Патент на изобретение №2170741
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ ЦИКЛОСПОРИНА
(57) Реферат: Описывается способ получения циклоспорина формулы (II) 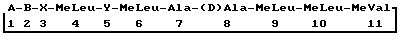 где А – остаток 3′-дезокси-3′-оксо-MeBmt; B – –  Abu-, -Val- или -Nva-; Х – -Sar- или остаток оптически-активной -N-метилированной -аминокислоты в (D)-конфигурации; Y – -Val- или, когда В является Nva, Nva, который включает селективное окисление промежуточного циклоспорина формулы (II), где А является MeBmt, диметилсульфоксидом в комбинации с N,N’-дициклогексилкарбодиимидом или додецилметилсульфоксидом в комбинации с N,N’-дициклогексилкарбодиимидом. Технический результат – упрощение процесса, улучшение его экологичности и повышение выхода целевого продукта. 2 с. и 6 з.п. ф-лы. Abu-, -Val- или -Nva-; Х – -Sar- или остаток оптически-активной -N-метилированной -аминокислоты в (D)-конфигурации; Y – -Val- или, когда В является Nva, Nva, который включает селективное окисление промежуточного циклоспорина формулы (II), где А является MeBmt, диметилсульфоксидом в комбинации с N,N’-дициклогексилкарбодиимидом или додецилметилсульфоксидом в комбинации с N,N’-дициклогексилкарбодиимидом. Технический результат – упрощение процесса, улучшение его экологичности и повышение выхода целевого продукта. 2 с. и 6 з.п. ф-лы.
Изобретение относится к способу получения [3′-дезокси-3′-оксо-MeBmt] 1циклоспорина, в частности к способу получения [3′-дезокси-3′-оксо-MeBmt] 1-[Nva]2-циклоспорина или [3′-дезокси-3′-оксо-MeBmt]1-[Val]2-циклоспорина. Согласно настоящему изобретению предложен способ получения [3′-дезокси-3′-оксо-MeBmt]1циклоспорина формулы 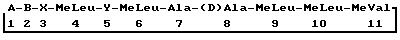 где A представляет собой остаток 3′-дезокси-3′-оксо-MeBmt; B представляет собой – Abu-, -Thr-, -Val- или -Nva-;X представляет собой -Sar- или остаток оптически активной -N-метилированной -аминокислоты в (D)-конфигурации; иY представляет собой -Val- или, когда B является Nva, Nva, при котором культивируют организм с получением промежуточного диклоспорина формулы II, где A является MeBmt, выделяют указанный промежуточный циклоспорин и селективно окисляют указанный промежуточный циклоспорин с получением циклоспорина формулы II, где A является остатком 3′-дезокси-3′-оксо-MeBmt. Соединения формулы II, где A является остатком 3′-дезокси-3′-оксо-MeBmt, полезны для увеличения чувствительности (или увеличения эффективности) к химиотерапевтической лекарственной терапии и, в частности, полезны при борьбе с химиотерапевтической лекарственной устойчивостью различных типов (например, приобретенной или врожденной) или к усилению или восстановлению чувствительности к назначенной лекарственной терапии. Формы химиотерапевтической лекарственной терапии, при которой эти соединения могут быть применены, включают в себя, например, противопаразитическую, в частности антивирусную, антибактериальную или антипротозойную химиотерапию, а также антинеоплазменную и цитостатическую химиотерапию. Конкретными предпочтительными циклоспоринами, которые могут быть получены способом по изобретению, являются [3′-деэокси-3′-оксо-MeBmt] 1-[Nva] 2-циклоспорин или особенно [3′-дезокси-3′-оксо-MeBmt]1-[Val]2-циклоспорин. Эти соединения получают путем окисления [Nva] 2-циклоспорина и [Val] 2-циклоспорина, сами способы окисления являются частью данного изобретения. Соответственно следующий аспект изобретения предусматривает способ получения [3′-дезокси-3′-оксо-MeBmt]1-[B’]2-циклоспорина, где B является Nva или Val, путем окисления [B’]2-циклоспорина. [Val]2-циклоспорин известен также как циклоспорин D. [B’]2-циклоспорин, используемый в процессе окисления, может быть получен путем культивирования организмов, продуцирующих [B’]2-циклоспорин, либо путем частичного или полного синтеза, включая получение производных другого циклоспорина или подходящего продукта ферментации. Любой подходящий окисляющий агент, растворитель, катализатор, а также условия реакции и