Патент на изобретение №2174946
|
||||||||||||||||||||||||||
(54) СПОСОБ ПРОИЗВОДСТВА АЗОТНОЙ КИСЛОТЫ
(57) Реферат: Способ относится к получению оксидов азота и переработке их в азотную кислоту. Сущность способа: процесс каталитического окисления молекулярного азота осуществляют под давлением, одинаковым со стадией абсорбции полученных оксидов азота водой, а энергию для эндотермического процесса каталитического окисления молекулярного азота подводят с потоком газов непосредственно в зону реакции. Процесс каталитического окисления молекулярного азота проводится при температурах ниже 1000oC и при содержании окислителя в газовой фазе перед катализатором ниже 10 об.% паров HNO3 + NOx и давлении в системе до 25 атм. Катализаторами окисления молекулярного азота используются сплавы платины с металлами платиновой группы или катализаторы на основе оксидов железа, кобальта, хрома, алюминия с промотирующими добавками тугоплавких металлов. Для образования потока газа с окислителем может быть использован не только атмосферный воздух, но и газовая смесь, получающаяся при отдуве продукционной кислоты. Технический результат – сокращение энергетических и капитальных затрат, а также упрощение технологической схемы производства азотной кислоты. 7 з.п.ф-лы, 1 ил., 2 табл. Способ относится к химической технологии и касается методов получения оксидов азота и переработки их в азотную кислоту (АК). Широко известен способ производства АК путем водной абсорбции оксидов азота, полученных конверсией аммиака воздухом. Он состоит из стадий окисления аммиака кислородом воздуха на катализаторах при температурах 800-900oC; регенерации тепла нитрозных газов; выделения реакционной воды; абсорбции оксидов азота водой с получением АК и очистки выхлопных газов (ВГ) от оксидов азота (Атрощенко В.И., Каргин С.И. Технология азотной кислоты, М.: Химия, 1970 г.). Недостатком данного способа являются высокие сырьевые (аммиак, платина) и материальные затраты (удельные капитальные вложения в производствах аммиака и кислоты), а также значительные выбросы вредных веществ в атмосферу. В способе, описанном в патенте РФ N 2070865 от 27.12.1996 г., предлагается: “в полученные при окислении аммиака на первой ступени катализатора нитрозные газы (НГ) вводить чистые газы, содержащие кислородные соединения азота, и осуществлять окисление ими молекулярного азота НГ на второй ступени катализатора”. Недостатком этого способа является то, что в качестве сырья для получения НГ на 1-ой ступени катализатора используется дорогостоящий аммиак, за счет энергии окисления которого может быть окислено количество азота (N2), позволяющего получить только 15-35% продукционной кислоты. Таким образом, указанное решение только частично снижает сырьевые и капитальные затраты против первого случая. Наиболее близким по технологической сущности и достигаемому эффекту является “Способ получения азотной кислоты” по патенту N 2151736 от 27 июня 2000 г. (приоритет от 15.10.1998 г.). В данном случае оксиды азота получаются только за счет каталитического окисления азота его кислородными соединениями при 850-450oC под давлением, близким к атмосферному, абсорбцию оксидов азота проводят под повышенным давлением, а энергию, необходимую для процесса окисления азота, подводят с потоком ВГ, содержащих оксиды азота и подогретых сторонним энергоносителем. Целью предлагаемого изобретения является повышение эффективности процесса за счет сокращения энергетических и капитальных затрат, а также упрощения технологической схемы производства АК. Новизна способа состоит в том, что процесс каталитического окисления молекулярного азота осуществляется под давлением одинаковым (единым) со стадией абсорбции водой, полученных оксидов азота, а энергия для процесса окисления подводится с потоком газа непосредственно в зону реакции от посторонних источников. Процесс каталитического окисления азота производится при температурах ниже 1000oC и при содержании окислителя в газовой фазе перед катализатором ниже 10 об.