Патент на изобретение №2170232
|
||||||||||||||||||||||||||
(54) АМИДЫ ФОСФИНОВЫХ КИСЛОТ И СПОСОБ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ, СВЯЗАННОГО С НЕЖЕЛАТЕЛЬНОЙ АКТИВНОСТЬЮ МЕТАЛЛОПРОТЕАЗЫ
(57) Реферат: Изобретение относится к амидам фосфиновых кислот ф-лы (I), где R1 – водород, алкил, фенилалкил, пиридинил, пиридинилалкил, алкоксиалкил, фенилалкоксиалкил; R2 – водород, алкил, фенилалкил, индолил, фенилалкоксиалкил, алкилтиоалкил, алкиламиноалкил; R3 – алкил или фенил; R4 – алкил, фенил или замещенный фенил, пиридил, тиенил или фурил, к их оптическим изомерам, диастереомерам, энантиомерам, фармацевтически приемлемым солям или биогидролизуемым сложным эфирам, которые могут быть использованы в качестве ингибиторов матриксной металлопротеазы при лечении состояний, характеризуемых чрезмерной активностью указанных ферментов. 3 с. и 7 з.п. ф-лы, 2 табл. 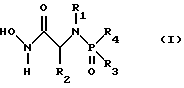
Изобретение относится к соединениям, полезным для лечения заболеваний, связанных с активностью металлопротеазы, в частности активностью цинковой металлопротеазы. Ряд структурно родственных металлопротеаз [MPs, МП] расщепляет структурные белки. Эти металлопротеазы часто воздействуют на межклеточный материал и, следовательно, участвуют в разрушении и ремоделировании ткани. Такие белки называют металлопротеазами или МП. Существует несколько различных групп МП, классифицированных по гомологии последовательностей. Некоторые группы МП и их примеры уже известны. Указанные МП включают матриксные металлопротеазы [ММП], цинковые металлопротеазы, многие из металлопротеаз, связанных с мембранами, TNF-превращающие ферменты, ангиотензин-превращающие ферменты (ACEs, АПФ), расщепители, включая ADAMs (смотри Wolfsberg et al, 131 J. Cell Bic. 275-78, октябрь 1995), и энкефалиназы. Примеры МП включают коллагеназу фибробласта кожи человека, желатиназу фибропласта кожи человека, коллагеназу, агреканазу и желатиназу мокроты человека и стромелизин человека. Считается, что коллагеназа, стромелизин, агреканаза и родственные ферменты важны в опосредовании симптоматологии многих заболеваний. Возможные терапевтические показания ингибиторов МП уже описаны в литературе. Смотри, например, патент США N 5506242 (Ciba Geigy Corp.); патент США N 5403952 (Merck & Co.); PCT опубликованную заявку WO 96/06074 (British Bio Tech Ltd); PCT публикацию WO 96/00214 (Ciba Geigy); WO 95/35275 (British Bio Tech Ltd); WO 95/35276 (British Bio Tech Ltd); WO 95/33731 (Hoffman-LaRoche); WO 95/33709 (Hoffman-LaRoche); WO 95/32944 (British Bio Tech Ltd); WO 95/26989 (Merck); WO 95/29892 (DuPont Merck); WO 95/24921 (Inst. Ophthalmology); WO 95/23790 (Smith Kline Beecham); WO 95/22966 (Sanofi Winthrop); WO 95/19965 (Glycomed); WO 95/19956 (British Bio Tech Ltd); WO 95/19957 (British Bio Tech Ltd); WO 95/19961 (British Bio Tech Ltd); WO 95/13289 (Chiroscience Ltd.); WO 95/12603 (Syntex); WO 95/09633 (Florida State Univ. ); WO 95/09620 (Florida State Univ.); WO 95/04033 (Celltech); WO 94/25434 (Celltech); WO 94/25435 (Celltech); WO 93/14112 (Merck); WO 94/0019 (Glaxo); WO 93/21942 (British Bio Tech Ltd); WO 92/22523 (Res. Corp. Tech. Inc.); WO 94/10990 (British Bio Tech Ltd); WO 93/09090 (Yamanouchi); и патенты Британии GB 2282598 (Merck) и GB 2268934 (British Bio Tech Ltd); опубликованные заявки на европейский патент ЕР 95/684240 (Hoffman LaRoche); ЕР 574758 (Hoffman LaRoche); ЕР 575874 (Hoffman LaRoche); опубликованные заявки на патент Японии JP 08053403 (Fujusowa Pharm. Co. Ltd); JP 7304770 (Kanebo Ltd. ); и Bird et al, J. Med. Chem. том 37, стр. 158-69 (1994). Примеры возможных терапевтических применений МП ингибиторов включают ревматоидный артрит (Mullins D.E., et al., Biochim. Biophys. Acta (1983) 695: 117-214); остеоартрит (Henderson, В. , et al. Drugs of the Future (1990) 15: 495-508); метастаз опухолевых клеток (там же, Broadhurst, M.J., заявка N 276436 на европейский патент (опубликована в 1987 г), Rech, R., et al., 48 Cancer Res. 3307-3312 (1988)); и различные язвы или язвенные состояния ткани. Например, язвенные состояния могут возникнуть в роговице в результате ожога щелочью или в результате инфекции бактериями Pseudomonas aeruginosa, Acanthamoeba, вирусами герпеса простого и вакцинии. Другие примеры состояний, характеризуемых нежелательной активностью металлопротеаз, включают периодонтальное заболевание, врожденный буллезный эпидермолиз, лихорадку, воспаление и склерит (ср. DeCicco et al., WO 95/29892, опубликованная 9 ноября 1995 г.). Ввиду участия таких металлопротеаз в создании многих болезненных состояний были предприняты попытки получения ингибиторов указанных ферментов. Целый ряд таких ингибиторов описан в литературе. Примеры включают патент США N 5183900, выданный 2 февраля 1993 г. на имя Galardy; патент США N 4996358, выданный 26 февраля 1991 г. на имя Handa et. al.; патент США N 4771038, выданный 13 сентября 1988 г. на имя Wolanin, et al.; патент США N 4743587, выданный 10 мая 1988 г. на имя Dickens, et al.; европейская патентная публикация N 575844, опубликованная 29 декабря 1993 г. by Broadhurst, et al., международная патентная публикация N WO 93/09090, опубликованная 13 мая 1993 г. by Isomura et al.; международная патентная публикация 92/17460, опубликованная 15 октября 1992 г. by Markwell et al.; и европейская патентная публикация N 498665, опубликованная 12 августа 1992 г. by Beckett et al. В качестве способа лечения заболеваний, связанных с нежелательной активностью металлопротеаз, было бы целесообразным ингибирование этих металлопротеаз. Хотя уже было получено много разных ингибиторов, тем не менее продолжает оставаться потребность в мощных ингибиторах матриксных металлопротеаз, пригодных для лечения таких заболеваний. Целью настоящего изобретения является создание мощных ингибиторов металлопротеаз. Другой целью настоящего изобретения является создание фармацевтических композиций, содержащих такие ингибиторы. Следующей целью настоящего изобретения является создание способа лечения заболеваний, связанных с металлопротеазами. В соответствии с настоящим изобретением предлагаются соединения, полезные для применения в качестве ингибиторов металлопротеаз и эффективные в лечении состояний, характеризуемых чрезмерной активностью указанных ферментов. В частности, настоящее изобретение относится к соединению, имеющему структуру, соответствующую формуле (I) 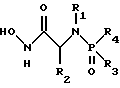 где R1i представляет водород, алкил, арилалкил, гетероциклоалкил, алкоксиалкил, арилалкоксиалкил или алкилтиоалкил; R2 представляет водород, алкил, арилалкил, гетероциклоалкил, алкоксиалкил, арилалкоксиалкил или алкилтиоалкил; R3 представляет алкил, циклоалкил, карбоциклический или гетероциклический арил, гидроксиалкил, алкоксиалкил или аминоалкил; и R4 представляет карбоциклический или гетероциклический арил; его оптическому изомеру, диастереомеру или энантиомеру или его фармацевтически приемлемой соли или биогидролизуемому алкоксиамиду, сложному эфиру, ацилоксиамиду или имиду. Предпочтительный R4 включает фенил и замещенный фенил. Предпочтительное замещение на R4 производят рядом с местом присоединения или напротив него (т. е., если R4 представляет фенил, то в положениях 2 и/или 4). Предпочтительные заместители фенила включают галоген, алкил, алкокси, нитро, циано и тому подобное. Предпочтительным R3 является алкил, а более предпочтительным – C1-C2 алкил. Предпочтительным R2 является H или алкил, а более предпочтительным – H или C1-C4 алкил. Предпочтительным R1 является H или алкил, арилалкил, а более предпочтительным – C1-C6 алкил или арил(C1-C2) алкил. Указанные соединения обладают способностью ингибировать по крайней мере одну матриксную металлопротеазу млекопитающего. Поэтому в других аспектах настоящее изобретение касается фармацевтических композиций, содержащих соединения формулы (I), и способов лечения заболеваний, характеризуемых активностью матриксных металлопротеаз, с использованием указанных соединений или фармацевтических композиций, содержащих эти соединения. Матриксные металлопротеазы, активные в особенно неподходящем месте (например, органе или некоторых типах клеток), могут быть сделаны мишенью путем сопряжения соединений по настоящему изобретению с нацеливающим лигандом, специфическим по отношению к маркеру в этом месте, таким, как антитело или его фрагмент, или лиганд рецептора. Методы сопряжения известны в данной области химии. Настоящее изобретение касается также многих других способов использования преимущества уникальных свойств указанных соединений. Так, в еще одном аспекте настоящее изобретение относится к соединениям формулы (I), объединенным с твердыми носителями. Эти конъюгаты можно использовать в качестве обладающих сродством реагентов для очистки требуемой матриксной металлопротеазы. И в еще одном аспекте настоящее изобретение касается соединений формулы (I) с введенной меткой. Когда соединения по настоящему изобретению связываются с по крайней мере одной матриксной металлопротеазой, метка может быть использована для обнаружения относительно высокого уровня матриксной металлопротеазы in vivo или клеточной культуры in vitro. Кроме того, соединения формулы (I) могут быть сопряжены с носителями, позволяющими использование этих соединений в схемах иммунизации для получения антител, специфически иммунореактивных с соединениями по настоящему изобретению. Типичные методы сопряжения известны в данной области. Указанные антитела в этом случае полезны и для терапии, и для контроля дозирования ингибиторов. Соединения по настоящему изобретению являются ингибиторами матриксных металлопротеаз млекопитающих. Соединения предпочтительно представляют собой соединение формулы (I) или его фармацевтически приемлемую соль, или биогидролизуемый алкоксиамид, ацилоксиамид или имид. Используемые термины и определения “Ацил” или “карбонил” представляет собой радикал, который мог быть образован путем удаления гидроксигруппы из карбоновой кислоты (т.е., R-C(=0)-). Предпочтительные ацильные группы включают, например, ацетил, формил и пропионил. “Ацилокси” представляет собой оксильный радикал, имеющий ацильный заместитель (т.е., -О- ацил), например -O-C(=O)-алкил. “Алкоксиацил” – это ацильный радикал (-C(=O)-), имеющий алкоксильный заместитель (т. е., -O-R), например -C(=O)-O-алкил. Этот радикал можно назвать сложным эфиром. “Ациламино” представляет собой амино-радикал, имеющий ацильный заместитель (т.е., -N-ацил), например -NH-C(=O)-алкил. “Алкенил” представляет собой незамещенный или замещенный углеводородный радикал, имеющий 2-15, предпочтительно 2-10 и более предпочтительно 2-8 углеродных атомов, если не указано иное. Алкенильные заместители имеют по крайней мере одну олефиновую двойную связь (включая, например, винил, аллил и бутенил). “Алкинил” представляет собой незамещенный или замещенный углеводородный радикал, имеющий 2-15, предпочтительно 2-10 и более предпочтительно 2-8 углеродных атомов, если не указано иное. Цепь имеет по крайней мере одну тройную углерод-углеродную связь. “Алкокси” – это кислородный радикал, имеющий углеводородный заместитель, где углеводородная цепь представляет собой алкил или алкенил (т.е., -О-алкил или -О-алкенил). Предпочтительные алкоксигруппы включают, например, метокси, этокси, пропокси и аллилокси. “Алкоксиалкил” – это незамещенный или замещенный алкильный фрагмент, замещенный алкокси-фрагментом (т. е. , -алкил-О-алкил). Является предпочтительным, когда алкил имеет 1-6 углеродных атомов (более предпочтительно 1-3 углеродных атома) и алкокси имеет 1-6 углеродных атомов (более предпочтительно 1-3 углеродных атомов). “Алкил” представляет собой незамещенный или замещенный насыщенный углеводородный радикал, имеющий 1-15, предпочтительно 1-10 и более предпочтительно 1-4 углеродных атомов, если не указано иное. Предпочтительные алкильные группы включают, например, замещенные или незамещенные метил, этил, пропил, изопропил и бутил. При упоминании в данном описании термин “спироцикл” или “спироциклический” относится к циклическому фрагменту, имеющему общий углеродный атом с другим кольцом. Такой циклический фрагмент может быть по своей природе карбоциклическим или гетероциклическим. Предпочтительные гетероатомы, входящие в основу гетероциклического спироцикла, включают кислород, азот и серу. Спироциклы могут быть незамещенными или замещенными. Предпочтительные заместители включают оксо, гидрокси, алкил, циклоалкил, арилалкил, алкокси, амино, гетероалкил, арилокси, конденсированные кольца (например, бензотиол, циклоалкил, гетероциклоалкил, бензимидазолы, пиридилтиол и т.д., которые могут быть также замещенными) и тому подобное. Кроме того, гетероатом гетероцикла может быть замещенным, если позволяет валентность. Предпочтительные по размерам спироциклические кольца включают 3-7-членные кольцевые системы. Алкилен относится к алкилу, алкенилу или алкинилу, который представляет собой не радикал, а дирадикал. Аналогичным образом “гетероалкилен” можно определить как (дирадикал)-алкилен, имеющий в своей цепи гетероатом. “Алкиламино” – это амино-радикал, имеющий один (вторичный амин) или два (третичный амин) алкильных заместителя (т.е., -N-алкил); например, метиламино (-NHCH3), диметиламино (-N(CH3)2), метилэтиламино (-N(CH3)CH2CH3). “Аминоацил” представляет собой ацильный радикал, имеющий амино-заместитель (т. е. , -C(=O)-N); например, -C(=O)-NH2. Аминогруппа аминоацильного фрагмента может быть незамещенной (т.