Патент на изобретение №2170228
|
||||||||||||||||||||||||||
(54) 1-ФЕНИЛАЛКАНОНЫ – НОВЫЕ ЛИГАНДЫ 5-НТ4-РЕЦЕПТОРА
(57) Реферат: Изобретение относится к 1-фенилалканонам – новым лигандам 5-НТ4-рецепторам формулы I, где R1 – галоген; R2 – Н, C1-C4алкокси; R3 – C1-C4алкокси, фенил C1-C4алкокси, где фенил необязательно замещен 1-3 заместителями, независимо выбранными из C1-C4алкила, C1-C4алкилокси, 3,4-метилендиокси; R2 и R3 вместе обозначают метилендиокси, этилендиокси; R4 обозначает группу формулы (а) или (b), где n = 3, 4, 5; р = 0; q = 1 или 2; R5 и R6 каждый C1-C4алкил или вместе – (CH2)4 – , – (CH2)6 -, – (CH2)2O(CH2)2 -, -CHR8CH2CR9R10CHR11CH2-, где R8 и R11 каждый – Н или вместе – (CH2)t-, где t =1; R9 – Н, -ОН, C1-C8алкил, C1-C4алкилокси; R10 – Н, C1-C8алкил, фенил, – (СН2)x R12, где х = 0, 1, 2, 3; R12 – ОН, C1-C4алкилокси, – C(O)NR13R14, – NR13C(O)OR14, -SO2NR13R14, -NR13SO2R14, -NR13SO2NR14R15, -NR13C(O)NR14R15; R13, R14, R15 – независимо – Н, C1-C4алкил, CF3; R7 – Н, C1-C8алкил, C3-C8алкенил, фенил C1-C4алкил, где фенил необязательно замещен C1-C4алкилокси, метилендиокси, этилендиокси; или R7 – (СН2)z – R12, где z = 2, 3. Соединения I являются новыми лигандами 5-НТ4-рецептора, что позволяет использовать их в фармацевтической композиции и в способе лечения состояний, выбранных из ЦНС, расстройств желудочно-кишечного тракта, сердечно-сосудистой системы, мочевых путей. 4 с. и 34 з.п.ф-лы. 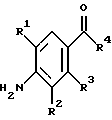 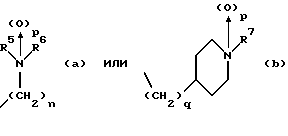
Область техники, к которой относится изобретение Настоящее изобретение относится к 1-фенилалкан-1-онам, новым лигандам 5-НТ4-рецептора, и способам применения и получения таких лигандов. Описание уровня техники Серотонин, нейромедиатор со смешанными и сложными фармакологическими характеристиками, был впервые открыт в 1948 г. и впоследствии стал предметом важных исследований. Серотонин, также называемый 5-гидрокси-триптамином (5-НТ), действует и на центральные, и на периферические дискретные 5-НТ-рецепторы. В настоящее время 5-НТ-рецептор подразделяется на четыре основных подкласса: 5-НТ1-, 5-НТ2-, 5-НТ3– и 5-НТ4-рецепторы, каждый из которых также может быть гетерогенным. 5-НТ4-рецептор был обнаружен в широком разнообразии тканей и видов. Например, 5-НТ4-рецепторы были обнаружены в центральной нервной системе (например, авторадиографические исследования показывают высокоспецифическое связывание лигандов с высоким сродством к 5-НТ4-рецептору в обонятельных бугорках, стратуме, черной субстанции, в верхнем колликулюсе и в дорзальном, медиальном и вентральном гиппокампе). Таким образом, полагают, что 5-НТ4-рецептор должен участвовать в деятельности областей центральной нервной системы, влияющих на страх, депрессию, умственные способности, зависимость, шизофрению, аппетит и т. д. ; и лекарства, которые взаимодействуют с 5-НТ4-рецепторами (т. е. лиганды 5-НТ4-рецептора), оказывают различные терапевтические воздействия на расстройства центральной нервной системы (ЦНС). 5-НТ4-рецепторы также обнаружены в пищеварительных системах у широкого разнообразия видов животных, включая человека, и обнаружено, что они модулируют двигательную функцию желудочно-кишечной системы (см. Prokinetic Agents: A Key in the Future of Gastroenterology. Reynolds R.C. Gastroenterology Clinics of North America, 1989, 18, 437-457). Кроме того, 5-НТ4-рецепторы модулируют тонус гладких мышц мочевого пузыря (см., например, Corsi М., Pietra C., Toson G., Trist D., Tuccito G., Artibani W. Br., J.Pharmacol., 1991, 104, 719-725), а также выполняет функцию медиатора вызванной 5-НТ положительной хронотропии в ткани правого предсердия (см., например, Kaumann A.; Sanders L.; Brown A.; Murray К.; Brown М. ; Brown M. Naunyn-Schmiedeberg’s, 1991, 344, 150-159). Описание этих и других упомянутых в данной заявке документов (например, в разделе “Фармакология” в “Подробном описании изобретения”) включены в нее в качестве ссылки. Краткое описание сущности изобретения Первым предметом настоящего изобретения является соединение формулы I: 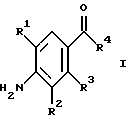 где R1 обозначает галоген; R2 обозначает водород или C1-C4алкилокси и R3 обозначает C1-C4алкилокси или фенил C1-C4алкилокси (где фенил необязательно замещен одним-тремя заместителями, независимо выбранными из галогена, гидроксила, C1-C4алкила, C1-C4алкилокси, нитро, амино, аминокарбонила, C1-C4алкиламино, диC1-C4алкиламино, C1-C4алканоиламино и 3,4-метилендиокси) или R2 и R3 вместе обозначают метилендиокси или этилендиокси; и R4 обозначает группу формулы (а) или (b) 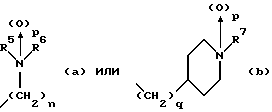 где n равно 3, 4 или 5; p равно 0 или 1; q равно 1 или 2; R5 и R6 каждый обозначают C1-C4алкил или вместе образуют -(CH2)4-, -(CH2)6-, -(CH2)2O(CH2)2– или -CHR8CH2CR9R10CHR11CH2-, где R8 и R11 каждый обозначает водород или вместе обозначают -(CH2)t, где t равно 1, 2 или 3, R9 обозначает водород, гидроксил, C1-C8алкил, C3-C8алкенил или C1-C4алкилокси и R10 обозначает водород, C1-C8алкил либо C3-C8алкенил или фенил, тиенил, пирролил либо фурил (необязательно замещенный одним-двумя заместителями, независимо выбранными из C1-C4алкила, C1-C4алкилокси, трифторметила и галогена) или -(CH2)xR12, где x равно 0, 1, 2 или 3 и R12 обозначает гидроксил, C1-C4алкилокси, -C(O)NR13R14, -NR13C(O)R14, -NR13C(O)OR14, -SO2NR13R14, -NR13SO2R14, -NR13SO2NR14R15 или – NR13C(O)NR14R15, где R13, R14 и R15 независимо обозначают водород, C1-C4алкил, трифторметил или арил; и R7 обозначает водород, C1-C8алкил или C3-C8алкенил, или фенилC1-C4алкил (где фенил необязательно замещен одним-тремя заместителями, независимо выбранными из C1-C4алкилокси, метилендиокси, этилендиокси или галогена), или -(CH2)zR12, где z равно 2 или 3 и R12 имеет указанные выше значения; и его фармацевтически приемлемые соли, отдельные изомеры и смеси изомеров. Вторым предметом настоящего изобретения является фармацевтическая композиция, включающая терапевтически эффективное количество соединения формулы I или его отдельного изомера, смеси изомеров либо фармацевтически приемлемой соли или солей в сочетании с одним или более фармацевтически приемлемыми инертными наполнителями. Третьим предметом настоящего изобретения является способ лечения состояния, способного улучшиться с помощью лекарства, взаимодействующего с 5-НТ4-рецепторами, у животного, если это необходимо, причем способ включает введение такому животному терапевтически эффективного количества соединения формулы I или его отдельного изомера, смеси изомеров или фармацевтически приемлемой соли или солей. Пятым предметом настоящего изобретения является способ получения соединений формулы I, и он представлен ниже в “Подробном описании изобретения”. Подробное описание изобретения Определения Если не указано иное, то следующие термины, используемые в описании изобретения и в формуле изобретения, имеют приведенные ниже значения. “Алкил” означает прямой или разветвленный насыщенный углеводородный радикал, имеющий от одного до указанного количества атомов углерода (например, C1-C4алкил включает радикалы: метил, этил, проп-1-ил, проп-2-ил, бут-1-ил, бут-2-ил, 2-метилпропил и 2-метилпроп-2-ил). “Алкенил” означает прямой или разветвленный ненасыщенный углеводородный радикал, имеющий от трех до указанного числа атомов углерода и в котором атом водорода со свободной валентностью является насыщенным (например, C3-C4алкенил включает радикалы: 2-пропенил, 2-бутенил, 3-бутенил, 1-метил-2-пропенил и 2-метил-2-пропенил). “Алкилокси” означает радикал -OR, где R обозначает алкил, имеющий от одного до указанного числа атомов углерода (например, C1-C4алкилокси включает радикалы: метокси, этокси, проп-1-илокси, проп-2-илокси, бут-1-илокси, бут-2-илокси, 2-метилпроп-1-илокси и 2-метилпроп-2-илокси). “Алканоил” означает радикал алкилкарбонила, имеющий от одного до указанного числа атомов углерода (например, C1-C4алканоил включает радикалы: метаноил, этаноил, пропаноил, бутаноил и 2-метилпропаноил). “Алкановая кислота или галоидангидрид” означает прямую насыщенную карбоновую кислоту или галоидангидрид, имеющий от 2 до 6 атомов углерода (например, уксусная кислота, пропионовая кислота, масляная кислота, валериановая кислота, гексановая кислота, ацетилхлорид, пропионилхлорид и т.д.), и может относиться к их замещенным производным. “Алканон или алкан-1-он” означает замещенный прямой насыщенный 1-кетон, имеющий от двух до указанного числа атомов углерода (например, C2-C6алкан-1-он включает замещенные этан-1-он, пропан-1-он, бутан-1-он, пентан-1-он и гексан-1-он). “Арил” означает органический радикал, образованный из ароматического углеводорода, и включает моноциклические или конденсированные карбоциклические ароматические группы, имеющие от 6 до 20 атомов углерода (например, фенил, нафтил и т.п.). “Галоген” означает фтор, хлор, бром или йод. “Уходящая группа” имеет значение, которое обычно используется по отношению к ней в синтетической органической химии, т.е. обозначает атом или группу, замещаемые в условиях алкилирования, и включает галоген и алкан- или арен-сульфонилокси, такие как мезилокси, этансульфонилокси, бензолсульфонилокси и тозилокси, и алкансульфониламино, алканкарбониламино, аминосульфониламино, аминокарбониламино и т. п. “Защитная группа” имеет значение, которое обычно используется по отношению к ней в синтетической органической химии, т.е. обозначает группу, которая селективно блокирует одно реактивное место в многофункциональном соединении, так что химическая реакция может селективно осуществляться в другом незащищенном реактивном месте. “Снятие защиты” или “процесс снятия защиты” обозначает процесс, при котором защитная группа удаляется после окончания селективной реакции. Определенные процессы по настоящему изобретению основаны на том, что защитные группы блокируют реактивные атомы азота, присутствующие в этих реагентах. Приемлемые аминозащитные группы включают ацетил и трет-бутоксикарбонил, которые могут быть легко удалены путем кислотного гидролиза. “Животное” включает людей, других млекопитающих, например собак, кошек, кроликов, крупный рогатый скот, лошадей, овец, коз, свиней и оленей, и не млекопитающих, например птиц и т.п. “Болезнь” особо включает любое нездоровое состояние животного или его части и включает нездоровое состояние, которое может быть вызвано или присуще медицинской или ветеринарной терапии, примененной к этому животному, т. е. “побочными эффектами” такой терапии. “Необязательный” или “необязательно” означает, что последовательно описываемое событие или обстоятельство может иметь место или его может не быть и что описание включает случаи, когда событие или обстоятельство имеет место, и случаи, когда его нет. Например, фраза “необязательно замещенный одним-двумя заместителями” означает, что эти заместители могут присутствовать и могут не присутствовать для того, чтобы описываемое соединение было включено в данное изобретение, и изобретение включает те соединения, где один-два заместителя присутствуют, и те соединения, в которых заместители не присутствуют. “Фармацевтически приемлемый” означает, что что-либо, пригодное для получения фармацевтической композиции, является полностью безопасным, нетоксичным и не является нежелательным ни с биологической, ни с другой точки зрения и включает что-либо, приемлемое для использования в ветеринарии или в фармацевтике для человека. “Фармацевтически приемлемые соли” означают соли, которые являются фармацевтически приемлемыми в том смысле, как это определено выше, и которые обладают желаемой фармацевтической активностью. Такие соли включают кислотно-аддитивные соли, образованные с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п.; или с органическими кислотами, такими как уксусная кислота, пропионовая кислота, гексановая кислота, гептановая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензольная кислота, о-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этандисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, р-хлорбензолсульфоновая кислота, 2-нафталенсульфоновая кислота, р-толуолсульфоновая кислота, камфорсульфоновая кислота, 4-метилбицикло [2.2.2] окт-2-ен-1-карбоновая кислота, 4,4′-метиленбис(3-гидрокси-2-ен-1-карбоновая кислота), 3-фенилпропионовая кислота, триметилуксусная кислота, трет-бутилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтойная кислота, салициловая кислота, стеариновая кислота, муконовая кислота и т.