реакционная схема могут быть использованы на стадии окисления способа по изобретению или способа согласно дальнейшему аспекту. Подходящие реагенты для реакции окисления включают в себя диметилсульфид в комбинации с N-хлор-сукцинимидом или предпочтительно диметилсульфоксид в комбинации с N,N’-дициклогексилкарбодиимидом, предпочтительно в неполярном органическом растворителе, таком как толуол. Додецилметилсульфоксид предпочтительнее как диметилсульфида, так и диметилсульфоксида, так как диметилсульфид нежелателен по причинам безопасности и экологическим причинам, а диметилсульфоксид повышает выход диметилсульфида в качестве побочного продукта реакции. Также существуют менее нежелательные проблемы запаха, связанные с использованием додецилметилсульфоксида. Предпочтительно реакцию окисления осуществляют при низких температурах, например от -15 до 20oC. Далее изобретение описывают путем иллюстрации в следующем далее примере, который описывает получение [3′-дезокси-3′-оксо-MeBmt]1-[Val]2-циклоспорина. Пример. а) Получение циклоспорина D Циклоспорин D (также известный как [Val]2-циклоспорин) получают путем ферментации подходящего организма, такого, как Tolypocladium inflatum GAMS (депонирован в US Northern Regional Research Laboratories Culture Collection под номером NRRL 8044), выделяют из ферментационного бульона и затем очищают как описано ниже. 500 л питательного раствора, содержащего на 1 л: 40 г глюкозы, 2 г казеината натрия, 2,5 г фосфата аммония, 5 г MgSO4  7H2O, 2 г KH2PO4, 3 г NaNO3, 0,5 г KCl, 0,01 г FeSO4 и деминерализованной воды до 1 л, инокулируют 50 л предварительной культуры штамма NRRL 8044 и инкубируют в стальном ферментере при перемешивании (170 мин-1) и аэрации (1 л воздуха/мин/1 л питательного раствора) в течение 13 дней пpи 27oC.
Культуральную жидкость перемешивают с таким же количеством n-бутилацетата, концентрируют выпариванием в вакууме после отделения органической фазы и грубый экстракт обезжиривают путем 3-стадийного разделения в смеси метанол/вода (9:1) и петролейном эфире. Метанольную фазу отделяют, концентрируют путем выпаривания в вакууме и грубый продукт осаждают путем добавления воды. Материал, полученный после фильтрации, подвергают хроматографии на силикагеле, используя в качестве элюента смесь гексан/ацетон (2:1). Фракции, элюируемые сначала, преимущественно содержат циклоспорин A и циклоспорин D, фракции, элюируемые позднее, преимущественно содержат циклоспорин C. Для дальнейшей очистки циклоспорин A- и D-содержащие фракции кристаллизуют из 2-2,5-кратного количества ацетона при температуре -15oC, Кристаллат дважды подвергают хроматографии на силикагеле, первые фракции, элюируемые насыщенным водой этилацетатом, значительно обогащены циклоспорином D. Их растворяют в двойном количестве ацетона и оставляют для кристаллизации при 15oC. Полученный грубый продукт циклоспорина D растворяют для дальнейшей очистки в 10-кратном количестве ацетона, добавляют 2% по весу активного древесного угля и нагревают в течение 5 мин до 60oC. После фильтрации через тальк получают прозрачный и почти бесцветный фильтрат, концентрируют его выпариванием до одной трети его объема и оставляют остывать при комнатной температуре, в результате чего спонтанно кристаллизуется циклоспорин D. Кристаллизацию завершают, помещая на -17oC. Полученные после фильтрации кристаллы промывают небольшим количеством охлажденного на льду ацетона и затем высушивают в высоком вакууме при 80oC в течение 2 ч.
Характеристики циклоспорина D: бесцветные призматические кристаллы, Т. пл. 148-151oC; []2D0 = -245o (c = 0,52 в хлороформе); []2D0 = -211oC (c = 0,51 в метаноле).
б) Получение додецилметилсульфоксида 7H2O, 2 г KH2PO4, 3 г NaNO3, 0,5 г KCl, 0,01 г FeSO4 и деминерализованной воды до 1 л, инокулируют 50 л предварительной культуры штамма NRRL 8044 и инкубируют в стальном ферментере при перемешивании (170 мин-1) и аэрации (1 л воздуха/мин/1 л питательного раствора) в течение 13 дней пpи 27oC.