е., первичный амин) или замещенной одной (вторичный амин) или двумя (т.е., третичный амин) алкильными группами. “Арил” представляет собой ароматический карбоциклический кольцевой радикал. Предпочтительные арильные группы включают, например, фенил, толил, ксилил, куменил, нафтил, дифенил и флуоренил. Такие группы могут быть замещенными или незамещенными. “Арилалкил” представляет собой алкильный радикал, замещенный арильной группой. Предпочтительные арилалкильные группы включают бензил, фенилэтил и фенилпропил. Такие группы могут быть замещенными или незамещенными. “Арилалкиламино” – это амино-радикал, замещенный арил-алкильной группой (например, -NH-бензил). Такие группы могут быть замещенными или незамещенными. “Ариламино” – это амино-радикал, замещенный арильной группой (например, -NH-арил). Такие группы могут быть замещенными или незамещенными. “Арилокси” – это кислородный радикал, имеющий арильный заместитель (т.е. , -О-арил). Такие группы могут быть замещенными или незамещенными. “Карбоциклическое кольцо” представляет собой незамещенный или замещенный, насыщенный, ненасыщенный или ароматический углеводородный кольцевой радикал. Карбоциклические кольца являются моноциклическими или конденсированными, снабженными мостиком или спирополициклическими кольцевыми системами. Моноциклические карбоциклические кольца обычно содержат 4-9, предпочтительно 4-7 атомов. Полициклические карбоциклические кольца содержат 7-17, предпочтительно 7-12 атомов. Предпочтительные полициклические системы содержат 4-, 5-, 6- или 7-членные кольца, сконденсированные с 5-, 6- или 7-членными кольцами. “Карбоциклоалкил” представляет собой незамещенный или замещенный алкильный радикал, замещенный карбоциклическим кольцом. Если не указано иное, карбоциклическое кольцо предпочтительно является арилом или циклоалкилом, более предпочтительно арилом. Предпочтительные карбоцикло-алкильные группы включают бензил, фенилэтил и фенилпропил. “Карбоциклогетероалкил” – это незамещенный или замещенный гетероалкильный радикал, замещенный карбоциклическим кольцом. Если не указано иное, карбоциклическое кольцо предпочтительно является арилом или циклоалкилом, более предпочтительно арилом. Гетероалкил предпочтительно представляет собой 2-оксапропил, 2-оксаэтил, 2-тиапропил или 2-тиаэтил. “Карбоксиалкил” – это незамещенный или замещенный алкильный радикал, замещенный карбокси (-C(=O)ОН) фрагментом. Например, – CH2-C(=O)OH. “Циклоалкил” представляет собой насыщенный карбоциклический кольцевой радикал. Предпочтительные циклоалкильные группы включают, например, циклопропил, циклобутил и циклогексил. “Циклогетероалкил” представляет собой насыщенное гетероциклическое кольцо. Предпочтительные циклогетеро-алкильные группы включают, например, морфолинил, пиперидинил, пиперазинил, тетрагидрофурил и гидантоинил. “Конденсированные кольца” – это кольца совмещенные друг с другом так, что они имеют два общих кольцевых атома. Данное кольцо может быть сконденсировано с более чем одним другим кольцом. Имеется в виду, что конденсированные кольца представляют собой гетероарильные, арильные и гетероциклические радикалы или тому подобное. “Гетероциклоалкил” – это алкильный радикал, замещенный гетероциклическим кольцом. Гетероциклическое кольцо является предпочтительно гетероарилом или циклогетероалкилом, а более предпочтительно гетероарилом. Предпочтительный гетероциклоалкил включает C1-C4 алкил с присоединенным к нему предпочтительным гетероарилом. Более предпочтительным является, например, пиридилалкил и т.п. “Гетероциклогетероалкил” представляет собой незамещенный или замещенный гетероалкильный радикал, замещенный гетероциклическим кольцом. Гетероциклическое кольцо является предпочтительно арилом или циклогетероалкилом, а более предпочтительно арилом. “Гетероатом” представляет собой атом азота, серы или кислорода. Группы, имеющие один или несколько гетероатомов, могут содержать разные гетероатомы. “Гетероалкенил” представляет собой незамещенный или замещенный ненасыщенный радикал, имеющий 3-8 членов, содержащих атомы углерода и один или два гетероатома. Цепь имеет по крайней мере одну двойную углерод-углеродную связь. “Гетероалкил” представляет собой незамещенный или замещенный насыщенный радикал, имеющий 2-8 членов, содержащих атомы углерода и один или два гетероатома. “Гетероциклическое кольцо” – это незамещенный или замещенный, насыщенный, ненасыщенный или ароматический кольцевой радикал, состоящий из углеродных атомов и одного или нескольких гетероатомов в кольце. Гетероциклические кольца являются моноциклическими или конденсированными, снабженными мостиком или спирополициклическими кольцевыми системами. Моноциклические гетероциклические кольца содержат 3-9, предпочтительно 4-7 атомов. Полициклические кольца содержат 7-17, предпочтительно 7-13 атомов. “Гетероарил” представляет собой ароматическое гетероциклическое кольцо или моноциклический или бициклический радикал. Предпочтительные гетероарильные группы включают, например, тиенил, фурил, пирролил, пиридинил, пиразинил, тиазолил, пиримидинил, хинолинил и тетразолил, бензотиазолил, бензофурил, индолил и тому подобное. Такие группы могут быть замещенными или незамещенными. “Гало”, “галоген” или “галогенид” – это радикал в виде атома хлора, брома, фтора или йода. Предпочтительными являются радикалы брома, хлора и фтора. Кроме того, при упоминании в данном описании “низший” углеводородный фрагмент (например, “низший” алкил) представляет собой углеводородную цепь, состоящую из 1-6, предпочтительно 1-4 углеродных атомов. “Фармацевтически приемлемая соль” представляет собой катионную соль, образованную по любой кислотной (например, карбоксильной) группе, или анионную соль, образованную по любой основной (например, амино) группе. Многие такие соли известны в данной области техники, например те, что описаны в международной патентной публикации 87/05297 (Johnston et al.), опубликованной 11 сентября 1987 г. (включена в данное описание путем ссылки). Предпочтительные катионные соли включают соли щелочных металлов (таких, как натрий и калии) и соли щелочноземельных металлов (таких, как магний и кальций) и органические соли. Предпочтительные анионные соли включают галогениды (такие, как хлориды). “Биогидролизуемый алкоксиамид” или “биогидролизуемый ацилоксиамид” – это амиды гидроксамовой кислоты, которые не мешают ингибирующей активности соединения или легко преобразуются in vivo у человека или низшего животного в активную гидроксамовую кислоту. “Биогидролизуемый гидроксиимид” – это имид соединения формулы (I), который не мешает ингибирующей активности соединения в отношении металлопротеазы или легко преобразуется in vivo у человека или низшего животного в активное соединение формулы (I). Такие гидроксиимиды включают те, что не мешают биологической активности соединений формулы (I). “Биогидролизуемый сложный эфир” – это сложный эфир соединения формулы (I), который не мешает ингибирующей активности указанных соединений в отношении металлопротеазы или легко преобразуется в организме животного в активное соединение формулы (I). “Сольват” представляет собой комплекс, образованный путем сочетания растворенного вещества (например, гидроксамовой кислоты) и растворителя (например, воды). Смотри J. Honig et al. The Van Nostrand Chemist’s Dictionary, стр. 650 (1953). Фармацевтически приемлемые растворители, используемые в соответствии с настоящим изобретением, включают те, которые не мешают биологической активности гидроксамовой кислоты (например, воду, этанол, уксусную кислоту, N,N-диметилформамид и другие известные или легко определяемые квалифицированным специалистом). “Оптический изомер”, “стереоизомер”, “диастереомер” при упоминании в данном описании имеют известные стандартные значения (Ср., Hawley’s Condensed Chemical Dictionary, 11-ое издание). Иллюстрацию конкретных защищенных форм и других производных соединений формулы (I) не следует считать ограничивающей. Применение других полезных защитных групп, видов солей и т.д. находится в компетенции квалифицированного специалиста. Как было указано выше, при использовании в настоящем изобретении замещающие группы могут сами быть замещенными. Такое замещение может быть произведено одним или несколькими заместителями. Такие заместители включают те, которые перечислены в публикации C. Hansch and A. Leo, Substituent Constants for Correlation Analysis in Chemistry and Biology (1979), включенной в данное описание путем ссылки. Предпочтительные заместители включают, например, алкил, алкенил, алкокси, гидрокси, оксо, нитро, амино, аминоалкил (например, аминометил и т. д. ), циано, галоген, карбокси, алкоксиацетил (например, карбоэтокси и т.д.), тиол, арил, циклоалкил, гетероарил, гетероциклоалкил (например, пиперидинил, морфолинил, пирролидинил и т.д.), имино, тиоксо, гидроксиалкил, арилокси, арилалкил и их сочетания. При использовании в данном описании “матриксная металлопротеаза млекопитающего” означает металлсодержащий фермент, найденный у млекопитающих, который способен катализировать расщепление коллагена, желатина или протеогликана при подходящих условиях анализа. Подходящие условия анализа можно найти, например, в патенте США N 4743587, где дана ссылка на методику Cawston, et al. Anal. Biochem. (1979) 99: 340-345, а применение синтетического субстрата описано у Weingarten, Н., et, al., Biochem. Biophy. Res. Comm. (1984) 139: 1184-1187. Разумеется, может быть применен любой стандартный метод анализа расщепления указанных структурных белков. Все упоминаемые в данном описании матриксные металлопротеазы являются цинксодержащими протеазами, которые подобны по структуре, например, человеческому стромелизину или фибробластной коллагеназе в коже. Способность предлагаемых соединений ингибировать активность матриксной металлопротеазы можно, конечно, проверить описанными выше методами анализа. Для подтверждения ингибирующей активности соединений по настоящему изобретению можно использовать выделенные матриксные металлопротеазы или неочищенные экстракты, содержащие ряд ферментов, способных к разрушению ткани. Соединения Соединения по настоящему изобретению описаны в разделе “Краткое изложение сущности изобретения”. Предпочтительные соединения формулы (I) включают соединение 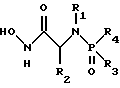 где R1 представляет водород, алкил, арилалкил, гетероциклоалкил, алкоксиалкил, арилалкоксиалкил или алкилтиоалкил; R2 представляет водород, алкил, арилалкил, гетероциклоалкил, алкоксиалкил, арилалкоксиалкил или алкилтиоалкил; R3 представляет алкил, циклоалкил, карбоциклический или гетероциклический арил, гидроксиалкил, алкоксиалкил или аминоалкил; и R4 представляет карбоциклический или гетероциклический арил; его оптический изомер, диастереомер или энантиомер, или его фармацевтически приемлемую соль или, биогидролизуемый алкоксиамид, сложный эфир, ацилоксиамид или имид. Получение соединений Гидроксамовые соединения формулы (I) могут быть получены разнообразными способами. Общие схемы включают схему I (см. в конце описания). Далее описаны представительные примеры получения конкретных соединений. Соединения формулы (I) легко получают из соединений формулы (A), R2 аминокислот, R2 2-галогензамещенных сложных эфиров и тому подобного. В соединении A Y представляет предпочтительно амино и соединение подвергают взаимодействию с соединением B, где Y представляет галоген или подходящую уходящую группу. Когда в соединении A Y представляет галоген, специалисту сразу же понятно, что соединение B имеет Y в виде амино. Когда R1 и R2 не образуют единой цепи, R1 фрагмент (B) вводят, используя традиционные способы. Например, при использовании 2-галогенэфира R1 первичное аминосоединение при основных условиях замещает галогенид или, если используют аминокислоту, он может быть обработан R1 карбонильным соединением, таким, как альдегид, после чего окси фрагмент может быть восстановлен традиционными способами с получением соединения C. Соединения формулы A могут быть произведены от известных аминокислот, включающих 20 распространенных аминокислот, их производные (например, саркозингидроксипролин, 2-аминомасляная кислота, пипиколиновая кислота и тому подобное) или любые такие d-аминокислоты. Многие из них известны или коммерчески доступны из таких, например, источников, как Sigma (St. Louis, МО) или Aldrich (Milwaukee, WI). Когда они не являются легко доступными, различные R2 аминокислоты могут быть получены любым из многих способов, известных в данной области химии. Когда более выгодным является получение соединений формулы I с использованием галогензамещенного сложного эфира или галогензамещенной кислоты, такие галогенэфиры и галогенкислоты известны в данной области химии или могут быть получены известными способами (см., например, March, Advanced Organic Chemistry, Wiley Interscience). Соединение R3R4POZ получают стандартными методами. Например, PCl3 может быть алкилирован и/или арилирован с образованием RPCl2 или R2RPCl и затем обработан короткоцепочечным алканолом с образованием R3R4POZ. В соответствии с другим вариантом, когда R3 и R1 образуют кольцо, соединение XC(O)CHR2NH2 может быть подвергнуто реакции при стандартных