 3 Н, СН3), 2,23 (с, 3 Н, СН3), 6,53 (с, 1 Н, Н-6′), 6,79 (д, J=7,4 Гц, 1 Н, Н-4′), 7,02 (т, J=8,0 Гц, 1 Н, Н-5), 7,11 (д, J=7,7 Гц, 1 Н, Н-3′), 8,03 (дд, J=8,1, 1,4 Гц, 1 Н, Н-6), 8,22 (дд, J= 7,7, 1,5 Гц, 1 Н, Н-4), 9,84 (шир. с, 1 Н, NH), 13,8 (шир. с, 1 Н, СO2Н). Элементный анализ (C15H14N2O4) С, Н, N.
Восстановительное замыкание цикла вышеуказанной кислоты при помощи NaOC2H5/NaBH4 давало 6,9-диметилфеназин-1-карбоновую кислоту (64%), т. пл. (МеОН) 246-247oС. 1H ЯМР [(СD3)2SO] 2,78 (с, 3 Н, СН3), 2,83 (с, 3 Н, СН3), 7,80 (д, J=7,0 Гц, 1Н, Н-7 или Н-8), 7,84 (сд, J=7,0 Гц, 1 Н, Н-7 или Н-8), 8,12 (дд, J=8,5, 7,2 Гц, 1 Н, Н-3), 8,56 (д, J=8,7 Гц, 1 Н, Н-4), 8,66 (д, J= 7,0 Гц, 1 Н, Н-2), 15,24 (шир. с, 1H, СO2Н). Элементный анализ (C15H12N2O2) С, Н, N.
Активирование и сочетание вышеуказанной кислоты давало бис[3-(6,9-диметилфеназин-1-карбоксамидо) пропил ]метиламин (65) в виде ярко-желтого твердого продукта (53%), т. пл. 97-101oС (СН2Сl2/н-гексан). 1H ЯМР (CDCl3) 2,02 (м, 4 Н, 2СН2СН2СН2), 2,34 (с, 3 Н, NСН3), 2,60-2,68 (шир., м, 4 Н, СН2N(СН3)СН2), 2,68 (с, 6 Н, 2АrСН3), 2,78 (с, 6 Н, 2АrСН3), 3,70 (к, J= 6,6 Гц, 4 Н, 2СН2NH), 7,32-7,40 (м, 4 Н, Н-7 и Н-8), 7,86 (дд, J=8,6, 7,2 Гц, 2 Н, Н-3), 8,28 (дд, J=8,7, 1,5 Гц, 2 Н, Н-4), 8,90 (дд, J=7,1, 1,5 Гц, 2 Н, Н-2) и 11,00 (шир. с, 2 Н, 2CONH); HRMS (FAB+) m/z вычислено для С37Н40N7O2 614,3243 (МН+), найдено 614,3237. Элементный анализ (С37Н40N7O2 0,5 Н2О) С, Н, N.
Пример 66: Получение соединения 66 таблицы 1.
Смесь 5-хлор-2-метиланилина (8,63 г, 61,0 ммоль), 2-бром-3-нитробензойной кислоты (10,0 г, 41,0 ммоль), CuCl (0,5 г), медного порошка (0,1 г) в бутан-2,3-диоле (25 мл) и N-этилморфолине (15 мл) перемешивали и нагревали в течение 18 ч при 70oС. Реакционную смесь разбавляли 0,5 М NH4OH (500 мл), затем фильтровали через целит. Оранжевый фильтрат затем медленно добавляли к перемешиваемому раствору 2 н. НСl, и получаемый желтый осадок собирали фильтрованием, сушили и перекристаллизовывали, получая 2-[(5-хлор-2 метил)фениламино] 3-бензойную кислоту в виде ярко-желтого кристаллического твердого продукта (70%), т. пл. 228-230oС (EtOAc/н-гексан). 1H ЯМР (СDСl3) 2,35 (с, 3 Н, СН3), 6,79 (д, J=2,1 Гц, 1 Н, Н-6′), 6,96-7,00 (м, 2 Н, Н-4′ и Н-5′), 7,15 (д, J=8,0 Гц, 1 Н, Н-3′), 8,07 (дд, J=8,1, 1,8 Гц, 1 Н, Н-4 или Н-6), 8,24 (дд, J=1,9, 1,7 Гц, 1 Н, Н-6 или Н-4) и 9,51 (с, 1 Н, СООН). Элементный анализ (C14H11ClN2O4) С, Н, N.
Раствор 2-[(5-хлор-2-метил)фениламино]-3-бензойной кислоты (3,59 г, 11,7 ммоль) и NaBH4 (2,62 г, 68,8 ммоль) в 2 М NaOH кипятили с обратным холодильником в течение 8 ч. Реакционную смесь затем охлаждали и подкисляли АсОН для осаждения сырой феназинкислоты. Этот твердый продукт собирали и перекристаллизовывали, получая 6-хлор-9-метилфеназин-1-карбоновую кислоту в виде горчично-желтых игл (45%), т. пл. 255-257oС (ацетон). 1H ЯМР [(СD3)2SO] 2,86 (с, 3 Н, СН3), 7,90 (дд, J=7,4, 1,1 Гц, 2 Н, ArH), 8,11-8,18 (м, 2 Н, АrН), 8,57-8,61 (м, 2 Н, ArH) и 14,52 (шир. с, 1 Н, СООН). Элементный анализ (С14Н9СlN2O2) С, Н, N, Cl.
Вышеуказанную 6-хлор-9-метилфеназин-1-карбоновую кислоту активировали и сочетали, получая бис-[(6-хлор-9-метилфеназин -1-карбоксамидо)пропил] метиламин (66) в виде зелено-желтого твердого продукта (84%), т. пл. 200-202oС (СН2Cl2/н-гексан). 1H ЯМР (CDCl3) 1,97 (квинтет, J=7,2 Гц, 4 Н, 2СН2СН2СН2), 2,31 (с, 3 Н, NСН3), 2,59 (т, J=7,1 Гц, 4 Н, СН2N(СН3)СН2), 2,76 (с, 6 Н, 2АrСН3), 3,69 (к, J=6,7 Гц, 4 Н, 2СН2NH), 7,50 (дд, J=7,6, 1,0 Гц, 2 Н, Н-8), 7,78 (д, J=7,5 Гц, 2 Н, Н-7), 7,93 (дд, J=8,7, 7,2 Гц, 2 Н, Н-3), 8,41 (дд, J=8,7, 1,5 Гц, 2 Н, Н-2), 8,94 (дд, J=7,1, 1,5 Гц, 2 Н, Н-4) и 10,72 (шир. с, 2 Н, 2CONH); HRMS (FAB+) m/z вычислено для C35H34 35Cl2N7O2 654,2151 (МН+), найдено 654,2159. Элементный анализ (С35Н34Сl2N7O2 0,5 Н2O) С, Н, N.
Пример 67: Получение соединения 67 таблицы 1.
Активирование и сочетание известной [Rewcastle et al., J. Med. Chem. 1987, 30, 843] 4-метилфеназин-1-карбоновой кислоты давало бис[(4-метилфеназин-1-карбоксамидо) пропил] метиламин (67) в виде ярко-желтого твердого продукта (78%), т. пл. 218-220oС (СН2Сl2/н-гексан). 1H ЯМР (СDСl3) 2,04 (квинтет, J= 7,0 Гц, 2 Н, 2СН2СН2CH2), 2,38 (с, 3 Н, NСН3), 2,75 (т, J=7,3 Гц, 4 Н, СН2N(СН3)СН2), 2,90 (с, 6Н, 2СН3), 3,71 (к, J=6,3 Гц, 4 Н, 2СН2NH), 7,58 (ддд, J=8,6, 6,7, 1,3 Гц, 2 H, ArH), 7,65 (ддд, J=8,6, 6,6, 1,4 Гц, 2 H, ArH), 7,70 (дд, J=7,2, 1,0 Гц, 2 H, ArH), 7,94 (дд, J=8,6, 0,9 Гц, 2 H, ArH), 8,00 (д, J=8,7 Гц, 2 H, ArH), 8,77 (д, J=7,3 Гц, 2 H, ArH) и 10,88 (шир. с, 2 H, 2CONH). HRMS (FAB+) m/z вычислено для C35H36N7O2 586,2930 (МН+), найдено 586,2922. Элементный анализ (C33H36N7O2 2,5 Н2O) С, Н, N.
