Патент на изобретение №2179969
|
||||||||||||||||||||||||||
(54) ПРОИЗВОДНЫЕ КАРБАМИНОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ
(57) Реферат: Изобретение относится к новым производным карбаминовой кислоты общей формулы 1, где R1 и R2 независимо друг от друга обозначают алкил, арил, аралкил, гетероарил, циклоалкил или гетероциклил, R3 и R4 независимо друг от друга обозначают водород, алкил, галоген, гидроксил или арил, R5 обозначает -COOR6, R6 обозначает водород или алкил, А обозначает алкилен или алкенилен, В обозначает -O(СН2)m– или -(СН2)n-, m обозначает целое число от 1 до 8 включительно, n обозначает целое число от 0 до 8 включительно, или к их рацематам, или индивидуальных изомерам, или к их новым фармацевтически приемлемым солям, или сольватам. Изобретение может быть использовано в качестве терапевтических лекарственных средств. Также раскрыты способ получения этих соединений, фармацевтическая композиция на основе новых производных карбаминовой кислоты и способ лечения болезненного состояния, связанного с перемежающейся хромотой. 4 с. и 16 з.п. ф-лы, 2 табл. 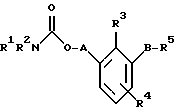
Настоящее изобретение относится к модуляторам I2-(IP-) рецептора простагландина, прежде всего к агонистам IP-рецептора, в частности к некоторым арилкарбоновым кислотам и к арилтетразоловым производным, к содержащим их фармацевтическим композициям и к способам их применения в качестве терапевтических лекарственных средств. Предпосылки создания изобретения Простациклин (PGI2) является представителем семейства простагландинов и представляет собой эндогенный агонистический лиганд для IP-рецептора. PGI2 обуславливает многочисленные физиологические и фармакологические воздействия на весь организм и оказывает заметное влияние на сердечнососудистую систему, в особенности на кровеносные сосуды, различные кровяные клетки, включая кровяные пластинки, почки, вегетативные нервы и компоненты воспалительной и иммунной систем. Так, например, в сердечно-сосудистой системе PGI2 вызывает сильное расширение сосудов, следствием которого является гипотензия. Он также оказывает действие на несосудистую гладкую мускулатуру, вызывая увеличение просвета бронхов, релаксацию матки и сокращение гладкой мускулатуры желудочно-кишечного тракта. Кроме того, он уменьшает значение рН, содержание пепсина и общую секрецию желудочного сока. В крови PGI2 ингибирует агрегацию кровяных пластинок и усиливает антитромбогенные свойства неповрежденной стенки сосуда. Предполагается также, что применение стабильных аналогичных имитаторов PGI2 позволяет ингибировать осаждение кровяных пластинок на тромбогенных поверхностях, таких, как атеросклеротические бляшки. В почках PGI2 вызывает диурез, натрийурез, калийурез и вызывает секрецию ренина из коркового вещества почки. Вследствие неустойчивости PGI2 было создано множество химически уникальных аналогов, но у них отсутствует селективность в отношении рецептора и/или они быстро разлагаются вследствие биотрансформации. В настоящее время существует потребность в эффективных хорошо переносимых и высоко селективных агонистах IP-рецептора с фармакокинетикой, пригодной для длительного удобного (т. е. ежедневно, два раза в день, три раза в день) перорального дозирования. Соединения по настоящему изобретению и содержащие их композиции предназначены для решения этой задачи и могут быть использованы для лечения самых разнообразных расстройств с незначительными побочными эффектами. В патенте US 3649637 (Howes и др.) описаны некоторые фенокси-тетразоловые производные, которые пригодны для использования при лечении воспалительных заболеваний. В патенте US 4878942 (Motegi и др.) описаны некоторые бензамидные производные, которые обладают гербицидным и регулирующим рост растений действием. В US 5378716, US 5536736, US 5703099, US 5935985 (Hamaka и др.) и ЕР 558062 В1 описаны некоторые производные феноксиуксусной кислоты, которые обладают способностью ингибировать действие IP-рецептора, вызывающее агрегацию кровяных пластинок. В патенте US 5763489 (Taniguchi и др.) и WO 95/24393 описаны некоторые нафталиновые производные, которые обладают агонистическим действием в отношении IP-рецептора и используются при лечении артериальной непроходимости, рестеноза, артериосклероза, цереброваскулярных заболеваний или ишемических болезней сердца. В заявке GB 1079414 (права на которую принадлежат фирме Smith & Nephew) описаны некоторые производные N-фенил-о-карбамоилфеноксиуксусной кислоты, которые проявляют болеутоляющее и противовоспалительное действие. В заявке DE 2432560 (права на которую принадлежат фирме Boehringer Mannheim) описаны некоторые производные 2-(4-карбанилоилалкил)феноксиалкановой кислоты, которые могут использоваться при лечении атеросклероза и в качестве промежуточных продуктов для получения антибиотиков  –лактамового строения.
В заявке WO 99/24397 (права на которую принадлежат фирме Fujisawa) описаны некоторые бензоциклогептеновые производные, которые обладают агонистическим действием в отношении IP-рецептора и используются при лечении артериальной непроходимости, цереброваскулярных заболеваний, цирроза печени, артериосклероза, ишемических болезней сердца, рестеноза после подкожной и внутрисосудистой коронарной ангиопластики, гипертензии и дерматоза.
В заявке WO 99/32435 (права на которую принадлежат фирме Fujisawa) описаны некоторые нафталиновые производные, которые обладают агонистическим действием в отношении IP-рецептора и могут использоваться при лечении артериосклероза, цереброваскулярных заболеваний, ишемических болезней сердца, дерматоза, воспалительных заболеваний пищевого тракта и для подавления метастазирования рака.
У Vavayannis и др. в Eur. J. Med. Chem. Chim. Ther. 1985, 20, 37-42, описаны некоторые диметилкарбаматные производные, которые проявляют антихолинэстеразную активность.
У Marsh и др. в J. Chem. Soc. Chem. Commun. 1996, 8, 941-942, описаны некоторые твердофазные полиаминовые линкеры, которые пригодны для использования при синтезе и получении библиотек, направленных против трипанотионредуктазы.
Все публикации, патенты и заявки на патент, упомянутые выше или ниже в настоящем описании, в полном объеме включены в настоящее описание в качестве ссылки.
По настоящему изобретению предлагаются соединения формулы I: –лактамового строения.
В заявке WO 99/24397 (права на которую принадлежат фирме Fujisawa) описаны некоторые бензоциклогептеновые производные, которые обладают агонистическим действием в отношении IP-рецептора и используются при лечении артериальной непроходимости, цереброваскулярных заболеваний, цирроза печени, артериосклероза, ишемических болезней сердца, рестеноза после подкожной и внутрисосудистой коронарной ангиопластики, гипертензии и дерматоза.
В заявке WO 99/32435 (права на которую принадлежат фирме Fujisawa) описаны некоторые нафталиновые производные, которые обладают агонистическим действием в отношении IP-рецептора и могут использоваться при лечении артериосклероза, цереброваскулярных заболеваний, ишемических болезней сердца, дерматоза, воспалительных заболеваний пищевого тракта и для подавления метастазирования рака.
У Vavayannis и др. в Eur. J. Med. Chem. Chim. Ther. 1985, 20, 37-42, описаны некоторые диметилкарбаматные производные, которые проявляют антихолинэстеразную активность.
У Marsh и др. в J. Chem. Soc. Chem. Commun. 1996, 8, 941-942, описаны некоторые твердофазные полиаминовые линкеры, которые пригодны для использования при синтезе и получении библиотек, направленных против трипанотионредуктазы.
Все публикации, патенты и заявки на патент, упомянутые выше или ниже в настоящем описании, в полном объеме включены в настоящее описание в качестве ссылки.
По настоящему изобретению предлагаются соединения формулы I: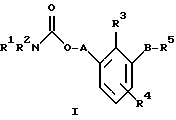 в которой R1 и R2 каждый независимо друг от друга в каждом случае обозначает алкил, арил, аралкил, гетероарил, циклоалкил или гетероциклил, R3 и R4 каждый независимо друг от друга в каждом случае обозначает водород, алкил, алкокси-, аминогруппу, галоген, галоидалкил, гидроксиалкил, нитрогруппу, арил, аралкил или гетероциклил, R5 в каждом случае независимо обозначает -COOR6 или тетразолил, R6 в каждом случае независимо обозначает водород или алкил, А в каждом случае независимо обозначает алкилен или алкенилен, В в каждом случае независимо обозначает -О(СН2)m– или -(СН2)n-, m в каждом случае независимо обозначает целое число от 1 до 8 включительно, n в каждом случае независимо обозначает целое число от 0 до 8 включительно, или их индивидуальные изомеры, рацемические и нерацемические смеси изомеров либо их фармацевтически приемлемые соли и сольваты. Далее настоящее изобретение относится к фармацевтическим композициям, включающим терапевтически эффективное количество по меньшей мере одного соединения формулы I, его индивидуального изомера, рацемической или нерацемической смеси изомеров, фармацевтически приемлемой соли или сольвата, в смеси с по меньшей мере одним приемлемым носителем. В предпочтительном варианте эти фармацевтические композиции могут быть использованы для введения в организм субъекта, страдающего заболеванием, которого может быть облегчено лечением с помощью модулятора IP-рецептора, в частности агониста IP-рецептора. Кроме того, настоящее изобретение относится к фармацевтическим композициям, пригодным для введения субъекту и включающим терапевтически эффективное количество по меньшей мере одного соединения формулы I, его индивидуального изомера, рацемической или нерацемической смеси изомеров, фармацевтически приемлемой соли или сольвата, в смеси с по меньшей мере одним приемлемым носителем. Настоящее изобретение относится также к способам лечения, включающим введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества по меньшей мере одного соединения формулы I, его индивидуального изомера, рацемической или нерацемической смеси изомеров, фармацевтически приемлемой соли или сольвата. В предпочтительном варианте субъект, нуждающийся в таком лечении, страдает заболеванием, связанным с неправильным заживанием раны, некрозом ткани, преждевременным сокращением матки, язвой желудка, половой дисфункцией у мужчин и женщин (самцов и самок), сильной менструальной болью, нарушением иммунорегуляции, нарушением агрегации кровяных пластинок или нарушением нейтрофильной функции. В другом предпочтительном варианте соединение формулы I, его индивидуальный изомер, рацемическая или нерацемическая смесь изомеров, фармацевтически приемлемая соль или сольват представляет собой модулятор IP-рецептора, в частности агонист IP-рецептора. Далее, настоящее изобретение относится к способам лечения, включающим введение в организм субъекта, страдающего заболеванием, связанным с нарушением кровотока, терапевтически эффективного количества по меньшей мере одного соединения формулы I, его индивидуального изомера, рацемической или нерацемической смеси изомеров, фармацевтически приемлемой соли или сольвата. В предпочтительном варианте терапии подвергают субъекта, страдающего сердечно-сосудистым заболеванием, гипертензивным заболеванием, ишемической болезнью или почечной недостаточностью. В более предпочтительном варианте терапии подвергают субъекта, страдающего сердечнососудистым заболеванием, которое представляет собой окклюзионное поражение периферической артерии (ОППА), перемежающуюся хромоту, критическую ишемию конечности, тромботическое заболевание, атеросклероз, облитерирующий тромбангиит (болезнь Бюргера), синдром Рейно, болезнь Такаясу, мигрирующий тромбофлебит поверхностных вен, острую артериальную окклюзию, поражение коронарной артерии, рестеноз после ангиопластики, приступ или возвратный инфаркт миокарда. В другом предпочтительном варианте соединение формулы I, его индивидуальный изомер, рацемическая или нерацемическая смесь изомеров, фармацевтически приемлемая соль или сольват представляет собой модулятор IP-рецептора, в частности агонист IP-рецептора. Во всех случаях, если не указано иное, следующие термины, использованные в настоящем описании, включая формулу изобретения, имеют приведенные ниже значения. Необходимо отметить, что во всех случаях, если из контекста не очевиден другой смысл, в тексте описания и формуле изобретения приведенные в единственном числе ссылки использованы в собирательном смысле. Термин “алкил” означает одновалентный разветвленный или неразветвленный насыщенный углеводородный радикал, включающий только углеродные и водородные атомы и содержащий, если не указано иное, от одного до двенадцати углеродных атомов включительно. Примеры алкильных радикалов включают, но не ограничиваются только ими, метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, н-гексил, октил, додецил и т.п. Понятие “алкилен” обозначает двухвалентный линейный или разветвленный насыщенный углеводородный радикал, включающий только углеродные и водородные атомы и содержащий, если не указано иное, от одного до восьми углеродных атомов включительно. Примеры алкиленовых радикалов включают, но не ограничиваются только ими, метилен, этилен, триметилен, пропилен, тетраметилен, пентаметилен, этилэтилен и т.п. Понятием “алкенилен” обозначают двухвалентный линейный или разветвленный ненасыщенный углеводородный радикал, включающий по меньшей мере одну двойную связь и содержащий, если не указано иное, от двух до восьми углеродных атомов включительно. Алкениленовый радикал включает цис- и транс-изомерные группы [(Е-) и (Z-)], а также их смеси, обусловленные асимметричными углеродными атомами. Примеры алкениленовых радикалов включают, но не ограничиваются только ими, этенилен, 2-пропенилен, 1-пропенилен, 2-бутенил, 2-пентенилен и т.п. Понятием “алкоксигруппа” обозначают радикал -OR, где R означает алкил, который описан выше. Примеры алкоксирадикалов включают, но не ограничиваются только ими, метокси-, этокси-, изопропокси-, бутокси-, втор-бутокси-, изобутоксигруппы и т.п. Термином “аралкил” обозначают радикал R’R”-, где R’ означает арильный радикал, который описан ниже, а R” обозначает алкильный радикал, который описан выше. Примеры аралкильных радикалов включают, но не ограничиваются только ими, бензил, фенилэтил, 3-фенилпропил и т.п. Понятие “арил” использовано для обозначения одновалентного моноциклического ароматического углеводородного радикала, состоящего из одного или нескольких сконденсированных колец, из которых по меньшей мере одно кольцо по своей природе является ароматическим, которое во всех случаях, если не указано иное, необязательно может быть замещено гидрокси-, цианогруппой, (низш.)алкилом, (низш.)алкоксигруппой, тиоалкилом, галогеном, галоидалкилом, гидроксиалкилом, нитрогруппой, алкоксикарбонилом, амино-, алкиламино-, диалкиламиногруппой, аминокарбонилом, карбониламиногруппой, аминосульфонилом, сульфониламиногруппой и/или трифторметилом. Примеры арильных радикалов включают, но не ограничиваются только ими, фенил, нафтил, дифенил, инданил, антрахинолил и т.п. Понятием “циклоалкил” обозначен одновалентный насыщенный карбоциклический радикал, который состоит из одного или нескольких колец и который во всех случаях, если не указано иное, необязательно может быть замещен гидроксилом, цианогруппой, алкилом, алкоксигруппой, тиоалкилом, галогеном, галоидалкилом, гидроксиалкилом, нитрогруппой, алкоксикарбонилом, амино-, алкиламино-, диалкиламиногруппой, аминокарбонилом, карбониламиногруппой, аминосульфонилом, сульфониламиногруппой и/или трифторметилом. Примеры циклоалкильных радикалов включают, но не ограничиваются только ими, циклопропил, циклобутил, 3-этилциклобутил, циклопентил, циклогексил, циклогептил и т.п. Понятием “гетероарил” обозначен одновалентный ароматический карбоциклический радикал, состоящий из одного или нескольких колец, включающих по одному, два или три гетероатома внутри кольца (выбранных из азота, кислорода и серы), причем этот радикал во всех случаях, если не указано иное, необязательно может быть замещен гидрокси-, цианогруппой, (низш.)алкилом, (низш. )алкоксигруппой, тиоалкилом, галогеном, галоидалкилом, гидроксиалкилом, нитрогруппой, алкоксикарбонилом, амино-, алкиламино-, диалкиламиногруппой, аминокарбонилом, карбониламиногруппой, аминосульфонилом, сульфониламиногруппой и/или трифторметилом. Примеры гетероарильных радикалов включают, но не ограничиваются только ими, имидазолил, оксазолил, пиразинил, тиофенил, хинолил, бензофурил, пиридиил, индолил, пирролил, пиранил, нафтиридинил и т.п. Термином “гетероциклил” обозначен одновалентный насыщенный карбоциклический радикал, состоящий из одного или нескольких колец, включающих по одному, два или три гетероатома (выбранных из азота, кислорода и серы), причем этот радикал во всех случаях, если не указано иное, необязательно может быть замещен гидроксилом, цианогруппой, низшим алкилом, низшей алкоксигруппой, тиоалкилом, галогеном, галоидалкилом, гидроксиалкилом, нитрогруппой, алкоксикарбонилом, амино-, алкиламино-, диалкиламиногруппой, аминокарбонилом, карбониламиногруппой, аминосульфонилом, сульфониламиногруппой и/или трифторметилом. Примеры гетероциклических радикалов включают, но не ограничиваются только ими, морфолинил, пиперазинил, пиперидинил, пирролидинил, тетрагидропиранил, тиоморфолинил и т.п. Термин “галоген” использован для обозначения атома фтора, брома, хлора и/или иода. Термин “галоидалкил” использован для обозначения алкила, описанного выше, замещенного в любом положении одним или несколькими атомами галогена, которые указаны выше. Примеры галоидалкильных радикалов включают, но не ограничиваются только ими, 1,2-дифторпропил, 1,2-дихлор-пропил, трифторметил, 2,2,2-трифторэтил, 2,2,2-трихлорэтил и т.п. Понятие “гидроксиалкил” использовано для обозначения алкила, описанного выше, замещенного одной или несколькими гидроксильными группами. Примеры гидроксиалкильных радикалов включают, но не ограничиваются только ими, гидроксиметил, 2-гидроксиэтил, 2-гидроксипропил, 3-гидроксипропил, 2-гидроксибутил, 3-гидроксибутил, 4-гидроксибутил, 2,3-дигидроксипропил, 1-(гидроксиметил)-2-гидроксиэтил, 2,3-дигидроксибутил, 3,4-дигидроксибутил, 2-(гидроксиметил)-3-гидроксипропил и т.п. Термином “изомеры” обозначают различные соединения, которые отвечают одной и той же молекулярной формуле, но различаются природой или последовательностью связи их атомов или пространственным расположением их атомов. Изомеры, которые различаются пространственным расположением их атомов, называют “стереоизомерами”. Стереоизомеры, которые являются зеркальными отражениями друг друга и обладают оптической активностью, называют “энантиомерами”, а стереоизомеры, которые не являются зеркальными отражениями друг друга, называют “диастереоизомерами”. Понятием “хиральный изомер” обозначают соединение с одним хиральным центром. Оно имеет две энантиомерные формы с противоположной хиральностью и может существовать либо как индивидуальный энантиомер, либо как смесь энантиомеров. Смесь, содержащая равные количества индивидуальных энантиомерных форм с противоположной хиральностью, называют “рацемической смесью”. Соединения с более чем одним хиральным центром может существовать либо как индивидуальный диастереоизомер, либо как смесь диастереоизомеров и называется “диастереоизомерной смесью”. В случае наличия одного хирального центра стереоизомер может быть охарактеризован абсолютной конфигурацией (R или S) этого хирального центра. Абсолютной конфигурацией называют пространственное расположение заместителей, связанных с этим хиральным центром. Заместители, связанные с хиральным центром, о котором идет речь, классифицируют в соответствии с Правилами последовательности Кана, Ингольда и Прелога (Cahn и др., Angew. Chem. Inter. Edit. 1966, 5, 385; список опечаток 511; Cahn и др., Angew. Chem. 1966, 78, 413; Cahn и Ingold, J. Chem. Soc. (Лондон) 1951, 612; Cahn и др., Experientia 1956, 12, 81; Cahn, J. Chem. Educ. 1964, 41, 116). Термином “геометрические изомеры” обозначают диастереоизомеры, существование которых обусловлено заторможенным вращением вокруг двойных связей. Названия этих конфигураций различают с помощью приставок цис- и транс- или Z- и Е-, которые указывают на то, что в соответствии с правилами Кана, Ингольда и Прелога группы находятся с одной и той же или с противоположных сторон относительно двойной связи в молекуле. “Атропическими изомерами” называют изомеры, существование которых обусловлено ограниченным вращением из-за затруднения вращения больших групп вокруг центральной связи. Термином “уходящая группа” обозначают группу, назначение которой обычно связано с химией органического синтеза, т.е. атом или группу, которая в условиях алкилирования способна к замещению. Примеры уходящей группы включают, но не ограничиваются только ими, атом галогена, алкан- и ариленсульфонилоксигруппы, такие, как метансульфонилокси-, этансульфонилоксигруппы, тиометил, бензолсульфонилокси-, тозилокси- и тиенилоксигруппы, дигалоидфосфиноилокси-, необязательно замещенные бензилокси-, изопропилокси-, ацилоксигруппы и т.п. Значение термина “защитная группа” или “защищающая группа” обычно связано с химией органического синтеза, т.е. он обозначает группу, которая селективно блокирует реакционноспособный участок соединения с несколькими функциональными группами, благодаря чему химическую реакцию можно селективно проводить по другому, незащищенному реакционноспособному участку. Некоторые способы по настоящему изобретению основываются на том, что защитные группы блокируют реакционноспособные кислородные атомы, содержащиеся в реагентах. Приемлемые защитные группы для спиртовых и фенольных гидроксильных групп, которые можно успешно и селективно удалять, включают группы, защищенные в форме ацетатов, галоидалкилкарбонатов, бензиловых эфиров, простых алкилсилиловых эфиров, простых гетероциклических эфиров, простых метиловых или других алкиловых эфиров и т. п. Защитные или блокирующие группы для карбоксильных групп аналогичны тем, которые описаны для гидроксильных групп, а среди них предпочтительны остатки третбутилового, бензилового или метилового сложных эфиров. Понятием “удаление защитной группы” или “деблокирование” обозначают процесс, в ходе проведения которого по завершении селективной реакции удаляют защитную группу. Некоторые защитные группы могут быть предпочтительнее других благодаря удобству или относительной простоте их удаления. Деблокирующие реагенты для защищенных гидроксильных и карбоксильных групп включают карбонаты калия и натрия, гидроксид лития в спиртовых растворах, цинк в метаноле, уксусную кислоту, трифторуксусную кислоту, палладиевые катализаторы, трибромид бора и т.