условиях с образованием XC(O)CHR2NA(R1R3)POCl, которое затем замыкают с образованием: 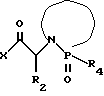 (R1R) предпочтительно представляет оксиметилен или оксиэтилен. Когда R4 представляет гетероцикл, способы получения его фосфинильных или фосфонильных производных известны в данной области химии. Предпочтительные гетероциклические радикалы 4 включают 2- или 3-тиенил, 2- или 3-фурил, 2-, 3- или 4-пиридил, пиримидил и тому подобное. (R3R4)PO фрагмент (D) вводят с использованием стандартных методов химии фосфонамидов, таких, как обработка амина фосфорилхлоридом в инертном растворителе и тому подобное. Обычно гидроксамовую кислоту получают в окончательном виде на конечной стадии путем обработки гидроксиламином по известной методике. Указанные стадии могут быть изменены для увеличения выхода целевого продукта. Квалифицированному специалисту будет также понятно, что здравый выбор реагентов, растворителей и температур является важной составляющей успешного синтеза. Определение оптимальных условий и т. д. является обычным делом, но понятно, что разнообразные соединения можно получить аналогичным образом, руководствуясь приведенной выше схемой. Исходные материалы, используемые для получения соединений по настоящему изобретению, известны, могут быть получены известными способами или коммерчески доступны. Понятно, что квалифицированный специалист в области органической химии может легко осуществить стандартные преобразования органических соединений без дополнительных указаний, то есть осуществление таких преобразований полностью находится в пределах компетенции специалиста. Эти преобразования включают (но не ограничиваются ими) восстановление карбонильных соединений до соответствующих спиртов, окисление гидроксилов и тому подобного, ацилирование, ароматические замещения (электрофильное и нуклеофильное), этерификацию, эстерификацию и омыление и тому подобное. Примеры этих преобразований описаны в стандартной литературе, такой, как March, Advanced Organic Chemistry (Wiley), Carey and Sundberg, Advanced Organic Chemistry (том 2) и Keeting, Heterocyclic Chemistry (все 17 томов). Специалист может легко понять, что некоторые реакции лучше всего осуществлять с обеспечением защиты других функциональных групп в молекуле, чтобы тем самым избежать нежелательных побочных реакций и/или повысить выход реакции. Чтобы получить такие повышенные выходы или избежать нежелательных реакций, специалист часто использует защитные группы. Эти реакции описаны в литературе, а также хорошо известны квалифицированному специалисту. Примеры многих таких преобразований можно найти, например, у T.Green, Protecting Groups in Organic Synthesis. Разумеется, используемые в качестве исходных материалов аминокислоты с реакционноспособными боковыми цепями предпочтительно блокируют (защищают) для предотвращения нежелательных побочных реакций. Соединения по настоящему изобретению могут иметь один или несколько хиральных центров. В результате, можно избирательно получать один оптический изомер (включая диастереомер и энантиомер) за другим, например, используя хиральные исходные материалы, катализаторы и растворители, или можно получать оба стереоизомера или оба оптических изомера (включая диастереомеры и энантиомеры) одновременно (рацемическая смесь). Поскольку соединения по настоящему изобретению могут существовать в виде рацемических смесей, смеси оптических изомеров (включая диастереомеры и энантиомеры) или стереоизомеров, могут быть разделены известными методами, такими, как метод хиральных солей, метод хиральной хроматографии и тому подобное. Кроме того, понятно, что один оптический изомер (включая диастереомер и энантиомер) или стереоизомер может иметь более подходящие свойства, чем другой. Поэтому со всей очевидностью подразумевается, что когда в описании и формуле изобретения раскрывается только рацемическая смесь, то раскрываются и заявляются также оба оптических изомера (включая диастереомеры и энантиомеры) или стереомера, по существу отдельных друг от друга. Способы применения Металлопротеазы (МП), обнаруженные в организме, участвуют в разрушении межклеточного матрикса, содержащего межклеточные белки и гликопротеины. Указанные белки и гликопротеины играют важную роль в поддержании размера, формы, структуры и стабильности ткани в теле. Ингибиторы металлопротеаз полезны для лечения заболеваний, вызываемых, по крайней мере частично, расщеплением таких белков. Известно, что МП близко вовлекаются в ремодулирование ткани. В результате такого действия они, как уже было сказано, принимают активное участие во многих нарушениях, влекущих за собой одно из двух: – разрушение ткани; в том числе дегенеративные заболевания, такие, как артрит, рассеянный склероз и тому подобное; метастазирование или изменчивость ткани в теле; – ремоделирование ткани, в том числе фиброзное заболевание, рубцевание, доброкачественную гиперплазию и тому подобное. Соединения по настоящему изобретению лечат нарушения, заболевания и/или нежелательные состояния, которые характеризуются нежелательной или повышенной активностью протеаз этого класса. Например, соединения могут быть использованы для ингибирования протеаз, которые – расщепляют структурные белки (т.е. белки, обеспечивающие стабильность и структуру ткани); – мешают межклеточной и внутриклеточной передаче сигналов, включая те, которые участвуют в регуляции цитокинов, и/или переработке цитокинов, и/или воспалении, разрушении ткани и других болезнях [Mohler KM, et al, Nature 370 (1994) 218-220, Gearing AJH, et al. Nature 370 (1994) 555-557, McGeehan GM, et al. Nature 370 (1994) 558-561], и/или содействуют процессам, которые являются нежелательными у субъекта, проходящего курс лечения, например процессам созревания спермы, оплодотворения яйцеклетки и т.п. При использовании в данном описании выражение “нарушение, связанное с МП” или “заболевание, связанное с МП”, означает участие нежелательной или повышенной активности МП в биологическом проявлении заболевания или нарушения, в биологическом каскаде, ведущем к нарушению или в виде симптома нарушения. Это “участие” МП включает: – нежелательную или повышенную активность как “причину” нарушения или биологического проявления независимо от того, обусловлена ли повышенная активность генетически, инфекцией, аутоиммунной реакцией, травмой, биомеханическими причинами, стилем жизни [например, ожирением] или какой-нибудь другой причиной; – МП как часть наблюдаемого проявления заболевания или нарушения. То есть степень заболевания или нарушения можно определять по повышенной активности МП или, с клинической точки зрения, нежелательные или повышенные уровни МП свидетельствуют о заболевании. МП не следует считать “отличительным признаком” заболевания или нарушения; нежелательная или повышенная активность МП является частью биохимического или клеточного каскада, что приводит к заболеванию или нарушению или связано с ним. В этом отношении ингибирование активности МП прерывает каскад и тем самым регулирует заболевание. Хорошо то, что многие МП распределены по телу неравномерно. Поэтому МП в тканях с ярко выраженной их локализацией часто специфичны к этим тканям. Например, распределение металлопротеаз, вовлеченных в процесс разрушения тканей в суставах, отличается от распределения металлопротеаз, найденных в других тканях. Поэтому, хотя это и несущественно в отношении активности или эффективности, некоторые нарушения предпочтительно лечат соединениями, которые действуют на специфические МП, найденные в пораженных тканях или участках тела. Например, соединение, проявляющее более высокую степень аффинитета и ингибирования по отношению к МП, найденной в суставах (например, хондроцитах), было бы предпочтительным для лечения заболевания, обнаруженного там же, в сравнении с другими соединениями, которые являются менее специфическими. Кроме того, некоторые ингибиторы более биологически доступны для определенных тканей, чем другие, и потому правильный выбор ингибитора с избирательностью, описанной выше, обеспечивает специфическое лечение нарушения, заболевания или нежелательного состояния. Например, соединения по настоящему изобретению различны по способности проникать в центральную нервную систему. Поэтому соединения могут быть выбраны с возможностью обеспечения эффектов через посредство МП, найденных определенно вне центральной нервной системы. Определение специфичности ингибитора определенной МП находится в пределах компетенции специалиста в данной области. Подходящие условия анализа можно найти в литературе. Известны, в частности, методы анализа для стромелизина и коллагеназы. Например, в патенте США N 4743587 имеется ссылка на методику Cawston, et al., Anal. Biochem. (1979) 99:340-345. Применение синтетического субстрата в анализе описано у Weingarten, Н. et, al., Biochem. Biophy. Res. Comm. (1984) 139:1184-1187. Можно, разумеется, использовать любой стандартный метод анализа расщепления структурных белков металлопротеазами. Способность соединений по настоящему изобретению ингибировать активность металлопротеаз можно, конечно, проверить путем анализов методами, найденными в литературе, или их вариантами. Для подтверждения ингибирующей активности соединений по настоящему изобретению можно использовать выделенные металлопротеазы или неочищенные экстракты, содержащие ряд ферментов, способных разрушать ткани. Благодаря их ингибирующему действию на МП соединения по настоящему изобретению полезны также для лечения следующих далее нарушений, обусловленных активностью металлопротеаз. Соединения по настоящему изобретению полезны также для профилактики или экстренного лечения. Их вводит по потребности любым способом специалист в области медицины или фармакологии. Для квалифицированного специалиста совершенно очевидно, что предпочтительные способы введения зависят от состояния заболевания, которое нужно лечить, и выбранной лекарственной формы. Предпочтительные способы системного введения включают пероральное или парентеральное введение. Однако специалист легко поймет преимущество введения ингибитора МП непосредственно в пораженную зону для многих нарушений. Например, целесообразном вводить ингибиторы МП непосредственно в зону поражения, такую, как зона хирургической травмы (например, пластической операции на сосудах, т.е. ангиопластики), зона рубцевания или ожога (например, местно в кожу). Поскольку ремоделирование кости вовлекает в процесс металлопротеазы, соединение по настоящему изобретению полезно для предотвращения ослабления крепления протеза. Известно, что с течением времени протезы начинают болтаться, вызывать боль и могут привести к дальнейшему повреждению кости, что требует их замены. Потребность замены таких протезов возникает для суставных протезов (например, протезов бедренного, коленного и плечевого суставов), стоматологических протезов, включая зубные протезы, мосты и протезы, закрепленные на верхней и/или нижней челюсти. Металлопротеазы активны также в ремоделировании сердечно-сосудистой системы (например, в застойной сердечной недостаточности). Предполагалось, что одним из соображений проведения ангиопластики является более высокая, чем можно было ожидать, степень продолжительной недостаточности (чрезмерное по времени перекрытие) и что активность МП становится нежелательной или повышается в ответ на то, что может быть опознано организмом как “повреждение” базальной мембраны сосуда. Поэтому регуляция активности МП при таких показаниях как дилатированная кардиомиопатия, застойная сердечная недостаточность, атеросклероз, разрыв бляшки, реперфузионное повреждение, ишемия, хроническая обструктивная болезнь легких, рестеноз после ангиопластики и аневризма аорты, может увеличивать продолжительность успеха какого-либо другого лечения или может быть лечебной сама по себе. При уходе за кожей МП принимают участие в ремоделировании или “обновлении” кожи. Поэтому регуляция МП улучшает лечение состояний кожи, включая (но не ограничиваясь ими) удаление, регуляцию и предупреждение морщин и устранение поражения кожи ультрафиолетовыми лучами. Такое лечение включает профилактическое лечение или лечение до появления очевидных физиологических проявлений. Например, МП может быть применена для обработки перед облучением для предотвращения поражения ультрафиолетовыми лучами и/или во время или после облучения для предупреждения или сведения к минимуму поражения после облучения. Кроме того, МП принимают участие в нарушениях и заболеваниях кожи, связанных с анормальными тканями, являющимися результатом анормального обновления, в котором принимают участие металлопротеазы, таких, как врожденный буллезный эпидермолиз, псориаз, склеродермия и атопический дерматит. Соединения по настоящему изобретению полезны также для лечения последствий “обычного” повреждения кожи, включающего рубцевание или “сокращение” ткани, например, после ожогов. Ингибирование МП полезно также при хирургических операциях на коже для предотвращения рубцевания и способствования нормального роста ткани в случаях, например, реплантации конечности и рефракторной хирургической операции (лазерной ли или резательной). Кроме того, МП связаны с нарушениями, включающими неупорядоченное ремоделирование других тканей, таких, как кость, например при отосклерозе и/или остеопорозе, или (для конкретных