Пример 68: Получение соединения 68 таблицы 1.
Активирование 9-метилфеназин-1-карбоновой кислоты и сочетание с N,N’-бис-(3-аминопропил)этилендиамином давало бис-[3-(9-метилфеназин-1-карбоксамидо) пропил] -1,2-этилендиамин (68) в виде смолы, которую превращали в дигидрохлоридную соль (10%), т. пл. (МеОН) 276oС. 1H ЯМР (D2O) 2,07 (квинтет, J= 6,7 Гц, 4 Н, 2СН2), 2,82 (с, 6 Н, 2АrСН3), 3,17 (м, 4 Н, 2СН2), 3,31 (шир. с, 4 Н, 2СН2), 3,65 (т, J=6,6 Гц, 4 Н, 2СН3), 7,87 (м, 4 Н, 4ArH), 7,96-8,00 (м, 4 Н, 4ArH), 8,27-8,30 (м, 2 Н, 2ArH), 8,60 (д, J= 7,2 Гц, 2 Н, 2ArH). HRMS (FAB+) m/z вычислено для C36H38N8O2 615,3196 (МН+), найдено 615,3196.
Пример 69. Получение соединения 69 таблицы 1.
Активирование 6,9-диметилфеназин-1-карбоновой кислоты и сочетание с триэтилентетрамином давали бис[2-(6,9-диметилфеназин-1-карбоксамидо)этил]-1,2-этилендиамин (69) (99%), т. пл. (дигидрохлоридная соль из МеОН) 299oС (разложение). 1H ЯМР (CF3CO2D) и 3,06 (с, 6 Н, 2СН3), 3,09 (с, Н, 2СН3), 3,87 (шир. с, 4 Н, 2СН3), 3,91 (шир. с, 4 Н, 2СН2), 4,27 (шир. с, 4 Н, 2СН2), 8,20 (д, J=7,3 Гц, 2 Н, Н-7 или Н-8), 8,24 (д, J=7,3 Гц, 2 Н, Н-7 или Н-8), 8,43 (т, J=8,1 Гц, 2 Н, Н-3), 8,96 (д, J=8,8 Гц, 2 Н, Н-4), 9,02 (д, J=7,3 Гц, 2 Н, Н-2). Элементный анализ (C36H40Cl2N8O2) С, Н, N.
Пример 70: Получение соединения 70 таблицы 1.
Активирование 9-метилфеназин-1-карбоновой кислоты и сочетание с N,N’-бис-(3-аминопропил)бутандиамином давало бис[2-(9-метилфеназин-1-карбоксамидо) пропил]-1,4-бутандиамин (70) (73%), т. пл. (СН2Сl2/гексан) 86-90,5oС. 1H ЯМР (СDСl3) 1,53 (квинтет, J=3,2 Гц, 4 Н, 2СН2), 1,97 (квинтет, J=7,0 Гц, 4 Н, СН2), 2,62 (т, J=6,2 Гц, 4 Н, 2СН2), 2,79 (т, J=7,0 Гц, 4 Н, 2СН2), 2,88 (с, 6 Н, 2СН3), 3,74 (к, J= 6,6 Гц, 4 Н, 2СН3), 7,71-7,78 (м, 4 Н, ArH), 7,93 (дд, J=8,7, 7,2 Гц, 2 Н, Н-3), 8,08 (д, J=7,9, 0,8 Гц, 2 Н, ArH), 8,34 (дд, J=8,7, 1,5 Гц, 2 Н, Н-4), 8,96 (дд, J=7,1, 1,5 Гц, 2 Н, Н-2), 11,05 (т, J=5,2 Гц, 2 Н, 2CONH). Элементный анализ (С38Н42N8O2 1,5 Н2O) С, Н, N.
Пример 71: Получение соединения 71 таблицы 1.
Активирование 5-метилакридин-4-карбоновой кислоты и сочетание с триэтилентетрамином давало бис[3-(5-метилакридин-4-карбоксамидо) этил]-1,2-этилендиамин (71) (76%), т. пл. (СН2Cl2, гексан) 167-170oС. 1H ЯМР (CDCl3) 2,08 (с, 6 Н, 2СН3), 2,85 (с, 4 Н, 2СН2), 2,99 (т, J=6,2 Гц, 4 Н, 2СН2), 3,74 (к, J=6,1 Гц, 4 Н, 2СН3), 7,39 (дд, J=8,4, 6,8 Гц, 2 Н, Н-2), 7,57-7,61 (м, 4 Н, Н-6 и Н-7), 7,75 (д, J=8,7 Гц, 2 Н, Н-8), 8,02 (дд, J=8,4, 1,5 Гц, 2 Н, Н-1), 8,68 (с, 2 Н, Н-9), 8,91 (дд, J=7,2, 1,5 Гц, 2 Н, Н-3), 11,81 (т, J=5,5 Гц, 2 Н, 2CONH). Элементный анализ (С36Н36N6О2) С, Н, N.
Пример 72: Получение соединения 72 таблицы 1.
Активирование акридин-4-карбоновой кислоты и сочетание с триэтилентетрамином давало бис[3-(акридин-4-карбоксамидо) этил]-1,2-этилендиамин (72) (72%), т. пл. (СН3Сl2/гексан) 170-171oС. 1H ЯМР (CDCl3) 2,91 (с, 8 Н, 2СН3), 3,53 (к, J=5,4 Гц, 4 Н, 2СН2), 7,53 (т, J=7,4 Гц, 2 Н, АrН), 7,68 (дд, J=8,3, 7,1 Гц, 2 Н, АrН), 7,81-7,85 (м, 2 Н, АrН), 8,03 (д, J=8,3 Гц, 2 Н, АrН), 8,22 (д, J=8,9 Гц, 2 Н, АrН), 8,26 (дд, J=8,5, 1,4 Гц, 2 Н, АrН), 8,64 (дд, J=7,1, 1,6 Гц, 2 Н, Н-3), 9,87 (с, 2 Н, Н-9), 11,56 (т, J=5,0 Гц, 2 Н, 2CONH). Элементный анализ (C34H32N6O2 2H2O) С, Н, N.
Пример 73: Получение соединения 73 таблицы 1.
Активирование известной [Rewcastle et al., J. Med. Chem. 1987, 30, 843] 9-метилфеназин-1-карбоновой кислоты и последующее сочетание с N,N’-бис-(2-аминоэтил)-1,3-пропандиамином давало бис[(9-метилфеназин-1-карбоксамидо) этил] -1, 3-пропандиамин (73) в виде желтого твердого продукта (68%), т. пл. 194-195oС (СН2Сl2/н-гексан). 1H ЯМР (CDCl3) 1,73 (квинтет, J=6,9 Гц, 2 Н, СH2СH2СН2), 2,79 (т, J=6,9 Гц, 4 Н, 2СН2), 2,88 (с, 6 Н, 2АrСН3), 2,97 (т, J= 6,2 Гц, 4 Н, 2СН2), 3,75 (к, J=6, 0 Гц, 4 Н, 2СН2), 7,64-7,69 (м, 2 Н, АrН), 7,72 (дд, J= 8,6, 6,8 Гц, 2 Н, АrН), 7,93 (дд, J=8,7, 7,2 Гц, 2 Н, АrН), 8,04 (дд, J= 8,7, 0,9 Гц, 2 Н, АrН), 8,33 (дд, J=8,7, 1,5 Гц, 2 Н, АrН), 8,96 (дд, J= 7,2, 1,5 Гц, 2 Н, АrН) и 11,06 (шир. т, J=5,3 Гц, 2 Н, 2CONH); HRMS (FAB+) m/z вычислено для C35H37N8O2 601,3039 (МН+), найдено 601,3043. Элементный анализ (C35H36N8O2 0,5 Н2O) С, Н, N.
