Патент на изобретение №2170230
|
||||||||||||||||||||||||||
(54) НОВЫЕ ПРОИЗВОДНЫЕ ПИРАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
(57) Реферат: Изобретение относится к производным пиразола формулы I, где 1 означает группу – NRR1R2 или группу -OR2, g2 – g6 одинаковые или разные и независимо друг от друга означают водород, галоген, С1-4 алкил, С1-4алкоксил, трифторметил или C1-4 алкилтиогруппу; w2-w6 одинаковые или разные и независимо друг от друга означают водород, галоген, С1-4алкил, C1-4алкоксил или трифторметил, при условии, что, по крайней мере, один из заместителей g2-g6 и один из заместителей w2-w6 отличаются от водорода; R1 означает водород или С1-4алкил; R2 – неароматический С3-15-карбоциклический радикал, незамещенный или одно- или многократно замещенный С1-4алкилом; R3 – водород или группа CH2-R6; R4 и R5 каждый независимо друг от друга означают водород или С1-4 алкил; или R4 означает водород и R5 и w6 вместе образуют этиленовый радикал; R6 означает водород, или когда заместители g2 g3, g4, g5 и/или g6 отличаются от С1-4 алкила, R6 означает водород, С1-4 алкил или С1-5 алкоксил, и их солям. Объектами изобретения также являются способ получения производных пиразола формулы I и фармацевтическая композиция их содержащая. Технический результат – получение новых соединений, которые являются антагонистами рецепторов СВ2 и фармацевтической композиции, имеющей сродство к рецепторам СВ2. Они могут быть использованы для лечения ауотоиммунных заболеваний. 3 с. и 14 з.п.ф-лы, 2 табл. 
Изобретение относится к новым производным пиразола и их возможным солям, способу их получения и содержащим их фармацевтическим композициям. Особенно настоящее изобретение относится к новым производным пиразола, обладающим высоким сродством к периферическим рецепторам каннабиноидов, называемых рецепторами CB2 и пригодны в терапевтических областях, где содержатся рецепторы CB2.  9-THC является основным активным компонентом Cannabis Sativa (Tuner, 1985; в Marijuana, 1984; изд. Harvey, DY, IRL Press, Oxford).
Характеризация рецепторов каннабиноидов возможна за счет включения синтетических лигандов, таких как агонисты WIN-55212-2 (J.Pharmacol. Exp. Ther. , 264, 1352-1363 (1993)) или CP-55940 (J.Pharmacol. Exp. Ther., 247, 1046-1051 (1988)).
В многочисленных статьях описываются не только психотропные эффекты каннабиноидов, но и также влияние этих последних на относящуюся к иммунитету функцию (Hollister L.E., Psychoact. Drugs, 24, 159-164 (1992)). Большинство исследований ин витро показывают иммуносупрессорные воздействия каннабиноидов: ингибирование пролиферативных ответных реакций T-лимфоцитов и B-лимфоцитов, индуцированных митогенами (Luo Y.D. и др. Int. J. Immunopharmacol., 14, 49-56 (1992); Schwartz Н. и др., J. Neuroimmunol., 55, 107-115 (1994)), ингибирование активности цитотоксических T-клеток (Klein и др., J. Toxicol. Environ. Health, 32, 465-477 (1991)), ингибирование бактерицидной активности макрофагов и синтеза 9-THC является основным активным компонентом Cannabis Sativa (Tuner, 1985; в Marijuana, 1984; изд. Harvey, DY, IRL Press, Oxford).
Характеризация рецепторов каннабиноидов возможна за счет включения синтетических лигандов, таких как агонисты WIN-55212-2 (J.Pharmacol. Exp. Ther. , 264, 1352-1363 (1993)) или CP-55940 (J.Pharmacol. Exp. Ther., 247, 1046-1051 (1988)).
В многочисленных статьях описываются не только психотропные эффекты каннабиноидов, но и также влияние этих последних на относящуюся к иммунитету функцию (Hollister L.E., Psychoact. Drugs, 24, 159-164 (1992)). Большинство исследований ин витро показывают иммуносупрессорные воздействия каннабиноидов: ингибирование пролиферативных ответных реакций T-лимфоцитов и B-лимфоцитов, индуцированных митогенами (Luo Y.D. и др. Int. J. Immunopharmacol., 14, 49-56 (1992); Schwartz Н. и др., J. Neuroimmunol., 55, 107-115 (1994)), ингибирование активности цитотоксических T-клеток (Klein и др., J. Toxicol. Environ. Health, 32, 465-477 (1991)), ингибирование бактерицидной активности макрофагов и синтеза  -ФНО (-фактора некроза опухоли) (Arata S. и др. Life Sci. , 49, 473-479 (1991); Fisher-Stenger и др., J. Pharm. Exp. Ther., 267, 1558-2565 (1993)), ингибирование цитолитической активности и продуцирования -ФНО некоторых лимфоцитов (Kusher и др., Cell. Immun., 154, 99-108 (1994)). Наоборот, в некоторых исследованиях наблюдают эффекты амплификации: повышение биологической активности интерлейкина-1 за счет гистиоцитов мыши или дифференцированных макрофагальных линий клеток вследствие повышенных уровней -ФНО (Zhu и др., J, Pharm. Exp. Ther., 270, 1334-1339 (1994); Shivers S.C. и др. Life Sci., 54, 1281-1289 (1994)).
Эффекты каннабиноидов возникают вследствие взаимодействия со специфическими рецепторами высокого сродства, присутствующими на центральном (Devane и др., Molecular Pharmacology, 34, 605-613 (1988)) и периферическом уровне (Nye и др. The Journal of Pharmacology and Experimental Theraupetics, 234, 784-791 (1985); Kaminski и др. Molecular Pharmacology, 42, 736-742 (1992); Munro и др., Nature, 365, 61-65 (1993)).
Центральные эффекты зависят от первого типа рецептора каннабиноидов (CB1), который находится в головном мозге. Кроме того, Munro и др., Nature, 365, 61-65 (1993) клонировали второй рецептор каннабиноидов, соединенный с протеинами G, называемый CB2, который присутствует только на периферии и в особенности в клетках иммунного происхождения. Наличие рецепторов каннабиноидов СВ2 в лимфоидных клетках может объяснять иммуномодуляцию, осуществляемую агонистами рецепторов каннабиноидов, как указано выше.
Многочисленные производные пиразола описаны в литературе: более конкретно, в заявке на европейский патент 268554 и в описании изобретения к выложенной акцептованной заявке на патент ФРГ 3910248 заявляются пиразолы, обладающие гербицидными свойствами; в заявке на европейский патент 430186 и в заявке на патент Японии 3031840 описываются соединения, пригодные для фотографии; и в заявке на европейский патент 418845 предметом изобретения являются пиразолы, обладающие противовоспалительной, анальгезирующей и антитромботической активностью.
Также описаны производные пиразолкарбоксамида, особенно в заявках на европейские патенты 0289879 и 0492125: эти соединения обладают инсектицидными свойствами.
Кроме того, в заявке на европейский патент 0477049 описываются производные пиразол-3-карбоксамида формулы (I); -ФНО (-фактора некроза опухоли) (Arata S. и др. Life Sci. , 49, 473-479 (1991); Fisher-Stenger и др., J. Pharm. Exp. Ther., 267, 1558-2565 (1993)), ингибирование цитолитической активности и продуцирования -ФНО некоторых лимфоцитов (Kusher и др., Cell. Immun., 154, 99-108 (1994)). Наоборот, в некоторых исследованиях наблюдают эффекты амплификации: повышение биологической активности интерлейкина-1 за счет гистиоцитов мыши или дифференцированных макрофагальных линий клеток вследствие повышенных уровней -ФНО (Zhu и др., J, Pharm. Exp. Ther., 270, 1334-1339 (1994); Shivers S.C. и др. Life Sci., 54, 1281-1289 (1994)).
Эффекты каннабиноидов возникают вследствие взаимодействия со специфическими рецепторами высокого сродства, присутствующими на центральном (Devane и др., Molecular Pharmacology, 34, 605-613 (1988)) и периферическом уровне (Nye и др. The Journal of Pharmacology and Experimental Theraupetics, 234, 784-791 (1985); Kaminski и др. Molecular Pharmacology, 42, 736-742 (1992); Munro и др., Nature, 365, 61-65 (1993)).
Центральные эффекты зависят от первого типа рецептора каннабиноидов (CB1), который находится в головном мозге. Кроме того, Munro и др., Nature, 365, 61-65 (1993) клонировали второй рецептор каннабиноидов, соединенный с протеинами G, называемый CB2, который присутствует только на периферии и в особенности в клетках иммунного происхождения. Наличие рецепторов каннабиноидов СВ2 в лимфоидных клетках может объяснять иммуномодуляцию, осуществляемую агонистами рецепторов каннабиноидов, как указано выше.
Многочисленные производные пиразола описаны в литературе: более конкретно, в заявке на европейский патент 268554 и в описании изобретения к выложенной акцептованной заявке на патент ФРГ 3910248 заявляются пиразолы, обладающие гербицидными свойствами; в заявке на европейский патент 430186 и в заявке на патент Японии 3031840 описываются соединения, пригодные для фотографии; и в заявке на европейский патент 418845 предметом изобретения являются пиразолы, обладающие противовоспалительной, анальгезирующей и антитромботической активностью.
Также описаны производные пиразолкарбоксамида, особенно в заявках на европейские патенты 0289879 и 0492125: эти соединения обладают инсектицидными свойствами.
Кроме того, в заявке на европейский патент 0477049 описываются производные пиразол-3-карбоксамида формулы (I);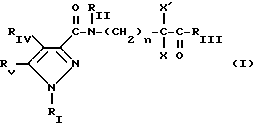 в которой, например, RI означает различным образом замещенную арильную группу; RII означает водород или алкил с 1-4 атомами углерода; RIII означает гидроксил, алкоксил с 1-6 атомами углерода, аминогруппу; RIV означает водород, галоген или алкил с 1-6 атомами углерода; RV означает различным образом замещенную фенильную группу; n означает 0, 1, 2 или 3. Эти соединения оказывают воздействие на центральную нервную систему и в особенности путем взаимодействия с рецептором нейротензина. Кроме того, в заявках на европейские патенты 576357 и 658546 описываются производные пиразола, обладающие сродством к рецепторам каннабиноидов. Сверх того, в заявке на европейский патент 656354 описывается N-пиперидино-5-(4-хлорфенил)-1-(2,4-дихлорфенил) -4-метилпиразол-3-карбоксамид или SR 141716 и его фармацевтически приемлемые соли, которые обладают высоким сродством к центральным рецепторам каннабиноидов. В настоящее время получены новые производные пиразола, которые обладают повышенным сродством к рецептору CB2 человека и специфичностью к вышеуказанному рецептору и которые являются сильнодействующими имммуномодуляторами. В настоящем описании под выражением “повышенное сродство к человеческому рецептору CB1>> понимают сродство, характеризующееся константой сродства обычно ниже 100 нМ, вплоть до 0,1 нМ, и термином “специфический” обозначают соединения, константа сродства которых к рецептору CB2 обычно, по крайней мере, в десять раз меньше константы сродства к рецептору CB1. Согласно одному из аспектов настоящего изобретения, предметом изобретения являются соединения формулы (I): 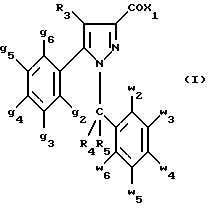 в которой X1 означает группу -NR1R2 или группу -OR2; g2, g3, g4, g5, g6 и w2, w3, w4, w5, w6 являются одинаковыми или разными, и каждый из них, независимо друг от друга, означает водород, атом галогена, алкил с 1-4 атомами углерода, алкоксил с 1-4 атомами углерода, трифторметил, нитрогруппу, алкилтиогруппу с 1-4 атомами углерода; при условии, что по крайней мере один из заместителей g2, g3, g4, g5, g6 и по крайней мере один из заместителей w2, w3, w4, w5, w6 отличаются от водорода; R1 означает водород или алкил с 1-4 атомами углерода; R2 означает неароматический карбоциклический радикал с 3-15 атомами углерода, незамещенный или одно- или многократно замещенный заместителем, выбираемым среди атома галогена, алкила с 1-4 атомами углерода или алкоксила с 1-4 атомами углерода; R3 означает водород или группу -CH2-R6; R4 и R5, каждый, независимо друг от друга, означают водород, алкил с 1-4 атомами углерода или трифторметил; или R4 означает водород и R5 и w6 вместе образуют этиленовый или триметиленовый радикал; R6 означает водород или, когда заместители g2, g3, g4, g5 и/или g6 являются другими, чем алкил с 1-4 атомами углерода, R6 означает водород, алкил с 1-4 атомами углерода, фтор, гидроксил, алкоксил с 1-5 атомами углерода, алкилтиогруппу с 1-5 атомами углерода, гидроксиалкоксил с 1-5 атомами углерода, цианогруппу, алкилсульфинил с 1-5 атомами углерода, алкилсульфонил с 1-5 атомами углерода; также как их возможные соли. Когда соединение формулы (I) согласно изобретению включает один или несколько асимметрических атомов углерода, различные оптические изомеры, также как рацематы, составляют часть изобретения. Возможные соли соединения формулы (I) включают соли присоединения фармацевтически приемлемых кислот, такие как гидрохлорид, гидробромид, сульфат, гидросульфат, дигидрофосфат, метансульфонат, метилсульфат, малат, оксалат, фумарат, нафталинсульфонат, глюконат, гликонат, цитрат, изоэтионат, п-толуолсульфонат, мезитиленсульфонат или бензолсульфонат. Неароматические карбоциклические радикалы с 3-15 атомами углерода включают моно- или полициклические, конденсированные, мостиковые или спирановые, насыщенные или ненасыщенные, при случае терпеновые радикалы. Эти радикалы незамещены или однократно или многократно замещены группой, выбираемой среди алкила с 1-4 атомами углерода, алкоксила с 1-4 атомами углерода или галогена, при условии, что в случае терпенов или терпеновых радикалов, как, например, борнил, ментил или ментенил, алкильные группы терпена не рассматриваются как заместители. Моноциклические радикалы включают циклоалкилы, как, например, циклопропил, циклопентил, циклогексил, циклогептил, циклооктил, циклододецил, которые незамещены или однократно или многократно замещены алкилом с 1-4 атомами углерода, алкоксилом с 1-4 атомами углерода или галогеном, как, например, 2-метилциклогекс-1-ил, 2,6- диметилциклогекс-1-ил, 2,2,6,6-тетраметилциклогекс-1-ил. Ди- или трициклические, конденсированные, мостиковые или спирановые, при случае терпеновые, радикалы включают, например, бицикло[2.2.1]гептил или норборнил, борнил, изоборнил, норадамантил, адамантил, бицикло[3.2.1]октил, бицикло[2.2.2] октил, трицикло[5.2.1.02,6] децил, спиро[5,5]-ундецил, бицикло[2.2.2]окт-2-ен-5-ил, трицикло[2.2.1.02,6]-гепт-3-ил, причем вышеуказанные радикалы незамещены или однократно или многократно замещены алкилом с 1-4 атомами углерода, галогеном или алкоксилом с 1-4 атомами углерода, как, например, 1,3,3- триметилбицикло[2.2.1]гепт-2-ил или фенхил. В настоящем описании алкильные или алкоксильные группы являются линейными или разветвленными. Под атомом галогена понимают атом хлора, брома, фтора или иода. Согласно настоящему изобретению, предпочтительны соединения формулы (I), в которой X1 означает группу -NR1R2; g2, g3, g4, g5, g6 и w2, w3, w4, w5, w6 являются одинаковыми или разными и каждый из них, независимо друг от друга, означает водород, атом галогена, алкил с 1-4 атомами углерода, алкоксил с 1-4 атомами углерода, трифторметил, нитрогруппу, алкилтиогруппу с 1-4 атомами углерода; при условии, что по крайней мере один из заместителей g2, g3, g4, g5, g6 и по крайней мере один из заместителей w2, w3, w4, w5, w6 являются отличными от водорода; R1 означает водород или алкил с 1-4 атомами углерода; R2 означает неароматический карбоциклический радикал с 3-15 атомами углерода, незамещенный или однократно или многократно замещенный заместителем, выбираемым среди атома галогена, алкила с 1-4 атомами углерода или алкоксила с 1-4 атомами углерода; R3 означает водород или группу -CH2-R6; R4 и R5, каждый, независимо друг от друга, означают водород, алкил с 1-4 атомами углерода или трифторметил; R6 означает водород, метил или этил; также как их возможные соли. Из соединений формулы (I), в которой X1 означает группу -NR1R2, предпочтительными являются такие, в которых R1 означает водород. Из соединений формулы (I), в которой X1 означает группу -NR1R2 или группу -OR2, предпочтительными являются такие, в которых R2 означает 1,3,3-триметилбицикло[2.2.1]гепт-2-ил или бицикло [3.2.1] окт-3-ил. Из соединений формулы (I) предпочтительными являются такие, в которых R3 означает водород или группу -CH2-R6, в которой R6 означает водород. Из соединений формулы (I) предпочтительными являются такие, в которых либо каждый из R3 и R5 означает водород, либо R4 означает водород, a R5 означает алкил с 1-4 атомами углерода. Из соединений формулы (I) предпочтительными являются такие, в которых g2, g5 и g6 означают водород и g3 и g4 имеют значения, указанные выше для соединений формулы (I). Из соединений формулы (I) предпочтительными являются такие, в которых w5 и w6, означают водород, w4 означает атом галогена, алкил с 1-4 атомами углерода, алкоксил с 1-4 атомами углерода, трифторметил или алкилтиогруппу с 1-4 атомами углерода, и либо каждый из w2 и g3 означают водород, либо один из них означает водород, а другой означает атом галогена, алкил с 1-4 атомами углерода или трифторметил. Одной группой предпочтительных соединений согласно настоящему изобретению является группа соединений формулы (Ia): 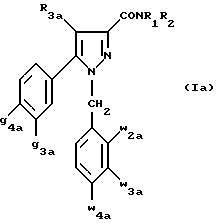 в которой R1, R2 имеют значения, указанные для соединения формулы (I); R3a означает водород или группу -CH2-R6a; R6a означает водород или при условии, что заместители g3a и g4a являются другими, чем алкил с 1-4 атомами углерода, R6a означает водород, метил или этил; g3a означает водород, атом галогена, алкил с 1-4 атомами углерода или трифторметил; g4a означает атом галогена, алкил с 1-4 атомами углерода или трифторметил; w4a означает атом галогена, алкил с 1-4 атомами углерода или трифторметил; каждый из w2a и w3a означает водород или один из них означает водород, а другой означает атом галогена, алкил с 1-4 атомами углерода или трифторметил; также как их возможные соли. Из этих соединений особенно предпочтительными являются соединения формулы (I’a): 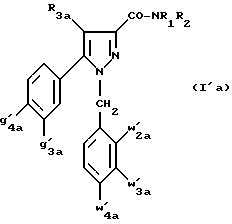 в которой R1, R2 имеют значения, указанные для соединений формулы (I); R3a имеет значение, указанное для соединений формулы (Ia); g’3a означает водород, атом хлора, атом фтора, метил или трифторметил; g’4a означает атом хлора, атом фтора, метил или трифторметил; w’4a означает атом хлора, атом фтора, метил или трифторметил; каждый из w’2a и w’3a означает водород или один из них означает водород, а другой означает атом хлора, атом фтора, метил или трифторметил; и их возможные соли. Еще более предпочтительными соединениями являются соединения формулы (I’a), в которой: R1 означает водород; R2 означает 1.3.3-триметилбицикло[2.2.1] гепт-1-ил или бицикло[3.2.1] окт-3-ил; R3a имеет значение, указанное для соединений формулы (Ia); w’2a, w’3a, w’4a, g’3a и g’4a имеют значения, указанные для соединений формулы (I’a); также как их возможные соли. В высшей степени предпочтительными являются соединения формулы (I’a), в которой; g’3a означает водород, атом хлора, атом фтора или метил; g’4a означает атом хлора, атом фтора или метил; w’4a означает атом хлора, атом фтора или метил; каждый из w’2a и w’3a означает водород или один из них означает водород, а другой означает атом хлора, атом фтора или метил; R1, R2 и R3 имеют значения, указанные для соединений формулы (I’a); также как их возможные соли. Другой группой предпочтительных соединений согласно изобретению является группа соединений формулы (Ib): 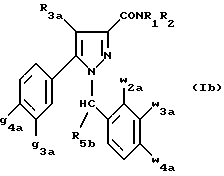 в которой R1, R2 имеют значения, указанные для соединений формулы (I); R3a, w2a, w3a, w4a, g3a и g4a имеют значения, указанные для соединений формулы (Ia); R5b означает алкил с 1-4 атомами углерода; также как их возможные соли. Из этих соединений особенно предпочтительными являются соединения формулы (I’b): 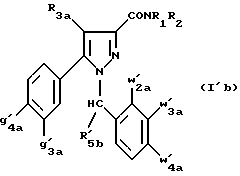 в которой R1, R2 имеют значения, указанные для соединений формулы (I); R3a имеет значение, указанное для соединений формулы (Ia); w’2a, w’3a, w’4a, g’3a и g’4a имеют значения, указанные для соединений формулы (Ia); R’5b означает метил; также как их возможные соли. Еще более предпочтительными соединениями являются соединения формулы (I’b) в которой R1 означает водород; R2 означает 1,3,3-триметилбицикло[2.2.1] гепт-2-ил или бицикло[3.2.1] окт-3-ил; R3a имеет значение, указанное для соединений формулы (Ia); R’5b означает метил; w’2a, w’3a, w’4a, g’3a и g’4a имеют значения, указанные для соединений формулы (I’a); также как их соли. В высшей мере предпочтительными являются соединения формулы (I’b), в которой: g’3a означает водород, атом хлора, атом фтора или метил; g’4a означает атом хлора, атом фтора или метил; w’4a означает атом хлора, атом фтора или метил; каждый из w’2a и w’3a означает водород или один из них означает водород, а другой означает атом хлора атом фтора или метил; R1, R2, R2 и R’5b имеют значения, указанные для соединений формулы (I’b); также как их возможные соли. Следующей группой предпочтительных соединений согласно изобретению является группа соединений формулы (Ic): 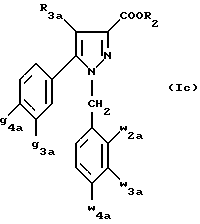 в которой: R2 имеет эначение, указанное для соединения формулы (I); R3a, w2a, w3a, w4a, g3a и g4a имеют значения, указанные для соединений формулы (Ia); так же, как их возможные соли. Согласно другому из аспектов настоящего изобретения, оно относится к способу получения соединений формулы (I) и их солей, отличающемуся тем, что: 1) функциональное производное пиразол-3-карбоновой кислоты формулы (II) 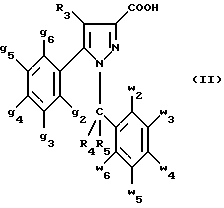 в которой w2, w3, w4, w5, w6, g2, g3, g4, g5, g6, R3, R4 и R5 имеют значения, указанные для соединений формулы (I), обрабатывают соединением формулы (XXIV): H-X1 (XXIV), в которой X1 имеет значение, указанное для соединений формулы (I); 2) и, в случае необходимости, таким образом полученное соединение превращают в одну из его солей. Один из способов получения согласно изобретению (способ А) пригоден для синтеза соединений формулы (I), в которой X1 означает группу – NR1R2. Этот способ отличается тем, что: 1) функциональное производное пиразол-3-карбоновой кислоты вышеприведенной формулы (II) обрабатывают амином формулы (III): HNR1R2 (III), в которой R1 и R2 имеют значения, указанные для соединений формулы (I); 2) и, в случае необходимости, таким образом полученное соединение превращают в одну из его солей. В качестве функционального производного кислоты формулы (II) можно использовать хлорангидрид кислоты; ангидрид; смешанный ангидрид; сложный алкиловый эфир с 1-4 атомами углерода в алкильной части, в котором алкил является линейным или разветвленным; активированный сложный эфир, как, например, п-нитрофениловый сложный эфир, или свободную кислоту, в свою очередь активированную, например, с помощью N,N-дициклогексилкарбодиимида или с помощью бензотриазол-1- илокситрис(диметиламино)фосфонийгексафторфосфата (ВОР). Таким образом, в способе А, согласно изобретению, хлорангидрид пиразол-3-карбоновой кислоты, полученный путем взаимодействия тионилхлорида с кислотой формулы (II), можно вводить во взаимодействие с амином формулы HNR1R2, в инертном растворителе, таком как хлорированный растворитель (например, дихлорметан, дихлорэтан, хлороформ), простой эфир (например, тетрагидрофуран, диоксан) или амид (например, N, N-диметилформамид) в инертной атмосфере, при температуре от 0oC до комнатной температуры, в присутствии третичного амина, такого как триэтиламин, N-метилморфолин или пиридин. Один вариант осуществления способа А состоит в получении смешанного ангидрида кислоты формулы (II) путем реакции этилхлорформиата с кислотой формулы (II) в присутствии основания, такого как триэтиламин, и во введении его во взаимодействие с амином HNR1R2 в растворителе, таком как дихлорметан, в инертной атмосфере, при комнатной температуре и в присутствии основания, такого как триэтиламин. Другой способ получения (способ Б) согласно изобретению пригоден для синтеза соединений формулы (I), в которой X1 означает группу -OR2. Этот способ отличается тем, что: 1) функциональное производное пиразол-3-карбоновой кислоты формулы (II) обрабатывают спиртом формулы (XIV): HO-R2 (XIV), в которой R2 имеет значение, указанное для соединений формулы (I); 2) и в случае необходимости таким образом попорченное соединение превращают в одну из его солей. В качестве функционального производного кислоты формулы (II) можно использовать хлорангидрид кислоты; ангидрид; смешанный ангидрид или свободную кислоту, в свою очередь активированную, например, с помощью N,N-дициклогексилкарбодиимида или с помощью бензотриазол- 1-илокситрис(диметиламино)фосфонийгексафторфосфата (ВОР). Таким образом, в способе Б, согласно изобретению, хлорангидрид пиразол-3-карбоновой кислоты, полученный путем взаимодействия тионилхлорида с кислотой формулы (II), можно вводить во взаимодействие со спиртом формулы HO-R2, либо в инертном растворителе, таком как хлорированный растворитель (например, дихлорметан, дихлорэтан, хлороформ), простой эфир (например, тетрагидрофуран, диоксан) или амид (например, N, N- диметилформамид), в инертной атмосфере, при температуре от 0oC до комнатной температуры, в присутствии третичного амина, такого как триэтиламин, N-метилморфолин или пиридин, либо в пиридине при комнатной температуре, в присутствии 4-диметиламинопиридина. Один вариант осуществления способа Б состоит в получении смешанного ангидрида кислоты формулы (II) путем реакции этилхлорформиата с кислотой формулы (II), в присутствии основания, такого как триэтиламин, и во введении его во взаимодействие со спиртом HO-R2 в растворителе, таком как дихлорметан, в инертной атмосфере, при комнатной температуре и в присутствии основания, такого как триэтиламин. В процессе какой-либо из стадий получения соединений формулы (I), и преимущественно при получении промежуточного соединения формулы (II), может оказаться необходимой и/или желательной защита функциональных реакционноспособных или чувствительных групп, таких как аминогруппа, гидроксил или карбоксил, присутствующих в какой-либо из вводимых во взаимодействие молекул. Эту запрету можно осуществлять, используя обычные защитные группы, такие как группы, описанные в Руководстве “Защитные группы в органической химии” J.F. W. McOmie, изд. Plenum Press, 1973, и в Руководстве “Защитные группы в органическом синтезе” T.W.Greene и P.G.M.Wutts, изд. John Wiley et Sons, 1991. Удаление защитных групп можно осуществлять в надлежащей последующей стадии, используя известные специалисту способы, которые не затрагивают остальной части соответствующей молекулы. Таким образом, полученное соединение формулы (I) выделяют обычными способами. В зависимости от природы заместителей, соединение формулы (I) в случае необходимости может быть переведено в соль. Соль получают путем обработки с помощью выбранной кислоты в органическом растворителе. Путем обработки свободного основания, растворенного, например, в простом эфире, таком как диэтиловый эфир, или в спирте, таком как пропан-2-ол, или в ацетоне, или в дихлорметане, с помощью раствора выбранной кислоты в том же самом растворителе получают соответствующую соль, которую выделяют классическими способами. Таким образом получают, например, гидрохлорид, гидробромид, сульфат, гидросульфонат, дигидрофосфат, метансульфонат, оксалат, малеат, фумарат, нафталинсульфонат, бензолсульфонат. Соединения формулы (II) получают по различным методикам. Соединения формулы (II), в которой R3=R’3 и означает водород или группу CH2-R6, где R6 означает водород или алкил с 1- 4 атомами углерода, получают согласно Схеме 1 (см. в конце описания). Первая стадия  состоит в получении соли щелочного металла производного ацетофенона формулы (IV), в которой R’3 означает водород или группу CH2-R6, где R6 означает водород или алкил с 1-4 атомами углерода, и g2 g3=, g4, g5 и g6 имеют указанное для формулы (I) значение, к которой затем добавляют эквимолярное количество диэтилоксалата (стадия b1) для получения соли сложного кетоэфира формулы (V).