органов) такими, как цирроз печени и пневмофиброз. Аналогичным образом, при таких заболеваниях, как рассеянный склероз, МП могут быть вовлечены в неупорядоченное моделирование гематоэнцефалического барьера и/или миелиновых оболочек нервной ткани. Поэтому регуляционная активность МП может быть использована стратегически при лечении, предупреждении и регулировании таких заболеваний. По-видимому, МП принимают также участие во многих инфекциях, включающих цитомегаловирусный [CMV, ЦМВ) ретинит, HIV (ВИЧ) и результирующий синдром AIDS (СПИД). МП могут также участвовать в избыточной васкуляризации, когда окружающие ткани нужно разрушить, чтобы дать возможность образоваться новым кровеносным сосудам, таким, как ангиофиброма и гемангиома. Поскольку МП расщепляют внеклеточный матрикс, можно предполагать, что ингибиторы указанных ферментов можно использовать как средства регулирования рождаемости, обеспечивающие, например, предотвращение овуляции, предотвращение проникновения спермы в и через внеклеточную среду яйцеклетки и имплантации оплодотворенной яйцеклетки и предотвращение созревания спермы. Кроме того, они полезны для предотвращения или прекращения преждевременных родов и родоразрешения. Так как МП участвуют в воспалительной реакции и в переработке цитокинов, соединения полезны также в качестве противовоспалительных средств для применения в лечении болезней с преимущественным воспалением, включающих воспалительное заболевание кишечника, болезнь Крона, неспецифический язвенный колит, панкреатит, дивертикулит, астму, или родственное заболевание легких, ревматоидный артрит, подагру и синдром Рейтера. Когда причиной заболевания является аутоиммунная реакция, иммунная реакция часто инициирует активность МП и цитокинов. Регуляция МП при лечении таких аутоиммунных нарушений является полезным методом лечения. Таким образом, ингибиторы МП могут быть использованы для лечения нарушений, включающих красную волчанку, анкилозирующий спондилит и аутоиммунный кератит. Иногда побочные эффекты терапии аутоиммунитета приводят к обострению других состояний, опосредованных МП, и тогда проводят также терапию МП ингибиторами, например, при фиброзе, вызванном терапией аутоиммунитета. Кроме того, для такого типа терапии подходят и другие фиброзные заболевания, включающие пневмонию, бронхит, эмфизему, кистозный фиброз, острый респираторный дистресс-синдром (в частности, острофазная реакция). Когда МП участвуют в нежелательном разрушении ткани внешними средствами, это можно лечить ингибиторами МП. Например, они эффективны как противоядие при укусе гремучей змеи, как противонарывное средство, при лечении аллергического воспаления, септицемии и шока. Кроме того, они полезны как антипаразитарные средства (например, при малярии) и антиинфекционные средства. Например, они, по-видимому, полезны для лечения вирусной инфекции, включая инфекцию, которая приводит к герпесу, “простуде” (например, риновирусной инфекции), менингиту, гепатиту, ВИЧ-инфекции и СПИДу. Предполагается, что МП ингибиторы полезны также для лечения болезни Альцгеймера, бокового амиотрофического склероза (ALS), мышечной дистрофии, диабетических осложнений, в частности включающих потерю жизнеспособности ткани, коагуляции, гомологичной болезни (“трансплантат против хозяина”), лейкоза, кахексии, анорексии, протеинурии и, вероятно, регуляции роста волос. Для некоторых заболеваний, состояний или нарушений ингибирование МП предположительно является предпочтительным способом лечения. Такие заболевания, состояния или нарушения включают артрит (в том числе остеоартрит и ревматоидный артрит), рак (в частности, предупреждение или купирование роста и метастаза опухоли), глазные нарушения (в частности, изъязвление роговицы, плохое заживление роговицы, дегенерация желтого пятна и птеригий) и заболевание десен (в частности, периодонтальное заболевание и гингивит). Соединениями, предпочтительными (но неограничиваемыми этим) для лечения артрита (включая остеоартрит и ревматоидный артрит), являются те соединения, которые избирательны по отношению к металлопротеазам и дезинтегрин-металлопротеазам. Соединениями, предпочтительными (но неограничиваемыми этим) для лечения рака (в частности, предотвращения или купирования роста и метастаза опухоли), являются те соединения, которые преимущественно ингибируют желатиназу или коллагеназу типа IV. Соединениями, предпочтительными (но неограничиваемыми этим), для лечения глазных нарушений (в частности, изъязвления роговицы, плохого заживления роговицы, дегенерации желтого пятна и птеригия), являются те соединения, которые широко ингибируют металлопротеазы. Эти соединения предпочтительно вводят местно, более предпочтительно в виде капель или геля. Соединениями, предпочтительными (но неограничиваемыми этим) для лечения заболевания десен (в частности, периодонтального заболевания и гингивита), являются те соединения, которые преимущественно ингибируют коллагеназы. Композиции Композиции по настоящему изобретению содержат: (а) безопасное и эффективное количество соединения формулы (I) и (b) фармацевтически приемлемый носитель. Как описано выше, известны многочисленные заболевания, опосредованные избыточной или нежелательной матриксразрушающей активностью металлопротеаз. Они включают метастаз опухоли, остеоартрит, ревматоидный артрит, воспаление кожи, язвы (в частности, роговицы), реакцию на инфекцию, периодонтит и тому подобное. Таким образом, соединения по настоящему изобретению полезны для терапии состояний с участием этой нежелательной активности. Предлагаемые соединения могут, таким образом, быть изготовлены в виде фармацевтических композиций для применения в лечении или профилактике указанных состояний. Используют стандартные технологии изготовления фармацевтических препаратов, такие, как описанные в Remington’s Pharmaceutical Sciences, Mack Publishing Company, Easton, Pa., самое последнее издание. “Безопасное и эффективное количество” соединения формулы (I) – это количество, которое является эффективным в ингибировании металлопротеаз в месте (ах) активности у млекопитающего субъекта без чрезмерных вредных побочных эффектов (таких, как токсичность, раздражение или аллергическая реакция) соизмеримо с разумным соотношением выгода/риск при использовании в соответствии с настоящим изобретением. Конкретное “безопасное и эффективное количество”, очевидно, будет колебаться в соответствии с такими факторами, как конкретное состояние, которое нужно лечить, физическое состояние больного, продолжительность лечения, характер совместной терапии (если она есть), конкретная используемая лекарственная форма, используемый носитель, растворимость в нем соединения формулы (I) и необходимая схема дозирования композиции. Кроме предлагаемого соединения, композиции по настоящему изобретению содержат фармацевтически приемлемый носитель. При использовании в данном описании термин “фармацевтически приемлемый носитель” означает один или несколько совместимых твердых или жидких наполнителей или веществ для изготовления капсул, подходящих для введения млекопитающему. Используемый здесь термин “совместимый” означает, что компоненты композиции способны смешиваться с предлагаемым соединением и друг с другом без всякого взаимодействия, которое могло бы существенно снизить фармацевтическую эффективность композиции при обычных условиях применения. Фармацевтически приемлемые носители должны, конечно, иметь достаточно высокую чистоту и достаточно низкую токсичность, чтобы сделать их пригодными для введения животному, предпочтительно млекопитающему, проходящему курс лечения. Некоторыми примерами веществ, которые могут служить в качестве фармацевтически приемлемых носителей или их компонентов, являются сахара, такие, как лактоза, глюкоза и сахароза; крахмалы, такие, как кукурузный и картофельный крахмалы; целлюлоза и ее производные, такие, как натрий-карбоксиметилцеллюлоза, этилцеллюлоза и метилцеллюлоза; порошкообразный трагакант; солод; желатин; тальк; твердые смазывающие вещества, такие, как стеариновая кислота и стеарат магния; сульфат кальция; растительные масла, такие, как арахисовое масло, хлопковое масло, сезамовое масло, оливковое масло, кукурузное масло и масло теоброма; полиолы, такие, как пропиленгликоль, глицерин, сорбит, маннит и полиэтиленгликоль; альгиновая кислота; эмульгаторы, такие, как ТВИНы; смачивающие вещества, такие, как лаурилсульфат натрия; красящие вещества; корригирующие вещества; средства, способствующие таблетированию; стабилизаторы; антиокислители; консерванты; апирогенная вода; изотонический солевой раствор и фосфатные буферные растворы. Выбор фармацевтически приемлемого носителя, используемого вместе с предлагаемым соединением, в основном определяется способом введения соединения. Если предлагаемое соединение должно быть введено путем инъекции, предпочтительным фармацевтически приемлемым носителем является стерильный физиологический солевой раствор (с совместимым с кровью суспендирующим агентом), pH которого доведен до примерно 7,4. В частности, фармацевтически приемлемые носители для системного введения включают сахара, крахмалы, целлюлозу и ее производные, солод, желатин, тальк, сульфат кальция, растительные масла, синтетические масла, полиолы, альгиновую кислоты, фосфатные буферные растворы, эмульгаторы, изотонический солевой раствор и апирогенную воду. Предпочтительные носители для парентерального введения включают полиэтиленгликоль, этилолеат, пирролидон, этанол и сезамовое масло. Фармацевтически приемлемый носитель в композициях для парентерального введения предпочтительно составляет по крайней мере около 90% массы всей композиции. Композиции по настоящему изобретению предпочтительно изготавливают в виде лекарственной формы. В данном описании термин “лекарственная форма” – это композиция по настоящему изобретению, содержащая соединение формулы (I) в количестве, подходящем, согласно медицинской практике, для введения животному, в частности млекопитающему, в виде единичной (разовой) дозы. Эти композиции предпочтительно содержат примерно от 5 до 1000 мг, предпочтительно от 10 до 500 мг и более предпочтительно от 10 до 300 мг соединения формулы (I). Композиции по настоящему изобретению могут быть получены в любой из множества форм, пригодной, например, для перорального, ректального, местного, назального или парентерального введения. В зависимости от желательного конкретного способа введения могут быть использованы самые разнообразные фармацевтически приемлемые носители, известные в данной области техники. Они включают твердые или жидкие наполнители, разбавители, гидротропы, поверхностно-активные вещества и вещества для капсулирования. Могут быть введены необязательные фармацевтически активные вещества, по существу не влияющие на ингибирующую активность соединения формулы (I). Количество носителя, используемого вместе с соединением формулы (I), достаточно для обеспечения практичного количества материала для введения на единичную дозу соединения формулы (I). Методы и составы для изготовления лекарственных форм, пригодных для способов по настоящему изобретению, описаны в следующих ссылочных материалах, которые все включены в данное описание путем ссылки: Modern Pharmaceutics, главы 9 и 10 (издатели Banker & Rhodes, 1979); Lieberman et al. Pharmaceutical Dosage Forms, Tablets (1981); и Ansel, Introduction to Pharmaceutical Dosage Forms: 2-ое издание (1976). Кроме предлагаемого соединения, композиция по настоящему изобретению содержит фармацевтически приемлемый носитель. При использовании в данном описании термин “фармацевтически приемлемый носитель” означает один или несколько совместимых твердых или жидких наполнителей или веществ для изготовления капсул, подходящих для введения человеку или низшему животному. Используемый здесь термин “совместимый” означает, что компоненты композиции способны смешиваться с предлагаемым соединением и друг с другом без всякого взаимодействия, которое могло бы существенно снизить фармацевтическую эффективность композиции при обычных условиях применения. Фармацевтически приемлемые носители должны, конечно, иметь достаточно высокую чистоту и достаточно низкую токсичность, чтобы сделать их пригодными для введения человеку или низшему животному, проходящему курс лечения. Некоторыми примерами веществ, которые могут служить в качестве фармацевтически приемлемых носителей или их компонентов, являются сахара, такие, как лактоза, глюкоза и сахароза; крахмалы, такие, как кукурузный и картофельный крахмалы; целлюлоза и ее производные, такие, как натрийкарбоксиметилцеллюлоза, этилцеллюлоза и метилцеллюлоза; порошкообразный трагакант; солод; желатин; тальк; твердые смазывающие вещества, такие, как стеариновая кислота и стеарат магния; сульфат кальция; растительные масла, такие, как арахисовое масло, хлопковое масло, сезамовое масло, оливковое масло, кукурузное масло и масло теоброма; полиолы, такие, как пропиленгликоль, глицерин, сорбит, маннит и полиэтиленгликоль; альгиновая кислота; эмульгаторы, такие, как ТВИНы; смачивающие вещества, такие, как лаурилсульфат натрия; красящие вещества; корригирующие вещества; средства, способствующие таблетированию; стабилизаторы; антиокислители; консерванты; апирогенная вода; изотонический солевой раствор и фосфатные буферные растворы. Выбор фармацевтически