Патент на изобретение №2172316
|
||||||||||||||||||||||||||
(54) 42-ОКСИМЫ И ГИДРОКСИЛАМИНЫ РАПАМИЦИНА, СПОСОБ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЙ ПРОДУКТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ
(57) Реферат: Описываются новые соединения формулы I, где Х-Y является С=NОR1 или СНNНОR2, в которых R1 обозначает водород, алкил с 1 – 6 атомами углерода, алкенил с 2 – 7 атомами углерода, алкинил с 2 – 7 атомами углерода, группа формулы -(СН2)mAr, в которой Ar представляет фенил или пиридинил и m = 1 – 6; R2 представляет водород или радикал формулы -(СН2)mAr, в которой Ar представляет пиридинил, или их фармацевтически приемлемые соли. Описывается также способ их получения, промежуточный продукт, фармацевтическая композиция и способ лечения заболевания. Новые соединения полезны в качестве иммунодепрессивных, противовоспалительных, противогрибковых, антипролиферативных и противоопухолевых средств. 5 с. и 13 з.п.ф-лы. 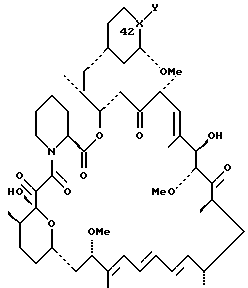
Данное изобретение относится с 42-оксимам и гидроксиламинам рапамицина и к способу их использования для индуцирования иммуносупрессии и в лечении отторжения трансплантата, реакции “трансплантат против хозяина”, аутоиммунных заболеваний, воспалительных заболеваний, Т-клеточного лейкоза/лимфомы у взрослых, твердых опухолей, микозов или грибковых инфекций и гиперпролиферативных сосудистых нарушений. Рапамицин является макроциклическим триеновым антибиотиком, продуцируемым Streptomyces hygroscopicus, у которого было обнаружено противогрибковое действие, в частности, против Candida albicans, как in vitro, так и in vivo [C. Vezina et al., J. Antibiot. 28, 721 (1975); S.N. Sehgal et al., J. Antibiot. 28, 727 (1975); H.A. Baker et al., J. Antibiot. 31, 539 (1978); Патент США 3929992 и Патент США 3993749]. Было показано, что рапамицин, используемый один (однокомпонентно) (Патент США 4885171) или в сочетании (комбинации) с пицибанилом (Патент США 4401653), обладает противоопухолевой активностью. Р. Мартел и др. (R. Martel et al.) [Can. J. Physiol. Pharmacol. 55, 48 (1977)] обнаружили, что рапамицин эффективен при использовании на экспериментальной модели аллергического энцефаломиелита, модели множественного склероза, модели адьювантного артрита, модели ревматоидного артрита и эффективно ингибирует или подавляет образование иммуноглуболин Е-подобных антител. Иммуносупрессивное (иммунодепрессивное) действие рапамицина было описано в FASEB 3, 3411 (1989). Было также показано, что циклоспорин А и ФК-506, другие макроциклические молекулы эффективны в качестве иммуносупрессивных средств и, таким образом, полезны в предотвращении отторжения трансплантата [FASEB 3, 3411 (1989); FASEB 3, 5256 (1989); R.Y. Calne et al., Lancet 1183 (1978); Патент США 5100899]. Было также показано, что рапамицин эффективен в предотвращении или лечении системной красной волчанки (Патент США 5078999), воспаления легких (Патент США 5080899), инсулинзависимого сахарного диабета [Fifth Int. Conf. Inflamm. Res. Assoc. 121 (Abstract), (1930)], пролиферации клеток гладких мыщц и утолщения интимы после сосудистых травм [Morris, R.J. Heart Lung Transplant 11 (pt. 2): 197 (1992)], Т-клеточного лейкоза/лимфомы [европейская заявка на патент 525960 A1] и воспаления глаз (европейская заявка на патент 532862 A1). Было показано, что моно- диацилированные производные рапамицина (этерифицированные в положениях 28 и 43) эффективны в качестве противогрибковых препаратов (Патент США 4316885) и могут использоваться для получения водорастворимых аминоацилированных пролекарств рапамицина (Патент США 4650803). Недавно было изменено соглашение о нумерации для рапамицина, поэтому, в соответствии с номенклатурой Chemical abstracts, образование вышеописанных сложных эфиров следует считать в положениях 31 и 42. В Патенте США 5023263 описано получение и использование 42-оксорапамицина, а в Патенте США 5023264 описано получение и применение 27-оксимов рапамицина. Описание Изобретение относится к производным рапамицина, которые могут использоваться в качестве иммунодепрессивных, противовоспалительных, противогрибковых, антипролиферативных и противовопухолевых средств, имеющим структуру 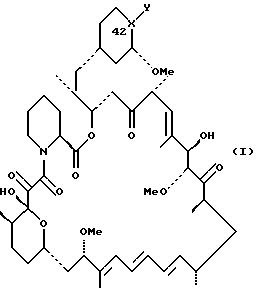 где X-Y представляет C=NOR1 или CHNHOR2; каждый из R1 и R2 независимо обозначает водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, аминоалкил с 1-6 атомами углерода, алкиламиноалкил с 1-6 атомами углерода в каждой алкильной группе, диалкиламиноалкил с 1-6 атомами углерода в каждой алкильной группе, циклоалкил с 3-8 атомами углерода, алкилокси с 1-6 атомами углерода, алкоксиалкил с 1-6 атомами углерода в каждой алкильной группе, циклоалкиламиноалкил с 4-14 атомами углерода, цианоалкил с 2-7 атомами углерода, фторалкил с 1-6 атомами углерода, трифторметилалкил с 2-7 атомами углерода, трифторметил, 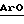 -, -(CH2)mAr или -COR3; -, -(CH2)mAr или -COR3;R3 обозначает алкил с 1-6 атомами углерода, -NH2, -NHR4, -NR4R5, -OR4 или 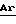 , ,каждый из R4 и R5 независимо обозначает алкил с 1-6 атомами углерода, Ar, или, в случае наличия обоих, они могут быть взяты вместе с образованием 4-7-членного кольца; Ar обозначает арильный или гетероарильный радикал, который может быть необязательно моно-, ди- или тризамещенным группой, выбранной из алкила с 1-6 атомами углерода, алкенила с 2-7 атомами углерода, алкинила с 2-7 атомами углерода, арилалкила с 7-10 атомами углерода, алкокси с 1-6 атомами углерода, циано, галогена, гидрокси, нитро, карбалкокси с 2-7 атомами углерода, трифторметила, трифторметокси, амино, диалкиламино с 1-6 атомами углерода в каждой алкильной группе, диалкиламиноалкила с 3-12 атомами углерода, гидроксиалкила с 1-6 атомами углерода, алкоксиалкила с 2-12 атомами углерода, алкилтио с 1-6 атомами углерода, -SO3H и -CO2H; и m = 0-6; или их фармацевтически приемлемым солям. Фармацевтически приемлемыми солями являются соли, образованные из таких неорганических катионов, как натрий, калий и т.п., органических оснований, таких как моно-, ди- и триалкиламины с 1-6 атомами углерода в каждой алкильной группе и моно-, ди- и тригидроксиалкиламины с 1-6 атомами углерода в каждой алкильной группе и т.п., и органических и неорганических кислот, таких как: уксусная, молочная, лимонная, винная, янтарная, малеиновая, малоновая, глюконовая, соляная, бромистоводородная, фосфорная, азотная, серная кислоты, метансульфокислота и другие, также известные приемлемые кислоты. Термины: алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода и алкинил с 2-7 атомами углерода включают как неразветвленную цепь, так и разветвленные углеродные цепи. Примерами алкила как группы или части группы, например, арилалкила, алкокси или карбалкокси, являются неразветвленные или разветвленные цепи с 1-6 атомами углерода, предпочтительно с 1-4 атомами углерода, например метил, этил, пропил, изопропил и н-бутил. Благодаря наличию двойной связи, оксимы по настоящему изобретению обладают цис-транс изометрией, поэтому данное изобретение охватывает не только смеси геометрических изомеров, но и индивидуальные E и Z изомеры, которые могут быть разделены методами, известными специалистам в данной области. Аналогично, гидроксиламины по настоящему изобретению состоят из смеси эпимеров при C-42, и поэтому данное изобретение охватывает не только смеси изомеров, но и индивидуальные изомеры, которые могут быть разделены методами, известными специалистам в данной области. Термин “арил” в качестве группы или части группы, например, арилалкила, включает любую моно- или бикарбоциклическую ароматическую группу с 6-10 атомами углерода. Термин “гетероарил” включает любой моно- или бициклический гетероциклический ароматический радикал, состоящий из 5-10 атомов в гетероцикле, из которых до трех атомов в гетероцикле, одинаковых или различных, являются гетероатомами, выбранными из атомов кислорода, азота и серы. Арильными и гетероарильными радикалами Ar предпочтительно являются фенил, пиридил, фурил, пирролил, тиофенил, имидазолил, оксазолил или тиазолил, которые необязательно могут быть моно-, ди- или тризамещенными группой, выбранной из алкила с 1-6 атомами углерода, алкенила с 2-7 атомами углерода, алкинила с 2-7 атомами углерода, арилалкила с 7-10 атомами углерода, алкокси с 1-6 атомами углерода, циано, галогена, гидрокси, нитро, карбалкокси с 2-7 атомами углерода, трифторметила, трифторметокси, амино, диалкиламино