п. Фармацевтически приемлемые соли также включают соли присоединения оснований, которые могут быть образованы, когда присутствующие кислотные протоны способны реагировать с неорганическими или органическими основаниями. Приемлемые неорганические основания включают гидроксид натрия, карбонат натрия, гидроксид калия, гидроксид алюминия и гидроксид кальция. Приемлемые органические основания включают этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин и т.п. “Терапевтически эффективное количество” означает такое количество, которое, будучи введенным животному для лечения болезни, является достаточным для осуществления такого лечения этой болезни. “Процесс лечения” или “лечение” болезни включает: (1) предупреждение наступления болезни у животного, которое может быть предрасположенным к болезни, но еще не испытывает и не проявляет симптомов болезни, (2) подавление болезни, т.е. задержка ее развития, или (3) ослабление болезни, т.е. вызов регрессии болезни. Соединения формулы I названы в соответствии с принятыми правилами номенклатуры, обычно применяемыми в “Chemical Abstracts”. Например, соединение формулы I, где R1 обозначает хлор, R3 обозначает метокси и R4 обозначает группу формулы (а), где n равно 4, p равно 0 и R5 и R6 каждый обозначают метил, 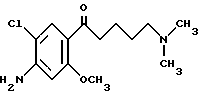 названо 1-(4-амино-5-хлор-2-метоксифенил)-5-диметиламинопентан-1-он. Предпочтительные варианты выполнения изобретения Соединения по настоящему изобретению являются производными 2-C6алкан-1-онов, имеющими замещенную фенильную часть в положении 1 и азотсодержащую часть в положении с самым большим номером. Азотсодержащая часть может представлять собой (N, N-дизамещенный)амино, (1-замещенный)пиперидин-4-ил, морфолин-1-ил или пирролидин-1-ил или необязательно замещенный пиперидин-1-ил, азациклогепт-1-ил, азабицикло[2.2.1]гепт-3-ил, азабицикло[2.2.2]окт-3-ил или азабицикло[3.2.2]нон-3-ил. Соединения формулы I, где R2 обозначает водород, R3 обозначает C1-C4алкилокси и R4 обозначает группу формулы (а), обозначают соединения формулы I(а): 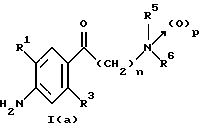 где n равно 3, 4 или 5; p равно 0 или 1; R1 обозначает галоген и R5 и R6 каждый обозначает C1-C4алкил или вместе образуют -(CH2)4-, -(CH2)6-, -(CH2)2O(CH2)2– или -CHR8CH2CR9R10CHR11CH2-, где R8 и R11 каждый обозначает водород или вместе обозначают -(CH2)t, где t равно 1, 2 или 3, R9 обозначает водород, гидроксил, C1-C8алкил, C3-C8алкенил или C1-C4алкилокси и R10 обозначает водород, C1-C8алкил или C3-C8алкенил или фенил, тиенил, пирролил или фурил (необязательно замещенный одним-двумя заместителями, независимо выбранными из C1-C4алкила, C1-C4алкилокси, трифторметила и галогена), или -(CH2)xR12, где x равно 0, 1, 2 или 3 и R12 обозначает гидроксил, C1-C4алкилокси, -C(O)NR13R14, -NR13C(O)R14, -NR13C(O)OR14, -SO2NR13R14, -NR13SO2R14, -NR13SO2NR14R15 или -NR13C(O)NR14R15, где R13, R14 и R15 независимо обозначают водород, C1-C4алкил или трифторметил; и их фармацевтически приемлемые соли, отдельные изомеры и смеси изомеров. Соединения формулы I, где R2 обозначает водород или C1-C4алкилокси и R3 обозначает C1-C4алкилокси или R2 и R3 вместе обозначают метилендиокси или этилендиокси и R4 обозначает группу формулы (b), обозначают соединения формулы I(b): 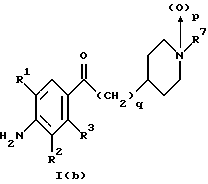 где p равно 0 или 1; q равно 1 или 2; R1 обозначает галоген и R7 обозначает водород, C1-C8алкил, C3-C8алкенил или фенилC1-C4алкил (где фенил необязательно замещен одним-тремя заместителями, независимо выбранными из C1-C4алкилокси, метилендиокси, этилендиокси или галогена), или -(CH2)zR12, где z равно 2 или 3 и R12 обозначает гидроксил, C1-C4алкилокси, -C(O)NR13R14, -NR13C(O)R14, NR13C(O)OR14, -SO2NR13R14, -NR13SO2R14, -NR13SO2NR14R15 или -NR13C(O)NR14R15, где R13, R14 и R15 независимо обозначают водород, C1-C4алкил, трифторметил или арил; и их фармацевтически приемлемые соли, отдельные изомеры и смеси изомеров. Соединения формулы I, где R2 обозначает водород и R3 обозначает C1-C4алкилфенилокси (где фенил необязательно замещен одним-тремя заместителями, независимо выбранными из галогена, гидроксила, C1-C4алкила, C1-C4алкилокси, нитро, амино, аминокарбонила, C1-C4алкиламино, диC1-C4алкиламино, C1-C4алканоиламино и 3,4-метилендиокси), обозначают соединения формулы I(с): 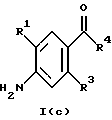 где R1 обозначает галоген и R4 обозначает группу формулы (а) или (b): 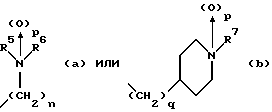 где n равно 3, 4 или 5; p равно 0 или 1; q равно 1 или 2; R5 и R6 каждый обозначает C1-C4алкил или вместе образуют -(CH2)4-, -(CH2)6-, -(CH2)2O(CH2)2– или -CHR8CH2CR9R10CHR11CH2-, где R8 и R11 каждый обозначает водород или вместе обозначают -(CH2)t, где t равно 1, 2 или 3, R9 обозначает водород, гидроксил, C1-C8алкил, C3-C8алкенил или C1-C4алкилокси и R10 обозначает водород, C1-C8алкил или C3-C8алкенил или фенил, тиенил, пирролил или фурил (необязательно замещенный одним-двумя заместителями, независимо выбранными из C1-C4алкила, C1-C4алкилокси, трифторметила и галогена), или -(CH2)xR12, где x равно 0, 1, 2 или 3 и R12 обозначает гидроксил, C1-C4алкилокси, -C(O)NR13R14, -NR13C(O)R14, -NR13C(O)OR14, -SO2NR13R14, -NR13SO2R14, -NR13SO2NR14R15 или -NR13C(O)NR14R15, где R13, R14 и R15 независимо обозначают водород, C1-C4алкил или трифторметил; и R7 обозначает водород, C1-C8алкил или C3-C8алкенил, или фенилC1-C4алкил (где фенил необязательно замещен одним-тремя заместителями, независимо выбранными из C1-C4алкилокси, метилендиокси, этилендиокси или галогена), или -(CH2)zR12, где z равно 2 или 3 и R12 имеет указанные выше значения; и их фармацевтически приемлемые соли, отдельные изомеры и смеси изомеров. Хотя весь спектр соединений, которые включены в объем изобретения, приведен выше в “Кратком описании сущности изобретения”, предпочтительны некоторые соединения. Например, предпочтительны соединения, которые являются соединениями формулы I(а), где n равно 4 и R5 и R6 вместе образуют -CHR8CH2CR9R10CHR11CH2-; и соединения формулы I(b), где q равно 2; и соединения формулы I(c), где R4 обозначает группу формулы (а), где n равно 4 и R5 и R6 вместе образуют -CHR8CH2CR9R10CHR11CH2-, или где R4 обозначает группу формулы I(b), где q равно 2. Особенно предпочтительными являются соединения формулы I(а), где n равно 4, R1 обозначает хлор, R3 обозначает метокси и R5 и R6 вместе образуют -CH2CH2CHR10CH2CH2-, где R10 обозначает водород, C1-C8алкил или -(CH2)xR12; и соединения формулы I(b), где q равно 2, R1 обозначает хлор, R3 обозначает метокси и R7 обозначает водород, C1-C8алкил или -(CH2)zR12; и соединения формулы I(с), где R1 обозначает хлор, R3 обозначает 3,5-диметоксибензилокси и R4 обозначает группу формулы (а), где n равно 4 и R5 и R6 вместе образуют -CH2CH2CHR10CH2CH2-, где R10 обозначает водород, C1-C8алкил или -(CH2)xR12, или R5 обозначает группу формулы (b), где q равно 2 и R7 обозначает водород, C1-C8алкил или -(CH2)zR12. Наиболее предпочтительными являются следующие соединения формулы I(а): 1-(4-амино-5-хлор-2-метоксифенил)-5-(пиперидин-1-ил)пентан-1-он, 1-(4-амино-5-хлор-2-метоксифенил)-5-(4-метилпиперидин-1-ил)пентан-1-он, 1-(4-амино-5-хлор-2-метоксифенил)-5-(4-аминокарбонилпиперидин-1-ил)пентан-1-он, 1-(4-амино-5-хлор-2-метоксифенил)-5-{ 4-[(метилсульфонил)амино]пиперидин-1-ил}пентан-1-он, и следующие соединения формулы I(b): 1-(4-амино-5-хлор-2-метоксифенил)-3-(1-метилпиперидин-4-ил)пропан-1-он, 1-(4-амино-5-хлор-2-метоксифенил)-3-(1-этилпиперидин-4-ил)пропан-1-он, 1-(4-амино-5-хлор-2-метоксифенил)-3-[1-(проп-1-ил)пиперидин-4-ил]пропан- 1-он, 1-(4-амино-5-хлор-2-метоксифенил)-3-[1-(бут-1-ил)пиперидин-4-ил] пропан- 1-он, 1-(4-амино-5-хлор-2-метоксифенил)-3-[1-(пент-1-ил)пиперидин-4-ил] пропан-1-он, 1-(4-амино-5-хлор-2-метоксифенил)-3-{ 1-[2-(метилсульфонил)аминоэтил] пиперидин-4-ил)пропан-1-он, 1-(4-амино-5-хлор-2-метоксифенил)-3-{ 1-[2-(диметиламиносульфонил) аминоэтил]пиперидин-4-ил}пропан -1-он, 1-(4-амино-5-хлор-2-метоксифенил)-3-{ 1-[3-(метилсульфонил)аминопроп- 1-ил]пиперидин-4-ил}пропан-1-он, 1-(4-амино-5-хлор-2,3-диметоксифенил)-3-[1-(3-бутил-1-ил)пиперидин- 4-ил]пропан-1-он, 1-(4-амино-5-хлор-2,3-этилендиоксифенил)-3-[1-(3-бутил-1-ил)пиперидин- 4-ил]пропан-1-он, 1-(4-амино-5-хлор-2-метоксифенил)-3-{ 1-[3-(4-метоксифенил)проп-1-ил] пиперидин-4-ил}пропан-1-он, 1-(4-амино-5-хлор-2-метоксифенил)-3-{ 1-[3-(2,3,4-триметоксифенил)проп-1-ил] пиперидин-4-ил}пропан-1-он, 1-(4-амино-5-хлор-2-метоксифенил)-3-{ 1-[3-(3,4-метилендиоксифенил)проп- 1-ил]пиперидин-4-ил}пропан-1-он, 1-(4-амино-5-хлор-2-метоксифенил)-3-{ 1-[3-(3,4-этилендиоксифенил)проп- 1-ил]пиперидин-4-ил}пропан-1-он, 1-(4-амино-5-хлор-2-метоксифенил)-3-{1-[3-(3,4-диметоксифенил)проп-1-ил] пиперидин-4-ил}пропан-1-он, 1-(4-амино-5-хлор-2-метоксифенил)-3-{1-[3-(3,5-диметоксифенил)проп-1-ил] пиперидин-4-ил}пропан-1-он, 1-(4-амино-5-хлор-2,3-этилендиоксифенил)-3-{ 1-[3-(4-метоксифенил) проп-1-ил]пиперидин-4-ил}пропан-1-он; и следующие соединения формулы I(с): 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил] -5-(пиперидин-1-ил) пентан-1-он, 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил]-5-(4-метилпиперидин- 1-ил)пентан-1-он, 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил] -5-[4- (проп-1-ил)пиперидин-1-ил]пентан-1-он, 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил]-5-{4-[2- (метилсульфонил)аминоэтил]пиперидин-1-ил}пентан-1-он, 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил]-5-[4- (метилсульфонил)аминометилпиперидин-1-ил]пентан-1-он, 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил]-5-[4- (аминокарбонил)аминометилпиперидин-1-ил]пентан-1-он, 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил] -5-(4- аминокарбонилпиперидин-1-ил]пентан-1-он, 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил] -5- (4-гидроксипиперидин-1-ил]пентан-1-он, 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил] -5-(4-метоксипиперидин-1-ил]пентан-1-он, 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил]-5- [4-(аминокарбонил)аминопиперидин-1-ил]пентан-1-он, 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил]-5-[4-(метилсульфонил) аминопиперидин-1-ил]пентан-1-он, 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил] -3-[1-(бут-1-ил) пиперидин-4-ил]пропан-1-он, 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил] -3-[1-(пент-1-ил) пиперидин-4-ил]пропан-1-он, 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил]-3-{1-[2- (метилсульфонилил)аминоэтил]пиперидин-4-ил}пропан-1-он; или их фармацевтически приемлемые соли, предпочтительно гидрохлориды. Фармакология и использование Соединения по настоящему изобретению взаимодействуют с 5-НТ4-рецепторами (т. е. соединения обладают сродством к 5-НТ4-рецепторам и проявляют агонистические или антагонистические свойства). Свойства испытываемых соединений взаимодействовать с 5-НТ4-рецептором определяют путем количественного анализа с использованием выделенной грудной мышцы пищевода крысы (т.