% паров HNO3 + NO2, и давлении в системе до 25 атм. В виде катализаторов окисления азота используют сплавы платины с металлами платиновой группы или катализаторы на основе оксидов железа, кобальта, хрома, алюминия с промотирующими добавками тугоплавких металлов. Катализаторы могут располагаться в реакторе в несколько слоев, причем окислитель подводится к каждому слою в заданных количествах, а энергетический поток – по необходимости. Для образования потока газа с окислителем может быть использован не только воздух, но и газовая смесь, получающаяся при отдуве продукционной кислоты. Существенной новизной предлагаемого патента по сравнению с прототипом является то, что процессы окисления азота и абсорбции оксидов азота проводятся под единым давлением, отсутствует циркуляция выхлопных газов, а носителем энергии является воздух (газ с высоким содержанием кислорода). Принципиальная технологическая схема представлена на чертеже, где 1 – турбокомпрессор воздуха, 2 – насытитель парокислотовоздушной смеси (ПКВС), 3 – обогревающая труба-теплообменник, 4 – реактор для окисления азота, 5, 14, 15 – котлы-утилизаторы, 6 – теплообменник, подогреватель теплоносителя, 7 – подогреватель выхлопных газов (ВГ), 8, 8* – холодильник-конденсатор нитрозных газов (НГ), 9 – абсорбционная колонна, 10 – отбелочная колонна 11 – камера сгорания, 12 – реактор каталитической очистки ВГ, 13 – газовая турбина, 16 – подогреватель воздуха, 17 – емкость для теплоносителя, 18 – емкость для кислоты-окислителя. Предлагаемый способ производства АК осуществляется следующим образом: воздух в заданном количестве сжимается до необходимого давления компрессором 1 и распределяется: – в насытитель 2 и затем в виде ПКВС – в реактор 4 для обеспечения реакции окисления кислородом; – для окисления оксида азота II в НГ при выходе из реактора 4; – для улучшения процесса кислотообразования в колонну 9 через отбелочную колонну 10; – для подогрева воздуха-носителя энергии в подогреватель 16. Насытитель 2 полочного типа орошается кислотой из емкости 18. Кислота в емкость передается из абсорбционной колонны 9. На полках насытителя располагаются змеевики, через которые подводится энергия для испарения кислоты – получения потока ПКВС. Температура кислоты определяется давлением и заданной концентрацией паров АК в ПКВС и будет находится в пределах 130-190oC. При выходе из насытителя 2 ПКВС в обогревающей трубе-теплообменнике 3 подогревается до 150-250oC во избежание конденсации паров кислоты и воды и подается в реактор 4 перед каждым слоем катализатора. Здесь ПКВС смешивается с воздухом (носителем энергии), который подается из подогревателя 16. Образовавшаяся смесь при температуре ниже 800oC поступает на катализатор. Количество вводимых перед полкой газов зависит от ряда факторов – в основном от заданного содержания окислителя в смеси и температуры перед слоем катализатора. Катализатор располагается в несколько слоев, что также определяется поставленными параметрами, активностью и селективностью катализатора. Однако нет необходимости температуру НГ на выходе из реактора иметь ниже 450oC, т. к. реакция окисления азота 2HNO3 + N2 = 3NO + NO2 + 2H2O – 355 кДж (1) практически прекращается, а преобладает при этой температуре реакция разложения паров HNO3: 4HNO3 = 4NO2 + 2H2O + O2 – 191,0 кДж. (2) При температурах ниже 600oC (зависит от избранного давления) протекает реакция (3) 2NO + O2 = 2NO2 + 113,9 кДж, (3) которая благоприятна с точки зрения энергетики для основного процесса. При выходе из реактора 4 в поток НГ вводится воздух в количествах, необходимых для окисления кислородом оксида азота II и переработки NOx в кислоту. Затем поток НГ разделяется. Меньшая часть направляется на обогрев ПКВС в обогревающую трубу 2, выходя из которой смешивается с общим потоком НГ перед холодильником-конденсатором 8. Основной поток НГ проходит котел-утилилизатор 5, подогреватели теплоносителя 6 и выхлопных газов 7 и также поступает в холодильники-конденсаторы 8. В конденсаторах 8, 8* НГ охлаждается до температуры около 40oC и вместе с образовавшимся раствором азотной кислоты поступает в абсорбционную колонну 9. В абсорбционной колонне 9 протекает обычный процесс кислотообразования. Выходящие из колонны (9) ВГ подогреваются в теплообменнике 7, камере сгорания 11, подвергаются высокотемпературной или селективной каталитической очистке от оксидов азота 12, проходят турбину 13, паровой котел 14 и выбрасываются в атмосферу. Образующаяся в колонне 9 азотная кислота через отбелочную колонну 10 выдается, как продукционная и окислитель. Воздух с оксидами азота, парами кислоты и воды передается в нижнюю часть абсорбционной колонны 9. При желании на агрегате может выдаваться кислота 2-х концентраций. При высоком содержании оксидов азота в НГ кислота концентрацией выше азеотропной легко может быть получена при конденсации реакционной воды в конденсаторах (8, 8*), а продукционная, до 62,0% HNO3 – в колонне 9. В то же время при высоких PNO2 кислота до 70% может быть получена и в колонне 9. В этом случае ее следует отводить с 3-5 тарелок колонны. В любом случае кислота более высокой концентрации должна использоваться, как окислитель. Концентрация продукционной кислоты всегда может быть отрегулирована в зависимости от потребности. Для обеспечения энергией процесса окисления азота (реакция 1) устанавливается специальный подогреватель носителя энергии 16. В нем за счет сторонней энергии (например, сжигания природного газа) воздух подогревается до температуры 800oC и подается в реактор 4 перед слоями катализатора. Энергия дымовых газов после подогревателя 16 используется для получения пара 15. Режим и параметры работы подогревателя определяются заданным режимом работы зоны катализа реактора. Предлагаемая схема позволяет достаточно широко разнообразить параметры ее работы. Это прежде всего относится к стадии переработки НГ. Здесь могут быть использованы различные варианты утилизации энергии НГ, вывода реакционной воды, изменения концентрации получаемой азотной кислоты и т.