В частном случае, где R’3=H, щелочным металлом предпочтительно является натрий (M= Na) и соль сложного кетоэфира (формулы (V), Alk=CH3) получают по способу, описанному в Bull. Soc.Chim.Fr., 14, 1098 (1947), используя метилат натрия в метаноле для осуществления стадии состоит в получении соли щелочного металла производного ацетофенона формулы (IV), в которой R’3 означает водород или группу CH2-R6, где R6 означает водород или алкил с 1-4 атомами углерода, и g2 g3=, g4, g5 и g6 имеют указанное для формулы (I) значение, к которой затем добавляют эквимолярное количество диэтилоксалата (стадия b1) для получения соли сложного кетоэфира формулы (V).
В частном случае, где R’3=H, щелочным металлом предпочтительно является натрий (M= Na) и соль сложного кетоэфира (формулы (V), Alk=CH3) получают по способу, описанному в Bull. Soc.Chim.Fr., 14, 1098 (1947), используя метилат натрия в метаноле для осуществления стадии  В стадии В стадии 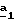 также можно воздействовать трет. -бутилатом калия в этаноле на производное формулы (IV), затем добавлять диэтилоксалат, как описано выше. Реакцию проводят при температуре кипения с обратным холодильником растворителя. Таким образом получают соединение формулы (V), в которой М=K и Alk = CH2CH3.
В частном случае, где R’3=CH3, щелочным металлом предпочтительно является литий (M=Li) и соль сложного кетоэфира (формулы (V), Alk=CH2CH3) получают по способу, описанному в J.Heterocyclic Chem., 26, 1389-1392 (1989), используя литиевую соль гексаметилдисилазана в инертном растворителе, таком как диэтиловый эфир или циклогексан, для осуществления стадии также можно воздействовать трет. -бутилатом калия в этаноле на производное формулы (IV), затем добавлять диэтилоксалат, как описано выше. Реакцию проводят при температуре кипения с обратным холодильником растворителя. Таким образом получают соединение формулы (V), в которой М=K и Alk = CH2CH3.
В частном случае, где R’3=CH3, щелочным металлом предпочтительно является литий (M=Li) и соль сложного кетоэфира (формулы (V), Alk=CH2CH3) получают по способу, описанному в J.Heterocyclic Chem., 26, 1389-1392 (1989), используя литиевую соль гексаметилдисилазана в инертном растворителе, таком как диэтиловый эфир или циклогексан, для осуществления стадии 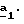 В стадии 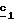 таким образом полученное соединение формулы (V) и избыток гидразина (гидразинмоногидрат или водный раствор гидразина) кипятят с обратным холодильником в уксусной кислоте. Путем осаждения в воде со льдом таким образом получают пиразол-3-карбоксилаты формулы (VI).
В стадии таким образом полученное соединение формулы (V) и избыток гидразина (гидразинмоногидрат или водный раствор гидразина) кипятят с обратным холодильником в уксусной кислоте. Путем осаждения в воде со льдом таким образом получают пиразол-3-карбоксилаты формулы (VI).
В стадии 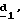 таким образом полученное соединение формулы (VI) обрабатывают сильным основанием, таким как гидрид натрия или амид натрия, в растворителе для получения аниона, который вводят во взаимодействие с соединением формулы (VII), в которой Hal означает галоген, предпочтительно хлор, бром или иод, R4 означает водород и w2, w3, w4, w5, w6 и R5 имеют значения, указанные для соединений формулы (I), для получения соединения формулы (IX). Реакцию проводят предпочтительно в толуоле при температуре от комнатной до температуры кипения с обратным холодильником растворителя для получения преобладающего целевого соединения формулы (IX). Когда реакцию осуществляют в N, N-диметилформамиде при температуре от 0oC до комнатной, то наблюдают преобладающее образование геометрического изомера формулы (XI): таким образом полученное соединение формулы (VI) обрабатывают сильным основанием, таким как гидрид натрия или амид натрия, в растворителе для получения аниона, который вводят во взаимодействие с соединением формулы (VII), в которой Hal означает галоген, предпочтительно хлор, бром или иод, R4 означает водород и w2, w3, w4, w5, w6 и R5 имеют значения, указанные для соединений формулы (I), для получения соединения формулы (IX). Реакцию проводят предпочтительно в толуоле при температуре от комнатной до температуры кипения с обратным холодильником растворителя для получения преобладающего целевого соединения формулы (IX). Когда реакцию осуществляют в N, N-диметилформамиде при температуре от 0oC до комнатной, то наблюдают преобладающее образование геометрического изомера формулы (XI):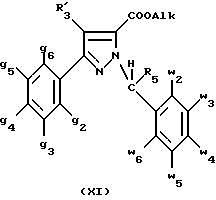 Альтернативно, согласно стадии  соединение формулы (V) вместе с избытком производного гидразина формулы (VIII), в которой R4, R5, w2, w3, w4, w5, w6 имеют значения, указанные для соединений формулы (I), кипятят с обратным холодильником в уксусной кислоте; путем осаждения в смеси воды со льдом получают соединения формулы (IX).
В стадии соединение формулы (V) вместе с избытком производного гидразина формулы (VIII), в которой R4, R5, w2, w3, w4, w5, w6 имеют значения, указанные для соединений формулы (I), кипятят с обратным холодильником в уксусной кислоте; путем осаждения в смеси воды со льдом получают соединения формулы (IX).
В стадии  путем гидролиза в щелочной среде соединений формулы (IX), затем подкисления получают целевые соединения формулы (II). Гидролиз проводят при использовании, например, гидроксида щелочного металла, такого как гидроксид калия, гидроксид натрия или гидроксид лития, в инертном растворителе, таком как вода, метанол, этанол, диоксан или смесь этих растворителей, при температуре от 0oC до температуры кипения с обратным холодильником растворителя.
Соединения формулы (IX), в которой R4 означает водород, предпочтительно получают через вышеописанные стадии путем гидролиза в щелочной среде соединений формулы (IX), затем подкисления получают целевые соединения формулы (II). Гидролиз проводят при использовании, например, гидроксида щелочного металла, такого как гидроксид калия, гидроксид натрия или гидроксид лития, в инертном растворителе, таком как вода, метанол, этанол, диоксан или смесь этих растворителей, при температуре от 0oC до температуры кипения с обратным холодильником растворителя.
Соединения формулы (IX), в которой R4 означает водород, предпочтительно получают через вышеописанные стадии  затем затем 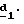 Соединения формулы (IX), в которой R4 и R5 отличаются от водорода, предпочтительно получают через стадию 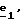 описанную выше.
Соединения формулы (IX), в которой R’3 означает CH2-(C1-C4)-алкил, предпочтительно получают, либо когда R4 и R5 отличаются от водорода, из самих соединений формулы (IX), либо когда R4=H, из соединений формулы (VI), согласно Схеме 2 (см. в конце описания).
Стадия описанную выше.
Соединения формулы (IX), в которой R’3 означает CH2-(C1-C4)-алкил, предпочтительно получают, либо когда R4 и R5 отличаются от водорода, из самих соединений формулы (IX), либо когда R4=H, из соединений формулы (VI), согласно Схеме 2 (см. в конце описания).
Стадия 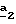 состоит в получении 4-бромметилпиразол-3- карбоксилата формулы (X) путем воздействия N-бромсукцинимида на соединение формулы (VI) или формулы (IX), где R’3 означает метил. Реакцию проводят в инертном растворителе, как тетрахлорид углерода, в присутствии дибензоилпероксида и при температуре кипения с обратным холодильником растворителя.
Когда используют соединение формулы (VI), то предпочтительно осуществляют бромирование согласно стадии состоит в получении 4-бромметилпиразол-3- карбоксилата формулы (X) путем воздействия N-бромсукцинимида на соединение формулы (VI) или формулы (IX), где R’3 означает метил. Реакцию проводят в инертном растворителе, как тетрахлорид углерода, в присутствии дибензоилпероксида и при температуре кипения с обратным холодильником растворителя.
Когда используют соединение формулы (VI), то предпочтительно осуществляют бромирование согласно стадии 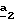 соединения, пиразольный азот которого защищен (Y=N-защитная группа). В качестве N-защитной группы используют хорошо известные специалисту классические N-защитные группы, как, например, трет.- бутоксикарбонил.
Стадия соединения, пиразольный азот которого защищен (Y=N-защитная группа). В качестве N-защитной группы используют хорошо известные специалисту классические N-защитные группы, как, например, трет.- бутоксикарбонил.
Стадия 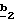 состоит в получении соединения формулы (VI) или (IX), в которой R’3 означает группу -CH2-(C1-C4)-алкил, путем воздействия органокупрата (Alk’)2CuLi, в котором Alk’ означает алкил с 1-4 атомами углерода. Реакцию проводят по способу, описанному в заявке на европейский патент 0658546.
В случае необходимости, когда используют соединение формулы (VI), защищенное по пиразольному азоту, после стадии состоит в получении соединения формулы (VI) или (IX), в которой R’3 означает группу -CH2-(C1-C4)-алкил, путем воздействия органокупрата (Alk’)2CuLi, в котором Alk’ означает алкил с 1-4 атомами углерода. Реакцию проводят по способу, описанному в заявке на европейский патент 0658546.
В случае необходимости, когда используют соединение формулы (VI), защищенное по пиразольному азоту, после стадии 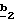 N-защитную группу удаляют известными специалисту способами.
Соединения формулы (II), в которой R4 означает водород и R3=R”3 и означает группу -CH2-R6, где R6 отличается от водорода или отличается от алкила с 1-4 атомами углерода, получают согласно Схеме 3 (см. в конце описания).
В стадии N-защитную группу удаляют известными специалисту способами.
Соединения формулы (II), в которой R4 означает водород и R3=R”3 и означает группу -CH2-R6, где R6 отличается от водорода или отличается от алкила с 1-4 атомами углерода, получают согласно Схеме 3 (см. в конце описания).
В стадии 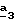 атом азота соединения формулы (VI) (R’3=CH3) защищают с помощью N-защитной группы, такой как трет.-бутоксикарбонил (Boc), согласно известным специалисту способам.
Стадия атом азота соединения формулы (VI) (R’3=CH3) защищают с помощью N-защитной группы, такой как трет.-бутоксикарбонил (Boc), согласно известным специалисту способам.
Стадия 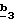 заключается в получении 4-бромметилпиразол-3- карбоксилата формулы (XVI) по способу, описанному выше в стадии заключается в получении 4-бромметилпиразол-3- карбоксилата формулы (XVI) по способу, описанному выше в стадии 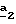 Схемы 2.
В стадии Схемы 2.
В стадии 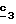 соединение формулы (XVI) обрабатывают соединением формулы R6-A, в которой R6, имеющий указанное для формулы (I) значение, отличается от водорода или отличается от алкила с 1-4 атомами углерода и A означает водород или катион, такой как катион щелочного или щелочноземельного металла или четвертичная аммониевая группа, такая как тетраэтиламмониевая группа.
Для получения соединения формулы (XVIII), в которой R6 означает алкоксил с 1-5 атомами углерода или гидроксиалкоксил с 1-5 атомами углерода, в качестве реагента формулы (XVII) используют спирт с 1-5 атомами углерода или двухатомный спирт с 1-5 атомами углерода в присутствии не нуклеофильного основания, такого как гидрид металла, как, например, гидрид натрия или калия. В зависимости от значений R6 в стадии соединение формулы (XVI) обрабатывают соединением формулы R6-A, в которой R6, имеющий указанное для формулы (I) значение, отличается от водорода или отличается от алкила с 1-4 атомами углерода и A означает водород или катион, такой как катион щелочного или щелочноземельного металла или четвертичная аммониевая группа, такая как тетраэтиламмониевая группа.
Для получения соединения формулы (XVIII), в которой R6 означает алкоксил с 1-5 атомами углерода или гидроксиалкоксил с 1-5 атомами углерода, в качестве реагента формулы (XVII) используют спирт с 1-5 атомами углерода или двухатомный спирт с 1-5 атомами углерода в присутствии не нуклеофильного основания, такого как гидрид металла, как, например, гидрид натрия или калия. В зависимости от значений R6 в стадии 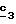 способа можно получать смесь сложных эфиров, которую омыляют в стадии способа можно получать смесь сложных эфиров, которую омыляют в стадии 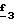 для получения кислоты формулы (II).
Для получения соединения формулы (XVIII), в которой R6 означает алкилтиогруппу с 1-5 атомами углерода, в качестве реагента формулы (XVII) используют тиоспирт с 1-5 атомами углерода в присутствии не нуклеофильного основания, такого как гидрид металла, как гидрид натрия или калия.
В желательном случае полученный в стадии для получения кислоты формулы (II).
Для получения соединения формулы (XVIII), в которой R6 означает алкилтиогруппу с 1-5 атомами углерода, в качестве реагента формулы (XVII) используют тиоспирт с 1-5 атомами углерода в присутствии не нуклеофильного основания, такого как гидрид металла, как гидрид натрия или калия.
В желательном случае полученный в стадии 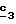 сложный эфир формулы (XVIII), в котором R6 означает алкилтиогруппу с 1-5 атомами углерода, путем воздействия на него окислителя, такого как пероксид водорода или м-хлорнадбензойная кислота, можно превращать в соединение формулы (XVIII), где R6 означает алкилсульфонил с 1-5 атомами углерода или алкилсульфинил с 1-5 атомами углерода.
Для получения соединения формулы (XVIII), в котором R6 означает цианогруппу, в качестве реагента формулы (XVII) можно использовать цианид четвертичного аммония, например тетраэтиламмоний цианид, или цианид металла, такой как цианид натрия; в этом последнем случае реакцию нуклеофильного замещения согласно стадии сложный эфир формулы (XVIII), в котором R6 означает алкилтиогруппу с 1-5 атомами углерода, путем воздействия на него окислителя, такого как пероксид водорода или м-хлорнадбензойная кислота, можно превращать в соединение формулы (XVIII), где R6 означает алкилсульфонил с 1-5 атомами углерода или алкилсульфинил с 1-5 атомами углерода.
Для получения соединения формулы (XVIII), в котором R6 означает цианогруппу, в качестве реагента формулы (XVII) можно использовать цианид четвертичного аммония, например тетраэтиламмоний цианид, или цианид металла, такой как цианид натрия; в этом последнем случае реакцию нуклеофильного замещения согласно стадии 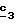 осуществляют в присутствии межфазного катализатора.
Для получения соединения формулы (XVIII), в котором R6 означает фтор, в качестве реагента формулы (XVII) можно использовать фторирующий агент; в качестве фторирующего агента можно использовать фторид металла, как, например, фторид калия, применяемый в присутствии комплексообразующего агента, как Kryptofix осуществляют в присутствии межфазного катализатора.
Для получения соединения формулы (XVIII), в котором R6 означает фтор, в качестве реагента формулы (XVII) можно использовать фторирующий агент; в качестве фторирующего агента можно использовать фторид металла, как, например, фторид калия, применяемый в присутствии комплексообразующего агента, как Kryptofix . .Для получения соединения формулы (XVIII), в котором R6 означает гидроксил, в качестве реагента формулы (XVII) используют гидроксид щелочного или щелочноземельного металла, такой как гидроксид натрия или калия. В стадии 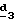 удаляют N-защитную группу известными специалисту способами.
В стадии удаляют N-защитную группу известными специалисту способами.
В стадии 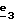 таким образом полученное соединение формулы (XIX) обрабатывают сильным основанием, таким как гидрид натрия или амид натрия, в растворителе для получения аниона, который вводят во взаимодействие с соединением формулы (VII), в которой Hal означает галоген, предпочтительно хлор, бром или иод; R4 означает водород и w2, w3, w4, w5, w6 и R5 имеют значения, указанные для соединений формулы (I), для получения соединения формулы (XX). Реакцию проводят предпочтительно в толуоле при температуре от комнатной до температуры кипения с обратным холодильником растворителя с целью получения в большинстве своем целевого соединения формулы (XX). Когда реакцию проводят в N,N-диметилформамиде при температуре от 0oC до комнатной, наблюдают преобладающее образование геометрического изомера формулы (XXIII): таким образом полученное соединение формулы (XIX) обрабатывают сильным основанием, таким как гидрид натрия или амид натрия, в растворителе для получения аниона, который вводят во взаимодействие с соединением формулы (VII), в которой Hal означает галоген, предпочтительно хлор, бром или иод; R4 означает водород и w2, w3, w4, w5, w6 и R5 имеют значения, указанные для соединений формулы (I), для получения соединения формулы (XX). Реакцию проводят предпочтительно в толуоле при температуре от комнатной до температуры кипения с обратным холодильником растворителя с целью получения в большинстве своем целевого соединения формулы (XX). Когда реакцию проводят в N,N-диметилформамиде при температуре от 0oC до комнатной, наблюдают преобладающее образование геометрического изомера формулы (XXIII):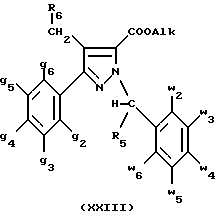 В стадии 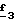 путем гидролиза в щелочной среде соединений формулы (XX), затем подкисления получают целевые соединения формулы (II). Гидролиз осуществляют при использовании, например, гидроксида щелочного металла, такого как гидроксид калия, гидроксид натрия или гидроксид лития, в инертном растворителе, таком как вода, метанол, этанол, диоксан или смесь этих растворителей, при температуре от 0oC до температуры кипения с обратным холодильником растворителя.
Соединения формулы (II), в которой R3=R”3 и означает группу -CH2-R6, в которой R6 отличается от водорода или отличается от алкила с 1-4 атомами углерода, и R4 и R5 отличны от водорода, получают согласно Схеме 4, где Alk означает метил или этил (см. в конце описания).
Стадия путем гидролиза в щелочной среде соединений формулы (XX), затем подкисления получают целевые соединения формулы (II). Гидролиз осуществляют при использовании, например, гидроксида щелочного металла, такого как гидроксид калия, гидроксид натрия или гидроксид лития, в инертном растворителе, таком как вода, метанол, этанол, диоксан или смесь этих растворителей, при температуре от 0oC до температуры кипения с обратным холодильником растворителя.
Соединения формулы (II), в которой R3=R”3 и означает группу -CH2-R6, в которой R6 отличается от водорода или отличается от алкила с 1-4 атомами углерода, и R4 и R5 отличны от водорода, получают согласно Схеме 4, где Alk означает метил или этил (см. в конце описания).
Стадия 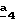 состоит в получении 4-бромметилпиразол-3- карбоксилата формулы (XXI) по способу, описанному выше в стадии состоит в получении 4-бромметилпиразол-3- карбоксилата формулы (XXI) по способу, описанному выше в стадии 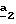 Схемы 2.
В стадии Схемы 2.
В стадии 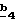 соединение формулы (XXI) обрабатывают соединением формулы R6-A (XVII), таким, как описанное выше, и по методикам, описанным в стадии соединение формулы (XXI) обрабатывают соединением формулы R6-A (XVII), таким, как описанное выше, и по методикам, описанным в стадии 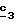 Схемы 3.
В стадии Схемы 3.
В стадии 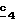 путем гидролиза в щелочной среде соединений формулы (XXII), затем подкисления получают целевые соединения формулы (II). Гидролиз проводят по способам, описанным в стадии путем гидролиза в щелочной среде соединений формулы (XXII), затем подкисления получают целевые соединения формулы (II). Гидролиз проводят по способам, описанным в стадии 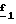 Схемы 1.
В стадии Схемы 1.
В стадии  Схемы 1 или в стадии Схемы 1 или в стадии  Схемы 3, во время реакции соединения формулы (VI) или соединения формулы (XIX) с галогенированным производным формулы (VII) можно получать смесь в изменяемых соотношениях соединения формулы (IX) или соединения формулы (XX) с их соответствующими изомерами формулы (XI) или (XXIII): Схемы 3, во время реакции соединения формулы (VI) или соединения формулы (XIX) с галогенированным производным формулы (VII) можно получать смесь в изменяемых соотношениях соединения формулы (IX) или соединения формулы (XX) с их соответствующими изомерами формулы (XI) или (XXIII):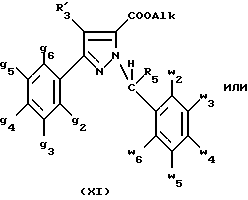 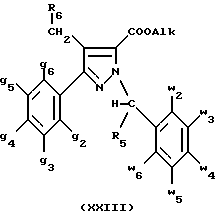 Оба изомера формул (IX) и (XI) или оба изомера формул (XX) и (XXIII) могут быть разделены путем хроматографии на силикагеле согласно классическим способам. Оба изомера формул (IX) и (XI) или формул (XX) и (XXIII) охарактеризовывают их ЯМР-спектром, особенно путем изучения эффекта Оверхаузера (N.O.E.). Стадию 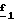 способа или стадию способа или стадию 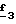 способа, такие как описанные в Схеме 1 или Схеме 3, можно осуществлять при использовании смеси изомеров с целью получения смеси кислот формулы (II) с ее изомером формулы (XII): способа, такие как описанные в Схеме 1 или Схеме 3, можно осуществлять при использовании смеси изомеров с целью получения смеси кислот формулы (II) с ее изомером формулы (XII):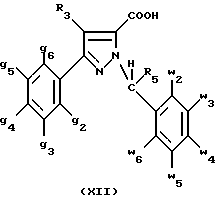 В таком случае для получения смеси двух изомеров формулы (II) и (XII) применяют способ согласно изобретению, описанный выше для получения смеси соединения формулы (I), в которой R4 означает водород, с его изомером формулы (XIII): 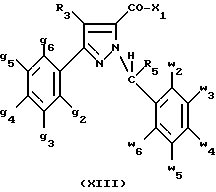 Тогда оба изомера разделяют согласно классическим методам, таким как, например, хроматография на силикагеле или кристаллизация, и, наконец, получают соединение формулы (I) согласно изобретению. Согласно другому из аспектов настоящего изобретения, предметом изобретения являются соединения, представляющие собой побочные продукты способа получения соединений формулы (I), формулы (XIII): 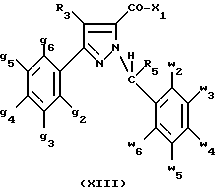 в которой X1, g2, g3, g4, g5, g6, w2, w3, w4, w5, w6, R3 и R5 имеют значения, указанные для соединений формулы (I); также как их возможные соли. Согласно следующему из аспектов настоящего изобретения, оно относится к способу получения промежуточных соединений формулы (II) и соединений формулы (XII), пригодных для получения соединений формулы (I), в которых R4 означает водород, и соединений формулы (XIII). Этот способ отличается тем, что: 1) соединение формулы (XXV): 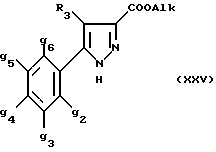 в которой g2, g3, g4, g5, g6 и R6 имеют значения, указанные для соединений формулы (I) и Alk означает метил или этил, обрабатывают сильным основанием в растворителе, затем таким образом полученный анион вводят во взаимодействие с соединением формулы (VII): 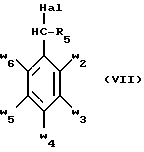 в которой w2, w3, w4, w5, w6 и R5 имеют значения, указанные для соединений формулы (I), и Hal означает атом галогена, для получения: – либо, когда реакцию проводят в толуоле при температуре от комнатной до температуры кипения с обратным холодильником растворителя, соединения формулы (XXVI): 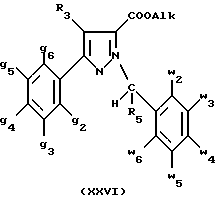 – либо, когда реакцию проводят в N,N-диметилформамиде при температуре от 0oC до комнатной, соединения формулы (XXVII): 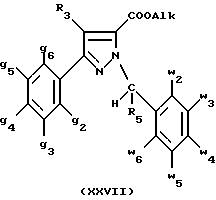 2) гидролизуют в щелочной среде либо соединение формулы (XXVI), либо соединение формулы (XXVII), для получения, соответственно, – либо соединения формулы (II: R4=H): 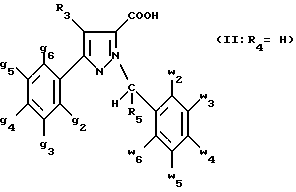 – либо соединения формулы (XII): 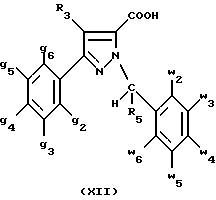 Согласно следующему из аспектов настоящего изобретения, оно относится к способу получения соединений формулы (XIII) и их солей, отличающемуся тем, что: 1) функциональное производное кислоты формулы (XII): 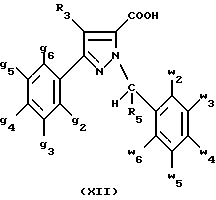 в которой g2, g3, g4, g5, g6, w2, w3, w4, w5, w6, R3 и R5 имеют значения, указанные для соединений формулы (I), обрабатывают соединением формулы (XXIV): H-X1 (XXIV), в которой X1 имеет значение, указанное для соединений формулы (I), для получения соединения формулы (XIII): 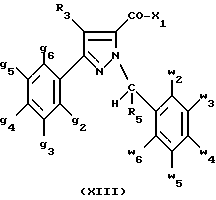 2) и в случае необходимости таким образом полученное соединение превращают в одну из его солей. Бензилгалогениды формулы (VII) известны или их получают известными способами. Как правило, соединения формулы (VII), в которой Hal означает атом брома, могут быть получены путем воздействия N-бромсукцинимида на соответствующие метилбензольные производные в присутствии дибензоилпероксида. Бензилбромид можно также получать из соответствующего бензилового спирта путем воздействия бромоводорода в виде раствора в воде или в уксусной кислоте. Можно также использовать воздействие трибромида фосфора на соответствующий бензиловый спирт для получения соединения формулы (VII), в которой Hal означает атом брома. Соединения формулы (VII), в которой Hal означает атом иода, могут быть получены путем воздействия иодида натрия на соединение формулы (VII), в которой Hal означает атом хлора, в растворителе, таком как ацетон или бутан-2-он. Соединения формулы (VII), в которой Hal означает атом хлора, могут быть получены путем воздействия тионилхлорида на соответствующий бензиловый спирт. Соединения формулы (VII), в которой R5 означает трифторметил и Hal означает атом хлора, предпочтительно можно получать по способу, описанному в J. Fluorine Chem., 32(4), 361-366 (1986). Соединения формулы (VII), в которой R5 означает трифторметил, также можно получать из соответствующих – (трифторметил)бензиловых спиртов согласно вышеописанным способам. – (Трифторметил) бензиловые спирты можно получать согласно методике, описанной в Tetrahedron, 45 (5), 1423 (1989), или согласно методике, описанной в J.Org.Chem., 56(1) 2 (1991).