приемлемого носителя, используемого вместе с предлагаемым соединением, в основном определяется способом введения соединения. Если предлагаемое соединение должно быть введено путем инъекции, предпочтительным фармацевтически приемлемым носителем является стерильный физиологический солевой раствор вместе с совместимым с кровью суспендирующим агентом, pH которого доводят примерно до 7,4. Могут быть использованы различные пероральные лекарственные формы, включающие такие твердые формы, как таблетки, капсулы, гранулы и нерасфасованные порошки. Эти пероральные формы содержат безопасное и эффективное количество, обычно по крайней мере примерно 5%, а предпочтительно от примерно 25% до примерно 50%, соединения формулы (I). Таблетки могут быть прессованными, тритурационными, с энтеросолюбильным покрытием, с сахарным покрытием, с пленочным покрытием или многослойными и могут содержать подходящие связывающие вещества, смазывающие вещества, наполнители, разрыхляющие вещества, красящие вещества, корригирующие вещества, вещества, обеспечивающие сыпучесть, и вещества, способствующие растаиванию. Жидкие пероральные лекарственные формы включают водные растворы, эмульсии, суспензии, растворы и/или суспензии, полученные из нешипучих гранул, и шипучие препараты, полученные из шипучих гранул, и содержат подходящие растворители, консерванты, эмульгирующие вещества, суспендирующие вещества, разбавители, подслащиватели, тающие вещества, красящие вещества и корригенты. Фармацевтически приемлемый носитель, пригодный для приготовления лекарственных форм для перорального введения, хорошо известен в данной области техники. Таблетки обычно содержат традиционные фармацевтически совместимые вспомогательные вещества в виде инертных наполнителей, таких, как карбонат кальция, карбонат натрия, маннит, лактоза и целлюлоза; связывающих веществ, таких, как крахмал, желатин и сахароза; разрыхлителей, таких, как крахмал, альгиновая кислота и кроскармелоза; смазывающих веществ, таких, как стеарат магния, стеариновая кислота и тальк. Для улучшения характеристик сыпучести порошкообразной смеси могут быть использованы скользящие вещества, такие, как диоксид кремния. Для внешнего вида могут быть добавлены красящие вещества, такие, как красители FD&C. Подслащиватели и корригенты, такие, как аспартам, сахарин, ментол, мята перечная и фруктовые корригенты, являются полезными вспомогательными веществами для жевательных таблеток. Капсулы обычно содержат один или несколько твердых разбавителей, описанных выше. Выбор компонентов носителя зависит от второстепенных соображений, таких, как вкус, стоимость и стабильность при хранении, которые не имеют существенного значения для целей настоящего изобретения, и потому может быть легко сделан специалистом в этой области техники. Пероральные композиции включают также жидкие растворы, эмульсии, суспензии и тому подобное. Фармацевтически приемлемые носители, пригодные для получения таких композиций, хорошо известны в данной области техники. Типичные компоненты носителей для сиропов, эликсиров, эмульсий и суспензий включают этанол, глицерин, пропиленгликоль, полиэтиленгликоль, жидкую сахарозу, сорбит и воду. Для суспензии типичные суспендирующие вещества включают метилцеллюлозу, натрий-карбоксиметилцеллюлозу, AVICEL RC-591, трагакант и альгинат натрия, типичные смачивающие вещества включают лицитин и полисорбат 80 и типичные консерванты включают метилпарабен и бензоат натрия. Пероральные жидкие композиции могут также содержать один или несколько компонентов, таких, как подслащиватели, корригенты и красители, описанные выше. Такие композиции могут быть также покрыты традиционными способами и содержать покрытия, зависящие, от pH или времени, в результате чего соединение высвобождается в желудочно-кишечном тракте вблизи желаемого места применения или обладает пролонгированным действием. Такие лекарственные формы обычно включают (но не ограничиваются ими) один или несколько компонентов из группы, состоящей из ацетатфталата целлюлозы, поливинилацетатфталата, фталата гидроксипропилметилцеллюлозы, этилцеллюлозы, евдрагитовых покрытий, восков и шеллака. Композиции по настоящему изобретению могут, необязательно, включать другие активные лекарственные вещества. Другие композиции, полезные для обеспечения системной доставки предлагаемых соединений, включают лекарственные формы подъязычные, для введения в рот и для введения в нос. Такие композиции обычно содержат одно или больше растворимых наполнителей, таких, как сахароза, сорбит и маннит, и связывающие вещества, такие, как аравийская камедь, микрокристаллическая целлюлоза, карбоксиметилцеллюлоза и гидроксипропилметилцеллюлоза. Они могут также включать скользящие вещества, смазывающие вещества, подслащиватели, красители, антиоксиданты и корригенты, описанные выше. Композиции по настоящему изобретению можно также вводить субъекту местно, например, путем непосредственного наложения на эпидермальную или эпителиальную ткань субъекта или размазывания по ней или чрескожно с помощью пластыря. Такие композиции включают, например, лосьоны, кремы, растворы, гели и твердые вещества. Эти местные вещества предпочтительно содержат безопасное и эффективное количество, по крайней мере около 0,1%, и предпочтительно, примерно от 1 до 5% соединения формулы (I). Предпочтительные носители для местного введения остаются на коже в виде сплошной пленки и сопротивляются удалению при потовыделении или погружении в воду. Обычно носитель является по своей природе органическим и способен к диспергированию или растворению в нем соединения формулы (I). Носитель может включать фармацевтически приемлемые мягчители, эмульгаторы, загустители, растворители и им подобные вещества. Способы введения В соответствии с настоящим изобретением предлагаются способы лечения или предупреждения нарушений, связанных с чрезмерной или нежелательной активностью матриксных металлопротеаз у человека или животного путем введения ему безопасного и эффективного количества соединения формулы (I). При использовании в данном описании “нарушение, связанное с чрезмерной или нежелательной активностью матриксных металлопротеаз” представляет собой всякое нарушение, характеризуемое расщеплением белков. Способы, заявленные в настоящем изобретении, пригодны для лечения таких нарушений, как, например, остеоартрит, периодонтит, изъязвление роговицы, прорастание опухолью и ревматоидный артрит. Соединения формулы (I) и композиции по настоящему изобретению могут быть введены местно или системно. Системное введение включает всякий способ введения соединения формулы (I) в ткани тела, например внутрисуставное (в частности, при лечении ревматоидного артрита), внутриоболочечное, эпидуральное, внутримышечное, чреcкожное, внутривенное, внутрибрюшинное, подкожное, подъязычное, ректальное и пероральное введение. Предпочтительно соединения формулы (I) по настоящему изобретению вводят перорально. Конкретное дозирование вводимого ингибитора, а также продолжительность лечения, местного ли или системного, взаимозависимы. Схемы дозирования и лечения зависят также от таких факторов, как конкретно используемое соединение формулы (I), показание к лечению, способность соединения формулы (I) достигать минимальной ингибирующей концентрации в месте, где нужно ингибировать матриксную металлопротеазу, личные показатели субъекта (такие, как масса), способность соблюдения режима и схемы лечения и наличие и тяжесть побочных эффектов лечения. Обычно взрослому человеку (с массой тела примерно 70 кг) вводят за сутки при системном введении примерно от 5 до 3000 мг, предпочтительно примерно от 5 до 1000 мг, а более предпочтительно от 10 до 100 мг соединения формулы (I). Понятно, что эти пределы дозирования приведены лишь в качестве примера и что суточное введение можно регулировать в зависимости от перечисленных выше факторов. Предпочтительным способом введения для лечения ревматоидного артрита является пероральное введение или парентеральное введение путем внутрисуставной инъекции. Как известно из теории и практики медицины, все препараты для парентерального введения должны быть стерильными. Для млекопитающих, в частности людей (предполагаемая масса тела приблизительно 70 кг), предпочтительными являются индивидуальные дозы примерно от 10 до 1000 мг. Предпочтительным способом системного введения является пероральное введение. Предпочтительными являются индивидуальные дозы примерно от 10 до 1000 мг, предпочтительно от 10 до 300 мг. Местное введение можно использовать для системной доставки соединения формулы (I) или для локального лечения субъекта. Количество соединения формулы (I) для местного введения зависит от таких факторов, как чувствительность кожи, тип и положение участка ткани, подлежащего лечению, вводимая композиция и носитель (если он есть), конкретно вводимое соединение формулы (I), а также конкретное расстройство и требуемая степень распространения системных (в отличие от местных) эффектов. Ингибиторы по настоящему изобретению могут быть нацелены на конкретные места, где аккумулирована матриксная металлопротеаза, путем применения нацеливающих лигандов. Например, чтобы мишенью ингибиторов стала матриксная металлопротеаза, находящаяся в опухоли, ингибитор конъюгируют с антителом (или его фрагментом), которое является иммунореактивным по отношению к маркеру опухоли, как это обычно понимают в отношении получения иммунотоксинов. Нацеливающий лиганд может быть также лигандом, подходящим для рецептора, присутствующего на опухоли. Можно использовать любой нацеливающий лиганд, который специфично взаимодействует с маркером для предполагаемой ткани-мишени. Способы связывания соединения по настоящему изобретению с нацеливающим лигандом хорошо известны и подобны тем, что описаны ниже для связывания с носителем. Конъюгаты составляют и вводят так, как описано выше. Для локализованных состояний предпочтительным является местное введение. Например, для лечения изъязвленной роговицы можно использовать прямое нанесение на пораженный глаз препарата в виде глазных капель или аэрозоля. Для лечения роговицы соединения по настоящему изобретению могут быть также приготовлены в виде гелей или мазей или могут быть включены в коллагеновое или из гидрофильного полимера защитное приспособление. Вещества могут быть также введены в виде контактных линз или резервуара, или в виде субконъюнктивального препарата. Для лечения воспаления кожи соединение наносят локально в определенном месте в виде геля, пасты или мази. Режим лечения отражает, таким образом, природу состояния, и для любого выбранного режима существуют подходящие лекарственные формы. Разумеется, что во всех описанных выше случаях соединения по настоящему изобретению могут быть введены в отдельности или в смесях, и композиции могут дополнительно содержать другие лекарственные средства или наполнители в соответствии с показанием. Некоторые из соединений по настоящему изобретению ингибируют также бактериальные металлопротеазы, хотя обычно на более низком уровне, чем металлопротеазы млекопитающих. Некоторые бактериальные металлопротеазы, по-видимому, менее зависимы от стереохимии ингибитора, тогда как в отношении способности диастереомеров дезактивировать протеазы млекопитающих между ними найдены существенные различия. Таким образом, эта особенность активности может быть использована для установления различия между ферментами млекопитающих и ферментами бактерий. Получение и применение антител Соединения по настоящему изобретению могут быть использованы в схемах иммунизации для получения антисывороток, иммуноспецифичных по отношению к соединениям по настоящему изобретению. Поскольку предлагаемые соединения относительно малы, то является целесообразным связывать их с антигенно нейтральными носителями, такими, как традиционно используемые носители типа гемоцианина KLH или сывороточного альбумина. Для тех предлагаемых соединений, которые имеют карбоксильные функциональные группы, связывание с носителем может быть осуществлено способами, уже известными в данной области. Например, карбоксильный остаток может быть восстановлен до альдегида и присоединен к носителю через реакцию с аминогруппами в боковых цепях носителей на основе белков с необязательным последующим восстановлением образовавшейся иминной связи. Карбоксильный остаток может быть также подвергнут взаимодействию с боковыми аминогруппами с использованием конденсирующих агентов, таких, как дициклогексилкарбодиимид, или других карбодиимидных дегидратирующих агентов. Для связывания можно также использовать связывающие соединения; как гомобифункциональные, так гетеробифункциональные связывающие соединения производит компания Pierce Chemical, Rockford, III. Полученный иммуногенный комплекс можно затем инъецировать подходящим млекопитающим субъектам, таким, как мыши, кролики и тому подобное. Подходящие схемы включают многократную инъекцию иммуногена в присутствии адьювантов по программе, которая способствует продукции антител в сыворотке. Титры иммунной сыворотки можно легко измерить методами иммунологического анализа (теперь стандартными в данной области) с использованием соединений по настоящему изобретению в качестве антигенов. Полученные антисыворотки можно использовать непосредственно или можно получить моноклональные антитела путем сбора лимфоцитов периферической крови или селезенки иммунизированного животного и иммортализации клеток, продуцирующих антитела, с последующей идентификацией подходящих продуцентов антител с помощью стандартной методики иммунологического анализа. Поликлональные или моноклональные препараты можно затем использовать для контроля режимов лечения или профилактики с использованием соединений по настоящему изобретению. Подходящие пробы, такие, как пробы, полученные из крови, сыворотки, мочи или слюны, можно тестировать на присутствие введенного ингибитора в различные моменты в ходе лечения, пользуясь методами стандартного иммунологического анализа с использованием содержащих антитела препаратов по настоящему изобретению. Соединения по настоящему изобретению можно также соединять с метками, такими, как сцинтиграфические метки, например технеций 99 или I-131, с использованием стандартных методов связывания. Меченые соединения вводят субъектам для определения мест нахождения избыточных количеств одной или нескольких металлопротеаз in vivo. Способность ингибиторов избирательно связываться с металлопротеазой выгодна в данном случае тем, что позволяет составить карту распределения этих ферментов на месте (in situ). Эту методику можно также использовать в гистологических процедурах, а меченые соединения по настоящему изобретению можно использовать в сравнительных иммунологических анализах. Следующие далее примеры служат только для иллюстраций соединения, композиции и применения по настоящему изобретению. Получение соединений Соединения получают так, как показано на приведенной схеме II синтеза. 