е. испытываемые соединения, которым свойственно вызывать релаксацию, характеризуются как агонисты 5-НТ4-рецептора, в то время как испытываемые соединения, которые ингибируют вызванные агонистом реакции релаксации, где функцию медиатора выполняет 5-НТ4-рецептор, характеризуются как антагонисты 5-НТ4-рецептора). Известно, что выделенная грудная мышца пищевода крысы является общепринятой моделью для идентификации и характеризации соединений, которые взаимодействуют с 5-НТ4-рецепторами (например, см. Baxter G.S.; Craig D.A.; Clarke D.E. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1991, 343, 439-446), что описано в примере 17. В качестве лигандов 5-НТ4-рецептора соединения по настоящему изобретению полезны для лечения состояний, которые могут улучшиться при взаимодействии с 5-НТ4-рецепторами. Такие состояния включают расстройства ЦНС, расстройства желудочно-кишечного тракта, расстройства сердечно-сосудистой системы, расстройства мочевых путей. Конкретные расстройства ЦНС включают разнообразные неврологические и психические нарушения, такие как расстройства познавательной способности, психозы и навязчиво/компульсивное и тревожно/депрессивное поведение. Расстройства познавательной способности включают недостаток внимания или памяти, состояния помешательства (включая старческий маразм типа болезни Альцгеймера и старение), церебральную сосудистую недостаточность и болезнь Паркинсона. Психозы, которые могут быть подвергнуты лечению с помощью соединений по настоящему изобретению, включают паранойю, шизофрению и аутизм. Навязчиво/компульсивное поведение включает нарушения питания (например, булимию, состояние, при котором присутствует ненормальное и постоянное стремление потреблять пищу). Тревожно/депрессивные состояния включают преждевременный страх (например, перед хирургической операцией, лечением зубов и т.п.), депрессию, манию, сезонные аффективные расстройства (CAP), конвульсии и страх, вызванные отменой наркотических веществ (абстиненция), таких как опиаты, бензодиазапины, никотин, алкоголь, кокаин и другие лекарства, которыми злоупотребляли. Конкретные пищеварительные расстройства включают болезни, которые прямо или косвенно связаны с двигательной функцией желудка, пищевода и как толстого, так и тонкого кишечников. Специфические болезни включают, но не ограничиваются ими, диспепсию (например, неязвенную диспепсию), желудочный стаз, пептическую язву, пищеводный рефлюкс, метеоризм, гастрит, вызванный билиарным рефлюксом, синдром псевдонепроходимости, синдром раздражимости толстой кишки (который может приводить к хроническому запору и поносу), болезнь дивертикула, нарушение моторики желчного пузыря (которая может приводить к дисфункции сфинктера Одди и “осадку” или микроскопическим кристаллам в желчном пузыре), гастропарез (например, диабетический, послеоперационный или идиопатический), синдром раздражения кишечника и замедленное опорожнение желудка. Другие использования включают кратковременный прокинез для осуществления диагностической радиологии и кишечной интубации и для лечения поноса, в частности поноса, вызванного холерой и раковым синдромом. Конкретные сердечно-сосудистые расстройства включают болезни, которые связаны прямо или косвенно с сердечными аритмиями. Конкретные расстройства мочевых путей включают болезни, связанные прямо или косвенно с дисфункцией гладкой мышцы мочевой системы или иннервацией, приводящей к неадекватному накоплению мочи или ее контролю или мочевому стазу, который может приводить к инфекции, камням или почечным повреждениям. В целом, конкретное состояние, для лечения которого может быть полезно соединение по настоящему изобретению, будет зависеть от того, проявляет ли данное соединение качества агониста или качества антагониста. Например, определенные соединения по настоящему изобретению, которые являются агонистами 5-НТ4-рецептора, полезны в качестве прокинетических агентов при лечении болезней, при которых ослаблена двигательная функция желудка, или для увеличения тонуса гладкой мышцы мочеточника при лечении болезней мочевой системы, при которых наблюдается гипотония мочевого пузыря, или в качестве агентов, повышающих умственную способность, при лечении состояний, прямо или косвенно связанных с нарушением познавательных способностей. В противоположность этому соединения, являющиеся антагонистами 5-НТ4-рецептора, могут блокировать перистальтику, где функцию медиатора выполняет 5-НТ4-рецептор, и полезны при лечении болезней, включающих состояние гиперподвижности, или могут блокировать сокращения гладкой мышцы мочеточника, где функцию медиатора выполняет 5-НТ4-рецептор, и полезны при лечении болезней, связанных со спастичностью мочевого пузыря, или могут блокировать позитивную хронотропию, где функцию медиатора выполняет 5-НТ4-рецептор, и полезны в качестве антиаритмических агентов. Поскольку состояние, для которого может быть полезно любое данное соединение по настоящему изобретению, сильно зависит от антагонистических или агонистических свойств данного соединения, то могут иметь место определенные индивидуальные различия. Так, лиганды 5-НТ4-рецептора по настоящему изобретению могут в дальнейшем быть испытаны в опытах in vivo или in vitro, предназначенных для определения терапевтической активности. Например, прокинетическая активность соединений по настоящему изобретению может быть определена путем измерения увеличения скорости опорожнения желудка у крыс после орального введения испытываемого соединения. Прокинетический анализ на крысах является хорошо известной моделью для идентификации соединений, обладающих прокинетической активностью (например, см. Droppleman D.; Gregory R.; Alphin R.S. J. Pharmacol. Methods, 1980, 4(3), 227-30), и описан в примере 18. Свойства соединений по настоящему изобретению повышать познавательные способности могут быть определены с использованием теста водного лабиринта Морриса, в котором оцениваются изменения умственной способности крыс. Тест водного лабиринта Морриса является хорошо известной моделью для демонстрации увеличения познавательной способности (например, см. Morris R.G.M., Garrud P. , Rawlins J.N.P., O’Keefe, J.Nature, 1982, 297, 681-683) и описан в примере 21. Подавляющую патологический страх активность определяют на общепризнанной в данной области техники исследовательской двухкамерной модели Crowley и Goodwin (см. , например, Kilfoil Т.; Michel A.; Montgomery D.; Whiting R.L. Neuropharmacology, 1989, 28(9), 901-905). Коротко говоря, в этом методе оценивается степень влияния соединения на естественный страх мышей в новой ярко освещенной зоне. Исследование поведения, подавляющего патологический страх, описано в примере 19. Обладающую успокаивающим действием активность при отмене лекарств после их чрезмерного употребления определяют принятым методом с помощью теста на страх при отмене (например, см. Carboni Е.; Acquas Е.; Leone P.; Perezzani L.; Di Chiara, G. Eur. J. Pharmacol, 1988, 151, 159-160). В этом методе используется описанная выше исследовательская модель для измерения степени уменьшения с помощью соединения симптомов синдрома отмены, которые возникают после постоянного лечения наркотическим веществом и затем резкого прекращения этого лечения. Исследование страха при отмене описано в примере 20. В целом, соединения по настоящему изобретению полезны для лечения состояний, которые могут быть улучшены путем взаимодействия с 5-НТ4-рецепторами. Такие состояния включают расстройства ЦНС, расстройства желудочно-кишечного тракта, расстройства сердечно-сосудистой системы и расстройства мочевых путей. В частности, соединения пригодны для лечения состояний, связанных с расстройствами познавательной способности, гипокинезией желудка, синдромом раздражимости кишечника, аритмией, гипотонусом мочевого пузыря или спастичностью мочевого пузыря. Введение и фармацевтическая композиция Обычно соединения по изобретению можно вводить в терапевтически эффективных количествах с помощью любых обычных и приемлемых способов, известных в данной области техники, либо по отдельности, либо в сочетании с другим соединением формулы I, либо с другим терапевтическим агентом. Терапевтически эффективное количество может сильно варьироваться в зависимости от серьезности заболевания, возраста и состояния здоровья субъекта, действенности используемого соединения и других факторов. Терапевтически эффективное количество может составлять от 0,01 миллиграмма на килограмм веса тела (мг/кг) в день до 10 мг/кг веса тела в день. Предпочтительно количество должно составлять приблизительно от 0,1 до 1 мг/кг/день. Следовательно, терапевтически эффективное количество для человека весом 70 кг может составлять от 0,7 до 700 мг/день, предпочтительно от 7 до 70 мг/день. Любой обычный специалист в области лечения таких болезней способен без проведения длительных экспериментов и полагаясь на собственные знания и описание настоящего изобретения установить для данной болезни терапевтически эффективное количество соединения формулы I. Обычно соединения по настоящему изобретению можно вводить в виде фармацевтических композиций одним из следующих путей: орально, системно (например, трансдермально, через нос или с помощью суппозитория) или парентерально (например, внутримышечно, внутривенно или подкожно). Композиции могут иметь форму таблеток, пилюль, капсул, быть полутвердыми, порошками, формами с непрерывным высвобождением, растворами, суспензиями, эликсирами, аэрозолями или любой другой пригодной композицией, и они обычно включают соединения формулы I в комбинации по крайней мере с одним фармацевтически приемлемым инертным наполнителем. Приемлемые инертные наполнители нетоксичны, способствуют введению и не оказывают вредного воздействия на терапевтическую пользу соединения формулы I. Такой инертный наполнитель может быть любым твердым, жидким, полутвердым или, в случае применения аэрозольной композиции, газообразным инертным наполнителем, который имеется в распоряжении специалиста в данной области техники. Твердые фармацевтические инертные наполнители включают крахмал, целлюлозу, тальк, глюкозу, лактозу, сахарозу, желатин, солод, рис, муку, мел, силикагель, стеарат магния, стеарат натрия, моностеарат глицерина, хлорид натрия, сухое обезжиренное молоко и т.п. Жидкие и полутвердые инертные наполнители могут быть выбраны из воды, этанола, глицерина, пропиленгликоля и различных масел, включающих масла, полученные из нефти, животного или растительного происхождения или синтетические (например, арахисовое масло, соевое масло, минеральное масло, кунжутное масло и т.