п. Понятие “необязательный” или “необязательно” означает, что описываемое событие или обстоятельство может иметь место или его может не быть и что описание охватывает примеры, когда такое событие или обстоятельство произошло или возникло, и примеры, когда его не было. Так, например, выражение “необязательная связь” означает, что связь может присутствовать или может отсутствовать и что описание охватывает одинарную, двойную и тройную связи. “Инертным органическим растворителем” или “инертным растворителем” называют растворитель, который инертен в условиях реакции, описываемой в связи с ним, включая, например, бензол, толуол, ацетонитрил, тетрагидрофуран, N,N-диметилформамид, хлороформ, метиленхлорид, дихлорметан, дихлорэтан, диэтиловый эфир, этилацетат, ацетон, метилэтилкетон, метанол, этанол, пропанол, изопропанол, трет-бутанол, диоксан, пиридин и т.п. Во всех случаях, если не указано иное, растворители, используемые во время реакций по настоящему изобретению, являются инертными. Понятие “фармацевтически приемлемый” означает возможность использования при приготовлении фармацевтической композиции, т.е. это понятие используется, как правило, в сочетании с веществом, обычно безвредным, нетоксичным и не являющимся нежелательным ни с биологической, ни с какой-либо другой точек зрения и пригодным для использования в ветеринарии, а также с фармацевтическими целями для терапии человека. Выражение “фармацевтически приемлемый носитель” обозначает носитель, который может быть использован для приготовления фармацевтической композиции, т. е. обычно совместимый с другими компонентами композиции, безвредный для реципиента и ни с биологической, ни с какой-либо другой точек зрения не являющийся нежелательным, и включает носители, которые пригодны для использования в ветеринарии, а также в фармацевтических целях для терапии человека. Понятие “фармацевтически приемлемый носитель” использован в описании и формуле изобретения для обозначения как одного, так и нескольких таких носителей. Словосочетание “фармацевтически приемлемая соль” соединения обозначает соль, которая приемлема с фармакологической точки зрения и которая обладает целевым фармакологическим действием основного соединения. К таким солям относятся, например, следующие соединения: (1) кислотно-аддитивные соли, образуемые с минеральными кислотами, такими, как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., или образуемые с органическими кислотами, такими, как уксусная кислота, пропионовая кислота, капроновая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировино-градная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этандисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, 2-нафталинсульфоновая кислота, 4-метилбицикло [2.2.2] окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 4,4′-метиленбис-(3-гидрокси-2-ен-1-карбоновая) кислота, 3-фенилпропионовая кислота, триметилуксусная кислота, трет-бутилуксусная кислота, лаурилсульфоновая кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтойная кислота, салициловая кислота, стеариновая кислота, муконовая кислота и т.п.; (2) соли, образуемые в условиях, когда кислотный протон, содержащийся в основном соединении, либо замещается ионом металла, например ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия, либо образует координационную связь с органическим основанием. Приемлемые органические основания включают этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин и т.п. Приемлемые неорганические основания включают гидроксид алюминия, гидроксид кальция, гидроксид калия, карбонат натрия, гидроксид натрия и т.п. Следует отметить, что ссылка на фармацевтически приемлемую соль охватывает ее формы с присоединением растворителя или кристаллические формы, в частности сольваты и полиморфы. Сольваты содержат либо стехиометрические, либо нестехиометрические количества растворителя и часто образуются в процессе кристаллизации. Когда растворителем служит вода, образуются гидраты, а когда растворителем является спирт, образуются алкоголяты. Полиморфы включают кристаллические упаковки различных структур одинакового элементного состава соединения. Полиморфы обычно характеризуются различными дифракционными рентгенограммами, инфракрасными спектрограммами, точками плавления, значениями плотности, твердости, формами кристаллов, оптическими и электрическими свойствами, стабильностью и растворимостью. Различные факторы, такие, как растворитель при перекристаллизации, скорость кристаллизации и температура хранения, могут обусловить доминирование одной кристаллической формы. Понятием “субъект” обозначают млекопитающее и немлекопитающее. Примеры млекопитающих включают, но не ограничиваются только ими, любых представителей класса млекопитающих: людей, приматов, отличных от человека, таких, как шимпанзе и обезьяны других видов, сельскохозяйственных животных, таких, как крупный рогатый скот, лошади, овцы, козы, свиньи, домашних животных, таких, как кролики, собаки и кошки, лабораторных животных, включая грызунов, таких, как крысы, мыши, морские свинки и т.п. Примеры немлекопитающих включают, но не ограничиваются только ими, птиц и т.п. Этот термин не обозначает конкретный возраст или пол. Термин “лечение” заболевания включает: (1) профилактику заболевания, т.е. предотвращение развития клинических симптомов заболевания у субъекта, который может находиться или уже находился под воздействием болезненного состоянию, но все еще не испытывает или не проявляет симптомов этого болезненного состояния, (2) подавление болезненного состояния, т.е. купирование развития болезненного состояния или клинических симптомов, или (3) ослабление болезненного состояния, т.е. обеспечение временного или постоянного регресса болезненного состояния или клинических симптомов. “Болезненное состояние” или “заболевание” означает любое заболевание, болезненное состояние, симптом или показание. Выражение “терапевтически эффективное количество” означает такое количество соединения, которого, когда его вводят субъекту для лечения болезненного состояния, достаточно для достижения эффекта такого лечения болезненного состояния. Это “терапевтически эффективное количество” обычно варьируется в зависимости от конкретного соединения, болезненного состояния, при котором проводят лечение, серьезности заболевания, при котором проводят лечение, возраста и относительного здоровья субъекта, пути и формы введения в организм, оценки практикующего врача, оказывающего помощь больному, и других факторов. Термином “модулятор” обозначена молекула такого соединения, которая взаимодействует с мишенью. Объектами взаимодействия являются, но не ограничиваются только ими, агонист, антагонист и т.п., которые представлены в настоящем описании. Под “агонистом” подразумевают молекулу такого соединения, лекарственного средства, активатора фермента или гормона, которая усиливает активность другой молекулы или участка рецептора. Термином “антагонист” обозначают молекулу такого соединения, лекарственного средства, ингибитора фермента или гормона, которая ослабляет или предотвращает действие другой молекулы или участка рецептора. Понятие “фармакологическое действие” включает действие, оказываемое на субъекта для достижения поставленной терапевтической цели. В предпочтительном варианте фармакологическим действием называют лечение субъекта, который нуждается в таком лечении. Так, например, фармакологическим действием является такое воздействие на субъекта, нуждающегося в лечении, результатом которого оказывается профилактика, улучшение самочувствия или облегчение при болезненном состоянии, связанном с нарушением кровотока, неправильным заживанием раны, некрозом ткани, преждевременным сокращением матки, язвой желудка, половой дисфункцией у мужчин и женщин (самцов и самок), ослабление сильной менструальной боли или улучшение при нарушении нейтрофильной функции. В другом предпочтительном варианте понятие “фармакологическое действие” означает, что активацию IP-рецептора связывают с благоприятным терапевтическим действием на субъекта, находящегося в болезненном состоянии, в котором его можно лечить введением модулятора IP-рецептора, в частности агониста IP-рецептора. Название и нумерация положений в молекуле соединений по настоящему изобретению проиллюстрированы ниже. 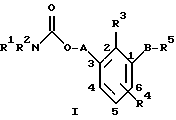 В целом номенклатура, которая использована в настоящем описании, основана на системе AutoNom, которая представляет собой компьютеризованную систему Института Бейлыптейна для формирования систематической номенклатуры ИЮПАК. Однако, поскольку жесткая привязка к ее рекомендациям привела бы к существенному изменению названия при изменении всего лишь одного заместителя, соединения получили названия в такой форме, при которой сохраняется постоянство названия для базовой структуры молекулы. Так, например, соединение формулы I, в которой R1 и R2 каждый обозначает фенил, R3 обозначает метил, R4 обозначает водород, R5 обозначает группу -СООН, А обозначает метилен, В обозначает группу -O(CH2)m-, a m обозначает 1, называют [3-[(дифенилкарбамоилокси)метил]-2-метилфенокси}уксусной кислотой. Соединение формулы I, в которой R1 обозначает фенил, R2 обозначает бензил, R3 и R4 каждый обозначает водород, R5 обозначает группу -СООН, А обозначает пропенилен, В обозначает группу -(СН2)n-, а n обозначает 2, называют 3-{3-[3-(бензилфенилкарбамоилокси) пропенил]фенил]пропионовой кислотой. Среди семейства соединений по настоящему изобретению, представленного в разделе “Краткое изложение сущности изобретения”, предпочтительны некоторые соединения формулы I, в которых: R1 и R2 каждый независимо друг от друга в каждом случае в предпочтительном варианте обозначает арил или аралкил, более предпочтительно фенил или бензил, наиболее предпочтительно фенил, R3 и R4 каждый независимо друг от друга в каждом случае в предпочтительном варианте обозначает водород, алкил, арил, аралкил или галоген, более предпочтительно водород, метил, этил, н-пропил, изопропил, бутил, фенил, бензил, бром или хлор, наиболее предпочтительно водород или метил, R5 в предпочтительном варианте в каждом случае независимо обозначает -COOR6, R6 в предпочтительном варианте в каждом случае независимо обозначает водород или алкил, более предпочтительно водород, А в предпочтительном варианте в каждом случае независимо обозначает алкилен или алкенилен, В в предпочтительном варианте в каждом случае независимо обозначает группу -O(СН2)m-, m в предпочтительном варианте в каждом случае независимо обозначает целое число от 1 до 5 включительно, n в предпочтительном варианте в каждом случае, независимо обозначает целое число от 0 до 5 включительно. Необходимо отметить, что предпочтительные соединения формулы I включают также изомеры соединений формулы I, в частности цис- и транс-изомеры, а также рацемические и нерацемические смеси изомеров, фармацевтически приемлемые соли и сольваты. Примерами особенно предпочтительных соединений являются следующие соединения формулы I, их индивидуальные изомеры, рацемические и нерацемические смеси изомеров, фармацевтически приемлемые соли и сольваты: 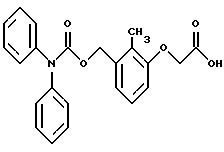 {3-[(дифенилкарбамоилокси)метил]-2-метилфенокси}уксусная кислота, 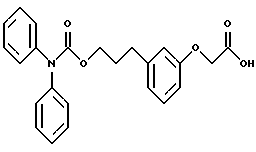 [3-(3-дифенилкарбамоилоксипропил)фенил] уксусная кислота, 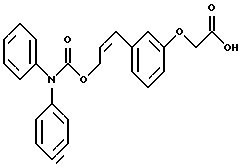 цис-[3-(3-дифенилкарбамоилоксипропенил)фенокси]уксусная кислота, 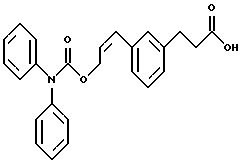 цис-3-[3-(3-дифенилкарбамоилоксипропенил)фенил]пропионовая кислота, 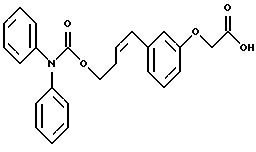 цис-[3-(4-дифенилкарбамоилоксибут-1-енил)фенокси]уксусная кислота, 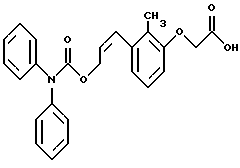 цис-[3-(3-дифенилкарбамоилоксипропенил)-2-метилфенокси] уксусная кислота, 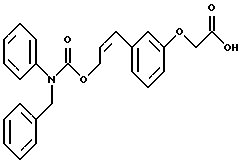 цис-{3-[3-(бензилфенилкарбамоилоксипропенил)фенокси]} уксусная кислота, 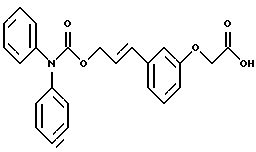 транс-[3-(3-дифенилкарбамоилоксипропенил)фенокси]уксусная кислота и 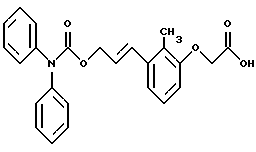 транс-[3-(3-дифенилкарбамоилоксипропенил)-2-метилфенокси] уксусная кислота. Группа предпочтительных соединений формулы I включает те соединения, в которых А обозначает алкилен, В обозначает -O(CH2)m-, a m обозначает целое число от 1 до 5 включительно. Равным образом предпочтительными соединениями являются те соединения, в которых А обозначает алкилен, В обозначает -(СН2)n-, а n обозначает целое число от 0 до 5 включительно. Предпочтительные соединения формулы I включают те соединения, в которых А обозначает алкенилен, В обозначает -O(CH2)m-, a m обозначает целое число от 1 до 5 включительно. Кроме того, предпочтительными соединениями являются те соединения, в которых А обозначает алкенилен, В обозначает -(CH2)n-, a n обозначает целое число от 0 до 5 включительно. Особенно предпочтительны следующие соединения: { 3-[(дифенилкарбамоилокси)метил]-2-метилфенокси} уксусная кислота, [3-(3-дифенилкарбамоилоксипропил)фенил] уксусная кислота, их индивидуальные изомеры, рацемические и нерацемические смеси изомеров, фармацевтически приемлемые соли и сольваты. Еще одну группу особенно предпочтительных соединений формулы I образуют следующие соединения: цис-{3-[3-(бензилфенилкарбамоилоксипропенил)фенокси]} уксусная кислота, цис-[3-(3-дифенилкарбамоилоксипропенил)фенокси]уксусная кислота, цис-[3-(4-дифенилкарбамоилоксибут-1-енил)фенокси]уксусная кислота, цис-[3-(3-дифенилкарбамоилоксипропенил)-2-метилфенокси] уксусная кислота, транс-[3-(3-дифенилкарбамоилоксипропенил)фенокси] уксусная кислота и транс-[3-(3-дифенилкарбамоилоксипропенил)-2-метилфенокси] уксусная кислота, а также их индивидуальные изомеры, рацемические и нерацемические смеси изомеров, фармацевтически приемлемые соли и сольваты. Также особенно предпочтительными являются цис-[3-(3-дифенилкарбамоилоксипропенил)фенил] пропионовая кислота, ее индивидуальные изомеры, рацемические и нерацемические смеси изомеров, фармацевтически приемлемые соли и сольваты. Предпочтительным является также способ получения соединения в соответствии с формулой I, причем этот способ включает взаимодействие соединения одной из следующих формул 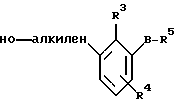 или 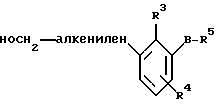 с соединением в соответствии с формулой 5 в которой R1, R2, R3, R4, R5, R6 А, В, m и n имеют значения, указанные в п.1, а Х обозначает галоген. В объем настоящего изобретения включены также фармацевтическая композиция, включающая терапевтически эффективное количество по меньшей мере одного соединения в соответствии с формулой I в смеси с по меньшей мере одним приемлемым носителем. Объектом изобретения является, кроме того, упомянутая выше фармацевтическая композиция, по меньшей мере одно соединение формулы I которой пригодно для введения в организм субъекта, страдающего заболеванием, состояние которого облегчают лечением с помощью модулятора IP-рецептора. Еще одним объектом изобретения является применение соединения формулы I при приготовлении лекарственных средств, содержащих любое из соединений формулы I. Кроме того, объектом настоящего изобретения является применение соединения формулы I при приготовлении лекарственных средств, включающих любое из соединений формулы I, для лечения болезненного состояния, связанного с неправильным заживанием раны, некрозом ткани, преждевременным сокращением матки, язвой желудка, половой дисфункцией у мужчин и женщин (самцов и самок), сильной менструальной болью, нарушением иммунорегуляции, нарушением агрегации кровяных пластинок или нарушением нейтрофильной функции. Еще одним объектом настоящего изобретения является применение соединения в соответствии с любым из пп.1-15 при приготовлении лекарственных средств, включающих любое из соединений формулы I, для лечения болезненного состояния, связанного с окклюзионным поражением периферической артерии (ОППА), перемежающейся хромотой, критической ишемией конечности, тромботическим заболеванием, атеросклерозом, облитерирующим тромбангиитом (болезнь Бюргера), синдромом Рейно, болезнью Такаясу, мигрирующим тромбофлебитом поверхностных вен, острой артериальной окклюзией, поражением коронарной артерии, рестенозом вследствие пластической операции на сосудах, приступом, возвратным инфарктом миокарда, легочной гипертензией, повышенным глазным давлением, шумом в ушах, обусловленным гипертензией, ишемией, связанной с пересадкой трансплантацией аллотрансплантата, почечной недостаточностью, нарушением диуреза, нарушением натрийуреза или нарушением калийуреза. Равным образом объектом изобретения являются соединения формулы I, когда их получают в соответствии с описанными способами. В настоящем изобретении предлагается также способ лечения болезненного состояния, связанного с IP-рецептором, причем этот способ включает введение эффективного количества соединения в соответствии с формулой I. Кроме того, объектом изобретения является способ лечения болезненного состояния, связанного с неправильным заживанием раны, некрозом ткани, преждевременным сокращением матки, язвой желудка, половой дисфункцией у мужчин и женщин (самцов и самок), сильной менструальной болью, нарушением иммунорегуляции, нарушением агрегации кровяных пластинок или нарушением нейтрофильной функции, окклюзионным поражением периферической артерии (ОППА), перемежающейся хромотой, критической ишемией конечности, тромботическим заболеванием, атеросклерозом, облитерирующим тромбангиитом (болезнь Бюргера), синдромом Рейно, болезнью Такаясу, мигрирующим тромбофлебитом поверхностных вен, острой артериальной окклюзией, поражением коронарной артерии, рестенозом вследствие пластической операции на сосудах, приступом, возвратным инфарктом миокарда, легочной гипертензией, повышенным глазным давлением, шумом в ушах, обусловленным гипертензией, при ишемии, связанной с пересадкой аллотрансплантата, почечной недостаточностью, нарушением диуреза, нарушением натрийуреза или нарушением калийуреза, причем этот способ включает введение эффективного количества соединения в соответствии с формулой I. Объектом настоящего изобретения является, кроме того, пригодная для введения субъекту фармацевтическая композиция, включающая терапевтически эффективное количество по меньшей мере одного соединения формулы I в смеси с по крайней мере одним фармацевтически приемлемым носителем. Еще одним объектом настоящего изобретения является способ лечения, включающий введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества по меньшей мере одного соединения формулы I. Равным образом объектом настоящего изобретения является вышеописанный способ, при осуществлении которого терапии подвергают субъекта, страдающего от болезненного состояния, связанного с неправильным заживанием раны, некрозом ткани, преждевременным сокращением матки, язвой желудка, половой дисфункцией у мужчин и женщин (самцов и самок), сильной менструальной болью, нарушением иммунорегуляции, нарушением агрегации кровяных пластинок или нарушением нейтрофильной функции. Объектом изобретения является также вышеописанный способ, при осуществлении которого соединением является модулятор IP-рецептора, в частности агонист IP-рецептора. Еще одним объектом изобретения является способ лечения, включающий введение терапевтически эффективного количества по меньшей мере одного соединения формулы I субъекту, страдающему болезненным состоянием, связанным с нарушением кровотока, в особенности когда нарушением кровотока является сердечно-сосудистое заболевание, в частности в том случае, когда сердечно-сосудистое заболевание представляет собой окклюзионное поражение периферической артерии (ОППА), перемежающуюся хромоту, критическую ишемию конечности, тромботическое заболевание, атеросклероз, облитерирующий тромбангиит (болезнь Бюргера), синдром Рейно, болезнь Такаясу, мигрирующий тромбофлебит поверхностных вен, острую артериальную окклюзию, поражение коронарной артерии, рестеноз вследствие пластической операции на сосудах, приступ или возвратный инфаркт миокарда. Объектом изобретения является также вышеописанный способ, при осуществлении которого соединение представляет собой модулятор IP-рецептора, в частности агонист IP-рецептора. Объектом изобретения является также вышеописанный способ, при осуществлении которого болезненное состояние, связанное с нарушением кровотока, представляет собой гипертоническую болезнь, в частности легочную гипертензию, повышенное глазное давление или шум в ушах, связанный с гипертензией. Далее, объектом изобретения является вышеописанный способ, при осуществлении которого болезненное состояние, связанное с нарушением кровотока, представляет собой ишемическое заболевание, в частности ишемию, связанную с пересадкой аллотрансплантата. Кроме того, объектом изобретения является вышеописанный способ, при осуществлении которого болезненное состояние, связанное с нарушением кровотока, представляет собой почечную недостаточность, в частности нарушение диуреза, нарушение натрийуреза или нарушение калийуреза. Соединения по настоящему изобретению могут быть получены способом, различные варианты осуществления которого представлены на приведенных ниже иллюстративных схемах реакций синтеза. Исходные материалы и реагенты, которые используют при получении этих соединений, либо имеются в продаже и производятся такими фирмами, как, например, фирма Aldrich Chemical Co., либо могут быть получены способами, известными специалистам в данной области техники в соответствии с методами, изложенными в таких источниках информации, как Fieser and Fieser’s Reagents for Organic Synthesis, Wiley & Sons: Нью-Йорк, 1991, тт. 1-15; Rodd’s Chemistry of Carbon Compounds, Elsevier Science Publishers, 1989, тт. 1-5 и дополнения, и Organic Reactions, Wiley & Sons: Нью-Йорк, 1991, тт. 1-40. Следующие схемы представлены только с целью проиллюстрировать некоторые варианты осуществления способа, согласно которым могут быть синтезированы соединения по настоящему изобретению, и поэтому специалист в данной области техники, основываясь на настоящем описании, может вносить в эти схемы различные модификации. Исходные материалы и промежуточные продукты этих реакционных схем могут быть выделены и очищены при необходимости с применением обычной технологии, включая, но не ограничиваясь этим, фильтрование, перегонку, кристаллизацию, хроматографию и т. п. Такие материалы могут быть охарактеризованы с применением обычных средств, включая физические константы и спектральные данные. Во всех случаях, если не указано иное, представленные в настоящем описании реакции протекают под атмосферным давлением в температурном интервале от примерно -78 до примерно 150oС, более предпочтительно от примерно 0 до примерно 125oС, наиболее предпочтительно при комнатной температуре (или температуре окружающей среды), в частности при примерно 20oС. На схемах А, Б, В и Г представлены альтернативные способы получения соединений формулы I (см. в конце описания). Исходное соединение формулы 1а имеется в продаже или является известным специалистам в данной области техники или может быть легко синтезировано. Так, например, синтез соединения 1а, у которого R3 обозначает бром, а R4 обозначает водород, описано у Beijer P.H., Rec. Trav. Chim. Pays-Bas. 1929, 48, 1010, а соединения, у которого R3 обозначает хлор, а R4 обозначает водород, описано у Beuhler и др. в J. Amer. Chem. Soc. 1946, 68, 574-577. Соединение формулы 1а может быть также получено замещением 2-метоксигруппы 2-(2,3-диметоксифенил)-4,4-диметил-4,5-дигидрооксазола алкильной группой в присутствии реактива Гриньяра или литийорганического реагента по методам, которые известны специалистам в данной области техники. В результате дальнейшего гидролиза 4,5-дигидрооксазоловой группы соединения под действием сильной кислоты, такой, как водная серная кислота, в модифицированных условиях реакции Мейерса, и последующего расщепления 3-метоксигруппы с образованием гидроксильной группы приемлемым простым эфиром в качестве расщепляющего агента, таким, как трибромид бора или концентрированные кислоты, такие, как бромистоводородная кислота, предпочтительно трибромид бора, получают конечное соединение формулы 1а. Пригодные для этой реакции растворители включают апротонные растворители, такие, как тетрагидрофуран, бензол, толуол и т.