Пример 74: Получение соединения 74 таблицы 1.
Активирование 6-хлор-9-метилфеназин-1-карбоновой кислоты, как указано выше, и последующее сочетание с триэтилентетрамином давало бис[2-(6-хлор-9-метилфеназин-1-карбоксамидо)этил] -1,2-этилендиамин (74) в виде желтого твердого продукта, 6%, т. пл. 301oС (разложение) (соль НСl) (MeOH/EtOAc). 1H ЯМР (СF3СO2D) 3,87 (шир. с, 4 Н, 2СН2NH), 4,04 (шир. с, 4 Н, 2СН2NH), 4,09 (с, 6 Н, 2СН3), 4,29 (шир. с, 4 Н, 2CONHСН2), 8,25 (шир. д, J=7,9 Гц, 2 Н, АrН), 8,44 (шир. д, J=7,7 Гц, 2 Н, АrН), 8,53 (шир. с, 2 Н, АrН), 9,03 (шир. д, J= 8,7 Гц, 2 Н, АrН) и 9,09 (шир. с, 2 Н, АrH); HRMS (FAB+) m/z вычислено для C34H33 35Cl2N8O2 655,2104 (МН+), найдено 655,2075.
Пример 74а: Получение соединения 74а таблицы 1.
N, N’-Диметил-N, N’-бис(цианометил) этилендиамин синтезировали описанным способом [Alcock et al., J. Chem. Soc. Dalton Trans. 1987, 2543]. Это соединение (5,0 г, 100 ммоль) затем гидрировали над никелем Ренея в абсолютном EtOH, насыщенном сухим аммиаком, в течение 5 дней при 20o С (если требовалось, добавляли дополнительный никель Ренея). Катализатор удаляли фильтрованием через подушку целита, и выпаривание растворителей давало по существу чистый N, N’-диметил-N, N’-бис-(2-аминоэтил) этилендиамин (4,6 г, 88%). 1Н ЯМР (СDСl3) 2,24 (с, 6 Н, 2СН3), 2,43 (шир. с, 4 Н, 2СН2), 2,49 (с, 2 Н, 2CH2), 2,73 (шир. с, 2 Н, 2СН2), который использовали непосредственно.
9-Метилфеназин-1-карбоновую кислоту (1,0 г, 4,2 ммоль) и CDI (1,36 г, 8,4 ммоль) в ДМФ (10 мл) перемешивали при 50-60 oС в течение 1 ч, затем охлаждали до 20oС. Добавляли сухой бензол (20 мл) и сефадекс LH-20 (2 г), и смесь перемешивали при 20oС в течение 1 ч, затем фильтровали через подушку целита для удаления сефадекса. Фильтрат выпаривали досуха при пониженном давлении, и остаток растворяли в сухом ТГФ (15 мл) и охлаждали в смеси лед/соль. Добавляли N,N’-диметил-N,N’-бис-(2-аминоэтил)этилендиамин (0,36 г, 2,1 ммоль) и смесь перемешивали, в то же время давая ей нагреться до 20oС. Добавляли воду (50 мл) и ТГФ выпаривали при пониженном давлении. Полученный осадок фильтровали и промывали водным Nа2СО3 (350 мл) и водой, затем растворяли в СН2Cl2 (100 мл) и сушили (Na2SO4). Выпаривание растворителей давало сырой продукт, который хроматографировали на силикагеле с элюированием градиентом 1-6% МеОН в СН2Cl2, получая 74а в виде масла (1,1 г, 78%). 1H ЯМР (СDСl3) 2,33 (с, 6 Н, 2NСН3), 2,63 (шир. с, 4 Н, СН2NСН2СН2NСН2), 2,74 (т, J=6,5 Гц, 4 Н, CONHСН2СН2), 2,83 (с, 6 Н, 2АrСН3), 3,73 (к, J=6,2 Гц, 4 Н, 2CONHСН2), 7,61 (д, J=6,7 Гц, 2 Н, Н-8), 7,68 (дд, J=8,7, 6,8 Гц, 2 Н, Н-7), 7,89 (дд, J=8,7, 7,2 Гц, 2 Н, Н-3), 7,98 (д, J=8,7 Гц, 2 Н, Н-6), 8,26 (дд, J= 8, 1,5 Гц, 2 Н, Н-4), 8,89 (дд, J=7,0, 1,5 Гц, 2 Н, Н-2), 10,85 (т, J=5,2 Гц, 2 Н, 2CONH).
Пример 75: Получение соединения 75 таблицы 1.
N-(трет-Бутокси-карбонил)-3,3′-диамино-N-метилдипропиламин получали, как описано [Huang, T.L, Dredar, S.A., Manneh, V.A., Blankenship, J.W., Fries, D.S., J. Med. Chem., 1992, 35, 2414-1418]. Раствор ди-трет-бутил-дикарбоната (2,51 г, 11,5 ммоль) в ТГФ (15 мл) в течение 1,5 ч добавляли к раствору 3,3′-диамино-N-метилдипропиламина (5,00 г, 34,4 ммоль) в ТГФ (15 мл), который выдерживали при 0oС (смесь лед/вода). Реакционную смесь перемешивали в течение дополнительных 18 ч при комнатной температуре, затем растворитель удаляли при пониженном давлении и полученный остаток распределяли между NaCl (насыщенный) (100 мл) и СН2Сl2 (200 мл). Слой СН2Сl2 промывали следующей порцией раствора NaCl (100 мл), затем сушили (NaSO4), и растворитель удаляли при пониженном давлении, получая N-(трет-бутоксикарбонил) -3,3′-диамино-N-метилдипропиламин (2,58 г, 46%) в виде светлого вязкого масла. 1H ЯМР (CDCl2) 1,44 [шир. с, 9 Н, С(СН3)3], 1,58-1,67 (м, 6 Н, 2СН2CH2СН2 и NH2), 2,22 (с, 3 Н, NCH3), 2,34-2,40 (м, 4 Н, 2СН2NCH3), 2,74 (т, J=6,9 Гц, 2 Н, СН3NН2), 3,12-3,21 (шир. м, 2 Н, СН2NHBOC) и 5,37 (шир. с, 1 Н, NHBOC).