с 1-6 атомами углерода в каждой алкильной группе, диалкиламиноалкила с 3-12 атомами углерода, гидроксиалкила с 1-6 атомами углерода, алкоксиалкила с 2-12 атомами углерода, алкилтио с 1-6 атомами углерода, -SO3H и CO2Н. Если R3 обозначает -NR4R5, то группы R4 и R5 могут быть одинаковыми или различными (как определено выше) или могут объединяться с образованием насыщенного гетероцикла, состоящего из 4-7 атомов, в кольце которого 1 атом является азотом, а 0-2 других атома в гетероцикле могут быть атомами азота, кислорода или серы. В том случае, когда R4 и R5 взяты вместе, предпочтительно, чтобы R4R5 представлял собой углеродную цепь, образующую азетидиновое, пирролидиновое, пиперидиновое или гомопиперидиновое кольцо. Предпочитаемыми соединениями по настоящему изобретению являются соединения, в которых X-Y обозначает C=NOR1; соединения, в которых X-Y обозначает C= NOR1 и R1 обозначает водород, алкил с 1-6 атомами углерода, алкенил с 2-7 атомами углерода, алкинил с 2-7 атомами углерода, алкилокси с 1-6 атомами углерода, -(CH2)mAr или -COR3; соединения, в которых X-Y обозначает CHNHOR2; соединения, в которых X-Y обозначает CHNHOR2 и R2 обозначает водород или -(CH2)mAr. В данном изобретении также описаны способы получения соединений рапамицина по настоящему изобретению. В частности, в данном изобретении описываются способы получения 42-оксимов и гидроксиламиновых производных рапамицина, которые включают один из следующих способов: а) взаимодействие 42-оксорапамицина (необязательно защищенного в гидроксильной функциональной группе в положении 31 силильной группой формулы R6, определенной ниже) с гидроксиламином формулы: H2NOR1, где R1 имеет данные выше значения, кроме содержащих карбонил частей с образованием соответствующего оксима формулы I, где X-Y обозначает C=NOR1, и удаление указанной защитной группы R6, или б) восстановление оксима формулы I (необязательно защищенного в гидроксильной функциональной группе в положении 31 силильной группой формулы R6, определенной ниже), где X-Y обозначает C=NOR1, где R1 имеет данные выше значения, за исключением содержащих карбонил радикалов, с образованием гидроксиламина формулы I, где X-Y обозначает CHNHOR2, или в) взаимодействие соединения формулы I (необязательно защищенного в гидроксильной функциональной группе в положении 31 силильной группой формулы R6, определенной ниже), в которой X-Y обозначает C=NOR1, где R1 обозначает водород, с цианатом, например, цианатом щелочного металла, таким как цианат натрия, с образованием соединения формулы I, где X-Y обозначает C=NOR1, где R1 обозначает -CONH2, или г) взаимодействие соединения формулы I, где X-Y обозначает C=NOR1 или CHNHOR2, где R1 и R2 являются водородом (когда X-Y обозначает C=NOR1, необязательно защищенный в гидроксильной функциональной группе в положении 31 силильной группой формулы R6, определенной ниже), с изоцианатом или галогенациламином формулы R4NCO, R4NCOгал или R4R5NCOгал, где hal обозначает галоген, такой как хлор с образованием соответствующего соединения формулы I, в которой R1 или R2 обозначают COR3 и R3 обозначает NHR4 или NR4R5, или д) взаимодействие соединения формулы I, где X-Y обозначает C=NOH или CHNHOH (когда X-Y обозначает C=NOH, необязательно защищенный по 31-й гидроксильной функциональной группе силильной группой формулы R6, определенной ниже), с алкил- или арилхлорформиатом с получением карбоната формулы I, где R1 или R2 обозначает COR3, где R3 обозначает алкил или арил. Соединения по настоящему изобретению могут быть получены из 42-оксорапамицина, который может быть получен с умеренным выходом путем избирательного (селективного) окисления рапамицина в 42-положении с помощью окисления, опосредованного рутением, как утверждается в Патенте США 5023263, указанном здесь для сведения. Альтернативно, примерно 50%-ный выход 42-оксорапамицина может быть получен с использованием смеси перрутенат тетрапропиламмония/N-оксида N- метилморфолина, как указано Холтом в Публикации PCT США 93/0668. Затем 42-оксорапамицин можно обработать соответствующим образом замещенным гидроксиламином с получением смеси 42-(Е) и (Z) оксимов (X-Y является C=NOR1), которая может быть разделена по стандартной методике. Далее 42-оксимы могут вступать в реакцию с соответствующим восстановителем, например, со смесью цианоборводород натрия /ТГФ/ диоксан/pH 3,5 с образованием гидроксиаминов по настоящему изобретению (X-Y является CHNHOR2). Соединения по настоящему изобретению, в которых X-Y обозначает C=NOR1 и R2 обозначает содержащую карбонил часть молекулы, могут быть получены из 42-оксима рапамицина (X-Y является C=NOR1 и R1 является водородом). Например, 42-оксим рапамицина может вступать в реакцию с цианатом натрия с образованием соединения, в котором R1 обозначает COR3 и R3 обозначает NH2. Аналогично, 42-оксим рапамицина можно обработать подходящим образом замещенным изоцианатом или галогенациламином (например, R4R5NC(O)Cl) для получения соединений, в которых R1 обозначает -COR3 и R3 обозначает -NR4R5. Аналогичным способом можно получить соединения, в которых 1 является -COR3 и R3 является -NHR4. Кроме того, обработка 42-оксима рапамицина соответствующим алкил- или арилхлорформиатом в пиридине и таком растворителе, как метиленхлорид, позволяет получить карбонаты оксима, в которых R1 является -COR3 и R3 является алкилом или Ar. Аналогичная функционализация может быть достигнута, когда X-Y является CHNHOR2, с использованием исходного соединения, в котором R1 является водородом (полученного путем восстановления 42-оксима рапамицина цианоборводородом). В качестве альтернативы окислительному методу получения 42-оксорапамицина с использованием рутения, данное изобретение описывает также синтетический путь получения оксимов и гидроксиламинов по настоящему изобретению посредством окисления периодинаном Десс-Мартина 31-O-защищенного рапамицина. Защита 31-положения рапамицина была описана в Патенте США 5120842, включенном здесь для сведения. Например, обработка рапамицина подходящим защитным реагентом, таким как смесь триэтилсилил трифлат /2,6-лутидин/ метиленхлорид, а затем уксусной кислотой /ТГФ/ водой обеспечивает количественный выход 31-O-триэтилсилилрапамицина. Окисление периодинаном Десс-Мартина обеспечивает приблизительно 65%-ный выход 31-O-триэтилсилил-42-оксорапамицина, который может быть подвергнут дальнейшей обработке подходящим гидроксиламином (с последующим получением производных), как описано выше. Для оксимов по настоящему изобретению триэтилсилильная защитная группа может быть удалена в условиях слабой кислотности (уксусная кислота /ТГФ/ вода), как описано в Патенте США 5120842. При использовании данного способа гидроксиламины по настоящему изобретению могут быть также получены из соответствующих оксимов восстановлением цианоборводородом в кислой среде с одновременным удалением силильной защитной группы. На основании вышеописанной методики Десс-Мартина с применением периодинана следующие соединения являются промежуточными продуктами, полезными для приготовления оксимов по настоящему изобретению. 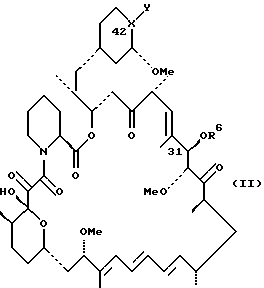 где R6 обозначает -SiR7R8R9 и каждый из R7, R8 и R9 независимо обозначает алкил с 1-8 атомами углерода, алкенил с 1-8 атомами углерода, фенилалкил с 7-10 атомами углерода, трифенилметил или фенил. Соединение примера 1 является предпочтительным промежуточным продуктом из вышеописанных промежуточных продуктов. Соответственно, данное изобретение представляет способ получения промежуточного продукта формулы II, описанной выше, который заключается в окислении 31-O-защищенного рапамицина (в котором защитной группой является R6, определенный выше) в условиях, подходящих для проведения окисления Десс-Мартина периодинаном и, при желании, удалении вышеуказанной защитной группы. Реагенты, используемые для получения соединений по настоящему изобретению, могут быть или приобретены или получены с помощью стандартных способов, описанных в литературе. Иммуносупрессивную активность типичных соединений по настоящему изобретению оценивали при помощи стандартной методики фармакологических испытаний in vitro по измерению степени подавления пролиферации лимфоцитов (фактора активации лимфоцитов/LAF/) и двух стандартных процедур фармакологического испытания in vivo. С помощью процедуры испытаний с использованием кусочка кожного трансплантата измеряют иммуносупрессивную активность тестируемого соединения, а также способность тестируемого соединения тормозить (ингибировать) или лечить отторжение трансплантата. С помощью процедуры стандартного фармакологического испытания с использованием адьювантного артрита измеряют способность тестируемого соединения ингибировать иммуноопосредованное воспаление. Испытание с использованием адьювантного артрита представляет собой стандартную методику фармакологического испытания ревматоидного артрита. Процедуры данных стандартных способов фармакологического испытания описаны ниже. Процедуру пролиферации тимоцитов, вызванной комитогенами (LAF), использовали для оценки иммуносупрессивного действия типичных соединений in vitro. Вкратце клетки тимуса нормальных мышей линии BALB/c культивировали в течение 72 ч с ФГА и интерлейкином-1 и подвергали импульсному воздействию меченного тритием тимидина в течение последних 6 ч. Клетки культивируют как с различными концентрациями рапамицина, циклоспорина или тестируемого соединения, так и без них. Осуществляют сбор клеток и определяют включенную радиоактивность. Подавление лимфопролиферации оценивают как процентное изменение количества импульсов в минуту, по сравнению с контрольными группами, которые не получали лекарственное средство. С целью сравнения параллельно с оценкой действия каждого соединения оценивали также действие рапамицина. Показатель IC50 был получен как для каждого тестируемого соединения, так и для рапамицина. При оценке рапамицина как вещества для сравнения с типичными соединениями по настоящему изобретению его показатель IC50 находился в диапазоне от 0,5 до 3,3 наномоль. Полученные результаты представлены показателем IC50 и процентным выражением ингибирования пролиферации Т-клеток при 0,1 мкмоль. Результаты, полученные для типичных соединений по настоящему изобретению, выражали также в виде отношения, в сравнении с рапамицином. Положительное отношение свидетельствует об иммуносупрессивной активности. Если величина отношения больше единицы, то это означает, что при использовании тестируемого соединения степень ингибирования пролиферации тимоцитов была выше, чем при использовании рапамицина. Расчет отношения показан ниже. 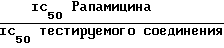 Типичные соединения по настоящему изобретению оценивали также по методике испытания in vivo, предназначенной для определения времени выживания (жизни) кусочков кожных трансплантатов, пересаженных от доноров-самцов линии BALA/c самцам, получавшим C3H(H-2K). Данный способ был заимствован из Billingham R. T. and Medawar P.В., J. Exp. Biol. 28: 385-402, (1951). Вкратце кожный транспантат в виде кусочков, полученный от донора, пересаживали на спину реципиента как аллотрансплантат, а изотрансплантат использовали в качестве контроля в той же области. Реципиентам вводили внутрибрюшинно или перорально различные концентрации тестируемых соединений. Рапамицин использовали в качестве контроля. Реципиенты, не получившие тестируемых соединений, служили в качестве контроля на отторжение. Осуществляли ежедневный мониторинг трансплантата, и данные наблюдений регистрировали до тех пор, пока трансплантат не высыхал и не превращался в почерневшую корку. Данный момент считался днем отторжения. Среднее время жизнеспособности трансплантата (количество дней  стандартное отклонение) в группе, получавшей тест-соединение, сравнивали с аналогичным показателем, полученным в контрольной группе. Результаты выражены как среднее время жизнеспособности трансплантата в днях. Кусочковые кожные трансплантаты в не получавшей тест-соединение (контрольной) группе обычно отторгались в течение 6-7 дней. Соединения исследовались при дозе 4 мг/кг, вводимой внутрибрюшинно. При внутрибрюшинном введении рапамицина в дозе 4 мг/кг время жизнеспособности трансплантата составило 11,6710,63 дня.
В стандартном фармакологическом испытании с использованием адъювантного артрита оценивают способность тестируемых соединений предотвращать иммуноопосредованное воспаление и подавлять или излечивать ревматоидный артрит. Ниже кратко описана используемая методика испытания. Соединение предварительно вводили группе крыс (инбредных крыс-самцов линии Вистар Льюис) для испытания (за 1 ч до введения антигена), а затем в правую заднюю лапку этих крыс впрыскивали полный адъювант Фрейнда (FСА) для индуцирования артрита. Затем определенные дозы вводили крысам перорально по схеме введения препарата в понедельник, среду и пятницу с 0 по 14 день при общем количестве 7 доз. Обе задние лапки измеряли на 16-й, 23-й и 30-й день. Определяли различие в объеме лапок (мл) от 16 до 0 дня и рассчитывали процентное изменение по сравнению с контролем. Воспаление левой задней лапки (не подвергавшейся инъекции) обусловлено воспалением, опосредованным Т-клетками, и регистрируется в виде процентного изменения по сравнению с контролем. С другой стороны, воспаление правой задней лапки обусловлено неспецифическим воспалением. Соединения испытывали при дозе 2 мг/кг. Результаты представлены в виде процентного изменения в лапке, не подвергавшейся инъекции, на 16-й день по сравнению с контролем, чем больше отрицательное процентное изменение, тем эффективнее соединение. Введение рапамицина обеспечило 70%-ное изменение по сравнению с контролем. Это означает, что у крыс, получивших рапамицин, наблюдалось на 70% меньшее иммуноопосредованное воспаление, чем у крыс в контрольной группе.
Результаты, полученные в этих стандартных фармакологических испытаниях, представлены после описания методики получения специфических соединений, которые были испытаны.
Результаты в этих стандартных фармакологических испытаниях свидетельствуют об иммуносупрессивном действии соединений по настоящему изобретению как in vivo, так и in vitro. Результаты, полученные в LAF-тесте, указывают на подавление пролиферации Т-клеток, свидетельствуя, таким образом, об иммуносупрессивной активности соединений по настоящему изобретению. Эффективность соединений по настоящему изобретению в качестве иммунодепрессивных веществ была далее доказана результатами, полученными в стандартных фармакологических тестах с использованием кожных трансплантатов и адъювантного артрита. Кроме того, результаты, полученные в тесте с использованием кожного трансплантата, также свидетельствуют о способности данных соединений полностью предотвращать или тормозить отторжение трансплантата. Результаты, полученные в стандартном фармакологическом тесте с использованием адъювантного артрита, также свидетельствуют о способности соединений по настоящему изобретению излечивать ревматоидный артрит или задерживать его прогрессирование.
На основании результатов этих стандартных фармакологических тестов можно сделать вывод, что указанные соединения являются эффективными в лечении или подавлении отторжения трансплантатов, таких как почка, сердце, печень, легкое, костный мозг, поджелудочная железа (островковые клетки), роговица, тонкая кишка, а также кожных аллотрансплантатов и ксенотрансплантатов клапанов сердца; в лечении и подавлении “трансплантат болезни против хозяина”, в лечении и подавлении аутоиммунных заболеваний, например, волчанки, ревматоидного артрита, сахарного диабета, миастении gravis и рассеянного склероза, а также воспалительных заболеваний, например, псориаза, дерматита, экземы, себореи, воспалительных заболеваний кишечника, воспаления легких (включая астму, хроническое обструктивное заболевание легких, эмфизему, острый респираторный дистресс-синдром, бронхит и другие подобные заболевания) и увеита.
Благодаря обнаруженному профилю действия, соединения по настоящему изобретению могут рассматриваться как имеющие противоопухолевую, противогрибковую и антипролиферативную активности. Отсюда следует, что соединения по настоящему изобретению также применимы при лечении твердых опухолей, Т-клеточного лейкоза/лимфомы у взрослых, микозов и гипер-пролиферативных сосудистых заболеваний, таких как рестеноз и атеросклероз. В случае использования при лечении рестеноза, соединения по настоящему изобретению предпочтительно применяют для лечения рестеноза, возникающего после пластической операции на сосудах. В случае использования для данной цели, соединения по настоящему изобретению могут вводиться до операции, во время операции, после операции или при любой комбинации вышеуказанных условий.
При использовании для лечения или торможения вышеуказанных заболеваний соединения по настоящему изобретению могут вводиться млекопитающему перорально, парентерально, интраназально, внутрибронхиально, чрескожно, локально, интравагинально или ректально.