п. Обычно исходное соединение формулы 1b имеется в продаже, например, производится фирмой Aldrich Chemical Co., известно или может быть легко синтезировано специалистом в данной области техники. На стадии 1а гидроксиметилфенол 2а получают восстановлением карбоксильной группы соединения 1а до спиртовой группы по обычным методам. Приемлемые условия восстановления включают использование литийалюминийгидрида, борана или борановых производных в апротонном органическом растворителе, таком, как диэтиловый эфир, диоксан, тетрагидрофуран и т.п. Соединение 2а может быть также получено отщеплением фталоильной группы 3-гидроксифталевого ангидрида и последующим восстановлением продукта до диола. Приемлемые условия расщепления ангидрида и восстановления включают использование литийалюминийгидрида, боранов или борановых комплексов в апротонном органическом растворителе, таком, как тетрагидрофуран, диэтиловый эфир, диоксан, простые гликолевые эфиры и т.п. На стадии 2а эфир 4 гидроксиметилфеноксикарбоновой кислоты может быть получен алкилированием гидроксильной группы соединения 2а пригодным алкилирующим агентом 3 формулы R5-(CH2)m-X, где R5 обозначает защищенную карбоксильную группу, а Х обозначает галоген, в частности бром или хлор. Реакция протекает в присутствии слабого основания, такого, как карбонат калия, карбонат цезия, карбонат натрия и т.п., в условиях синтеза Вильямсона. Приемлемые для этой реакции инертные органические растворители включают апротонные органические растворители, такие, как ацетон, диоксан, тетрагидрофуран и т. п. Алкилирующие агенты 3 имеются в продаже или могут быть синтезированы специалистом в данной области техники. В другом варианте на стадии 1б путем алкилирования гидроксильной группы соединения 1b пригодным алкилирующим агентом 3 формулы R5-(CH2)m-Х, как это указано в описании упомянутой выше стадии 2а, получают эфир 2b формилфеноксикарбоновой кислоты. Кроме того, соединение 2b может быть синтезировано по методам, которые в данной области техники известны, по таким, как окисление первичной спиртовой группы до соответствующей альдегидной группы приемлемыми окислителями, такими, как диметилсульфоксид, уксусный ангидрид, оксалилхлорид, тозилхлорид и т.п. Так, например, синтез соединения формулы 2b, в которой R3 обозначает метил, R4 обозначает водород, R5 обозначает остаток трет-бутилового эфира карбоновой кислоты, a m обозначает 1, описан у Marx М. и Tidwell Т. в J. Org. Chem, 1984, 49, 788-793. В другом варианте на стадии 2б эфир 4 гидроксиметилфеноксикарбоновой кислоты может быть получен восстановлением альдегидной группы соединения 2b до спиртовой группы по общепринятым методам. Приемлемые для восстановления альдегида условия включают восстановление с использованием литийсодержащих гидридов, таких, как литийалюминийгидрид, или боргидридов, таких, как натрийборгидрид, или гидрогенизацию с применением платинового или палладиевого катализатора в приемлемом протонном растворителе. Кроме того, соединение формулы 4, где А обозначает разветвленную алкиленовую группу, может быть получено обработкой соединения 2b металлорганическим реагентом, таким, как реагент Гриньяра, или алкиллитиевым реагентом. Пригодные для этой реакции растворители включают апротонные органические растворители, такие, как тетрагидрофуран, диэтиловый эфир и т.п. На стадии 3 соединение формулы I может быть получено различными методами, известными специалисту в данной области техники. Так, например, соединение формулы I может быть получено ацилированием соединения 4 ацилирующим агентом 5 формулы R1R2NC(O)X, где Х обозначает галоген, в частности бром или хлор. Реакция протекает в присутствии сильного основания, такого, как алкиламиды лития, литийалкилы и бис(триметилсилил)амид калия. Инертные органические растворители, пригодные для этой реакции, включают апротонные органические растворители, такие, как диэтиловый эфир, тетрагидрофуран и т.п. Ацилирующий агент 5 имеется в продаже, известен или может быть легко синтезирован специалистами в данной области техники. Так, например, синтез соединения 5 с различными радикалами R1 и R2 можно проводить путем обработки соответствующего амина формулы R1R2NH ацилгалогенидом, таким, как оксалилхлорид, фосген, фосгеновые эквиваленты и т.п. Соединение формулы I, где R5 обозначает группу -COOR6 или тетразолил, обычно получают с использованием защитной группы, которую затем деблокируют по общепринятым методам, получая конечный продукт. Так, например, соединение формулы I, где R5 обозначает группу -COOR6, получают с защищенной карбоксильной группой в форме такого соединения, как алкиловый эфир, с последующим удалением защитной группы и получением карбоновой кислоты. Эта реакция протекает в присутствии сильного основания, такого, как водный гидроксид лития, гидроксид натрия и гидроксид калия, в протонном органическом растворителе, таком, как метанол, этанол, вода и их смеси. Соединение формулы I, где R5 обозначает тетразолил, может быть получено с защищенным тетразолилом в форме такого соединения, как трифенилметилтетразолил (тритилтетразолил), с последующим деблокированием. Соединение 2а можно обрабатывать алкилирующим агентом формулы N  C-(CH2)m-X, где Х обозначает галоген, в частности бром или хлор. Эта реакция протекает в присутствии слабого основания, такого, как карбонат калия, карбонат цезия или карбонат натрия, в апротонном органическом растворителе, таком, как ацетон, диоксан, тетрагидрофуран, N, N-диметилформамид и т.п. На следующей стадии проводят реакцию цианового продукта с ацилирующим агентом 5 формулы R1R2NC(O)X, где Х обозначает галоген, в частности бром или хлор, а затем с азидом натрия, который присоединяется к цианогруппе с последующими циклизацией и образованием тетразолиловой группы. Реакция протекает в присутствии катализатора, такого, как аммонийхлорид, в апротонном органическом растворителе, таком, как ацетон, диоксан, тетрагидрофуран, N,N-диметилформамид и т. п. В другом варианте для введения азидной группы без катализа может быть использован триметилсилил- или триметилоловоазид.
Некоторые процессы получения соединения формулы 1а приведены в примере 1 получения. Различные процессы получения соединений формулы I с созданием реакционных условий, представленных на схеме А, приведены в примерах 1-5. (см. в конце описания).
Альтернативные исходные соединения, гидроксибензальдегид 1b или галоидированный фенол 1с, где Х обозначает галоген, предпочтительно бром или иод, имеются в продаже, например, выпускаются фирмой Aldrich Chemical Co., известны или могут быть легко синтезированы специалистами в данной области техники.
На стадии 1 эфир 6 транс-гидроксифенилалкиленилкарбоновой кислоты, где R обозначает (С1-С4)алкил, а Аa обозначает связь, алкилен или алкенилен, может быть получен в условиях, известных специалисту в данной области техники. Так, например, соединение 6 может быть получено реакцией альдегида 1b с алкилидентрифенилфосфораном или алкилиденфосфонатом, который получают in situ обработкой фосфониевой соли или фосфоната, такого, как алкилфосфонацетат, сильным основанием, таким, как гидрид лития или гидрид натрия, в условиях реакции Виттига или Хорнера. Соединение 6 может быть также получено обработкой альдегида 1b 1,8-диазабицикло(5.5.0)ундец-7-еном (ДБУ) и литийгалогенидом в реакционных условиях, описанных у Blanchette М.А. и др. в Tetrhedron Letters, 1984, 25, 2183. Растворители, пригодные для проведения реакции олефинилирования, включают инертные апротонные растворители, такие, как ацетонитрил, тетрагидрофуран и т.п.
На альтернативной стадии 1 эфир 6 транс-гидроксифенилалкиленилкарбоновой кислоты, где R обозначает (С1-С4)алкил, может быть получен реакцией галоидированного фенола 1с с акриловым эфиром, таким, как этилакрилат, в присутствии фосфинового лиганда, такого, как три(о-толил)фосфин в сочетании с палладиевой солью, такой, как ацетат палладия(II). Реакция протекает в присутствии основания, такого, как триэтиламин, в инертной атмосфере, например, в условиях реакции сочетания типа реакции Хека. Пригодные для этой реакции растворители включают апротонные растворители, такие, как ацетонитрил, тетрагидрофуран и т.п.
На стадии 2 селективным восстановлением карбоксильной группы эфира соединения 6 до соответствующей спиртовой группы получают транс-гидроксифенилалкилениловый спирт 7. Приемлемые условия восстановления карбоксильной группы эфира включают условия, создаваемые с использованием литийборгидрида, литийалюминийгидрида, диизобутилалюминийгидрида (ДИБАЛ-Г), борана или борановых производных. Предпочтительные условия восстановления описаны у Trost В. М. и др. в J. Org. Chem., 1980, 45, 1838, и включают применение соли, называемой ate-комплексом, образуемым ДИБАЛ-Г и алкиллитиевым соединением, таким, как н-бутиллитий. Апротонные растворители, пригодные для этой реакции, включают тетрагидрофуран, гексан, диметоксиэтан, диоксан и т.п.
На стадии 3 эфир 8 транс-гидроксиметилалкиленилфеноксикарбоновой кислоты получают аналогично тому, как это представлено на схеме А, стадия 2а, например, алкилированием гидроксильной группы соединения 7 пригодным алкилирующим агентом 3 формулы R5-(CH2)m-X, где R5 обозначает защищенную карбоксильную группу.
На стадии 4 транс-изомер соединения формулы I получают аналогично тому, как это представлено на схеме А, стадия 3, например, ацилированием соединения 8 ацилирующим агентом 5 формулы R1R2NC(O)X, в которой Х обозначает галоген, в частности бром или хлор.
Соединение формулы I, где А обозначает алкилен, необязательно может быть получено селективным гидрированием углерод-углеродной двойной связи продукта или любого из промежуточных соединений, синтезированных перед конечным продуктом, с получением соответствующих насыщенных соединений. Приемлемые условия селективного восстановления включают условия каталитического восстановления, такие, как создаваемые с помощью никеля Ренея, палладия на угле, борида никеля, металлической платины, ее оксида и т.п., предпочтительно металлического палладия или его оксида. Растворители, пригодные для этой реакции, включают инертные органические растворители, такие, как этилацетат, метанол и т.п. В предпочтительном варианте соединения 6, 7 или 8 подвергают селективному гидрированию с получением соединений формулы I, где А обозначает алкилен.
Некоторые процессы получения транс-изомера соединения формулы I с созданием реакционных условий, представленных на схеме Б, приведены в примерах 6 и 7. Различные процессы получения соединения формулы I, в которой А обозначает алкилен, с созданием реакционных условий, представленных на схеме В, приведены в примерах 11 и 12 (см. в конце описания).
Исходный галоидированный фенол формулы 1с, где Х обозначает галоген, предпочтительно бром или иод, имеется в продаже, например, производится фирмой Aldrich Chemical Co. , известен или может быть легко синтезирован обычными специалистами в данной области техники.
На стадии 1 эфир 9 галоидфеноксикарбоновой кислоты получают аналогично тому, как это проиллюстрировано на схеме А, стадия 2а, например, алкилированием гидроксильной группы соединения 1с пригодным алкилирующим агентом 3 формулы R5-(CH2)m-Х, где R5 обозначает защищенную карбоксильную группу.
На стадии 2 эфир 10 гидроксиметилалкинилфеноксикарбоновой кислоты, где Аa обозначает связь, алкилен или алкенилен, получают реакцией соединения 9 с алкиниловым спиртом, таким, как пропаргиловый спирт, в условиях реакции сочетания ацетилена. Эта реакция протекает в присутствии палладийорганического катализатора, такого, как, например, тетракис(трифенилфосфин)палладий(0) и бис(трифенилфосфин)палладий(II)хлорид, необязательно в присутствии в качестве катализатора галогенида меди, такого, как иодид меди(I). Пригодным для реакции растворителем является пирролидин, который дополнительно служит в качестве реагента. В другом варианте эту реакцию можно проводить в присутствии диизопропиламина, необязательно в присутствии в качестве катализатора галогенида меди, в приемлемом апротонном растворителе, таком, как тетрагидрофуран.
На стадии 3 получают эфир 11 алкинилфеноксикарбоновой кислоты аналогично тому, как это проиллюстрировано на схеме А, стадия 3, например, ацилированием соединения 10 ацилирующим агентом 5 формулы R1R2NC(O)X, где Х обозначает галоген, в частности бром или хлор.
На стадии 4 цис-изомер соединения формулы 1 получают селективным превращением тройной связи в молекуле соединения 11 в двойную цис-связь в условиях частичного гидрирования. Пригодными для селективного частичного гидрирования алкинов до цис-алкенов катализаторами являются диизобутилалюминийгидрид (ДИБАЛ-Г) или водород с палладиевым катализатором, таким, как катализатор Линдлара. Эту реакцию проводят с добавлением повышающего селективность агента, такого, как хинолин, в протонном органическом растворителе, таком, как метанол.
На альтернативной стадии 3 эфир 12 цис-гидроксиметилалкенилфеноксикарбоновой кислоты получают селективным превращением тройной связи в молекуле соединения 10 в двойную цис-связь в условиях частичного гидрирования, представленных в вышеприведенном описании стадии 3.
На альтернативной стадии 4 цис-изомер соединения формулы 1 получают аналогично тому, как это проиллюстрировано на схеме А, стадия 4, например, ацилированием соединения 12 ацилирующим агентом 5 формулы R1R2NC(O)X, где Х обозначает галоген, в частности бром или хлор.
Соединение формулы I, где А обозначает алкилен, необязательно может быть получено селективным гидрированием углерод-углеродной двойной связи продукта или любого из промежуточных соединений, синтезированных перед конечным продуктом, с получением соответствующих насыщенных соединений. Приемлемые условия селективного восстановления включают условия каталитического восстановления, такие, как создаваемые с помощью никеля Ренея, палладия на угле, борида никеля, металлической платины, ее оксида и т.п., предпочтительно металлического палладия или его оксида. Растворители, пригодные для этой реакции, включают инертные органические растворители, такие, как этилацетат, метанол и т. п. В предпочтительном варианте соединения 10, 11 или 12 подвергают селективному гидрированию с получением соединений формулы I, где А обозначает алкилен.
Некоторые процессы получения цис-изомера соединения формулы I с созданием реакционных условий, представленных на схеме В, приведены в примерах 8 и 10 (см. в конце описания).
Цис-изомер соединения формулы Iа, в которой В обозначает группу -(СH2)n-, синтезируют аналогично способу, который проиллюстрирован на схеме В, но с использованием других исходных соединений, получая целевой конечный продукт.
Исходное соединение формулы 1d, где Х обозначает галоген, предпочтительно бром или иод, a Z обозначает галоген, предпочтительно бром или иод, или группу -СНО или -(СН2)nСООН, где n имеет значения, приведенные в разделе “Краткое изложение сущности изобретения”, имеется в продаже, например, выпускается фирмой Aldrich Chemical Co., известно или может быть легко синтезировано специалистами в данной области техники.
На стадии 1 эфир 13 галоидфенилкарбоновой кислоты, где Аa обозначает связь, алкилен или алкенилен, может быть получено по различным методам. Так, например, соединение 13, у которого n обозначает 3, получают сочетанием соединения 1с, у которого каждый из Х и Z обозначает галоген, с винилкарбоновым эфиром, таким, как метил-3-бутеноат, в присутствии гидроборирующего агента, такого, как 9-борабицикло[3.3.1]нонановый димер (9-ББН). Эту реакцию проводят в присутствии катализаторов сочетания, таких, как палладийхлорид и трикалийфосфат, в апротонном растворителе, таком, как дихлорметан, N,N-диметилформамид, тетрагидрофуран и т. п. Переэтерификацию получаемого в качестве продукта эфира карбоновой кислоты проводят обработкой спиртом, таким, как 2-метил-2-пропанол, и основанием, таким, как н-бутиллитий, в инертной атмосфере.
По другому варианту соединение 13, где n обозначает 2, может быть получено обработкой соединения 1с, где Х обозначает галоген, а Z обозначает группу -СНО, алкилидентрифенилфосфораном или алкилиденфосфонатом, который получают in situ в присутствии фосфониевой соли или фосфоната, такого, как алкилфосфонацетат, сильным основанием, таким, как гидрид лития или гидрид натрия, в условиях реакции Виттига или Хорнера. Получаемый в качестве продукта алкениленкарбоновый эфир селективно гидрируют, получая соответствующее насыщенное соединение. Приемлемые условия селективного восстановления включают условия каталитического восстановления, такие, как создаваемые с помощью никеля Ренея, палладия на угле, борида никеля, металлической платины, ее оксида и т.п., в инертном органическом растворителе, таком, как этилацетат, метанол и т.п.
На стадии 2 эфир 14 гидроксиметилалкинилфенилкарбоновой кислоты получают аналогично тому, как это проиллюстрировано на схеме В, стадия 2, например, обработкой соединения 13 алкиниловым спиртом, таким, как пропаргиловый спирт. В другом варианте соединение 14, у которого n обозначает О или 1, можно получать непосредственно реакцией исходного соединения 1d, у которого Х обозначает галоген, a Z обозначает группу -(СН2)nСООН, где n обозначает соответственно 0 или 1, алкиниловым спиртом, таким, как пропаргиловый спирт.
Далее на стадиях 3 и 4 или на альтернативных стадиях 3 и 4 аналогично тому, как это проиллюстрировано на схеме В, получают цис-изомер соединения формулы 1а.
Соединение формулы 1а, в которой А обозначает алкилен, а В обозначает группу -(СН2)n-, необязательно может быть получено селективным гидрированием углерод-углеродной двойной связи продукта или любого из промежуточных соединений, синтезированных перед конечным продуктом, с получением соответствующих насыщенных соединений. Приемлемые условия селективного восстановления включают условия каталитического восстановления, такие, как создаваемые с помощью никеля Ренея, палладия на угле, борида никеля, металлической платины, ее оксида и т.п., предпочтительно металлического палладия или его оксида. Растворители, пригодные для этой реакции, включают инертные органические растворители, такие, как этилацетат, метанол и т.п. В предпочтительном варианте соединения 14, 15 или 16 подвергают селективной гидрогенизации с получением соединений формулы 1а, в которой А обозначает алкилен.
Некоторые процессы получения соединения формулы 1а с созданием реакционных условий, представленных на схеме Г, приведены в примерах 13-15.
Промышленная применимость C-(CH2)m-X, где Х обозначает галоген, в частности бром или хлор. Эта реакция протекает в присутствии слабого основания, такого, как карбонат калия, карбонат цезия или карбонат натрия, в апротонном органическом растворителе, таком, как ацетон, диоксан, тетрагидрофуран, N, N-диметилформамид и т.п. На следующей стадии проводят реакцию цианового продукта с ацилирующим агентом 5 формулы R1R2NC(O)X, где Х обозначает галоген, в частности бром или хлор, а затем с азидом натрия, который присоединяется к цианогруппе с последующими циклизацией и образованием тетразолиловой группы. Реакция протекает в присутствии катализатора, такого, как аммонийхлорид, в апротонном органическом растворителе, таком, как ацетон, диоксан, тетрагидрофуран, N,N-диметилформамид и т. п. В другом варианте для введения азидной группы без катализа может быть использован триметилсилил- или триметилоловоазид.
Некоторые процессы получения соединения формулы 1а приведены в примере 1 получения. Различные процессы получения соединений формулы I с созданием реакционных условий, представленных на схеме А, приведены в примерах 1-5. (см. в конце описания).
Альтернативные исходные соединения, гидроксибензальдегид 1b или галоидированный фенол 1с, где Х обозначает галоген, предпочтительно бром или иод, имеются в продаже, например, выпускаются фирмой Aldrich Chemical Co., известны или могут быть легко синтезированы специалистами в данной области техники.
На стадии 1 эфир 6 транс-гидроксифенилалкиленилкарбоновой кислоты, где R обозначает (С1-С4)алкил, а Аa обозначает связь, алкилен или алкенилен, может быть получен в условиях, известных специалисту в данной области техники. Так, например, соединение 6 может быть получено реакцией альдегида 1b с алкилидентрифенилфосфораном или алкилиденфосфонатом, который получают in situ обработкой фосфониевой соли или фосфоната, такого, как алкилфосфонацетат, сильным основанием, таким, как гидрид лития или гидрид натрия, в условиях реакции Виттига или Хорнера. Соединение 6 может быть также получено обработкой альдегида 1b 1,8-диазабицикло(5.5.0)ундец-7-еном (ДБУ) и литийгалогенидом в реакционных условиях, описанных у Blanchette М.А. и др. в Tetrhedron Letters, 1984, 25, 2183. Растворители, пригодные для проведения реакции олефинилирования, включают инертные апротонные растворители, такие, как ацетонитрил, тетрагидрофуран и т.п.