Феназин-1-карбоновую кислоту (494 мг, 2,24 ммоль) получали по способу, описанному в литературе [Rewcastle, G.W., Denny, W.A., Baguley, B.C., J. Med. Chem. , 1987, 30, 843-857] и затем подвергали реакции с CDI (544 мг, 3,36 ммоль) в сухом ДМФ (15 мл) в течение 2,5 ч при 30oС. ДМФ удаляли при пониженном давлении, и полученный желтый твердый продукт растворяли в смеси петролейного эфира и СН2Сl2 (40 мл, 3:1). При охлаждении имидазолид кристаллизовался, и этот сырой материал использовали в следующей реакции сочетания. Сырой имидазолид суспендировали в ТГФ (20 мл), охлаждали до 0oС (лед/вода), затем добавляли раствор в ТГФ (20 мл) вышеуказанного N- (трет-бутоксикарбонил)-3,3′-диамино-N-метилдипропиламина (659 мг, 2,69 ммоль). Реакционную смесь оставляли для перемешивания в течение еще 2 ч при 0oС, затем растворитель удаляли при пониженном давлении, и полученное желтое масло распределяли между СН2Cl2 (200 мл) и 1 М (Na2CO3). Слой CH2Cl2 сушили Na2SO4, растворитель удаляли при пониженном давлении, и полученное желто-зеленое масло очищали хроматографией на оксиде алюминия (0,25% МеОН в СН2Cl2), получая N-1-(3-{ N-метил-N-[N-(трет-бутоксикарбонил) -3-аминопропил] } аминопропил)феназин-1-карбоксамид (992 мг, 98%) в виде желто-зеленого масла, которое использовали непосредственно. 1H ЯМР (СDСl3) 1,41 (шир. с, 9 Н, С(СН3)3), 1,44 (шир. с, 2 Н, СН2СН2СН2NHBOC), 1,95-2,04 (м, 2 Н, СН2СН2СН2NHCOAr), 2,18 (с, 3 Н, NСН3), 2,46 (т, J=6,7 Гц, 2 Н, СН2СН2CH2NHBOC), 2,56 (т, J=7,2 Гц, 2 H, СН2СН2СН2NHCOAr), 3,20 (м, 2 H, СН2NHBOC), 3,74 (к, J=6,2 Гц, 2 Н, СН2NHCOAr), 5,45 (шир. с, 1 H, NHBOC), 7,89-8,00 (м, 3 Н, АrН), 8,22-8,26 (м, 1Н, ArH), 8,28-8,32 (м, 1Н, ArH), 8,40 (дд, J=8,7, 1,5 Гц, 1Н, ArH), 9,02 (дд, J=7,1, 1,5 Гц, 1H, ArH) и 11,03 (шир. с, 1 Н, CONH).
К раствору вышеуказанного БОК-защищенного амина (545 мг, 1,21 ммоль) в СН2Cl2 (8 мл) добавляли трифторуксусную кислоту (8 мл). Эту смесь перемешивали при комнатной температуре в течение 2 ч, в этой точке по данным ТСХ реакция была завершена. Все растворители удаляли при пониженном давлении, и масляный остаток распределяли между CH2Cl2 (100 мл) и 1 М (Nа2СО3) (100 мл). Водный слой экстрагировали дополнительным СН2Cl2 (4100 мл), и все экстракты в CH2Cl2 объединяли и сушили Na2SO4. Растворитель удаляли при пониженном давлении, получая N-1-{3-|N-метил-N-(3-аминопропил)]аминопропил}феназин-1-карбоксамид (392 мг, 92%) в виде зелено-желтого масла, которое использовали непосредственно. 1H ЯМР (СDСl3) 1,61-1,67 (м, 4 Н, CH2CH2СН2NH2), 2,00 (квинтет, J=7, 1 Гц, 2 H, CH2СН2СН2HCOAr), 2,29 (с, 3 Н, NСН3), 2,46 (т, J= 7,2 Гц, 2 H, СН2СН2СН2NH2), 2,59 (т, J=7,2 Гц, 2 H, СН2CH2СН2NHCOAr), 2,75 (т, J= 6,8 Гц, 2 H, CH2NH2), 3,72 (к, J=6, 5 Гц, 2 H, СН2NHCOAr), 7,89-7,99 (м, 3 H, ArH), 8,22-8,25 (м, 1 H, ArH), 8,28-8,32 (м, 1 H, ArH), 8,40 (дд, J= 8,6, 1,5 Гц, 1 H, ArH), 9,01 (дд, J=7,1, 1,5 Гц, 1 H, ArH) и 11,01 (шир. с, 1 H, CONH).
Известную [Atwell, G.J,, Rewcastle, G.W., Baguley, B.C., Denny, W.A., J. Med. Chem. , 1987, 30, 664-669] акридин-4-карбоновую кислоту (274 мг, 1,23 ммоль) подвергали реакции с CDI (300 мг, 1,85 ммоль) с образованием имидазолида, который выделяли, как описано выше. Имидазолид диспергировали в ТГФ (15 мл), суспензию охлаждали до 0oС (вода/лед), затем медленно добавляли раствор вышеуказанного амина (392 мг, 1,12 ммоль) в ТГФ (10 мл). Реакционную смесь перемешивали в течение 2 ч при 0oС, затем 18 ч при комнатной температуре. Растворитель удаляли при пониженном давлении, и остаток распределяли между СН2Сl2 (100 мл) и 1 М (Nа2СО3) (100 мл). Слой СН2Cl2 сушили Na2SO4, растворитель удаляли при пониженном давлении, получая оранжевый твердый продукт, который очищали хроматографией на оксиде алюминия (0,5% МеОН в CH2Cl2 в качестве элюента) на диоксиде кремния (1% МеОН и 0,25% триэтиламин в СН2Cl2 в качестве элюента), получая N-1-{ 3-[{3-[(акридинил-4-карбонил) амино]пропил} (метил)амино]пропил} феназин-1-карбоксамид (75) (в виде бледно-желтого твердого продукта); т. пл. 171-173oС (СН2Сl2/н-гексан), 1H ЯМР (CDCl3) 1,88 (квинтет, J=5,6 Гц, 2 H, СН2CH2СН2), 2,02 (квинтет, J=6,0 Гц, СН2СН2СН2), 2,38 (с, 3 H, NCH3), 2,65-2,70 (м, 4 H, 2СН2NСН3), 3,62-3,68 (м, 2 H, СН2NHCO), 3,76 (к, J=6,5 Гц, 2 H, СН2NHCO), 7,11 (т, J=7,7 Гц, 1 H, ArH), 7,22-7,29 (м, 1 H, ArH), 7,36 (д, J=8,3 Гц, 1 H, ArH), 7,65 (ддд, J= 8,3, 6,9, 1,5 Гц, 1 H, ArH), 7,85-7,93 (м, 3 Н, ArH), 8,00 (дд, J=7,5, 0,9 Гц, 1 H, ArH), 8,09-8,13 (м, 1 H, ArH), 8,23-8,27 (м, 1 H, ArH), 8,36 (дд, J=8,6, 1,5 Гц, 1 H, ArH), 8,42 (д, J=8,1 Гц, 1 H, ArH), 8,53 (дд, J=8,0, 1,2 Гц, 1 H, ArH), 8,88 (дд, J=7,2, 1,5 Гц, 1 H, ArH), 9,15 (с, 1 H, H-9), 11,03 [шир. с, 1 H, NH (феназин)] и 12,55 [шир. с, 1 H, NH (акридин)]. Элементный анализ (C34H32N6O2 2 H2O) С, H, N.