д.). Предпочтительные жидкие носители, в частности, для инъецируемых растворов включают воду, физиологический раствор, водную декстрозу и гликоли. Для распыления соединения по изобретению в аэрозольной форме могут быть использованы сжатые газы. Пригодными для этой цели инертными газами являются азот, двуокись углерода, закись азота и т.д. Другие фармацевтически пригодные носители и их формы описаны A.R. Alfonso в Remington’s Pharmaceutical Sciences, 1985, 17-е издание, Easton, Pa.: Mack Publishing Company. Количество соединения по изобретению в композиции может сильно варьироваться в зависимости от типа композиции, размера стандартной дозы, типа инертных наполнителей и других факторов, известных специалистам в данной области фармацевтических наук. Обычно готовая композиция может содержать от 25 вес. % до 75 вес.% соединения формулы I, предпочтительно от 30 вес.% до 50 вес. % с дополнением недостающего количества за счет инертного наполнителя или наполнителей. Предпочтительно фармацевтическую композицию вводят в форме однократной стандартной дозы для непрерывного лечения или по желанию в форме однократной стандартной дозы, когда специально требуется облегчение симптомов. Характерные фармацевтические композиции, содержащие соединение формулы I, описаны в примере 16. Способы получения соединений по изобретению Способ получения соединений формулы I представлен на реакционной схеме I. Соединения формулы I могут быть получены с помощью процесса, который включает взаимодействие соединения формулы II или его защищенного производного с соединением формулы III, подкисление, декарбоксилирование и, если необходимо, удаление любых защитных групп. Реакцию между соединениями формул II и III осуществляют в присутствии сильного основания (например, диизопропиламида лития) и в пригодном растворителе (например, тетрагидрофуране (ТГФ) и гексаметилфосфотриамиде (ГМФА), предпочтительно в смеси 50/50 ТГФ/ГМФА) при температуре от -40o до 0oC. Реакционную смесь затем подкисляют и экстрагируют подходящим растворителем (например, метиленхлоридом). Затем может быть осуществлено декарбоксилирование с помощью стандартных методов для образования соединения формулы I. Получение соединения формулы I с помощью процесса, изображенного на реакционной схеме I, описано в примере 3. Соединения формулы II, где R4 обозначает группу формулы (а), могут быть получены взаимодействием галогенC4-C6алкионата с соединением формулы NHR5R6 для получения аминированного C4-C6алкионата и последующего гидролиза и превращения в кислотно-аддитивную соль. Реакцию с соединением формулы NHR5R6 проводят в пригодном растворителе (например, 1-метил-2-пирролидиноне, ДМФ и т.д.) при 25-50oC, предпочтительно приблизительно при 35oC и в течение 12-48 часов. Гидролиз может быть выполнен с помощью кислоты (например, соляной кислоты, серной кислоты и т.д.) при 90-110oC, предпочтительно приблизительно при 100oC и в течение 2-4 часов. Кислотно-аддитивная соль может быть восстановлена путем кристаллизации из растворителя (например, путем разбавления ацетоном). Соединения формулы II, где R4 обозначает группу формулы (b), могут быть получены из соответственно 1-замещенного 4-гидроксипиперидина или 4-гидроксиметилпиперидина. 4-Пиперидиновый спирт превращают в соответствующий сульфонат (т. е. в 4-(п-толуолсульфонилокси)пиперидин или 4-[(п-толуолсульфонилокси) метил] пиперидин путем взаимодействия с р-толуолсульфонилхлоридом). Затем осуществляют взаимодействие сульфоната с диэтилмалонатом в присутствии основания для получения 4-[ди(этоксикарбонил)алкил] пиперидина. Затем 4-[ди(этоксикарбонил)алкил] пиперидин превращают в дикарбоновую кислоту, из которой в результате декарбоксилирования получают соединение формулы II. Защищенные производные соединений формулы II получают из защищенных производных пиперидиновых спиртов (например, 1-трет-бутоксикарбонил- 4-пиперидинметанола). Получение защищенных производных формулы II, где R4 обозначает формулу (b), описано в примере 1. Соединения формулы III могут быть получены путем взаимодействия подходящей 4-амино-5-галоидбензойной кислоты с гидрохлоридом N,O-диметилгидроксиамина. Реакцию проводят в присутствии карбонилдиимидазола в пригодном растворителе (например, N,N-диметилформамиде (ДМФ)). Получение соединения формулы III описано в примере 2. В противоположность этому соединения формулы I, где R4 обозначает группу формулы (b) и R7 не является водородом, могут быть получены путем алкилирования соответствующего соединения формулы I, где R7 обозначает водород, с соединением формулы L-R20, где L обозначает уходящую группу и R20 обозначает C1-C8алкил, C3-C8алкенил, фенилC1-C4алкил (где фенил замещен аналогично описанному в “Кратком описании сущности изобретения”) или -(CH2)zR12 (где z и R12 имеют значения, указанные в “Кратком описании сущности изобретения”). Алкилирование проводят в стандартных для амидного алкилирования условиях (Luh Т. ; Fung S. H. Synth. Commun. 1979, 9, 757) в инертном растворителе (например, ацетонитриле, ДМФ, ТГФ и т.д.) при температуре реакции 20-100oC и в течение 1-48 часов. Алкилирование можно проводить также с помощью системы катализатора фазового переноса (КФП), т.е. с катализатором в двухфазной системе растворителей жидкость-жидкость (Gajda Т. ; Zwierzak A. Synthesis, Communications 1981, 1005), или предпочтительно в системе твердая фаза-жидкость (Yamawaki J.; Ando Т.; Hanafusa, Т. Chem. Lett. 1981, 1143; Koziara A.; Zawaszki S.; Zwierzak A. Synthesis 1979, 527, 549). Двухфазная система жидкость-жидкость включает водную фазу, состоящую из концентрированного раствора гидроксида щелочи (например, 50% водный гидроксид натрия), органическую фазу, включающую инертный, не смешивающийся с водой органический растворитель и подходящий катализатор. Система твердая фаза-жидкость включает порошкообразный гидроксид щелочи/карбонат щелочи, суспендированный в органическом растворителе, и катализатор. Реакцию выполняют, медленно добавляя алкилирующий агент к МФК-системе, содержащей соединение формулы I, до тех пор, пока не будет достигнут 10-50% избыток алкилирующего агента. Реакционную смесь выдерживают при температуре кипения с обратным холодильником до завершения реакции. Затем смесь охлаждают до комнатной температуры и продукт выделяют с помощью обычных способов. Пригодные органические растворители включают бензол, толуол и т.п. Подходящие катализаторы включают окись алюминия, покрытую фторидом калия, и четвертичные сульфаты аммония, такие как бисульфат тетра-н-бутиламмония и трикаприлилметиламмонийхлорид. Алкилирование соединения формулы I(b), где R7 обозначает водород, описано в примере 10. Соединения формулы I, где R4 обозначает группу формулы (b) и R7 обозначает -(CH2)2NHC(O)R14, -(CH2)2NHSO2R14, -(CH2)2NHSO2R14R15 или -(CH2)2NHC(O)NR14R15 (где R14 и R15 имеют значения, указанные в “Кратком описании сущности изобретения”), могут быть получены путем алкилирования соединения формулы I, где R7 обозначает водород, соединением формулы X-R21, где X обозначает азациклопроп-1-ил и R21 обозначает -C(O)R9, -SO2R14, -SO2NR14R15 или -C(O)NR14R15. Алкилирование проводят в пригодном растворителе (например, ТГФ) при 0-20oC. Получение соединения формулы I(b), где R7 обозначает 2-[(метилсульфонил)амино]этил, описано в примере 11. Способ получения соединений формулы I, где R2 обозначает водород и R4 обозначает группу формулы (а), представлен на реакционной схеме II. Соединения формулы I, где R2 обозначает водород и R4 обозначает группу формулы (а) (формула IV), могут быть получены с помощью процесса, который включает взаимодействие соединения формулы V с соединением формулы VI; алкилирование соединением формулы L-R3; взаимодействие с соединением формулы HNR5R6 или его N-оксидом и удаление защитной группы. Реакцию между соединением формулы V и соединением формулы VI проводят в присутствии кислоты Льюиса (например, хлорида алюминия (AlCl3), трехфтористого бора, фтористого водорода, фосфорной кислоты и т.д., предпочтительно AlCl3) и в пригодном растворителе (например, этилендихлориде (ЭДХ), метиленхлориде, сероуглероде и т. д., предпочтительно в ЭДХ) для образования 2-гидроксифенилалканона. Алкилирование 2-гидроксифенилалканона соединением формулы L-R3 проводят в присутствии карбоната калия в пригодном растворителе (например, метилэтилкетоне (МЭК), ДМФ, этаноле, ТГФ и т.д., предпочтительно в МЭК) при температуре 40-200oC. Реакцию с соединением формулы HNR5R6 проводят в пригодном растворителе (например, ДМФ, этаноле, ТГФ, толуоле и т.д., предпочтительно в ДМФ) при температуре 40-200oC. Снятие защиты может быть проведено любыми способами, которые селективно удаляют защитную группу. Получение соединения формулы I(а) с помощью вышеуказанного процесса описано в примере 4. Соединения формулы VI либо являются коммерчески доступными (например, 5-хлорвалериановая кислота и 5-хлорвалерилхлорид), или же могут быть легко получены галоидированием соответствующей алкановой кислоты. N-ацетилгалоген-5-метоксианилины формулы V либо являются коммерчески доступными, или же могут быть получены из N-ацетил-3-метоксианилина. Например, соединение формулы V, где R1 обозначает бромид, может быть получено взаимодействием N-ацетил-3-метоксианилина с бромином в пригодном растворителе (например, дихлорэтане). Соединения формулы HNR5R6 (например, амины, такие как диметиламин, диэтиламин, дипроп-1-иламин и т. д., и азациклоалканы, такие как пирролидин, пиперидин, морфолин, 4-фенилпиперидин, 4-метилпиперидин и т.д.) известны и являются коммерчески доступными или же могут быть синтезированы способами, известными обычным специалистам в данной области техники. В вариантах реакционной схемы II соединения формулы I могут быть получены путем взаимодействия в присутствии кислоты Льюиса соединения формулы V с соединением формулы VI; взаимодействия с соединением формулы HNR5R6 или его N-оксидом; алкилирования соединением формулы L-R3 и удаления защитной группы или путем взаимодействия соединения формулы VI с соединением формулы HNR5R6 или его N-оксидом; взаимодействия в присутствии кислоты Льюиса с соединением формулы V; алкилирования соединением формулы L-R3 и удаления защитной группы. Способ получения соединений формулы I, где R4 обозначает группу формулы (b), где q равно 2 и R7 обозначает водород, изображен на реакционной схеме III. Соединения формулы I, где R4 обозначает группу формулы (b), где p равно 0, q равно 2 и R7 обозначает водород, могут быть получены с помощью процесса, который включает взаимодействие соединения формулы VIII с соединением формулы IX для получения производного 3-пиридин-4-ил-3-гидроксипропан-1-она, дегидратацию 3-гидроксипропан-1-она для получения производного 3-пиридин-4-илпропен-1-она и последующую гидрогенизацию для получения соответствующего 3-пиперидин-4-илпропан-1-она. Реакцию между соединениями формул VIII и IX проводят в присутствии сильного основания (например, диизопропиламида лития) в пригодном растворителе (например, ТГФ) при температуре от -50 до 50oC. Дегидратацию 3-пиперидин-4-илпропан-1-она проводят стандартными методами (например, катализируемой кислотой дегидратацией). Если реакцию между соединением формул VIII и IX проводят в присутствии гидроксида калия, дегидратация происходит спонтанно, и реакция приводит к получению за одну стадию 3-пиридин-4-ил-2-пропен-1-она. Гидрогенизацию 3-пиридин-4-ил-2-пропен-1-она проводят с подходящим катализатором (например, с 5% родием на окисноалюминиевом носителе, 10% палладием на угле, диоксидом платины, дигидроксидом палладия и т.