Предполагается, что в тех случаях, когда соединения по настоящему изобретению используются в качестве иммунодепрессивных или противовоспалительных средств, они могут вводиться в комбинации с другими (одним или более) иммунорегуляторными агентами. К таким иммунорегуляторным агентам относятся (но не исчерпывают возможного перечня): азатиоприн, кортикостероиды, такие как преднизон и метилпреднизолон, циклофосфамид, рапамицин, циклоспорин А, ФК-506, ОКТ-3 и АТГ (антитимоцитный глобулин). Если соединения по настоящему изобретению применяются в комбинации с подобными другими лекарственными средствами или агентами для индуцирования иммуносупрессии или лечения воспалительных состояний, то для достижения желаемого эффекта требуются меньшие количества каждого из этих средств. Обоснование такой комбинаторной терапии было дано Степковским (Stepkowski), результаты исследований которого показали, что применение комбинации рапамицина и циклоспорина А в субтерапевтических дозах значительно продлевает время жизнеспособности аллотрансплантата сердца /Transplantation Proc. 23: 507 (1991)].
Соединения по настоящему изобретению могут быть приготовлены в чистом виде с фармацевтическим носителем для млекопитающих, нуждающихся в них. Фармацевтический носитель может быть твердым или жидким. В случае приготовления пероральной формы было обнаружено, что 0,01% Твин 80 /Tween 80/ в ФОСАЛ ПГ-50 /PHOSAL PG-50/ (фосфолипидный концентрат с 1,2-пропиленгликолем, A. Nattermann & Cie. GmbH) обеспечивает приемлемую пероральную форму препарата.
Твердый носитель может включать одно или несколько веществ, которые могут также выполнять функцию корригентов, смазывающих веществ, веществ, повышающих растворимость, суспендирующих веществ, наполнителей, скользящих веществ, улучшающих прессование средств, связующих или дезинтегрирующих таблетку веществ; он также может быть инкапсулирующим материалом. В порошках носителем является тонко измельченное твердое вещество, которое находится в смеси с тонко измельченным активным ингредиентом. В таблетках активный ингредиент смешан в необходимых пропорциях с носителем, имеющим необходимые компрессионные свойства, и спрессован в нужную форму необходимого размера. Предпочтительно, порошки и таблетки содержат до 99% активного ингредиента. К соответствующим твердым носителям относятся, например, фосфат кальция, стеарат магния, тальк, сахара, лактоза, декстрин, крахмал, желатин, целлюлоза, метилцеллюлоза, натрийкарбоксиметилцеллюлоза, поливинилпирролидин, низкоплавкий воск и ионообменные смолы.
Жидкие носители используют для приготовления растворов, суспензий, эмульсий, сиропов, эликсиров и находящихся под давлением композиций. Активный ингредиент может быть растворен или суспендирован в фармацевтически приемлемом жидком носителе, таком как вода, органический растворитель, их смесь или фармацевтически приемлемые масла или жиры. Жидкий носитель может содержать другие пригодные фармацевтические добавки, например, солюбилизаторы, эмульгаторы, буферы, консерванты, подслащивающие вещества и вещества, изменяющие вкус и запах препарата, суспендирующие вещества, загустители, красители, регуляторы вязкости, стабилизаторы или осморегуляторы. К примерам приемлемых жидких носителей для перорального или парентерального введения можно отнести воду (частично содержащую вышеуказанные добавки, такие как производные целлюлозы, предпочтительно, раствор натрийкарбоксиметилцеллюлозы), спирты (включая одноатомные спирты и многоатомные спирты, например, гликоли) и их производные, лецитины и масла (например, фракционированное кокосовое масло и арахисовое масло). Для парентерального введения носителем может также служить масляный эфир, такой как этилолеат и изопропилмиристат. Стерильные жидкие носители применимы для приготовления стерильных жидких форм композиций для парентерального введения. В качестве жидкого носителя для композиций, находящихся под давлением, может использоваться галогенированный углеводород или другой фармацевтически приемлемый газ-вытеснитель.
Жидкие фармацевтические композиции в виде стерильных растворов или суспензий могут вводиться путем, например, внутримышечной, внутрибрюшинной или подкожной инъекции. Стерильные растворы могут также вводиться внутривенно. Жидкая или твердая лекарственная форма соединения может также вводиться перорально.
Соединения по настоящему изобретению могут вводиться ректально в форме обычного суппозитория. Для введения путем интраназальной или интрабронхиальной ингаляции или инсуффляции соединения по настоящему изобретению могут быть приготовлены в форме водных или частично водных растворов, которые затем могут использоваться в форме аэрозоля. Соединения по настоящему изобретению могут также вводиться чрескожно путем использования чрескожного пластыря, содержащего активное соединение и носитель, инертный по отношению к активному соединению, нетоксичный по отношению к коже и обеспечивающий системную абсорбцию препарата в кровоток через кожу. Носитель может быть в любой из многочисленных форм, например, в форме кремов и мазей, паст, гелей и окклюзивных присоединений. Кремы и мази могут быть вязкими, жидкими или полутвердыми эмульсиями типа “масло в воде” либо типа “вода в масле”. Также могут использоваться пасты, состоящие из абсорбирующих порошков, диспергированных в вазелине или гидрофильном вазелине, содержащем активный ингредиент. Существуют разнообразные варианты окклюзивных приспособлений, которые могут использоваться для выделения активного ингредиента в кровоток, например, полупроницаемая мембрана, покрывающая резервуар, содержащий активный ингредиент с носителем или без носителя, или матрикс, содержащий активный ингредиент. В литературе известны другие окклюзивные приспособления.
Кроме того, соединения по настоящему изобретению могут использоваться в виде раствора, крема или лосьона, имеющих в своем составе фармацевтически приемлемые носители, содержащие 0,1-5%, желательно 2%, активного соединения, которые могут наноситься на область, пораженную грибком.
Требования к дозировке варьируются в зависимости от конкретных используемых композиций, способа введения, тяжести присутствующих симптомов и конкретного объекта лечения. Согласно результатам, полученным в стандартных фармакологических испытаниях, предполагаемые суточные дозы активного соединения должны быть 0,1 мкг/кг – 100 мг/кг, предпочтительно между 0,001-25 мг/кг и более предпочтительно между 0,01-5 мг/кг. Как правило, лечение начинают с введения низких доз, меньших, чем оптимальная доза соединения. С этого момента дозу увеличивают, пока не достигается оптимальный эффект, возможный при данных условиях, точные дозы для перорального, парентерального, назального или внутрибронхиального введения определяются лечащим врачом на основании опыта лечения индивидуального пациента. Фармацевтическая композиция предпочтительно находится в форме единичных доз, например, в виде таблеток или капсул. В такой форме композиция разделена на единичные дозы, содержащие подходящие количества активного ингредиента, формами единичных доз могут быть композиции в упаковках, например, порошки в пакетиках, ампулы, заранее заполненные шприцы или пакеты-саше, содержащие жидкости. Лекарственной формой единичной дозы может быть, например, сама капсула или таблетка или это может быть соответствующее количество любой из таких композиций в упакованной форме.
Следующие примеры иллюстрируют получение и биологические активности типичных соединений по настоящему изобретению.
Пример 1 стандартное отклонение) в группе, получавшей тест-соединение, сравнивали с аналогичным показателем, полученным в контрольной группе. Результаты выражены как среднее время жизнеспособности трансплантата в днях. Кусочковые кожные трансплантаты в не получавшей тест-соединение (контрольной) группе обычно отторгались в течение 6-7 дней. Соединения исследовались при дозе 4 мг/кг, вводимой внутрибрюшинно. При внутрибрюшинном введении рапамицина в дозе 4 мг/кг время жизнеспособности трансплантата составило 11,6710,63 дня.
В стандартном фармакологическом испытании с использованием адъювантного артрита оценивают способность тестируемых соединений предотвращать иммуноопосредованное воспаление и подавлять или излечивать ревматоидный артрит. Ниже кратко описана используемая методика испытания. Соединение предварительно вводили группе крыс (инбредных крыс-самцов линии Вистар Льюис) для испытания (за 1 ч до введения антигена), а затем в правую заднюю лапку этих крыс впрыскивали полный адъювант Фрейнда (FСА) для индуцирования артрита. Затем определенные дозы вводили крысам перорально по схеме введения препарата в понедельник, среду и пятницу с 0 по 14 день при общем количестве 7 доз. Обе задние лапки измеряли на 16-й, 23-й и 30-й день. Определяли различие в объеме лапок (мл) от 16 до 0 дня и рассчитывали процентное изменение по сравнению с контролем. Воспаление левой задней лапки (не подвергавшейся инъекции) обусловлено воспалением, опосредованным Т-клетками, и регистрируется в виде процентного изменения по сравнению с контролем. С другой стороны, воспаление правой задней лапки обусловлено неспецифическим воспалением. Соединения испытывали при дозе 2 мг/кг. Результаты представлены в виде процентного изменения в лапке, не подвергавшейся инъекции, на 16-й день по сравнению с контролем, чем больше отрицательное процентное изменение, тем эффективнее соединение. Введение рапамицина обеспечило 70%-ное изменение по сравнению с контролем. Это означает, что у крыс, получивших рапамицин, наблюдалось на 70% меньшее иммуноопосредованное воспаление, чем у крыс в контрольной группе.