Пример 76: Получение соединения 76 таблицы 1 способом по схеме 3Активирование и сочетание 9-метилфеназин-1-карбоновой кислоты [Rewcastle, G. W. , Denny, W. A., Baguley, B.C., J. Med. Chem., 1987, 30, 843-857] с N-(трет-бутоксикарбонил)-3,3′-диамино-N-метилдипропиламином, как в примере 75, давало N-1-(3-{N-метил-N[N-(трет-бутоксикарбонил)-3-аминопропил]} аминопропил)-9-метилфеназин-1- карбоксамид в виде желто-зеленого масла (89%), которое использовали непосредственно. 1H ЯМР (СDСl3): 1,41 (шир. с, 9 H, С(СН3)3), 1,65 (квинтет, J=6,6 Гц, 2 H, CH2СН2CH2NHBOC), 1,98 (квинтет, J=7,2 Гц, 2 H, СН2СН2СН2NHCOAr), 2,24 (с, 3 H, NСН3), 2,42 (т, J= 6,7 Гц, 2 H, СН2CH2СН2NHBOC), 2,53 (т, J=7,3 Гц, 2 H, СН2СН2СН2NHCOAr), 2,94 (с, 3 H, АrСН3), 3,12 3,23 (шир. с, 2 H, СН2NHBOC), 3,73 (к, J=6,7 Гц, 2 H, СН2NHCOAr), 5,39 (шир. с, 1 H, NHBOC), 7,77 (дт, J=6,5, 1,1 Гц, 1 H, ArH), 7,81 (дд, J=8,5, 6,8 Гц, 1 H, ArH), 7,97 (дд, J=8,7, 7,2 Гц, 1 H, ArH), 8,14 (д, J= 8,4 Гц, 1 H, ArH), 8,39 (дд, J=8,6, 1,5 Гц, 1 H, ArH), 9,02 (дд, J= 7,2, 1,5 Гц, 1 H, ArH) и 11,13 (шир. т, J=5,2 Гц, 1 H, CONH).
Удаление защитной группы у вышеуказанного БОК-защищенного амина, как в примере 75, давало N-1-{ 3-[N-метил-N-(3-аминопропил)] аминопропил}-9-метилфеназин-1- карбоксамид в виде зелено-желтого масла, (85%), который использовали непосредственно. 1H ЯМР (СDСl3) 1,62 (квинтет, J=7,0 Гц, 2Н, СН2СН2СН2NH2), 1,98 (квинтет, J=7,3 Гц, 2 Н, СН2СН2СН2NHCOAr), 2,26 (с, 3 Н, NСН3), 2,43 (т, J= 7,2 Гц, 2 Н, СН2СН2СН2NH2), 2,53 (т, J=7,3 Гц, 2 H, СН2СН2СН2NHCOAr), 2,75 (м, 2 Н, СН2NH2), 2,93 (с, 3 Н, АrСН3), 3,73 (к, J= 6,7 Гц, 2 H, СН2NHCOAr), 7,76 (дт, J=6,7, 1,3 Гц, 1 H, ArH), 7,81 (дд, J= 8,4, 6,9 Гц, 1 H, ArH), 7,97 (дд, J=8,6, 7,1 Гц, 1 H, 79 ArH), 8,13 (дд, J= 8,7, 1,0 Гц, 1 H, ArH), 8,38 (дд, J=8,7, 1,5 Гц, 1 H, ArH), 9,00 (дд, J=7,1, 1,5 Гц, 1 H, ArH) и 11,11 (шир. с, 1 H, CONH).
Активирование и сочетание 5-метилакридин-4-карбоновой кислоты [Atwell, D. J., Rewcastle, G.W., Baguley, B.C., Denny, W.A., J. Med. Chem., 1987, 30, 664-669] с вышеуказанным защищенным амином, как в примере 75, давало N-1-{ 3-[{ 3-[(5-метилакридинил-4 карбонил)амино] пропил} (метил)амино] пропил}-9 метилфеназин-1-карбоксамид (76) в виде бледно-желтого твердого продукта (66%), т. пл. 116-121oС (СН2Cl2/н-гексан). 1H ЯМР (CDCl3) 1,94-2,02 (м, 4 Н, 2СН2СН2СН2), 2,32 (с, 3 Н, NСН3), 2,58-2,63 (м, 4 Н, 2СН2NCH3), 2,73 (с, 3 Н, АrСН3), 2,80 (с, 3 Н, АrСН3), 3,66-3,74 (м, 4 Н, 2СН2NHCO), 7,31 (дд, J=8,4, 6,8 Гц, 1 Н, ArH), 7,52 (м, 2 H, ArH), 7,59 (дд, J=8,2, 7,2 Гц, 1 H, ArH), 7,63-7,69 (м, 2 H, ArH), 7,90 (дд, J=8,7, 7,2 Гц, 1 H, ArH), 7,95-8,00 (м, 2 H, ArH), 8,28 (дд, J=8,7, 1,5 Гц, 1 H, ArH), 8,61 (c, 1 H, Н-9), 8,90-8,95 (м, 2 H, ArH), 10,87 [шир. с, 1 H, NH (феназин) ] и 11,78 [шир. с, 1 Н, NH (акридин)].
Анализ для N-1-{ 3-[{ 3-[(5-метилакридинил-4-карбонил) амино] пропил} (метил)амино] пропил}-9-метилфеназин-1- карбоксамида (76). Элементный анализ (C36H36N6O2 H2O) С, Н, N.
Пример 77: Получение соединения 77 таблицы 1 способом по схеме 3.
Активирование и сочетание феназин-1-карбоновой кислоты [Rewcastle, G.W., Denny, W. A., Baguley, B.C., J. Med. Chem., 1987, 30,843-857] с N-1-{3-[N-метил-N-(3-аминопропил)] аминопропил} -9-метилфеназин-1- карбоксамидом [для получения см. пример 76], как указано выше, давали N-1-{3-[{3-[(феназинил-1-карбонил) амино]пропил} (метил)амино] пропил}-9-метилфеназин-1- карбоксамид (77) в виде желтого твердого продукта, 77%, т.пл. 120oС (разложение) (СН2Сl2/н-гексан). 1H ЯМР (СDСl3) 1,96-2,07 (м, 4 Н, 2СН2CH2CH2), 2,36 (с, 3Н, NСН3), 2,65-2,73 (м, 7 Н, 2CH2NСН3 и АrСН3), 3,68-3,78 (м, 4Н, 2СН2NHCO), 7,47 (дт, J=6,9, 1,1 Гц, 1 Н, ArH), 7,57 (ддд, J=8,7, 6,6, 1,2 Гц, 1 Н, ArH), 7,63 (дд, J=8,7, 6,8 Гц, 1Н, ArH), 7,71 (ддд, J=8,7, 6,7, 1,5 Гц, 1Н, ArH), 7,87 (дд, J=8,7, 7,2 Гц, 1Н, ArH), 7,90 (дд, J=8,6, 7,1 Гц, 1Н, ArH), 7,94 (д, J=9,2 Гц, 1Н, ArH), 8,00 (д, J=8,6 Гц, 1Н, ArH), 8,08 (д, J= 8,5 Гц, 1Н, ArH), 8,21 (дд, J=8,7, 1,5 Гц, 1Н, ArH), 8,31 (дд, J=8,7, 1,5 Гц, 1H, ArH), 8,89 (дд, J=7,1, 1,5 Гц, 1 Н, ArH), 8,92 (дд, J=7,2, 1,5 Гц, 1H, ArH) и 10,87 (шир. с, 2 Н, 2NH).
Анализ для N-1-{3-[{3-[(феназинил-1-карбонил) амино]пропил} (метил)амино]пропил} -9-метилфеназин-1- карбоксамида (77). Элементный анализ (С34Н33N7O2 Н2О) С, Н, N.
Пример 78: Получение соединения 78 таблицы 1 способом по схеме 3.
Триэтилентетрамин подвергали реакции с ди-трет-бутилдикарбонатом по способу Blagbrough et al (Pharm. Sciences, 1997, в печати, частное сообщение), получая, после очистки колоночной хроматографией (20% МеОН в CH2Cl2 в качестве элюента), N-аминоэтил-N,N’-бис(трет-бутоксикарбонил) -N-{(N-трет-бутоксикарбонил) аминоэтил] этилендиамин в виде бледно-желтого вязкого масла (59%). 1ЯMP (СDСl3) 1,39-1,50 [м, 29 Н, 3С(СН3)3, NH2], 3,00-3,62 (М, 12 Н, 6СН2), 4,45 (шир. с, 1 Н, NHBOC).