Соединения формулы (VIII) известны или их получают известными способами, такими как описанные в J.Org.Chem., 53, 1768-1774 (1988) или в J.Am.Chem. Soc., 80, 6562-6568 (1958).
Амины формулы HNR1R2 либо имеются в продаже, либо описаны в литературе, либо их получают известными способами согласно нижеуказанным методикам получения:эндо- и экзо-бицикло[3.2.1]октан-2-иламины получают согласно H.Maskill и др., J.Chem.Soc., Perkin II, 119 (1984): 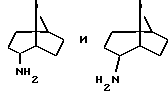 бицикло[2.2.2] октан-2-иламин получают согласно R.Seka и др., Ber., 1379 (1942): 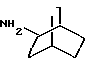 эндо- и экзо-бицикло[3.2.1]октан-3-иламины получают согласно H.Maskill и др., J.Chem.Soc., Perkin II, 1369 (1984): 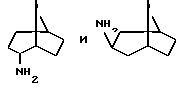 эндо-трицикло[5.2.1.02,6]декан-8-иламин получают согласно G. Buchbauer и др. Arch. Pharm., 323, 367 (1990):  эндо- и экзо-, 1R- и 1S- 1,3,3-триметилбицикло[2.2.1]- гептан-2-иламины получают согласно Ingersoll и др., J.Am.Chem.Soc., 73, 3360 (1951) и J.A. Suchocki и др., J.Med.Chem., 34, 1003-1010 (1991):  3-метилциклогексиламин получают согласно Smith и др., J.Org.Chem., 17, 294 (1952):  2,6-диметилциклогексиламин получают согласно Cornubert и др. Bull. Soc. Chim. Fr., 12, 367 (1945):  2-метoкcициклoгекcилaмин получают согласно Noyce и др., J.Am.Chem.Soc., 76, 768 (1954): 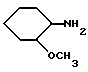 4-этилциклогексиламин получают согласно A. Shirachata и др., Biochem. Pharmacol., 41, 205 (1991): 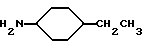 бицикло[2.2.2]окт-2-ен-5-амин получают согласно H.L.Goering и др., J.Am. Chem.Soc., 83, 1391 (1961): 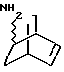 N-этил-1-адамантиламин получают согласно V. L.Narayanan и др., J.Med. Chem., 15, 443 (1972): 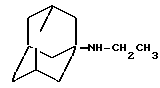 трицикло [2.2.1.02,6] гептан-3-иламин получают согласно G.Muller и др., Chem. Ber., 98, 1097 (1965): 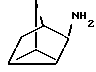 N-метил-экзo-бициклo[2.2.1]гептaн-2-илaмин получают согласно W.G.Kabalka и др., Synth.Commun., 20, 231 (1991): 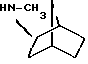 2,2,6,6-тетраметилциклогексиламин получают согласно J. Chem. Soc., С, 1845 (1970): 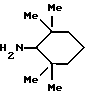 Спирты формулы HO-R2 либо имеются в продаже, либо описаны в литературе, либо их получают известными способами. Например, для получения спиртов формулы (XIV) можно осуществлять восстановление соответствующих кетонов. Восстановление осуществляют с помощью восстановителя, такого, как боргидрид натрия в растворителе, таком как метанол или литийалюминийгидрид в растворителе, таком как тетрагидрофуран или диэтиловый эфир, при температуре от комнатной до температуры кипения с обратным холодильником растворителя. 2,2,6,6-Тетраметилциклогексанол в особенности получают согласно Compterendu hebdomadaire des seances de 1’Academic des Sciences, 156, 1201. Использование соединений формулы (III) в форме индивидуальных энантиомеров в стадии 1) способа А, или использование соединений формулы (XIV) в форме индивидуальных энантиомеров в стадии 1) способа Б, и использование соединений формулы (VII) или формулы (VIII) в форме индивидуальных энантиомеров в стадиях  Схемы 1 или в стадии Схемы 1 или в стадии 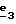 Схемы 3 получения соединения формулы (II), позволяют получать соединения формулы (I) в форме индивидуальных энантиомеров.
Расщепление рацемических смесей соединений формулы (III), (VII), (VIII) или (XIV) осуществляют хорошо известными специалисту методами.
Соединения формулы (I) обладают высоким сродством ин витро к рецепторам CB2 в экспериментальных условиях, описанных Bouaboula и др., Eur.J.Biochem., 214, 173-180 (1993).
В особенности соединения согласно настоящему изобретению и их возможные соли являются сильными и селективными лигандами рецепторов CB2, Ki-значение которых обычно составляет от 0,1 до 100 нМ. Они обычно в 10-1000 раз более активны по отношению к рецепторам CB2, чем к рецепторам CB1, и эффективны при введении перорально.
Соединения формулы (I) согласно изобретению являются антагонистами рецепторов CB2. Антагонистическую активность этих соединений по отношению к рецептору CB2 определяют на различных моделях. Известно, что агонисты рецепторов каннабиноидов (9-THC, WIN 55212-2 или CP-55940) способны ингибировать активность аденилатциклазы, индуцированную форсколином, как описывается M. Rinaldi-Carmona и др., Journal of Pharmacology and Experimental Therapeutics, 278, 871-878 (1996). В случае этой модели соединения формулы (I) согласно изобретению способны полностью блокировать воздействие агонистов рецепторов каннабиноидов.
С другой стороны, известно, что в наномолярных концентрациях агонисты рецепторов каннабиноидов (WIN 55212-2 или CP-55940) способны повышать синтез ДНК в случае B-клеток человека, костимулированных антителами против иммуноглобулина: увеличение примерно на 40% включения тимидина (J.M.Decorcq и др., FEBS Letters, 369, 177-182 (1995)). Когда используют соединения формулы (I) согласно изобретению или одну из их возможных солей в широкой концентрационной области, от 10-10 моль до 10-5 моль, то обнаруживают, что они блокируют увеличение синтеза ДНК в случае B-клеток человека, стимулированных, как описано выше, индуцируемого агонистами рецепторов каннабиноидов (WIN 55212-2 или CP-55940).
Кроме того, агонисты рецепторов каннабиноидов (C3-55940 или WIN 55212-2) индуцируют активацию протеинкиназ, активируемых митогенами (MARKs: “митоген-активируемые протеинкиназы”) в клетках, экспримирующих рецептор CB2. Соединения формулы (I) согласно изобретению специфически блокируют эту активацию MAPKs, индуцируемую агонистами рецепторов каннабиноидов (CP-55940 или WIN 55212-2).
Соединения согласно изобретению или их возможные соли также обладают сродством ин виво к рецепторам каннабиноидов CB2, присутствующих в селезенке мыши, когда их вводят внутривенно, внутрибрюшинно или перорально. Их активность выявляют путем экспериментов на связывание ex vivo [3H]-CP-55940. Опыты проводят в экспериментальных условиях, описанных M.Rinaldi-Carmona и др. Life Sciences, 56, 1941-1947 (1995).
Соединения согласно настоящему изобретению обладают токсичностью, приемлемой для их использования в качестве лекарственных средств.
Благодаря своим замечательным свойствам, особенно своему высокому сродству и своей селективности к периферическим рецепторам CB2, соединения формулы (I) как таковые или в форме фармацевтически приемлемых солей могут быть использованы в качестве действующих начал лекарственных средств.
Заболеваниями, для лечения которых могут быть использованы соединения формулы (I) и в случае необходимости их фармацевтически приемлемые соли, являются патологии, в которых принимают участие клетки иммунной системы или относящиеся к иммунитету нарушения, как, например, аутоиммунные заболевания, связанные с трансплантацией органов заболевания, инфекционные болезни, аллергические заболевания, заболевания желудочно-кишечного тракта, например болезнь Крона. В особенности можно назвать следующие аутоиммунные заболевания: системная красная волчанка, заболевания соединительной ткани, синдром Шегрена, болезнь Бехтерева, реактивный артрит, ревматоидный полиартрит, недифференцированный спондилоартрит, болезнь Бехчета, аутоиммунные гемолитические анемии, рассеянный склероз, псориаз. Излечимые аллергические заболевания могут быть, например, такими, как аллергическая реакция немедленного типа или астма. Точно так же соединения и их возможные фармацевтически приемлемые соли могут быть использованы для лечения васкулитов, паразитарных инфекций, вирусных инфекций, бактериальных инфекций, амилоидоза, заболеваний, поражающих лимфогематопоэтическую систему.
Таким образом, согласно другому из аспектов настоящего изобретения, оно относится к способу лечения вышеуказанных заболеваний, который состоит во введении пациенту, нуждающемуся в этом, эффективного количества соединения формулы (I) или одной из его фармацевтически приемлемых солей.
Согласно следующему из аспектов настоящего изобретения, оно также относится к применению соединений формулы (I) для получения лекарственных средств, предназначенных для лечения расстройств, связанных с рецепторами каннабиноидов CB2, в особенности относящихся к иммунитету нарушений, и также заболеваний, в которых принимает участие иммунная система.
Более того, соединения формулы (I) или формулы (XIII) согласно изобретению, как таковые или в меченной радиоактивным изотопом форме, могут быть использованы в качестве фармакологических средств для человека или животного для определения и маркировки периферических рецепторов CB2 каннабиноидов. Это составляет следующий аспект настоящего изобретения.
Соединения согласно настоящему изобретению обычно вводятся в виде разовой дозы. Вышеуказанные разовые дозы предпочтительно включают в состав фармацевтических композиций, в которых действующее начало смешано с фармацевтическим эксципиентом.
Таким образом, согласно дальнейшему из аспектов настоящего изобретения, оно относится к фармацевтическим композициям, включающим в качестве действующего начала соединение формулы (I) или одну из его фармацевтически приемлемых солей.
Соединения вышеприведенной формулы (I) и их фармацевтически приемлемые соли могут быть использованы в суточных дозах от 0,01 до 100 мг на килограмм массы тела излечиваемого млекопитающего, предпочтительно в суточных дозах от 0,1 до 50 мг/кг. В случае человека дозу можно изменять предпочтительно в пределах от 0,5 до 4000 мг в день, в особенности от 2,5 до 1000 мг, в зависимости от возраста излечиваемого субъекта или типа обработки: профилактика или лечение.
В фармацевтических композициях настоящего изобретения для введения перорально, подъязычно, путем ингаляции, подкожно, внутримышечно, внутривенно, чрескожно, локально или ректально, действующие начала могут вводиться в унитарных формах введения, в смеси с классическими фармацевтическими носителями, животным и людям. Соответствующие унитарные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы с лекарством, порошки, гранулы и пероральные растворы или суспензии; подъязычные и щечные формы введения; аэрозоли; имплантаты; подкожные, внутримышечные, внутривенные, внутриносовые или внутриглазные формы введения и ректальные формы введения.
Когда готовят твердую композицию в форме таблеток, то к действующему началу, микронизированному или нет, можно добавлять смачиватель, такой как лаурилсульфат натрия, и все смешивают с фармацевтическим эксципиентом, таким как диоксид кремния, желатина, крахмал, лактоза, стеарат магния, тальк, гуммиарабик или аналогичные компоненты. Таблетки можно покрывать сахарозой, различными полимерами или другими соответствующими веществами или еще обрабатывать их таким образом, чтобы они обладали пролонгированной или замедленной активностью и непрерывно высвобождали заданное количество действующего начала.
Препарат в виде желатиновых капсул с лекарством получают путем смешения действующего начала с разбавителем, таким как гликоль или сложный эфир глицерина, и введения полученной смеси в мягкие или твердые желатиновые капсулы.
Препарат в форме сиропа или эликсира может содержать действующее начало вместе с подслащивающим, предпочтительно некалорийным средством, метилпарабеном и пропилпарабеном в качестве антисептика, также как придающим вкус агентом и соответствующим красителем.
Диспергируемые в воде порошки или гранулы могут содержать действующее начало в смеси с диспергаторами, смачивателями или суспендирующими агентами, как поливинилпирролидон, точно так же, как с подслащивающими средствами или корректирующими вкус веществами.
Для ректального введения используют суппозитории, которые готовят со связующими, плавящимися при ректальной температуре, как, например, масло какао или полиэтиленгликоли.
Для парентерального, внутриносового или внутриглазного введения используют водные суспензии, изотонические солевые растворы или стерильные растворы для инъекций, которые содержат диспергаторы и/или фармакологически приемлемые солюбилизирующие агенты, как, например, пропиленгликоль или полиэтиленгликоль.
Таким образом, для приготовления водного раствора для инъекции внутривенно можно использовать сорастворитель: спирт, такой как этанол; гликоль, такой как полиэтиленгликоль или пропиленгликоль; и гидрофильный поверхностно-активный агент, такой как, ТВИН 80. Для приготовления масляного раствора для инъекции внутримышечно можно солюбилизировать действующее начало с помощью триглицерида или сложного эфира глицерина.
Для локального введения можно использовать кремы, мази, гели.
Для чрескожного введения можно использовать пэтчи в мультиламинированной или резервуарной форме, в которых действующее начало может находиться в виде спиртового раствора. Для введения путем ингаляции используют аэрозоль, содержащий, например, сорбитантриолеат или олеиновую кислоту, так же как трихлорфторметан, дихлорфторметан, дихлортетрафторэтан или любой другой, биологически приемлемый пропеллент; также можно использовать систему, содержащую действующее начало индивидуально или в сочетании с эксципиентом, в виде порошка.
Действующее начало также может быть использовано для приготовления препаративной формы в виде микрокапсул или микросфер, в случае необходимости с одним или несколькими носителями или одной или несколькими добавками.
Действующее начало также может находиться в виде комплекса с циклодекстрином, как, например, -, Схемы 3 получения соединения формулы (II), позволяют получать соединения формулы (I) в форме индивидуальных энантиомеров.
Расщепление рацемических смесей соединений формулы (III), (VII), (VIII) или (XIV) осуществляют хорошо известными специалисту методами.
Соединения формулы (I) обладают высоким сродством ин витро к рецепторам CB2 в экспериментальных условиях, описанных Bouaboula и др., Eur.J.Biochem., 214, 173-180 (1993).
В особенности соединения согласно настоящему изобретению и их возможные соли являются сильными и селективными лигандами рецепторов CB2, Ki-значение которых обычно составляет от 0,1 до 100 нМ. Они обычно в 10-1000 раз более активны по отношению к рецепторам CB2, чем к рецепторам CB1, и эффективны при введении перорально.
Соединения формулы (I) согласно изобретению являются антагонистами рецепторов CB2. Антагонистическую активность этих соединений по отношению к рецептору CB2 определяют на различных моделях. Известно, что агонисты рецепторов каннабиноидов (9-THC, WIN 55212-2 или CP-55940) способны ингибировать активность аденилатциклазы, индуцированную форсколином, как описывается M. Rinaldi-Carmona и др., Journal of Pharmacology and Experimental Therapeutics, 278, 871-878 (1996). В случае этой модели соединения формулы (I) согласно изобретению способны полностью блокировать воздействие агонистов рецепторов каннабиноидов.
С другой стороны, известно, что в наномолярных концентрациях агонисты рецепторов каннабиноидов (WIN 55212-2 или CP-55940) способны повышать синтез ДНК в случае B-клеток человека, костимулированных антителами против иммуноглобулина: увеличение примерно на 40% включения тимидина (J.M.Decorcq и др., FEBS Letters, 369, 177-182 (1995)). Когда используют соединения формулы (I) согласно изобретению или одну из их возможных солей в широкой концентрационной области, от 10-10 моль до 10-5 моль, то обнаруживают, что они блокируют увеличение синтеза ДНК в случае B-клеток человека, стимулированных, как описано выше, индуцируемого агонистами рецепторов каннабиноидов (WIN 55212-2 или CP-55940).
Кроме того, агонисты рецепторов каннабиноидов (C3-55940 или WIN 55212-2) индуцируют активацию протеинкиназ, активируемых митогенами (MARKs: “митоген-активируемые протеинкиназы”) в клетках, экспримирующих рецептор CB2. Соединения формулы (I) согласно изобретению специфически блокируют эту активацию MAPKs, индуцируемую агонистами рецепторов каннабиноидов (CP-55940 или WIN 55212-2).
Соединения согласно изобретению или их возможные соли также обладают сродством ин виво к рецепторам каннабиноидов CB2, присутствующих в селезенке мыши, когда их вводят внутривенно, внутрибрюшинно или перорально. Их активность выявляют путем экспериментов на связывание ex vivo [3H]-CP-55940. Опыты проводят в экспериментальных условиях, описанных M.Rinaldi-Carmona и др. Life Sciences, 56, 1941-1947 (1995).
Соединения согласно настоящему изобретению обладают токсичностью, приемлемой для их использования в качестве лекарственных средств.
Благодаря своим замечательным свойствам, особенно своему высокому сродству и своей селективности к периферическим рецепторам CB2, соединения формулы (I) как таковые или в форме фармацевтически приемлемых солей могут быть использованы в качестве действующих начал лекарственных средств.
Заболеваниями, для лечения которых могут быть использованы соединения формулы (I) и в случае необходимости их фармацевтически приемлемые соли, являются патологии, в которых принимают участие клетки иммунной системы или относящиеся к иммунитету нарушения, как, например, аутоиммунные заболевания, связанные с трансплантацией органов заболевания, инфекционные болезни, аллергические заболевания, заболевания желудочно-кишечного тракта, например болезнь Крона. В особенности можно назвать следующие аутоиммунные заболевания: системная красная волчанка, заболевания соединительной ткани, синдром Шегрена, болезнь Бехтерева, реактивный артрит, ревматоидный полиартрит, недифференцированный спондилоартрит, болезнь Бехчета, аутоиммунные гемолитические анемии, рассеянный склероз, псориаз. Излечимые аллергические заболевания могут быть, например, такими, как аллергическая реакция немедленного типа или астма. Точно так же соединения и их возможные фармацевтически приемлемые соли могут быть использованы для лечения васкулитов, паразитарных инфекций, вирусных инфекций, бактериальных инфекций, амилоидоза, заболеваний, поражающих лимфогематопоэтическую систему.
Таким образом, согласно другому из аспектов настоящего изобретения, оно относится к способу лечения вышеуказанных заболеваний, который состоит во введении пациенту, нуждающемуся в этом, эффективного количества соединения формулы (I) или одной из его фармацевтически приемлемых солей.
Согласно следующему из аспектов настоящего изобретения, оно также относится к применению соединений формулы (I) для получения лекарственных средств, предназначенных для лечения расстройств, связанных с рецепторами каннабиноидов CB2, в особенности относящихся к иммунитету нарушений, и также заболеваний, в которых принимает участие иммунная система.
Более того, соединения формулы (I) или формулы (XIII) согласно изобретению, как таковые или в меченной радиоактивным изотопом форме, могут быть использованы в качестве фармакологических средств для человека или животного для определения и маркировки периферических рецепторов CB2 каннабиноидов. Это составляет следующий аспект настоящего изобретения.
Соединения согласно настоящему изобретению обычно вводятся в виде разовой дозы. Вышеуказанные разовые дозы предпочтительно включают в состав фармацевтических композиций, в которых действующее начало смешано с фармацевтическим эксципиентом.
Таким образом, согласно дальнейшему из аспектов настоящего изобретения, оно относится к фармацевтическим композициям, включающим в качестве действующего начала соединение формулы (I) или одну из его фармацевтически приемлемых солей.
Соединения вышеприведенной формулы (I) и их фармацевтически приемлемые соли могут быть использованы в суточных дозах от 0,01 до 100 мг на килограмм массы тела излечиваемого млекопитающего, предпочтительно в суточных дозах от 0,1 до 50 мг/кг. В случае человека дозу можно изменять предпочтительно в пределах от 0,5 до 4000 мг в день, в особенности от 2,5 до 1000 мг, в зависимости от возраста излечиваемого субъекта или типа обработки: профилактика или лечение.
В фармацевтических композициях настоящего изобретения для введения перорально, подъязычно, путем ингаляции, подкожно, внутримышечно, внутривенно, чрескожно, локально или ректально, действующие начала могут вводиться в унитарных формах введения, в смеси с классическими фармацевтическими носителями, животным и людям. Соответствующие унитарные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы с лекарством, порошки, гранулы и пероральные растворы или суспензии; подъязычные и щечные формы введения; аэрозоли; имплантаты; подкожные, внутримышечные, внутривенные, внутриносовые или внутриглазные формы введения и ректальные формы введения.
Когда готовят твердую композицию в форме таблеток, то к действующему началу, микронизированному или нет, можно добавлять смачиватель, такой как лаурилсульфат натрия, и все смешивают с фармацевтическим эксципиентом, таким как диоксид кремния, желатина, крахмал, лактоза, стеарат магния, тальк, гуммиарабик или аналогичные компоненты. Таблетки можно покрывать сахарозой, различными полимерами или другими соответствующими веществами или еще обрабатывать их таким образом, чтобы они обладали пролонгированной или замедленной активностью и непрерывно высвобождали заданное количество действующего начала.
Препарат в виде желатиновых капсул с лекарством получают путем смешения действующего начала с разбавителем, таким как гликоль или сложный эфир глицерина, и введения полученной смеси в мягкие или твердые желатиновые капсулы.
Препарат в форме сиропа или эликсира может содержать действующее начало вместе с подслащивающим, предпочтительно некалорийным средством, метилпарабеном и пропилпарабеном в качестве антисептика, также как придающим вкус агентом и соответствующим красителем.
Диспергируемые в воде порошки или гранулы могут содержать действующее начало в смеси с диспергаторами, смачивателями или суспендирующими агентами, как поливинилпирролидон, точно так же, как с подслащивающими средствами или корректирующими вкус веществами.
Для ректального введения используют суппозитории, которые готовят со связующими, плавящимися при ректальной температуре, как, например, масло какао или полиэтиленгликоли.
Для парентерального, внутриносового или внутриглазного введения используют водные суспензии, изотонические солевые растворы или стерильные растворы для инъекций, которые содержат диспергаторы и/или фармакологически приемлемые солюбилизирующие агенты, как, например, пропиленгликоль или полиэтиленгликоль.
Таким образом, для приготовления водного раствора для инъекции внутривенно можно использовать сорастворитель: спирт, такой как этанол; гликоль, такой как полиэтиленгликоль или пропиленгликоль; и гидрофильный поверхностно-активный агент, такой как, ТВИН 80. Для приготовления масляного раствора для инъекции внутримышечно можно солюбилизировать действующее начало с помощью триглицерида или сложного эфира глицерина.
Для локального введения можно использовать кремы, мази, гели.
Для чрескожного введения можно использовать пэтчи в мультиламинированной или резервуарной форме, в которых действующее начало может находиться в виде спиртового раствора. Для введения путем ингаляции используют аэрозоль, содержащий, например, сорбитантриолеат или олеиновую кислоту, так же как трихлорфторметан, дихлорфторметан, дихлортетрафторэтан или любой другой, биологически приемлемый пропеллент; также можно использовать систему, содержащую действующее начало индивидуально или в сочетании с эксципиентом, в виде порошка.
Действующее начало также может быть использовано для приготовления препаративной формы в виде микрокапсул или микросфер, в случае необходимости с одним или несколькими носителями или одной или несколькими добавками.
Действующее начало также может находиться в виде комплекса с циклодекстрином, как, например, -, – или – или  – циклодекстрин, 2-гидроксипропил- – циклодекстрин или метил- – циклодекстрин.
Из форм с пролонгированным высвобождением, пригодных для непрерывных обработок, можно использовать имплантаты. Их можно приготовлять в виде масляной суспензии или в форме суспензии микросфер в изотонической среде.
В каждой разовой дозе действующее начало формулы (I) находится в количествах, адаптированных к предусматриваемым суточным дозам. Обычно каждую разовую дозу надлежащим образом подбирают, в зависимости от дозировки и предусматриваемого типа введения, как, например, таблетки, желатиновые капсулы с лекарством и подобные формы, пакетики, ампулы, сиропы и подобные формы, капли, таким образом, что такая разовая доза содержит 0,5-1000 мг действующего начала, предпочтительно 2,5-250 мг, для введения 1-4 раза в день.
Нижеследующие примеры иллюстрируют изобретение, однако не ограничивая объема его охраны.
Температуру плавления или разложения продуктов, т.пл., определяют в капилляре с помощью прибора Тоттоли.
1H-ЯМР-спектры снимают при 200 МГц в гексадейтеродиметилсульфоксиде.
Используемый для препаративной высокоэффективной жидкостной хроматографии (ВЭЖХ) прибор представляет собой таковой модели Prochrom LC 50 с диаметром колонки 50 мм и максимальной высотой слоя 35-40 см.
Используемыми условиями являются следующие: – циклодекстрин, 2-гидроксипропил- – циклодекстрин или метил- – циклодекстрин.
Из форм с пролонгированным высвобождением, пригодных для непрерывных обработок, можно использовать имплантаты. Их можно приготовлять в виде масляной суспензии или в форме суспензии микросфер в изотонической среде.
В каждой разовой дозе действующее начало формулы (I) находится в количествах, адаптированных к предусматриваемым суточным дозам. Обычно каждую разовую дозу надлежащим образом подбирают, в зависимости от дозировки и предусматриваемого типа введения, как, например, таблетки, желатиновые капсулы с лекарством и подобные формы, пакетики, ампулы, сиропы и подобные формы, капли, таким образом, что такая разовая доза содержит 0,5-1000 мг действующего начала, предпочтительно 2,5-250 мг, для введения 1-4 раза в день.
Нижеследующие примеры иллюстрируют изобретение, однако не ограничивая объема его охраны.
Температуру плавления или разложения продуктов, т.пл., определяют в капилляре с помощью прибора Тоттоли.