д. Возможны разнообразные варианты переработки нитрозных газов, однако они не затронут основные технологические решения, определяющие процесс окисления молекулярного азота. Исследования и отработка процесса производилась на опытной установке, изготовленной из нержавеющей стали и титана. Установка имела: компрессор воздуха давлением до 30 атм (имел циркуляцию газа); насытитель для получения парокислотовоздушной смеси (ПКВС); реактор, имеющий две полки для катализатора и стадию переработки оксидов азота – теплообменники, конденсаторы, абсорбционную колонну и каталитическую очистку ВГ от NOx. Заданные температуры в реакторе и насытителе поддерживались внешним обогревом. В зависимости от этапа исследований, схема подачи газов в реактор могла изменяться. Количество подаваемых газов, температура и давление непрерывно измерялись приборами КИПиА; содержание оксидов азота определялось химическими анализами, а в отдельных случаях спектрофотометрическим. Содержание кислорода в ВГ при необходимости определялось химическими анализами. В исследованиях расход по воздуху составлял 350-1500 л/час н.у.; давление от 1 до 20 атм, температура перед катализатором изменялась от 600 до 800oC. На выходе из катализаторного слоя, за счет протекания эндотермической реакции окисления азота она снижалась до 450-550oC (в зависимости от заданного режима). На основании расхода воздуха и результатов химических анализов ПКВС и выходящих НГ на компьютере по специальной программе рассчитывался материальный баланс опыта. По изменению объемов компонентов, поступающих на катализатор и отходящих газов, рассчитывали степень прироста соединений азота,  0 (степень полезного использования паров HNO3, реакция 1) и степень термического разложения паров HNO3 (по реакции 2).
На основании данных баланса могли быть рассчитаны и другие показатели процесса, например степень использования азота, кислорода и др.
Пример 1. (Таблица 1, п.п.4). Проверена герметичность установки. Электрообогревом поднята температура в реакторе до 750oC в насытителе, заполненном 70% HNO3, до 80oC и линии подачи ПКВС до 240oC, запускается компрессор, воздух сжимается до 7,4 атм и постепенно поднимается давление в аппаратах установки. Корректируется температура в реакторе и поднимается в насытителе до 140oC. После достижения заданной температуры в реакторе и насытителе из основного потока нагнетания компрессора отбирается очищенный от масла воздух и установка пускается на проток. Из компрессора воздух направляется в насытитель, где насыщается парами азотной кислоты и воды. Получаемая парокислотовоздушная смесь (ПКВС) по обогреваемой линии поступает в реактор, проходит слой катализатора и далее все аппараты переработки оксидов азота.
На установленном расходе воздуха (1000 л/час) вновь корректируется температура раствора азотной кислоты – 140oC и газовой смеси перед катализаторным слоем – 750oC.
Установленный режим выдерживался 2 часа, после чего одновременно (до и после слоя катализатора) отбирались пробы газов на содержание соединений азота. Поглощающей жидкостью являлась перекись водорода. Отсутствие закиси азота в газах подтверждено спектральным анализом.
Отобрано по 4 параллельных пробы газа.
Содержание оксидов азота составило: перед катализаторным слоем (ПКВС) – 16,03 + 0,23%; на выходе из слоя – 25,50 + 0,2%; абсолютный прирост соединений азота – 9,5 об.%.
Имея расход воздуха, концентрацию и температуру кислоты в насытителе, равновесное давление паров HNO3 и H2O для этих условий, а также зная содержание паров АК в ПКВС по специальной программе на компьютере, рассчитывали материальный баланс реактора.
По полученным объемам компонентов газов до и после слоя катализатора рассчитывали степень превращения паров HNO3 – по реакции (1) 0 (степень полезного использования паров HNO3, реакция 1) и степень термического разложения паров HNO3 (по реакции 2).
На основании данных баланса могли быть рассчитаны и другие показатели процесса, например степень использования азота, кислорода и др.