д.) в пригодном растворителе (например, уксусной кислоте, этаноле, ДМФ, ТГФ и т.д.) при давлении 5-60 фунт/кв.дюйм и в течение 1-48 часов. Получение соединения формулы I(b) с помощью вышеописанного процесса приведено в примере 7. В противоположность этому гидрогенизацию 3-пиридин-4- ил-2-пропен-1-она проводят путем (I) гидрогенизации 2-пропен-1-она до тех пор, пока не завершится превращение в соответствующий 3-пиридин-4-илпропан-1-он, (II) необязательно путем алкилирования пропан-1-она соединением формул L-R20 или X-R21 (где R20 и R21 имеют вышеуказанные значения) и затем (III) путем продолжения гидрогенизации для получения соответствующего 3-пипиридин-4-илпропан-1-она. Гидрогенизацию пропенона в 3-пиридин-4-илпропан-1-он проводят с помощью подходящего катализатора (5% палладия на угле, 20% гидроксида палладия и т.д.) в пригодном растворителе (например, ТГФ, уксусной кислоте, ДМФ, этаноле и т. д. ) при давлении 5-60 фунт/кв.дюйм и в течение 1-48 часов. Получение соединения формулы I(b), где R7 обозначает 3-(4-метоксифенил)проп-1-ил, описано в примере 13. Необязательную реакцию с соединением формулы L-R20 или X-R21 проводят аналогично описанному выше для алкилирования соединений формулы I, где R4 обозначает группу формулы (b) и R7 обозначает водород. Гидрогенизацию 3-пиридин-4-илпропан-1-она проводят с подходящим катализатором (например, с 5% родием на окисноалюминиевом носителе, 10% палладием на углероде, диоксидом платины, дигидроксидом палладия и т.д.) в пригодном растворителе (например, уксусной кислоте, этаноле, ДМФ, ТГФ и т.д.) при давлении 5-60 фунт/кв.дюйм и в течение 1-48 часов. Соединения формулы VIII могут быть получены метилированием соответствующего N-метокси-N-метилбензамида. Метилирование проводят путем взаимодействия N-метокси-N-метилбензамида с метилирующим агентом (например, метиллитием, метилмагнийбромидом и т.д.) в пригодном растворителе (например, ТГФ, простом эфире и т.д.) при температуре от -20 до 20oC и в течение 1-24 часов. Получение соединения формулы VIII, где R1 обозначает хлор и R3 обозначает метокси, описано в примере 5. В противоположность этому соединения формулы VIII, где R2 и R3 вместе обозначают этилендиокси, могут быть получены путем галоидирования защищенного производного 4′-амино-2′,3′-этилендиоксиацетофенона и последующего снятия защиты. Галоидирование может быть проведено путем взаимодействия ацетофенона с галоидирующим агентом (например, N-хлорсукцинимидом, хлорином и т.д.) в пригодном растворителе (например, ДМФ, ацетонитриле и т.д.) при температуре от -20 до 80oC и в течение 1-24 часов. Снятие защиты может быть проведено с основанием (например, гидроксидом натрия и т.п.) в пригодном растворите (например, метаноле и т.п.). 4′-Амино-2′, 3′-этилендиоксиацетофенон может быть получен путем взаимодействия 5′, 6′-дихлор-2′,3′-этилендиоксиацетофенона с дымящей азотной кислотой для получения 5′,6′-дихлор-2′,3′-этилендиокси-4′- нитроацетофенона и последующего восстановления. Реакцию с азотной кислотой проводят при температуре 0-25oC в течение 0,5-2 часов. Восстановление проводят в присутствии катализатора (например, 10% палладия на угле) в пригодном растворителе (например, этаноле, уксусной кислоте, этилацетате и т.д.) при комнатной температуре и в течение 1-24 часов. Защита 4′-амино-2′,3′- этилендиоксиацетофенона может быть осуществлена путем взаимодействия с пригодным защитным агентом. Например, 2′, 3′- этилендиокси-4′-(метилкарбониламино)ацетофенон получают путем взаимодействия незащищенного ацетофенона с уксусным ангидридом в пиридине. Реакцию с уксусным ангидридом проводят при температуре 0-30oC в течение 1-8 часов. 5′, 6′-Дихлор-2′, 3′-этилендиокси-4′-нитроацетофенон может быть получен путем хлорирования 1,4-бензодиоксана для получения 6,7-дихлор-1,4-бензодиоксана и последующего ацетилирования. Хлорирование может быть проведено путем обработки бензодиоксана газообразным хлором в уксусной кислоте при 0-20oC и в течение 1-8 часов. Ацетилирование может быть осуществлено путем взаимодействия 6,7-дихлор-1,4-бензодиоксана с хлористым ацетилом в присутствии кислоты Льюиса в пригодном растворителе (например, 1,2-дихлорэтане, сероуглероде, нитробензоле и т.д.) при температуре 20-40oC и в течение 24-48 часов. Способ получения соединений формулы I, где R4 обозначает группу формулы (а), изображен на реакционной схеме IV. Соединения формулы I, где R4 обозначает группу формулы (а), могут быть получены с помощью процесса, который включает обработку соединения формулы XI магнием для получения реактива Гриньяра, взаимодействие реактива Гриньяра с соединением формулы XII для получения соответствующего защищенного C4-C6алканона, снятие защиты и последующее взаимодействие незащищенного алканона с соединением формулы NHR5R6. Обработку магнием проводят путем добавления соединения формулы XI в атмосфере азота к суспензии магния в пригодном растворителе (например, ТГФ, диэтиловом эфире и т.д.) с такой скоростью, чтобы контролировать выделение тепла, и затем дают протекать реакции при 20-25oC в течение 1-2 часов. Реакцию с реактивом Гриньяра проводят путем охлаждения раствора соединения формулы XII в пригодном растворителе (например, ТГФ, диэтиловом эфире и т.д.) до температуры между -10 и -20oC, предпочтительно приблизительно до -15oC, и последующего добавления суспензии, содержащей реактив Гриньяра, охлажденный до приблизительно такой же температуры, и дают реакции протекать при температуре 0-15oC, предпочтительно приблизительно при 10oC в течение 1-2 часов. Снятие защиты может быть осуществлено с помощью любых способов, позволяющих удалить защитную группу и дающих желаемый незащищенный алканон с хорошим выходом. Реакцию с незащищенным алканоном проводят в атмосфере азота с 3-6 эквивалентами, предпочтительно с 3-4 эквивалентами NHR5R6 в пригодном растворителе (например, ДМФ, N-метилпирролидоне (NMП) и т.д.) при температуре 55-85oC, предпочтительно при 60oC и в течение 4-8 часов. Получение соединения формулы I с помощью вышеуказанного способа описано в примере 14. Соединения формулы XII могут быть получены путем взаимодействия соединения формулы III с пригодным защитным агентом. Детальное описание методик, применяемых к защитным группам и для их удаления, можно найти у T.W.Greene в Protective Groups in Organic Synthesis, John Wiley & Sons, Inc. 1981. Например, соединения формулы XII, где защитные группы представляют собой триметилсилил, могут быть получены путем взаимодействия соединения формулы III с хлортриметилсиланом. Реакцию проводят в присутствии сильного основания (например, третбутилмагнийхлорида, литийбис(триметилсилил)амида (ЛиТМСА) и т. д. ) в атмосфере азота в пригодном растворителе (например, ТГФ, простом эфире и т.д.) при температуре от -15 до 15oC, предпочтительно приблизительно при 10oC и в течение 15-30 мин. Снятие защиты соединения формулы XII, где защитные группы являются триметилсилилом, может быть осуществлено с помощью кислоты (например, соляной кислоты, трифторуксусной кислоты и т.д.). Способ получения соединений формулы I, где R4 обозначает группу формулы (а), изображен на реакционной схеме V. Соединения формулы I, где R4 обозначает группу формулы (а), могут быть получены путем взаимодействия кислотно-аддитивной соли соответствующего соединения формулы II (формулы II(а)) с соединением формулы XIII в присутствии сильного основания (например, ЛиТМСА, натрийбис(триметилсилил)амида, диизопропиламида лития, предпочтительно ЛиТМСА) и последующего снятия защиты. Реакция между соединениями формул II(а) и XIV может быть проведена путем охлаждения смеси, содержащей соединение формулы II(а), в пригодном растворителе (например, ТГФ, диметиловом эфире этиленгликоля, диэтиловом эфире и т. д. ) до температуры между 10 и 30oC, предпочтительно приблизительно до 15oC; добавления минимум 3 эквивалентов сильного основания и выдерживания 0,5-2 часа при температуре 10-30oC, предпочтительно приблизительно при 20oC; охлаждения смеси до температуры между 0 и 10oC, предпочтительно приблизительно до 5oC; и последующего добавления соединения формулы XIII, после чего реакции дают протекать при температуре 40-55oC в течение приблизительно 4 часов. Снятие защиты может быть осуществлено любым способом, который позволяет удалить защитную группу и получить желаемый продукт с хорошим выходом. Пригодной защитной группой для соединения формулы XIII является триметилсилил, который может быть легко удален кислотой. Получение соединения формулы I с помощью вышеописанного процесса приведено в примере 15. Соединения формулы XIII могут быть получены путем взаимодействия соответствующей бензойной кислоты с метанолом в присутствии тионилхлорида и последующего добавления защитных групп. Реакцию с бензойной кислотой проводят при температуре 0-25oC, предпочтительно приблизительно при 15oC и в течение 1-4 часов. Защита может быть проведена с помощью хлортриметилсилана в присутствии сильного основания (например, трет-бутилмагнийхлорида, ЛиТМСА и т. д.) в пригодном растворителе. Дополнительные процессы Соединения формулы I, где R3 обозначает C2-C4алкилокси или фенилC1-C4алкилокси, могут быть получены путем деметилирования соединения формулы I, где R3 обозначает метокси, и последующего алкилирования соединением формулы L-R22, где R22 обозначает C2-C4алкил или фенилC1-C4алкил (где фенил необязательно замещен одним-тремя заместителями, независимо выбранными из галогена, гидроксила, C1-C4алкила, C1-C4алкилокси, нитро, амино, аминокарбонила, C1-C4алкиламино, диC1-C4алкиламино, C1-C4алканоиламино и 3,4-метилендиокси). Деметилирование проводят стандартными способами с подходящим деметилирующим агентом (например, трибромидом бора, трихлоридом бора и т.д.) и в апротонном растворителе (например, метиленхлориде, дихлорэтане и т.д.) или кислотой (например, водной бромистоводородной кислотой) для образования соответствующего 2-гидроксифенилалкан-1-она. Алкилирование проводят в присутствии карбоната калия в пригодном растворителе (например, метилэтилкетоне (МЭК), ДМФ, этаноле, ТГФ и т.д., предпочтительно в МЭК) при 40-200oC. Получение соединения формулы 1 с помощью приведенного выше процесса описано в примере 12. Соединения формулы I, где p равно 1, (соединения формулы I в форме N-оксида) могут быть получены путем окисления соединения формулы I, где p равно 0. Окисление проводят при температуре реакции приблизительно 0oC с подходящим окислителем и в пригодном инертном органическом растворителе. Пригодные окислители включают перекись водорода или надкислоты, такие как трифторперуксусная кислота, пермалеиновая кислота, пербензойная кислота, перуксусная кислота и м-хлорпероксибензойная кислота. Пригодные растворители включают галоидированные углеводороды, например метиленхлорид, и спирты. В противоположность этому соединения формулы I, где p равно 1, могут быть получены с использованием производных N-оксидов в качестве исходных материалов или промежуточных продуктов. Соединения формулы I, где p равно 0, могут быть получены путем восстановления соединения формулы I, где p равно 1. Восстановление проводят в стандартных условиях с подходящим восстановителем в пригодном растворителе. Смесь изредка перемешивают, в то время как температура реакции постепенно повышается в диапазоне от 0oC до 80oC. Подходящие восстановители включают серу, диоксид серы, триарилфосфины (например, трифенилфосфин), боргидриды щелочных металлов (например, боргидрид лития, боргидрид натрия и т.