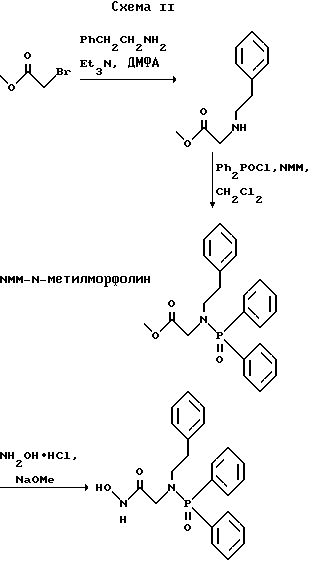 Пример 1. Метиловый эфир N-(2-фенил) глицина: Раствор фенилэтилаланина (6,63 мл, 52,8 ммоль) и триэтиламина (7,39 мл, 53 ммоль) в безводном N,N-диметилформамиде (80 мл) охлаждают до 0oC и к полученной смеси прибавляют по каплям раствор метилбромацетата (5 мл, 52,8 ммоль) в безводном N,N- диметилформамиде (40 мл). Реакционной смеси дают перемешиваться в течение 20 минут при 0oC. Смесь вливают в 250 мл этилацетата и промывают водой (3 раза), сушат над сульфатом натрия и выпаривают с получением бесцветного масла. Путем растворения неочищенного масла в 75 мл эфира получают гидрохлорид. Отдельно к 2,5 мл метанола при 0oC прибавляют по каплям 3,8 мл ацетилхлорида. Этот раствор прибавляют по каплям к эфирному раствору. Выпавшие в осадок твердые частицы собирают путем фильтрования с получением 9,2 г (76%) гидрохлорида метилового эфира N-(2-фенетил) глицина в виде бесцветного твердого вещества. Метиловый эфир N-(дифенилфосфинил)-N-(2-фенилэтил) глицина: Дифенилфосфинхлорид (0,42 мл, 2,2 ммоль) растворяют в дихлорметане (5 мл) и охлаждают до 0oC. К полученному раствору прибавляют раствор метилового эфира N-(2-фенетил)глицина (500 мг, 2,2 ммоль) и N-метилморфолина (0,73 мл, 6,6 ммоль) в дихлорметане (5 мл). Реакционную смесь перемешивают 16 часов при комнатной температуре, промывают водой и рассолом, сушат над сульфатом натрия и концентрируют с получением метилового эфира N-(дифенилфосфинил)-N- (2-фенилэтил) глицина в виде бесцветного твердого вещества. N-Гидрокси-2-[[дифенилфосфинил](2-фенилэтил)амино] ацетамид: Метиловый эфир N-(дифенилфосфинил)-N-(2-фенилэтил) глицина (160 мг, 0,41 ммоль) растворяют в метаноле (2,5 мл). К полученному раствору прибавляют гидроксиламингидрохлорид (57 мг, 0,81 ммоль) и затем 2 ммоль 25%-ного метанолового раствора метоксида натрия. Реакционную смесь перемешивают 16 часов, нейтрализуют 1 н. хлороводородной кислотой и концентрируют. Неочищенный продукт очищают путем флэш-хроматографии на силикагеле с получением 41,6мг (26%) N-гидрокси-2-[[дифенилфосфинил] (2- фенилэтил)амино]ацетамида в виде бесцветного твердого вещества: MC (IS): m/z 395 [М+Н)+, 417 [M+Na]+, 433 [M+K]+. (R1 = фенилэтил, R2 = H, R3 = фенил, R4 = фенил). Пример 2 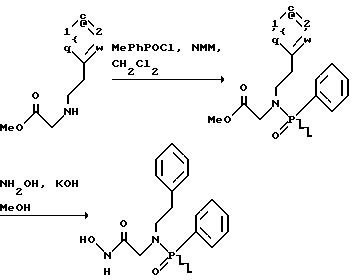 Метиловый эфир N-(метилфенилфосфинил)-N-(2-фенилэтил) глицина: Метилфенилфосфинхлорид (0,45 мл, 3,26 ммоль) растворяют в дихлорметане (5 мл) и охлаждают до 0oC. К полученному раствору прибавляют по каплям раствор 750 мг (3,27 ммоль) метилового эфира N-(2-фенетил)глицина и N-метилморфолина (1,1 мл, 10 ммоль) в дихлорметане (5 мл). Реакционную смесь перемешивают 1 час, подогревая до комнатной температуры. Смесь разбавляют этилацетатом, органическую фазу промывают водой и затем рассолом и сушат над безводным сульфатом натрия. Неочищенный продукт очищают путем флэш-хроматографии на силикагеле с получением метилового эфира N- (метилфенилфосфинил)-N-(2-фенилэтил) глицина в виде бесцветного масла. N-Гидрокси-2-[[метилфенилфосфинил](2-фенилэтил)амино] ацетамид: Метиловый эфир N-(метилфенилфосфинил)-N-(2-фенилэтил) глицина (300 мг, 0,905 ммоль) обрабатывают 0,77 мл NH2OK (1,76 М в метаноле, раствор получен так, как описано у Fieser and Fieser, том 1, стр. 478). Смесь перемешивают 3 часа при комнатной температуре, нейтрализуют муравьиной кислотой и концентрируют. Неочищенный продукт очищают путем флэш-хроматографии на силикагеле (этилацетат: этанол=85:15) с получением продукта с небольшими примесями. Препаративная ТСХ (этилацетат: этанол= 90: 10) дает N-гидрокси-2-[[метил-фенилфосфинил](2-фенилэтил)амино]ацетамид в виде бесцветного твердого вещества: МС (IS):m/z 333 [М+Н)+, 350 [M+NH4]+, 355 [M+Na]+, 371 [M+K]+. (R1 = фенилэтил, R2 = H, R3 = метил, R4 = фенил). Пример 3 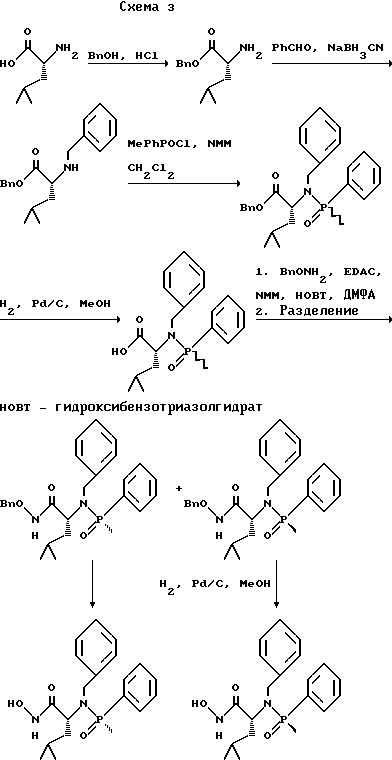 Бензиловый эфир D-лейцина: D-лейцин (10 г, 76,23 ммоль) суспендируют в бензиловом спирте (157 мл) и нагревают до 55oC. Через смесь барботируют газообразный хлороводород и реакционная смесь становится очень вязкой. Затем прибавляют 150 мл бензола при интенсивном перемешивании. Через 30 минут барботирования с нагревом смесь разжижается и перемешивание облегчается. Реакционную смесь продолжают перемешивать при 55oC в течение часа, пока раствор не становится гомогенным. Подачу хлороводорода прекращают и реакционной смеси дают перемешиваться при 55oC еще 30 минут. Дают реакционной смеси остыть до комнатной температуры и разбавляют ее этилацетатом. Продукт экстрагируют с использованием 1 М HCl (3 раза) и органику удаляют в отходы. Промывные воды объединяют, до pH 8 50%-ным водным раствором NaOH. Несколько раз экстрагируют амин этилацетатом. Органику сушат над сульфатом натрия и выпаривают. К полученному маслу прибавляют эфир и барботируют HCl с осаждением бензилового эфира D-лейцина в виде хлороводородной соли. Бензиловый эфир N-бензил-D-лейцина: Метиловый эфир D-лейцина (3 г, 11,66 ммоль) растворяют в метаноле и к полученному раствору прибавляют ацетат натрия (1,9 г, 23,3 ммоль) и затем бензальдегид (1,2 мл, 11,66 ммоль). Смеси дают перемешиваться в течение 10 минут, после чего прибавляют по каплям раствор цианборогидрида натрия (427 мг, 6,8 ммоль) в метаноле (4 мл). Реакционная смесь перемешивается 3 часа, после чего с помощью ТСХ определяют завершение реакции. К реакционной смеси прибавляют при перемешивании 10%-ный водный раствор NaHCO3. Затем удаляют летучие вещества и продукт экстрагируют эфиром (3 раза). Органику промывают водой (2 раза), сушат над сульфатом натрия и выпаривают с получением бензилового эфира N-бензил-D-лейцина в виде бесцветного масла. Бензиловый эфир N-(R/S-метилфенилфосфинил)-N-бензил-D- лейцина: Метилфенилфосфинхлорид (0,89 мл, 6,42 ммоль) растворяют в дихлорметане и охлаждают до 0oC. К полученному раствору прибавляют раствор метилового эфира N-бензил-D-лейцина (2 г, 6,42 ммоль) и N-метилморфолина (1,5 мл, 13,48 ммоль) в дихлорметане. Прибавляют каталитическое количество 4-диметиламинопиридина и дают реакционной смеси перемешиваться в течение 22 часов. Реакционную смесь концентрируют и к остатку прибавляют этилацетат. Полученную смесь промывают водой и рассолом, сушат над сульфатом натрия и концентрируют. Путем флэш-хроматографии на силикагеле (100%-ный этилацетат) выделяют диастереомеры. N-((R/S)-Метилфенилфосфинил)-N-бензил-D-лейцин: Колбу, содержащую бензиловый эфир N-(R/S-метилфенилфосфинил)-N-бензил-D- лейцина (2,04 г, 4,53 ммоль) и 10%-ный Pd/C (500 мг), вакуумируют и затем вводят в нее метанол. Создают среду водорода и дают реакционной смеси перемешиваться в течение 45 минут. Смесь фильтруют через целит и фильтрат собирают и концентрируют с получением N-((R/S)-метилфенилфосфинил)-N-бензил-D-лейцина в виде белого стекловидного вещества. N-Бензилокси-2-(R)-[[(R)-метилфенилфосфинил]бензиламино]-4 метилпентанамид и N-бензилокси-2-(R)-[[(S)-метилфенилфосфинил] бензиламино]-4-метилпентанамид: N-((R/S)-метилфенилфосфинил)-N-бензил-D-лейцин (1,5 г, 4,17 ммоль), растворяют в N-диметилформамиде и охлаждают до 0oC. К полученному раствору последовательно прибавляют гидроксибензотриазолгидрат (1,69 г, 12,5 ммоль), N-метилморфолин (1,37 мл, 12,5 ммоль) и 1-этил-З-(3- диметиламинопропил)карбодиимид (EDAC, 959 мг, 5 ммоль). После перемешивания в течение 10 минут, прибавляют О-бензилгидроксиламин гидрохлорид (666 мг, 4,17 ммоль) и дают реакционной смеси перемешиваться 3 часа с согреванием до комнатной температуры. ТСХ показывает наличие двух диастереомеров. Прибавляют к смеси воду и экстрагируют смесь этилацетатом. Органику объединяют промывают водой и рассолом, сушат над сульфатом натрия и концентрируют с получением масла. Затем выделяют диастереомеры путем флэш-хроматографии на силикагеле (гексан: этилацетат= 1: 1) с получением N-бензилокси-2-(R)-[[(R) -метилфенилфосфинил] бензиламино] -4-метилпентанамида: Rf= 0,25 (гексан:этилацетат=1:1); 31P ЯМР (CD3OD) d 43,89; и N-бензилокси-2-(R)-[[(S)-метилфенилфосфинил]бензиламино] -4-метилпентанамида: Rf=0,15 (гексан:этилацетат – 1:1). N-Гидрокси-2-(R)-[[(S)-метилфенилфосфинил]бензиламино]- 4-метилпентанамид: N-Бензилокси-2-(R)-[[(S)- метилфенилфосфинил]-бензиламино]-4-метилпентанамид (334 мг, 0,719 ммоль) и 80 мг 10%-ного Pd/C вакуумируют в колбе. Добавляют 10 мл метанола и вводят в колбу водород. Реакционной смеси дают перемешиваться при комнатной температуре в течение 2 часов, после чего путем ТСХ определяют, что реакция завершена. Смесь фильтруют через целит и фильтрат собирают и выпаривают с получением белого стекловидного твердого вещества. Продукт растворяют в этилацетате и к полученному раствору прибавляют по каплям гексан до выпадения продукта в осадок, в результате чего получают N-гидрокси-2-(R)- [[(S)-метилфенилфосфинил]бензиламино]-4-метил-пентанамид в виде бесцветного твердого вещества: MC (IS): m/z 375 [М+Н)+, 397 [M+Na]+, 413 [М+K]+. N-Гидрокси-2-(R)-[[(R)-метилфенилфосфинил] бензиламино] -4- метилпентанамид: N-Бензилокси-2-(R)-[[(R)-метилфенилфосфинил]- бензиламино]-4-метилпентанамид (460 мг, 0,99 ммоль) и 10%-ный Pd/C (100 мг) вакуумируют в колбе. Добавляют метанол (10 мл) и вводят в колбу водород. Реакционной смеси дают перемешиваться при комнатной температуре в течение 2 часов, после чего путем ТСХ определяют, что реакция завершена. Смесь фильтруют через целит и фильтрат собирают и выпаривают с получением белого стекловидного твердого вещества. Гидроксамовую кислоту кристаллизуют путем растворения в этилацетате и прибавления по каплям гексана, пока раствор не станет мутным. В результате получают N-гидрокси-2-(R)-[[(R)-метилфенилфосфинил] бензиламино]-4-метилпентанамид в виде белого кристаллического твердого вещества: МС (IS): m/z 375 [M+H)+, 397 [M+Na]+, 413 [М+К]+. (R2 = изобутил, R1 = бензил, R3 = метил, R4 = фенил). Пример 4 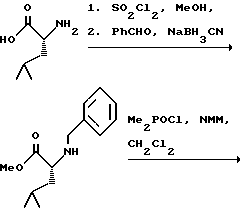 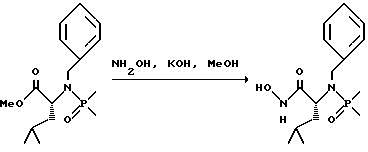 Гидрохлорид метилового эфира D-лейцина: D-лейцин (43,63 г, 333 ммоль) растворяют в метаноле (400 мл) и охлаждают до 0oC. К полученному раствору прибавляют по каплям тионилхлорид (25,5 мл, 350 ммоль). Реакционной смеси дают перемешиваться при комнатной температуре в течение 16 часов, после чего удаляют летучие вещества с получением не совсем белого твердого вещества. Продукт перекристаллизовывают из смеси этилацетат-метанол с получением гидрохлорида метилового эфира D-лейцина в виде рыхлого белого твердого вещества. Метиловый эфир N-бензил-D-лейцина: Гидрохлорид метилового эфира D-лейцина растворяют в метаноле. К раствору прибавляют ацетат натрия (39,4 г, 480 ммоль) и затем бензальдегид (19,8 мл, 195 ммоль). Полученную смесь перемешивают 15 минут и прибавляют к ней в течение 15 минут раствор цианборогидрида натрия (7,1 г, 113 ммоль) в метаноле (50 мл). Через 3 часа реакция заканчивается. Прибавляют при перемешивании 10%-ный водный раствор бикарбоната натрия и через 10 минут удаляют летучие вещества. Продукт экстрагируют эфиром и промывают водой (2 раза). Эфирную смесь сушат над сульфатом натрия и выпаривают с получением 39,9 г метилового эфира N-бензил-D-лейцина в виде бесцветного масла. Метиловый эфир N-(диметилфосфинил)-N-бензил-D-лейцина: Диметилфосфонхлорид (200 мг, 1,78 ммоль) растворяют в дихлорметане (5 мл) и охлаждают до 0oC. К полученному раствору прибавляют раствор метилового эфира N-бензил-D-лейцина (412 мг, 1,75 ммоль) и N-метилморфолина (0,44 мл, 4 ммоль) в дихлорметане (5 мл). Прибавляют каталитическое количество 4-диметиламинопиридина и дают реакционной смеси перемешиваться в течение 16 часов при комнатной температуре. После этого отфильтровывают твердые частицы и фильтрат собирают и выпаривают. Неочищенный продукт очищают путем флэш-хроматографии на силикагеле (этилацетат:метанол=96:4) с