Результаты, полученные в этих стандартных фармакологических испытаниях, представлены после описания методики получения специфических соединений, которые были испытаны.
Результаты в этих стандартных фармакологических испытаниях свидетельствуют об иммуносупрессивном действии соединений по настоящему изобретению как in vivo, так и in vitro. Результаты, полученные в LAF-тесте, указывают на подавление пролиферации Т-клеток, свидетельствуя, таким образом, об иммуносупрессивной активности соединений по настоящему изобретению. Эффективность соединений по настоящему изобретению в качестве иммунодепрессивных веществ была далее доказана результатами, полученными в стандартных фармакологических тестах с использованием кожных трансплантатов и адъювантного артрита. Кроме того, результаты, полученные в тесте с использованием кожного трансплантата, также свидетельствуют о способности данных соединений полностью предотвращать или тормозить отторжение трансплантата. Результаты, полученные в стандартном фармакологическом тесте с использованием адъювантного артрита, также свидетельствуют о способности соединений по настоящему изобретению излечивать ревматоидный артрит или задерживать его прогрессирование.
На основании результатов этих стандартных фармакологических тестов можно сделать вывод, что указанные соединения являются эффективными в лечении или подавлении отторжения трансплантатов, таких как почка, сердце, печень, легкое, костный мозг, поджелудочная железа (островковые клетки), роговица, тонкая кишка, а также кожных аллотрансплантатов и ксенотрансплантатов клапанов сердца; в лечении и подавлении “трансплантат болезни против хозяина”, в лечении и подавлении аутоиммунных заболеваний, например, волчанки, ревматоидного артрита, сахарного диабета, миастении gravis и рассеянного склероза, а также воспалительных заболеваний, например, псориаза, дерматита, экземы, себореи, воспалительных заболеваний кишечника, воспаления легких (включая астму, хроническое обструктивное заболевание легких, эмфизему, острый респираторный дистресс-синдром, бронхит и другие подобные заболевания) и увеита.
Благодаря обнаруженному профилю действия, соединения по настоящему изобретению могут рассматриваться как имеющие противоопухолевую, противогрибковую и антипролиферативную активности. Отсюда следует, что соединения по настоящему изобретению также применимы при лечении твердых опухолей, Т-клеточного лейкоза/лимфомы у взрослых, микозов и гипер-пролиферативных сосудистых заболеваний, таких как рестеноз и атеросклероз. В случае использования при лечении рестеноза, соединения по настоящему изобретению предпочтительно применяют для лечения рестеноза, возникающего после пластической операции на сосудах. В случае использования для данной цели, соединения по настоящему изобретению могут вводиться до операции, во время операции, после операции или при любой комбинации вышеуказанных условий.
При использовании для лечения или торможения вышеуказанных заболеваний соединения по настоящему изобретению могут вводиться млекопитающему перорально, парентерально, интраназально, внутрибронхиально, чрескожно, локально, интравагинально или ректально.
Предполагается, что в тех случаях, когда соединения по настоящему изобретению используются в качестве иммунодепрессивных или противовоспалительных средств, они могут вводиться в комбинации с другими (одним или более) иммунорегуляторными агентами. К таким иммунорегуляторным агентам относятся (но не исчерпывают возможного перечня): азатиоприн, кортикостероиды, такие как преднизон и метилпреднизолон, циклофосфамид, рапамицин, циклоспорин А, ФК-506, ОКТ-3 и АТГ (антитимоцитный глобулин). Если соединения по настоящему изобретению применяются в комбинации с подобными другими лекарственными средствами или агентами для индуцирования иммуносупрессии или лечения воспалительных состояний, то для достижения желаемого эффекта требуются меньшие количества каждого из этих средств. Обоснование такой комбинаторной терапии было дано Степковским (Stepkowski), результаты исследований которого показали, что применение комбинации рапамицина и циклоспорина А в субтерапевтических дозах значительно продлевает время жизнеспособности аллотрансплантата сердца /Transplantation Proc. 23: 507 (1991)].
Соединения по настоящему изобретению могут быть приготовлены в чистом виде с фармацевтическим носителем для млекопитающих, нуждающихся в них. Фармацевтический носитель может быть твердым или жидким. В случае приготовления пероральной формы было обнаружено, что 0,01% Твин 80 /Tween 80/ в ФОСАЛ ПГ-50 /PHOSAL PG-50/ (фосфолипидный концентрат с 1,2-пропиленгликолем, A. Nattermann & Cie. GmbH) обеспечивает приемлемую пероральную форму препарата.
Твердый носитель может включать одно или несколько веществ, которые могут также выполнять функцию корригентов, смазывающих веществ, веществ, повышающих растворимость, суспендирующих веществ, наполнителей, скользящих веществ, улучшающих прессование средств, связующих или дезинтегрирующих таблетку веществ; он также может быть инкапсулирующим материалом. В порошках носителем является тонко измельченное твердое вещество, которое находится в смеси с тонко измельченным активным ингредиентом. В таблетках активный ингредиент смешан в необходимых пропорциях с носителем, имеющим необходимые компрессионные свойства, и спрессован в нужную форму необходимого размера. Предпочтительно, порошки и таблетки содержат до 99% активного ингредиента. К соответствующим твердым носителям относятся, например, фосфат кальция, стеарат магния, тальк, сахара, лактоза, декстрин, крахмал, желатин, целлюлоза, метилцеллюлоза, натрийкарбоксиметилцеллюлоза, поливинилпирролидин, низкоплавкий воск и ионообменные смолы.
Жидкие носители используют для приготовления растворов, суспензий, эмульсий, сиропов, эликсиров и находящихся под давлением композиций. Активный ингредиент может быть растворен или суспендирован в фармацевтически приемлемом жидком носителе, таком как вода, органический растворитель, их смесь или фармацевтически приемлемые масла или жиры. Жидкий носитель может содержать другие пригодные фармацевтические добавки, например, солюбилизаторы, эмульгаторы, буферы, консерванты, подслащивающие вещества и вещества, изменяющие вкус и запах препарата, суспендирующие вещества, загустители, красители, регуляторы вязкости, стабилизаторы или осморегуляторы. К примерам приемлемых жидких носителей для перорального или парентерального введения можно отнести воду (частично содержащую вышеуказанные добавки, такие как производные целлюлозы, предпочтительно, раствор натрийкарбоксиметилцеллюлозы), спирты (включая одноатомные спирты и многоатомные спирты, например, гликоли) и их производные, лецитины и масла (например, фракционированное кокосовое масло и арахисовое масло). Для парентерального введения носителем может также служить масляный эфир, такой как этилолеат и изопропилмиристат. Стерильные жидкие носители применимы для приготовления стерильных жидких форм композиций для парентерального введения. В качестве жидкого носителя для композиций, находящихся под давлением, может использоваться галогенированный углеводород или другой фармацевтически приемлемый газ-вытеснитель.
Жидкие фармацевтические композиции в виде стерильных растворов или суспензий могут вводиться путем, например, внутримышечной, внутрибрюшинной или подкожной инъекции. Стерильные растворы могут также вводиться внутривенно. Жидкая или твердая лекарственная форма соединения может также вводиться перорально.
Соединения по настоящему изобретению могут вводиться ректально в форме обычного суппозитория. Для введения путем интраназальной или интрабронхиальной ингаляции или инсуффляции соединения по настоящему изобретению могут быть приготовлены в форме водных или частично водных растворов, которые затем могут использоваться в форме аэрозоля. Соединения по настоящему изобретению могут также вводиться чрескожно путем использования чрескожного пластыря, содержащего активное соединение и носитель, инертный по отношению к активному соединению, нетоксичный по отношению к коже и обеспечивающий системную абсорбцию препарата в кровоток через кожу. Носитель может быть в любой из многочисленных форм, например, в форме кремов и мазей, паст, гелей и окклюзивных присоединений. Кремы и мази могут быть вязкими, жидкими или полутвердыми эмульсиями типа “масло в воде” либо типа “вода в масле”. Также могут использоваться пасты, состоящие из абсорбирующих порошков, диспергированных в вазелине или гидрофильном вазелине, содержащем активный ингредиент. Существуют разнообразные варианты окклюзивных приспособлений, которые могут использоваться для выделения активного ингредиента в кровоток, например, полупроницаемая мембрана, покрывающая резервуар, содержащий активный ингредиент с носителем или без носителя, или матрикс, содержащий активный ингредиент. В литературе известны другие окклюзивные приспособления.