д.), треххлористый фосфор и трибромид. Пригодные растворители включают ацетонитрил, этанол и водный диоксан. Соединения формулы I могут быть получены в виде фармацевтически приемлемых кислотно-аддитивных солей путем взаимодействия соединения формулы I в форме свободного основания с фармацевтически приемлемой неорганической или органической кислотой. Альтернативно, фармацевтически приемлемые соли присоединения основания соединений формулы I могут быть получены путем взаимодействия соединений формулы I в форме свободной кислоты с фармацевтически пригодными неорганическими или органическими основаниями. Неорганические и органические кислоты и основания, пригодные для получения фармацевтически приемлемых солей соединений формулы I, приведены выше в разделе “Определения” данного описания. Альтернативно, соединения формулы I в форме солей могут быть получены с использованием солей в качестве исходных материалов или промежуточных продуктов. Соединения формулы I в форме свободной кислоты или свободного основания могут быть получены из соответствующей соли присоединения основания или кислотно-аддитивной соли. Например, соединения формулы I в форме кислотно-аддитивной соли могут быть превращены в соответствующее свободное основание путем обработки пригодным основанием (например, раствором гидроксида аммония, гидроксидом натрия и т.д.). Соединения формулы I в форме соли присоединения основания могут быть превращены в соответствующую свободную кислоту путем обработки пригодной кислотой (например, соляной кислотой и т.д.). В целом, способы получения соединений по настоящему изобретению представляют собой: (А) для получения соединения формулы I взаимодействие в присутствии сильного основания соединения формулы II: 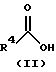 или его защищенного производного, где R4 имеет значения, указанные в “Кратком описании сущности изобретения”, с соединением формулы III: 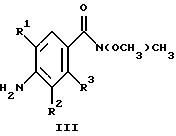 где каждый из R1, R2 и R3 имеет значения, указанные в “Кратком описании сущности изобретения”; подкисление; декарбоксилирование и, если необходимо, удаление любых защитных групп; или (Б) для получения соединения формулы I, где R2 обозначает водород и R4 обозначает группу формулы (а), взаимодействие в присутствии кислоты Льюиса соединения формулы V: 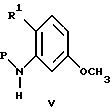 где P обозначает защитную группу и R1 обозначает галоген, с соединением формулы VI: 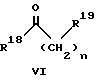 где R18 обозначает галоген или гидроксил, R19 обозначает галоген и n равно 3, 4 или 5; алкилирование соединением формулы L-R3, где L обозначает уходящую группу и R3 имеет указанные выше значения; взаимодействие с соединением формулы HNR5R6 или его N-оксидом, где R5 и R6 имеют указанные выше значения; и снятие защиты; или (В) для получения соединения формулы I, где R4 обозначает группу формулы (b), где p равно 0 и q равно 2, взаимодействие соединения формулы VIII: 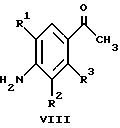 где R1, R2 и R3 имеют значения, указанные в “Кратком описании сущности изобретения”, с соединением формулы IX: 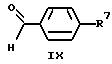 где R7 имеет значения, указанные в “Кратком описании сущности изобретения”; дегидратацию и последующую гидрогенизацию; или (Г) для получения соединения формулы I, где R4 обозначает группу формулы (а), обработку соединения формулы XI: 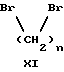 где n имеет значения, указанные в “Кратком описании сущности изобретения”, магнием для получения соответствующего реактива Гриньяра, взаимодействие реактива Гриньяра с соединением формулы XII: 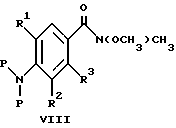 где каждый P обозначает защитную группу и R1, R2 и R3 имеют значения, указанные в “Кратком описании сущности изобретения”, снятие защиты и последующее взаимодействие с соединением формулы HNR5R6; или (Д) для получения соединения формулы I, где R4 обозначает группу формулы (а), взаимодействие в присутствии сильного основания соединения формулы II(а): 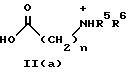 где R5 и R6 имеют значения, указанные в “Кратком описании сущности изобретения”, с соединением формулы XIII: 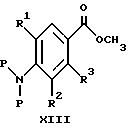 где каждый P обозначает защитную группу и значения R1, R2 и R3 имеют значения, указанные в “Кратком описании сущности изобретения”; или (Е) необязательно алкилирование соединения формулы I, где R4 обозначает группу формулы (b) и R7 обозначает водород, соединением формулы L-R20, где L обозначает уходящую группу и R20 обозначает C1-C8алкил, C3-C8алкенил или фенилC1-C4алкил (где фенил необязательно замещен одним-тремя заместителями, независимо выбранными из C1-C4алкилокси, метилендиокси, этилендиокси или галогена), или L-(CH2)zR12 (где z и R12 имеют значения, указанные в “Кратком описании сущности изобретения”) для получения соединения формулы I, где R4 обозначает группу формулы (b), где R7 не является водородом; или (Ж) необязательно алкилирование соединения формулы I, где R4 обозначает группу формулы (b) и R7 обозначает водород, соединением формулы X-R21, где X обозначает азациклопроп-1-ил и R21 обозначает -C(O)R14, -SO2R14, – SO2NR14R15 или -CONR14R15 (где R14 и R15 имеют значения, указанные в “Кратком описании сущности изобретения”) для получения соединения формулы I, где R4 обозначает группу формулы (b), где R7 обозначает -CH2CH2NHC(0)R14, -CH2CH2NHSO2R14, -CH2CH2NHSO2NR14R15 или -CH2CH2NHCONR14R15; или (З) необязательно деметилирование соединения формулы I, где R3 обозначает метокси, и последующее алкилирование соединением формулы L-R22, где R22 обозначает C2-C4алкил или фенилC1-C4алкил (где фенил необязательно замещен одним-тремя заместителями, независимо выбранными из галогена, гидроксила, C1-C4алкила, C1-C4алкилокси, нитро, амино, аминокарбонила, C1-C4алкиламино, диC1-C4алкиламино, C1-C4алканоиламино и 3,4-метилендиокси) для получения соединения формулы I, где R3 обозначает C2-C4алкилокси или фенилC1-C4алкилокси (где фенил необязательно замещен аналогично указанному в “Кратком описании сущности изобретения”); или (И) необязательно окисление соединения формулы I, где p равно 0, для получения соединения формулы I, где p равно 1; или (К) необязательно восстановление соединения формулы 1, где p равно 1, для получения соединения формулы I, где p равно 0; или (Л) необязательно взаимодействие соответствующей несолевой формы соединения формулы I с фармацевтически приемлемыми неорганической или органической кислотой или основанием для получения фармацевтически приемлемой соли; или (М) необязательно взаимодействие соответствующей кислотно-аддитивной соли или соли присоединения основания соединения формулы I с пригодными основанием или кислотой соответственно для получения свободной кислоты или свободного основания. В любом из вышеперечисленных процессов заключительной стадии ссылка на формулы I, II, II(а), III, IV, V, VI, VII, VIII, IX, X, XI, XII и XIII относится к таким формулам, где 1, R2, R3, R4, R5 и R7 имеют самые широкие значения, которые определены в “Кратком описании сущности изобретения”, причем эти процессы прежде всего наилучшим образом применимы к наиболее предпочтительным вариантам выполнения изобретения. ПРИМЕР 1 3-[1-(Трет-бутоксикарбонил)пиперидин-4-ил]пропионовая кислота Ниже описано получение защищенного производного соединения формулы II, где R4 обозначает группу формулы (b), где q равно 2. Стадия (а) 4-Гидроксиметилпиперидин (27,0 г, 262 ммоля) и ди-трет-бутилдикарбонат (51,2 г, 235 ммолей) растворяли в ТГФ (300 мл) и раствор перемешивали 12 часов при комнатной температуре. Смесь концентрировали в вакууме и остаток растворяли в простом эфире. Раствор эфира промывали водой и затем соляным раствором и сушили над сульфатом натрия. Путем выпаривания растворителей получали остаток (51,0 г) в виде масла. Масляный остаток растворяли в пиридине (200 мл) и раствор охлаждали до 0oC. Затем к раствору добавляли п-толуолсульфонилхлорид (47,5 г, 250 ммолей) и смесь выдерживали при 5oC в течение 12 часов. Смесь сливали в воду и экстрагировали этилацетатом. Этилацетатный экстракт промывали 5% HCl, водой и затем соляным раствором. Затем этилацетат сушили над сульфатом натрия, после чего выпаривали. Путем кристаллизации из этилацетата-гексана получали 1-(трет-бутоксикарбонил)-4-[(п- толуолсульфонилокси)метил]пиперидин (50 г, 136 ммолей), tпл 71-72oC. Стадия (б) Диэтилмалонат (10,4 г, 65 ммолей) добавляли к раствору этоксида натрия (4,42 г, 65 ммолей) в этаноле (75 мл). Добавляли 1-(трет-бутоксикарбонил)-4-[(п-толуолсульфонилокси)метил] пиперидин (25 г, 68 ммолей), полученный в соответствии со стадией (а) примера 1, и смесь нагревали с обратным холодильником в течение 4 часов. Смесь охлаждали и разделяли между этилацетатом и водой. Этилацетатный слой отделяли, промывали водой и затем соляным раствором, сушили над сульфатом натрия и выпаривали. Путем очистки остатка с помощью хроматографии на силикагеле (25% этилацетат-гексан) получали 1-(трет-бутоксикарбонил)-4-[2,2-ди(этоксикарбонил)этил] пиперидин (16 г, 45 ммолей) в виде масла. Стадия (в) 1-(Трет-бутоксикарбонил)-4- [2,2-ди(этоксикарбонил)этил]пиперидин (12,2 г, 34 ммоля), полученный в соответствии со стадией (б) примера 1, и гидроксид калия (4,2 г, 75 ммолей) смешивали в этаноле (10 мл), ТГФ (20 мл) и воде (50 мл) и смесь нагревали с обратным холодильником в течение 3 часов. Смесь охлаждали и промывали простым эфиром, подкисляли серной кислотой и экстрагировали эфиром. Эфир сушили над сульфатом натрия и выпаривали. Путем кристаллизации остатка из простого эфира получали 1-(трет-бутоксикарбонил)-4-[2,2- ди(карбокси)этил] пиперидин (8,5 г, 28 ммолей), который нагревали в масляной бане до 165oC, пока не прекращалось выделение CO2 (приблизительно 10 минут). Остаток охлаждали и путем кристаллизации получали 3-[1-(трет-бутоксикарбонил)пиперидин-4- ил] пропионовую кислоту (7,2 г, 28 ммолей), tпл 81-83oC. ПРИМЕР 2 N-метокси-N-метил-4-амино-5-хлор-3-метоксибензамид Ниже описано получение соединения формулы III, где R1 обозначает хлор и R3 обозначает метокси. 