На альтернативной стадии 1 эфир 6 транс-гидроксифенилалкиленилкарбоновой кислоты, где R обозначает (С1-С4)алкил, может быть получен реакцией галоидированного фенола 1с с акриловым эфиром, таким, как этилакрилат, в присутствии фосфинового лиганда, такого, как три(о-толил)фосфин в сочетании с палладиевой солью, такой, как ацетат палладия(II). Реакция протекает в присутствии основания, такого, как триэтиламин, в инертной атмосфере, например, в условиях реакции сочетания типа реакции Хека. Пригодные для этой реакции растворители включают апротонные растворители, такие, как ацетонитрил, тетрагидрофуран и т.п.
На стадии 2 селективным восстановлением карбоксильной группы эфира соединения 6 до соответствующей спиртовой группы получают транс-гидроксифенилалкилениловый спирт 7. Приемлемые условия восстановления карбоксильной группы эфира включают условия, создаваемые с использованием литийборгидрида, литийалюминийгидрида, диизобутилалюминийгидрида (ДИБАЛ-Г), борана или борановых производных. Предпочтительные условия восстановления описаны у Trost В. М. и др. в J. Org. Chem., 1980, 45, 1838, и включают применение соли, называемой ate-комплексом, образуемым ДИБАЛ-Г и алкиллитиевым соединением, таким, как н-бутиллитий. Апротонные растворители, пригодные для этой реакции, включают тетрагидрофуран, гексан, диметоксиэтан, диоксан и т.п.
На стадии 3 эфир 8 транс-гидроксиметилалкиленилфеноксикарбоновой кислоты получают аналогично тому, как это представлено на схеме А, стадия 2а, например, алкилированием гидроксильной группы соединения 7 пригодным алкилирующим агентом 3 формулы R5-(CH2)m-X, где R5 обозначает защищенную карбоксильную группу.
На стадии 4 транс-изомер соединения формулы I получают аналогично тому, как это представлено на схеме А, стадия 3, например, ацилированием соединения 8 ацилирующим агентом 5 формулы R1R2NC(O)X, в которой Х обозначает галоген, в частности бром или хлор.
Соединение формулы I, где А обозначает алкилен, необязательно может быть получено селективным гидрированием углерод-углеродной двойной связи продукта или любого из промежуточных соединений, синтезированных перед конечным продуктом, с получением соответствующих насыщенных соединений. Приемлемые условия селективного восстановления включают условия каталитического восстановления, такие, как создаваемые с помощью никеля Ренея, палладия на угле, борида никеля, металлической платины, ее оксида и т.п., предпочтительно металлического палладия или его оксида. Растворители, пригодные для этой реакции, включают инертные органические растворители, такие, как этилацетат, метанол и т.п. В предпочтительном варианте соединения 6, 7 или 8 подвергают селективному гидрированию с получением соединений формулы I, где А обозначает алкилен.
Некоторые процессы получения транс-изомера соединения формулы I с созданием реакционных условий, представленных на схеме Б, приведены в примерах 6 и 7. Различные процессы получения соединения формулы I, в которой А обозначает алкилен, с созданием реакционных условий, представленных на схеме В, приведены в примерах 11 и 12 (см. в конце описания).
Исходный галоидированный фенол формулы 1с, где Х обозначает галоген, предпочтительно бром или иод, имеется в продаже, например, производится фирмой Aldrich Chemical Co. , известен или может быть легко синтезирован обычными специалистами в данной области техники.
На стадии 1 эфир 9 галоидфеноксикарбоновой кислоты получают аналогично тому, как это проиллюстрировано на схеме А, стадия 2а, например, алкилированием гидроксильной группы соединения 1с пригодным алкилирующим агентом 3 формулы R5-(CH2)m-Х, где R5 обозначает защищенную карбоксильную группу.
На стадии 2 эфир 10 гидроксиметилалкинилфеноксикарбоновой кислоты, где Аa обозначает связь, алкилен или алкенилен, получают реакцией соединения 9 с алкиниловым спиртом, таким, как пропаргиловый спирт, в условиях реакции сочетания ацетилена. Эта реакция протекает в присутствии палладийорганического катализатора, такого, как, например, тетракис(трифенилфосфин)палладий(0) и бис(трифенилфосфин)палладий(II)хлорид, необязательно в присутствии в качестве катализатора галогенида меди, такого, как иодид меди(I). Пригодным для реакции растворителем является пирролидин, который дополнительно служит в качестве реагента. В другом варианте эту реакцию можно проводить в присутствии диизопропиламина, необязательно в присутствии в качестве катализатора галогенида меди, в приемлемом апротонном растворителе, таком, как тетрагидрофуран.
На стадии 3 получают эфир 11 алкинилфеноксикарбоновой кислоты аналогично тому, как это проиллюстрировано на схеме А, стадия 3, например, ацилированием соединения 10 ацилирующим агентом 5 формулы R1R2NC(O)X, где Х обозначает галоген, в частности бром или хлор.
На стадии 4 цис-изомер соединения формулы 1 получают селективным превращением тройной связи в молекуле соединения 11 в двойную цис-связь в условиях частичного гидрирования. Пригодными для селективного частичного гидрирования алкинов до цис-алкенов катализаторами являются диизобутилалюминийгидрид (ДИБАЛ-Г) или водород с палладиевым катализатором, таким, как катализатор Линдлара. Эту реакцию проводят с добавлением повышающего селективность агента, такого, как хинолин, в протонном органическом растворителе, таком, как метанол.
На альтернативной стадии 3 эфир 12 цис-гидроксиметилалкенилфеноксикарбоновой кислоты получают селективным превращением тройной связи в молекуле соединения 10 в двойную цис-связь в условиях частичного гидрирования, представленных в вышеприведенном описании стадии 3.
На альтернативной стадии 4 цис-изомер соединения формулы 1 получают аналогично тому, как это проиллюстрировано на схеме А, стадия 4, например, ацилированием соединения 12 ацилирующим агентом 5 формулы R1R2NC(O)X, где Х обозначает галоген, в частности бром или хлор.
Соединение формулы I, где А обозначает алкилен, необязательно может быть получено селективным гидрированием углерод-углеродной двойной связи продукта или любого из промежуточных соединений, синтезированных перед конечным продуктом, с получением соответствующих насыщенных соединений. Приемлемые условия селективного восстановления включают условия каталитического восстановления, такие, как создаваемые с помощью никеля Ренея, палладия на угле, борида никеля, металлической платины, ее оксида и т.п., предпочтительно металлического палладия или его оксида. Растворители, пригодные для этой реакции, включают инертные органические растворители, такие, как этилацетат, метанол и т. п. В предпочтительном варианте соединения 10, 11 или 12 подвергают селективному гидрированию с получением соединений формулы I, где А обозначает алкилен.
Некоторые процессы получения цис-изомера соединения формулы I с созданием реакционных условий, представленных на схеме В, приведены в примерах 8 и 10 (см. в конце описания).
Цис-изомер соединения формулы Iа, в которой В обозначает группу -(СH2)n-, синтезируют аналогично способу, который проиллюстрирован на схеме В, но с использованием других исходных соединений, получая целевой конечный продукт.
Исходное соединение формулы 1d, где Х обозначает галоген, предпочтительно бром или иод, a Z обозначает галоген, предпочтительно бром или иод, или группу -СНО или -(СН2)nСООН, где n имеет значения, приведенные в разделе “Краткое изложение сущности изобретения”, имеется в продаже, например, выпускается фирмой Aldrich Chemical Co., известно или может быть легко синтезировано специалистами в данной области техники.
На стадии 1 эфир 13 галоидфенилкарбоновой кислоты, где Аa обозначает связь, алкилен или алкенилен, может быть получено по различным методам. Так, например, соединение 13, у которого n обозначает 3, получают сочетанием соединения 1с, у которого каждый из Х и Z обозначает галоген, с винилкарбоновым эфиром, таким, как метил-3-бутеноат, в присутствии гидроборирующего агента, такого, как 9-борабицикло[3.3.1]нонановый димер (9-ББН). Эту реакцию проводят в присутствии катализаторов сочетания, таких, как палладийхлорид и трикалийфосфат, в апротонном растворителе, таком, как дихлорметан, N,N-диметилформамид, тетрагидрофуран и т. п. Переэтерификацию получаемого в качестве продукта эфира карбоновой кислоты проводят обработкой спиртом, таким, как 2-метил-2-пропанол, и основанием, таким, как н-бутиллитий, в инертной атмосфере.
По другому варианту соединение 13, где n обозначает 2, может быть получено обработкой соединения 1с, где Х обозначает галоген, а Z обозначает группу -СНО, алкилидентрифенилфосфораном или алкилиденфосфонатом, который получают in situ в присутствии фосфониевой соли или фосфоната, такого, как алкилфосфонацетат, сильным основанием, таким, как гидрид лития или гидрид натрия, в условиях реакции Виттига или Хорнера. Получаемый в качестве продукта алкениленкарбоновый эфир селективно гидрируют, получая соответствующее насыщенное соединение. Приемлемые условия селективного восстановления включают условия каталитического восстановления, такие, как создаваемые с помощью никеля Ренея, палладия на угле, борида никеля, металлической платины, ее оксида и т.п., в инертном органическом растворителе, таком, как этилацетат, метанол и т.п.
На стадии 2 эфир 14 гидроксиметилалкинилфенилкарбоновой кислоты получают аналогично тому, как это проиллюстрировано на схеме В, стадия 2, например, обработкой соединения 13 алкиниловым спиртом, таким, как пропаргиловый спирт. В другом варианте соединение 14, у которого n обозначает О или 1, можно получать непосредственно реакцией исходного соединения 1d, у которого Х обозначает галоген, a Z обозначает группу -(СН2)nСООН, где n обозначает соответственно 0 или 1, алкиниловым спиртом, таким, как пропаргиловый спирт.
Далее на стадиях 3 и 4 или на альтернативных стадиях 3 и 4 аналогично тому, как это проиллюстрировано на схеме В, получают цис-изомер соединения формулы 1а.
Соединение формулы 1а, в которой А обозначает алкилен, а В обозначает группу -(СН2)n-, необязательно может быть получено селективным гидрированием углерод-углеродной двойной связи продукта или любого из промежуточных соединений, синтезированных перед конечным продуктом, с получением соответствующих насыщенных соединений. Приемлемые условия селективного восстановления включают условия каталитического восстановления, такие, как создаваемые с помощью никеля Ренея, палладия на угле, борида никеля, металлической платины, ее оксида и т.п., предпочтительно металлического палладия или его оксида. Растворители, пригодные для этой реакции, включают инертные органические растворители, такие, как этилацетат, метанол и т.п. В предпочтительном варианте соединения 14, 15 или 16 подвергают селективной гидрогенизации с получением соединений формулы 1а, в которой А обозначает алкилен.
Некоторые процессы получения соединения формулы 1а с созданием реакционных условий, представленных на схеме Г, приведены в примерах 13-15.
Промышленная применимостьСоединения по настоящему изобретению представляют собой модуляторы IP-рецептора, в частности агонисты IP-рецептора, и как таковые проявляют селективное агонистическое действие в отношении IP-рецептора. Полагают, что эти соединения (и включающие их композиции) могут оказаться эффективными при профилактике и лечении самых разнообразных заболеваний, прямо или косвенно связанных с нарушением кровотока у млекопитающих, в частности у людей. Так, в частности, полагают, что соединения по настоящему изобретению могут найти применение для лечения при болезненных состояниях, связанных с сердечно-сосудистыми заболеваниями, включающими, но не ограниченными только ими, окклюзионные поражения периферических артерий (ОППА), такие, которые вызывают перемежающуюся хромоту, критическую ишемию конечности, тромботические заболевания, атеросклероз, облитерирующие тромбангииты (болезнь Бюргера), синдром Рейно, болезнь Такаясу, мигрирующий тромбофлебит поверхностных вен, острую артериальную окклюзию, поражение коронарной артерии, рестеноз вследствие пластической операции на сосудах, приступ или возвратный инфаркт миокарда. Соединения по изобретению могут также применяться при лечении гипертензивных болезненных состояний, включающих, но не ограниченных только ими, общую гипертензию, легочную гипертензию, повышенное глазное давление и шум в ушах, связанный с гипертензией. Кроме того, соединения по изобретению могут быть использованы для лечения болезненных состояний, связанных с ишемией, включающих, но не ограниченных только ими, ишемию, связанную с пересадкой аллотрансплантата, такой, как пересадка почки и пересадка других органов. Соединения по изобретению могут быть также использованы для лечения почечных заболеваний, включающих, но не ограниченных только ими, почечную недостаточность, нарушение диуреза, нарушение натрийуреза и нарушение калийуреза. Соединения по изобретению могут быть использованы для лечения других болезненных состояний, включающих, но не ограниченных только ими, неправильное заживание раны, некроз ткани, преждевременные сокращения матки, язву желудка, половую дисфункцию у мужчин и женщин, сильную менструальную боль, нарушение иммунорегуляции, нарушение агрегации кровяных пластинок и нарушение нейтрофильной функции. Улучшения при лечении заболеваний, обусловленных нарушением кровотока, могут проявляться в ослаблении или устранении боли, обусловленной этим болезненным состоянием. Так, например, применение соединений по изобретению может обеспечить ослабление периферических заболеваний нервной системы, связанных, например, с диабетической невропатией, посттравматической невропатией, постхирургической болью, болью, обусловленной химиотерапией, и т.д. Эти и другие области терапевтического применения описаны, например, в Goodman & Gilman’s, The Pharmacological Basis of Therapeutics, 9-изд., McGraw-Hill, Нью-Йорк, 1966, глава 26, 601-616; у Coleman R.A., Pharmacological Reviews, 1994, 46: 205-229; в Harrison’s Principals of Internal Medicine, 14-е изд., McGraw-Hill, Нью-Йорк, 1998, 1398-1403; в Handbook of Phase I/II Clinical Drug Trials, под ред. J. O’Grady и Р.Н. Joubert, CRC Press, Нью-Йорк, 1997, 249-278. Тестирование Агонистическое сродство соединений по настоящему изобретению к IP-рецептору можно определить по методу радиолигандного замещения из мембран тромбоцита человека, которые экспрессируют эндогенный IP-рецептор, или клеток яичника китайского хомячка, экспрессирующих рекомбинантный IP-рецептор крысы. Это последнее испытание более подробно описано в примере 23. Агонистическую эффективность соединений по настоящему изобретению в отношении IP-рецептора можно определить по накоплению циклического АМФ в опыте с использованием либо тромбоцитов человека, либо клеток яичника хомячка китайского, экспрессирующих рекомбинантный IP-рецептор крысы. Это последнее испытание более подробно описано в примере 24. Предполагаемую эффективность соединений по изобретению в отношении IP-рецептора можно определить на модели заболевания периферических сосудов у собаки. В основу этого испытания была положена модель перемежающейся хромоты у животного; испытание более подробно описано в примере 25. Применение и фармацевтические композиции В изобретении предлагается также фармацевтическая композиция, содержащая соединение по изобретению, включая его изомеры, рацемические и нерацемические смеси изомеров, или фармацевтически приемлемые соли и сольваты совместно с одним или несколькими фармацевтически приемлемыми носителями и необязательными другими терапевтическими и/или профилактическими компонентами. Обычно соединения по изобретению вводят в терапевтически эффективном количестве любым из пригодных для этой цели методов введения агентов, для чего служат аналогичные средства. Приемлемая ежедневная доза варьируется от 1 до 2500 мг, предпочтительно от 1 до 1500 мг, наиболее предпочтительно от 1 до 500 мг ежедневно, что зависит от многочисленных факторов, таких, как серьезность заболевания, при котором необходимо лечение, возраст и относительное состояние здоровья субъекта, эффективность используемого соединения, путь введения и препаративная форма для него, показание, в соответствии с которым предписано введение, а также предпочтения, свойственные лечащему врачу, и его опыт. Специалист в области лечения таких заболеваний обычно может самостоятельно установить терапевтически эффективное количество соединений по изобретению для данного заболевания без проведения излишних экспериментов, опираясь на личные знания и описание настоящего изобретения. Соединения по настоящему изобретению обычно применяют в виде фармацевтических композиций, включающих те вещества, которые пригодны для перорального (включая оральное и подъязычное), ректального, назального, локального, легочного, вагинального или парентерального (включая внутримышечное, внутриартериальное, интратекальное, подкожное и внутривенное) введения, или в форме, пригодной для введения путем ингаляции или инсуффляции. Предпочтительным путем введения является пероральный с применением удобного режима приема ежедневной дозы, который можно регулировать в соответствии со степенью заболевания. Из соединений по изобретению совместно с обычным адъювантом, носителем или разбавителем можно приготавливать фармацевтические композиции и унифицированные дозы. Фармацевтические композиции и унифицированные дозы могут включать обычные компоненты в обычных пропорциях вместе с дополнительными активными веществами или основами или без них, и такие унифицированные дозы могут содержать любое приемлемое эффективное количество активного компонента, соразмеримое с предусматриваемым для приема диапазоном ежедневных доз. Такая фармацевтическая композиция может быть использована в виде твердых препаратов, таких, как таблетки или наполненные капсулы, полутвердых препаратов, порошков, лекарственных препаратов с непрерывным высвобождением или жидких препаратов, таких, как растворы, суспензии, эмульсии, эликсиры и наполненные капсулы для перорального применения, или в форме суппозиториев для ректального или вагинального введения, или в форме стерильных растворов для инъекций при парентеральном применении. Таким образом, типичным примером унифицированной дозы является композиция, содержащая одну тысячу (1000) миллиграммов активного вещества или в более широком диапазоне – от одной сотни (100) миллиграммов до пятисот (500) миллиграммов на таблетку. С использованием соединений по настоящему изобретению можно готовить самые разнообразные лекарственные препараты для перорального введения. Фармацевтические композиции и лекарственные препараты в качестве активного компонента могут включать соединения по изобретению или их фармацевтически приемлемые варианты в виде солей или кристаллов. Фармацевтически приемлемые носители могут представлять собой либо твердые вещества, либо жидкости. Твердые препаративные формы включают порошки, таблетки, пилюли, капсулы, крахмальные облатки, суппозитории и диспергируемые гранулы. Твердым носителем может служить одно или несколько веществ, которые могут также выполнять функции разбавителей, корригентов, солюбилизаторов, смазывающих веществ, суспендирующих агентов, связующих веществ, консервантов, разрыхлителей для таблеток или инкапсулирующего материала. В порошках носитель представляет собой тонкоизмельченное твердое вещество, которое смешивают с тонкоизмельченным активным компонентом. При изготовлении таблеток активный компонент смешивают с носителем, обладающим в приемлемых пропорциях необходимой связующей способностью, и прессуют, придавая целевые форму и размеры. Предпочтительные порошки и таблетки включают от одного до примерно семидесяти процентов активного вещества. Пригодными носителями являются карбонат магния, стеарат магния, тальк, сахар, лактоза, пектин, декстрин, крахмал, желатина, трагакант, метилцеллюлоза, натрийкарбоксиметилцеллюлоза, низкоплавкий воск, масло какао и т.п. Термин “приготовление” подразумевает объединение активного вещества в композиции с инкапсулирующим материалом в качестве носителя с получением капсулы, в которой активный компонент с носителем или без него окружен носителем, связанным с ним. Подобным же образом сюда относят крахмальные облатки и лепешки. Таблетки, порошки, капсулы, пилюли, крахмальные облатки и лепешки могут быть приготовлены в твердой препаративной форме, припригодной для перорального введения. Другие препаративные формы, пригодные для перорального введения, включают жидкие препараты, к которым относятся эмульсии, сиропы, эликсиры, водные растворы, водные суспензии, и твердые препаративные формы, которые предназначены для перевода незадолго до употребления в жидкие препаративные формы. Эмульсии могут быть приготовлены в виде растворов, водных пропиленгликолевых растворов или могут содержать эмульгаторы, такие, как лецитин, сорбитанмоноолеат и гуммиарабик. Водные растворы могут быть приготовлены растворением действующего компонента в воде и добавлением приемлемых красителей, корригентов, стабилизаторов и загустителей. Водные суспензии могут быть приготовлены диспергированием тонкоизмельченного активного компонента в воде с вязким материалом, таким, как природные и синтетические камеди, смолы, метилцеллюлоза, натрийкарбоксиметилцеллюлоза и другие хорошо известные суспендирующие агенты. Препараты в жидкой форме включают растворы, суспензии и эмульсии и могут содержать в дополнение к активному компоненту красители, корригенты, стабилизаторы, буферы, искусственные и природные подслащивающие вещества, диспергаторы, загустители, солюбилизаторы и т.