Культуральную жидкость перемешивают с таким же количеством n-бутилацетата, концентрируют выпариванием в вакууме после отделения органической фазы и грубый экстракт обезжиривают путем 3-стадийного разделения в смеси метанол/вода (9:1) и петролейном эфире. Метанольную фазу отделяют, концентрируют путем выпаривания в вакууме и грубый продукт осаждают путем добавления воды. Материал, полученный после фильтрации, подвергают хроматографии на силикагеле, используя в качестве элюента смесь гексан/ацетон (2:1). Фракции, элюируемые сначала, преимущественно содержат циклоспорин A и циклоспорин D, фракции, элюируемые позднее, преимущественно содержат циклоспорин C. Для дальнейшей очистки циклоспорин A- и D-содержащие фракции кристаллизуют из 2-2,5-кратного количества ацетона при температуре -15oC, Кристаллат дважды подвергают хроматографии на силикагеле, первые фракции, элюируемые насыщенным водой этилацетатом, значительно обогащены циклоспорином D. Их растворяют в двойном количестве ацетона и оставляют для кристаллизации при 15oC. Полученный грубый продукт циклоспорина D растворяют для дальнейшей очистки в 10-кратном количестве ацетона, добавляют 2% по весу активного древесного угля и нагревают в течение 5 мин до 60oC. После фильтрации через тальк получают прозрачный и почти бесцветный фильтрат, концентрируют его выпариванием до одной трети его объема и оставляют остывать при комнатной температуре, в результате чего спонтанно кристаллизуется циклоспорин D. Кристаллизацию завершают, помещая на -17oC. Полученные после фильтрации кристаллы промывают небольшим количеством охлажденного на льду ацетона и затем высушивают в высоком вакууме при 80oC в течение 2 ч.
Характеристики циклоспорина D: бесцветные призматические кристаллы, Т. пл. 148-151oC; []2D0 = -245o (c = 0,52 в хлороформе); []2D0 = -211oC (c = 0,51 в метаноле).
б) Получение додецилметилсульфоксидаСмешивают 478 г (2,2 моль) додецилметилсульфида, 52,5 г ледяной уксусной кислоты и 1 л безводного этанола и нагревают до 70oC при перемешивании. В течение 90 мин при перемешивании добавляют раствор 215 г (2,2 моль) перекиси водорода в 540 мл воды и продолжают перемешивание около 1 ч, до тех пор пока реакция не завершится по меньшей мере на 90%, что определяют с помощью газовой хроматографии. Затем охлаждают смесь до 20-25oC и добавляют раствор 27 r метабисульфита натрия в 41 мл воды, перемешивают полученную смесь в течение 20 мин, поддерживая температуру 20-25oC. Затем добавляют 3 л толуола и раствор 93 г бикарбоната натрия в 1,07 л воды, смесь перемешивают и позволяют разделиться фазам. Водную фазу удаляют, а органическую фазу промывают 1 л воды, промывочную воду также удаляют и концентрируют органическую фазу при пониженном давлении до объема около 1,25 л. Нагревают концентрат до 50-55oC и добавляют 1,5 л гексана. В образовавшийся раствор вносят кристаллы додецилметилсульфоксида, охлаждают в течение 1 ч до 20-25oC и затем перемешивают при этой температуре. Затем охлаждают полученный раствор до 0-5oC в течение примерно 30 мин, перемешивают при этой температуре около 3 ч и собирают твердый осадок путем центрифугирования. Затем промывают твердое вещество 400 мл холодного гексана (0oC) и высушивают при температуре, не превышающей 60oC при пониженном давлении с получением 462 г додецилметилсульфоксида (около 90% теоретического выхода). в) Окисление циклоспорина D до [3′-дезокси-3′-оксо-MeBmt] 1-[Val] 2-циклоспорина 269 г (1,22 моль) чистого циклоспорина D растворяют в 2,95 л толуола, добавляют 283 г (1,22 моль) додецилметилсульфоксида, полученного как описано выше, смесь охлаждают до 0-5oC и добавляют 78 г дихлоруксусной кислоты вместе с 50 мл толуола. Температуру реакционной смеси поддерживают при 0-5oC около 45 мин, после чего добавляют раствор 205 г (0,99 моль) N,N’-дициклогексилкарбодиимида в 290 мл толуола. Реакционную смесь перемешивают около 30 мин, до тех пор пока реакция не завершится на 95%, что определяют путем ЖХВД хроматографии. Добавляют раствор 81 г (0,64 моль) щавелевой кислоты в 630 мл воды и перемешивают образовавшуюся смесь при 0-5oC в течение 3 ч. Образовавшееся твердое вещество отделяют путем центрифугирования, промывают 440 мл холодного толуола (5oC), после чего твердое вещество выбрасывают. Жидкую