получением метилового эфира N-(диметилфосфинил)-N-бензил-D-лейцина в виде бесцветного твердого вещества. N-Гидрокси-2(R)-[[диметилфосфинил]бензиламино]-4-метил-пентанамид: Метиловый эфир N-(диметилфосфинил)-N-бензил-D-лейцина (158 мг, 0,51 ммоль) обрабатывают раствором NH2OK (2,8 мл, 1,76 М в метаноле), полученным так, как описано у Fieser and Fieser, том 1, стр. 478. Дают реакционной смеси перемешиваться в течение 3 часов при комнатной температуре, после чего путем ТСХ определяют, что реакция закончилась. Реакционную смесь нейтрализуют 1 М водным раствором HCl, удаляют летучие вещества до получения масла. Прибавляют метанол и затем воду по каплям, пока раствор не станет мутным. Собирают путем фильтрования кристаллы, что дает N-гидрокси-2(R)-[[диметилфосфинил]бензиламино]-4-метилпентанамид в виде бесцветного твердого вещества: МС (IS): m/z 313 [М+Н]+, 335 [M+Na]+, 357 [M+K]+. (R2 = изобутил, R1 = бензил, R3 = метил, R4 = фенил). Пример 5 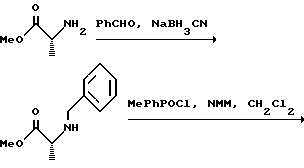 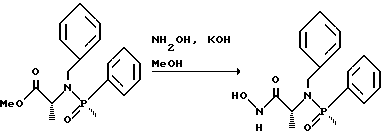 Метиловый эфир N-бензил-D-аланина: Метиловый эфир D-аланина (4 г, 28,66 ммоль) растворяют в метаноле (100 мл). К раствору прибавляют ацетат натрия (5,88 г, 71,65 ммоль) и бензальдегид (2,9 мл, 28,66 ммоль). Полученную смесь перемешивают 15 минут и прибавляют к ней по каплям раствор цианборогидрида натрия (1,08 г, 17,2 ммоль) в метаноле (5 мл). После перемешивания в течение 2 часов выпаривают при пониженном давлении метанол и продукт экстрагируют эфиром и промывают водой (2 раза). Неочищенный продукт очищают путем флэш-хроматографии на силикагеле (гексан-этилацетат=8:2) с получением 3,3 г метилового эфира N-бензил-D-аланина в виде масла. Метиловый эфир N-((R)-метилфенилфосфинил)-N-бензил-D-алакина: Метилфенилфосфинхлорид (361 мг, 2,07 ммоль) растворяют в дихлорметане (2,5 мл) и охлаждают до 0oC. К полученному раствору прибавляют раствор метилового эфира N-бензил-D-аланина (400 мг, 2,07 ммоль) и N-метилморфолина (0,51 мл, 4,6 ммоль) в дихлорметане (2,5 мл). К перемешиваемой смеси прибавляют каталитическое количество 4-диметиламинопиридина. Реакционную смесь перемешивают в течение 16 часов при комнатной температуре, промывают водой и рассолом, сушат над сульфатом натрия и концентрируют. Неочищенный продукт очищают путем флэш-хроматографии на силикагеле (100%-ный этилацетат) с получением метилового эфира N-((R)-метилфенилфосфинил)-N-бензил-D- аланина в виде масла. N-Гидрокси-2(R)-[[(R)-метилфенилфосфинил]бензиламино] пропионамид: Метиловый эфир N-((R)-метилфенилфосфинил)-N-бензил- D-аланина (181 мг, 0,55 ммоль) обрабатывают раствором NH2OK (2,2 мл, 1,76 М в метаноле), полученным так, как описано у Fieser and Fieser, том 1, стр. 478. Реакционную смесь перемешивают в течение 16 часов, после чего путем ТСХ определяют, что реакция закончилась. Реакционную смесь нейтрализуют 1 М водным раствором HCl и удаляют летучие вещества. Целевой продукт очищают на флэш-диоксиде кремния с использованием ТГФ в качестве элюента. Полученный остаток кристаллизуют путем растворения в этилацетате и прибавления гексана до помутнения раствора. В результате получают N-гидрокси-2(R)-[[(R)-метилфенилфосфинил]бензиламино] пропионамид в виде твердых и плотных бесцветных кристаллов: MC (IS): m/z 333 [М+H]+, 355 [M+Na]+. (R1 = бензил, R2 = метил, R3 = фенил, R4 = фенил). Пример 6 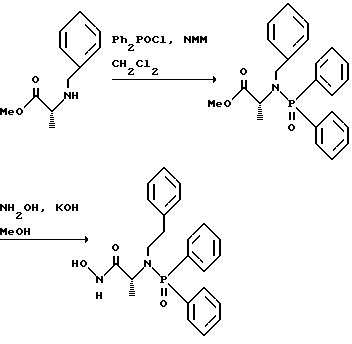 Метиловый эфир N-(дифенилфосфинил)-Н-бензил-D-аланина: Дифенилфосфинхлорид (0,2 мл) растворяют в дихлорметане (5 мл) и охлаждают до 0oC. К полученному раствору прибавляют раствор метилового эфира N-бензил-D-аланина (243 мг, 1,26 ммоль) и триэтиламина (0,39 мл, 2,8 ммоль) в дихлорметане (2,5 мл). К реакционной смеси прибавляют каталитическое количество 4-диметиламинопиридина. Реакционную смесь перемешивают в течение 48 часов при комнатной температуре. Дихлорметановый раствор разбавляют еще 20 мл дихлорметана и затем промывают 1 М водным раствором HCl (2 раза). Продукт очищают путем флэш-хроматографии на силикагеле (этилацетат: гексан=8:2) с получением метилового эфира N-(дифенилфосфинил)-N-бензил-D-аланина в виде масла. N-Гидрокси-2(R)-[[дифенилфосфинил]бензиламино] пропионамид: К метиловому эфиру N-(дифенилфосфинил)-N-бензил-D- аланина (100 мг, 0,25 ммоль) прибавляют раствор NH2OK (0,88 мл, 1,76 М в метаноле), полученный так, как описано у Fieser and Fieser, том 1, стр. 478. Реакционную смесь перемешивают в течение 16 часов, после чего путем ТСХ определяют, что реакция закончилась. Реакционную смесь нейтрализуют 1 М водным раствором HCl и удаляют летучие вещества. Продукт очищают путем флэш-хроматографии на силикагеле (100%-ный этилацетат) с получением N-гидрокси-2(R)-[[дифенилфосфинил] бензиламино]пропионамида в виде масла: МС (IS): m/z 395 [М+H]+, 417 [M+Na]+. (R1 = бензил, R2 = метил, R3 = фенил, R4 = фенил). Пример 7 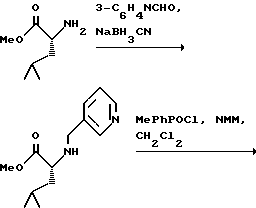 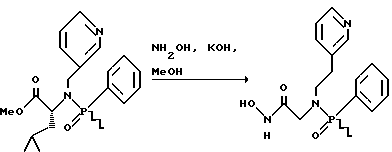 Метиловый эфир N-(3-пиколил)-D-лейцина: Гидрохлорид метилового эфира D-лейцина (20 г, 110,43 ммоль) растворяют в метаноле. К полученному раствору прибавляют ацетат натрия (22,64 г, 276 ммоль) и затем 3-пиридинкарбоксальдегид (10,9 мл, 115,5 ммоль). Дают смеси перемешиваться при комнатной температуре в течение 15 минут и затем медленно прибавляют в течение 15 минут цианборогидрид натрия (4,15 г, 66 ммоль). После перемешивания в течение 16 часов при комнатной температуре метанол выпаривают при пониженном давлении и полученное масло растворяют в этилацетате и промывают водой (2 раза). Органику сушат над сульфатом натрия и концентрируют до масла. Продукт очищают путем флэш-хроматографии на силикагеле (100%-ный этилацетат) с получением метилового эфира N-(3-пиколил)-D-лейцина в виде масла. N-Гидрокси-2(R)-[[(R/S)-метилфенилфосфинил] -3- пиколиламино] -4-метилпентанамид: Метилфенилфосфинхлорид (12,23 г, 70 ммоль) растворяют в дихлорметане (100 мл) и охлаждают до 0oC. К полученному раствору прибавляют раствор метилового эфира N-(3-пиколил)-D-лейцина (15,5 г, 65,6 ммоль) и N-метилморфолина (19,24 мл, 175 ммоль) в дихлорметане (100 мл). Прибавляют каталитическое количество 4-диметиламинопиридина и дают реакционной смеси перемешиваться в течение 16 часов при комнатной температуре. Прибавляют дополнительный метилфенилфосфинхлорид (2 г, 11,46 ммоль). Реакционная смесь продолжает перемешиваться в течение 24 часов, после чего реакция заканчивается. ТСХ не дает разделения диастереомеров. Продукт очищают путем флэш-хроматографии на силикагеле (этанол:этилацетат=5:95) с получением метилового эфира N-((R/S)-метилфенилфосфинил)-N-(3-пиколил)-D- лейцина в виде масла. К этому эфиру прибавляют раствор NH2OK (250 мл, 1,76 М в метаноле), полученный так, как описано у Fieser and Fieser, том 1, стр. 478. Реакционную смесь перемешивают в течение 16 часов, после чего путем ТСХ определяют, что реакция закончилась. Реакционную смесь нейтрализуют 1 М водным раствором НС1 и удаляют летучие вещества. Продукт очищают путем флэш-хроматографии на силикагеле (этанол: этилацетат= 10: 90) с получением 8,6 г N-гидрокси-2(R)-[[(R/S)-метилфенилфосфинил]-3- пиколиламино]-4-метилпентанамида в виде смеси диастереомеров (60R/40S):МС (IS): m/z 376 [М+H]+, 398 [M+Na]+, (R1 = 3-пиридилметил, R2= 2-изобутил, R3 = метил, R4 = фенил). Пример 8 Схема IV 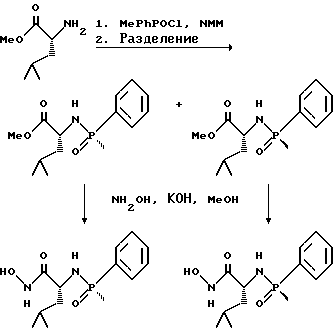 Метиловый эфир N-((R и S)-метилфенилфосфинил)-D-лейцина: Метилфенилфосфинхлорид (113 мг, 0,65 ммоль) растворяют в дихлорметане (5 мл) и охлаждают до 0oC. К полученному раствору прибавляют раствор гидрохлорида метилового эфира D-лейцина (100 мг, 0,55 ммоль) и N-метилморфолина (0,18 мл, 1,65 ммоль) в дихлорметане (3 мл). После перемешивания в течение 16 часов при комнатной температуре на пластинке для ТСХ наблюдают два пятна. Эти соединения разделяют путем флэш-хроматографии на силикагеле (этилацетат:метанол=95:5) с получением двух диастереомерных продуктов: метилового эфира N-((R)-метилфенилфосфинил)-D-лейцина, Rf= 0,25 (100%-ный этилацетат) и метилового эфира N-((S)-метилфенилфосфинил)-D-лейцина, Rf = 0,14 (100%-ный этилацетат). N-Гидрокси-2(R)-[[(R)-метилфенилфосфинил] амино] -4- метилпентанамид: Метиловый эфир N-((R)-метилфенилфосфинил)-D- лейцина (60 мг, 0,21 ммоль) обрабатывают раствором NH2OK (0,57 мл, 1,76 М в метаноле), полученным так, как описано у Fieser and Fieser, том 1, стр. 478. Реакционную смесь перемешивают в течение 7 часов, после чего путем ТСХ определяют, что реакция завершена. Реакционную смесь нейтрализуют 1 М водным раствором HCl и удаляют летучие вещества. Остаток очищают путем флэш-хроматографии на силикагеле (этилацетат: этанол = 95:5) с получением N-гидрокси-2(R)-[[(R)-метилфенилфосфинил]амино]-4- метилпентанамида в виде бесцветного твердого вещества: МС (IS): m/z 285 [М+H]+. N-Гидрокси-2(R)-[[(S)-метилфенилфосфинил] амино] -4-метилпентaнaмид: Метиловый эфир N-((S)-метилфенилфосфинил)-D-лейцина (55 мг, 0,19 ммоль) обрабатывают раствором NH2OK (0,57 мл, 1,76 М в метаноле), полученным так, как описано у Fieser and Fieser, том 1, стр. 478. Реакционную смесь перемешивают в течение 6 часов, после чего путем ТСХ определяют, что реакция завершена. Реакционную смесь нейтрализуют 1 М водным раствором HCl и удаляют летучие вещества. Остаток очищают путем флэш-хроматографии на силикагеле (этилацетат: этанол =80:20) с последующей кристаллизацией из смеси этилацетат-гексан и в результате получают N-гидрокси-2(R)-[[(S)-метилфенилфосфинил] амино] -4- метилпентанамида в виде бесцветного твердого вещества: МС (IS): m/z 285 [М+H]+. (R1 = H, R2 = изобутил, R3 = метил, R4 = фенил). Пример 9 Схема V 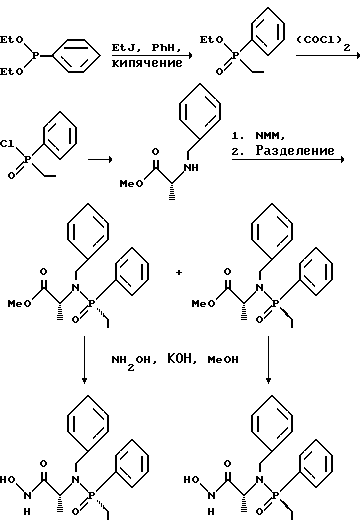 Этил этилфенилфосфинат: Смесь диэтилфенилфосфонита (4,5 22,70 ммоль), этилиодида (0,24 мл, 3 ммоль) и бензола (100 мл) перемешивают и нагревают при 85oC в течение 24 часов. ТСХ показывает, что реакция осуществлена на 30%. Прибавляют другую порцию этилиодида (0,30 мл, 3,75 ммоль) и реакционную смесь перемешивают еще 36 часов при 85oC, после чего ТСХ показывает, что реакция закончена. Удаляют на роторном испарителе летучие вещества, в результате чего получают этил этилфенилфосфинат в виде масла. Этилфенилфосфинхлорид: К раствору этил этилфенилфосфината (2 г, 10 ммоль) в бензоле (200 мл) прибавляют оксалилхлорид (1,3 мл, 15 ммоль). Смесь перемешивают 3 часа при комнатной температуре. На роторном испарителе удаляют летучие вещества и продукт сушат в вакууме в течение 12 часов с получением этилфенилфосфинхлорида в виде масла. Метиловый эфир N-((R и S)-этилфенилфосфинил)-N-бензил-D- аланина: К раствору этилфенилфосфинхлорида (1,04 г, 5,5 ммоль) в дихлорметане (15 мл) прибавляют раствор метилового эфира N-бензил-D-аланина (1,37 г, 7,1 ммоль) и N-метилморфолина (1,36 мл, 12,4 ммоль) в дихлорметане (15 мл). Добавляют каталитическое количество 4-диметиламинопиридина и реакционную смесь перемешивают 90 часов при комнатной температуре. На пластинке для ТСХ наблюдают два пятна. Эти соединения разделяют путем флэш-хроматографии на силикагеле (этилацетат:метанол=95:5) с получением двух диастереомерных продуктов: метилового эфира N-((S)-этилфенилфосфинил)-N-бензил-D-аланина, Rf = 0,25 (100%-ный этилацетат) и метилового эфира N-((R)-этилфенилфосфинил)-N- бензил-D-аланина, Rf = 0,35 (100%-ный этилацетат). N-Гидрокси-2(R)-[[(S)-этилфенилфосфинил] амино] пропионамид: Метиловый эфир N-((S)-этилфенилфосфинил)-N-бензил-D-аланина (105 мг, 0,30 ммоль) обрабатывают раствором NH2OK {1,0 мл, 1,76 М в метаноле), полученным так, как описано у Fieser and Fieser, том 1, стр. 478. Реакционную смесь перемешивают 16 часов, после чего путем ТСХ определяют, что реакция завершена. Реакционную смесь нейтрализуют 1 М водным раствором HCl и удаляют летучие вещества. Остаток очищают путем флэш-хроматографии на силикагеле (этилацетат: метанол = 95:5) с получением N-гидрокси-2(R)-[[(S)-этилфенилфосфинил]амино] пропионамида в виде бесцветного твердого вещества: МС (IS): m/z 347 [М+H]+ 369 [M+Na]+. N-Гидрокси-2(R)-[[(R)-этилфенилфосфинил] амино]пропионамид: Метиловый эфир N-((R)-этилфенилфосфинил)-N- бензил-D-аланина (333 мг, 0,96 ммоль) обрабатывают раствором NH2OK (3,3 мл, 1,76 М в метаноле), полученным так, как описано у Fieser and Fieser, том 1, стр. 478. Реакционную смесь перемешивают 16 часов, после чего путем ТСХ определяют, что реакция завершена. Реакционную смесь нейтрализуют 1 М водным раствором HCl и удаляют летучие вещества. Остаток очищают путем флэш-хроматографии на силикагеле (этилацетат: метанол = 95: 5) с получением 110 мг (33%) N-гидрокси-2(R)-[[(R)-этилфенилфосфинил] амино]пропионамида в виде бесцветного твердого вещества: МС (IS): m/z 347 [М+H]+, 369 [M+Na]+. (R1 = бензил, R2 = метил, R3 = этил, R4 = фенил). Пример 10 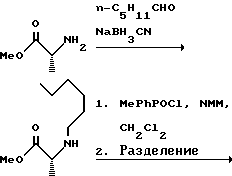 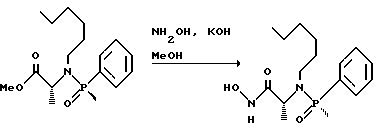 Метиловый эфир N-гексил-D-аланина: Метиловый эфир D-аланина (1,5 г, 10,75 ммоль) растворяют в 50 мл метанола и охлаждают до 0oC. К раствору прибавляют гексаналь (1,3 мл, 10,75 ммоль) и затем ацетат натрия (2,62 г, 32 ммоль). После перемешивания в течение 15 минут при 0oC прибавляют цианборогидрид натрия (440 мг, 7 ммоль) и смесь перемешивают еще 16 часов при комнатной температуре. Метанол выпаривают и полученный остаток растворяют в эфире и переносят в делительную воронку, промывают водой (2 раза), сушат над