п. На основе соединений по настоящему изобретению могут быть приготовлены композиции для парентерального введения (например, инъекцией, в частности инъекцией ударной дозы вещества или непрерывным вливанием), которые могут представлять собой препараты в унифицированных дозах в ампулах, предварительно заполненных шприцах, контейнерах для вливания небольшого объема или на несколько доз с добавкой консерванта. Эти композиции можно также приготавливать в виде таких препаративных форм, как суспензии, растворы или эмульсии в масляных или водных носителях, например растворы в водном полиэтиленгликоле. Примеры масляных и неводных носителей, разбавителей, растворителей и носителей включают пропиленгликоль, полиэтиленгликоль, растительные масла (например, оливковое масло) и пригодные для инъекций органические сложные эфиры (например, этилолеат) и могут содержать добавки для композиций, такие, как консерванты, смачивающие агенты, эмульгаторы, суспендирующие агенты, стабилизаторы и/или диспергирующие агенты. В другом варианте активный компонент может находиться в порошкообразной форме, в которой его получают выделением в асептических условиях стерильного твердого вещества или лиофилизацией из раствора для придания соответствующей структуры перед использованием с пригодным носителем, например, со стерильной, не содержащей пирогенов воды. На основе соединений по настоящему изобретению могут быть приготовлены композиции для локального введения нанесением на эпидермис в виде мазей, кремов и лосьонов или бляшки для чрескожного введения. Мази и кремы можно, например, готовить с использованием водной или масляной основы и добавлением приемлемых загустителей и/или гелеобразователей. Лосьоны могут быть приготовлены с использованием водной или масляной основы и обычно содержат также один или несколько эмульгаторов, стабилизаторов, диспергирующих агентов, суспендирующих агентов, загустителей и красителей. Композиции, пригодные для локального введения через рот, включают лепешки, содержащие активные вещества в обработанной корригентом основе, обычно в сахарозе и гуммиарабике или трагаканте, пастилки, содержащие активное вещество в инертной основе, такой, как желатин и глицерин или сахароза и гуммиарабик, и жидкости для полоскания рта, содержащие активное вещество в пригодном жидком носителе. На основе соединений по настоящему изобретению могут быть приготовлены композиции для введения в виде суппозиториев. Вначале плавят низкоплавкий воск, такой, как глицериды жирных кислот, или масло какао и равномерно диспергируют, например, путем перемешивания, активный компонент. Далее расплавленную однородную смесь выливают в формы соответствующего размера и оставляют для остывания и застывания. На основе соединений по настоящему изобретению могут быть приготовлены препараты для вагинального введения. К ним относятся вагинальные суппозитории, тампоны, кремы, гели, пасты, пенки и аэрозоли, содержащие, помимо активного компонента, такие носители, которые известны в данной области техники как приемлемые. На основе соединений по настоящему изобретению могут быть приготовлены препараты для назального введения. Растворы и суспензии вводят непосредственно в носовую полость с помощью обычных средств, например, капельницы, пипетки или аэрозольного баллончика. Эти композиции могут быть приготовлены в форме препарата на одну дозу или несколько доз. В последнем случае при наличии капельницы и пипетки этой цели можно добиться введением пациенту соответствующего заданного объема раствора или суспензии. В случае аэрозольного препарата такой цели можно, например, добиться с помощью дозировочного насоса для распыления аэрозоля. На основе соединений по настоящему изобретению могут быть приготовлены препараты для введения в форме аэрозоля, в частности через дыхательные пути, включая интраназальное введение. Соединение обычно находится в форме мелких частиц размером, в частности, примерно 5 мкм или менее. Частицы такого размера могут быть получены с помощью средств, известных в данной области техники, например, путем очень тонкого измельчения. Действующий компонент содержится в упаковке под давлением совместно с пригодным пропеллентом, таким, как хлорфторуглерод (ХФУ), например, дихлордифторметан, трихлорфторметан или дихлортетрафторэтан, диоксид углерода или другой приемлемый газ. Приемлемый аэрозоль может также включать поверхностно-активное вещество, такое, как лецитин. Дозу лекарственного средства можно контролировать с помощью дозирующего клапана. В другом варианте на основе активных компонентов можно готовить препараты в форме сухого порошка, например порошкообразной смеси соединения с приемлемой порошкообразной основой, такой, как лактоза, крахмал, производные крахмала, такие, как гидроксипропилметилцеллюлоза и поливинилпирролидон (ПВП). В носовой полости порошкообразный носитель образует гель. Порошкообразная композиция может представлять собой препарат в унифицированной дозе, например в виде капсул или кассет, например в желатиновой или блистерной упаковке, содержащийся в которой порошок можно вводить с помощью ингалятора. При необходимости препараты на основе композиций можно снабжать энтеросолюбильными покрытиями, рассчитанными на введение благодаря длительному или регулируемому высвобождению действующего вещества. Предпочтительны фармацевтические препараты, которые представлены в форме унифицированных доз. В такой форме препарат разделен на дозы, рассчитанные на один прием и содержащие соответствующие количества активного вещества. Препарат в дозах на один прием может быть соответствующим образом упакован, причем такая упаковка содержит раздельные порции препарата, такие, как упакованные таблетки, капсулы и порошки во флаконах или ампулах. Кроме того, препарат в дозах на один прием может представлять собой саму капсулу, таблетку, крахмальную облатку или лепешку или же им может служить соответствующее число любых из таких упакованных форм. Другие приемлемые фармацевтические носители и включающие их композиции описаны в Remington: The Science and Practice of Pharmacy 1995, под ред. E. W. Martin, Mack Publishing Company, 19-е изд., Easton, шт. Пенсильвания. Типичные фармацевтические композиции, включающие соединения по настоящему изобретению, описаны в примерах 16-22. ПРИМЕРЫ Ниже изобретение проиллюстрировано на примерах, которые не ограничивают объем изобретения. ПРОЦЕСС 1 ПОЛУЧЕНИЯ Получение соединений формулы 1а 1А. Получение 3-гидроксиметил-2-метилбензойной кислоты Раствор 43,1 г (285 ммолей) 3-амино-2-метилбензойной кислоты, 700 мл воды и 55 мл серной кислоты кратковременно выдерживали при повышенной температуре с получением прозрачного раствора, а затем охлаждали до температуры ниже oС. Отдельными порциями добавляли раствор 21 г (300 ммолей) нитрита натрия в минимальном количестве воды, одновременно поддерживая внутреннюю температуру ниже 7oС. После перемешивания в ммолей) нитрита натрия в минимальном количестве воды, одновременно поддерживая внутреннюю температуру ниже 7oС. После перемешивания в течение примерно 30 мин на ледяной бане для устранения избытка нитрила добавляли 7 г мочевины. В диазониевый раствор добавляли охлажденный раствор 240 мл 98%-ной серной кислоты в 1000 мл воды и объединенную реакционную смесь медленно нагревали до 90oС. Медленно добавляли 410 мг твердого гидроксида натрия. Смесь экстрагировали этилацетатом, органический слой сушили над сульфатом натрия и упаривали с получением в виде желтого твердого вещества 44,4 г (100%-ный выход) 3-гидроксиметил-2-метилбензойной кислоты. 1Б. Получение 3-гидрокси-2-изопропилбензойной кислоты 2-(2,3-диметоксифенил)-4,4-диметил-4,5-дигидрооксазол получали по методу, описанному A.I. Meyers и др. в J. Org. Chem., 1978, 43, 1372-1379. В раствор 7,06 г (30,0 ммоля) 2-(2,3-диметоксифенил)-4,4-диметил-4,5-дигидрооксазола в свежеперегнанном тетрагидрофуране при комнатной температуре по каплям вводили 18,75 мл (37,5 ммоля) 2,0 М раствора изопропилмагнийхлорида в диэтиловом эфире. Смесь перемешивали в течение ночи, реакцию прекращали добавлением воды и разбавляли диэтиловым эфиром. Слои разделяли и водный слой дважды промывали диэтиловым эфиром. Объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали под вакуумом. В результате очистки экспресс-хроматографией на силикагеле с элюированием смесью гексан/этилацетат в виде прозрачного масла получили 7,08 г (выход: 95,4%) 2-(2-изопропил-3-метоксифенил)-4,4-диметил-4,5-дигидрооксазола. Раствор 6,18 г (25 ммолей) 2- (2-изопропил-3-метоксифенил) -4,4-диметил-4,5-дигидрооксазола в 40 мл 35%-ного раствора серной кислоты кипятили с обратным холодильником и перемешивали в течение 48 ч. При охлаждении медленно добавляли 18,15 г твердого гидроксида натрия. Реакционную смесь экстрагировали диэтиловым эфиром и объединенные органические экстракты экстрагировали раствором бикарбоната натрия. Далее бикарбонатные экстракты подкисляли 3 н. соляной кислотой, фильтровали и сушили, получая в виде белого кристаллического твердого вещества 1,73 г (выход: 35,7%) 2-изопропил-3-метоксибензойной кислоты. 1 М раствор трибромида бора в 15 мл (15 ммолей) дихлорметана по каплям вводили в раствор 2-изопропил-3-метоксибензойной кислоты в дихлорметане. После перемешивания в течение 2 ч реакционную смесь охлаждали до 0oС, реакцию прекращали добавлением воды и слои разделяли. Водный слой экстрагировали дихлорметаном. Объединенные органические экстракты сушили над безводным сульфатом натрия и концентрировали под вакуумом. В результате очистки остатка экспресс-хроматографией на силикагеле с элюированием смесью дихлорметан/метанол и 1% уксусной кислоты в виде белого твердого кристаллического вещества получили 1,32 г (выход: 97,5%) 3-гидрокси-2-изопропилбензойной кислоты. 1В. Получение 3-гидрокси-2-фенилбензойной кислоты 10 мл 2 М водного раствора бикарбоната в атмосфере азота вводили в суспензию 2,06 г (7,7 ммоля) этилового эфира 2-бром-3-метоксибензойной кислоты, 1,11 г бензолборной кислоты и 0,30 г тетракис(трифенилфосфин)палладия(0) в 35 мл толуола. Смесь перемешивали при 80oС в течение 2 ч, сливали в воду и экстрагировали диэтиловым эфиром. Органический слой упаривали и остаток хроматографировали, элюируя смесью гексан/ацетон в соотношении 3:1, с получением полутвердого масла. В результате перекристаллизации из диэтилового эфира/гексана в виде бесцветных кристаллов получили 0,69 г (выход: 34%) этилового эфира 3-метокси-2-фенилбензойной кислоты. В атмосфере азота объединяли 0,53 г этилового эфира 3-метокси-2-фенилбензойной кислоты и 6 г пиридингидрохлорида, а затем в течение 1 ч выдерживали при 180oС. Расплав выливали в разбавленную водную соляную кислоту и экстрагировали этилацетатом. В результате очистки хроматографией на силикагеле с элюированием смесью гексан/ацетон в соотношении 2:1 и 0,5% уксусной кислоты в виде белого твердого вещества получили 0,25 г (выход: 58%) 3-гидрокси-2-фенилбензойной кислоты. ПРИМЕР 1 {2-хлор-3-[(дифенилкарбамоилокси)метил]фенокси}уксусная кислота 1А. Получение 2-хлор-3-гидроксиметилфенола 2-Хлор-3-гидроксибензойную кислоту синтезировали по методу, описанному Buehler и др. в J. Amer. Chem. Soc., 1946, 68, 574-577. Раствор 2,59 г (15 ммолей) 2-хлор-3-гидроксибензойной кислоты в тетрагидрофуране при температуре 0oС по каплям вводили в 150 мл раствора (48 ммолей) борана в тетрагидрофуране. Далее реакционную смесь подогревали до 60oС и оставляли перемешиваться в течение ночи. При охлаждении реакцию прекращали добавлением воды и подщелачивали 3 М гидроксидом натрия. Водный слой выделяли, подкисляли 3 М соляной кислотой и экстрагировали дихлорметаном. Объединенные органические экстракты сушили над сульфатом натрия и концентрировали под вакуумом. В результате перекристаллизации из этанола/гексана в виде белого твердого кристаллического вещества получили 1,63 г (выход: 64,8%) 2-хлор-3-гидроксиметилфенола. 1Б. Получение метилового эфира (2-хлор-3-гидроксиметилфенокси) уксусной кислоты 3,42 г (10,5 ммоля) карбоната цезия при комнатной температуре вводили в раствор 1,59 г (10 ммолей) 2-хлор-3-гидроксиметилфенола в ацетоне и реакционную смесь оставляли перемешиваться в течение 30 мин. Далее добавляли 1,68 г (11 ммолей) метилбромацетата. Смесь перемешивали в течение ночи, фильтровали и фильтрат концентрировали под вакуумом. В результате очистки остатка экспресс-хроматографией на силикагеле с элюированием смесью дихлорметан/метанол в виде белого твердого кристаллического вещества получили 1,94 г (выход: 84,1%) метилового эфира (2-хлор-3-гидроксиметилфенокси) уксусной кислоты. 1В. Получение {2-хлор-3-[(дифенилкарбамоилокси)метил]фенокси} уксусной кислоты Раствор 1,94 г (8,41 ммоля) метилового эфира (2-хлор-3-гидроксиметилфенокси) уксусной кислоты в тетрагидрофуране охлаждали до -78oС, обрабатывали бис(триметилсилил)амидом калия (10,1 ммоля) и оставляли перемешиваться при -78oС в течение примерно часа. По каплям добавляли раствор 2,92 г (12,6 ммоля) дифенилкарбамилхлорида в тетрагидрофуране, образовывавшейся смеси давали нагреться до комнатной температуре и ее перемешивали в течение ночи. Реакцию прекращали добавлением воды, разбавляли диэтиловым эфиром и слои разделяли. Водный слой еще дважды экстрагировали диэтиловым эфиром. Объединенные органические экстракты сушили над сульфатом натрия и концентрировали под вакуумом. В результате очистки остатка экспресс-хроматографией на силикагеле с элюированием смесью гексан/ацетон в соотношении 4:1 в виде белого твердого кристаллического вещества получили 1,72 г (выход: 48,0%) метилового эфира {2-хлор-3-[(дифенилкарбамоилокси)метил]фенокси} уксусной кислоты. 0,64 г (1,5 ммоля) метилового эфира {2-хлор-3-[(дифенилкарбамоилокси)метил]фенокси} уксусной кислоты растворяли в смеси 20 мл метанола, 5 мл воды и 2 мл тетрагидрофурана. Добавляли водный раствор 0,07 г (1,65 ммоля) гидроксида лития и смесь оставляли перемешиваться в течение ночи. Смесь концентрировали под вакуумом и остаток разбавляли водой и промывали диэтиловым эфиром. Водный слой подкисляли 3 М соляной кислотой, вследствие чего образовывался белый осадок, который отфильтровывали и сушили. В результате перекристаллизации из метанола в виде белых игольчатых кристаллов получили 0,47 г (выход: 76,5%) {2-хлор-3-[(дифенилкарбамоилокси)метил] фенокси} уксусной кислоты; tпл 182,2-182,9oС; 1Н-ЯМР: 4,66 (s, 2H), 5,30 (s, 2H), 6,83 (dt, J=7,5 Гц, 2H), 7,11 (t, J=8,0 Гц), 7,28 (m, 10Н); 13С-ЯМР: 64,98 (t), 66,02 (t), 112,92 (d), 121,62 (d), 126,27 (d), 126,93 (d), 127,02 (d), 128,94 (d), 135,79 (s), 142,34 (s), 153,68 (s), 154,27 (s), 170,27 (s), Данные элементного анализа для С22Н18ClNО5: рассч. : С 64,16; Н 4,41; N 3,40; найдено: С 64,30; Н 4,39; N 3,56. ПРИМЕР 2 {3-[(дифенилкарбамоилокси)метил]-2-метилфенокси} уксусная кислота 2А. Получение 3-гидроксиметил-2-метилфенола Раствор 44,4 г (285 ммолей) 3-гидрокси-2-метилбензойной кислоты (полученной аналогично процессу 1 получения, п. 1А) в 200 мл сухого тетрагидрофурана при температуре 0oC вводили с одновременным перемешиванием в 1 М раствор борана в 800 мл тетрагидрофурана. Образовывавшуюся полутвердую смесь нагревали в сосуде с защитным кожухом и обратным холодильником почти до кипения и выдерживали при повышенной температуре в течение примерно 15 ч. В реакционную смесь добавляли 200 мл метанола. Образовывавшийся прозрачный раствор упаривали под вакуумом и затем упаривали с дополнительными порциями метанола с получением 30,3 г (выход: 99%) 3-гидроксиметил-2-метилфенола. 2Б. Получение трет-бутилового эфира (3-гидроксиметил-2-метилфенокси)уксусной кислоты 75 г (385 ммолей) трет-бутилбромацетата вводили в раствор/суспензию 46,6 г (337 ммолей) 3-гидроксиметил-2-метилфенола и 60 г карбоната калия в виде тонкодисперсного порошка в 500 мл ацетона. Смесь кипятили с обратным холодильником в течение примерно 40 ч или до завершения реакции. Твердое вещество отфильтровывали и жидкий остаток упаривали. В результате очистки остатка хроматографией на силикагеле, элюируя смесью ацетон/гексан, в виде бледно-желтого масла получили 80,1 г (выход: 96%) трет-бутилового эфира (3-гидроксиметил-2-метилфенокси)уксусной кислоты. 2В. Получение [3-[(дифенилкарбамоилокси) метил]-2-метилфенокси] уксусной кислоты Раствор 80,1 г (318 ммолей) трет-бутилового эфира (3-гидроксиметил-2-метилфенокси) уксусной кислоты в 400 мл тетрагидрофурана охлаждали до -50oС и обрабатывали диизопропиламидом лития (325 ммолей), свежеполученным из 49 мл диизопропиламина и 130 мл 2,5М н-бутиллития в 100 мл тетрагидрофурана. По истечении примерно 10 мин выдержки при -50oС добавляли раствор 80,8 г (350 ммолей) дифенилкарбамилхлорида, растворенного в 100 мл тетрагидрофурана. Всю реакционную смесь перемешивали в течение еще 30 мин и затем ей давали нагреться до комнатной температуры в течение 3 ч. Раствор упаривали под вакуумом, выливали в разбавленную соляную кислоту и экстрагировали диэтиловым эфиром. Органический слой сушили над рассолом, упаривали и очищали хроматографией на силикагеле, элюируя смесью ацетон/гексан (с изменением соотношения 1: 5–>1: 3), с получением в виде белого твердого вещества 104,1 г (выход: 73%)трет-бутилового эфира {3-[(дифенилкарбамоилокси)метил]-2-метилфенокси} уксусной кислоты. 104,1 г (233 ммолей) трет-бутилового эфира [3- [(дифенилкарбамоилокси)метил]-2-метилфенокси} уксусной кислоты и 10,3 г (245 ммолей) гидроксида лития растворяли в смеси 100 мл воды, 300 мл метанола и 300 мл тетрагидрофурана. Смесь становилась твердой, но при выдержке при температуре кипения она вновь переходила в жидкое состояние. Прозрачный раствор перемешивали при комнатной температуре в течение примерно 16 ч и упаривали под вакуумом. Остаток выливали в разбавленную соляную кислоту и экстрагировали дихлорметаном. В результате выпаривания растворителя и перекристаллизации остатка из смеси гексан/ацетон в виде не беловатых гранул получали 87,0 г (выход: 95%) {3-[(дифенилкарбамоилокси)метил]-2-метилфенокси} уксусной кислоты; tпл 180,1-180,7oС. Данные элементного анализа для С23Н21NО5: рассч.: С 70,58; Н 5,41; N 3,58; найдено: С 70,51; Н 5,37; N 3,76. ПРИМЕР 3 3А. Аналогично примеру 2, но при замене 3-гидрокси-2-метилбензойной кислоты, указанной в примере 2А, на другие соединения формулы 1а и внесении соответствующих изменений в процесс были получены следующие соединения формулы I: {2,6-диметил-3-[(дифенилкарбамоилокси)метил]фенокси] уксусная кислота tпл 177,6-178,1oС. 1Н-ЯМР: 2,21 (s, 3Н), 2,27 (s, 3H), 4,62 (s, 2H), 5,30 (s, 2H), 6,65 (d, J=8,4 Гц, 1Н), 6,93 (d, J=8,4 Гц, 1Н), 7,22 (m, 10 Н). 13С-ЯМР: 11,83 (q), 19,25 (q), 62,53 (t), 65,67 (t), 112,19 (d), 126,14 (d), 126,90 (d), 128,02 (d), 128,07 (d), 128,88 (d), 131,96 (s), 134,02 (s), 142,51 (s), 153,88 (s), 154,79 (s), 172,66 (s). Данные элементного анализа для С24Н23NО5  0,1Н2О: рассч.: С 70,78; Н 5,74; N 3,44; найдено: С 70,52; Н 5,67; N 3,52.
{2-бром-3-[(дифенилкарбамоилокси)метил]фенокси} уксусная кислота 0,1Н2О: рассч.: С 70,78; Н 5,74; N 3,44; найдено: С 70,52; Н 5,67; N 3,52.
{2-бром-3-[(дифенилкарбамоилокси)метил]фенокси} уксусная кислотаtпл 188,2-189,0oС. Данные элементного анализа для С20Н18ВrNO5: рассч.: С 57,91; Н 3,98; N 3,07; найдено: С 57,91; Н 3,98; N 3,13. {3-[(дифенилкарбамоилокси)метил]-2-фенилфенокси}уксусная кислота tпл 181,0-181,5oС. Данные элементного анализа для С28Н23NО5: рассч.: С 74,16; Н 5,11; N 3,09; найдено: С 73,84; Н 5,15; N 3,19. {3-[(дифенилкарбамоилокси) метил]-2-этилфенокси} уксусная кислота tпл 160,9-161,7oС. 1Н-ЯМР: 0,95 (t, J=7,4 Гц, 3Н), 2,57 (q, J=7,4 Гц, 2Н), 4,69 (s, 2H), 5,17 (s, 2Н), 6,79 (d, J=7,9 Гц, 1Н), 6,81 (d, J=7,9 Гц, 1H), 7,08 (t, J=7,9 Гц, 1Н), 7,30 (m, 10 Н), 12,98 (широкий s, 1H). 13С-ЯМР: 13,79 (q), 18,58 (t), 64,60 (t), 64,92 (t), 111,27 (d), 121,35 (d), 126,28 (d), 127,07 (d), 128,91 (d), 130,80 (s), 134,65 (s), 142,26 (s), 153,74 (s), 155,45 (s), 170,19 (s). Данные элементного анализа для С24Н23NO5: рассч. : С 71,10; Н 5,72; N 3,45; найдено: С 70,98; Н 5,71; N 3,61. {3-[(дифенилкарбамоилокси)метил]-2-пропилфенокси} уксусная кислота tпл 51,0-54,0oС. 1Н-ЯМР: 0,858 (t, J=7,4 Гц, 3Н), 1,41 (секстет, J=7,4 Гц, 2H), 2,56 (t, J=7,7 Гц, 2H), 4,33 (s, 2H), 5,17 (s, 2H), 6,76 (t, J=8,8 Гц, 2H), 7,02 (t, J=8,0 Гц, 1H), 7,29 (m, 10 Н). 13С-ЯМР: 14,44 (q), 22,33 (t), 27,58 (t), 65,18 (t), 66,50 (t), 111,49 (d), 120,71 (d), 126,12 (d), 126,31 (d), 127,12 (d), 128,97 (d), 129,29 (s), 134,64 (s), 142,34 (s), 153,84 (s), 156,43 (s), 170,43 (s). Данные элементного анализа для 25H25NO5 0,55H2O: рассч. : С 69,83; Н 6,12; N 3,39; найдено: С 69,93; Н 6,13; N 3,26.