1H-ЯМР-спектры снимают при 200 МГц в гексадейтеродиметилсульфоксиде.
Используемый для препаративной высокоэффективной жидкостной хроматографии (ВЭЖХ) прибор представляет собой таковой модели Prochrom LC 50 с диаметром колонки 50 мм и максимальной высотой слоя 35-40 см.
Используемыми условиями являются следующие:– неподвижная фаза: Kromasil C 18 – 100 Ангстрем, 10 мкм – подвижная фаза: элюирующее средство А: вода элюирующее средство Б: метанол/вода (90/10 по объему); – расход: 114 мл/мин; позиция насосов 8 мм; – градиент элюирования: Время (мин) – %А %Б 0 – 10 90 5 – 10 90 80 – 4 96 – УФ-детектирование при  =230 нм; длина ячейки…0; поглощение = 0,5 AUFS.
Используемый для аналитической высокоэффективной жидкостной хроматографии (ВЭЖХ) прибор представляет собой ВЭЖХ-анализатор фирмы Хьюлетт Паккард. Используемыми условиями являются следующие: =230 нм; длина ячейки…0; поглощение = 0,5 AUFS.
Используемый для аналитической высокоэффективной жидкостной хроматографии (ВЭЖХ) прибор представляет собой ВЭЖХ-анализатор фирмы Хьюлетт Паккард. Используемыми условиями являются следующие:– колонка: неподвижная фаза: Kromasil (Вотерс) C 18 – 100 Ангстрем, 10 мкм; – подвижная фаза: элюирующее средство А: вода элюирующее средство Б: метанол – градиент элюции: Время (мин) – %А %Б 0 – 20 80 5 – 20 80 50 – 7 93 – расход: 1 мл/мин. – УФ-детектирование при =230 нм; поглощение = 8;– объем введенной пробы: 30 мкл. В Препаративных примерах и в Примерах используют следующие аббревиатуры: Me, OMе: метил, метокси Et, OEt: этил, этокси EtOH: этанол MeOH: метанол эфир: диэтиловый эфир изоэфир: диизопропиловый эфир ДМФА: диметилформамид ДМСО: диметилсульфоксид ДХМ: дихлорметан CCl4: тетрахлорид углерода ТГФ: тетрагидрофуран AcOEt: этилацетат K2CO3: карбонат калия Na2CO3: карбонат натрия KHCO3: гидрокарбонат калия NaHCO3: гидрокарбонат натрия NaCl: хлорид натрия Na2SO4: сульфат натрия MgSO4: сульфат магния NaOH: гидроксид натрия КОН: гидроксид калия AcOH: уксусная кислота H2SO4: серная кислота HCl: соляная кислота HBr: бромоводородная кислота солянокислый эфир: насыщенный раствор хлороводорода в эфире BOP: бензотриазол-1-илокситрис(диметиламино)фосфоний-гексахлорат DBU: 1,8-диазабицикло [5.4.0]ундец-7-ен NH4Cl: хлорид аммония т.пл.: температура плавления т.кип.: температура кипения ТК: комнатная температура диоксид кремния H: силикагель 6OH, выпускаемый в продажу фирмой Мерк (Дармштадт) ВЭЖХ: высокоэффективная жидкостная хроматография TR: время удерживания ЯМР: ядерный магнитный резонанс  : химический сдвиг, выраженный в миллионных долях (м.д.) : химический сдвиг, выраженный в миллионных долях (м.д.)s: с (синглет); se: уш.с (уширенный синглет); sd: дс (двойной синглет); d: д (дублет); dd: (двойной дублет); t: т (триплет); qd кд (квадруплет); sept: септуплет; mt: мультиплет; т: массив. Препаративные примеры Препаративный пример 1.1 1-(3,4-Дихлорбензил)-5-(4-метилфенил)пиразол-3-карбоновая кислота А) Натриевая соль метил-4-(4-метилфенил)-2-оксо-4- оксидобут-3-еноата 6.24 г Натрия растворяют в 150 мл метанола. После охлаждения до комнатной температуры добавляют 36,4 мл 4′-метилацектофенона, затем раствор 37 мл диэтилоксалата в 50 мл метанола. После этого добавляют 100 мл метанола для разбавления реакционной смеси и перемешивают 2 часа при комнатной температуре. Добавляют 500 мл диэтилового эфира и перемешивают 30 минут при комнатной температуре. Отсасывают выпавший осадок, промывают его диэтиловым эфиром и высушивают его. Получают 57,4 г целевого продукта. Б) Метиловый эфир 5-(4-метилфенил)пиразол-3-карбоновой кислоты На бане со льдом охлаждают смесь 30 г полученного в предыдущей стадии соединения в 100 мл уксусной кислоты и прикапывают 7,94 мл 55% -ного раствора гидразина в воде. Затем реакционную смесь кипятят с обратным холодильником в течение 5 часов и перемешивают в течение ночи при комнатной температуре. Выпавший осадок отсасывают, промывают его водой и таким образом получают первую порцию продукта. Фильтрат выливают в смесь воды со льдом, отсасывают выпавший осадок, промывают его водой, высушивают его и получают вторую порцию продукта. Первую и вторую порции продукта объединяют, промывают этилацетатом и сушат в вакууме. Получают 21,44 г целевого продукта. В) Метиловый эфир 1-(3,4- дихлорбензил)-5-(4- метилфенил) пиразол-3-карбоновой кислоты К суспензии 5 г полученного в предыдущей стадии соединения в 50 мл толуола при комнатной температуре и порциями добавляют 2,03 г гидрида натрия в виде 60%-ной дисперсии в масле, затем реакционную смесь нагревают при 65oC в течение 1 часа. После охлаждения до комнатной температуры прикапывают 5,82 г 3,4 – дихлорбензилбромида, затем кипятят с обратным холодильником в течение 20 часов. Реакционную смесь охлаждают до комнатной температуры и прикапывают 100 мл 50%-ного раствора хлорида аммония в воде. После декантации органическую фазу промывают насыщенным раствором хлорида натрия, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексан/этилацетат (75:25 по объему). Получают 5,28 г целевого продукта, т.пл. = 98,6oC. Г) 1-(3,4-Дихлорбензил)-5-(4-метилфенил)пиразол-3- карбоновая кислота К раствору 5,2 г полученного в предыдущей стадии соединения в 100 мл этанола при комнатной температуре добавляют раствор 1,16 г КОН в 20 мл воды, затем кипятят с обратным холодильником в течение 5 часов. Реакционную смесь перемешивают при комнатной температуре в течение ночи, добавляют 200 мл водного 1 н. раствора соляной кислоты, выпавший осадок отсасывают и высушивают его в вакууме. Получают 5,12 г целевого продукта, т.пл. = 171,5oC. ЯМР-спектр, (м. д.): 2,4 (с, 3H); 5,5 (с, 2H); 6,9 (с, 1H); 7,0 (дд, 1H); 7,3 (д, 1H); 7,35 (система AA’-BB’, 4H); 7,65 (д, 1H); 1,30 (уш.с, 1H).
Наблюдают эффект Оверхаузера (N.O.E.) между бензильными протонами (R4= R5=H) и протонами g2=g6=H.
Препаративный пример 1.21-(3-Хлор-4-метилбензил)-5-(4-метилфенил)пиразол-3-карбоновая кислота А) Метиловый эфир 1-(3-хлор-4-метилбензил)-5-(4-метилфенил) пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии В Препаративного примера 1.1, исходя из 5 г соединения, полученного в стадии Б Препаративного примера 1.1, 2,03 г гидрида натрия в виде 60%-ной дисперсии в масле, 100 мл толуола и 4,22 г 3-хлор-3-метилбензилхлорида. Получают 1,52 г целевого продукта. Б) 1-(3-Хлор-4-метилбензил)-5-(4-метилфенил)пиразол-3- карбоновая кислота К раствору 1,52 г полученного в предыдущей стадии соединения в 50 мл этанола при комнатной температуре добавляют раствор 0,36 г КОН в 10 мл воды, затем кипятят с обратным холодильником в течение ночи. Реакционную смесь концентрируют в вакууме, остаток обрабатывают водой, водную фазу промывают эфиром, водную фазу подкисляют до pH 2 путем добавления 6 н. раствора соляной кислоты, выпавший осадок отсасывают, промывают его водой и высушивают его в вакууме. Получают 1,42 г целевого продукта, т.пл. = 108,5oC. ЯМР-спектр, (м.д.): 2,1-2,45 (2с, 6H); 5,4 (с, 2H); 6,7-7,5 (м, 8H); 12,85 (уш.с, 1H).
Препаративный пример 1.31-(3-Фтор-4-метилбензил)-5-(4-метилфенил)пиразол-3- карбоновая кислота и 1-(3-фтор-4-метилбензил)-3-(4-метилфенил) пиразол-5-карбоновая кислота А) Метиловый эфир 1-(3-фтор-4-метилбензил)-5-(4- метилфенил)пиразол-3-карбоновой кислоты и метиловый эфир 1- (3-фтор-4-метилбензил)-3-(4-метилфенил)пиразол-5-карбоновой кислоты К суспензии 2,5 г полученного в стадии Б Препаративного примера 1.1 соединения в 100 мл толуола при комнатной температуре и порциями добавляют 1,01 г гидрида натрия в виде 60%-ной дисперсии в масле, затем реакционную смесь нагревают в течение 1 часа при 65oC. После охлаждения до комнатной температуры прикапывают 2,45 г 3-фтор-4-метилбензилбромида (Препаративный пример 3.1), затем кипятят с обратным холодильником в течение 48 часов. После охлаждения до комнатной температуры прикапывают 50 мл 50%-ного раствора хлорида аммония в воде. После декантации органическую фазу промывают насыщенным раствором хлорида натрия, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью этилацетата с циклогексаном (50:50 по объему). Получают 1,29 г смеси целевых продуктов. Б) 1-(3-Фтор-4-метилбензил)-5-(4-метилфенил)пиразол-3- карбоновая кислота и 1-(3-фтор-4-метилбензил)-3-(4-метилфенил) пиразол-5-карбоновая кислота К раствору 1,2 г смеси соединений, полученных в предыдущей стадии, в 50 мл этанола при комнатной температуре добавляют раствор 0,3 г КОН в 10 мл воды, затем кипятят с обратным холодильником в течение 5 часов и перемешивают в течение ночи при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток обрабатывают водой, водную фазу промывают этилацетатом, водную фазу подкисляют до pH 2 путем добавления 6 н. раствора соляной кислоты, выпавший осадок отсасывают, промывают его водой и высушивают его в вакууме. Получают 0,95 г смеси целевых продуктов. Препаративный пример 1.4 1-(3,4-Дихлорбензил)-5-(4-метоксифенил)пиразол-3-карбоновая кислота А) Натриевая соль метил-4-(4-метоксифенил)-2-оксо-4- оксидобут-3-еноата 3,9 г натрия растворяют в 100 мл метанола. После охлаждения до комнатной температуры добавляют 24,4 г 4′-метоксиацетофенона, затем раствор 23 мл диэтилоксалата в 50 мл метанола и перемешивают 2 часа при комнатной температуре. Добавляют 500 мл диэтилового эфира и перемешивают 30 минут при комнатной температуре. Выпавший осадок отсасывают, промывают его диэтиловым эфиром и высушивают его. Получают 33,4 г целевого продукта. Б) Метиловый эфир 5-(4-метоксифенил)пиразол-3-карбоновой кислоты На бане со льдом охлаждают смесь 9 г полученного в предыдущей стадии соединения в 100 мл уксусной кислоты и прикапывают 2,2 мл гидразинмоногидрата. Затем реакционную смесь кипятят с обратным холодильником в течение 5 часов и перемешивают в течение ночи при комнатной температуре. Реакционную смесь выливают в смесь воды со льдом, отсасывают выпавший осадок и промывают его водой. Осадок обрабатывают дихлорметаном, отфильтровывают нерастворимую часть, фильтрат сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 7.1 г целевого продукта. В) Метиловый эфир 1-(3,4-дихлорбензил)-5-(4-метоксифенил) пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии В Препаративного примера 1.1, исходя из 3,5 г полученного в предыдущей стадии соединения, 1,32 г гидрида натрия в виде 60%-ной дисперсии в масле, 150 мл толуола и 3,77 г 3,4-дихлорбензилбромида. Продукт хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (60:40 по объему). Получают 2.4 г целевого продукта; т.пл. = 95,5oC. ЯМР-спектр (м.д.): 3,8 (2с, 6H); 5,45 (с, 2H); 6,8-7,1 (м, 4H); 7,25 (д, 1H); 7.35 (д, 2H); 7,55 (д, 1H)Г) 1-(3,4-дихлорбензил)-5-(4-метоксифенил)пиразол-3- карбоновая кислота Это соединение получают по методике, описанной в стадии Б Препаративного примера 1.2, исходя из 2,4 г полученного в предыдущей стадии соединения, 50 мл этанола, 0,51 г КОН и 10 мл воды. Получают 2,23 г целевого продукта. Препаративный пример 1.5 1-(3-Хлор-4-метилбензил)-5-(4-метоксифенил)пиразол-3- карбоновая кислота и 1-(3-хлор-4-метилбензил)-3-(4-метоксифенил) пиразол-5-карбоновая кислота А) Метиловый эфир 1-(3-хлор-4-метилбензил)-5-(4- метоксифенил)пиразол-3-карбоновой кислоты и метиловый эфир 1-(3-хлор-4-метилбензил)-3-(4-метоксифенил)пиразол-5-карбоновой кислоты Смесь этих двух соединений получают по методике, описанной в стадии А Препаративного примера 1.3, исходя из 3,5 г полученного в стадии Б Препаративного примера 1.4 соединения, 1,32 г гидрида натрия в виде 60%-ной дисперсии в масле, 100 мл толуола и 2,75 г 3-хлор-4-метилбензилхлорида. Получают 1,93 г смеси целевых продуктов. Б) 1-(3-Хлор-4-метилбензил)-5-(4-метоксифенил) пиразол-3-карбоновая кислота и 1-(3-хлор-4-метилбензил)-3-(4- метоксифенил)пиразол-5-карбоновая кислота Смесь этих двух соединений получают по методике, описанной в стадии Б Препаративного примера 1.3, исходя из 1,9 г смеси соединений, полученных в предыдущей стадии, 50 мл этанола, 0.43 г КОН и 10 мл воды. Получают 1,73 г смеси целевых продуктов. Препаративный пример 1.6 1-(3-Хлор-4-метилбензил)-5-(4-фторфенил)пиразол-3-карбоновая кислота А) Натриевая соль метил-4-(4-фторфенил)-3-оксо-4- оксидобут-3-еноата Это соединение получают по методике, описанной в стадии А Препаративного примера 1.1, исходя из 4,16 г натрия в 100 мл метанола, 21,87 мл 4′-фторацетофенона и 24,72 мл диэтилоксалата в 50 мл метанола. Получают 42,68 г целевого продукта. Б) Метиловый эфир 5-(4-фторфенил)пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии Б Препаративного примера 1.1, исходя из 15 г полученного в предыдущей стадии соединения, 100 мл уксусной кислоты и 3,73 мл 55%-ного раствора гидразина в воде. После перемешивания в течение ночи при комнатной температуре реакционную смесь выливают в смесь воды со льдом, отсасывают выпавший осадок, промывают его водой и высушивают его. Получают 10,74 г целевого продукта. В) Метиловый эфир 1-(3-хлор-4-метилбензил)-5-(4-фторфенил) пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии В Препаративного примера 1.1, исходя из 3,5 г полученного в предыдущей стадии соединения, 1,4 г гидрида натрия в виде 60%-ной дисперсии в масле, 100 мл толуола и 4,45 г 3-хлор-4-метилбензилиодида (Препаративный пример 3.2) в 50 мл толуола. Хроматографируют на диоксиде кремния, элюируя смесь циклогексана с этилацетатом (70:30 по объему). Получают 1,57 г целевого продукта; т.пл. = 110oC. Г) 1-(3-Хлор-4-метилбензил)-5-(4-фторфенил)пиразол-3- карбоновая кислота Это соединение получают по методике, описанной в стадии Б Препаративного примера 1.2, исходя из 1,55 г полученного в предыдущей стадии соединения, 50 мл этанола, 0,36 г КОН и 10 мл воды. Получают 1,46 г целевого продукта; т. пл. = 120oC. Препаративный пример 1.7 1-(2,4-Дихлорбензил)-5-(4-метоксифенил)пиразол-3- карбоновая кислота А) Натриевая соль метил-4-(4-хлорфенил)-2-оксо-4- оксидобут-3-еноата Это соединение получают по методике, описанной в стадии А Препаративного примера 1.1, исходя из 12,66 г натрия в 270 мл метанола, 68,4 мл 4′-хлорацетофенона и 71,8 мл диэтилоксалата в 110 мл метанола. Получают 97 г целевого продукта. Б) Метиловый эфир 5-(4-хлорфенил)пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии Б Препаративного примера 1.4, исходя из 15 г полученного в предыдущей стадии соединения, 65 мл уксусной кислоты, 3,05 мл гидразинмоногидрата. Полученный осадок порошкуют в смеси 100 мл дихлорметана с 50 мл этилацетата, отсасывают его и высушивают. Получают 8,13 г целевого продукта; т.пл. = 215oC. В) Метиловый эфир 1-(2,4-дихлорбензил)-5-(4-хлорфенил) пиразол-3-карбоновой кислоты К суспензии 1,02 г гидрида натрия (60%-ный в масле) в 100 мл толуола при комнатной температуре прикапывают раствор 5,07 г полученного в предыдущей стадии соединения в 100 мл толуола, затем нагревают при 65oC в течение 1 часа. После этого добавляют 3,12 мл 2,4-диихлорбензилхлорида и кипятят с обратным холодильником в течение 20 часов. Реакционную смесь охлаждают до комнатной температуры, добавляют 100 мл 50%-ного раствора хлорида аммония в воде. После декантации органическую фазу промывают насыщенным раствором хлорида натрия, сушат над сульфатом магния и растворитель выпаривают в вакууме, остаток хроматографируют на диоксиде кремния, элюируя смесью толуол/дихлорметан/этилацетат (80: 10: 10 по объему). Получают 3,68 г целевого продукта; т.пл. = 105oC. Г) 1-(2,4-Дихлорбензил)-5-(4-хлорфенил)пиразол-3- карбоновая кислота Это соединение получают по методике, описанной в стадии Г Препаративного примера 1.1, исходя из 3,6 г полученного в предыдущей стадии соединения, 60 мл метанола, 1,27 г КОН и 6 мл воды. Получают 3,52 г целевого продукта; т. пл. = 185oC. Препаративный пример 1.8 1-(3-Хлор-4-метилбензил)-5-(3,4-диметилфенил)пиразол-3- карбоновая кислота А) Натриевая соль метил-4-(3,4-диметилфенил)-2-оксо-4- оксидобут-3-еноата Это соединение получают по методике, описанной в стадии А Препаративного примера 1.1, исходя из 3,9 г натрия в 100 мл метанола, 25 г 3′,4′-диметилацетофенона и 23 мл диэтилоксалата в 50 мл метанола. Получают 39,42 г целевого продукта. Б) Метиловый эфир 5-(3,4-диметилфенил)пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии Б Препаративного примера 1.6, исходя из 10 г полученного в предыдущей стадии соединения в 150 мл уксусной кислоты и 2,2 мл 55%-ного раствора гидразина в воде. Получают 9 г целевого продукта. В) Метиловый эфир 1-(3-хлор-4-метилбензил)-5-(3,4- диметилфенил)пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии В Препаративного примера 1.1, исходя из 1,52 г гидрида натрия в виде 60%-ной дисперсии в масле, 4 г полученного в предыдущей стадии соединения, 70 мл толуола и 5,8 г 3-хлор-4-метилбензилиодида (Препаративный пример 3.2). Полученный продукт очищают путем порошкования в гексане, затем отсасывания и промывки гексаном. Получают 2,84 г целевого продукта; т.пл. = 88oC. ЯМР-спектр, (м. д. ): 2,0-2,35 (м, 9H); 3,95 (с, ЗH); 5,3 (с, 2H); 6,6-7,4 (м, 7H).
Г) 1-(3-Хлор-4-метилбензил)-5-(3,4-диметилфенил)пиразол-3- карбоновая кислотаЭто соединение получают по методике, описанной в стадии Б Препаративного примера 1.2, исходя из 1,8 г полученного в предыдущей стадии соединения в 30 мл этанола и 0,392 г КОН в 30 мл воды. Получают 1,6 г целевого продукта; т. пл. = 163oC. Препаративный пример 1.9 1-(3-Хлор-4- фторбензил)-5-(3,4-диметилфенил)пиразол-3- карбоновая кислота А) Метиловый эфир 1-(3-хлор-4-фторбензил)-5-(3,4-диметилфенил) пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии В Препаративного примера 1.1, исходя из 2,5 г соединения, полученного в стадии Б Препаративного примера 1.8, 50 мл толуола, 0,88 г гидрида натрия в виде 60%-ной дисперсии в масле и 2,5 г 3- хлор-4-фторбензилбромида (Препаративный пример 3.3). Продукт очищают путем порошкования в этилацетате, затем отсасывания и высушивания. Получают 3,6 г целевого продукта. Б) 1-(3-Хлор-4-фторбензил)-5-(3,4-диметилфенил)пиразол-3- карбоновая кислота Это соединение получают по методике, описанной в стадии Б Препаративного примера 1.2, исходя из 1,6 г полученного в предыдущей стадии соединения в 25 мл этанола и 0,481 г КОН в 10 мл воды. Получают 1,22 г целевого продукта. ЯМР-спектр, (м.д.): 2,2 (2с, 6H); 5,35 (с, 2H); 6,6-7,4 (м, 7H).
Препаративный пример 1.101-(4-Метилбензил)-5-(4-хлор-3-метилфенил)пиразол-3- карбоновая кислота А) Натриевая соль метил-4-(4-хлор-3-метилфенил)-2-оксо-4- оксидобут-3-еноата Это соединение получают по методике, описанной в стадии А Препаративного примера 1.1, исходя из 7,6 г натрия в 100 мл метанола, 55,6 г 4′-хлор-3′-метилацетофенона и 45 мл диэтилоксалата в 100 мл метанола. Получают 85,8 г целевого продукта. Б) Метиловый эфир 5-(4-хлор-3-метилфенил)пиразол-3- карбоновой кислоты Это соединение получают по методике, описанной в стадии Б Препаративного примера 1-4, исходя из полученного в предыдущей стадии соединения в 150 мл уксусной кислоты и 2,9 мл гидразинмоногидрата. После перемешивания в течение ночи при комнатной температуре реакционную смесь выливают в воду со льдом, отсасывают выпавший осадок и промывают его водой. После высушивания получают 13 г целевого продукта. В) Метиловый эфир 1- (4-метилбензил)-5-(4-хлор-3-метилфенил) пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии В Препаративного примера 1-1, исходя из 5 г полученного в предыдущей стадии соединения в 50 мл толуола, 1,8 г гидрида натрия в виде 60%-ной дисперсии в масле и 4,07 г 4-метилбензилбромида. Получают 4,6 г целевого продукта; т.пл. = 98oC. ЯМР-спектр, (м.д.): 2,0-2,4 (2с, 6H); 3,8 (с, 3H); 5,4 (с, 2H); 6,7-7,6 (м, 8H).
Г) 1-(4-Метилбензил)-5-(4-хлор-3-метилфенил)пиразол-3- карбоновая кислотаЭто соединение получают по методике, описанной в стадии Б Препаративного примера 1.2, исходя из 4 г полученного в предыдущей стадии соединения в 100 мл этанола и 0,95 г КОН в 20 мл воды. Получают 3,4 г целевого продукта; т. пл. = 180oC. ЯМР-спектр, (м.д.): 2,25 (с, 3H); 2,35 (с, 3H); 5,4 (с, 2H); 6,7-7,15 (м, 5H); 7,25 (дд, 1Н); 7,4-7,6 (м, 2Н).
Следуя методикам, описанным в стадии В (исходя из соединения, полученного в стадии Б Препаративного примера 1.10 и соответствующих бензилгалогенидов), затем в стадии Г Препаративного примера 1.10, получают сложные эфиры, затем кислоты, описанные в таблице 1. Препаративный пример 1.11: Z= OMe; ЯМР-спектр, (м.д.) 2,35 (с, Н); 3,85 (с, Н); 5,5 (с, 2Н); 6,8-7,6 (м, 8Н).
Препаративный пример 1.141-[1-(2,4-Дихлорфенил)этил] -5-(4-хлорфенил) пиразол-3- карбоновая кислота А) Метиловый эфир 1-[1-(2,4-дихлорфенил)этил]-5-(4- хлорфенил)пиразол-3-карбоновой кислоты К суспензии 1,5 г соединения, полученного в стадии Б Препаративного примера 1.7, в 50 мл толуола при комнатной температуре и порциями добавляют 0,3 г гидрида натрия в виде 60%-ной дисперсии в масле, затем нагревают при 65oC в течение 30 минут. После охлаждения до комнатной температуры прикапывают 1,77 г 1-(1-бромэтил)-2,4-дихлорбензола (Препаративный пример 3.4), затем кипятят с обратным холодильником в течение 5 дней. После охлаждения до комнатной температуры реакционную смесь выливают в 100 мл 50%-ного раствора хлорида аммония в охлажденной до 0oC воде. Экстрагируют этилацетатом, органическую фазу промывают водой, насыщенным раствором хлорида натрия, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью толуола с этилацетатом (95:5 по объему). Получают 1,02 г целевого продукта. Б) 1-[1-(2,4-Дихлорфенил)этил]-5-(4-хлорфенил)пиразол-3- карбоновая кислота К раствору 1,02 г полученного в предыдущей стадии соединения в 20 мл метанола при комнатной температуре добавляют раствор 0,35 г КОН в 5 мл воды, затем кипятят с обратным холодильником в течение двух часов. Реакционную смесь выливают в 100 мл охлажденного до 0oC 5%-ного раствора соляной кислоты, отсасывают выпавший осадок, промывают его водой и высушивают его в вакууме. Получают 0,74 г целевого продукта; т.пл. = 80oC. Препаративный пример 1.15 1-(3,4-Дихлорбензил)-5-(2,6-диметоксифенил)пиразол-3- карбоновая кислота А) Калиевая соль этил-4-(2,6-диметоксифенил)-2- оксо-4-оксидобут-3-еноата Смесь 18 г 2′,6′-диметоксиацетофенона в 54 мл этанола нагревают до 50oC и в течение 5 минут добавляют раствор 13,4 г трет-бутилата калия в 72 мл этанола. Реакционную смесь нагревают до температуры кипения с обратным холодильником, в течение 10 минут добавляют 16.3 мл диэтилоксалата и продолжают кипятить с обратным холодильником в течение 1 часа. Отгоняют 40 мл этанола, затем перемешивают в течение 2,5 часов при снижении температуры до комнатной, отсасывают выпавший осадок, промывают его этанолом и высушивают его в вакууме при 60oC. Получают 31 г целевого продукта. Б) Метиловый эфир 5-(2,6-диметоксифенил)пиразол-3- карбоновой кислоты Это соединение получают по методике, описанной в стадии Б Препаративного примера 1.4, исходя из 4 г полученного в предыдущей стадии соединения, 50 мл уксусной кислоты и 0,7 мл гидразинмоногидрата. После перемешивания в течение ночи при комнатной температуре реакционную смесь выливают в смесь воды со льдом, экстрагируют этилацетатом, органическую фазу промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель частично удаляют в вакууме. Выпавший осадок отсасывают и высушивают его. Получают 2,53 г целевого продукта. В) 1-(3,4-Дихлорбензил)-5-(2,6-диметоксифенил)пиразол- 3-карбоновая кислота Это соединение получают по методике, описанной в стадии В Препаративного примера 1.1, исходя из 2,5 г полученного в предыдущей стадии соединения, 50 мл толуола, 0,88 г гидрида натрия в виде 60%-ной дисперсии в масле и 2,16 г 3,4-дихлорбензилбромида. После кипячения с обратным холодильником в течение ночи реакционную смесь охлаждают до комнатной температуры, прикапывают 50 мл 50%-ного раствора хлорида аммония в воде, отсасывают выпавший осадок и высушивают его. Получают 1,1 г целевого продукта. ЯМР-спектр, (м. д. ): 3,5 (с, 6H); 4,8 (очень уширенный сигнал, 1H); 4,95 (с, 2H); 6,25 (с, 1H); 6,6 (д, 2H); 6,85 (дд, 1H); 6,95 (д, 1H); 7,3 (т, 1H), 7,4 (д, 1H).