Кроме того, соединения по настоящему изобретению могут использоваться в виде раствора, крема или лосьона, имеющих в своем составе фармацевтически приемлемые носители, содержащие 0,1-5%, желательно 2%, активного соединения, которые могут наноситься на область, пораженную грибком.
Требования к дозировке варьируются в зависимости от конкретных используемых композиций, способа введения, тяжести присутствующих симптомов и конкретного объекта лечения. Согласно результатам, полученным в стандартных фармакологических испытаниях, предполагаемые суточные дозы активного соединения должны быть 0,1 мкг/кг – 100 мг/кг, предпочтительно между 0,001-25 мг/кг и более предпочтительно между 0,01-5 мг/кг. Как правило, лечение начинают с введения низких доз, меньших, чем оптимальная доза соединения. С этого момента дозу увеличивают, пока не достигается оптимальный эффект, возможный при данных условиях, точные дозы для перорального, парентерального, назального или внутрибронхиального введения определяются лечащим врачом на основании опыта лечения индивидуального пациента. Фармацевтическая композиция предпочтительно находится в форме единичных доз, например, в виде таблеток или капсул. В такой форме композиция разделена на единичные дозы, содержащие подходящие количества активного ингредиента, формами единичных доз могут быть композиции в упаковках, например, порошки в пакетиках, ампулы, заранее заполненные шприцы или пакеты-саше, содержащие жидкости. Лекарственной формой единичной дозы может быть, например, сама капсула или таблетка или это может быть соответствующее количество любой из таких композиций в упакованной форме.
Следующие примеры иллюстрируют получение и биологические активности типичных соединений по настоящему изобретению.
Пример 131-O-(Триэтилсилил)-42-оксорапамицин Стадия А. 31-O-(Триэтилсилил) рапамицин К раствору рапамицина (15,39 г, 16,84 ммоль) и 2,6-лютидина (7,65 г, 75,76 мл) в дихлорметане (100 мл) при oC добавляли по каплям триэтилсилилтрифторметансульфонат (10 г, 37,88 ммоль) в течение 30 мин. Полученную смесь перемешивали при 0oC в течение еще 90 мин и фильтровали. Фильтрат разбавляли этилацетатом (500 мл), промывали водой (3х250 мл) и рассолом (1х100 мл), высушивали (MgSO4) и выпаривали досуха. Полученный материал повторно растворяли в безводном ТГФ (40 мл), охлаждали до 0oC и обрабатывали ледяной уксусной кислотой, охлажденной до температуры льда, (150 мл) и водой (80 мл). Смесь перемешивали в течение 3 ч при 0oC, разбавляли этилацетатом (500 мл) и осторожно доводили до pH 7-8 NaHCO3 при 0oC. Органический слой промывали водой (2х250 мл), рассолом (1х100 мл), сушили (MgSO4) и выпаривали досуха с получением продукта, указанного в заголовке с количественным выходом 1H ЯМР (CDCl3, 400 МГц):  1,66 (3H,6-CH3C=C), 1,75 (3H,30-CH3C=C), 3,14 (3H,41-OCH3), 3,27 (3H,7-CH3O), 3,40 (3H,32-CH3O), 4,12 (м, 1H,31-CH).
МС (отрицат. ион FAB, m/z): 1027,4 [M]–, 589,3.
Стадия Б. 31-O-(Триэтилсилил)-42-оксорапамицин 1,66 (3H,6-CH3C=C), 1,75 (3H,30-CH3C=C), 3,14 (3H,41-OCH3), 3,27 (3H,7-CH3O), 3,40 (3H,32-CH3O), 4,12 (м, 1H,31-CH).
МС (отрицат. ион FAB, m/z): 1027,4 [M]–, 589,3.
Стадия Б. 31-O-(Триэтилсилил)-42-оксорапамицинРаствор 31-O-(триэтилсилил)рапамицина (17,32 г, 16,84 ммоль) и периодинана Десс-Мартина (8,65 г, 20,35 ммоль) в безводном дихлорметане (150 мл) перемешивали в атмосфере азота в течение 5 ч. Полученную смесь фильтровали, фильтрат разбавляли этилацетатом (500 мл), промывали водой (3х250 мл) и рассолом (1х100 мл), высушивали (MgSO4) и выпаривали досуха. Неочищенный материал предварительно абсорбировали на колонке с силикагелем Merck-60 и мгновенно испаряли с гексан-этилацетатом 95:5 и 7:2, с получением чистого продукта, указанного в заголовке, с 64%-ным выходом. 1H ЯМР (CDCl3, 400 МГц): 0,42-0,48 (м, 9Н), 0,80-0,83 (м, 6H), 1,61 (3H, 6-CH3C= C), 1,77 (3H,30-CH3C=C), 3,04 (3H,41-OCH3), 3,16 (3H,7-OCH3), 3,28 (3H,32-CH3O), 3,90 (м, 1H,41-CH).
МС (отрицат. ион FAB, m/z): 1025,3 [M]–.
Пример 242-Деоксо-42-(гидроксиимино) рапамицин Способ получения А Смесь 42-оксорапамицина (0,183 г, 0,22 ммоль, полученных согласно способу, описанному в Патенте США 5023263), гидрохлорида гидроксиламина (0,0143 г, 0,22 ммоль) и ацетата натрия (0,025 г, 0,3 ммоль) в метаноле (5 мл) перемешивали в атмосфере азота в течение 15 мин. Смесь упаривали досуха и остаток очищали с помощью флэш-хроматографии (на силикагеле Merck 60, элюент 50% ТГФ в гексане). Чистые фракции объединяли, выпаривали, и полученное масло перекристаллизовывали из смеси изопропиловый эфир/циклогексан при соотношении 20: 80, с получением указанного в заголовке продукта в виде смеси E/Z изомеров (0,075 г, 40%-ный выход). 1H ЯМР (CDCl3, 400 МГц): 1,65 (3H,6-CH3C=C), 1,75 (3H,30-CH3C=C), 3,13 (3H,41-OCH3), 3,34 (3H,7-OCH3), 3,41 (3H,32-OCH3).
13C ЯМР (CDCl3, 400 МГц): 215,28; 208,17; 169,26; 166,72; 158,83; 140,60; 140,07; 135,95; 135,67, 133,56; 130,18; 129,47; 126,62; 126,43; 98,48; 86,33; 83,73; 84,30; 78,90; 67,17; 59,29, 59,13; 57,51; 55,93; 55,88; 51,234; 46,58; 46,04; 44,19; 41,47; 40,65; 40,19; 38,93; 38,41; 37,41; 35,08; 33,78; 33,24; 32,05; 32,00, 31,82; 31,21; 30,92; 27,21; 27,05; 26,89; 25,27; 22,84; 21,97; 21,62; 21,46; 20,64; 16,32; 16,22; 16,09; 15,99; 14,74; 13,60; 13,18; 13,10; 10,33; 10,15.
МС (отрицат. ион FAB, m/z): 926 [M]–, 590, 334.
Анализ: Рассчитано для C51H78N2O13: С 66,07; H 8,48; N 3,02.
Найдено: C 66,25; H 8,67; N 3,03.
Способ получения БСтадия А. 31-O-(Триэтилсилил)-42-деоксо-42-(гидроксиимино) рапамицин При безводных условиях смесь 31-O-(триэтилсилил)-42-оксорапамицина из примера 1 (0,105 г, 0,102 ммоль), гидрохлорида гидроксиламина (7,6 мг, 0,109 ммоль) и ацетата натрия (12,5 мг, 0,153 ммоль) в безводном метаноле (5 мл) перемешивали в течение 30 мин. Смесь фильтровали и полученный фильтрат упаривали досуха. Остаток повторно растворяли в этилацетате (50 мл), промывали водой (2х50 мл) и рассолом (1х50 мл), высушивали (MgSO4) и выпаривали досуха с получением указанного в заголовке продукта в виде смеси E/Z изомеров (0,114 г, 94%-ный выход). 1H ЯМР (CDCl3, 400 МГц): 0,47-0,55 (м, 9Н), 0,84-0,88 (м, 6H), 1,65 (3H, 6-CH3C= C), 1,75 (3H,30-CH3C=C), 3,14 (3H, 41-OCH3), 3,26 (3H, 7-OCH3), 3,44 (3H, 32-CH3O).
МС (отрицат. ион FAB, M/z): 1040,7 [M]–.