трет-Бутиламиновая соль { 3-[(дифенилкарбамоилокси)метил]-2-изопропилфенокси} уксусной кислоты (0,49 г, выход: 58,4%)tпл 173,7-182,2oС. 1Н-ЯМР (част./млн,  ): 1,19 (s, 3Н), 1,21 (s, 9H), 3,05 (септет, J=6,8 Гц, 1H), 4,16 (s, 2H), 5,12 (s, 2H), 6,72 (d, J=7,6 Гц, 1H), 6,74 (d, J=8,4 Гц, 1H), 6,98 (t, J=7,9 Гц, 1H), 7,29 (m, 10 Н), 8,33 (широкий s, 3Н). 13С-ЯМР (част./млн, ): 20,50 (q), 27,41 (q), 28,06 (d), 50,13 (s), 66,32 (t), 67,89 (t), 113,09 (d), 121,59 (d), 126,02 (d), 126,34 (d), 127,23 (d), 129,00 (d), 133,49 (s), 134,37 (s), 142,39 (s), 153,84 (s), 156,15 (s), 171,13 (s). Данные элементного анализа для C25H25NO5C4H11N: рассч.: С 70,71; Н 7,37; N 5,69; найдено: С 70,75; Н 7,39; N 5,84.
3Б. Раствор 0,77 г (4,7 ммоля) 3-гидроксифталевого ангидрида в 15 мл тетрагидрофурана кипятили с обратным холодильником совместно с 20 мл борантетрагидрофуранового комплекса в течение 18 ч. Образовывавшуюся суспензию охлаждали и разлагали избытком метанола (выделение газа) и под вакуумом удаляли растворитель. Остаток упаривали с метанолом, получая в виде белого твердого вещества 0,72 г (выход: 98%) (2,3-бисгидроксиметил)фенола. Работая в дальнейшем аналогично примеру 2, но заменяя 3-гидроксиметил-2-метилфенол, упомянутый в примере 2Б, на (2,3-бисгидроксиметил)фенол и проводя процесс в соответствии с примером 2В, получили следующее соединение формулы I: ): 1,19 (s, 3Н), 1,21 (s, 9H), 3,05 (септет, J=6,8 Гц, 1H), 4,16 (s, 2H), 5,12 (s, 2H), 6,72 (d, J=7,6 Гц, 1H), 6,74 (d, J=8,4 Гц, 1H), 6,98 (t, J=7,9 Гц, 1H), 7,29 (m, 10 Н), 8,33 (широкий s, 3Н). 13С-ЯМР (част./млн, ): 20,50 (q), 27,41 (q), 28,06 (d), 50,13 (s), 66,32 (t), 67,89 (t), 113,09 (d), 121,59 (d), 126,02 (d), 126,34 (d), 127,23 (d), 129,00 (d), 133,49 (s), 134,37 (s), 142,39 (s), 153,84 (s), 156,15 (s), 171,13 (s). Данные элементного анализа для C25H25NO5C4H11N: рассч.: С 70,71; Н 7,37; N 5,69; найдено: С 70,75; Н 7,39; N 5,84.
3Б. Раствор 0,77 г (4,7 ммоля) 3-гидроксифталевого ангидрида в 15 мл тетрагидрофурана кипятили с обратным холодильником совместно с 20 мл борантетрагидрофуранового комплекса в течение 18 ч. Образовывавшуюся суспензию охлаждали и разлагали избытком метанола (выделение газа) и под вакуумом удаляли растворитель. Остаток упаривали с метанолом, получая в виде белого твердого вещества 0,72 г (выход: 98%) (2,3-бисгидроксиметил)фенола. Работая в дальнейшем аналогично примеру 2, но заменяя 3-гидроксиметил-2-метилфенол, упомянутый в примере 2Б, на (2,3-бисгидроксиметил)фенол и проводя процесс в соответствии с примером 2В, получили следующее соединение формулы I:{ 3-[(дифенилкарбамоилокси)метил] -2-гидроксиметилфенокси] уксусную кислоту; tпл 137,2-137,9oС. Данные элементного анализа для C23H21NO6: рассч.: С 67,81; Н 5,20; N 3,44; найдено: С 67,81; Н 5,24; N 3,57. 3В. Аналогично примеру 2, но с заменой дифенилкарбамилхлорида, упомянутого в примере 2В, на N-фенил-N-пиридин-3-илкарбамилхлорид или N-циклогексил-N-фенилкарбамилхлорид и при соответствующем проведении процесса были получены следующие соединения формулы I: { 2-метил-3-[(фенилпиридин-3-илкарбамоилокси)метил]фенокси} уксусная кислота tпл 180,1-180,7oС. Данные элементного анализа для С22Н20N2O5 0,35Н2O: рассч.: С 66,27; Н 5,23; N 7,03; найдено: С 66,41; Н 5,36; N 6,76.
{3-[(циклогексилфенилкарбамоилокси)метил]фенокси} уксусная кислотаtпл 175,5-176,2oС. 1H-ЯМР: 0,90 (m, 1Н), 1,10 (m, 2Н), 1,32 (m, 2Н), 1,53 (m, 1H), 1,71 (m, 2H), 1,89 (m, 2H), 2,10 (s, 3H), 4,15 (m, 1H), 4,58 (s, 2H), 5,08 (s, 2H), 6,70 (m, 2H), 7,06 (m, 3H), 7,32 (m, 3H). 13С-ЯМР: 11,04 (q), 25,28 (t), 25,83 (t), 31,96 (t), 56,85 (d), 65,37 (t), 65,73 (t), 111,23 (d), 121,60 (d), 125,90 (s), 125,98 (d), 127,45 (d), 128,61 (d), 130,05 (d), 136,45 (s), 138,20 (s), 156,20 (s), 171,21 (s). Данные элементного анализа для С23Н27N05: рассч. : С 69,50; Н 6,85; N 3,52; найдено: С 69,36; Н 6,85; N 3,70. ПРИМЕР 4 {3-[(дифенилкарбамоилокси)метил]фенокси} уксусная кислота 4А. Получение метилового эфира (3-формилфенокси) уксусной кислоты 5 г порошкообразного карбоната калия вводили в раствор 3,1 г (25 ммолей) 3-гидроксибензальдегида и 4,6 г (30 ммолей) метилбромацетата в 40 мл ацетона и смесь перемешивали при кипячении с обратным холодильником в течение 4 ч. Далее реакционную смесь выливали в избыток воды, экстрагировали дихлорметаном и упаривали. Полученный продукт в виде желтого масла перегоняли на трубке с шаровыми расширениями (1 мм рт.ст., 150oС) с получением в виде бесцветного масла 4,53 г (выход: 93%) метилового эфира (3-формилфенокси) уксусной кислоты. 4Б. Получение метилового эфира (3-гидроксиметилфенокси) уксусной кислоты Раствор 1 г боргидрида натрия в 10 мл воды быстро вводили в раствор 4,53 г метилового эфира (3-формилфенокси) уксусной кислоты в 50 мл метанола. По истечении 10 мин образовывавшийся прозрачный раствор подкисляли избытком 10%-ной водной соляной кислоты, выливали в воду, экстрагировали дихлорметаном и выпаривали растворители. В результате хроматографии на силикагеле с элюированием смесью 5% ацетона/дихлорметан в виде бесцветного масла получили 2,51 г (выход: 55%) метилового эфира (3-гидроксиметилфенокси) уксусной кислоты. 4В. Получение {3-[(дифенилкарбамоилокси)метил]фенокси} уксусной кислоты 400 мг (2 ммоля) метилового эфира (3-гидроксиметилфенокси) уксусной кислоты и 580 мг (2,5 ммоля) дифенилкарбамоилхлорида объединяли в 2 мл пиридина. Образовывавшийся желтый раствор выдерживали в течение 10 дней в атмосфере азота. Далее смесь выливали в воду, экстрагировали дихлорметаном и упаривали. В результате очистки остатка хроматографией на силикагеле с элюированием смесью 5% ацетона/дихлорметан в виде бесцветного твердого вещества получили 575 мг (выход: 73%) метилового эфира {3-[(дифенилкарбамоилокси)метил]фенокси} уксусной кислоты. Метиловый эфир {3-[(дифенилкарбамоилокси)метил]фенокси} уксусной кислоты гидролизовали аналогично примеру 2В с получением в виде белого твердого вещества { 3-[(дифенилкарбамоилокси)метил] фенокси} уксусной кислоты; пл 142,2-143,8oС. 1Н-ЯМР: 4,63 (s, 2H), 5,11 (m, 2H), 6,82 (m, 3Н), 7,30 (m, 11Н), 12,89 (широкий s, 1H). Данные элементного анализа для 22Н19NО5: рассч.: С 70,02; Н 5,07; N 3,71; найдено: С 70,06; Н 5,07; N 3,89. ПРИМЕР 5 {3-[(1-дифенилкарбамоилокси)этил]-2-метилфенокси} уксусная кислота 5А. Получение трет-бутилового эфира (3-формил-2-метилфенокси)уксусной кислоты По методу, описанному у Marx М. и Tidwell Т. в J. Org. Chem, 1984, 49, 788-793, получали трет-бутиловый эфир (3-гидроксиметил-2-метилфенокси) уксусной кислоты. 0,46 мл (5,25 ммоля) оксалилхлорида по каплям вводили в раствор 0,88 мл (12,50 ммоля) диметилсульфоксида в 40 мл дихлорметана. Смесь охлаждали до -78oС и оставляли перемешиваться в течение 1 ч. В реакционную смесь с помощью полой иглы добавляли раствор 1,26 г трет-бутилового эфира (3-гидроксиметил-2-метилфенокси)уксусной кислоты в дихлорметане и оставляли перемешиваться при -78oС в течение еще одного часа. Добавляли 3,49 мл (25 ммолей) триэтиламина. Реакционной смеси давали нагреться до комнатной температуры, выливали в воду и слои разделяли. Водный слой дважды экстрагировали дихлорметаном. Объединенные органические экстракты промывали 2н. соляной кислотой и насыщенным раствором бикарбоната натрия, сушили над безводным сульфатом натрия и концентрировали под вакуумом. В результате очистки хроматографией на силикагеле с элюированием смесью гексан/ацетон в виде желтоватого твердого кристаллического вещества получили 1,07 г (выход: 85,9%) трет-бутилового эфира (3-формил-2-метилфенокси) уксусной кислоты. 5Б. Получение трет-бутилового эфира [3-(1-гидроксиэтил)-2-метилфенокси] уксусной кислоты 0,94 г (3,75 ммоля) трет-бутилового эфира (3-формил-2-метилфенокси) уксусной кислоты растворяли в свежеперегнанном тетрагидрофуране и охлаждали до -78oС. По каплям добавляли 2,62 мл (7,88 ммоля) метилмагнийхлорида и смесь оставляли перемешиваться при -78oС в течение двух часов. Смеси давали нагреться до комнатной температуры, перемешивали еще в течение шести часов, а затем реакцию прекращали осторожным добавлением воды и экстрагировали этилацетатом. Органические экстракты сушили над безводным сульфатом магния и концентрировали под вакуумом. В результате очистки хроматографией на силикагеле с элюированием смесью гексан/ацетон в виде прозрачного масла получили 594 мг (выход: 59,5%) трет-бутилового эфира [3-(1-гидроксиэтил)-2-метилфенокси] уксусной кислоты. 5В. Получение {3-[(1-дифенилкарбамоилокси)этил]-2-метилфенокси} уксусной кислоты 553 мг (2,00 ммоля) трет-бутилового эфира [3-(1-гидроксиэтил)-2-метилфенокси] уксусной кислоты и 695 мг (3,00 ммоля) дифенилкарбамилхлорида растворяли в 30 мл свежеперегнанного тетрагидрофурана и охлаждали до -78oС. По каплям добавляли 1,20 мл (2,40 ммоля) диизопропиламида лития. Смеси давали нагреться до комнатной температуры и перемешивали в течение ночи, реакцию прекращали добавлением диэтилового эфира и слои разделяли. Органический слой сушили над безводным сульфатом натрия и концентрировали под вакуумом. В результате очистки хроматографией на силикагеле с элюированием смесью гексан/ацетон в соотношении 9: 1 в виде прозрачного масла получили 411 мг (выход: 44,5%) трет-бутилового эфира {3-[(1-дифенил-карбамоилокси)этил]-2-метилфенокси] уксусной кислоты. 346 мг (0,75 ммоля) трет-бутилового эфира [3-[(1-дифенилкарбамоилокси)этил] -2-метилфенокси} уксусной кислоты растворяли в 25 мл метанола и 10 мл воды. Добавляли водный раствор 69 мг (1,65 ммоля) гидроксида лития и смесь перемешивали при комнатной температуре в течение 6 ч. Смесь концентрировали под вакуумом, а остаток разбавляли водой и промывали диэтиловым эфиром. Водный слой подкисляли разбавленной соляной кислотой и экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и концентрировали под вакуумом с получением в виде прозрачной пены 293 мг (выход: 96,4%) {3-[(1-дифенилкарбамоилокси) этил]-2-метилфенокси} уксусной кислоты; tпл 57,0-69,0oС. 1H-ЯМР: 1,35 (d, J=6,5 Гц, 3Н), 2,13 (s, 3H), 4,67 (s, 2H), 5,96 (q, J=6,5 Гц, 1Н), 6,67 (d, J=7,8 Гц, 1H), 6,74 (d, J=8,2 Гц, 1Н), 7,08 (d, J=8,0 Гц, 1Н), 7,26 (m, 6H), 7,39 (m, 4H), 12,99 (широкий s, 1H). 13С-ЯМР: 9,56 (q), 20,44 (q), 63,82 (t), 69,83 (d), 109,33 (d), 116,41 (d), 121,56 (s), 125,20 (d), 125,28 (d), 125,99 (d), 127,87 (d), 140,33 (s), 141,21 (s), 154,46 (s), 169,16 (s). Данные элементного анализа для C22H18ClNO5: 0,4СH2Cl2 рассч.: С 69,47; Н 5,62; N 3,36; найдено: С 69,41; Н 5,62; N 3,78. ПРИМЕР 6 транс-[3-(3-Дифенилкарбамоилоксипропенил)фенокси] уксусная кислота 6А. Получение метилового эфира транс-3-(3-гидроксифенил)акриловой кислоты Раствор 9,94 мл (61,4 ммоля) триметилфосфонацетата, 2,60 г (12,2 ммоля) хлорида лития и 15,92 мл (106,5 ммоля) 1,8-диазабицикло(5.4.0)ундец-7-ена (ДБУ) в 80 мл ацетонитрила при комнатной температуре в атмосфере азота перемешивали в течение 30 мин. Добавляли 5,00 г (40,9 ммоля) 3-гидроксибензальдегида и смесь при комнатной температуре перемешивали еще в течение 3 ч. Добавляли воду и водную фазу экстрагировали диэтиловым эфиром. Объединенные органические фракции промывали водой и рассолом, сушили над сульфатом магния и упаривали досуха. В результате очистки экспресс-хроматографией с элюированием смесью гексан/этилацетат в виде белого твердого кристаллического вещества получили 6,30 г (выход: 86%) метилового эфира транс-3-(3-гидроксифенил)акриловой кислоты. 6Б. Получение транс-3-(3-гидроксипропенил)фенола Литий-н-бутилдиизобутилалюминийгидрид получали in situ введением при -5oС в атмосфере аргона 1,0 М диизобутилалюминийгидрида (ДИБАЛ-Г) в 16,8 мл (16,8 ммоля) толуола в 6,73 мл (16,8 ммоля) 2,5 М раствора н-бутиллития в гексане. Добавляли 70 мл тетрагидрофурана и смесь охлаждали до -78oС. В нее по каплям в течение 15 мин добавляли раствор 1,0 г (5,61 ммоля) метилового эфира транс-3-(3-гидроксифенил)акриловой кислоты в 70 мл тетрагидрофурана, объединенному раствору давали достичь температуры -20oС и перемешивали в течение 3 ч. Смесь охлаждали до -78oС и добавляли 6 г декагидрата сульфата натрия, после чего 150 мл насыщенного раствора хлорида аммония и концентрированной соляной кислоты до рН 2-3. Смесь экстрагировали дихлорметаном, экстракт промывали водой и рассолом, сушили над сульфатом натрия и концентрировали досуха. В результате очистки экспресс-хроматографией с элюированием смесью гексан/этилацетат в виде прозрачного твердого кристаллического вещества получили 0,58 г (выход: 68%) транс-3 -(3-гидроксипропенил) фенола. 6В. Получение трет-бутилового эфира транс-[3-(3-гидроксипропенил)фенокси] уксусной кислоты Раствор 0,60 мл (4,03 ммоля) трет-бутилбромацетата и 1,12 г (8,06 ммоля) карбоната калия при комнатной температуре в атмосфере азота вводили в раствор 0,58 г (3,84 ммоля) транс-3-(3-гидроксипропенил)фенола в 15 мл ацетона и перемешивали в течение примерно 20 ч. Смесь фильтровали и фильтрат концентрировали досуха. В результате очистки экспресс-хроматографией с элюированием смесью гексан/этилацетат в виде прозрачного масла получили 0,987 г (выход: 97%) трет-бутилового эфира транс-[3-(3-гидроксипропенил)фенокси] уксусной кислоты. 6Г. Получение транс-[3-(3-дифенилкарбамоилоксипропенил)фенокси] уксусной кислоты Раствор 0,50 г (1,89 ммоля) трет-бутилового эфира транс-[3-(3-гидроксипропенил) фенокси] уксусной кислоты в 15 мл тетрагидрофурана при -78oС в атмосфере аргона вводили в 1,04 мл (2,08 ммоля) 2,0 М раствора диизопропиламида лития в смеси гептан/тетрагидрофуран/этилбензол и 0,46 г (1,98 ммоля) дифенилкарбамилхлорида. Смеси давали достичь температуры 0-5oС и выдерживали при этой температуре в течение 16 ч. Добавляли насыщенный раствор хлорида аммония и продукт экстрагировали дихлорметаном. Экстракт промывали водой и рассолом, сушили над сульфатом натрия и концентрировали досуха. В результате очистки экспресс-хроматографией с элюированием смесью гексан/этилацетат в соотношении 7: 3 в виде белого твердого кристаллического вещества получили 0,56 г (выход: 65%) трет-бутилового эфира транс- [3-(3-дифенилкарбамоилоксипропенил)фенокси] уксусной кислоты. 1,46 мл (1,46 ммоля) 1,0 М гидроксида лития при комнатной температуре в атмосфере азота вводили в раствор 0,56 г (1,22 ммоля) трет-бутилового эфира транс-[3-(3-дифенилкарбамоилоксипропенил) фенокси] уксусной кислоты в 2 мл метанола и 2 мл тетрагидрофурана. Смесь перемешивали при комнатной температуре в течение 3 ч. Выпаривали растворитель, добавляли воду, а затем 2н. соляную кислоту до значения рН 1-2 и продукт экстрагировали этилацетатом. Экстракт промывали водой и рассолом, сушили над сульфатом магния и концентрировали досуха. В результате перекристаллизации из диэтилового эфира в виде белого твердого кристаллического вещества получили 0,38 г (выход: 78%) транс-[3-(3-дифенилкарбамоилоксипропенил)фенокси] уксусной кислоты, tпл 112,4-113,7oС. ПРИМЕР 7 транс-[3-(3-Дифенилкарбамоилоксипропенил)-2-метилфенокси] уксусная кислота 7А. Получение этилового эфира транс-3-(3-гидрокси-2-метилфенил) акриловой кислоты 1,16 мл (10,7 ммоля) этилакрилата, 60,1 мг (0,27 ммоля) ацетата палладия, 160 мг (0,54 ммоля) три-о-толилфосфина и 1,62 мл триэтиламина в атмосфере азота вводили в раствор 1 г (5,35 ммоля) 3-бром-2-метилфенола в 2,5 мл ацетонитрила. Смесь выдерживали при 82oС в течение 16 ч и охлаждали. Добавляли 1н. соляную кислоту и смесь экстрагировали этилацетатом. Экстракт промывали водой и рассолом, сушили над сульфатом натрия и концентрировали досуха. В результате очистки экспресс-хроматографией с элюированием смесью гексан/этилацетат в виде прозрачного масла получили 4850 мг (выход: 43,5%) этилового эфира транс-3-(3-гидрокси-2-метилфенил)акриловой кислоты. 7Б. Получение транс-[3-(3-дифенилкарбамоилоксипропенил)-2-метилфенокси] уксусной кислоты Работая аналогично примеру 6, но заменяя метиловый эфир транс-3-(3-гидроксифенил)акриловой кислоты, использованной в примере 6Б, на этиловый эфир транс-3-(3-гидрокси-2-метилфенил)акриловой кислоты и проводя процесс в соответствии с примером 6В, получили 141 мг (выход: 89%) транс-[3-(3-дифенилкарбамоилоксипропенил)-2-метилфенокси] уксусной кислоты в виде белого твердого вещества; tпл 118,0-118,6oС. ПРИМЕР 8 цис-[3-(3-Дифенилкарбамоилоксипропенил)-2-метилфенокси] уксусная кислота 8А. Получение трет-бутилового эфира (3-бром-2-метилфенокси)уксусной кислоты 1,08 мл (7,27 ммоля) трет-бутилбромацетата и 2,01 г (14,55 ммоля) карбоната калия вводили в раствор 1,29 г (6,92 ммоля) 3-бром-2-метилфенола в 17 мл ацетона и смесь перемешивали при комнатной температуре в атмосфере азота в течение 5 ч. Смесь фильтровали и фильтрат упаривали досуха с получением в виде бледно-желтого масла 2,12 г (выход: 100%) трет-бутилового эфира (3-бром-2-метилфенокси)уксусной кислоты. 8Б. Получение трет-бутилового эфира [3-(3-гидроксипроп-1-инил)-2-метилфенокси] уксусной кислоты 0,41 г (0,35 ммоля) тетракис(трифенилфосфин) палладия(0) и 0,83 мл (14,3 ммоля) пропаргилового спирта при комнатной температуре в атмосфере аргона вводили в раствор 2,13 г (7,08 ммоля) трет-бутилового эфира (3-бром-2-метилфенокси) уксусной кислоты в 21 мл пирролидина. Эту смесь выдерживали при 75-80oС в течение 2,5 ч. Добавляли избыток насыщенного раствора аммонийхлорида и смесь экстрагировали диэтиловым эфиром. Экстракт промывали рассолом, сушили над сульфатом натрия и концентрировали досуха. В результате очистки экспресс-хроматографией с элюированием смесью гексан/этилацетат в виде масла получили 0,38 г (выход: 19,5%) трет-бутилового эфира [3-(3-гидроксипроп-1-инил)-2-метилфенокси] уксусной кислоты. 8В. Получение трет-бутилового эфира [3-(3-дифенилкарбамоилоксипроп-1-инил)-2-метилфенокси] уксусной кислоты 0,77 мл (1,54 ммоля) 2,0 М диизопропиламида лития в смеси толуол/гептан/этилбензол при -78oС вводили в атмосфере аргона в перемешиваемый раствор 370 мг (1,34 ммоля) трет-бутилового эфира [3-(3-гидроксипроп-1-инил)-2-метилфенокси] уксусной кислоты в 4 мл тетрагидрофурана. Смесь перемешивали в течение 10 мин и добавляли 310 мг (1,34 ммоля) твердого дифенилкарбамоилхлорида. Смеси давали достичь температуры 0-5oС и перемешивали в течение 16 ч. Добавляли насыщенный раствор хлорида аммония и смесь экстрагировали этилацетатом. Экстракт промывали рассолом, сушили над сульфатом натрия и концентрировали досуха. В результате очистки экспресс-хроматографией с элюированием смесью гексан/этилацетат в виде бесцветного масла получили 220 мг (выход: 35%) трет-бутилового эфира [3-(3-дифенилкарбамоилоксипроп-1-инил) -2-метилфенокси] уксусной кислоты. 8Г. Получение цис-[3-(3-дифенилкарбамоилоксипропенил)-2-метилфенокси] уксусной кислоты 22 мкл хинолина и 22 мг катализатора Линдлара в атмосфере азота вводили в раствор 215 мг (0,46 ммоля) трет-бутилового эфира [3-(3-дифенилкарбамоилоксипроп-1-инил)-2-метилфенокси] уксусной кислоты в 2 мл метанола. Смесь перемешивали в течение 35 мин в баллоне, наполненном водородом. Смесь фильтровали и фильтрат упаривали досуха. В результате очистки экспресс-хроматографией с элюированием смесью гексан/этилацетат в соотношении 92,5:7,5 в виде бесцветного масла получили 150 мг (выход: 69,4%) трет-бутилового эфира цис-[3-(3-дифенилкарбамоилоксипропенил)-2-метилфенокси] уксусной кислоты. 13 мг (0,31 ммоля) моногидрата гидроксида лития при комнатной температуре и в токе азота вводили в перемешиваемый раствор 144 мг (0,30 ммоля) трет-бутилового эфира цис-[3-(3-дифенилкарбамоилоксипропенил)-2-метилфенокси] уксусной кислоты в 1,4 мл тетрагидрофурана, 0,31 мл метанола и 0,3 мл воды. Смесь перемешивали в течение 4 ч при комнатной температуре, добавляли 1н. соляную кислоту до значения рН 1-2, затем добавляли воду и экстрагировали этилацетатом. Экстракт промывали рассолом, сушили над сульфатом натрия и концентрировали досуха. В результате кристаллизации из смеси дихлорметан/гексан в виде белого твердого вещества получили 141 мг (выход: 89%) цис-[3-(3-дифенилкарбамоилокси-пропенил)-2-метилфенокси] уксусной кислоты; tпл 133,4-135,5oС. ПРИМЕР 9 цис-[3-(3-Дифенилкарбамоилоксипропенил)фенокси] уксусная кислота 9А. Получение трет-бутилового эфира (3-бромфенокси) уксусной кислоты 1,88 мл (12,69 ммоля) трет-бутилбромацетата и 3,5 г (25,38 ммоля) карбоната калия вводили в раствор 2,09 г (12,08 ммоля) 3-бромфенола в 30 мл ацетона и смесь при комнатной температуре перемешивали в атмосфере азота в течение 5 ч. Смесь фильтровали и фильтрат упаривали досуха с получением в виде бесцветного масла 3,53 г (выход: 100%) трет-бутилового эфира (3-бром-фенокси) уксусной кислоты. 