сульфатом натрия и выпаривают с получением 1,81 г метилового эфира N-гексил-D-аланина в виде бесцветного масла. Метиловый эфир N-((R)-метилфенилфосфинил)-N-гексил-D-аланина: Метилфенилфосфинхлорид (1,05 г, 6 ммоль) растворяют в дихлорметане (50 мл) и охлаждают до 0oC. К полученному раствору прибавляют раствор метилового эфира N-гексил-D-аланина (1 г, 5,34 ммоль) и триэтиламина (2,1 мл, 15 ммоль) в дихлорметане (10 мл). Прибавляют каталитическое количество 4-диметиламинопиридина и реакционную смесь перемешивают в течение 16 часов, промывают водой и рассолом, сушат над сульфатом натрия и концентрируют. Неочищенный продукт очищают путем флэш-хроматографии на силикагеле (100%-ный этилацетат) с получением метилового эфира N-((R)-метилфенил-фосфинил)-N-гексил-D-аланина в виде масла. N-Гидрокси-2(R)-[[(R)-метилфенилфосфинил]гексиламино] пропионамид: Метиловый эфир N-((R)-метилфенилфосфинил)-N-гексил-D- аланина (198 мг, 0,61 ммоль) обрабатывают раствором NH2OK (2 мл, 1,76 М в метаноле), полученным так, как описано у Fieser and Fieser, том 1, стр. 478. Реакционную смесь перемешивают в течение 16 часов, после чего путем ТСХ определяют, что реакция закончилась. Реакционную смесь нейтрализуют 1 М водным раствором HCl и удаляют летучие вещества. Неочищенный продукт очищают путем препаративной TCX (этилацетат: метанол= 95:5) с получением 110 мг N-гидрокси-2(R)-[[(R)-метилфенилфосфинил]гексиламино]пропионамида в виде бесцветного твердого вещества: МС (IS): m/z 327 [М+Н)+, 349 [M+Na]+. (R1 = 2-гексил, R2 = метил, R3 = метил, R4 = фенил). В табл. 1 представлены дополнительные примеры соединений, которые получают с использованием способов, описанных выше, и подходящих известных исходных материалов или исходных материалов, полученных известными способами: 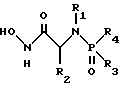 Указанные примеры являются для специалиста достаточным руководством для осуществления настоящего изобретения и ни в коей мере не ограничивают изобретение. Примеры композиций и способов применения Соединения по настоящему изобретению пригодны для изготовления композиций для лечения заболеваний и тому подобного. Следующие далее примеры композиций и способов не ограничивают изобретение, но дают специалисту указание, как получать и применять соединения, композиции и способы по настоящему изобретению. В каждом случае соединения формулы I могут быть заменены, например, соединением, показанным ниже, с аналогичными результатами. Примеры способов применения не ограничивают изобретение, а являются для специалиста руководством, как применять соединения, композиции и способы по настоящему изобретению. Квалифицированным практикующим врачам будет понятно, что примеры дают направление и могут быть изменены в зависимости от состояния и пациента. Пример A Композиция в виде таблеток для перорального введения имеет следующий состав, мг: Соединение примера 9 – 15 Лактоза – 120 Маисовый крахмал – 70 Тальк – 4 Стеарат магния – 1 Другие соединения, имеющие структуру, соответствующую формуле I, применяют, по существу, с аналогичными результатами. Женщину с массой тела 60 кг (132 фунта), страдающую ревматоидным артритом, лечили способом по настоящему изобретению. В частности, ей вводили таблетки перорально в течение 2 лет по схеме три таблетки в сутки. По окончании периода лечения больную исследовали и нашли, что воспаление уменьшилось и увеличилась подвижность без сопутствующей боли. Пример B Капсула для перорального введения в соответствии с настоящим изобретением имеет следующий состав, %/мм: Соединение примера 3 – 15 Полиэтиленгликоль – 85 Другие соединения, имеющие структуру, соответствующую формуле I, применяют по существу с аналогичными результатами. Мужчину с массой тела 90 кг (198 фунта), страдающего остеоартритом, лечили способом по настоящему изобретению. В частности, ему вводили в течение 5 лет ежесуточно капсулу, содержавшую 70 мг соединения примера 3. По окончании периода лечения больного исследовали с помощью ортоскопа и нашли, что прогрессирование эрозии и фибрилляции суставного хряща отсутствует. Пример C Композиция на основе солевого раствора для локального введения в соответствии с настоящим изобретением имеет следующий состав, % м/м: Соединение примера 13 – 5 Поливиниловый спирт – 15 Солевой раствор – 80 Другие соединения, имеющие структуру, соответствующую формуле I, применяют, по существу, с аналогичными результатами. Больному с сильным изъязвлением роговицы вводили каплю препарата в каждый глаз два раза в сутки. Лечение было успешным без видимых осложнений. Пример D Композиция для местного введения в соответствии с настоящим изобретением, имеет следующий состав, % м/м: Соединение примера 3 – 0,20 Хлорид бензалкония – 0,02 Тимеросал – 0,002 d-Сорбит – 5,00 Глицин – 0,35 Ароматизаторы – 0,075 Очищенная вода – По потреб. Итого: – 100,00 Любые другие соединения, структуру, соответствующую формуле I, применяют по существу с аналогичными результатами. Больному с химическими ожогами наносили композицию при каждой перевязке (дважды в сутки). Образование рубцов существенно уменьшилось. Пример E Аэрозольная композиция для ингаляции в соответствии с настоящим изобретением имеет следующий состав, % м/м: Соединение примера 2 – 5,0 Спирт – 33,0 Аскорбиновая кислота – 0,1 Ментол – 0,1 Натрий-сахарин – 0,2 Газ-вытеснитель (F12, F114) – По потреб. Итого: – 100,00 Любые другие соединения, имеющие структуру, соответствующую формуле I, применяют, по существу, с аналогичными результатами. Больной астмой с помощью ингалятора впрыскивал себе 0,01 мл препарата в рот при вдыхании. Симптомы астмы ослаблялись. Пример F Глазная композиция для местного применения в соответствии с настоящим изобретением имеет следующий состав, % м/м: Соединение примера 5 – 0,10 Хлорид бензалкония – 0,01 ЭДТА – 0,05 Гидроксиэтилцеллюлоза (NATROSOL М) – 0,50 Метабисульфит натрия – 0,10 Хлорид натрия (0,9%) – По потреб. Итого: – 100,0 Любые другие соединения, имеющие структуру, соответствующую формуле I, применяют, по существу, с аналогичными результатами. Мужчину с массой тела 90 кг (198 фунтов) с изъязвлениями роговицы лечили способом по настоящему изобретению. В частности, в его пораженный глаз вводили дважды в день в течение 2 месяцев солевой раствор, содержавший 10 мг соединения примера 5. Пример G Композиция для парентерального введения имеет следующий состав: Компонент – Количество Пример 4 – 100 мг/мл носителя Носитель: Натрийцитратный буфер с (процент по массе носителя): – 0,48 лецитином – 0,48 карбоксиметилцеллюлозой – 0,53 повидоном – 0,50 метилпарабеном – 0,11 пропилпарабеном – 0,011 Указанные компоненты смешивают с образованием суспензии. Приблизительно 2,0 мл суспензии вводят путем инъекции человеку с предметастатической опухолью. Место инъекции – рядом с опухолью. Указанную дозу вводят дважды в сутки в течение примерно 30 дней. Через 30 дней симптомы болезни ослабляются и дозу постепенно уменьшают до поддерживающего состояние больного уровня. Другие соединения, имеющие структуру, соответствующую формуле I, применяют по существу с аналогичными результатами. Пример H Композиция для полоскания рта, % м/о: Соединение примера 1 – 3,00 Спирт SDA 40 – 8,00 Корригент – 0,08 Эмульгатор – 0,08 Фторид натрия – 0,05 Глицерин – 10,00 Подслащиватель – 0,02 Бензойная кислота – 0,05 Гидроксид натрия – 0,20 Краситель – 0,04 Вода – До 100 Пациент с больными деснами применяет 1 мл жидкости для полоскания рта трижды в сутки для предотвращения дальнейшей внутриротовой дегенерации. Другие соединения, имеющие структуру, соответствующую формуле I, применяют, по существу, с аналогичными результатами. Пример 1 Композиция в виде пастилки, % м/о: Соединение примера 3 – 0,01 Сорбит – 17,50 Маннит – 17,50 Крахмал – 13,60 Подслащиватель – 1,20 Корригент – 11,70 Краситель – 0,10 Кукурузный сироп – До 100 Пациент применяет пастилку для предотвращения расшатывания имплантата в верхней челюсти. Другие соединения, имеющие структуру, соответствующую формуле I, применяют по существу с аналогичными результатами. Пример J Композиция в виде жевательной резинки, % м/о: Соединение примера 1 – 0,03 Кристаллы сорбита – 38,44 Основа в виде камеди Paloja-T – 20,00 Сорбит (70%-ный водный раствор) – 22,00 Маннит – 10,00 Глицерин – 7,56 Корригент – 1,00 Пациент жует резинку для предотвращения расшатывания зубов. Другие соединения, имеющие структуру, соответствующую формуле I, применяют, по существу, с аналогичными результатами. Пример К Компоненты, % м/о: Вода USP (фармацевтической чистоты согласно Фармакопеи США) – 54,656 Метилпарабен – 0,05 Пропилпарабен – 0,01 Ксантановая камедь – 0,12 Гуаровая камедь – 0,09 Карбонат кальция – 12,38 Противовспениватель – 1,27 Сахароза – 15,0 Сорбит – 11,0 Глицерин – 5,0 Бензиловый спирт – 0,2 Лимонная кислота – 0,15 Холодящее вещество – 0,00888 Корригент – 0,0645 Краситель – 0,0014 Композицию получают путем первоначального смешивания 80 кг глицерина и всего бензилового спирта и нагревания до 65oC, после чего медленно добавляют и смешивают метилпарабен, пропилпарабен, воду, ксантановую камедь и гуаровую камедь. Смешивают эти компоненты в течение примерно 12 минут с помощью соосной мешалки Сильверсона (Silverson). Затем медленно добавляют следующие компоненты в следующем порядке: оставшийся глицерин, сорбит, противовспениватель C, карбонат кальция, лимонную кислоту и сахарозу. Отдельно объединяют корригенты и красители и затем медленно добавляют их к другим компонентам. Смешивают в течение примерно 40 минут. Пациент принимает препарат для предотвращения резкого обострения колита. Все источники информации, упомянутые в данном описании, включены в него в виде ссылок. Были описаны конкретные варианты предлагаемого изобретения, но для специалистов в данной области очевидно, что в них могут быть внесены различные изменения и дополнения, не выходя за пределы сущности и объема изобретения. Предполагается, что прилагаемая формула изобретения охватывает все такие изменения и дополнения. ДАННЫЕ ПО БИОЛОГИЧЕСКОЙ АКТИВНОСТИ В табл. 2 приведены данные испытаний in vitro на биологическую активность соединений согласно изобретению. Результаты испытаний ингибирующего действия соединений на металлопротеиназы матрикса (ферменты ММР) даны в отношении следующих ферментов этого класса: ММР1 – коллагеназа 1 ММР2 – желатиназа A; ММР3 – стромелизин; ММР7 – матрилизин; ММР8 – нейтрофил-коллагеназа; ММР9 – желатиназа В; ММР13 – коллагеназа 3 крыс. Методика испытаний описана, в частности, в работе S. Pikul et al., J. Med.Chem., 1999, Vol. 42, No.1, pp. 87-94. Данные испытаний представлены в виде значений величины IC50, которая является общепринятым показателем ингибирующей активности. Формула изобретения
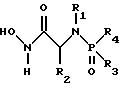 где R1 – водород, алкил, фенилалкил, пиридинил, пиридинилалкил, алкоксиалкил, фенилалкоксиалкил; R2 – водород, алкил, фенилалкил, индолил, фенилалкоксиалкил, алкилтиоалкил, алкиламиноалкил; R3 – алкил или фенил; R4 – алкил, фенил или замещенный фенил, пиридил, тиенил или фурил, их оптические изомеры, диастереомеры или энантиомеры, или их фармацевтически приемлемые соли, или биогидролизуемые сложные эфиры. 2. Соединения по п.1, общей формулы (I) 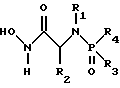 где R1 – водород, алкил, фенилалкил, пиридинил, алкоксиалкил, фенилалкоксиалкил; R2 – водород, алкил, фенилалкил, индолил, фенилалкоксиалкил, алкилтиоалкил; R3 – алкил или фенил; R4 – фенил, пиридил, тиенил или фурил, его стереоизомер или энантиомер, или его фармацевтически приемлемая соль, или биогидролизуемый сложный эфир. 3. Соединение по п.1 или 2, где R1 выбран из водорода, фенилалкила, алкила и пиридинила. 4. Соединение по пп.1-3, где R2 – водород или C1-C6 алкил, R4 – фенил или замещенный фенил. 5. Соединение по пп.1-4, где R4 – фенил, замещенный алкокси. 6. Соединение по пп.1-5, где R3 – C1-C6 низший алкил или фенил, R2 – водород, изобутил или метил. 7. Соединение по пп.1-6, где R1 выбран из фенила, фенилэтила, бензила, пиридинилметила или метила. 8. Соединение по любому из предшествующих пунктов, которое выбрано из N – гидрокси-2-[[дифенилфосфинил] (2-фенилэтил)амино] – ацетамида; N – гидрокси-2-[[метилфенилфосфинил] (2-фенилэтил)амино] – ацетамида; N – гидрокси-2(R)-[[(R)-метилфенилфосфинил] бензиламино] -4-метилпентанамида; N – гидрокси-2(R) – [[диметилфосфинил]бензиламино]-4-метилпентанамида; N – гидрокси-2(R)-[[(R)-метилфенилфосфинил]бензиламино]-пропионамида; N – гидрокси-2(R) – [[дифенилфосфинил] бензиламино] пропионамида; N – гидрокси-2(R)-[[(R/S)-метилфенилфосфинил] -3-пиколиламино] -4-метилпентанамида; N – гидрокси-2(R)-[[(S)-метилфенилфосфинил] -амино] -4-метил-пентанамида; N – гидрокси-2(R)-[[(R)-этилфенилфосфинил] амино] пропионамида или N – гидрокси-2(R)-[[(R)-метилфенилфосфинил]-гексиламино]-пропионамида. 9. Соединение по любому из пп.1-8, применимое для получения фармацевтической композиции. 10. Способ предупреждения или лечения заболевания, связанного с нежелательной активностью металлопротеазы у млекопитающего, включающий введение млекопитающему безопасного и эффективного количества соединения по любому из предшествующих пунктов. РИСУНКИ
MM4A Досрочное прекращение действия патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 23.08.2002
Извещение опубликовано: 20.11.2004 БИ: 32/2004
|
||||||||||||||||||||||||||