Препаративный пример 1.161-(4-Фторбензил)-5-(3,4-диметилфенил)пиразол-3-карбоновая кислота А) Метиловый эфир 1-(4-фторбензил)-5-(3,4-диметилфенил)- пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии В Препаративного примера 1.1, исходя из 2,3 г соединения, полученного в стадии Б Препаративного примера 1.8, 50 мл толуола, 0,88 г гидрида натрия в виде 60%-ной дисперсии в масле и 2,08 г 4- фторбензилбромида. Получают 1 г целевого продукта; т.пл. = 93oC. ЯМР-спектр, (м. д.): 2,25 (2с, 6H); 3,8 (с, 3H); 5,4 (с, 2H); 6,85 (с, 1H); 6,9-7,3 (м, 7H).
Б) 1-(4-Фторбензил)-5-(3,4-диметилфенил)пиразол-3- карбоновая кислотаЭто соединение получают по методике, описанной в стадии Б Препаративного примера 1.14, исходя из 1 г полученного в предыдущей стадии соединения, 15 мл метанола, 0,406 г КОН и 15 мл воды. Получают 0,94 г целевого продукта; т. пл. = 141oC. ЯМР-спектр, (м.д.): 2 (2с, 6H); 3,4 (уш.с, 1H); 5,35 (с, 2H); 6,75 (с, 1H); 6,8-7,3 (м, 7H).
Препаративный пример 1.171-(2,4-Дихлорбензил)-5-(4-метилфенил)пиразол-3-карбоновая кислота А) Метиловый эфир 1-(2,4-дихлорбензил)-5-(4-метилфенил)- пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии В Препаративного примера 1.1, исходя из 2.5 г соединения, полученного в стадии Б Препаративного примера 1.1, 1 г гидрида натрия в виде 60%-ной дисперсии в масле, 50 мл толуола и 2,3 г 2,4-дихлорбензилхлорида. Получают 2,53 г целевого продукта; т.пл. = 105oC. Б) 1-(2,4-Дихлорбензил)-5-(4-метилфенил)пиразол-3- карбоновая кислота К суспензии 1.5 г полученного в предыдущей стадии соединения в 15 мл метанола при комнатной температуре добавляют раствор 0,5 г КОН в 15 мл воды, затем кипятят с обратным холодильником в течение двух часов. Реакционную смесь концентрируют в вакууме, остаток выливают в смесь 1 н. соляной кислоты со льдом, выпавший осадок отсасывают, промывают его водой и высушивают его в вакууме. Получают 1,4 г целевого продукта. ЯМР-спектр, (м.д.): 2,3 (с, 3H); 5,45 (с, 2H); 6,6-7,7 (м, 8H); 12,85 (уш.с, 1H).
Препаративный пример 1.181-(4-Этилбензил)-5-(4-хлор-3-метилфенил)пиразол-3- карбоновая кислота А) Метиловый эфир 1-(4-этилбензил)-5-(4-хлор-3-метилфенил) пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии В Препаративного примера 1.1, исходя из 2,5 г соединения, полученного в стадии Б Препаративного примера 1.10, в 50 мл толуола, 0,88 г гидрида натрия в виде 60%-ной дисперсии в масле и 2 г 4-этилбензилбромида (Препаративный пример 3.5). После гидролиза с помощью 50%-ного водного раствора хлорида аммония, затем декантации, органическую фазу концентрируют в вакууме. Остаток экстрагируют этилацетатом, органическую фазу промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (80:20 по объему). Получают 2,63 г целевого продукта. Б) 1-(4-Этилбензил)-5-(4-хлор-3-метилфенил)пиразол-3- карбоновая кислота Это соединение получают по методике, описанной в стадии Б Препаративного примера 1.17, исходя из 2,5 г полученного в предыдущей стадии соединения в 20 мл метанола и 0,57 г КОН в 20 мл воды. Получают 2,2 г целевого продукта. ЯМР-спектр, (м. д.): 1.1 (т, 3H); 2,3 (с, 3H); 2,5 (мультиплет, 2H); 5,4 (с, 2H); 6,7-7,7 (м, 8H); 12,9 (уш.с, 1H).
Препаративный пример 1.191-(3,4-Дихлорбензил)-5-(4-хлор-3-метилфенил)пиразол-3- карбоновая кислота К суспензии 2,5 г соединения, полученного в стадии Б Препаративного примера 1.10, в 25 мл толуола при комнатной температуре и порциями добавляют 0,88 г гидрида натрия в виде 60%-ной дисперсии в масле, затем реакционную смесь нагревают при 65oC в течение 1 часа. После охлаждения до комнатной температуры прикапывают раствор 2,4 г 3,4-дихлорбензилбромида в 25 мл толуола, затем кипятят с обратным холодильником в течение ночи. Реакционную смесь охлаждают до комнатной температуры, отсасывают выпавший осадок и высушивают его. Получают 2 г целевого продукта, используемого без дальнейшей очистки. Препаративный пример 1.20 1-(2,4-Дихлорбензил)-5-(4-хлор-3-метилфенил)пиразол-3- карбоновая кислота А) Метиловый эфир 1-(2,4-дихлорбензил)-5-(4-хлор-3-метилфенил) пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии А Препаративного примера 1.18, исходя из 2,5 г соединения, полученного в стадии Б Препаративного примера 1.10, в 25 мл толуола, 0,88 г гидрида натрия в виде 60%-ной дисперсии в масле и 1,6 мл 2,4-дихлорбензилхлорида в 25 мл толуола. Получают 0,86 г целевого продукта. Б) 1-(2,4-Дихлорбензил)-5-(4-хлор-3-метилфенил)пиразол-3- карбоновая кислота Это соединение получают по методике, описанной в стадии Б Препаративного примера 1.17, исходя из 0,5 г полученного в предыдущей стадии соединения в 15 мл метанола и 0,205 г КОН в 15 мл воды. Получают 0,31 г целевого продукта. ЯМР-спектр, (м.д.): 2,25 (с, 3H); 5,4 (с, 2H); 6,6- 7,6 (м, 7H); 3,3 (с DOH:1H).
Наблюдают эффект Оверхаузера (N.O.E.) между бензильными протонами (R4= R5=H) и протонами g2=g6=Н.
Препаративный пример 1.211-(4-Метилбензил)-5-(3,4-дихлорфенил)пиразол-3-карбоновая кислота А) Натриевая соль метил-4-(3,4-дихлорфенил)-2-оксо-4- оксидобут-3-еноата 15,17 г натрия растворяют в 500 мл метанола. После охлаждения до комнатной температуры добавляют 124,97 г 3′, 4′-дихлорацетофенона, затем раствор 91 мл диэтилоксалата в 400 мл метанола и перемешивают в течение двух часов при комнатной температуре. Добавляют 1 л диэтилового эфира и перемешивают 30 минут при комнатной температуре. Выпавший осадок отсасывают, промывают его диэтиловым эфиром и высушивают его. Получают 140,77 г целевого продукта. Б) Метиловый эфир 5-(3,4-диихлорфенил)пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии Б Препаративного примера 1.1, исходя из 15 г полученного в предыдущей стадии соединения в 150 мл уксусной кислоты и 6 мл 55 %-ного водного раствора гидразина. После перемешивания в течение ночи при комнатной температуре реакционную смесь выливают в ледяную воду, выпавший осадок отсасывают и промывают его водой. После высушивания получают 12,9 г целевого продукта. В) Метиловый эфир 1-(4-метилбензил)-5-(3,4-дихлорфенил) пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии А Препаративного примера 1.18, исходя из 2,5 г полученного в предыдущей стадии соединения, в 25 мл толуола, 0,88 г гидрида натрия в виде 60%-ной дисперсии в масле и 1,9 г метилбензилбромида в 25 мл толуола. Получают 0,82 г целевого продукта. Г) 1-(4-Метилбензил)-5-(3,4-дихлорфенил)пиразол-3- карбоновая кислота Это соединение получают по методике, описанной в стадии Б Препаративного примера 1.17, исходя из 0,7 г полученного в предыдущей стадии соединения в 15 мл метанола и 0,252 г КОН в 15 мл воды. Получают 0,65 г целевого продукта. ЯМР-спектр, (м. д.): 2,2 (с, 3H); 5,35 (с, 2H); 6,7-7,2 (м, 5H); 7,4 (дд, 1H); 7,6-7,8 (м, 2H); 12,85 (уш.с, 1H).
Препаративный пример 1.221-(3-Хлор-4-метилбензил)-5-(3,4-дихлорфенил)пиразол- 3-карбоновая кислота А) Метиловый эфир 1-(3-хлор-4-метилбензил)-5-(3,4-дихлорфенил) пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии А Препаративного примера 1.18, исходя из 20,51 г соединения, полученного в стадии Б Препаративного примера 1.21, в 350 мл толуола, 2,18 г гидрида натрия в виде 60%-ной дисперсии в масле и 21,96 г 3-хлор-4-метилбензилиодида (Препаративный пример 3.2). После кристаллизации из гексана получают 21,79 г целевого продукта. Б) 1-(3-Хлор-4-метилбензил)-5-(3,4-дихлорфенил)пиразол-3- карбоновая кислота Это соединение получают по методике, описанной в стадии Б Препаративного примера 1.17, исходя из 21,79 г полученного в предыдущей стадии соединения в 59 мл метанола и 8,93 г КОН в 50 мл воды. Получают 17,43 г целевого продукта, используемого без дальнейшей очистки. Препаративный пример 1.23 1-(2,4-Дихлорбензил)-5-(4-метилтиофенил)пиразол-3- карбоновая кислота А) 4′-Метилтиоацетофенон При температуре от 0oC до 10oC, 11,8 мл ацетилхлорида прикапывают к суспензии 20,4 г хлорида алюминия в 85 мл хлороформа. Затем, при температуре 0-5oC, прикапывают 15 мл тиоанизола и перемешивают 1,5 часа при комнатной температуре. Реакционную смесь охлаждают до 0oC и гидролизуют за счет добавления 100 мл воды. Экстрагируют хлороформом, органическую фазу промывают водой, сушат над сульфатом натрия и растворитель выпаривают в вакууме. После кристаллизации из этанола и перекристаллизации из гептана получают 10,7 г целевого продукта. А) Натриевая соль метил-4-(4-метилтиофенил)-2-оксо-4- оксидобут-3-еноата 1,48 г Натрия растворяют в 35 мл метанола и этот раствор быстро добавляют к суспензии 10,7 г полученного в предыдущей стадии соединения и 9,8 мл диэтилоксалата в 80 мл охлажденного до 0oC метанола. Перемешивают 30 минут при комнатной температуре, кипятят с обратным холодильником в течение 1 часа, затем в течение двух часов перемешивают при комнатной температуре. Реакционную смесь выливают в 400 мл диэтилового эфира, перемешивают в течение 15 минут, выпавший осадок отсасывают, промывают его диэтиловым эфиром и высушивают его. Получают 12,8 г целевого продукта. В) Метиловый эфир 5-(4-метилтиофенил)пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии Б Препаративного примера 1.1, исходя из 15 г полученного в предыдущей стадии соединения в 140 мл уксусной кислоты и 7 мл 55%- ного водного раствора гидразина. После перемешивания в течение ночи при комнатной температуре реакционную смесь выливают в смесь воды со льдом, выпавший осадок отсасывают и промывают его водой. После высушивания в вакууме над КОН получают 12,96 г целевого продукта. Г) Метиловый эфир 1-(2,4-дихлорбензил)-5-(4-метилтиофенил) пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии А Препаративного примера 1.18, исходя из 5 г полученного в предыдущей стадии соединения в 100 мл толуола, 1,063 г гидрида натрия в виде 60%-ной дисперсии в масле и 3,4 мл 2,4-дихлорбензилхлорида. Получают 3,2 г целевого продукта. Д) 1-(2,4-Дихлорбензил)-5-(4-метилтиофенил)пиразол-3- карбоновая кислота Это соединение получают по методике, описанной в стадии Б Препаративного примера 1.14, исходя из 1,52 г полученного в предыдущей стадии соединения в 30 мл метанола и 0,7 г КОН в 25 мл воды. После высушивания в вакууме получают 1,4 г целевого продукта. ЯМР-спектр, (м. д.): 2,5 (с, 3H); 5,4 (с, 2H); 6,6-7,6 (м, 8H); 12,8 (уш.с, 1H).
Препаративный пример 1.241-(2,4-Дихлорбензил)-5-(4-трифторфенил)пиразол-3- карбоновая кислота А) Натриевая соль метил-4-(4-трифторметилфенил)-2-оксо-4- оксидобут-3-еноата Это соединение получают по методике, описанной в стадии А Препаративного примера 1.4, исходя из 4′-трифторметилацетофенона. Б) Метиловый эфир 5-(4-трифторметилфенил)пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии В Препаративного примера 1.23, исходя из 6,37 г полученного в предыдущей стадии соединения в 50 мл уксусной кислоты и 3 мл 55%- ного водного раствора гидразина. Получают 5,43 г целевого продукта. В) Метиловый эфир 1-(2,4-дихлорбензил)-5-(4- трифторфенил)пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии А Препаративного примера 1.18, исходя из 2,5 г полученного в предыдущей стадии соединения в 150 мл толуола, 0,44 г гидрида натрия в виде 60%-ной дисперсии в масле и 2 мл 2,4-дихлорбензилхлорида. Получают 1,82 г целевого продукта. Г) 1-(2,4-Дихлорбензил)-5-(4-трифторфенил)пиразол-3- карбоновая кислота Это соединение получают по методике, описанной в стадии Б Препаративного примера 1.14, исходя из 1,6 г полученного в предыдущей стадии соединения в 30 мл метанола и 0,63 г КОН в 30 мл воды. Получают 1,5 г целевого продукта. ЯМР-спектр, (м.д.): 5,45 (с, 2H); 6,85 (д, 1H); 6,95 (с, 1H); 7,3 (дд, 1H); 7,5 (д, 1H); 7,55-7,8 (система AA’- BB’, 4H); 12,9 (уш.с, 1H).
Препаративный пример 1.25 1-[1-(3,4-Дихлорфенил)этил] -5- (4-метилфенил)пиразол-3-карбоновая кислотаА) Метиловый эфир1-[1-(3,4-дихлорфенил)этил]-5-(4-метилфенил) пиразол-3-карбоновой кислоты К суспензии 2,5 г соединения, полученного в стадии Б Препаративного примера 1.1, в 25 мл толуола при комнатной температуре и порциями добавляют 1 г гидрида натрия в виде 60%-ной дисперсии в масле, затем нагревают при 65oC в течение 1 часа. После этого прикапывают раствор 3 г 1-(1-бромэтил)-3,4- дихлорбензола (Препаративный пример 3.6) в 25 мл толуола, потом кипятят с обратным холодильником в течение ночи. Охлаждают до 0oC и прикапывают 100 мл 50%-ного водного раствора хлорида аммония. После декантации органическую фазу промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (75:25 по объему). Получают 1,6 г целевого продукта. Б) 1-[1-(3,4-Дихлорфенил)этил]-5-(4-метилфенил)пиразол-3- карбоновая кислота К суспензии 1,4 г полученного в предыдущей стадии соединения в 15 мл метанола при комнатной температуре добавляют раствор 0,5 г КОН в 15 мл воды, затем кипятят с обратным холодильником в течение двух часов. После удаления в вакууме метанола реакционную смесь выливают в смесь 1 н. соляной кислоты со льдом, выпавший осадок отсасывают, промывают его водой и высушивают его в вакууме над КОН. Получают 1,25 г целевого продукта. ЯМР-спектр, (м.д.): 1,85 (д, 3H); 2,4 (с, 3H); 5,7 (кд, 1H); 6,85 (с, 1H); 7,05 (дд. 1H); 7,2-7.45 (м, 5H); 7,65 (д, 1H); 12,95 (уш.с, 1H).
Препаративный пример 1.261-[1-(4-Метилфенил)этил] -5-(4-хлор-3-метилфенил)пиразол- 3-карбоновая кислота А) Метиловый эфир 1-[1-(4-метилфенил)этил] -5-(4-хлор-3- метилфенил)пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии А Препаративного примера 1.25, исходя из 2,5 г соединения, полученного в стадии Б Препаративного примера 1.10, в 25 мл толуола, 0,88 г гидрида натрия в виде 60%-ной дисперсии в масле и 2,3 г 1- (1-бромэтил)-4-метилбензола (Препаративный пример 3.7) в 25 мл толуола. После добавления водного раствора хлорида аммония и декантации органическую фазу концентрируют в вакууме, остаток обрабатывают этилацетатом, полученный раствор промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (75:25 по объему). Получают 0,7 г целевого продукта. Б) 1-[1-(4-Метилфенил)этил]-5-(4-хлор-3-метилфенил) пиразол-3-карбоновая кислота Это соединение получают по методике, описанной в стадии Б Препаративного примера 1.25, исходя из 0,7 г полученного в предыдущей стадии соединения в 15 мл метанола и раствора 0,28 г КОН в 15 мл воды. Получают 0,63 г целевого продукта, используемого без дальнейшей очистки. Препаративный пример 1.27 1-[1-(3,4-Дихлорфенил)этил]-5-(4-хлор-3-метилфенил) пиразол-3-карбоновая кислота А) Метиловый эфир 1-[1-(3,4-дихлорфенил)этил] -5-(4-хлор-3- метилфенил)пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии А Препаративного примера 1.25, исходя из 3,0 г соединения, полученного в стадии Б Препаративного примера 1.10, в 25 мл толуола, 1 г гидрида натрия в виде 60%-ной дисперсии в масле и 3 г 1-(1-бромэтил)-3,4-дихлорбензола (Препаративный пример 3.6) в 25 мл толуола. После добавления водного раствора хлорида аммония и декантации органическую фазу концентрируют в вакууме, остаток обрабатывают этилацетатом, полученный раствор промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (80:20 по объему). Получают 1,4 г целевого продукта. Б) 1-[1-(3,4-Дихлорфенил)этил] -5-(4-хлор-3-метилфенил) пиразол-3-карбоновая кислота Это соединение получают по методике, описанной в стадии Б Препаративного примера 1.25, исходя из 1 г полученного в предыдущей стадии соединения в 20 мл метанола и раствора 0,40 г КОН в 20 мл воды. Получают 0,96 г целевого продукта. ЯМР-спектр, (м.д.): 1,8 (д, 3H); 2,35 (с, 3H); 5,7 (кд, 1H); 6,7-7,7 (м, 7H); 12,9 (уш.с 1H).
Наблюдают эффект Оверхаузера (N.O.E.) между бензильным протоном (R4=H) и протонами g2=g6=H.
Препаративный пример 1.281-[1-(4-Метилфенил)пропил] -5-(4-хлор-3-метилфенил) пиразол-3-карбоновая кислота А) N,N’- Дипропилиденгидразин 58 г Пропионового альдегида охлаждают до 0oC, добавляют 10 мл воды, затем прикапывают 19,9 мл гидразинмоногидрата, поддерживая температуру ниже 10oC. После этого порциями добавляют 67,5 г КОН в таблетках и реакционную смесь оставляют стоять в течение ночи при комнатной температуре. После декантации органическую фазу перегоняют при пониженном давлении. Получают 24,37 г целевого продукта; т. кип. = 48oC при 2400 Па. Б) N-Пропилиден-N’-[1-(4-метилфенил)пропил]гидразин 100 мл 1 М Раствора n-толилмагний бромида в диэтиловом эфире нагревают до температуры кипения с обратным холодильником, в атмосфере азота, затем прикапывают раствор 10 г полученного в предыдущей стадии соединения в 40 мл безводного диэтилового эфира и перемешивают в течение ночи при комнатной температуре. После охлаждения реакционной смеси до 5oC добавляют водный насыщенный раствор хлорида аммония, декантируют, экстрагируют диэтиловым эфиром, объединенные органические фазы сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 6 г целевого продукта; т. кип. = 102-103oC при 20 Па; nD20 = 1,5180. В) [1-(4-Метилфенил)пропил]гидразиноксалат К раствору 4,78 г щавелевой кислоты в 20 мл этанола и 20 мл диэтилового эфира добавляют раствор 6 г полученного в предыдущей стадии соединения в 5 мл диэтилового эфира и выдерживают в течение ночи при 0 – 5oC. Образовавшийся кристаллический продукт отсасывают и промывают его диэтиловым эфиром. Получают 1,84 г целевого продукта. Г) Метиловый эфир 1-[1-(4-метилфенил)пропил] -5-(4-хлор-3- метилфенил)пиразол-3-карбоновой кислоты К раствору 2 г соединения, полученного в стадии А Препаративного примера 1.10, в 100 мл воды и 100 мл этанола при комнатной температуре добавляют раствор 1,84 г полученного в предыдущей стадии соединения в 20 мл воды и 30 мл этанола и перемешивают 3 часа при комнатной температуре. Реакционную смесь экстрагируют диэтиловым эфиром, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток обрабатывают гексаном, отфильтровывают нерастворимую часть и фильтрат концентрируют в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (80:20 по объему). Получают 1,12 г целевого продукта. Д) 1-[1-(4-Метилфенил)пропил] -5-(4-хлор-3-метилфенил) пиразол-3-карбоновая кислота К раствору 1,1 г полученного в предыдущей стадии соединения в 40 мл этанола при комнатной температуре добавляют раствор 0,47 г КОН в 10 мл воды, затем кипятят с обратным холодильником в течение трех часов. Концентрируют в вакууме, остаток обрабатывают с помощью 50 мл воды, подкисляют до pH 2 путем добавления 6 н. раствора соляной кислоты, отсасывают выпавший осадок, промывают его водой и высушивают его. Получают 0,89 г целевого продукта. Препаративный пример 1.29 1-[1-Метил-1-(4-метилфенил)этил] -5-(4-хлор-3-метилфенил) пиразол-3-карбоновая кислота А) N,N’-Диизопропилиденгидразин Это соединение получают по методике, описанной в стадии А Препаративного примера 1.28, исходя из 92 г ацетона, 20 мл воды, 39,7 гидразинмоногидрата и 135 г КОН. Получают 66 г целевого продукта; т. кип. = 55oC при 2100 Па. Б) N-Изопропилиден-N’-[1-метил-1-(4-метилфенил)этил]гидразин 100 мл 1 М Раствора n-толилмагний бромида в диэтиловом эфире нагревают до температуры кипения с обратным холодильником, в атмосфере азота, затем прикапывают раствор 8,46 г полученного в предыдущей стадии соединения в 200 мл безводного диэтилового эфира и продолжают кипячение с обратным холодильником в течение 5 дней. После охлаждения до 5oC добавляют водный насыщенный раствор хлорида аммония, декантируют, экстрагируют диэтиловым эфиром, объединенные органические фазы сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток перегоняют при пониженном давлении. Получают 6,45 г целевого продукта; т. кип. =95oC при 266,6 Па. В) [1-Метил-1-(4-метилфенил)этил]гидразиноксалат К раствору 5,12 г щавелевой кислоты в 24 мл этанола и 24 мл диэтилового эфира добавляют раствор 6,45 г полученного в предыдущей стадии соединения в 5 мл диэтилового эфира и выдерживают в течение ночи при комнатной температуре. Образовавшийся кристаллический продукт отсасывают и промывают его гексаном. Получают 2,57 г целевого продукта. Г) Метиловый эфир 1-[1-метил-1-(4-метилфенил)этил]-5-(4- хлор-3-метилфенил)пиразол-3-карбоновой кислоты К раствору 2,66 г соединения, полученного в стадии А Препаративного примера 1.10, в 600 мл воды и 1 л этанола при комнатной температуре добавляют раствор 2,54 г полученного в предыдущей стадии соединения в 200 мл воды и 300 мл этанола и перемешивают 3 часа при комнатной температуре, затем оставляют стоять в течение 24-х часов. Выпавший осадок отсасывают, промывают его гексаном и высушивают его. Получают 3,46 г целевого продукта. Д) 1-[1-Метил-1-(4-метилфенил)этил]-5-(4-хлор-3- метилфенил)пиразол-3-карбоновая кислота К раствору 3,3 г полученного в предыдущей стадии соединения в 80 мл этанола при комнатной температуре добавляют раствор 1,44 г КОН в 20 мл воды, затем кипятят с обратным холодильником в течение трех часов. Отфильтровывают нерастворимую часть и фильтрат концентрируют в вакууме до объема 20 мл. Добавляют 30 мл ледяной воды, подкисляют до pH 3,5 путем добавления 1 н. соляной кислоты, декантируют, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 3,15 г целевого продукта. Препаративный пример 1.30 1-(2,4-Дихлорбензил)-5-(4-хлорфенил)-4-метилпиразол-3- карбоновая кислота А) Литиевая соль этил-4-(4-хлорфенил)-3-метил-4-оксидо-2- оксобут-3-еноата В атмосфере азота, 154 мл 1 М раствора литиевой соли гексаметилдисилазана в тетрагидрофуране добавляют к 120 мл охлажденного до 0oC циклогексана. Затем при температуре 0oC в течение 30 минут прикапывают раствор 21,6 г 4′-хлорпропиофенона в 60 мл циклогексана и перемешивают 3 часа при комнатной температуре. После этого быстро добавляют 21,4 г диэтилоксалата, поддерживая температуру ниже 25oC, и перемешивают 48 часов при комнатной температуре. Выпавший осадок отсасывают, промывают его циклогексаном и высушивают его. Получают 31,3 г целевого продукта. Б) Этиловый эфир 5-(4-хлорфенил)-4-метилпиразол-3-карбоновой кислоты К раствору 15 г полученного в предыдущей стадии соединения в 100 мл уксусной кислоты при комнатной температуре прикапывают 2,04 мл гидразинмоногидрата, затем кипятят с обратным холодильником в течение 5 часов и перемешивают в течение ночи при комнатной температуре. Реакционную смесь выливают в смесь воды со льдом, выпавший осадок отсасывают, промывают его водой, затем гексаном и высушивают его. Получают 11,47 г целевого продукта. В) Этиловый эфир 1-(2,4-дихлорбензил)-5-(4-хлорфенил)-4- метилпиразол-3-карбоновой кислоты К раствору 2,17 г полученного в предыдущей стадии соединения в 25 мл толуола при комнатной температуре и порциями добавляют 0,72 г гидрида натрия в виде 60%-ной дисперсии в масле, затем нагревают при 65oC в течение 1 часа. После охлаждения до комнатной температуры прикапывают раствор 2,28 г 2,4-дихлорбензилхлорида в 25 мл толуола, затем кипятят с обратным холодильником в течение 20 часов. После охлаждения до комнатной температуры добавляют водный насыщенный раствор хлорида аммония, декантируют, органическую фазу промывают насыщенным раствором хлорида натрия, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток экстрагируют этилацетатом, органическую фазу промывают насыщенным раствором гидрокарбоната натрия, буферным раствором с pH 2, водой, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (75: 25 по объему). Получают 1,10 г целевого продукта; т.пл. = 82oC. ЯМР-спектр, (м. д.): 1,2 (т, 3H); 2,0 (с, 3H); 4,2 (кд, 2H); 5,3 (с, 2H); 6,7 (д, 1H); 7,15-7,6 (м, 6H).