9Б. Получение трет-бутилового эфира [3-(3-гидроксипроп-1-инил)фенокси] уксусной кислоты 0,66 г (0,57 ммоля) тетракис(трифенилфосфин) палладия(0) и 1,34 мл (22,97 ммоля) пропаргилового спирта при комнатной температуре в атмосфере аргона вводили в раствор 3,3 г (11,49 ммоля) трет-бутилового эфира (3-бромфенокси) уксусной кислоты в 35 мл пирролидина. Эту смесь выдерживали при 75-80oС в течение 2,5 ч. Добавляли избыток насыщенного раствора хлорида аммония и смесь экстрагировали диэтиловым эфиром. Экстракт промывали рассолом, сушили над сульфатом натрия и концентрировали досуха. В результате очистки экспресс-хроматографией с элюированием смесью гексан/этилапетат в виде бледно-желтого масла получили 1,93 г (выход: 64%) трет-бутилового эфира [3-(3-гидроксипроп-1-инил)фенокси] уксусной кислоты. 9В. Получение трет-бутилового эфира цис-[3-(3-гидроксипропенил) фенокси] уксусной кислоты. 54 мкл хинолина и 54 мг катализатора Линдлара вводили в атмосфере азота в перемешиваемый раствор 547 мг (2,09 ммоля) трет-бутилового эфира [3-(3-гидроксипроп-1-инил)фенокси] уксусной кислоты в 4,3 мл метанола. Смесь перемешивали в течение 1 ч в баллоне, заполненном водородом, фильтровали и фильтрат упаривали досуха. В результате очистки остатка экспресс-хроматографией с элюированием смесью гексан/этилапетат, растворения в диэтиловом эфире и промывки 10 мл 2н. соляной кислоты и 10%-ным бикарбонатом натрия в виде бесцветного масла получили 420 мг (выход: 76%) трет-бутилового эфира цис-[3-(3-гидроксипропенил)фенокси] уксусной кислоты. 9Г. Получение цис-[3-(3-дифенилкарбамоилоксипропенил)фенокси] уксусной кислоты 0,9 мл (1,8 ммоля) 2,0 М раствора диизопропиламида лития в смеси тетрагидрофуран/гептан/этилбензол при -78oС вводили в атмосфере аргона в перемешиваемый раствор 421 мг (1,56 ммоля) трет-бутилового эфира цис-[3-(3-гидроксипропенил)фенокси] уксусной кислоты в 4,5 мл тетрагидрофурана. Смесь перемешивали в течение 10 мин и добавляли 361 мг (1,56 ммоля) твердого дифенилкарбамоилхлорида. Смеси давали достичь температуры 0-5oС и перемешивали в течение 16 ч. Добавляли насыщенный раствор хлорида аммония и смесь экстрагировали этилацетатом. Экстракт сушили над сульфатом натрия и концентрировали досуха. В результате очистки экспресс-хроматографией с элюированием смесью гексан/этилацетат в соотношении 9:1 получили 433 мг (выход: 60,4%) трет-бутилового эфира цис-[3-(3-дифенилкарбамоилоксипропенил)фенокси] уксусной кислоты. 48,2 мг (1,15 ммоля) моногидрата гидроксида лития при комнатной температуре и в токе азота вводили в перемешиваемый раствор 421 мг (1,04 ммоля) трет-бутилового эфира цис-[3-(3-гидроксипропенил)фенокси] уксусной кислоты в 4,9 мл тетрагидрофурана, 1,3 мл метанола и 1,2 мл воды и перемешивали в течение 24 ч при комнатной температуре. Добавляли 1н. соляную кислоту до значения рН 1-2, после чего добавляли воду. Смесь экстрагировали этилацетатом и экстракт промывали рассолом, сушили над сульфатом натрия и концентрировали досуха. Полученный в виде масла продукт растворяли в 3,5 мл диэтилового эфира и добавляли 94 мкл трет-бутиламина. Образовывавшуюся суспензию фильтровали и твердый материал промывали диэтиловым эфиром с получением 360 мг (выход: 82,5%) трет-бутиламиновой соли цис-[3-(3-дифенилкарбамоилоксипропенил)фенокси] уксусной кислоты; tпл 132,5-135oС. ПРИМЕР 10 10А. Аналогично либо примеру 8, либо примеру 9, но при необязательной замене 3-бром-2-метилфенола, использованного в примере 8А, или 3-бромфенола, использованного в примере 9А, на другие соединения формулы 1с, или замене трет-бутилбромацетата на другие соединения формулы 3, или замене пропаргилового спирта на другие алкиниловые спирты и при соответствующем проведении процесса были получены следующие соединения формулы I: трет-бутиламиновая соль цис-[3-(4-дифенилкарбамоилоксибут-1-енил)фенокси] уксусной кислоты; tпл 112,5-112,8oС; трет-бутиламиновая соль цис-[3-(5-дифенилкарбамоилоксипент-1-енил)фенокси] уксусной кислоты; данные элементного анализа для C26H25NO5: рассч.: С 71,41; Н 7,19; N 5,55; найдено: С 71,02; Н 7,12; N 5,54; трет-бутиламиновая соль цис-[3-(6-дифенилкарбамоилоксигекс-1-енил)фенокси] уксусной кислоты; tпл 87,0-88,5oС; трет-бутиламиновая соль цис-[4-(3-дифенилкарбамоилоксипропенил) фенокси] масляной кислоты; tпл 102-106oС; цис-5-[3-(3-дифенилкарбамоилоксипропенил)фенокси] валериановая кислота; tпл 56,3-56,7oС. 10Б. Аналогично либо примеру 8, либо примеру 9, но при необязательной замене 3-бром-2-метилфенола, использованного в примере 8А, или 3-бром-фенола, использованного в примере 9А, на другие соединения формулы 1с и при замене дифенилкарбамоилхлорида на другие соединения формулы 5 и при соответствующем проведении процесса были получены следующие соединения формулы I: трет-бутиламиновая соль цис-[3-(3-метилфенилкарбамоилоксипропенил)фенокси] уксусной кислоты; tпл 152,3-154,2oС; трет-бутиламиновая соль цис-[3-(3-бензилфенилкарбамоилоксипропенил)фенокси] уксусной кислоты; tпл 107,0-108,5oС. ПРИМЕР 11 [3- (3-дифенилкарбамоилоксипропил) фенил] уксусная кислота 11А. Получение метилового эфира 3-(3-гидроксифенил)пропионовой кислоты 0,31 г 10%-ного палладия на угле при комнатной температуре вводили в атмосфере азота в раствор 31,0 г (17,4 ммоля) метилового эфира транс-3-(3-гидроксифенил)акриловой кислоты (полученного согласно примеру 6А) в 30 мл этилацетата. Смесь гидрогенизовали под давлением в баллоне в течение 4 ч. Катализатор отфильтровывали и фильтрат концентрировали досуха. 3,16 г сырого метилового эфира 3-(3-гидроксифенил)пропионовой кислоты использовали непосредственно на следующей стадии. 11Б. Получение 3-(3-гидроксипропил) фенола 35,07 мл (35,07 ммоля) 1,0 М алюмогидрида лития в тетрагидрофуране при 0-5oС вводили в атмосфере аргона в раствор 3,16 г (17,5 ммоля) метилового эфира 3-(3-гидроксифенил)пропионовой кислоты в 30 мл тетрагидрофурана. Смесь перемешивали при комнатной температуре в течение 6 ч и в эту смесь добавляли 15 г декагидрата сульфата натрия с последующим охлаждением до 0-5oС и добавлением по каплям примерно 10 мл воды и концентрированной соляной кислоты до значения рН 2-3. Смесь экстрагировали этилацетатом и экстракт промывали водой и рассолом, сушили над сульфатом магния и концентрировали досуха. В результате очистки экспресс-хроматографией с элюированием смесью гексан/этилацетат в виде прозрачного масла получили 2,34 г (выход: 88%) 3-(3-гидроксипропил) фенола. 11В. Получение трет-бутилового эфира [3-(3-гидроксипропил)фенокси] уксусной кислоты 2,39 мл (16,2 ммоля) трет-бутилбромацетата и 4,47 г (32,3 ммоля) карбоната калия вводили в раствор 2,34 г (15,4 ммоля) 3-(3-гидроксипропил) фенола в ацетоне. Смесь при комнатной температуре перемешивали в течение 20 ч, фильтровали и экстракт концентрировали досуха. В результате очистки экспресс-хроматографией с элюированием смесью гексан/этилацетат в виде прозрачного масла получили 3,65 г (выход: 89%) трет-бутилового эфира [3-(3-гидроксипропил)фенокси) уксусной кислоты. 11Г. Получение [3-(3-дифенилкарбамоилоксипропил)фенил] уксусной кислоты 7,57 мл (15,1 ммоля) 2,0 М раствора диизопропиламида лития и раствор 3,35 г (14,5 ммоля) дифенилкарбамилхлорида в 100 мл тетрагидрофурана вводили в раствор трет-бутилового эфира [3-(3-гидроксипропил)фенокси) уксусной кислоты в 40 мл тетрагидрофурана. Смеси давали достичь температуры 0-5oС и выдерживали при этой температуре в течение 16 ч. Добавляли насыщенный раствор хлорида аммония и смесь экстрагировали этилацетатом. Органический слой сушили над рассолом, сушили над сульфатом натрия и концентрировали досуха. В результате очистки хроматографией на силикагеле с элюированием смесью ацетон/гексан в соотношении 7:3 в виде желтого масла получили 3,88 г (выход: 61%) трет-бутилового эфира [3-(3-дифенилкарбамоилоксипропил)фенил] уксусной кислоты. После последующих гидролиза этого эфира и перекристаллизации получали 0,47 г (выход: 54%) [3-(3-дифенилкарбамоилоксипропил)фенил] уксусной кислоты в виде белого твердого кристаллического вещества, tпл 133,7-134,6oС. ПРИМЕР 12 Аналогично примеру 11, но при замене метилового эфира транс-3-(3-гидроксифенил)акриловой кислоты, использованной в примере 11А, на другие соединения формулы 6 и при соответствующем проведении процесса были получены следующие соединения формулы I: [3-(2-дифенилкарбамоилоксиэтил)фенил] уксусная кислота, tпл 123,0-123,3oС; [3-(4-дифенилкарбамоилоксибутил)фенил] уксусная кислота, tпл 117,8-119,6oС; [3-(5-дифенилкарбамоилоксипентил)фенил] уксусная кислота, tпл 128,1-128,7oС; [3-(6-дифенилкарбамоилоксигексил)фенил] уксусная кислота, tпл 99,8-101,7oС; [3-(3-дифенилкарбамоилоксипропил)-2-метилфенил] уксусная кислота, tпл 128,7-129,2oС; [3-(3-дифенилкарбамоилоксипропил)-2-метилфенил] валериановая кислота, tпл 81,5-81,9oС. ПРИМЕР 13 цис-3-[3-(3-Дифенилкарбамоилоксипропенил)фенил] пропионовая кислота 13А. Получение трет-бутилового эфира 3-(3-бромфенил)акриловой кислоты Раствор 2,86 мл (12,2 ммоля) трет-бутилдиэтилфосфонацетата, 0,52 г (12,2 ммоля) хлорида лития и 1,57 мл (10,54 ммоля) 1,8-диазабицикло(5.4.0)ундец-7-ен (ДБУ) в 25 ацетонитрила перемешивали при комнатной температуре в атмосфере азота в течение 30 мин. Добавляли 0,95 мл (10,5 ммоля) 3-бромбензальдегида и смесь при комнатной температуре перемешивали еще в течение 3 ч. Добавляли воду и водную фазу экстрагировали диэтиловым эфиром. Объединенные органические фракции промывали водой и рассолом, сушили над сульфатом магния и упаривали досуха. В результате очистки экспресс-хроматографией с элюированием смесью гексан/этилацетат в виде прозрачного масла получили 2,24 г (выход: 97%) трет-бутилового эфира 3-(3-бромфенил)акриловой кислоты. 13Б. Получение трет-бутилового эфира 3-(3-бромфенил)пропионовой кислоты 0,002 г (0,032 ммоля) оксида платины (IV) при комнатной температуре вводили в атмосфере азота в раствор 2,24 г (7,90 ммоля) трет-бутилового эфира 3-(3-бромфенил)акриловой кислоты в 7 мл метанола и 1 мл тетрагидрофурана. Смесь гидрировали под давлением в баллоне в течение 10 ч. Далее смесь фильтровали и фильтрат концентрировали досуха. В результате очистки экспресс-хроматографией с элюированием смесью гексан/этилацетат в виде прозрачного масла получили 1,32 г (выход: 58%) трет-бутилового эфира 3-(3-бромфенил) пропионовой кислоты. 13В. Получение трет-бутилового эфира 3-[3-(3-гидроксипроп-1-инил) фенил] пропионовой кислоты 0,41 г (0,14 ммоля) тетракис(трифенилфосфин) палладия(0), 0,5 г (0,25 ммоля) иодида одновалентной меди и 0,29 мл (4,91 ммоля) пропаргилового спирта в атмосфере аргона при комнатной температуре вводили в раствор 2,13 г (7,08 ммоля) трет-бутилового эфира 3-(3-бромфенил) пропионовой кислоты в 8 мл пирролидина. Эту смесь выдерживали при 75-80oС в течение 5 ч. После охлаждения до комнатной температуры добавляли насыщенный раствор хлорида аммония и смесь экстрагировали диэтиловым эфиром. Экстракт промывали рассолом, сушили над сульфатом магния и концентрировали досуха. В результате очистки экспресс-хроматографией с элюированием смесью гексан/этилацетат в соотношении 85: 15 в виде желтого масла получили 0,21 г (выход: 33%) трет-бутилового эфира 3-[3-(3-гидроксипроп-1-инил)фенил] пропионовой кислоты. 13Г. Получение 3-[3-(3-дифенилкарбамоилоксипропенил)фенил] пропиновой кислоты Работая аналогично примеру 8, но заменяя трет-бутиловый эфир 3-[3-(3-гидроксилроп-1-инил)-2-метилфенокси] уксусной кислоты, использованной в примере 8В, на трет-бутиловый эфир [3-(3-гидроксипроп-1-инил)фенил] пропионовой кислоты и проводя процесс в соответствии с примером 8Г, 0,14 г (выход: 75%) 3-[3-(3-дифенилкарбамоилоксипропенил)фенил] пропионовой кислоты в виде прозрачного масла получили. В результате кристаллизации из диэтилового эфира совместно с трет-бутиламином получили 570 мг (выход: 75%) трет-бутиламиновой соли цис-3-[3-(3-дифенилкарбамоилоксипропенил)фенил] пропионовой кислоты. ПРИМЕР 14 14А. Аналогично примеру 13, но при замене 3-бромбензальдегида на 3-бромфенилуксусную кислоту, при замене пропаргилового спирта на другой алкиниловый спирт и при соответствующем проведении процесса были получено следующее соединение формулы I: трет-бутиламиновая соль цис-[3-(4-дифенилкарбамоилоксибут-1-енил)фенил] уксусной кислоты; tпл 151,2-153,1oС. 14Б. Аналогично примеру 13, но при замене 1,3-дибромбензола на 3-иодбензойную кислоту, использованную в примере 13А, при необязательной замене пропаргилового спирта на другие алкиниловые спирты и при соответствующем проведении процесса были получены следующие соединения формулы Iа: цис-[3-(3-дифенилкарбамоилоксипропенил)фенил] бензойная кислота, tпл 137,5-137,8oС; трет-бутиламиновая соль цис-3-(4-дифенилкарбамоилоксибут-1-енил)бензойной кислоты, пл 172,8-176,8oС; трет-бутиламиновая соль цис-3-(3-дифенилкарбамоилоксипропенил) бензойной кислоты, tпл 180,5-184,0oС. 14В. Аналогично примеру 13, но при замене 3-бромбензальдегида на 3-бромфенилуксусную кислоту, при замене пропаргилового спирта на другие алкиниловые спирты, путем гидрогенизации по углерод-углеродной двойной связи до соответствующей насыщенной связи и при соответствующем проведении процесса было получено следующее соединение формулы I: трет-бутиламиновая соль [3-(3-дифенилкарбамоилоксибутил)фенил] уксусной кислоты, tпл 112,3-113,1oС. ПРИМЕР 15 Цис-4-[3-(3-дифенилкарбамоилоксипропенил)фенил] масляная кислота 15А. Получение метилового эфира 4-(3-бромфенил) масляной кислоты 13,4 мл (6,68 ммоля) 9-борабицикло[3.3.1]нонанового димера (9-ББН-димер) вводили в атмосфере аргона при 0-5oС в раствор 0,71 мл (6,68 ммоля) метил-3-бутеноата в 2,7 мл тетрагидрофурана и смесь перемешивали в течение 4 ч. Добавляли 27 мл N,N-диметилформамида, хлорид палладия (II), 130 г (0,16 ммоля) дихлорметана, 0,77 мл (61,37 ммоля) 1,3-дибромбензола и 1,5 г (6,94 ммоля) порошкообразного фосфата калия. Смесь обрабатывали при 50oС в течение ночи. Добавляли воду и смесь экстрагировали диэтиловым эфиром. Экстракт промывали водой и рассолом, сушили над сульфатом натрия и концентрировали досуха. В результате очистки экспресс-хроматографией с элюированием смесью гексан/этилацетат в виде прозрачного масла получили 0,26 мг (выход: 16%) метилового эфира 4-(3-бромфенил) масляной кислоты. 15Б. Получение трет-бутилового эфира 4-(3-бромфенил) масляной кислоты 4,43 мл (11,07 ммоля) н-бутиллития при -10oС в атмосфере аргона вводили в раствор 1,0 мл (10,53 ммоля) 2-метил-2-пропанола в 21 мл тетрагидрофурана и смесь перемешивали в течение 10 мин. При -10oС по каплям добавляли раствор 1,35 г (5,27 ммоля) метилового эфира 4-(3-бромфенил) масляной кислоты в 5,3 мл тетрагидрофурана, смеси давали достичь комнатной температуры и перемешивали в течение 16 ч. Добавляли насыщенный раствор хлорида аммония и продукт экстрагировали этилацетатом. Экстракт сушили над сульфатом натрия и концентрировали досуха. В результате очистки экспресс-хроматографией с элюированием смесью гексан/этилацетат в соотношении 98:2 в виде желтого масла получили трет-бутиловый эфир 4-(3-бромфенил) масляной кислоты (выход: 74%). 15В. Получение цис-4-[3-(3-дифенилкарбамоилоксипропенил)фенил] масляной кислоты Работая аналогично примеру 8, но заменяя трет-бутиловый эфир [3-(3-гидроксипроп-1-инил)2-метилфенокси] уксусной кислоты, использованный в примере 8В, на трет-бутиловый эфир 4-(3-бромфенил) масляной кислоты и соответствующим образом проводя процесс, получили 0,14 г (выход: 75%) цис-4-[3-(3-дифенилкарбамоилоксипропенил)фенил] масляной кислоты в виде прозрачного масла. В результате кристаллизации из диэтилового эфира совместно с трет-бутиламином получили трет-бутиламиновую соль цис-4-[3-(3-дифенилкарбамоилоксипропенил)фенил] масляной кислоты, tпл 112,4-115,3oС. ПРИМЕР 16 Композиция для перорального введения, мас.%: Активный компонент – 20,0 Лактоза – 79,5 Стеарат магния – 0,5 Компоненты смешивают и распределяют по капсулам вместимостью по 100 мг каждая. Одна капсула приблизительно соответствует общей ежедневной дозе. ПРИМЕР 17 Композиция для перорального введения, мас.%: Активный компонент – 20,0 Стеарат магния – 0,5 Натрийкросскармеллоза – 2,0 Лактоза – 76,5 Поливинилпирролидон (ПВП) – 1,0 Компоненты смешивают и гранулируют с использованием растворителя, такого, как метанол. Далее композицию сушат и формуют с получением таблеток (содержащих примерно по 20 мг действующего вещества) в соответствующей таблетировочной машине. ПРИМЕР 18 Композиция для перорального введения: Активное соединение – 1,0 г Фумаровая кислота – 0,5 г Хлорид натрия – 2,0 г Метилпарабен – 0,15 г Пропилпарабен – 0,05 г Гранулированный сахар – 25,5 г Сорбит (70%-ный раствор) – 12,85 г Veegum К (фирмы Vanderbilt Co.) – 1,0 г Корригент – 0,035 мл Красители – 0,5 мг Дистиллированная вода – q.s. до 100 мл Компоненты смешивают с получением суспензии для перорального введения. ПРИМЕР 19 Композиция (IV) для парентерального введения Активный компонент – 0,25 г Хлорид натрия – q.s. для достижения изотоничности Вода для инъекций – 100 мл Активный компонент растворяют в части воды для инъекций. Далее с перемешиванием добавляют хлорид натрия в количестве, которого достаточно для получения изотонического раствора. Добавлением оставшейся части воды для инъекций массу раствора доводят до целевой, фильтруют через мембранный фильтр с размером пор 0,2 мкм и упаковывают в стерильных условиях. ПРИМЕР 20 Композиция для суппозитория, мас.%: Активный компонент – 20,0 Полиэтиленгликоль 1000 – 74,5 Полиэтиленгликоль 4000 – 24,5 Компоненты совместно плавят, смешивают на паровой бане и разливают по формам общей вместимости по массе 2,5 г. ПРИМЕР 21 Композиция для локального применения, г: Активное вещество – 0,2-2 Спан 60 – 2 Твин 60 – 2 Минеральное масло – 5 Вазелин – 10 Метилпарабен – 0,15 Пропилпарабен – 0,05 Бутилированный гидроксианизол (БГА) – 0,01 Вода – q.s. до 100 мл Все компоненты, за исключением воды, смешивают и при перемешивании нагревают до примерно 60oС. Далее при интенсивном перемешивании добавляют воду с температурой примерно 60oС в количестве, достаточном для эмульгирования компонентов, а затем воду добавляют в количестве, необходимом до массы примерно 100 г. ПРИМЕР 22 Композиции для интраназального распыления В качестве композиций для интраназального распыления готовят несколько водных суспензий, содержащих по 0,025-0,5% действующего вещества. Эти композиции включают необязательные неактивные компоненты, такие, как микрокристаллическая целлюлоза, натрийкарбоксиметилцеллюлоза, декстроза и т.п. Для регулирования рН можно добавлять соляную кислоту. Композиции для интраназального распыления можно дозировать через дозировочный насос для интраназального распыления, который при однократном нажатии выдает, как правило, 50-100 мкл композиции. Типичный режим дозирования составляет по 2-4 впрыскивания каждые 4-12 ч. ПРИМЕР 23 Испытание на связывание IP-рецептора Сродство соединений по настоящему изобретению к IP-рецептору определяли по методу радиолигандного замещения с использованием клеток яичника китайского хомячка, экспрессирующих рекомбинантный IP-рецептор крысы. Этот метод является модификацией общепризнанных методов, в которых в качестве меченого лиганда используют [3Н] -илопрост. Клетки яичника китайского хомячка, экспрессирующие рекомбинантный IP-рецептор крысы, инкубировали в среде Хэма F-12, содержавшей 10% фетальной сыворотки быка и 250 мкг/мл генетицина, в атмосфере 5% углекислого газа при 37oС. Клетки собирали надо льдом, используя 2 мМ ЭДТК в забуференном фосфатом физиологическом растворе (без кальция/магния, 4oС) и центрифугировали при 500 g. Определяли число клеток и осадок хранили при -70oС.