5-Метилакридин-4-карбоновой кислоте (1,00 г, 4,23 ммоль) давали реагировать с CDI (1,02 г, 6,33 ммоль) для получения имидазолида, как описано выше. Имидазолид суспендировали в ТГФ (80 мл) при комнатной температуре и медленно добавляли раствор указанного выше амина (2,04 г, 4,65 ммоль) в ТГФ (20 мл). Реакционную смесь затем перемешивали в течение 18 ч при 20oС. ТГФ удаляли при пониженном давлении, и полученный желтый твердый продукт распределяли между СН2Сl2 (200 мл) и 1 М Na2СО3 (200 мл). Слой СH2Сl2 сушили NaSO4, и растворитель удаляли при пониженном давлении, получая желтое масло, которое очищали колоночной хроматографией на оксиде алюминия (0,5% МеОН в СН2Сl2 в качестве элюента), получая три-БОК-аналог N-1-[2-(N-{2-[N-(2-аминоэтил)] аминоэтил} )аминоэтил] -5-метилакридин-4-карбоксамида в виде желтой пены (1,41 г, 51%). 1H ЯМР (СDСl3) 1,46 [м, 30 Н, 3С(СН3)3, АrСН3], 3,13-3,65 (м, 12 Н, 6СН2), 4,43 (шир. с, 1 Н, NHBOC), 7,48-7,55 (м, 1 H, ArH), 7,64-7,74 (м, 2 H, ArH), 7,88-7,94 (м, 1 H, ArH), 8,12-8,19 (м, 1 H, ArH), 8,85-8,90 (м, 1 H, ArH), 8,93-9,00 (м, 1 H, ArH), 12,23 (шир. с, 1 H, NH).
Трифторуксусную кислоту (10 мл) добавляли к раствору вышеуказанного три-БОК-амида (1,00 г, 1,52 ммоль) в СН2Cl2 (10 мл). Смесь перемешивали при 20oС в течение 2 ч, в этой точке по данным ТСХ реакция была завершена. Все растворители удаляли при пониженном давлении, и масляный остаток распределяли между СНСl3 (100 мл) и насыщенным Nа2СО3 (20 мл). Водный слой далее экстрагировали дополнительным СНСl3 (11100 мл), и все экстракты в СНСl3 объединяли и сушили Na2SO4. Растворитель удаляли при пониженном давлении, получая N-1-[2-(N-{ 2-[N-(2-аминоэтил)] аминоэтил} )аминоэтил]-5-метилакридин-4-карбоксамид (533 мг, 98%) в виде желтого масла. 1H ЯМР (СDСl3) 2,58-2,68 (м, 2 H, СН2), 2,70-2,76 (м, 4 H, 2СН2), 2,82-2,86 (м, 2 H, СН2), 2,93 (с, 3 H, АrСН3), 3,03-3,07 (м, 2 H, СН2), 3,82-3,88 (м, 2 H, СH2), 7,49-7,55 (м, 1 H, ArH), 7,65-7,75 (м, 2 H, ArH), 7,89-7,94 (м, 1 H, ArH), 8,13-8,18 (м, 1 H, ArH), 8,88 (шир. д, J=5,3 Гц, 1 H, ArH), 8,98 (тд, J=7,1, 1,5 Гц, 1 H, ArH), 12,04 (шир. с, 1 H, NH).
9-Метилфеназин-1-карбоновой кислоте (1,00 г, 4,20 ммоль) давали реагировать с CDI (1,02 г, 6,30 ммоль) с образованием имидазолида, который выделяли, как описано выше. Этот имидазолид суспендировали в ТГФ (20 мл), суспензию охлаждали до 0oС (лед/вода), затем медленно добавляли раствор вышеуказанного полиамина (529 мг, 1,48 ммоль) в ТГФ (20 мл). Реакционную смесь перемешивали в течение 2 ч при 0oС, затем 18 ч при комнатной температуре. Растворитель удаляли при пониженном давлении, и остаток распределяли между СН2Cl2 (100 мл) и 1 М Na2СО3 (100 мл). Слой СН2Cl2 сушили Na2SO4, и растворитель удаляли при пониженном давлении, получая желтый твердый продукт (553 мг, 61%), который очищали колоночной хроматографией на оксиде алюминия (2% МеОН в CH2Cl2 в качестве элюента), получая N-1(2-{[2-({2- [(5-метилакридинил-4-карбонил) амино]этил}амино)этил] амино}этил)-9-метилфеназин-1- карбоксамид (78) в виде желтого твердого продукта. 1H ЯМР (СDСl3) 2,77 (с, 3 Н, АrСН3), 2,79 (с, 3 Н, АrСН3), 2,82, 2,88 (м, 4 Н, 2СН2), 2,97 (т, J=6,0 Гц, 2 Н, СН2), 3,02 (т, J=6,0 Гц, 2 Н, СН2), 3,70 (к, J=5,9 Гц, 2 Н, СН2), 3,79 (к, J=6,0 Гц, 2 Н, СН2), 7,31 (дд, J=8,4, 6,8 Гц, 1 Н, АrН), 7,51-7,70 (м, 5 H, ArH), 7,87 (дд, J=8,5, 7,1 Гц, 1 Н, АrН), 7,91-7,96 (м, 2 Н, АrН), 8,26 (дд, J=8,7, 1,5 Гц, 1 Н, АrН), 8,57 [с, 1 Н, Н-9 (акридин)], 8,85-8,91 (м, 2 Н, АrН), 10,83 [шир. т, J=5,1 Гц, 1 Н, NH (феназин)], 11,83 [шир. т, J=5,4 Гц, 1 Н, NH (акридин)].
Пример 79: Получение соединения 79 таблицы 1 способом по схеме 3.
Этилендиамин алкилировали избытком хлорацетонитрила по способу Overman и Burk [Bradshaw et al, Tetrohedron, 1992, 48, 4475], и продукт очищали фильтрованием через подушку флэш-диоксида кремния (1% МеОН в СН2Cl3 в качестве элюента), получая N,N’-бис(цианометил)этилендиамин в виде желтого масла, которое отверждалось при охлаждении *88%, т. пл. 41-42oС. 1H ЯМР (СDСl3) 1,58 (с, 2 Н, 2NH), 2,90 (с, 4 Н, 2СН2), 3, 63 (с, 4 Н, 2СН2).
Вышеуказанный диамин (3,00 г, 21,7 ммоль) в смеси ТГФ (90 мл), воды (10 мл) и триэтиламина (10 мл) обрабатывали ди-трет-бутилдикарбонатом (19,0 г, 87,0 ммоль). Все растворители удаляли при пониженном давлении, и полученный остаток распределяли между водой (100 мл) и EtOAc (2100 мл). Объединенные слои EtOAc сушили (Na2SO4), растворитель удаляли, и полученный бледно-коричневый твердый продукт очищали флэш-хроматографией на диоксиде кремния (50% EtOAc/PE в качестве элюента), получая N,N’-бис(трет-бутоксикарбонил) -N, N’-бис(цианометил) этилендиамин в виде белого твердого продукта (6,59 г, 90%), т. пл. 112-113oС (EtOAc/петролейный эфир). 1H ЯМР (СDСl3) 1,50 [с, 18 Н, 2С(СН3)3], 3,52 (с, 4 Н, 2СН2), 4,13 (шир. с, 4 Н, 2СН2).
Гидрирование вышеуказанного соединения с никелем Ренея W-7 по описанной методике [Ravikumar, Syn. Commun., 1994, 24, 1767] давало N,N’-бис(аминоэтил)-N, N’-бис (трет-бутоксикарбонил) этилендиамин в виде белого твердого продукта (100%), т. пл. 81-82oС. 1H ЯМР (CDCl3) 1,46 [с, 18 Н, 2(СН3)3] , 1,57 (шир. с, 4 Н, 2NH2), 2,83 (шир. с, 4 Н, 2СН2), 3,30 (шир. м, 8 Н, 4СН2).