фракцию реакционной смеси и промывную жидкость объединяют, позволяют фазам разделиться, водную фазу отбрасывают и к органической фазе добавляют водный раствор бикарбоната натрия (80 г в 1,25 л) до тех пор, пока pH не достигнет 7-8. После перемешивания позволяют фазам разделиться, отбрасывают водную фазу, а органическую фазу промывают 650 мл воды. Промывную воду отбрасывают и выпаривают органическую фазу досуха при пониженном давлении при температуре, не превышающей 60oC. Остаток растворяют в смеси 560 мл изопропилацетата и 10 мл воды при нагревании до 55-60oC. В течение около 30 мин раствор охлаждают до около 40oC и вносят в него кристаллы додецилметилсульфоксида. Через примерно 90 мин смесь охлаждают до температуры от -10 до -15oC и перемешивают при этой температуре около 4 ч. Удаляют твердое вещество путем центрифугирования, промывают 390 мл изопропилацетата и отбрасывают. Жидкую фракцию реакционной смеси и промывную жидкость объединяют с получением 1,28 кг раствора, содержащего около 256 г [3′-дезокси-3′-оксо-MeBmt] 1-[Val]2-циклоспорина (около 95% от теоретического вы хода). Полученный как указано выше раствор обогащают с помощью хроматографии на силикагеле. Готовят две колонки, каждая из которых содержит 1 кг силикагеля, суспендированного в 2,5 л изопропилацетата, и каждую загружают 162 г указанного раствора. Колонки элюируют насыщенным водой изопропилацетатом со скоростью потока 500 мл/ч и собирают фракции в следующем порядке: первые 4 л (первый элюат), следующие 500 мл (второй элюат), следующие 4,5 л (третий элюат) и следующий 1 л (четвертый элюат). Как определяют путем ЖХВД, желаемый продукт находится в третьем элюате. Таким образом, первый элюат отбрасывают, а второй и четвертый оставляют для повторной обработки. Третий элюат из обеих колонок объединяют, концентрируют при пониженном давлении до объема около 320 мл и затем выпаривают досуха при пониженном давлении и температуре, не превышающей 60oC, с получением 40 г продукта. Продукт далее очищают путем кристаллизации из трет-бутилметилового эфира. Обнаружено, что образовавшийся очищенный [3′-дезокси-3′-оксо-MeBmt] 1-[Val] 2-циклоспорин имеет температуру плавления в пределах 195-198oC, оптическое вращение [ ]2D0 = -255,1o (c = 0,5 в CHCl3) и молекулярный вес 1214,65.
[3′-дезокси-3′-оксо-MeBmt] 1-[Nva] 2-циклоспорин может быть приготовлен аналогичным образом, путем окисления [Nva]2-циклоспорина по существу так же, как описано окисление [Val]2-циклоспорина.
Формула изобретения
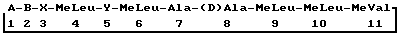 где А – остаток 3′-дезокси-3′-оксо-МеBmt; B – – Abu-, -Thr-, -Val- или -Nva-;X – -Sar- или остаток оптически активной -N-метилированной -аминокислоты в (D)-конфигурации;Y – -Val- или, когда В является Nva, Nva, который включает селективное окисление промежуточного циклоспорина формулы II, где А – MeBmt, диметилсульфоксидом в комбинации с N,N’-дициклогексилкарбодиимидом или додецилметилсульфоксидом в комбинации с N,N’-дициклогексилкарбодиимидом. 2. Способ по п.1, при котором промежуточный циклоспорин формулы II получают в результате культивирования организма, продуцирующего промежуточный циклоспорин, и извлечения указанного промежуточного продукта. 3. Способ по п.1 или 2, отличающийся тем, что получают [3′-дезокси-3′-оксо-MeBmt] 1-[Nva] 2-циклоспорин или [3′-дезокси-3′-оксо-MeBmt] 1-[Val] 2-циклоспорин. 4. Способ по п.3, отличающийся тем, что получают [3′-дезокси-3′-оксо-MeBmt]1-[Val]2-циклоспорина. 5. Способ по п.4, отличающийся тем, что реакцию окисления проводят при 0oC и 5oC. 6. Способ получения [3′-дезокси-3′-оксо-MeBmt]1-[B’]2-циклоспорина, где B’ – Nva или Val, при котором окисляют [B’]2-циклоспорин диметилсульфоксидом в комбинации с N,N’-дициклогексилкарбодиимидом или додецилметилсульфоксид в комбинации с N,N’-дициклогексилкарбодиимидом. 7. Способ по пп.1 – 6, отличающийся тем, что в качестве окислительного агента используют додецилметилсульфоксид в комбинации с N,N’-дициклогексилкарбодиимидом. 8. Способ по п.7, отличающийся тем, что его осуществляют при температуре от -15 до +20oC в неполярном органическом растворителе, таком, как толуол. MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 17.08.2009
Извещение опубликовано: 20.07.2010 БИ: 20/2010
|
||||||||||||||||||||||||||