Осадки в пробирках после центрифугирования размораживали при комнатной температуре, а затем разбавляли буфером для анализа (20 мМ трис-HCl, 5 мМ хлорид магния, рН 7,4) до требуемой концентрации и подвергали кратковременной гомогенизации. После этого в пробирки, содержащие буфер, тестируемое соединение и меченый лиганд, добавляли суспензию осадка из пробирки после центрифугирования. Пробирки инкубировали при 25oС в течение 1 ч, три раза промывали охлажденным льдом буфером для анализа и с помощью жидкостного сцинтилляционного счетчика определяли связанную радиоактивность.
Для каждого тестируемого соединения с использованием итеративной аппроксимации кривой определяли концентрацию, которая вызывала 50%-ное ингибирование связывания (ИК50) и угловой коэффициент Хилла. Константу ингибирования диссоциации (Кi) каждого тестируемого соединения определяли в соответствии с методом Ченга и Прусова (1973).
При оценке ряда соединений по изобретению было установлено, что в ходе проведения этого испытания они проявляли активность при значениях рК, в интервале от примерно 4,8 до примерно 7,2.
ПРИМЕР 24Оценка агонистической активности в отношении IP-рецептора Агонистическую активность соединений по изобретению в отношении IP-рецептора определяли по накоплению циклического АМФ, вызванного агонистом в ходе проведения испытания с использованием клеток яичника китайского хомячка, экспрессирующих рекомбинантный IP-рецептор крысы. Содержание циклического АМФ устанавливали с помощью технически доступного набора реагентов Adenylate Cyclase cAMP Flashplate Assay (фирма New England Nuclear), предназначенного для определения цАМФ. Клетки яичника китайского хомячка, экспрессирующие рекомбинантный IP-рецептор крысы, инкубировали в среде Хэма, содержавшей 10% фетальной сыворотки быка и 250 мкг/мл генетицина, в атмосфере 5% углекислого газа (95% О2) при 37oС. Клетки собирали при приблизительно 90%-ном слиянии с использованием забуференного фосфатом физиологического раствора Дульбекко, содержавшего 2 мМ ЭДТК, один раз промывали и центрифугировали при 1000 g и повторно суспендировали в промывном буфере. Из образца отбирали аликвоту для определения белка. Суспензию клеток центрифугировали при 1000g и содержание клеток доводили до 110-140103/50 мкл в “буфере для стимулирования и определения” из набора реагентов для анализа.
Тестируемые соединения или наполнитель инкубировали совместно с 50 мкл клеток (110-140103 клеток) в течение 5 мин при комнатной температуре. После инкубирования в лунки добавляли 100 мкл лизисно-индикаторного раствора и после инкубации в течение ночи с помощью сцинтилляционного счетчика для микропланшетов типа Packard Topcount определяли радиоактивность. Количество радиоактивного цАМФ, связанного с антителом, обратно пропорционально концентрации добавляемого нерадиоактивного цАМФ. Далее определяли значения рЕС50 и сравнивали со стандартными значениями для агониста.
При оценке ряда соединений по изобретению было установлено, что в ходе проведения этого испытания они активны и в реакции стимулирования внутриклеточного цАМФ значения их рЕС50 превышают 4,82.
ПРИМЕР 25Модель перемежающейся хромоты у собаки Предполагаемую эффективность агонистических соединений IP-рецептора можно определить с помощью модели заболевания периферических сосудов у собаки. Ослабление симптомов перемежающейся хромоты можно определить по способности собак переносить физическую нагрузку после вызванной хирургическим путем острой артериальной недостаточности задней конечности, используя модификацию метода, описанного Bohm E и др. в Arch. Pharmacol. 1990, 341, дополнение R61. В целом этот метод состоит в том, что перед хирургическим вмешательством, включавшем наложение лигатуры/удаление бедренной артерии и окклюзию мелких артериол сверхтонким сефадексом, собак дрессировали на выполнение упражнений при однообразной механической нагрузке и эту способность переносить физическую нагрузку (темп и скорость) для каждой собаки определяли, основываясь на времени до проявления клинических симптомов (хромота или неспособность продолжать выполнять упражнения при однообразной механической нагрузке). По истечении пятидесяти дней после хирургического вмешательства вновь определяли способность переносить физическую нагрузку до достижения предоперационной базовой линии для физической нагрузки. Конечной точкой испытания с физической нагрузкой являлось наблюдение клинических признаков перемежающейся хромоты, таких, как прихрамывание или неспособность продолжать упражнения при однообразной механической нагрузке. Перед и/или после упражнений при однообразной механической нагрузке 1-3 раза в неделю измеряли кровоток в нормальной и подвергнутой хирургической операции задних конечностей. С помощью парного t-критерия сравнивали реакцию в послеоперационный день с реакцией в предоперационный день. Для каждого дня испытаний, используя двухвыборочный t-критерий, определяли разницу результатов испытаний между группой, которую лечили, и контрольной группой. В этой модели соединения по настоящему изобретению увеличивали продолжительность пробега при однообразной механической нагрузке в течение конкретного промежутка времени после хирургического вмешательства в сравнении с достигаемой у контрольных животных. После хирургического вмешательства с удалением бедренной артерии собаки, дрессированные на выполнение упражнений при однообразной механической нагрузке, демонстрировали резкое уменьшение продолжительности пробега. Собакам, как правило, требовалось 21-28 дней после операции для того, чтобы восстановить нормальную продолжительность пробега при однообразной механической нагрузке, тогда как собаки, которых лечили соединениями по изобретению, восстанавливали способность к нормальной продолжительности пробега в течение 5-10 дней после лечения. Хотя настоящее изобретение описано выше на пример конкретных вариантов его выполнения, для специалистов в данной области техники очевидно, что в них можно вносить разнообразные изменения и одни эквиваленты можно заменять другими, не выходя при этом за объем изобретения. Кроме того, для адаптации конкретной ситуации, материала, состава материала, процесса, технологической стадии или стадий к целевым сущности и объему настоящего изобретения можно осуществить множество модификаций. Все такие модификации следует рассматривать как охватываемые объемом прилагаемой формулы изобретения. Накопление циклического АМФ в отношении IP-клона крысы, экспрессированного в СНО клетках. Цель исследования – определить агонистическую активность ряда соединений по накоплению циклического АМФ в отношении простаноидного рецептора в IP-клоне крысы, экспрессированном в СНО клетках. Методика: Определение белка: БЦА (бицинхониновая кислота) реагент для определения белка (Стандартный аналитический набор 23225) Использовались следующие буферы: Промывной буфер: фосфатный физиологический раствор Дульбекко, рН 7,4 при 25oС Буфер для стимулирования и буфер для определения: набор реагентов Adenylate Cyclase cAMP Flashplate Assay (фирма New England Nuclear), предназначенный для определения цАМФ 1. Методика культивирования клеток Инструкции по культивированию и питанию IP-клона крысы, экспрессированного в СНО клетках, соответствуют методике “The Standard Cell Core Facility SOP”. Клетки инкубировали в среде Хэма, содержавшей 10% фетальной сыворотки быка и 250 мкг/мл G-418, в атмосфере 5% углекислого газа (95% 02). Клетки собирали при приблизительно 90%-ном слиянии с использованием забуференного фосфатом физиологического раствора Дульбекко, содержавшего 2 м-М ЭДТК, один раз промывали промывным буфером и центрифугировали при 1000 g.
Клетки повторно суспендировали в промывном буфере. Из образца отбирали аликвоту для определения белка. Суспензию клеток центрифугировали при 1000g и содержание клеток доводили до 12510/50 мкл в “буфере для стимулирования и определения” из набора реагентов для анализа.
2. Определение циклического АМФ сцинтилляционным методом (Adenylate Cvclase cAMP Flashplate Assay)Использовалась следующая схема серийных разбавлений: Базисное, 1Е-4, 3Е-4, 1Е-5, 3Е-6, 1Е-6, 3Е-7, 1Е-7, 3Е-8, 1Е-8, 1Е-9, 1Е-10, 10 мкМ КПК (карба-простациклин использовали для контроля в концентрации 10 мкМ). Все разбавления были сделаны в промывном буфере. ДМСО контролировали при наивысшей концентрации (0,6%), но не более 1%. Содержание циклического АМФ устанавливали с помощью технически доступного набора реагентов Adenylate Cyclase cAMP Flashplate Assay (фирма New England Nuclear), предназначенного для определения цАМФ. Были произведены дополнительные определения, приведенные в табл.1. Тестируемые соединения или наполнитель инкубировали совместно с 50 мкл клеток в течение 5 мин при комнатной температуре. После инкубирования в лунки добавляли 100 мкл лизисно-индикаторного раствора и после инкубации в течение ночи с помощью сцинтилляционного счетчика для микропланшетов типа Packard Topcount определяли радиоактивность. Количество радиоактивного цАМФ, связанного с антителом, обратно пропорционально концентрации добавляемого нерадиоактивного цАМФ. Результаты испытаний представлены в табл.2. В таблице использована следующая аббревиатура: N: значение “n” или число сделанных определений рЕС50: молярная концентрация агониста, обеспечивающая 50%-ную активность от максимально возможной; “р”ЕС50 отрицательный десятичный логарифм ЕС50 SEM: стандартное отклонение значения рЕС50 и nН nH: коэффициент Хилла, он равен тангенсу угла наклона кривой концентрации агониста; единичные значения указывают на то, что агонист взаимодействует с единичным сайтом рецептора; значения nН, представленные в таблице, соответствуют значению единичного сайта0 Формула изобретения
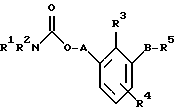 в которой R1 и R2 каждый независимо друг от друга в каждом случае обозначает алкил, арил, аралкил, гетероарил, циклоалкил или гетероциклил; R3 и R4 каждый независимо друг от друга в каждом случае обозначает водород, алкил, галоген, гидроксиалкил или арил; R5 в каждом случае независимо обозначает -COOR6; R6 в каждом случае независимо обозначает водород или алкил; А в каждом случае независимо обозначает алкилен или алкенилен; В в каждом случае независимо обозначает -O(СН2)m– или -(СН2)n-; m в каждом случае независимо обозначает целое число от 1 до 8 включительно; n в каждом случае независимо обозначает целое число от 0 до 8 включительно; в виде их рацемата или индивидуального изомера, или их фармацевтически приемлемые соли, или сольваты. 2. Соединение по п. 1, у которого R1 и R2 каждый независимо друг от друга обозначает арил или аралкил. 3. Соединение по п. 1 или 2, у которого R1 и R2 каждый независимо друг от друга обозначает фенил или бензил. 4. Соединение по любому из пп. 1-3, у которого R3 и R4 каждый независимо друг от друга обозначает водород, алкил, арил или галоген. 5. Соединение по любому из пп. 1-4, у которого R3 и R4 каждый независимо друг от друга обозначает водород, метил, этил, н-пропил, изопропил, бутил, фенил, бром или хлор. 6. Соединение по любому из пп. 1-5, у которого R5 обозначает -COOR6. 7. Соединение по любому из пп. 1-6, у которого R6 обозначает водород. 8. Соединение по любому из пп. 1-7, у которого А обозначает алкилен, В обозначает -O(CH2)m-, a m обозначает целое число от 1 до 5 включительно. 9. Соединение по п. 8, которое представляет собой: { 3-[(дифенилкарбамоилокси)метил] -2-метилфенокси} уксусную кислоту; [3-(3-дифенилкарбамоилоксипропил)фенил] уксусную кислоту, или ее индивидуальный изомер, рацемическую или нерацемическую смесь изомеров, либо фармацевтически приемлемую соль, или сольват. 10. Соединение по любому из пп. 1-7, у которого А обозначает алкилен, В обозначает -(СН2)n-, а n обозначает целое число от 0 до 5 включительно. 11. Соединение по любому из пп. 1-7, у которого А обозначает алкенилен, В обозначает -О(CH2)m-, a m обозначает целое число от 1 до 5 включительно. 12. Соединение по п. 11, которое представляет собой: цис-{ 3-[3-(бензилфенилкарбамоилоксипропенил)фенокси] } уксусную кислоту; цис-[3-(3-дифенилкарбамоилоксипропенил)фенокси] уксусную кислоту; цис-[3-(4-дифенилкарбамоилоксибут-1-енил)фенокси] уксусную кислоту; цис-[3-(3-дифенилкарбамоилоксипропенил)-2-метилфенокси] уксусную кислоту; транс-[3-(3-дифенилкарбамоилоксипропенил)фенокси] уксусную кислоту или транс-[3-(3-дифенилкарбамоилоксипропенил)-2-метилфенокси] уксусную кислоту, или ее индивидуальный изомер, рацемическую или нерацемическую смесь изомеров, либо фармацевтически приемлемую соль, или сольват. 13. Соединение по любому из пп. 1-7, у которого А обозначает алкенилен, В обозначает -(СН2)n-, а n обозначает целое число от 0 до 5 включительно. 14. Соединение по п. 13, которое представляет собой цис-3-[3-(3-дифенилкарбамоилоксипропенил)фенил] пропионовую кислоту или ее индивидуальный изомер, рацемическую или нерацемическую смесь изомеров, либо фармацевтически приемлемую соль, или сольват. 15. Соединение по любому из пп. 1-11 или его индивидуальный изомер, рацемическая или нерацемическая смесь изомеров, либо фармацевтически приемлемая соль, или сольват, выбранные из соединений формул 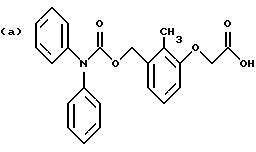 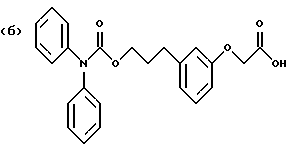 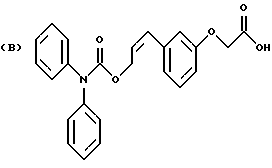 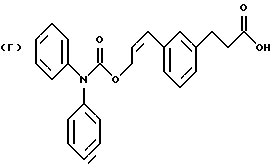 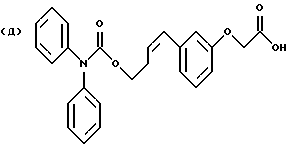 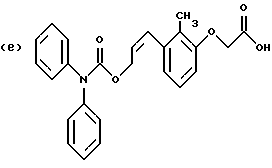 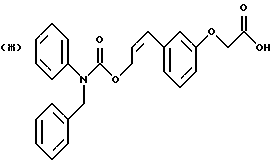 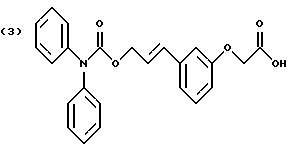 или 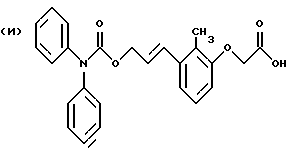 16. Способ получения соединения по любому из пп. 1-15, который включает взаимодействие соединения одной из следующих формул 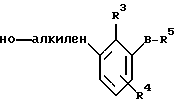 или 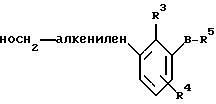 с соединением формулы 5 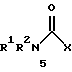 где R1, R2, R3, R4, R5, R6, В, m и n имеют значения, указанные в п. 1; Х обозначает галоген. 17. Фармацевтическая композиция, обладающая агонистической активностью в отношении IP-рецептора и включающая терапевтически эффективное количество по меньшей мере одного соединения по пп. 1-15 в смеси с по крайней мере одним приемлемым носителем. 18. Композиция по п. 17, в которой по меньшей мере одно соединение по пп. 1-15 пригодно для введения субъекту, находящемуся в болезненном состоянии, которое облегчают лечением с использованием модулятора IP-рецептора, в частности в болезненном состоянии, связанном с перемежающейся хромотой. 19. Соединения по любому из пп. 1-15, полученные способом по п. 16. 20. Способ лечения болезненного состояния, связанного с IP-рецептором, в частности в болезненного состояния, связанного с перемежающейся хромотой, предусматривающий введение в организм эффективного количества соединения по пп. 1-15. Приоритеты по пунктам: 23.12.1998 по пп. 1-20; 30.08.1999 по пп. 1-20 (уточнение признаков). РИСУНКИ
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 23.12.2006
Извещение опубликовано: 20.07.2010 БИ: 20/2010
|
||||||||||||||||||||||||||