Пример 1. (Таблица 1, п.п.4). Проверена герметичность установки. Электрообогревом поднята температура в реакторе до 750oC в насытителе, заполненном 70% HNO3, до 80oC и линии подачи ПКВС до 240oC, запускается компрессор, воздух сжимается до 7,4 атм и постепенно поднимается давление в аппаратах установки. Корректируется температура в реакторе и поднимается в насытителе до 140oC. После достижения заданной температуры в реакторе и насытителе из основного потока нагнетания компрессора отбирается очищенный от масла воздух и установка пускается на проток. Из компрессора воздух направляется в насытитель, где насыщается парами азотной кислоты и воды. Получаемая парокислотовоздушная смесь (ПКВС) по обогреваемой линии поступает в реактор, проходит слой катализатора и далее все аппараты переработки оксидов азота.
На установленном расходе воздуха (1000 л/час) вновь корректируется температура раствора азотной кислоты – 140oC и газовой смеси перед катализаторным слоем – 750oC.
Установленный режим выдерживался 2 часа, после чего одновременно (до и после слоя катализатора) отбирались пробы газов на содержание соединений азота. Поглощающей жидкостью являлась перекись водорода. Отсутствие закиси азота в газах подтверждено спектральным анализом.
Отобрано по 4 параллельных пробы газа.
Содержание оксидов азота составило: перед катализаторным слоем (ПКВС) – 16,03 + 0,23%; на выходе из слоя – 25,50 + 0,2%; абсолютный прирост соединений азота – 9,5 об.%.
Имея расход воздуха, концентрацию и температуру кислоты в насытителе, равновесное давление паров HNO3 и H2O для этих условий, а также зная содержание паров АК в ПКВС по специальной программе на компьютере, рассчитывали материальный баланс реактора.
По полученным объемам компонентов газов до и после слоя катализатора рассчитывали степень превращения паров HNO3 – по реакции (1)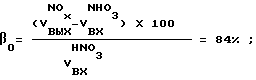 по реакции (2) – 16%, 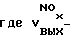 объем NOx на выходе; объем NOx на выходе;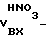 объем HNO3 на входе.
Степень окисленности NO ( объем HNO3 на входе.
Степень окисленности NO ( ) – 31,5% ) – 31,5%Скорости газа в рабочих условиях: Vр = 0,115 м/с, Wр = 16080 час, где Vр – линейная скорость в рабочих условиях; Wр – объемная скорость в рабочих условиях. Прирост соединений азота в опыте составил: 1082 л/час. на описанной в примере 1 установке (таблица 1, пп.4) проведены достаточно широкие исследования процесса в параметрах: давление – от 1 до 20 атм, температура в слое катализатора 850 – 500oC, объемная скорость от 7 до 12,5 тыс.ч под избыточным давлением и до 80 тыс.ч под атмосферным давлением. Изменялось также количество загружаемого катализатора. Часть опытов приведена в таблице 1. Степень полезного использования паров HNO3 по реакции (1) колеблется в пределах от 60 до 85% при содержании в газе до 17% кислорода и до 25% паров воды. Отличие данной схемы от прототипа состоит в проведении всех стадий процесса получения АК под единым давлением в отсутствии циркуляции ВГ, некоторых других технологических параметрах и решениях. Выполнена технико-экономическая проработка нескольких вариантов производства неконцентрированной азотной кислоты из оксидов азота, полученных окислением молекулярного азота. Агрегаты производства АК привязывались к одному месту строительства и рассчитывались на единую мощность аналога – реконструированного агрегата УКЛ-7 – 120 тыс.т HNO3 в год. Как следует из табл.2 все варианты производств, основанных на использовании НГ, полученных окислением азота, имеют значительно лучшие показатели, чем производства АК из аммиака. В частности, себестоимость АК всех вариантов (сх. 1, 2, 3) на 24-35% ниже чем современные схемы производства АК из аммиака (сх. 4). Агрегат производства АК по заявленному способу (сх. 2, прямоточный) имеет против прототипа (сх. 1, 3) ниже на 11,5-13,0% энергозатраты и на 5-8% себестоимость кислоты. Это объясняется в основном отсутствием циркуляции выхлопных газов в предлагаемой схеме. Формула изобретения
РИСУНКИ
MM4A Досрочное прекращение действия патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 28.12.2004
Извещение опубликовано: 10.03.2006 БИ: 07/2006
NF4A Восстановление действия патента Российской Федерации на изобретение
Извещение опубликовано: 10.07.2006 БИ: 19/2006
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 28.12.2006
Извещение опубликовано: 27.04.2008 БИ: 12/2008
NF4A Восстановление действия патента СССР или патента Российской Федерации на изобретение
Дата, с которой действие патента восстановлено: 27.11.2008
Извещение опубликовано: 27.11.2008 БИ: 33/2008
|
||||||||||||||||||||||||||