Указанному выше диамину давали реагировать с этилтрифторацетатом по способу Blagbrough et al (Pharm. Sciences, 1997, в печати; частное сообщение), получая N-аминоэтил-N, N’-бис(трет-бутоксикарбонил)-N'[(N-трифторацетамидо)аминоэтил] этилендиамин в виде светлого масла (39%). 1ЯМР (СDСl3) 1,46 [с, 18 Н, 2С(СН3)3] , 1,95 (шир. с, 2 Н, NH2), 2,84-2,96 (м, 2 Н, СН2), 3,20-3,48 (м, 10 Н, 5СН2), 7,99, 8,21, 8,51 (все шир. с, общий 1 Н, NH).
Феназин-1-карбоновую кислоту (212 мг, 0,95 ммоль) подвергали реакции с CDI (230 мг, 1,42 ммоль) для полученияимидазолида, который выделяли, как описано выше. Этот имидазолид затем суспендировали в ТГФ (10 мл), и к раствору медленно добавляли раствор вышеуказанного монотрифторацетамида (416 мг, 0,95 ммоль) в ТГФ (10 мл). Эту смесь оставляли при перемешивании при комнатной температуре в течение 18 ч, после чего растворитель удаляли при пониженном давлении, и остаток распределяли между CH2Cl2 (100 мл) и 1 М Nа2СО3 (50 мл). Слой СН2Сl2 затем сушили Nа2SO4, и растворитель удаляли при пониженном давлении, получая остаток, который очищали колоночной хроматографией на оксиде алюминия (0,5% МеОН в СН2Сl2 в качестве элюента), получая N-1-{2-[N’-трет-бутоксикарбонил-N’-(2-{ N-трет-бутоксикарбонил-N-[2-N-трифторацетамидо) аминоэтил] аминоэтил)] аминоэтил} феназин-1-карбоксамид в виде желтого масла (558 мг, 91%). 1H ЯМР (CDCl3) 1,39 [шир. с, 9 Н, С(СН3)3], 1,43 [шир. с, 9 Н, С(СН3)3], 3,31-3,68 (м, 10 Н, 5СН2), 3,82-3,89 (м, 2 Н, СН2), 7,90-8,44 [м, 7 Н, 6АrН, NНС(O)СF3], 8,99 (шир. с, 1 Н, АrН), 11,13 (шир. с, 1 Н, CONH).
Раствор вышеуказанного трифторацетамида (548 мг, 0,85 ммоль) в смеси МеОН (30 мл) и Н2O (20 мл) обрабатывали К2СО3 (584 мг, 4,23 ммоль). Эту реакционную смесь перемешивали и кипятили с обратным холодильником в течение 2 ч, затем перемешивали при комнатной температуре в течение 18 ч. Полное превращение трифторацетамида в свободное основание наблюдали методом ТСХ. МеОН удаляли при пониженном давлении, затем к остатку добавляли насыщенный Na2СО3 (20 мл), и эту водную часть экстрагировали СНСl3 (250 мл). Объединенные части СНСl3 сушили Na2SO4, затем растворитель удаляли при пониженном давлении, получая N-1-[2-(N’-трет-бутоксикарбонил -N’-{2-[N-(аминоэтил)-N-трет-бутоксикарбонил] аминоэтил} )аминоэтил] феназин-1-карбоксамид (470 мг, 100%). 1H ЯМР (СDСl3) 1,38 [шир. с, 9 Н, С(СН3)3], 1,43 [шир. с, 9 Н, С(СН3)3] , 2,78 (т, J=6,5 Гц, 2 Н, NH2), 3,18-3,70 (м, 10 Н, 5СН2), 3,81-3,90 (м, 2 Н, СН2), 7,89-8,00 (м, 3 Н, ArH), 8,21-8,43 (м, 3 Н, ArH), 9,00 (шир. с, 1 Н, ArH), 11,08 (шир. с, 1 Н, CONH).
Акридин-4-карбоновую кислоту (192 мг, 0,86 ммоль) подвергали реакции с CDI (210 мг, 1,29 ммоль) для получения имидазолида, который выделяли, как описано выше. Получаемый имидазолид затем суспендировали в ТГФ (20 мл), и к перемешиваемому раствору медленно добавляли раствор вышеуказанного амина (470 мг, 0,86 ммоль) в ТГФ (10 мл). Эту смесь оставляли для перемешивания при комнатной температуре в течение 18 ч, после чего растворитель удаляли при пониженном давлении, и остаток распределяли между СН2Сl2 (100 мл) и 1 М Nа2СО3 (100 мл). Слой СН2Сl2 затем сушили над Na2SO4, и растворитель удаляли при пониженном давлении, получая остаток, который очищали колоночной хроматографией на оксиде алюминия (0,5% МеОН в СН2Сl2 в качестве элюента), получая N-1-(2-{ [2-({2-[(акридинил-4-карбонил) амино]этил}-N-трет-бутоксикарбониламино) этил]-N’-трет-бутоксикарбониламино} этил) феназин-1-карбоксамид в виде желтого масла (568 мг, 89%). 1H ЯМР (СDСl3) 1,42 [шир. с, 9 Н, С(СН3)3], 1,46 [шир. с, 9 Н, С(СН3)3], 3,37-3,69 (м, 10 Н, 5СН2), 3,81-3,89 (м, 2 Н, СН2, 7,09-7,15 (м, 1 Н, ArH), 7,25 (т, J=7,4 Гц, 1 Н, ArH), 7,36 (д, J= 8,3 Гц, 1 Н, ArH), 7,64 (т, J=7,2 Гц, 1 Н, ArH), 7,83-7,97 (м, 4 Н, ArH), 8,11-8,27 (м, 3 Н, ArH), 8,31-8,45 (м, 2 H, ArH), 8,53-8,61 (шир. с, 1 H, ArH), 8,89-8,99 (шир. с, 1 H, ArH), 11,10 [с, 1 H, CONH (феназин)], 12,52 [с, 1 H, CONH (акридин)].
Газообразный НСl барботировали через МеОН до тех пор, пока раствор не становился сильно кислотным по индикаторной бумаге для определения рН. Вышеуказанный защищенный амин растворяли в этом растворе (20 мл) и перемешивали при комнатной температуре в течение 18 ч. МеОН удаляли при пониженном давлении, и остаток растворяли в насыщенном Nа2СО3 (50 мл), который затем экстрагировали СНСl3 (1350 мл). Объединенные экстракты СНСl3 сушили Na2SO4, затем растворитель удаляли при пониженном давлении, получая N-1-(2-{[2-({ 2-[(акридинил-4-карбонил)амино] этил}амино)-этил] амино}этил) феназин-1-карбоксамид (79) в виде желтого твердого продукта (159 мг, 96%), т. пл. 173-176oС (СН2Cl2/MeOH). 1H ЯМР (СDСl3) 2,83-2,96 (м, 6 H, 3СН2), 3,07 (т, J= 5,9 Гц, 2 H, СН2), 3,46 (к, J=5,3 Гц, 2 H, СН2), 3,80 (к, J=5,8 Гц, 2 H, СН2), 7,01 (т, J= 7,8 Гц, 1 H, ArH), 7,25 (ддд, J=8,1, 7,2, 0,9 Гц, 1 H, ArH), 7,32 (д, J=8,2 Гц, 1 H, ArH), 7,55 (шир. с, 2 H, ArH), 7,65 (ддд, J= 8,3, 7,0, 1,5 Гц, 1 H, ArH), 7,79-7,90 (м, 4 H, ArH), 8,12-8,16 (м, 2 H, ArH), 8,26 (дд, J= 8,7, 1,5 Гц, 1 H, ArH), 8,39 (дд, J=8,1, 1,3 Гц, 1 H, ArH), 8,43 (дд, J= 8,1, 1,4 Гц, 1 H, ArH), 8,89 (дд, J=7,1, 1,5 Гц, 1 H, ArH), 11,11 [c, 1 H, CONH (феназин)], 12,20 [c, 1 H, CONH (акридин)].