Стадия Б. 42-Деоксо-42-(гидроксиимино)рапамицинРаствор 31-O-(триэтилсилил)-42-деоксо-42-(гидроксиимино)рапамицина (1 г, 0,974 ммоль) в 20 мл 10% раствора п-толуолсульфокислоты в метаноле перемешивали в течение одного часа в присутствии азота при 0oC. Раствор разбавляли этилацетатом и гасили 5% водным NaHCO3. Органический слой промывали водой, рассолом, сушили (MgSO4) и выпаривали досуха, с получением указанного в заголовке продукта, идентичного материалу, описанному в способе получения A (0,812 г, выход 89,9%). Результаты, полученные в стандартных фармакологических тестах: LAF IC50: 3,88 нмоль. LAF отношение: 0,85. Время жизнеспособности кожного трансплантата: 10,8 0,4.
Пример 342-деоксо-42-(гидроксиимино) рапамицин К раствору 31-O-(триэтилсилил)-42-деоксо-42-(гидроксиимино)рапамицина примера 2, Способа получения Б, стадии А (2,08 г, 2 ммоль) в безводном метаноле (75 мл) в атмосфере азота при 0oC одновременно добавляли в течение 30 мин 1 н. раствор цианобороводорода натрия в тетрагидрофуране (2 мл) и 4 н. раствор HCl в диоксане так, чтобы поддерживать pH на уровне 3,5. Смесь перемешивали в течение 30 мин, разбавляли EtOAc и промывали 2,5% NaHCO3 (100 мл), водой (2х250 мл) и рассолом (1х250 мл), высушивали (MgSO4) и выпаривали досуха с получением указанного в заголовке продукта в виде смеси изомеров (0,763 г, 41%-ный выход). 1H ЯМР (CDCl3, 400 МГц): 1,60 и 1,63 (3H, 6-CH3C=C), 1,72 и 1,74 (3H, 30-CH3C= C), 3,09 и 3,11 (3H, 41-OCH3), 3,28; 3,32; 3,34; 3,36 (6Н, 7- и 32-OCH3).
МС (отрицат. ион FAB, m/z): 928,4 [M]–, 590,3, 336,6.
Анализ. Рассчитано для C51H80N2O13: C 65,92, H 8,68, N 3,01.
Найдено: C 65,36; H 8,53; N 2,82.
Результаты, полученные в стандартных фармакологических испытаниях:LAF IC50: 5,60 нмоль LAF отношение: 0,18 Время жизнеспособности кожного трансплантата: 9,6 0,0Процентное изменение в группе с адъювантным артритом в сравнении с контрольной группой: -84% Пример 4 42-Деокси-42-оксорапамицин-42-0-карбамоилоксим Смесь 42-деоксо-42-(оксиимино)рапамицина примера 2 (0,813 г, 0,876 ммоль), цианата натрия (0,228 г, 3,5), ледяной уксусной кислоты (8 мл) и воды (8 мл) перемешивали в течение 1,5 ч в атмосфере азота. Смесь разбавляли этилацетатом (100 мл) и гасили водным NaHCO3. Органический слой промывали водой, рассолом, сушили (MgSO4) и выпаривали досуха. Неочищенный продукт растворяли в дихлорметане, предварительно абсорбировали на силикагеле Merck 60 и очищали с помощью флэш-хроматографии ступенчатый градиент от 50% этилацетата в гексане до чистого этилацетата с получением указанного в заголовке продукта в виде смеси E/Z изомеров (0,289 г, 34%-ный выход). 1ЯМР (CDCl3, 400 МГц): 1,66 (3H,6-CH3C=C), 1,75 (3H, 30-CH3C=C), 3,14 (3H, 41-OCH3), 3,33 (3H, 7-OCH3), 3,43 (3H, 32-OCH3).
13C ЯМР (CDCl3, 400 МГц): d 215,17, 208,11, 169,67, 169,30, 166,73, 164,31, 164,25, 156,37, 140,05, 138,34, 137,28, 136,07, 135,78, 135,09, 133,51, 133,25, 132,17, 130,61, 130,22, 130,05, 129,40, 127,33, 126,81, 126,66, 126,57, 126,44, 125,76, 125,50, 125,22, 98,70, 98,50, 84,73, 84,34, 84,28, 82,70, 78,92, 68,91, 67,21, 59,29, 57,93, 57,82, 56,34, 56,31, 56,16, 56,09, 55,85, 55,56, 52,00, 51,25, 46,55, 46,06, 45,68, 44,54, 44,18, 41,92, 41,83, 41,48, 41,40, 41,26, 41,14, 41,01, 40,88, 40,57, 40,42, 40,35, 40,19, 39,77, 39,30, 39,17, 38,97, 38,83, 38,66, 38,59, 37,98, 37,45, 37,26, 35,53, 35,13, 34,88, 34,79, 34,72, 34,57, 33,83, 33,49, 33,37, 33,31, 33,24, 32,19, 32,03, 31,98, 31,85, 31,70, 31,64, 31,20, 27,35, 27,21, 27,03, 26,80, 25,23, 25,04, 24,45, 24,31, 21,62, 21,51, 21,37, 20,99, 20,74, 20,63, 16,69, 16,30, 16,18, 15,95, 15,80, 15,71, 15,03, 14,57, 13,88, 13,64, 13,18, 13,13, 13,07, 10,16.
МС (отрицат. ион FAB, m/z): 969,8 [M]–, 925,8 [M-CONH2]– 590,6.
Результаты, полученные в стандартных фармакологических испытаниях:LAF IC50: 2,00 нмоль. LAF отношение: 0,25 Время жизнеспособности кожного трансплантата: 9,5 1,1Пример 5 42-[O-(Пиридин-2-илметил)]-оксим 42-деокси-42-оксорапамицина Соединение, указанное в заголовке, получали согласно способу получения Б примера 2, при замене гидрохлорида гидроксиламина гидрохлоридом О-(пиридин-2-илметил)-гидроксиламина (выход 71,1%). 1H ЯМР (CDCl3, 400 МГц): 1,654 (3H, 6-CH3C=C), 1,751 (3H, 30-CH3C=C), 3,138 (3H, 41-OCH3), 3,334 (3H, 7-OCH3), 3,338 (3H, 32-OCH3), 7,17-8,58 (мм, 4H, Harom).
13C ЯМР (CDCl3, 400 МГц): 1,58,96 (42-C=NO-).
MC (отрицат. ион FAB, m/z): 1017,5 [M]–, 590,4, 425,3.
Результаты, полученные в стандартных фармакологических испытаниях:LAF IC50: 1,50 нмоль. LAF отношение: 0,40 Время жизнеспособности кожного трансплантата: 7,8 0,8Процентное изменение в группе с адъювантным артритом в сравнении с контрольной группой: -53%. Пример 6 42-[O-(Пиридин-4-илметил)]-оксим 42-деокси-42-оксорапамицина Соединение, указанное в заголовке, получали согласно Способу получения Б примера 2, при замене гидрохлорида гидроксиламина дигидрохлоридом О-(пиридин-4-илметил)- гидроксиламина (выход 34,5%). 1H ЯМР (CDCl3, 400 МГц): 1,653 (3H, 6-CH3C=C), 1,753 (3H, 30-CH3C=C), 3,153 (3H, 41-OCH3), 3,29 (2х3H, 7-OCH3 и 32-OCH3), 7,2 (м, 2H, Harom), 8,52 (m, 2H, Harom).
МС (отрицат. ион FAB, m/z): 1017,2 [M]–, 590,2, 425,1.
Результаты, полученные в стандартных фармакологических испытаниях:LAP IC50: 1,80 нмоль LAF соотношение: 0,31 Время жизнеспособности кожного трансплантата: 8,0 0,9Пример 7 42-[О-(трет-бутил)]-оксим 42-деокси-42-оксорапамицина Соединение, указанное в заголовке, получали согласно Способу получения Б примера 2, за исключением замены гидрохлорида гидроксиламина гидрохлоридом O-(трет-бутил)- гидроксиламина (выход 26,6%). 1H ЯМР (CDCl3, 400 МГц): 1,28 (9Н, трет-бутил), 1,654 (3H, 6-CH3C=C), 1,75 (3H, 30-CH3C= C), 3,136 (3H, 41-OCH3), 3,336 (3H, 7-OCH3), 3,37 (3H, 32-OCH3).
13C ЯМР (CDCl3, 400 МГц): 155,89 (42-C=NO-).
МС (отрицат. ион FAB, m/z): 982,5 [M]–, 590,3, 390,2.
Результаты, полученные в стандартных фармакологических испытаниях:LAF: 49%-ное подавление при 0,1 мкмоль. Пример 8 42-[O-(фенилметил)]-оксим 42-деокси-42- оксорапамицина Соединение, указанное в заголовке, получали согласно Способу получения Б примера 2, за исключением замены гидрохлорида гидроксиламина гидрохлоридом O-(фенилметил)-гидроксиламина (выход 29,9%). 1H ЯМР (CDCl3, 400 МГц): 1,652 (3H, 6-CH3C=C), 1,747 (3H, 30-CH3C=C), 3,136 (3H, 41-OCH3), 3,332 (3H, 7-OCH3), 3,35 (3H, 32- OCH3), 5,134 (2H, = NOCH2-, при C-42), 7,27-7,37 (м, 5H, Harom).