Наблюдают эффект Оверхаузера (N.O.E.) между бензильными протонами (R4= R5=H) и протонами g2=g6=Н.
Г) 1-(2,4-Дихлорбензил)-5-(4-хлорфенил)-4-метилпиразол- 3-карбоновая кислотаК раствору 0,8 г полученного в предыдущей стадии соединения в 50 мл этанола при комнатной температуре добавляют раствор 0,166 г КОН в 10 мл воды, затем кипятят с обратным холодильником в течение четырех часов. Концентрируют в вакууме, остаток обрабатывают водой, отфильтровывают нерастворимую часть, фильтрат подкисляют до pH 2 путем добавления 6 н. соляной кислоты, выпавший осадок отсасывают, промывают его водой и высушивают его. Получают 0,73 г целевого продукта. Препаративный пример 1.31 1-(3,4-Дихлорбензил)-5-(4-хлорфенил)-4-метилпиразол-3- карбоновая кислота А) Этиловый эфир 1-(3,4-дихлорбензил)-5-(4-хлорфенил)-4- метилпиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии В Препаративного примера 1.30, исходя из 2,17 г соединения, полученного в стадии Б Препаративного примера 1.30, в 25 мл толуола, 0,72 г гидрида натрия в виде 60%-ной дисперсии в масле и 2,99 г 3,4-дихлорбензилхлорида в 25 мл толуола. Получают 1,87 г целевого продукта; т.пл. = 117,5oC. ЯМР-спектр, (м.д.): 1,25 (т, 3H); 2,05 (с, 3H); 4,25 (кд, 2H); 5,3 (с, 2H); 6,8 (дд, 1H); 7,15 (с, 1H); 7,3 (д, 2H); 7,4-7,6 (м, 3H).
Наблюдают эффект Оверхаузера (N.O.E.) между бензильными протонами (R4= R5=H) и протонами g2=g6=H.
Б) 1-(3,4-Дихлорбензил)-5-(4-хлорфенил)-4-метилпиразол-3- карбоновая кислотаЭто соединение получают по методике, описанной в стадии Г Препаративного примера 1.30, исходя из 1,5 г полученного в предыдущей стадии соединения в 50 мл этанола и 0,3 г КОН в 10 мл воды. Получают 1,31 г целевого продукта. Препаративный пример 1.32 1-(3-Хлор-4-метилбензил)-5-(4-хлорфенил)-4-метилпиразол- 3-карбоновая кислота К раствору 2,17 г соединения, полученного в стадии Б Препаративного примера 1.30, в 100 мл толуола при комнатной температуре и порциями добавляют 0,72 г гидрида натрия в виде 60%-ной дисперсии в масле, затем нагревают при 65oC в течение 1 часа. После охлаждения до комнатной температуры прикапывают 2,2 г 3-хлор-4-метилбензилхлорида, затем кипятят с обратным холодильником в течение 20 часов. После охлаждения до комнатной температуры добавляют водный насыщенный раствор хлорида аммония, декантируют, органическую фазу промывают насыщенным раствором хлорида натрия, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток экстрагируют этилацетатом, органическую фазу промывают насыщенным раствором гидрокарбоната натрия, буферным раствором с pH 2, водой, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток обрабатывают смесью циклогексана с этилацетатом (75:25 по объему), осадок отсасывают и получают 1,38 г сырого продукта. 0,35 г таким образом полученного продукта растворяют в этилацетате, добавляют циклогексан вплоть до осаждения и осадок отсасывают. Получают 0,17 г очищенного целевого продукта; т.пл. = 162oC. ЯМР-спектр, (м.д.): 1,95-2,6 (м, 6H); 5,2 (с, 2H); 6,6- 7,7 (м, 7H); 12,85 (уш.с, 1H).
Наблюдают эффект Оверхаузера (N.O.E.) между бензильными протонами (R4= R5=H) и протонами g2=g6=H.
Препаративный пример 1.331-(4-Метилбензил)-5-(4-хлор-3-метилфенил)-4-метилпиразол- 3-карбоновая кислота А) 4′-Хлор-3′-метилпропиофенон К смеси 18 мл толуола и 22,7 г хлорида алюминия прикапывают 16 г пропионилхлорида, затем нагревают при 130oC в течение трех часов. После охлаждения до комнатной температуры реакционную смесь выливают в смесь 100 мл концентрированной соляной кислоты и льда, экстрагируют диэтиловым эфиром, органическую фазу промывают насыщенным раствором гидрокарбоната натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток перегоняют при пониженном давлении. Получают 1,27 г целевого продукта, который кристаллизуется; т. кип. = 95oC при 5 Па. Б) Литиевая соль этил-4-(4-хлор-3- метилфенил)-3-метил-4- оксидо-2-оксобут-3-еноата Это соединение получают по методике, описанной в стадии А Препаративного примера 1.30, исходя из 50 мл 1 М раствора литиевой соли гексаметилдисилазана в тетрагидрофуране, 40 мл циклогексана, 10 г полученного в предыдущей стадии соединения в 70 мл циклогексана и 9,8 г диэтилоксалата. После перемешивания в течение ночи при комнатной температуре реакционную смесь концентрируют в вакууме, остаток обрабатывают диэтиловым эфиром и растворитель выпаривают в вакууме. Остаток обрабатывают гептаном и выпавший осадок отсасывают. Получают 15 г целевого продукта. В) Этиловый эфир 5-(4-хлор-3-метилфенил)-4-метилпиразол- 3-карбоновой кислоты Раствор 15 г полученного в предыдущей стадии соединения в 150 мл уксусной кислоты охлаждают до 5oC, прикапывают 2,5 мл гидразинмоногидрата, затем кипятят с обратным холодильником в течение 5 часов и перемешивают в течение ночи при комнатной температуре. Реакционную смесь выливают в смесь воды со льдом, экстрагируют этилацетатом, органическую фазу промывают трижды насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток обрабатывают диизопропиловым эфиром, выпавший осадок отсасывают. Промывают его диизопропиловым эфиром и высушивают его. Получают 7,88 г целевого продукта. Г) Этиловый эфир 1-(4-метилбензил)-5-(4-хлор-3-метилфенил) -4-метилпиразол-3-карбоновой кислоты К суспензии 5 г полученного в предыдущей стадии соединения в 70 мл толуола при комнатной температуре и порциями добавляют 0,98 г гидрида натрия в виде 60%-ной дисперсии в масле, затем нагревают при 65oC в течение 1 часа. После охлаждения до комнатной температуры прикапывают 3,7 г 4-метилбензилбромида, затем кипятят с обратным холодильником в течение ночи. После охлаждения до комнатной температуры добавляют 50%-ный водный раствор хлорида аммония, декантируют и органическую фазу концентрируют в вакууме. Остаток экстрагируют этилацетатом, органическую фазу промывают дважды насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (80:20 по объему). Получают 2,1 г целевого продукта. Д) 1-(4-Метилбензил)-5-(4-хлор-3-метилфенил)-4- метилпиразол-3-карбоновая кислота К раствору 2 г полученного в предыдущей стадии соединения в 20 мл этанола при комнатной температуре добавляют раствор 0,439 г КОН в 20 мл воды, затем кипятят с обратным холодильником в течение трех часов. После охлаждения до комнатной температуры реакционную смесь выливают в смесь 1 н. соляной кислоты со льдом, выпавший осадок отсасывают, промывают его водой и высушивают его в вакууме. Получают 1,8 г целевого продукта. ЯМР-спектр, (м.д.): 2-2,4 (м, 9H); 5,2 (с, 2H); 6,7-7,6 (м, 7H); 12,65 (уш.с, 1H)Препаративный пример 1.34 1-(3,4-Дихлорбензил)-5-(4-хлор-3-метилфенил)-4- метилпиразол-3-карбоновая кислота А) Этиловый эфир 1-(3,4-дихлорбензил)-5-(4-хлор-3- метилфенил)-4- метилпиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии Г Препаративного примера 1.33, исходя из 2,5 г соединения, полученного в стадии В Препаративного примера 1.33, в 50 мл толуола, 0,8 г гидрида натрия в виде 60%-ной дисперсии в масле и 2,4 г 3,4-дихлорбензилбромида. После гидролиза за счет добавления водного 50%-ного раствора хлорида аммония органическую фазу декантируют, отфильтровывают нерастворимую часть и фильтрат концентрируют в вакууме. Остаток экстрагируют этилацетатом, органическую фазу промывают дважды насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (80:20 по объему). Получают 3 г целевого продукта. Б) 1-(3,4-Дихлорбензил)-5-(4-хлор-3-метилфенил)-4- метилпиразол-3-карбоновая кислота Это соединение получают по методике, описанной в стадии Д Препаративного примера 1.33, исходя из 3 г полученного в предыдущей стадии соединения в 20 мл этанола и 0,56 г КОН в 20 мл воды. Получают 2,7 г целевого продукта. ЯМР-спектр, (м.д.): 2 (с, 3H); 2,3 (м, 3H); 5,2 (с, 2H); 6,7-7,6 (м, 6H); 12,65 (уш.с, 1H).
Препаративный пример 1.351-(4-Хлор-3-метилбензил)-5-(4-хлорфенил)-4-(этоксиметил) пиразол-3-карбоновая кислота А) Этиловый эфир 1-(трет.-бутоксикарбонил)-5-(4-хлорфенил) -4-метилпиразол-3-карбоновой кислоты К раствору 6 г соединения, полученного в стадии Б Препаративного примера 1.30, в 150 мл диоксана добавляют 4,62 мл триэтиламина, 6,57 г ди-трет.бутилдикарбоната и 1,18 г гидрида натрия в виде 60%-ной дисперсии в масле, затем перемешивают в течение 72-х часов при комнатной температуре. Отфильтровывают нерастворимую часть и фильтрат концентрируют в вакууме. Остаток обрабатывают циклогексаном, выпавший осадок отсасывают, промывают его циклогексаном и высушивают. Получают 6,23 г целевого продукта. Б) Этиловый эфир 1-(трет.-бутоксикарбонил)-5-(4-хлорфенил)-4- (бромметил)пиразол-3-карбоновой кислоты К раствору 6,2 г полученного в предыдущей стадии соединения в 150 мл тетрахлоруглерода добавляют 3,18 г N-бромсукцинимида и 0,02 г дибензоилпероксида, затем кипятят с обратным холодильником в течение ночи. Отфильтровывают нерастворимую часть и фильтрат концентрируют в вакууме. Остаток обрабатывают циклогексаном, декантируют и растворитель выпаривают в вакууме. Получают 8,72 г целевого продукта в виде масла. В) Этиловый эфир 5-(4-хлорфенил)-4-(этоксиметил)пиразол-3- карбоновой кислоты К 100 мл этанола добавляют 0,23 г натрия, затем, после растворения, прикапывают 4,5 г полученного в предыдущей стадии соединения и кипятят с обратным холодильником в течение ночи. Реакционную смесь концентрируют в вакууме и остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (60: 40 по объему). Получают 2,26 г целевого продукта. Г) Этиловый эфир 1-(3-хлор-4-метилбензил)-5-(4-хлорфенил) -4-(этоксиметил)пиразол-3-карбоновая кислота К раствору 1,65 г полученного в предыдущей стадии соединения в 50 мл толуола при комнатной температуре и порциями добавляют 0,254 г гидрида натрия в виде 60%-ной дисперсии в масле, затем нагревают при 65oC в течение 1 часа. После охлаждения до комнатной температуры прикапывают 1,7 г 3-хлор-4-метилбензилиодида, затем кипятят с обратным холодильником в течение 20 часов. После охлаждения до комнатной температуры отфильтровывают нерастворимую часть и фильтрат концентрируют в вакууме. Остаток экстрагируют этилацетатом, органическую фазу промывают насыщенным раствором хлорида аммония, буферным раствором с pH 2, водой, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (75:25 по объему). Получают 0,90 г продукта, который повторно хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (80:20 по объему). Получают 0,24 г целевого продукта. Д) 1-(4-Хлор-3-метилбензил)-5-(4-хлорфенил)-4- (этоксиметил)пиразол-3-карбоновая кислота К раствору 0,23 г полученного в предыдущей стадии соединения в 10 мл этанола при комнатной температуре добавляют раствор 0,088 г КОН в 10 мл воды, затем кипятят с обратным холодильником в течение трех часов. Концентрируют в вакууме вплоть до объема 10 мл, добавляют 10 мл воды, подкисляют до pH 2,5 путем добавления 1 н. соляной кислоты, выпавший осадок отсасывают и высушивают его. Получают 0,21 г целевого продукта. Препаративный пример 1.36 1-(5-Хлориндан-1-ил)-5-(4-хлорфенил)пиразол-3-карбоновая кислота К суспензии 2,5 г соединения, полученного в стадии Б Препаративного примера 1.7, в 20 мл толуола порциями добавляют 0,88 г гидрида натрия в виде 60%-ной дисперсии в масле, затем нагревают при 65oC в течение 1 часа. После охлаждения до комнатной температуры прикапывают раствор 2,3 г 1-бром-5-хлориндана (Препаративный пример 3.8) в 10 мл толуола и кипятят с обратным холодильником в течение ночи. Реакционную смесь охлаждают до 5oC, прикапывают 20 мл водного 50%-ного раствора хлорида аммония, декантируют и органическую фазу концентрируют в вакууме. Остаток экстрагируют этилацетатом, органическую фазу промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 1,04 г целевого продукта, используемого без дальнейшей очистки. Препаративный пример 1.37 1-(4-Метоксибензил)-5-(4-хлор-3-метилфенил)пиразол-3- карбоновая кислота А) Метиловый эфир 1-(4-метоксибензил)-5-(4-хлор-3-метилфенил) пиразол-3-карбоновой кислоты К суспензии 2,5 г соединения, полученного в стадии Б Препаративного примера 1.10, в 25 мл толуола при комнатной температуре и порциями добавляют 0,53 г гидрида натрия в виде 60%-ной дисперсии в масле, затем нагревают при 65oC в течение 1 часа. После охлаждения до комнатной температуры прикапывают раствор 1,7 г 4-метоксибензилхлорида в 25 мл толуола, затем кипятят с обратным холодильником в течение 20 часов. После охлаждения до комнатной температуры добавляют 20 мл водного 50%- ного раствора хлорида аммония, декантируют и органическую фазу выпаривают в вакууме. Остаток экстрагируют этилацетатом, органическую фазу промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (80: 20 по объему). Получают 1,03 г целевого продукта, используемого без дальнейшей очистки. Б) 1-(4-Метоксибензил)-5-(4-хлор-3-метилфенил)пиразол-3- карбоновая кислота К раствору 1,03 г полученного в предыдущей стадии соединения в 20 мл метанола при комнатной температуре добавляют раствор 0,23 г КОН в 20 мл воды, затем кипятят с обратным холодильником в течение трех часов. После охлаждения до комнатной температуры реакционную смесь выливают в смесь 1 н. соляной кислоты со льдом, экстрагируя диэтиловым эфиром, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 0,8 г целевого продукта, используемого без дальнейшей очистки. Препаративный пример 1.38 1-(2,4-Дихлорбензил)-3-(4-хлорфенил)пиразол-5-карбоновая кислота А) Метиловый эфир 1-(2,4-дихлорбензил)-3-(4-хлорфенил) пиразол-5-карбоновой кислоты К раствору 0,8 г соединения, полученного в стадии Б Препаративного примера 1.7, в 15 мл диметилформамида при комнатной температуре и порциями добавляют 0,16 г гидрида натрия в виде 60%-ной дисперсии в масле и перемешивают в течение 30 минут при комнатной температуре. Реакционную смесь охлаждают до 0oC, прикапывают раствор 0,49 мл 2,4-дихлорбензилхлорида в 5 мл диметилформамида, после чего перемешивают в течение 16 часов при комнатной температуре. Реакционную смесь выливают в ледяную воду, водную фазу промывают дихлорметаном, отсасывают осадок белого цвета, промывают его водой и высушивают его. Получают 0,86 г целевого продукта; т.пл. = 120oC. Б) 1-(2,4-Дихлорбензил)-3-(4-хлорфенил)пиразол-5- карбоновая кислота К раствору 0,85 г полученного в предыдущей стадии соединения в 15 мл метанола добавляют раствор 0,3 г КОН в 5 мл воды, затем кипятят с обратным холодильником в течение трех часов. Реакционную смесь выливают в 100 мл ледяной воды, выпавший осадок отсасывают, промывают его водой и высушивают его в вакууме. Получают 0,79 г целевого продукта; т.пл. = 218oC. Препаративный пример 2.1 (1S)-2эндо-Амино-1,3,3-триметилбицикло[2.2.1]гептангидрохлорид A) (1S)-1,3,3-Триметилбицикло[2.2.1]гептан-2-он-оксим К раствору 38,1 г (1S)-(+)-фенхона в 120 мл метанола при комнатной температуре добавляют раствор 23 г гидроксиламингидрохлорида и 41 г ацетата натрия в 200 мл воды, затем кипятят с обратным холодильником в течение 48 часов. После охлаждения до комнатной температуры отсасывают выпавший осадок, промывают его водой и высушивают его в вакууме. Получают 41 г целевого продукта. Б) (1S)-2эндо-Амино-1,3,3-триметилбицикло [2.2.1] гептангидрохлорид В аппарате Парра, при комнатной температуре и давлении 6 бар в течение 48 часов гидрируют смесь 8 г полученного в предыдущей стадии соединения и 0,8 г оксида платины в 700 мл этанола и 20 мл хлороформа. Катализатор отфильтровывают на целите и фильтрат концентрируют в вакууме. Осадок обрабатывают диэтиловым эфиром и выпавший осадок отсасывают. Получают 2,61 г целевого продукта.
2D0= -4,6 (c = 1; этанол) (c = 1; этанол)Препаративный пример 2.2 (1S)-2эндо, экзо-Амино-1,3,3-Триметилбицикло[2.2.1]гептан Раствор соединения, полученного в стадии А Препаративного примера 2.1, в 70 мл уксусной кислоты охлаждают до 10oC, добавляют 25 г никеля Ренея и оставляют смесь стоять для повышения температуры до комнатной. Затем смесь гидрируют в течение 24-х часов при комнатной температуре и атмосферном давлении. Катализатор отфильтровывают на целите , к фильтрату добавляют смесь 100 мл воды со льдом, доводят pH-значение до 7 путем добавления концентрированного раствора гидроксида натрия, экстрагируют диэтиловым эфиром, органическую фазу сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 7,37 г целевого продукта в виде масла (эндо, экзо-смесь).
Препаративный пример 2.3(1R)-2эндо, экэо-Амино-1,3,3-триметилбицикло[2.2.1] гептан и (1R)-2-имино-1,3,3-триметилбицикло[2.2.1]гептан A) (1R)-1,3,3-Триметилбицикло[2.2.1]гептан-2-он-оксим Это соединение получают по методике, описанной в стадии А Препаративного примера 2.1, исходя из (1R)-(-)-фенхона. Б) (1R)-2эндо, экзо-Амино-1,3,3-триметилбицикло[2.2.1] гептан и (1R)-2-имино-1,3,3-триметилбицикло[2.2.1]гептан Раствор 14 г полученного в предыдущей стадии соединения в 100 мл уксусной кислоты охлаждают до 10oC, добавляют 35 г никеля Ренея и оставляют смесь стоять для повышения температуры до комнатной. Затем смесь гидрируют в течение 24-х часов при комнатной температуре и атмосферном давлении. Катализатор отфильтровывают на целите , добавляют к фильтрату смесь 100 мл воды со льдом, доводят pH-значение до 7 путем добавления концентрированного раствора гидроксида натрия, экстрагируют диэтиловым эфиром, органическую фазу сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 10,77 г смеси целевых продуктов.
Препаративный пример 2.42экзо-(Пропиламино)бицикло[2.2.1]гептангидрохлорид А) 2экзо-(Пропиониламино)бицикло[2.2.1]гептан На бане со льдом охлаждают раствор 15 г 2экзо- аминонорборнана и 20,5 мл триэтиламина в 80 мл дихлорметана, прикапывают раствор 11,2 мл пропионилхлорида в 80 мл дихлорметана и перемешивают в течение ночи при комнатной температуре. После фильтрации реакционной смеси фильтрат промывают водным насыщенным раствором гидрокарбоната натрия, водой, органическую фазу сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 23 г целевого продукта. Б) 2экзо-(Пропиламино)бицикло[2.2.1]гептангидрохлорид К суспензии 7,6 г литийалюминийгидрида в 100 мл тетрагидрофурана прикапывают раствор 23 г полученного в предыдущей стадии соединения в 100 мл тетрагидрофурана, затем кипятят с обратным холодильником в течение двух часов и перемешивают в течение ночи при комнатной температуре. Реакционную смесь гидролизуют путем добавления 10 мл воды, затем 5 мл 15%-ного раствора гидроксида натрия и 14 мл воды. После перемешивания в течение 15 минут отфильтровывают неорганические соли и фильтрат концентрируют в вакууме. Полученное масло обрабатывают диизопропиловым эфиром, добавляют вплоть до pH 1 насыщенный раствор хлороводорода в диэтиловом эфире и выпавший осадок отсасывают. Осадок обрабатывают этилацетатом, экстрагируют водой, водную фазу подщелачивают до pH 12 путем добавления 5 н. раствора гидроксида натрия, экстрагируют этилацетатом, органическую фазу промывают водой, сушат над сульфатом магния и растворитель выпаривают в вакууме. Полученный продукт обрабатывают диизопропиловым эфиром, добавляют вплоть до pH 1 насыщенный раствор хлороводорода в диэтиловом эфире и выпавший осадок отсасывают. Получают 14 г целевого продукта; т.пл. = 230oC (разложение). Препаративный пример 2.5 2эндо-Амино-бицикло[3.2.1]октангидрохлорид Это соединение получают по методике, описанной H.Maskill и др., J.Chem. Soc., Perkin Trans.II, 1369-1376 (1984). Препаративный пример 2.6 (1R)-2эндо-Амино-1,3,3-триметилбицикло[2.2.1] гептангидрохлорид В аппарате Парра, при комнатной температуре и давлении 8 бар, гидрируют смесь 15,5 г соединения, полученного в стадии А Препаративного примера 2.3, и 2 г оксида платины в 500 мл этанола и 14 мл хлороформа. Катализатор отфильтровывают на целите и фильтрат концентрируют в вакууме. Осадок обрабатывают насыщенным раствором хлороводорода в диэтиловом эфире и растворитель выпаривают в вакууме. Остаток обрабатывают гексаном, отфильтровывают нерастворимую часть и растворитель удаляют в вакууме. Получают 5,64 г масла, которое с течением времени кристаллизуется. Образовавшиеся кристаллы отсасывают, обрабатывают их гексаном, снова отсасывают и промывают их. Получают 0,85 г целевого продукта; 2D0= +1 (с = 1; этанол).
Препаративный пример 2.72,2,6,6-Тетраметилциклогексиламин A) 2,2,6,6-Тетраметилциклогексаноноксим К раствору 3,4 г 2.2.6.6-тетраметилциклогексанона в 20 мл метанола при комнатной температуре добавляют раствор 2.3 г гидроксиламингидрохлорида и 3,6 г ацетата натрия в 20 мл воды, затем кипятят с обратным холодильником в течение 48 часов. После охлаждения до комнатной температуры выпавший осадок отсасывают, промывают его водой и высушивают его в вакууме. Получают 1,2 г целевого продукта. Б) 2,2,6,6-Тетраметилциклогексиламин Охлаждают до 10oC, в атмосфере азота, раствор 1 г полученного в предыдущей стадии соединения в 30 мл уксусной кислоты, добавляют 2,5 г никеля Ренея и оставляют температуру повышаться до комнатной. Таким образом полученную смесь гидрируют при комнатной температуре и атмосферном давлении в течении 24-х часов. Катализатор отфильтровывают на целите , к фильтрату добавляют смесь воды со льдом, доводят pH-значение до 7 путем добавления концентрированного раствора гидроксида натрия, экстрагируют этилацетатом, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 0,63 г целевого продукта.
Препаративный пример 3.1.