4-Амино-5-хлор-2-метоксибензойную кислоту (10,1 г, 50 ммолей) растворяли в ДМФ (50 мл). К раствору добавляли карбонилдиимидазол (8,9 г, 55 ммолей) и смесь перемешивали в течение 15 минут. Добавляли триэтиламин (7 мл, 5,1 г, 50 ммолей) и гидрохлорид N,O-диметилгидроксиламина (6,3 г, 65 ммолей) и смесь перемешивали в течение 12 часов. Затем смесь разбавляли водой и экстрагировали этилацетатом. Этилацетатный экстракт промывали 5% HCl, водой и затем соляным раствором. Экстракт сушили над сульфатом натрия и затем выпаривали. Путем кристаллизации из этилацетата получали N-метокси-N-метил-4-амино-5-хлор-2- метоксибензамид (10 г, 41 ммоль), tпл 134-135oC. ПРИМЕР 3 1-(4-Амино-5-хлор-3-метоксифенил)-3-(1- пиперидин-4-ил)пропан-1-он Ниже описано получение соединения формулы I, где R1 обозначает хлор, R3 обозначает метокси, R4 обозначает группу формулы (b), где p равно 0, q равно 2 и R7 обозначает водород. Стадия (а) 3-[1-(Трет-бутоксикарбонил)пиперидин-4-ил]пропионовую кислоту (4,6 г, 18 ммолей), полученную в соответствии с примером 1, растворяли в ГМФА (10 мл) и ТГФ (10 мл) и этот раствор добавляли к раствору диизопропиламида лития (60 ммолей) с температурой -20oC. Смесь нагревали до 0oC, перемешивали в течение 30 минут и затем охлаждали до -40oC. N-метокси-N-метил-4-амино-5-хлор-2- метоксибензамид (4,9 г, 20 ммолей), полученный в соответствии с примером 2, растворяли в ГМФА (10 мл) и ТГФ (10 мл) и этот раствор добавляли к смеси. Смеси давали нагреться до 0oC в течение 1 часа, затем разбавляли водой, промывали простым эфиром, подкисляли соляной кислотой и экстрагировали метиленхлоридом. Метиленхлоридный экстракт концентрировали в вакууме и остаток нагревали в масляной бане до 140oC в течение 30 минут. Смесь охлаждали, разделяли между водой и этилацетатом. Этилацетатный слой отделяли, промывали 5% гидроксидом натрия и затем соляным раствором, сушили над сульфатом натрия и выпаривали. Путем очистки с помощью хроматографиии на силикагеле (40% этилацетат-гексан) получали 1-(4-амино-5-хлор-2-метоксифенил)-3-[1-(трет- бутоксикарбонил)пиперидин-4-ил] пропан-1-он (1,5 г, 3,8 ммоля), tпл 133-134oC. Стадия (б) 1-(4-Амино-5-хлор-2-метоксифенил)-3-[1-(трет-бутоксикарбонил) пиперидин-4-ил]пропан-1-он (1,5 г, 3,8 ммолей) растворяли в метиленхлориде (20 мл) и к раствору добавляли трифторуксусную кислоту (5 мл). Через 30 минут раствор промывали водным гидроксидом аммония и сушили над сульфатом натрия. Путем выпаривания растворителей получали 1-(4-амино-5-хлор-3- метоксифенил)-3-(1-пиперидин-4-ил)пропан-1-он (1,1 г, 3,8 ммолей), tпл 138-140oC. ПРИМЕР 4 1-(4-Амино-5-хлор-2-метоксифенил)-5-(пиперидин-1-ил) пентан-1-он Ниже описано получение соединения формулы I, где R1 обозначает хлор, R3 обозначает метокси, R4 обозначает группу формулы (а), где n равно 4, p равно 0 и R5 и R6 вместе обозначают пиперидин-1-ил. Стадия (а) N-ацетил-2-хлор-5-метоксианилин (10,0 г, 50 ммолей), хлорид алюминия (13,3 г, 100 ммолей) и 5-хлорпентаноилхлорид (11,7 г, 75 ммолей) смешивали в дихлорэтане (100 мл) и смесь перемешивали при комнатной температуре и в атмосфере азота в течение 7 дней. Затем смесь сливали в ледяную крошку и экстрагировали метиленхлоридом. Метиленхлоридный экстракт промывали водой и затем сушили. Растворитель выпаривали в вакууме. Путем кристаллизации остатка из этилацетата/гексана получали 1-(4-ацетиламино-5-хлор-2- гидроксифенил)-5-хлорпентан-1-он (10,18 г, 35,1 ммолей), tпл 126-128oC. Тем же способом, что и на стадии (а) примера 4, но заменяя 1,5-дихлорпентан-1-он на 4-хлорбутирилхлорид, получали 1-(4-ацетиламино-5-хлор-2-гидроксифенил)-4-хлорбутан-1-он, пл 103-106oC. Тем же способом, что и на стадии (а) примера 4, но заменяя 1,5-дихлорпентан-1-он на 1,5-дибромпентан-1-он и N-ацетил-2-хлор-5-метоксианилин на N-ацетил-2-бром-5-метоксианилин, получали 1-(4-ацетиламино-5-бром-2-гидроксифенил)-5-бромпентан-1-он. Стадия (б) 1-(4-Ацетиламино-5-хлор-2-гидроксифенил)-5-хлорпентан-1-он (1,07 г, 3,5 ммоля), карбонат калия (1,38 г, 10,0 ммолей) и йодметан (0,62 мл, 10,0 ммолей) смешивали в метилэтилкетоне (20 мл) и перемешивали при температуре кипения с обратным холодильником в течение 4 часов 15 минут. Смесь разделяли между этилацетатом и водой. Этилацетатный слой отделяли, промывали в соляном растворе, сушили над сульфатом натрия и затем выпаривали до получения твердого остатка. Путем кристаллизации остатка из этилацетата/гексанов получали 1-(4-ацетиламино-5-хлор-2-метоксифенил)-5- хлорпентан-1-он (0,720 г, 2,37 ммолей), tпл 98-100oC. Тем же способом, что и на стадии (б) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-гидроксифенил)-5-хлорпентан-1-он на 1-(4-ацетиламино-5-хлор-2-гидроксифенил)-4-хлорбутан-1-он, получали 1-(4-ацетиламино-5-хлор-2-метоксифенил)-4-хлорбутан-1-он в виде масла. Тем же способом, что и на стадии (б) примера 4, но заменяя йодметан на 4-метоксибензилхлорид, получали 1-[4-ацетиламино-5- хлор-2-(4-метоксибензилокси)фенил]-5-хлорпентан-1-он, tпл 147-149oC. Тем же способом, что и на стадии (б) примера 4, но заменяя йодметан на 3,4-диметоксибензилхлорид, получали 1-[4-ацетиламино- 5-хлор-2-(3,4-диметоксибензилокси)фенил]-5-хлорпентан-1-он, пл 147oC. Тем же способом, что и на стадии (б) примера 4, но заменяя йодметан на 3,5-диметоксибензилхлорид, получали 1-[4-ацетиламино- 5-хлор-2-(3,5-диметоксибензилокси)фенил]-5-хлорпентан-1-он, пл 135-137oC. Тем же способом, что и на стадии (б) примера 4, но заменяя йодметан на 3,5-диметоксибензилхлорид и 1-(4-ацетиламино-5-хлор-2- гидроксифенил)-5-хлорпентан-1-он на 1-(4-ацетиламино-5-бром-2- гидроксифенил)-5-бромпентан-1-он, получали 1-[4-ацетиламино-5- бром-2-(3,5-диметоксибензилокси)фенил]-5-бромпентан-1-он. Тем же способом, что и на стадии (б) примера 4, но заменяя йодметан на 3,4-метилендиоксибензилхлорид, получали 1-[4-ацетиламино-5-хлор- 2-(3,4-метилендиоксибензилокси)фенил]-5-хлорпентан-1-он, tпл 132-134oC. Тем же способом, что и на стадии (б) примера 4, но заменяя йодметан на 2-(4-метоксифенил)-1-йодэтан, получали 1-{4-ацетиламино-5-хлор-2-[2-(4-метоксифен)этокси]фенил}-5-хлорпентан-1-он, tпл 108-110oC. Тем же способом, что и на стадии (б) примера 4, но заменяя йодметан на 2-(3,4-диметоксифенил)-1-йодэтан, получали 1-{ 4-ацетиламино-5-хлор-2-[2-(3,4-диметоксифен)этокси]фенил}- 5-хлорпентан-1-он. Стадия (в) 1-(4-Ацетиламино-5-хлор-2-метоксифенил)-5-хлорпентан-1-он (0,72 г, 2,26 ммоля), йодистый натрий (0,15 г, 0,1 ммоля) и пиперидин (1,72 г, 20 ммолей) смешивали в ДМФ (8,0 мл) и смесь нагревали до 80oC в течение 4,5 часов. Затем добавляли воду с образованием осадка. Путем фильтрации получали 1-(4-ацетиламино-5-хлор-2-метоксифенил)-5- (пиперидин-1-ил)пентан-1-он (0,8 г, 2,18 ммоля), tпл 74-75oC. 1-(4-Ацетиламино-5-хлор-2-метоксифенил)-5-(пиперидин-1-ил) пентан-1-он растворяли в 2 н. соляной кислоте (25 мл) и раствор перемешивали в течение 30 минут. Раствор охлаждали в ледяной бане с образованием осадка. Осадок собирали путем фильтрации и промывали водой. Путем сушки в вакуумной печи при 70oC получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-5-(пиперидин-1-ил)пентан-1-она (0,65 г, 2 ммоля), tпл 220-221oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)- 5-хлорпентан-1-он на 1-(4-ацетиламино-5-хлор-2-метоксифенил)-4- хлорбутан-1-он и пиперидин на диэтиламин, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-4-диэтиламинобутан-1-она, пл 145-150oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)-5-хлорпентан-1-он на 1-(4-ацетиламино-5-хлор-2-метоксифенил)-4-хлорбутан-1-он и пиперидин на пирролидин, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-4- (пирролидин-ил)бутан-1-она, tпл 221-224oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)-5-хлорпентан-1-он на 1-(4-ацетиламино-5-хлор-2-метоксифенил)-4-хлорбутан-1-он, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-4-(пиперидин-1-ил) бутан-1-она, tпл 235-238oC. Тем же способом, что и на стадии (в) примера 4, но заменяя пиперидин на диметиламин, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-5-диметиламинопентан-1-она, tпл 219-220oC. Тем же способом, что и на стадии (в) примера 4, но заменяя пиперидин на диэтиламин, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-5-диэтиламинопентан- 1-она, tпл 178-179oC. Тем же способом, что и на стадии (в) примера 4, но заменяя пиперидин на ди(проп-1-ил)амин, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-5-ди(проп-1-ил)аминопентан-1-она, tпл 162-165oC. Тем же способом, что и на стадии (в) примера 4, но заменяя пиперидин на пирролидин, получали гидрохлорид 1-(4-амино-5-хлор-2- метоксифенил)-5-(пирролидин-1-ил)пентан-1-она, tпл 203-205oC. Тем же способом, что и на стадии (в) примера 4, но заменяя пиперидин на 4-метилпиперидин, получали гидрохлорид 1-(4-амино-5-хлор-2- метоксифенил)-5-(4-метилпиперидин-1-ил) пентан-1-она, tпл 195-197oC. Тем же способом, что и на стадии (в) примера 4, но заменяя пиперидин на 4,4-диметилпиперидин, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-5-(4,4-диметилпиперидин-1-ил) пентан-1-она, tпл 239-241oC. Тем же способом, что и на стадии (в) примера 4, но заменяя пиперидин на 4-этилпиперидин, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-5-(4-этилпиперидин- 1-ил)пентан-1-она, tпл 197-198oC. Тем же способом, что и на стадии (в) примера 4, но заменяя пиперидин на 4-(проп-1-ил)пиперидин, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-5-[4- (проп-1-ил)пиперидин-1-ил]пентан-1-она, tпл 212-213oC. Тем же способом, что и на стадии (в) примера 4, но заменяя пиперидин на 4-аминокарбонилпиперидин, получали гидрохлорид 1-(4-амино-5-хлор- 2-метоксифенил)-5-(4-аминокарбонилпиперидин-1-ил) пентан-1-она, tпл 230-235oC. Тем же способом, что и на стадии (в) примера 4, но заменяя пиперидин на 4-гидроксипиперидин, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-5-(4-гидроксипиперидин-1-ил) пентан-1-она, tпл 205-207oC. Тем же способом, что и на стадии (в) примера 4, но заменяя пиперидин на 4-метоксипиперидин, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-5-(4- метоксипиперидин-1-ил)пентан-1-она, tпл 193-195oC. Тем же способом, что и на стадии (в) примера 4, но заменяя пиперидин на 4-(метилсульфонил)аминопиперидин, получали гидрохлорид 1-(4-амино-5- хлор-2-метоксифенил)-5-[4-(метилсульфонил)аминопиперидин-1-ил] пентан-1-она, tпл 245-246oC. Тем же способом, что и на стадии (в) примера 4, но заменяя пиперидин на 4-(метилсульфонил)аминометилпиперидин, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-5-[4- (метилсульфонил)аминометилпиперидин-1-ил] пентан-1-она, tпл 231-232oC. Тем же способом, что и на стадии (в) примера 4, но заменяя пиперидин на 4-фенилпиперидин, получали гидрохлорид 1-(4-амино-5- хлор-2-метоксифенил)-5-(4-фенилпиперидин-1-ил)пентан-1-она, пл 257-259oC. Тем же способом, что и на стадии (в) примера 4, но заменяя пиперидин на морфолин, получали гидрохлорид 1-(4-амино-5- хлор-2-метоксифенил)-5-(морфол-1-ил)пентан-1-она, tпл 229-231oC. Тем же способом, что и на стадии (в) примера 4, но заменяя пиперидин на азациклогептан, получали гидрохлорид 1-(4-амино-5- хлор-2-метоксифенил)-5-(азациклогепт-1-ил)пентан-1-она, tпл 203-205oC. Тем же способом, что и на стадии (в) примера 4, но заменяя пиперидин на 3-азабицикло[2.2.1] гептан, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-5-(3-азабицикло[2.2.1]гепт-3- ил)пентан-1-она, tпл 158-160oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)- 5-хлорпентан-1-он на 1-[4-ацетиламино-5-хлор-2-(4- метоксибензилокси)фенил]-5-хлорпентан-1-он, получали гидрохлорид 1-[4-амино-5-хлор-2-(4-метоксибензилокси)фенил] -5-(пиперидин-1- ил)пентан-1-она, tпл 229-230oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)- 5-хлорпентан-1-он на 1-[4-ацетиламино-5-хлор-2-(3,4- диметоксибензилокси)фенил] -5-хлорпентан-1-он, получали 1-[4- амино-5-хлор-2-(3,4-диметоксибензилокси)фенил] -5-(пиперидин-1- ил)пентан-1-он, tпл 125-127oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)- 5-хлорпентан-1-он на 1-[4-ацетиламино-5-хлор-2-(3,4- метилендиоксибензилокси)фенил]-5-хлорпентан- 1-он, получали 1-[4-амино-5-хлор-2-(3,4- метилендиоксибензилокси)фенил] -5-(пиперидин-1-ил)пентан-1-он, tпл 120-122oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)-5-хлорпентан-1-он на 1-[4-ацетиламино-5-хлор-2-(3,5-диметоксибензилокси)фенил] -5- хлорпентан-1-он и пиперидин на