Пример 80: Биологическое испытание соединений изобретения in vitroЦитотоксичность in vitro соединений данного изобретения оценивали изучением лейкемии Р388 мышей, LLTC-линии последнего пассирования карциномы легких мышей Lewis и линии лейкемии дикого типа человека (Jurkat; JLC). Клетки выращивали, как описано в Finlay et al. в Oncol. Res. 1994, 6, 33-37 и в Eur. J. Cancer 1996, 32A, 708-714, и анализы ингибирования роста проводили путем культивирования клеток при 4 103 (Р388), 103 (LLTC) и 3,75103 (линии Jurkat) на лунку в планшетах для микрокультур (150 мкл на лунку) в течение 3 (Р388) или 4 дней в присутствии лекарственного средства. Рост клеток определяли по поглощению [H3] TdR (Р388), как описано в Marshall et al. в J. Natl. Cancer Inst. 1992, 84, 340-345, или анализом с использованием сульфородамина, как описано в Skehan et al. в J. Natl. Cancer Inst. 1990, 82, 1107-1112. Независимые анализы проводили в повторениях, и коэффициенты изменений составляли 12% (Р388), 12% (LLTC) и 6,3% (JLC).
Биологическая активность выбранных соединений формулы (I), приведенных в табл. 1, приведена в табл. 2.
Испытание in vivoВыбранные соединения общей формулы (I) проявляли также активность in vivo против подкожных опухолей 38 ободочной кишки у мышей. Эти данные проиллюстрированы на чертеже для соединений 13 (при 60 мг/кг) и 17 (при 90 мг/кг). В этом анализе опухоли ободочной кишки 38 выращивали подкожно из фрагментов 1 мм3, имплантированных подкожно в один бок анестезированных мышей (пентобарбитон 90 мг/кг). Обработку начинали сразу, как только опухоли достигали диаметра приблизительно 4 мм (обычно через 7 дней). Мышей делили на группу из 5 особей для контроля и группу, которую обрабатывают лекарственными средствами, с одинаковыми средними объемами опухолей для каждой из групп. Лекарственные средства растворяли либо в дистиллированной воде, либо в 15% водном этаноле, и инъецировали в объеме 0,01 мл на 1 г массы тела в виде разделенной дозы из двух равных инъекций, вводимых порознь, причем вторую вводят через один час после первой. Мышей тщательно обследовали и взвешивали через 7 дней для контролирования вызванной лекарственным средством потери массы. Мышей умерщвляли, когда средний диаметр опухоли достигал 20 мм. Диаметры опухолей измеряли кронциркулями три раза в неделю, и объем опухолей вычисляли как 0,52 а2b, где а и b представляют собой меньшую и большую оси опухолей. Данные наносили на 1/2 логарифмический чертеж (средний объем опухоли от времени после обработки), и вычисляли время, необходимое для достижения среднего объема опухолей, в четыре раза большего, чем их объем до обработки. Из этого вычисляли задержку роста опухолей относительно контрольных мышей. ПРИМЕР 81 Таблетки массой 0,15 г каждая, содержащие 25 мг активного ингредиента (соединение настоящего изобретения). Состав для 10000 таблеток, г: Активный ингредиент (любое из соединений настоящего изобретения) – 250 Лактоза – 800 Кукурузный крахмал – 415 Тальк (порошок) – 30 Стеарат магния – 5 Соединение изобретения, лактозу и половину требуемого количества кукурузного крахмала смешивают, смесь затем пропускают через сито с размером ячеек 0,5 мм. Кукурузный крахмал (10 г) суспендируют в теплой воде (90 мл). Полученную пасту используют для получения гранулированного порошка. Гранулят сушат, измельчают и пропускают через сито с размером ячеек 1,4 мм. Добавляют оставшееся количество крахмала, тальк и стеарат магния, тщательно смешивают и прессуют с получением таблеток. Формула изобретения
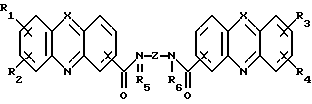 где каждый из X, которые могут быть одинаковыми или различными в данной молекуле, представляет -СН= или -N= ; каждый из R1-R4, которые могут быть одинаковыми или различными, представляет Н, С1-С4-алкил, NH2, С1-С4-алкокси, фенил, фенокси, NHR, N(R)2, СF3 или галоген, где R – С1-С4алкил; каждый из R5 и R6, которые могут быть одинаковыми или различными, представляет Н или С1-С4-алкил; Z – (CH2)nN(R7)(CH2)n, (CH2)nN(R7)(CH2)N(R7)(CH2)n или (CH2)nNH(CH2CH2)2NH(CH2)n, где R7 – Н или С1-С4-алкил; n и m, которые могут быть одинаковыми или различными, каждый равен целому числу от 1 до 4, или их фармацевтически приемлемые кислотно-аддитивные соли, за исключением соединений, где каждый Х – N, каждый из R1-R6 – Н, карбоксамидная часть присоединена в положении 1 каждого кольца феназина и Z – (CH2)2NH(CH2)2, (СН2)3NH(СН2)3, (CH2)3NH(CH2CH2)2NH(CH2)3, (CH2)2NH(CH2)2NH(CH2)2 или (CH2)3NH(СН2)2NH(СН2)3. 2. Соединение по п. 1, которое представляет собой производное бис(акридинкарбоксамида) формулы (Iа)  где каждый из R1 и R3, которые одинаковы или различны, представляет С1-С4-алкокси, С1-С4-алкил или галоген; каждый из R2 и R4, которые одинаковы или различны, представляет водород, С1-С4-алкокси, С1-С4-алкил или галоген; каждый из R5 и R6 – Н, или его фармацевтически приемлемую соль. 3. Соединение по п. 1, которое представляет собой производное бис(феназинкарбоксамида) формулы (Ib) 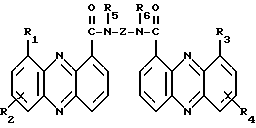 где каждый из R1 и R3, которые одинаковы или различны, представляет С1-С4-алкокси, С1-С4-алкил или галоген; каждый из R2 и R4, которые одинаковы или различны, представляет водород, С1-С4-алкокси, С1-С4-алкил или галоген; каждый из R5 и R6 – Н, или его фармацевтически приемлемую соль. 4. Соединение по п. 1, обладающее противоопухолевой активностью. 5. Способ получения соединения по п. 1, отличающийся тем, что 2 моль производного акридинкарбоновой кислоты или 9-азаакридинкарбоновой кислоты или эквимолярную смесь этих соединений общей формулы (II) 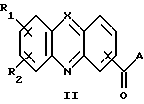 где R1, R2 и Х такие, как определено в п. 1; А – ОН, Сl или N-имидазолил, подвергают взаимодействию с 1 моль бис(амина) формулы (III) NHR5-Z-NHR6 где R5, R6 и Z такие, как определено в п. 1, и, если необходимо, превращение полученного соединения в его фармацевтически приемлемую кислотно-аддитивную соль. 6. Фармацевтическая композиция, обладающая противоопухолевой активностью, включающая фармацевтически приемлемый носитель или разбавитель и в качестве активного ингредиента соединение, определенное в п. 1. 7. Соединение по п. 1 в качестве противоопухолевого агента. РИСУНКИ
|
||||||||||||||||||||||||||