3-Фтор-4-метилбензилбромидА) Этиловый эфир 3-фтор-4-метилбензойной кислоты На бане со льдом охлаждают 150 мл этанола, медленно добавляют 10 мл тионилхлорида, затем добавляют 10 г 3-фтор-4-метилбензойной кислоты и перемешивают во время повышения температуры до комнатной. Реакционную смесь кипятят с обратным холодильником в течение двух часов, затем концентрируют в вакууме. Получают 10,45 г целевого продукта в виде масла. Б) 3-Фтор-4-метилбензиловый спирт Суспензию 3,26 г литийалюминийгидрида в 100 мл тетрагидрофурана охлаждают до 0oC, прикапывают раствор 10,45 г полученного в предыдущей стадии соединения в 50 мл тетрагидрофурана и перемешивают до тех пор, пока температура не повысится до комнатной. Затем реакционную смесь кипятят с обратным холодильником в течение трех часов, добавляют еще 1,5 г литийалюминийгидрида и продолжают кипячение с обратным холодильником в течение 48 часов. После охлаждения до комнатной температуры реакционную смесь гидролизуют путем добавления водного насыщенного раствора хлорида аммония, затем декантируют и органическую фазу сохраняют. Водную фазу экстрагируют тетрагидрофураном, затем объединенные органические фазы сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 7,78 г целевого соединения в виде масла. В) 3-Фтор-4-метилбензилбромид Смесь 7,8 г полученного в предыдущей стадии соединения в 112 мл 47%-ного водного раствора бромоводорода нагревают при 100oC в течение двух часов. После охлаждения до комнатной температуры реакционную смесь экстрагируют диэтиловым эфиром, органическую фазу сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 11,15 г целевого продукта в виде масла, который используют без дальнейшей очистки. Препаративный пример 3.2. 3-Хлор-4-метилбензилиодид К раствору 9,6 г 3-хлор-4-метилбензилхлорида в 15 мл ацетона при комнатной температуре прикапывают раствор 8,5 г иодида натрия в 25 мл ацетона и перемешивают в течение 48 часов при комнатной температуре. Выпавший в осадок хлорид натрия отфильтровывают, фильтрат сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 13 г целевого продукта в виде масла, которое используют без дальнейшей очистки. Препаративный пример 3.3 3-Хлор-4-фторбензилбромид К смеси 5 г 3-хлор-4-фтортолуола в 100 мл тетрахлорида углерода добавляют 0,05 г дибензоилпероксида, затем 6,1 г N-бромсукцинимида и кипятят с обратным холодильником в течение 12 часов. Отфильтровывают нерастворимую часть и промывают ее с помощью тетрахлорида углерода. Фильтрат промывают водой, насыщенным раствором хлорида натрия, органическую фазу сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 7,5 г целевого продукта в виде масла, которое используют без дальнейшей очистки. Препаративный пример 3.4 1-(1-Бромэтил)-2,4-дихлорбензол 15 мл 33%-ного раствора бромоводорода в уксусной кислоте охлаждают до 0oC, прикапывают 2 мл 2,4-дихлор-1-(1-гидроксиэтил)бензола и перемешивают 30 минут при 0oC, затем 3 часа при комнатной температуре. Реакционную смесь выливают в ледяную воду, экстрагируют этилацетатом, органическую фазу промывают водным насыщенным раствором гидрокарбоната натрия, водой, насыщенным раствором хлорида натрия, сушат над сульфатом магния и растворитель выпаривают в вакууме. Получают 3,4 г целевого продукта. Препаративный пример 3.5 4-Этилбензилбромид 15 мл 33%-ного раствора бромоводорода в уксусной кислоте охлаждают до 0oC, прикапывают 5 г 4-этилбензилового спирта и перемешивают 30 минут при 0oC, затем в течение ночи при комнатной температуре. Реакционную смесь выливают в 200 мл ледяной воды, экстрагируют этилацетатом, органическую фазу промывают водным насыщенным раствором гидрокарбоната натрия, насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 5,6 г целевого продукта. Препаративный пример 3.6 1-(1-Бромэтил)-3,4-дихлорбензол Это соединение получают по методике, описанной в Препаративном примере 3.4, исходя из 25 мл 33%-ного раствора бромоводорода в уксусной кислоте и 5 г 3,4-дихлор-1-(1-гидроксиэтил)бензола. Получают 6,4 г целевого продукта. Препаративный пример 3.7 1-(1-Бромэтил)-4-метилбензол Это соединение получают по методике, описанной в Препаративном примере 3.4, исходя из 30 мл 33%-ного раствора бромоводорода в уксусной кислоте и 7 мл 1-(1-гидроксиэтил)-4-метилбензола. Получают 9 г целевого продукта. Препаративный пример 3.8 1-Бром-5-хлориндан А) 1-Гидрокси-5-хлориндан Смесь 2,5 г 5-хлориндан-1-она в 30 мл тетрагидрофурана охлаждают до 0-5oC, добавляют 2,5 мл концентрированного раствора гидроксида натрия, затем порциями 0,88 г борогидрида натрия и перемешивают в течение ночи при комнатной температуре. Реакционную смесь выливают в 100 мл воды, подкисляют до pH 2 путем добавления концентрированной соляной кислоты, экстрагируют этилацетатом, органическую фазу промывают насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (80:20 по объему). Получают 2,1 г целевого продукта. Б) 1-Бром-5-хлориндан 12 мл 33%-ного раствора бромоводорода в уксусной кислоте охлаждают до 0oC, прикапывают 2 г полученного в предыдущей стадии соединения, перемешивают 30 минут при 0oC, затем 3 часа при комнатной температуре. Реакционную смесь выливают в 200 мл ледяной воды, экстрагируют этилацетатом, органическую фазу промывают насыщенным раствором гидрокарбоната натрия, насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Получают 2,8 г целевого продукта. Пример 1 N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] – 1-(3,4-дихлорбензил)-5-(4-метилфенил)пиразол-3-карбоксамид А) Хлорангидрид 1-(3,4-дихлорбензил)-5-(4-метилфенил)пиразол-3- карбоновой кислоты К раствору 5,1 г полученного в Препаративном примере 1.1 соединения в 50 мл толуола при комнатной температуре добавляют 1,52 мл тионилхлорида, затем кипятят с обратным холодильником в течение четырех часов и перемешивают в течение ночи при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток обрабатывают диэтиловым эфиром и растворитель выпаривают в вакууме. Получают 5,17 г целевого продукта в виде масла. Б) N-[(1S)-1,3,3-Триметилбицикло[2.2.1]гепт-2эндо- ил]-1-(3,4-дихлорбензил)-5-(4-метилфенил)пиразол-3- карбоксамид К раствору 0,38 г полученного в Препаративном примере 2.1 соединения и 0,554 мл триэтиламина в 50 мл дихлорметана при комнатной температуре добавляют 0,759 г полученного в предыдущей стадии соединения и перемешивают в течение ночи при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток экстрагируют этилацетатом, органическую фазу промывают насыщенным раствором гидрокарбоната натрия, буферным раствором с pH 2, водой, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток обрабатывают гексаном, выпавший осадок отсасывают и высушивают его. Получают 0,67 г целевого продукта; т.пл. = 137oC, 2D0= +1 (c = 1; этанол).
Пример 2N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2экзо-ил] – 1-(3,4-дихлорбензил)-5-(4-метилфенил)пиразол-3-карбоксамид К раствору 0,5 г полученного в Препаративном примере 2.2 соединения и 0,364 мл триэтиламина в 50 мл дихлорметана при комнатной температуре добавляют 1 г соединения, полученного в стадии А Примера 1, и перемешивают в течение ночи при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток экстрагируют этилацетатом, органическую фазу промывают водным насыщенным раствором гидрокарбоната натрия, буферным раствором с pH 2, водой, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (75:25 по объему). Выделяют два изомера: – наименее полярный, соединение Примера 1; – наиболее полярный, целевое соединение: получают 0,08 г, т.пл. 101oC. ЯМР-спектр, (м.д.): 0,6-2 (м, 16H); 2,3 (с, 3H); 3,45 (д, 1H); 5,4 (с, 2H); 6,8 (с, 1H); 6,9 (дд, 1H); 7,1-7,4 (м, 6H); 7,5 (д, 1H).
Пример 3 и Пример 4N-[(1R)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил]-1- (3,4-дихлорбензил)-5-(4-метилфенил)пиразол-3-карбоксамид (Пример З) и N-[(1R)-1,3,3-Триметилбицикло[2.2.1] гепт-2экзо-ил] -1-(3,4- дихлорбензил)-5-(4-метилфенил)пиразол-3-карбоксамид (Пример 4) К раствору 0,5 г смеси соединений, полученных в Препаративном примере 2.3, и 0,364 г триэтиламина в 50 мл дихлорметана при комнатной температуре добавляют 1 г соединения, полученного в стадии А Примера 1 и перемешивают в течение ночи при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток экстрагируют этилацетатом, органическую фазу промывают водным насыщенным раствором гидрокарбоната натрия, буферным раствором с pH 2, водой, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (75:25 по объему). Выделяют различные соединения: – наименее полярное, соединение Примера 3: получают 0,07 г; т.пл. = 130oC; ЯМР-спектр, (м.д.): 0,6-1,9 (м, 16H); 2,3 (с, 3H); 3,65 (д, 1H); 5,4 (с, 2H); 6,85 (с, 1H); 6,9 (дд, 1H); 7,0 (д, 1H); 7,15-7,4 (м, 5H); 7,5 (д, 1H);– соединение Примера 4: получают 0,09 г; т.пл. = 121oC; ЯМР-спектр, (м.д.): 0,6-2 (м, 16H); 2,3 (с, 3H); 3,4 (д, 1H); 5,4 (с, 2H); 6,8 (с, 1H); 6,9 (дд, 1H); 7,1-7,4 (м, 5H); 7,5 (д, 1H).
– и наиболее полярное соединение: N[(1R)-1,3,3-триметилбицикло [2.2.1] гепт-2-илиден] -1-(3,4-дихлорбензил)-5-(4-метилфенил)пиразол- 3-карбоксамид; получают 0,54 г.
Пример 5N-[Бицикло[2.2.1] гепт-2экзо-ил] -N-пропил-1-(3,4- дихлорбензил)-5-(4-метилфенил)пиразол-3-карбоксамид К раствору 0,266 г полученного в Препаративном примере 2.4 соединения и 0,48 мл триэтиламина в 50 мл дихлорметана при комнатной температуре добавляют 0,66 г соединения, полученного в стадии А Примера 1, и перемешивают в течение ночи при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток экстрагируют этилацетатом, органическую фазу промывают водным насыщенным раствором гидрокарбоната натрия, буферным раствором с pH 2, водой, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (70:30 по объему). Получают 0,5 г целевого продукта; т.пл.=108oC. Пример 6 N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1-(3-фтор-4-метилбензил)-5-(4-метилфенил)пиразол-3- карбоксамид А) Хлорангидрид 1-(3-фтор-4-метилбензил)-5-(4- метилфенил)пиразол-3-карбоновой кислоты и хлорангидрид 1-(3-фтор- 4-метилбензил)-3-(4-метилфенил)пиразол-5-карбоновой кислоты К раствору 0,9 г смеси соединений, полученных в Препаративном примере 1.3, в 50 мл толуола при комнатной температуре добавляют 0,3 мл тионилхлорида, затем кипятят с обратным холодильником в течение 5 часов и перемешивают в течение ночи при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток обрабатывают диэтиловым эфиром и растворитель выпаривают в вакууме. Получают 1 г смеси целевых продуктов. Б) N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил]-1- (3-фтор-4-метилбензил)-5-(4-метилфенил)пиразол-3-карбоксамид К раствору 0,33 г полученного в Препаративном примере 2.1 соединения и 0,48 мл триэтиламина в 50 мл дихлорметана при комнатной температуре добавляют 0,63 г смеси соединений, полученных в предыдущей стадии, и перемешивают в течение 48 часов при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток элюируют этилацетатом, органическую фазу промывают водным насыщенным раствором гидрокарбоната натрия, буферным раствором с pH, водой, сушат над сульфатом магния и растворитель выпаривают в вакууме. Выделяют два соединения: – наименее полярное, соединение Примера 6: получают 0,5 г; т.пл. = 53oC, 2D0= -3,3 (с = 1; этанол).
– наиболее полярное: N-[(1S)-1,3,3-триметилбицикло-[2.2.1] гепт-2эндо-ил]-1-(3-фтор-4-метилбензил)-3-(4-метил-фенил) пиразол-5-карбоксамид.
Пример 7N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1-(3- хлор-4-метилбензил)-5-(4-метоксифенил)пиразол-3-карбоксамид А) Хлорангидрид 1-(3-хлор-4-метилбензил)-5-(4- метоксифенил)пиразол-3-карбоновой кислоты и хлорангидрид 1- (3-хлор-4-метилбензил)-3-(4-метоксифенил)пиразол-5-карбоновой кислоты Смесь этих двух соединений получают по методике, описанной в стадии А Примера 6, исходя из 1,7 г смеси соединений, полученных в Препаративном примере 1.5, 0,54 мл тионилхлорида и 50 мл толуола. Получают 1,77 г смеси целевых продуктов. Б) N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1-(3-хлор-4-метилбензил)-5-(4-метоксифенил)пиразол-3- карбоксамид Это соединение получают по методике, описанной в стадии Б Примера 6, исходя из 0,531 г полученного в Препаративном примере 2.1 соединения, 0,775 мл триэтиламина, 100 мл дихлорметана и 1 г смеси соединений, полученных в предыдущей стадии. Хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (75:25 по объему). Выделяют два соединения: – наименее полярное, соединение Примера 7: получают 0,77 г; т.пл. = 116oC; 2D0= -1,1 (с = 1; этанол);– наиболее полярное: N [(1S)-1,3,3-триметилбицикло-[2.2.1] гепт-2эндо-ил]-1-(3-хлор-4-метилбензил)-3-(4-метоксифенил) пиразол-5-карбоксамид. Пример 8 N-(Адамант-2-ил)-1-(2,4-дихлорбензил)-5-(4- хлорфенил)пиразол-3-карбоксамид А) Хлорангидрид 1-(2,4-дихлорбензил)-5-(4-хлорфенил) пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии А Примера 1, исходя из 3,27 г полученного в Препаративном примере 1.7 соединения, 2,2 мл тионилхлорида и 60 мл толуола. Получают 3,25 г целевого продукта, который используют без дальнейшей очистки. Б) N-(Адамант-2-ил)-1-(2,4-дихлорбензил)-5-(4-хлорфенил) пиразол-3-карбоксамид Раствор 0,39 г 2-аминоадамантангидрохлорида и 0,58 мл триэтиламина в 15 мл дихлорметана охлаждают до 0oC, прикапывают раствор 0,8 г полученного в предыдущей стадии соединения в 15 мл дихлорметана и перемешивают в течение 16 часов при комнатной температуре. Реакционную смесь выливают в 50 мл ледяной воды, после декантации органическую фазу промывают водой, насыщенным раствором хлорида натрия, сушат над сульфатом магния и растворитель выпаривают в вакууме. После кристаллизации, затем перекристаллизации из смеси дихлорметана с диизопропиловым эфиром получают 0,42 г целевого продукта; т.пл. = 154oC. Пример 9 N-[эндо-Бицикло[3.2.1] окт-3-ил] -1-(2,4-дихлорбензил)-5- (4-хлорфенил)пиразол-3-карбоксамид Это соединение получают по методике, описанной в стадии Б Примера 8, исходя из 0,17 г полученного в Препаративном примере 2.5 соединения, 0,3 мл триэтиламина в 10 мл дихлорметана и 0,41 г соединения, полученного в стадии А Примера 8, в 10 мл дихлорметана. Полученный продукт очищают путем хроматографии на диоксиде кремния, элюируя градиентом смеси толуола с этилацетатом (от 97:3 до 95:5 по объему). Получают 0,43 г целевого продукта; т.пл.=130oC. Пример 10 N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] – 1-[(1R,S)-1-(2,4-дихлорфенил)этил]-5-(4-хлорфенил) пиразол-3-карбоксамид А) Хлорангидрид 1-[1-(2,4-дихлорфенил)этил]-5-(4-хлорфенил) пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии А Примера 1, исходя из 0,68 г полученного в Препаративном примере 1.14 соединения, 0,45 мл тионилхлорида и 30 мл толуола. Получают 0,86 г целевого продукта. Б) N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1- [(1R,S)-1-(2,4-дихлорфенил)этил]-5-(4-хлорфенил)пиразол-3- карбоксамид Это соединение получают по методике, описанной в стадии Б Примера 8, исходя из 0,34 г полученного в Препаративном примере 2.1 соединения и 0,5 мл триэтиламина в 15 мл дихлорметана и 0,72 г полученного в предыдущей стадии соединения в 15 мл дихлорметана. Полученный продукт очищают путем хроматографии на диоксиде кремния, элюируя смесью толуола с этилацетатом (96:4 по объему). Получают 0,84 г целевого продукта; т.пл. = 70oC. Пример 11 N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1-(3,4- дихлорбензил-5-(2,6-диметоксифенил)пиразол-3-карбоксамид А) Хлорангидрид 1-(3,4-дихлорбензил-5-(2,6-диметоксифенил) пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии А Примера 1, исходя из 1,1 г полученного в Препаративном примере 1.15 соединения, 0,6 мл тионилхлорида и 25 мл толуола. Получают 1 г целевого соединения. Б) N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1- (3,4-дихлорбензил-5-(2,6-диметоксифенил)пиразол-3-карбоксами Это соединение получают по методике, описанной в стадии Б Примера 1, исходя из 0,303 г полученного в Препаративном примере 2.1 соединения и 0,4 мл триэтиламина в 15 мл дихлорметана, 0,6 г полученного в предыдущей стадии соединения в 15 мл дихлорметана. Очищают путем кристаллизации из смеси циклогексана с этилацетатом (7: 25 по объему). Получают 0,53 г целевого продукта. 2D0= +1,2 (с = 1; этанол).
Следуя методикам, описанным в предыдущих примерах, исходя из хлорангидридов соответствующих кислот, которые в свою очередь получают из описанных в Препаративных примерах кислот, и полученного в Препаративном примере 2.1 соединения, получают соединения согласно изобретению, указанные в Таблице 2.
(a) Это соединение получают по методикам, описанным в стадиях А, затем Б Примера 1. Продукт очищают путем хроматографии на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (75:25 по объему).
(b) Это соединение получают по методикам, описанным в стадиях А, затем Б Примера 8. Продукт очищают путем хроматографии на диоксиде кремния, элюируя смесью толуола с этилацетатом (90:10 по объему).
(с) Это соединение получают по методикам, описанным в стадиях А, затем Б Примера 1. Продукт очищают путем хроматографии на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (80:20 по объему).
(d) Это соединение получают по методикам, описанным в стадиях А, затем Б Примера 1. Продукт очищают путем кристаллизации из этилацетата.
(е) Это соединение получают по методикам, описанным в стадиях А, затем Б Примера 1. Продукт очищают путем кристаллизации из диизопропилового эфира.
(f) Это соединение получают по методикам, описанным в стадиях А, затем Б Примера 32, используя полученное в Препаративном примере 2.1 соединение. Продукт очищают путем хроматографии на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (80:20 по объему).
(g) Это соединение получают по методикам, описанным в стадиях А, затем Б Примера 32, используя полученное в Препаративном примере 2.1 соединение. После высушивания над сульфатом натрия растворитель частично удаляют, выпавший осадок отсасывают, промывают его диэтиловым эфиром и высушивают его.
Пример 32N-[(1R)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1- (4-метилбензил-5-(4-хлор-3-метилфенил)пиразол-3-карбоксамид А) Хлорангидрид 1-(4-метилбензил-5-(4-хлор-3-метилфенил)- пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии А Примера 1, исходя из 2,8 г полученного в Препаративном примере 1.10 соединения, 1,7 мл тионилхлорида и 50 мл толуола. Получают 2,9 г целевого соединения. Б) N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1- (4-метилбензил-5-(4-хлор-3-метилфенил)пиразол-3-карбоксамид К раствору 0,765 г полученного в Препаративном примере 2.6 соединения и 1 мл триэтиламина в 20 мл дихлорметана при комнатной температуре прикапывают раствор 1,6 г полученного в предыдущей стадии соединения в 20 мл дихлорметана и перемешивают в течение ночи при комнатной температуре. Концентрируют в вакууме, остаток обрабатывают этилацетатом, отфильтровывают нерастворимую часть, фильтрат промывают водным насыщенным раствором гидрокарбоната натрия, буферным раствором с pH 2, водным насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (75:25 по объему). Получают 1,08 г целевого продукта; т.пл. = 108oC. 2D0= +0,3 с = 1; этанол).
Пример 33N-[(1R)-1,3,3-Триметилбицикло[2.2.1] гепт-2экзо-ил]-1- (4-метилбензил)-5-(4-хлор-3-метилфенил)пиразол- 3-карбоксамид К раствору 0,4 г смеси соединений, полученных в Препаративном примере 2.3, в 10 мл дихлорметана добавляют 0,6 мл триэтиламина, затем прикапывают раствор 0,66 г полученного в стадии А Примера 32 соединения в 10 мл дихлорметана и перемешивают в течение ночи при комнатной температуре. Концентрируют в вакууме, остаток экстрагируют этилацетатом, отфильтровывают нерастворимую часть, фильтрат промывают два раза водным насыщенным раствором гидрокарбоната натрия, два раза буферным раствором с pH 2, два раза водным насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (80: 20 по объему). Получают смесь, содержащую 58% соединения Примера 33 (форма экзо) и 41,6 % соединения Примера 32 (форма эндо) (определяют путем аналитической ВЭЖХ). Оба изомера разделяют путем препаративной ВЭЖХ: очищаемый образец массой 0,31 г растворяют в 13 мл исходного элюирующего средства (А/Б=10%/90%), добавляют 9 мл метанола и 4 мл ацетонитрила. После лиофилизации получают: – соединение Примера 33: масса= 0,084 г; чистота 100% (аналитическая ВЭЖХ) с временем удерживания = 41 минута; 2D0= +0,3 (с = 1; этанол).
ЯМР-спектр (м.д.): 0,8-1,15 (3с, 9H); 1,2-2 (м, 7H); 2,2-2,25 (м, 6H); 3,5 (д, 1H); 5,4 (с, 2H); 6,8-7,7 (м, 9H);– соединение Примера 32: масса = 0,056 г; чистота 98% (аналитическая ВЭЖХ) с временем удерживания = 43,9 минут. Пример 34 N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2экзо-ил]-1- (4-метилбензил)-5-(4-хлор-3-метилфенил)пиразол-3-карбоксамид К раствору 0,853 г полученного в Препаративном примере 2.2 соединения в 20 мл дихлорметана добавляют 1,5 мл триэтиламина, затем прикапывают раствор 1,6 г полученного в стадии А Примера 32 соединения в 20 мл дихлорметана и перемешивают в течение ночи при комнатной температуре. Концентрируют в вакууме, остаток экстрагируют этилацетатом, отфильтровывают нерастворимую часть, фильтрат промывают два раза водным насыщенным раствором гидрокарбоната натрия, два раза буферным раствором с pH 2, два раза водным насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (80:20 по объему). Получают смесь, содержащую 58% соединения Примера 34 (форма экзо) и 37% соединения Примера 18 (форма эндо) (определяют путем аналитической ВЭЖХ). Оба изомера разделяют путем препаративной ВЭЖХ: очищаемый образец массой 0,23 г растворяют в 13 мл исходного элюирующего средства (А/Б=10%/90%), добавляют 3 мл метанола и 9 мл ацетонитрила. После лиофилизации получают: – соединение Примера 34: масса=0,16 г; чистота 99,6% (аналитическая ВЭЖХ) с временем удерживания = 34,7 минут; т.пл. = 49oC; 2D0= -6,8 (с = 1; этанол).
ЯМР-спектр (м.д.): 0,8-1,15 (3с, 9H); 1,2-2 (м, 7H); 2,1-2,4 (м, 6H); 3,5 (д, 1H); 5.4 (с, 2H); 6,8-7,6 (м, 9H);– соединение Примера 18: масса = 0,066 г; чистота 94,7% (аналитическая ВЭЖХ) с временем удерживания = 37,2 минуты. Пример 35 N-(2,2,6,6-Тетраметилциклогекс-1-ил)-1-(4-метилбензил)-5- (4-хлор-3-метилфенил)пиразол-3-карбоксамид К раствору 0,25 г полученного в Препаративном примере 2.7 соединения в 15 мл дихлорметана добавляют 0,4 мл триэтиламина, затем прикапывают раствор 0,55 г полученного в стадии А Примера 32 соединения в 15 мл дихлорметана и перемешивают в течение ночи при комнатной температуре. Концентрируют в вакууме, остаток экстрагируют этилацетатом, фильтрат промывают два раза водным насыщенным раствором гидрокарбоната натрия, два раза буферным раствором с pH 2, два раза водным насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (80:20 по объему). Получают 0,1 г целевого продукта. Пример 36 N-[Бицикло[2.2.1] гепт-2экзо-ил] -1-(3-хлор-4-метилбензил)-5- (3,4-дихлорфенил)пиразол-3-карбоксамид А) Хлорангидрид 1-(3-хлор-4-метилбензил)-5-(3,4- дихлорфенил)-пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии А Примера 1, исходя из 10 г полученного в Препаративном примере 1.22 соединения, 5,5 мл тионилхлорида и 125 мл толуола. Получают 9,41 г целевого продукта. Б) N-[Бицикло[2.2.1] гепт-2экзо-ил] -1-(3-хлор-4-метилбензил) -5-(3,4-дихлорфенил)пиразол-3-карбоксамид К раствору 0,411 г 2экзо-аминонорборнана и 1 мл триэтиламина в 15 мл дихлорметана при комнатной температуре добавляют раствор 1,5 г полученного в предыдущей стадии соединения в 15 мл дихлорметана и перемешивают в течение ночи при комнатной температуре. Реакционную смесь концентрируют в вакууме, остаток экстрагируют этилацетатом, отфильтровывают нерастворимую часть, органическую фазу промывают насыщенным раствором гидрокарбоната натрия, буферным раствором с pH 2, водой, сушат над сульфатом магния и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (80:20 по объему). Получают 0,5 г целевого продукта; т.пл. = 145,6oC. Пример 37 N-[Бицикло[2.2.1] гепт-2экзо-ил] -N-пропил-1-(3-хлор-4- метилбензил)-5-(3,4-дихлорфенил)пиразол-3-карбоксамид Это соединение получают по методике, описанной в стадии Б Примера 36, исходя из 0,695 г полученного в Препаративном примере 2.4 соединения, 1 мл триэтиламина в 15 мл дихлорметана и 1,5 г соединения, полученного в стадии А Примера 36, в 15 мл дихлорметана. Получают 0,417 г целевого продукта. Пример 38 N-(2-Метилциклогекс-1-ил)-1-(3-хлор-4-метилбензил)-5- (3,4-дихлорфенил)пиразол-3-карбоксамид Это соединение получают по методике, описанной в стадии Б Примера 36, исходя из 1 мл 2-метилциклогексиламина, 1 мл триэтиламина в 15 мл дихлорметана и 1,5 г соединения, полученного в стадии А Примера 36, в 15 мл дихлорметана. Соединение очищают путем хроматографии на диоксиде кремния, элюируя смесью дихлорметана с метанолом (98:2 по объему). Получают 0,27 г целевого продукта; т.пл. = 165oC. Пример 39 N-(2,6-Диметилциклогекс-1-ил)-1-(3-хлор-4-метилбензил)- 5-(3,4-дихлорфенил)пиразол-3-карбоксамид Это соединение получают по методике, описанной в стадии Б Примера 36, исходя из 0,605 г 2,6-диметилциклогексиламингидрохлорида, 1 мл триэтиламина в 15 мл дихлорметана и 1,5 г соединения, полученного в стадии А Примера 36, в 15 мл дихлорметана. Получают 0,6 г целевого продукта; т.пл. = 59,9oC. Пример 40 N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1- [(1R,S)-1-(3,4-дихлорфенил)этил]-5-(4-метилфенил)пиразол-3- карбоксамид А) Хлорангидрид 1-[1-(3,4-дихлорфенил)этил]-5-(4- метилфенил)пиразол-3-карбоновой кислоты К суспензии 1,2 г полученного в Препаративном примере 1.25 соединения в 50 мл толуола при комнатной температуре добавляют 0,7 мл тионилхлорида, затем кипятят с обратным холодильником в течение двух часов. После охлаждения до комнатной температуры реакционную смесь концентрируют в вакууме, остаток обрабатывают толуолом и растворитель выпаривают в вакууме. Получают 1,3 г целевого продукта. Б) N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо- ил] -1-[(1R,S)-1-(3,4-дихлорфенил)этил]-5-(4-метилфенил) пиразол-3-карбоксамид К раствору 0,341 г полученного в Препаративном примере 2.1 соединения в 15 мл дихлорметана добавляют 0,6 мл триэтиламина, затем прикапывают раствор 0,7 г полученного в предыдущей стадии соединения в 15 мл дихлорметана и перемешивают в течение ночи при комнатной температуре. Концентрируют в вакууме, остаток экстрагируют этилацетатом, отфильтровывают нерастворимую часть, фильтрат промывают водным насыщенным раствором гидрокарбоната натрия, буферным раствором с pH 2, водным насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (80:20 по объему). Получают 0,54 г целевого продукта; т.пл. = 112oC. ЯМР-спектр (м. д. ): 0,6-1,9 (м, 19H); 2,3 (с, 3H); 3,6 (мультиплет, 1H); 5,55 (кд, 1H); 6,6-7,6 (м, 9H).