13C ЯМР (CDCl3, 400 МГц): 158,48 (42-C=NO-).
МС (отрицат. ион FAB, m/z): 1016,2 [M]–, 590,1, 424,1.
Результаты, полученные в стандартных фармакологических тестах:LAF IC50: 21,67 нмоль. LAF отношение: 0,03. Пример 9 42-[O-(аллил)]-оксим 42-деокси-42-оксорапамицина Соединение, указанное в заголовке, получали согласно Способу получения Б примера 2, за исключением замены гидрохлорида гидроксиламина гидрохлоридом O-(аллил)-гидроксиламина (выход 57,1%). 1H ЯМР (CDCl3, 400 МГц): 1,655 (3H, 6-CH3C=C), 1,751 (3H, 30-CH3C=C), 3,139 (3H, 41-OCH3), 3,336 (3H, 7-OCH3), 3,395 (3H, 32-OCH3), 4,60 (м, 2H, = NOCH2C= , при C-42), 5,17-5,31 (м, 2H, -C=CH2, при C-42), 5,94-6,03 (м, 1H, -CCH=C-, при C-42).
13C ЯМР (CDCl3, 400 МГц): 158,1 (42-C=N-0-).
МС (отрицат. ион FAB, m/z): 996,5 [M]–, 590,3, 374,2.
Результаты, полученные в стандартных фармакологических испытаниях:LAF: 43%-ное подавление при 0,1 мкмоль. Пример 10 42-[O-(проп-2-инил)]-оксим 42-деокси-42-оксорапамицина Соединение, указанное в заголовке, получали согласно Способу получения Б примера 2, за исключением замены гидрохлорида гидроксиламина гидрохлоридом O-(проп-2-инил)- гидроксиламина (выход 35,4%). 1H ЯМР (CDCl3, 400 МГц): 1,66 (3H, 6-CH3C=C), 1,75 (3H, 30-CH3C=C), 3,13 (3H, 41-OCH3), 3,33 (3H, 7-OCH3), 3,415 (3H, 32-OCH3), 4,69 (2H, = NOCH2C, при C-42).
13C ЯМР (CDCl3, 400 МГц): 159,5 (42-C=NO-).
MC (отрицат. ион FAB, m/z): 964,2 [M]–, 590,2, 372,1.
Результаты, полученные в стандартных фармакологических испытаниях:LAF IC50: 8,13 нмоль. LAF отношение: 0,08. Время жизнеспособности кожного трансплантата: 8,0 1,1Пример 11 42-Деоксо-42-[O-(пиридин-4-илметил)]-гидроксиаминорапамицин Соединение, указанное в заголовке, получали согласно примеру 3, за исключением замены 31-O-(триэтилсилил)-42- деоксо-42-гидроксииминорапамицина 42-[O-(пиридин-4-илметил)]- оксимом 42-деоксо-42-оксорапамицина примера 6. MC (отрицат. ион FAB, m/z): 1019,5 [M]–. Формула изобретения
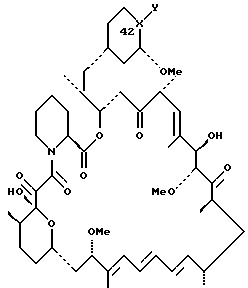 в которой Х-Y представляет С=NOR1 или CHNHOR2, в которых R1 представляет водород, алкил с 1 – 6 атомами углерода, алкенил с 2 – 7 атомами углерода, алкинил с 2 – 7 атомами углерода, группа формулы -(CH2)mAr, в которой Ar представляет фенил или пиридинил и m = 1 – 6; R2 представляет водород или радикал формулы -(CH2)mAr, в которой Ar представляет пиридинил, или их формацевтически приемлемые соли. 2. Соединение по п.1, в котором X-Y представляет С=NOR1, или его фармацевтически приемлемая соль. 3. Соединение по п.1, в котором X-Y является группой CHNHOR2, или его фармацевтически приемлемая соль. 4. Соединение по п.1, которое является 42-деоксо-42-(гидроксиимино)рапамицином или его фармацевтически приемлемой солью. 5. Соединение по п.1, которое является 42-деоксо-42-(гидроксиамино)рапамицином или его фармацевтически приемлемой солью. 6. Соединение по п.1, которое является 42-деокси-42-оксорапамицин-42-О-карбамоилоксимом или его фармацевтически приемлемой солью. 7. Соединение по п.1, которое является 42-деокси-42-оксорапамицин-42-[О-(пиридин-2-илметил)]-оксимом или его фармацевтически приемлемой солью. 8. Соединение по п.1, которое является 42-деокси-42-оксорапамицин-42-[О-(пиридин-4-илметил)]-оксимом или его фармацевтически приемлемой солью. 9. Соединение по п.1, которое является 42-деокси-42-оксорапамицин-42-[О-(трет-бутил)]-оксимом или его фармацевтически приемлемой солью. 10. Соединение по п.1, которое является 42-деокси-42-оксорапамицин-42-[О-(фенилметил)]-оксимом или его фармацевтически приемлемой солью. 11. Соединение по п.1, которое является 42-деокси-42-оксорапамицин-42-(О-аллил)-оксимом или его фармацевтически приемлемой солью. 12. Соединение по п.1, которое является 42-деокси-42-оксорапамицин-42-[О-(проп-2-инил)]-оксимом или его фармацевтически приемлемой солью. 13. Соединение по п.1, которое является 2-деоксо-42-[O-(пиридин-4-илметил)]-гидроксиаминорапамицином или его фармацевтически приемлемой солью. 14. Способ лечения заболевания, выбранного из группы, состоящей из отторжения трансплантата или болезни “трансплантат против хозяина” и ревматоидного артрита, у млекопитающего, нуждающегося в лечении, который включает введение указанному млекопитающему эффективного против заболевания количества соединения, имеющего структуру 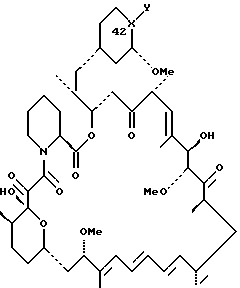 в которой X-Y является группой С=NOR1 или CHNHOR2, R1 представляет водород, алкил с 1 – 6 атомами углерода, алкенил с 2 – 7 атомами углерода, алкинил с 2 – 7 атомами углерода, группу формулы -(CH2)mAr, в которой Ar представляет фенил или пиридинил и m = 1 – 6; R2 представляет водород или радикал формулы -(CH2)mAr, в которой Ar представляет пиридинил, или его фармацевтически приемлемой соли. 15. Фармацевтическая композиция, проявляющая иммуносупрессивную активность, которая включает соединение структуры 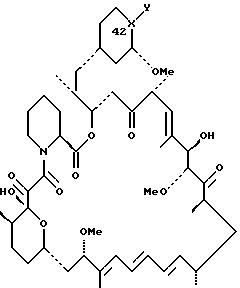 в которой X-Y является С= NOR1 или CHNHOR2, R1 представляет водород, алкил с 1 – 6 атомами углерода, алкенил с 2 – 7 атомами углерода, алкинил с 2 – 7 атомами углерода, группу формулы -(CH2)mAr, в которой Ar представляет фенил или пиридинил и m = 1 – 6; R2 представляет водород или радикал формулы -(CH2)mAr, в которой Ar представляет пиридинил, или его фармацевтически приемлемую соль и фармацевтический носитель. 16. Соединение структуры 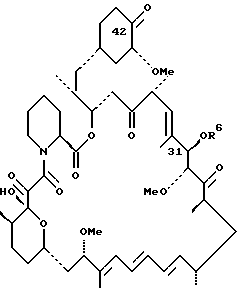 в которой R6 является группой SiR7R8R9, в которой каждый R7, R8 и R9 представляет независимо алкил с 1 – 8 атомами углерода. 17. Соединение по п.16, которое является 31-О-(три-этилсилил)-42-оксорапамицином. 18. Способ получения соединений формулы I, охарактеризованных в п.1, который включает взаимодействие 42-оксорапамицина (необязательно защищенного по гидроксильной функциональной группе в положении 31 силильной группой формулы R6, определенной в п.16 с гидроксиламином формулы H2NOR1, в которой R1 имеет указанные выше значения, с образованием соответствующего оксима формулы I, в котором X-Y является группой С=NOR1, и удаление указанной защитной группы R6 . PD4A – Изменение наименования обладателя патента Российской Федерации на изобретение
Номер и год публикации бюллетеня: 23-2002
(73) Новое наименование патентообладателя:
Извещение опубликовано: 20.08.2002
MM4A Досрочное прекращение действия патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 07.12.2003
Извещение опубликовано: 10.05.2005 БИ: 13/2005
|
||||||||||||||||||||||||||