Пример 41N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1- [(1R,S)-1-(4-метилфенил)этил]-5-(4-хлор-3-метилфенил) пиразол-3-карбоксамид А) Хлорангидрид 1-[1-(4-метилфенил)этил]-5-(4-хлор-3- метилфенил)пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии А Примера 40, исходя из 0,61 г полученного в Препаративном примере 1.26 соединения в 30 мл толуола и 0,4 мл тионилхлорида. Получают 0,7 г целевого продукта. Б) N-[(1S)-1,3,3-Триметилбицикло[2.2.1]гепт-2эндо-ил]-1- [(1R,S)-1-(4-метилфенил)этил]-5-(4-хлор-3-метилфенил)пиразол- 3-карбоксамид Это соединение получают по методике, описанной в стадии Б Примера 40, исходя из 0,36 г полученного в Препаративном примере 2.1 соединения в 15 мл дихлорметана, 0,45 г триэтиламина и раствора 0,7 г полученного в предыдущей стадии соединения в 15 мл дихлорметана. Соединение очищают путем хроматографии на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (75:25 по объему). Получают 0.13 г целевого продукта; т.пл. = 65oC. ЯМР-спектр (м. д.): 0,8-2,05 (м, 19H); 2,2-2,5 (м, 6H); 3,75 (д, 1H; 5,65 (кд, 1H); 6,8-7,7 (м, 9H).
Пример 42N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1- [(1R,S)-1-(3,4-дихлорфенил)этил]-5-(4-хлор-3-метилфенил) пиразол-3-карбоксамид А) Хлорангидрид 1-[1-(3,4-дихлорфенил)этил] -5-(4-хлор-3- метилфенил)пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии А Примера 40, исходя из 0,9 г полученного в Препаративном примере 1.27 соединения в 20 мл толуола и 0,5 мл тионилхлорида. Получают 0,95 г целевого продукта. Б) N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1- [(1R,S)-1-(3,4-дихлорфенил)этил]-5-(4-хлор-3-метилфенил) пиразол-3-карбоксамид Это соединение получают по методике, описанной в стадии Б Примера 40, исходя из 0,455 г полученного в Препаративном примере 2.1 соединения в 15 мл дихлорметана, 0,8 г триэтиламина и раствора 0,95 г полученного в предыдущей стадии соединения в 15 мл дихлорметана. После высушивания органической фазы над сульфатом натрия растворитель частично удаляют в вакууме и образовавшиеся кристаллы отсасывают. Получают 0,76 г целевого продукта; т.пл. = 156oC. ЯМР-спектр; (м. д. ): 0,6-2 (м, 19H); 2,35 (с, 3H); 3,7 (мультиплет, 1H); 5,7 (кд, 1H); 6,7-7,7 (м, 8H).
Пример 43N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1- [(1R,S)-1-(4-метилфенил)пропил]-5-(4-хлор-3-метилфенил) пиразол-3-карбоксамид А) Хлорангидрид 1-[1-(4-метилфенил)пропил] -5-(4-хлор-3- метилфенил)пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии А Примера 40, исходя из 0,87 г полученного в Препаративном примере 1.28 соединения в 50 мл толуола и 0,26 мл тионилхлорида. Получают 0,89 г целевого продукта. Б) N-[(1S)-1,3,3-Триметилбицикло[2.2.1]гепт-2эндо-ил]-1- [(1R,S)-1-(4-метилфенил)пропил]-5-(4-хлор-3-метилфенил) пиразол-3-карбоксамид К раствору 0,51 г полученного в предыдущей стадии соединения и 0,25 г полученного в Препаративном примере 2.1 соединения в 50 мл дихлорметана добавляют 0,363 мл триэтиламина и перемешивают 72 часа при комнатной температуре. Концентрируют в вакууме, остаток экстрагируют этилацетатом, отфильтровывают нерастворимую часть, фильтрат промывают водным насыщенным раствором гидрокарбоната натрия, буферным раствором с pH 2, насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (75:25 по объему). Получают 0,44 г целевого продукта; т.пл. = 136oC. ЯМР-спектр (м. д. ): 0,5-2,6 (м, 27H); 3,75 (д, 1H); 4,95 (дд, 1H); 6,6-7,4 (м, 9H).
Пример 44N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1-[1-метил-1-(4-метилфенил)этил]-5-(4-хлор-3-метилфенил) пиразол-3-карбоксамид А) Хлорангидрид 1-[1-метил-1-(4-метилфенил)этил]-5-(4-хлор-3- метилфенил)пиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии А Примера 40, исходя из 1,5 г полученного в Препаративном примере 1.29 соединения в 100 мл толуола и 0,44 мл тионилхлорида. Получают 1,55 г целевого соединения. Б) N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1- [1-метил-1-(4-метилфенил)этил]-5-(4-хлор-3-метилфенил) пиразол-3-карбоксамид К раствору 0,59 г полученного в предыдущей стадии соединения и 0,29 г полученного в Препаративном примере 2.1 соединения в 50 мл дихлорметана добавляют 0,424 мл триэтиламина и перемешивают в течение ночи при комнатной температуре. Концентрируют в вакууме, остаток экстрагируют этилацетатом, отфильтровывают нерастворимую часть, фильтрат промывают водным насыщенным раствором гидрокарбоната натрия, буферным раствором с pH 2, насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью дихлорметана с метанолом (97,5: 2,5 по объему). Получают 0,3 г целевого продукта; т.пл. = 195oC; 2D0= -6,5 (с = 0,64; дихлорметан).
Пример 45N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1- (2,4-дихлорбензил)-5-(4-хлорфенил)-4-метилпиразол-3-карбоксамид А) Хлорангидрид 1-(2,4-дихлорбензил)-5-(4-хлорфенил)-4- метилпиразол-3-карбоновой кислоты К раствору 0,71 г полученного в Препаративном примере 1.30 соединения в 50 мл толуола при комнатной температуре добавляют 0,191 мл тионилхлорида, затем кипятят с обратным холодильником в течение ночи. Концентрируют в вакууме, остаток обрабатывают толуолом и растворитель выпаривают в вакууме. Получают 0,72 г целевого продукта в виде масла, которое затвердевает. Б) N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1-(2,4-дихлорбензил)-5-(4-хлорфенил)-4-метилпиразол-3-карбоксамид Это соединение получают по методике, описанной в стадии Б Примера 43, исходя из 0,315 г полученного в Препаративном примере 2.1 соединения, 0,46 мл триэтиламина и 0,653 г полученного в предыдущей стадии соединения в 50 мл дихлорметана. Получают 0,68 г целевого продукта; т.пл. = 125oC; 2D0= -0,3 (с = 1; этанол).
Пример 46N-[(1S)-1,3,3-Триметилбицикло[2.2.1]гепт-2эндо-ил]-1-(3, 4-дихлорбензил)-5-(4-хлорфенил)-4-метилпиразол-3-карбоксамид А) Хлорангидрид 1-(3,4-дихлорбензил)-5-(4-хлорфенил)-4- метилпиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии А Примера 45, исходя из 1 г полученного в Препаративном примере 1.31 соединения в 50 мл толуола и 0,28 мл тионилхлорида. Получают 1,03 г целевого продукта в виде масла, которое затвердевает. Б) N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1-(3,4-дихлорбензил)-5-(4-хлорфенил)-4-метилпиразол-3- карбоксамид Это соединение получают по методике, описанной в стадии Б Примера 43, исходя из 0,315 г полученного в Препаративном примере 2.1 соединения, 0,46 мл триэтиламина и 0,687 г полученного в предыдущей стадии соединения в 50 мл дихлорметана. Получают 0.72 г целевого продукта; т.пл. = 69oC; 2D0= -1,8 (с = 1; этанол).
Пример 47N-[(1S)-1,3,3-Триметилбицикло[2.2.1]гепт-2эндо-ил]-1-(3- хлор-4-метилбензил)-5-(4-хлорфенил)-4-метилпиразол-3-карбоксамид А) Хлорангидрид 1-(3-хлор-4-метилбензил)-5-(4-хлорфенил)- 4-метилпиразол-3-карбоновой кислоты Это соединение получают по методике, описанной в стадии А Примера 45, исходя из 1 г неочищенного соединения, полученного в Препаративном примере 1.32, в 50 мл толуола и 0,29 мл тионилхлорида. Получают 1,05 г целевого продукта. Б) N-[(1S)-1,3,3-Триметилбицикло[2.2.1]гепт-2эндо-ил]-1- (3-хлор-4-метилбензил)-5-(4-хлорфенил)-4-метилпиразол-3- карбоксамид Это соединение получают по методике, описанной в стадии Б Примера 43, исходя из 0,29 г полученного в Препаративном примере 2.1 соединения, 0,24 мл триэтиламина и 0,6 г полученного в предыдущей стадии соединения в 50 мл дихлорметана. Получают 0,43 г целевого продукта; т.пл. = 58oC; 2D0= -1,7 (с = 1; этанол).
Пример 48N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил]- 1-(4-метилбензил)-5-(4-хлор-3-метилфенил)-4-метилпиразол-3- карбоксамид А) Хлорангидрид 1-(4-метилбензил)-5-(4-хлор-3-метилфенил) -4-метилпиразол-3-карбоновой кислоты К суспензии 1,5 г полученного в Препаративном примере 1.33 соединения в 40 мл толуола добавляют 1 мл тионилхлорида и кипятят с обратным холодильником в течение двух часов. Реакционную смесь концентрируют в вакууме, остаток обрабатывают толуолом и растворитель выпаривают в вакууме. Получают 1,5 г целевого продукта. Б) N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1-(4-метилбензил)-5-(4-хлор-3-метилфенил)-4-метилпиразол-3- карбоксамид Это соединение получают по методике, описанной в стадии Б Примера 43, исходя из 0,42 г полученного в Препаративном примере 2.1 соединения, 0,7 мл триэтиламина и 0,8 г полученного в предыдущей стадии соединения в 40 мл дихлорметана. Получают 0,62 г целевого продукта; т.пл. = 58oC; 2D0= -2,4 (с = 1; этанол).
Пример 49N-[(1S)-1,3,3-Триметилбицикло[2.2.1]гепт-2эндо-ил]-1-(3, 4-дихлорбензил)-5-(4-хлор-3-метилфенил)-4-метилпиразол-3- карбоксамид А) Хлорангидрид 1-(3,4-дихлорбензил)-5-(4-хлор-3- метилфенил)-4-метилпиразол-3-карбоновой кислоты К суспензии 2,7 г полученного в Препаративном примере 1.34 соединения в 50 мл толуола добавляют 1,5 мл тионилхлорида и кипятят с обратным холодильником в течение двух часов. Реакционную смесь концентрируют в вакууме, остаток обрабатывают толуолом и растворитель выпаривают в вакууме. Получают 2.8 г целевого продукта. Б) N-[(1S)-1,3,3-Триметилбицикло[2.2.1]гепт-2эндо-ил] -1-(3,4-дихлорбензил)-5-(4-хлор-3-метилфенил)-4-метилпиразол- 3-карбоксамид Это соединение получают по методике, описанной в стадии Б Примера 43, исходя из 0,683 г полученного в Препаративном примере 2.1 соединения, 1,2 мл триэтиламина и 1,5 г полученного в предыдущей стадии соединения в 40 мл дихлорметана. Получают 1,03 г целевого продукта; т.пл. = 69oC; 2D0= -2,1 (с = 1; этанол).
Пример 50N-[(1S)-1,3,3-Триметилбицикло[2.2.1]гепт-2эндо-ил]-1-(3- хлор-4-метилбензил)-5-(4-хлорфенил)-4-(этоксиметил)пиразол-3- карбоксамид А) Хлорангидрид 1-(3-хлор-4-метилбензил)-5-(4-хлорфенил)- 4-(этоксиметил)пиразол-3-карбоновой кислоты К раствору 0,2 г полученного в Препаративном примере 1.35 соединения в 30 мл толуола добавляют 0,053 мл тионилхлорида и кипятят с обратным холодильником в течение двух часов. Концентрируют в вакууме, остаток обрабатывают толуолом и растворитель выпаривают в вакууме. Получают 0,21 г целевого продукта в виде масла. Б) N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо- ил]-1-(3-хлор-4-метилбензил)-5-(4-хлорфенил)-4-(этоксиметил) пиразол-3-карбоксамид Это соединение получают по методике, описанной в стадии Б Примера 40, исходя из 0,086 г полученного в Препаративном примере 2.1 соединения в 25 мл дихлорметана, 0,125 мл триэтиламина и 0,2 г полученного в предыдущей стадии соединения в 25 мл дихлорметана. Получают 0,15 г целевого продукта; т.пл. = 50-51oC; 2D0= -2,8 (с = 1; этанол).
Пример 51N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил]-1-(5-хлориндан-1-ил)-5-(4-хлорфенил) пиразол-3-карбоксамид А) Хлорангидрид 1-(5-хлориндан-1-ил)-5-(4-хлорфенил) пиразол-3-карбоновой кислоты К суспензии 1 г полученного в Препаративном примере 1.36 соединения в 20 мл толуола добавляют 0,6 мл тионилхлорида и кипятят с обратным холодильником в течение двух часов. После охлаждения до комнатной температуры реакционную смесь концентрируют в вакууме, остаток обрабатывают толуолом и растворитель выпаривают в вакууме. Получают 1 г целевого продукта. Б) N-[(1S)-1,3,3-Триметилбицикло[2.2.1]гепт-2эндо-ил]-1- (5-хлориндан-1-ил)-5-(4-хлорфенил)пиразол-3-карбоксамид К раствору 0,493 г полученного в Препаративном примере 2.1 соединения и 1 мл триэтиламина в 15 мл дихлорметана при комнатной температуре прикапывают раствор 1 г полученного в предыдущей стадии соединения в 15 мл дихлорметана и перемешивают в течение ночи при комнатной температуре. Концентрируют в вакууме, остаток обрабатывают этилацетатом, отфильтровывают нерастворимую часть, фильтрат промывают водным насыщенным раствором гидрокарбоната натрия, буферным раствором с pH 2, водным насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом (80: 20 по объему). Получают 0,71 г целевого продукта; т.пл. = 86oC; 2D0= +0,5 (c=1; этанол).
ЯМР-спектр, (м. д. ): 0,4-1,8 (м, 16H); 2,5 (мультиплет, 2H); 2,95 (мультиплет, 2H); 3,5 (д, 1H); 5,8 (т, 1H); 6,6-7,7 (м. 9H).
Наблюдают эффект Оверхаузера (N.O.E.) между протонами в положении 1 индан-1-ильной группы и протонами g2=g6=H.
Пример 52[(1R)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1-(3,4- дихлорбензил)-5-(4-метилфенил)пиразол-3-карбоксилад В течение 72-х часов при комнатной температуре перемешивают смесь 1,5 г соединения, полученного в стадии А Примера 1, 0,73 г (1R)-эндо-(+)фенхилового спирта, 50 мл пиридина и 0,02 г 4-диметиламинопиридина. Реакционную смесь концентрируют в вакууме остаток хроматографируют на диоксиде кремния, элюируя дихлорметаном. Получают 0,23 г целевого продукта; т.пл. = 107oC 2D0= +6,8 (с = 1; этанол)Пример 53 [(1R)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1- (3-хлор-4-метилбензил)-5-(3,4-дихлорфенил)пиразол-3- карбоксилат К раствору 2,6 г соединения, полученного в стадии А Примера 36, в 50 мл пиридина добавляют 0,87 г (1R)-эндо-(+)фенхилового спирта и 0,02 г 4-диметиламинопиридина, затем перемешивают при комнатной температуре в течение 48 часов. Концентрируют в вакууме, остаток обрабатывают диэтиловым эфиром, выпавший осадок отфильтровывают и высушивают его. Осадок хроматографируют на диоксиде кремния, элюируя смесью циклогексана с этилацетатом и дихлорметаном (85:15:3 по объему). Получают 0,40 г целевого продукта; т.пл. = 161oC; 2D0= +3,9 (с = 1; дихлорметан).
Пример 54N-[(1S)-1,3,3-Триметилбицикло[2.2.1] гепт-2эндо-ил] -1-(2, 4-дихлорбензил)-3-(4-хлорфенил)пиразол-5-карбоксамид А) Хлорангидрид 1-(2,4-дихлорбензил)-3-(4-хлорфенил) пиразол-5-карбоновой кислоты К суспензии 0,79 г полученного в Препаративном примере 1.38 соединения в 20 мл толуола добавляют 0,54 мл тионилхлорида и кипятят с обратным холодильником в течение трех часов. Концентрируют в вакууме, остаток обрабатывают с помощью 20 мл толуола и растворитель выпаривают в вакууме. Получают 0,9 г целевого продукта. Б) N-[(1S)-1,3,3-Триметилбицикло[2.2.1]гепт-2эндо-ил]-1- (2,4-дихлорбензил)-3-(4-хлорфенил)пиразол-5-карбоксамид Раствор 0,21 г полученного в Препаративном примере 2.1 соединения и 0,3 мл триэтиламина в 10 мл дихлорметана охлаждают до 0oC, прикапывают раствор 0,45 г полученного в предыдущей стадии соединения в 15 мл дихлорметана и перемешивают в течение 16 часов при комнатной температуре. Реакционную смесь выливают в 100 мл ледяной воды, экстрагируют дихлорметаном, органическую фазу промывают водой, насыщенным раствором хлорида натрия, сушат над сульфатом натрия и растворитель выпаривают в вакууме. После кристаллизации из смеси дихлорметана с диизопропиловым эфиром получают 0,37 г целевого продукта; т.пл. = 171oC; 2D0= -3,2 (с = 0,5; этанол).
Пример 55Желатиновая капсула (с лекарством) Соединение Примера 18 – 1 мг Модифицированный кукурузный крахмал – 47 мг Моногидрат лактозы EFK – 150 мг Стеарат магния – 2 мг На одну белую непрозрачную желатиновую капсулу, размер 3 – Заполнение: 200 мг Пример 56 Желатиновая капсула (с лекарством) Соединение Примера 18 – 30 мг Модифицированный кукурузный крахмал – 30 мг Стеарат магния – 2 мг Моногидрат лактозы EFK – Достаточное количество На одну белую непрозрачную желатиновую капсулу, размер 3 – Заполнение: 200 мг Пример 57 Таблетка: Соединение Примера 18 – 10 мг Поливинилпирролидон КЗО – 4,5 мг Сшитая натриевая соль карбоксиметилцеллюлозы – 3 мг Стеарат магния – 1,5 мг Моногидрат лактозы, 200 меш – Достаточное количество Очищенная вода – Достаточное количество На одну конечную делимую таблетку: – 150 мг Пример 58 Желатиновая капсула (с лекарством) Соединение Примера 48 – 30 мг Гидроксипропилметилцеллюлоза 6mPas – 7,5 мг Моногидрат лактозы, 200 меш – Достаточное количество Стеарат магния – 2,5 мг Очищенная вода – Достаточное количество На одну желатиновую капсулу, размер 1 – Заполнение: 250 мг Пример 59 Таблетка: Соединение Примера 48 – 40 мг Кукурузный крахмал – 50 мг Поливинилпирролидон КЗО – 9 мг Натриевая соль карбоксиметилкрахмала – 9 мг Стеарат магния – 3 мг Моногидрат лактозы, 200 меш – Достаточное количество Ha одну конечную делимую таблетку: – 300 мгм Формула изобретения
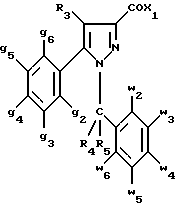 где Х1 означает группу -NR1R2 или группу -OR2; g2, g3, g4, g5, g6 являются одинаковыми или разными, и каждый из них, независимо друг от друга, означает водород, атом галогена, алкил с 1 – 4 атомами углерода, алкоксил с 1 – 4 атомами углерода, трифторметил или алкилтиогруппу с 1 – 4 атомами углерода; w2, w3, w4, w5, w6 являются одинаковыми или разными, и каждый из них, независимо друг от друга, обозначает водород, атом галогена, алкил с 1 – 4 атомами углерода, алкоксил с 1 – 4 атомами углерода, или трифторметил, при условии, что по крайней мере один из заместителей g2, g3, g4, g5, g6 и по крайней мере один из заместителей w2, w3, w4, w5, w6 отличаются от водорода; R1 означает водород или алкил с 1 – 4 атомами углерода; R2 означает неароматический карбоциклический радикал с 3 – 15 атомами углерода, незамещенный или одно- или многократно замещенный алкилом с 1 – 4 атомами углерода; R3 означает водород или группу -СН2-R6; R4 и R5, каждый, независимо друг от друга, означает водород или алкил с 1 – 4 атомами углерода; или R4 означает водород и R5 и w6 вместе образуют этиленовый радикал; R6 означает водород или, когда заместители g2, g3, g4, g5, и/или g6 отличаются от алкила с 1 – 4 атомами углерода, R6 означает водород, алкил с 1 – 4 атомами углерода или алкоксил с 1 – 5 атомами углерода, и их соли. 2. Соединение формулы I по п.1, где Х1 означает группу -NR1R2, в которой R1 означает водород, и его соли. 3. Соединение формулы I по п.1, где Х1 означает группу -NR1R2 или группу -OR2, в которой R2 означает 1,3,3-триметилбицикло [2.2.1]гепт-2-ил или бицикло[3.2.1]окт-3-ил, и его соли. 4. Соединение формулы I по п.1, где R3 означает водород или группу -СН2-R6, в которой R6 означает водород, и его соли. 5. Соединение формулы I по п.1, где либо каждый из R4 и R5 означает водород, либо R4 означает водород, а R5 означает алкил с 1 – 4 атомами углерода, и его соли. 6. Соединение формулы I по п.1, где каждый из g2, g5 и g6 означает водород, а g3 и g4 имеют значения, указанные выше для соединений формулы I в п.1, и его соли. 7. Соединение формулы I по п.1, где каждый из w5 и w6 означает водород, w4 означает атом галогена, алкил с 1 – 4 атомами углерода, алкоксил с 1 – 4 атомами углерода или трифторметил, и либо каждый из w2 и w3 означают водород, либо один из них означает водород, а другой означает атом галогена или алкил с 1 – 4 атомами углерода, и его соли. 8. Соединение по п.1 формулы Ia 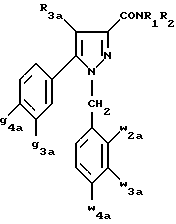 где R1, R2 имеют значения, указанные для соединений формулы I в п.1; R3a означает водород или группу -СН2-R6a; R6a означает водород или при условии, что заместители g3a и/или g4a отличаются от алкила с 1 – 4 атомами углерода, R6a означает водород, метил или этил; g3a означает водород, атом галогена, алкил с 1 – 4 атомами углерода или трифторметил; g4a означает атом галогена, алкил с 1 – 4 атомами углерода или трифторметил; w4a означает атом галогена, алкил с 1 – 4 атомами углерода или трифторметил; каждый из w2a и w3а означает водород или один из них означает водород, а другой означает атом галогена или алкил с 1 – 4 атомами углерода, и его соли. 9. Соединение по п.1 формулы Ib 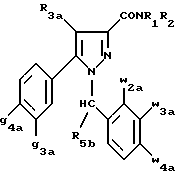 где R1, R2 имеют значения, указанные для соединений формулы I в п.1; R3a, w2a, w3a, g3a и g4a имеют значения, указанные для соединений формулы Ia в п.8; R5b означает алкил с 1 – 4 атомами углерода, и его соли. 10. Соединение по п.1 формулы Ic 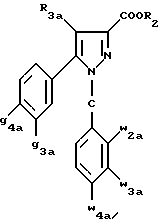 где R2 имеет значение, указанное для соединений формулы I в п.1; R3a, w2a, w3a, w4a, g3a и g4a имеют значения, указанные для соединений формулы Ia в п.8, и его соли. 11. Способ получения производных пиразола формулы I по п.1 и их солей, отличающийся тем, что: 1) функциональное производное пиразол-3-карбоновой кислоты формулы II 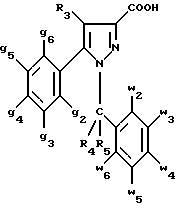 где w2, w3, w4, w5, w6, g2, g3, g4, g5, g6, R3, R4 и R5 имеют значения, указанные для соединений формулы I в п.1, обрабатывают соединением формулы XXIV H-X1 где Х1 имеет значение, указанное для соединений формулы I в п.1; 2) и, в случае необходимости, таким образом полученное соединение превращают в одну из его солей. 12. Способ по п. 11, получения соединений формулы I, где Х1 означает группу -NR1R2, и их солей, отличающийся тем, что: 1) функциональное производное пиразол-3-карбоновой кислоты формулы II по п.11 обрабатывают амином формулы III HNR1R2 где R1 и R2 имеют значения, указанные для соединений формулы I в п.1; 2) и, в случае необходимости, таким образом полученное соединение превращают в одну из его солей. 13. Способ по п. 11 получения соединений формулы I, где Х1 означает группу -OR2, и их солей, отличающийся тем, что: 1) функциональное производное пиразол-3-карбоновой кислоты формулы II по п.11 обрабатывают спиртом формулы XIV HO-R2 где R2 имеет значение, указанное для соединений формулы I в п.1; 2) и, в случае необходимости, таким образом полученное соединение превращают в одну из его солей. 14. Фармацевтическая композиция, имеющая сродство к рецепторам СВ2, содержащая активное начало и фармацевтически приемлемые эксципиенты, отличающаяся тем, что активное начало является соединением по пп.1 – 10 или одной из его фармацевтически приемлемых солей, содержащимися в эффективном количестве. 15. Фармацевтическая композиция по п.14, содержащая 0,5 – 1000 мг действующего начала. 16. Фармацевтическая композиция по п. 15, содержащая 2,5 – 250 мг действующего начала. 17. Соединения по любому из пп.1 – 10, или их фармацевтически приемлемые соли, отличающиеся тем, что указанные соединения являются антагонистами рецепторов СВ2. РИСУНКИ
PD4A – Изменение наименования обладателя патента Российской Федерации на изобретение
Номер и год публикации бюллетеня: 9-2002
(73) Новое наименование патентообладателя:
Извещение опубликовано: 27.03.2002
PD4A – Изменение наименования обладателя патента Российской Федерации на изобретение
(73) Новое наименование патентообладателя:
Извещение опубликовано: 27.01.2006 БИ: 03/2006
|
||||||||||||||||||||||||||