диметиламин, получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил]-5- диметиламинопентан-1-она, tпл 221-224oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2- метоксифенил)-5-хлорпентан-1-он на 1-[4-ацетиламино-5-хлор-2-(3,5- диметоксибензилокси)фенил] -5-хлорпентан-1-он и пиперидин на диэтиламин, получали 1-[4-амино-5-хлор-2-(3,5- диметоксибензилокси)фенил]-5-диэтиламинопентан-1-он, tпл 105-107oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)-5-хлорпентан-1-он на 1-[4-ацетиламино-5-хлор-2-(3,5-диметоксибензилокси)фенил] -5- хлорпентан-1-он и пиперидин на пирролидин, получали 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил] -5-(пирролидин-1-ил) пентан-1-он, tпл 125-127oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)-5- хлорпентан-1-он на 1-[4-ацетиламино-5-хлор-2-(3,5- диметоксибензилокси)фенил]-5-хлорпентан-1-он, получали 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил]-5-(пиперидин-1- ил)пентан-1-он, tпл 128-130oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)-5- хлорпентан-1-он на 1-[4-ацетиламино-5-бром-2-(3,5- диметокси)бензилоксифенил] -5-бромпентан-1-он, получали гидрохлорид 1-[4-амино-5-бром-2-(3,5-диметокси)бензилоксифенил] – 5-(пиперидин-1-ил)пентан-1-он, tпл 238-239oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор- 2-метоксифенил)-5-хлорпентан-1-он на 1-[4-ацетиламино-5-хлор-2- (3,5-диметоксибензилокси)фенил] -5-хлорпентан-1-он и пиперидин на 4-метилпиперидин, получали 1-[4-амино-5-хлор- 2-(3,5-диметоксибензилокси)фенил]-5-(4-метилпиперидин-1- ил)пентан-1-он, tпл 136-137oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)- 5-хлорпентан-1-он на 1-[4-ацетиламино-хлор-5-хлор-2-(3,5- диметоксибензилокси)фенил]-5-хлорпентан-1-он и пиперидин на 4- (проп-1-ил)пиперидин, получали 1-[4-амино-5-хлор-2-(3,5- диметоксибензилокси)фенил]-5-[4-(проп-1-ил)пиперидин-1-ил] пентан-1-он, tпл 119-120oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)-5- хлорпентан-1-он на 1-[4-ацетиламино-5-хлор-2-(3,5- диметоксибензилокси)фенил]-5-хлорпентан-1-он и пиперидин на 4,4-диметилпиперидин, получали 1-[4-амино-5-хлор-2-(3,5- диметоксибензилокси)фенил]-5-(4,4-диметилпиперидин-1-ил)пентан- 1-он, tпл 135-136oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)-5-хлорпентан-1- он на 1-[4-ацетиламино-5-хлор-2-(3,5-диметоксибензилокси)фенил] – 5-хлорпентан-1-он и пиперидин на 4-гидроксипиперидин, получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил]- 5-(4-гидроксипиперидин-1-ил)пентан-1-она, tпл 220-222oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4- ацетиламино-5-хлор-2-метоксифенил)-5-хлорпентан-1-он на 1-[4- ацетиламино-5-хлор-2-(3,5-диметоксибензилокси)фенил] -5- хлорпентан-1-он и пиперидин на 4-(2-гидроксиэтил)пиперидин, получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5- диметоксибензилокси)фенил] -5-[4-(2-гидроксиэтил)пиперидин-1- ил]пентан-1-она, tпл 180-183oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)-5- хлорпентан-1-он на 1-[4-ацетиламино-5-хлор-2-(3,5- диметоксибензилокси)фенил] -5-хлорпентан-1-он и пиперидин на 4-метоксипиперидин, получали гидрохлорид 1-[4-амино-5- хлор-2-(3,5-диметоксибензилокси)фенил] -5-(4- метоксипиперидин-1-ил)пентан-1-она, tпл 195-196oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4- ацетиламино-5-хлор-2-метоксифенил)-5-хлорпентан-1-он на 1-[4- ацетиламино-5-хлор-2-(3,5-диметоксибензилокси)фенил] -5- хлорпентан-1-он и пиперидин на 4-аминокарбонилпиперидин, получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил] – 5-(4-аминокарбонилпиперидин-1-ил)пентан-1-она, tпл 207-209oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4- ацетиламино-5-хлор-2-метоксифенил)-5-хлорпентан-1-он на 1-[4-ацетиламино-5-хлор-2-(3,5-диметоксибензилокси)фенил] -5- хлорпентан-1-он и пиперидин на 4-(аминокарбонил)аминопиперидин, получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси) фенил] -5-[4-(аминокарбонил)аминопиперидин-1-ил] пентан-1-она, tпл 220-224oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)-5-хлорпентан-1-он на 1-[4-ацетиламино-5-хлор-2-(3,5-диметоксибензилокси)фенил] -5- хлорпентан-1-он и пиперидин на 4-(аминокарбонил)аминометилпиперидин, получали гидрохлорид 1-[4- амино-5-хлор-2-(3,5-диметоксибензилокси)фенил]-5-[4- (аминокарбонил)аминометилпиперидин-1-ил]пентан-1-она, tпл 130-135oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)-5-хлорпентан-1-он на 1-[4-ацетиламино-5-хлор-2-(3,5-диметоксибензилокси)фенил] -5- хлорпентан-1-он и пиперидин на 4-(метилсульфонил)аминопиперидин, получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси) фенил] -5-[4-(метилсульфонил)аминопиперидин-1-ил] пентан-1-она, tпл 240-245oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)-5-хлорпентан-1-он на 1-[4-ацетиламино-5-хлор-2-(3,5-диметоксибензилокси) фенил] -5- хлорпентан-1-он и пиперидин на 4-(метилсульфонил)аминометилпиперидин, получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил]-5-[4- (метилсульфонил)аминометилпиперидин-1-ил]пентан-1-она, tпл 211-213oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)-5-хлорпентан-1-он на 1-[4-ацетиламино-5-хлор-2-(3,5-диметоксибензилокси)фенил] -5-хлорпентан- 1-он и пиперидин на 4-[2-(метилсульфонил)аминоэтил] пиперидин, получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5- диметоксибензилокси)фенил]-5-{4-[2- (метилсульфонил)аминоэтил] пиперидин-1-ил}пентан-1-она, tпл 205-206oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)-5-хлорпентан-1-он на 1-[4-ацетиламино-5-хлор-2-(3,5-диметоксибензилокси)фенил] -5- хлорпентан-1-он и пиперидин на 4-фенилпиперидин, получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил]- 5-(4-фенилпиперидин-1-ил)пентан-1-она, tпл 237-239oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)-5-хлорпентан-1-он на 1-[4-ацетиламино-5-хлор-2-(3,5-диметоксибензилокси)фенил] -5- хлорпентан-1-он и пиперидин на азациклогептан, получали 1-[4- амино-5-хлор-2-(3,5-диметоксибензилокси)фенил]-5-(азациклогепт-1- ил)пентан-1-он, tпл 137-139oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)-5- хлорпентан-1-он на 1-{ 4-ацетиламино-5-хлор-2-[2-(4- метоксифен)этокси]фенил}-5-хлорпентан-1-он, получали 1-{4-амино-5-хлор-2-[2-(4-метоксифен)этокси] фенил} -5-(пиперидин-1- ил)пентан-1-он, tпл 211-212oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)-5- хлорпентан-1-он на 1-{ 4-ацетиламино-5-хлор-2-[2-(3,4- диметоксифен)этокси]фенил}-5-хлорпентан-1-он, получали 1-{4- амино-5-хлор-2-[2-(3,4-диметоксифен)этокси] фенил} -5-(пиперидин- 1-ил)пентан-1-он, tпл 224-225oC. Тем же способом, что и на стадии (в) примера 4, но заменяя 1-(4-ацетиламино-5-хлор-2-метоксифенил)- 5-хлорпентан-1-он на 1-{ 4-ацетиламино-5-хлор-2-[2-(4- метоксифен)этокси]фенил}-5-хлорпентан-1-он и пиперидин на 4-метилпиперидин, получали гидрохлорид 1-{ 4- амино-5-хлор-2-[2-(4-метоксифен)этокси]фенил}-5-(4- метилпиперидин-1-ил)пентан-1-она, tпл 226-228oC. ПРИМЕР 5 4′-Амино-5′-хлор-2′-метоксиацетофенон Ниже описано получение соединения формулы VIII, где R1 обозначает хлор, R2 обозначает водород и R3 обозначает метокси. N-метокси-N-метил-4-амино-5-хлор-2-метоксибензамид (24,4 г, 100 ммолей) растворяли в ТГФ (400 мл) и раствор охлаждали до -40oC. Добавляли метиллитий в эфире (143 мл, 4,4 г, 200 ммолей) и смеси давали нагреться до 0oC. Добавляли водную соляную кислоту и смесь экстрагировали этилацетатом. Экстракт промывали соляным раствором, сушили над сульфатом натрия и выпаривали до получения твердого вещества. Путем кристаллизации из этилацетата-гексана получали 4′-амино-5′-хлор-2′-метоксиацетофенон (15,0 г, 75 ммолей), tпл 114-116oC. Тем же способом, что и в примере 5, но заменяя N-метокси-N-метил-4-амино-5-хлор-2-метоксибензамид на N-метокси-N-метил-4-амино-5-хлор-2,3-этилендиоксибензамид, получали 4-амино-5-хлор-2,3-этилендиоксифенилацетофенон, tпл 133-135oC. Тем же способом, что и в примере 5, но заменяя N-метокси-N-метил-4-амино-5-хлор-2-метоксибензамид на N-метокси-N-метил-4-амино-5-хлор-2,3-диметоксибензамид, получали 4-амино-5-хлор-2,3-диметоксифенилацетофенон, tпл 75-78oC. ПРИМЕР 6 4′-Амино-5′-хлор-2’3′-этилендиоксиацетофенон Ниже описано получение соединения формулы VI, где R1 обозначает хлор, и R2 и R3 вместе обозначают этилендиокси. Стадия (а) Раствор 1,4-бензодиоксана (8,4 М, 25 мл, 0,21 моля) в 15 мл уксусной кислоты обрабатывали газообразным хлором при 10-20oC, получая желтый осадок. Осадок выделяли с помощью фильтрации и промывали водой. Путем кристаллизации осадка из этанола получали 6,7-дихлор-1,4- бензодиоксан (25,5 г, 0,124 моля), tпл 147-148oC. Стадия (б) Смесь 6,7-дихлор-1,4-бензодиоксана (10,25 г, 0,05 моля), ацетилхлорида (13,5 М, 5 мл, 0,0675 моля) и хлористого алюминия (10 г, 0,075 моля) в 200 мл 1,2-дихлорэтана перемешивали в атмосфере азота в течение 24 часов. Смесь сливали в приблизительно 500 мл льда/разбавленной соляной кислоты и экстрагировали метиленхлоридом. Метиленхлоридный экстракт промывали бикарбонатом натрия (1 раз), водой (1 раз) и соляным раствором (1 раз) и затем сушили над сульфатом натрия. Метиленхлорид фильтровали и концентрировали с помощью роторного испарителя. Путем очистки остатка с помощью колоночной хроматографии (двуокись кремния 230-400 меш; 15% этилацетат/гексан) получали 5,6-дихлор-2,3-этилендиоксиацетофенон (8,1 г, 0,033 моля), tпл 83-86oC. Стадия (в) 5′,6′-Дихлор-2′,3′-этилендиоксиацетофенон (8,5 г, 0,0344 моля) добавляли порциями к 34,6 мл перемешиваемой дымящей азотной кислоты с такой скоростью, чтобы температура реакционной смеси оставалась ниже 10oC. Смесь перемешивали еще в течение 10 минут при 5oC и затем сливали в 250 г ледяной крошки, получая желтый осадок. Осадок выделяли путем фильтрации и промывали водой. Путем сушки получали 5′, 6′-дихлор-2′,3′-этилендиокси-4′-нитроацетофенон (8,9 г, 0,0341 моля), tпл 181-182oC. Стадия (г) Смесь 5′, 6′-дихлор-2′, 3′-этилендиокси-4′-нитроацетофенона (2 г, 7,63 ммоля) и 10% палладия на угле (800 г) гидрогенизировали в 17 мл 15% гидроксида натрия и 160 мл этанола при давлении 57 фунт/кв.дюйм в течение 7 часов. Смесь фильтровали и концентрировали с помощью роторного испарителя и остаток перемешивали с водой. Путем сушки получали 4′-амино-2′,3′- этилендиоксиацетофенон (970 мг, 5,04 ммоля), tпл 135-136oC. Стадия (д) Раствор 4′-амино-2′, 3′-этилендиоксиацетофенона (310 мг, 1,60 ммоля) в 2,3 мл пиридина охлаждали в атмосфере азота в водно/ледяной бане и по каплям добавляли 6,9 мл уксусного ангидрида. Смеси давали нагреться до комнатной температуры и затем перемешивали в течение 16 часов. Смесь концентрировали с помощью роторного испарителя и остаток перемешивали с водой. Остаток выделяли путем фильтрации и промывали водой. Путем сушки получали 2′,3′-этилендиокси-4′- (метилкарбониламино)ацетофенон (289 мг, 1,23 ммоля), tпл 142-144oC. Стадия (е) Раствор 2′,3′-этилендиокси-4′- (метилкарбониламино)ацетофенона (250 мг, 1,06 ммоля) в 6 мл диметилформамида охлаждали в атмосфере азота в водно/ледяной бане и добавляли N-хлорсукцинимид (156 мг, 1,17 ммоля). Смеси давали нагреться до комнатной температуры и затем выдерживали при 55oC в течение 1,5 часов. Затем смесь охлаждали до комнатной температуры и перемешивали в течение 16 часов. Смесь концентрировали с помощью роторного испарителя и остаток перемешивали с водой. Остаток выделяли путем фильтрации и промывали водой. Путем сушки получали 5′-хлор-2′, 3′-этилендиокси-4′-(метилкарбониламино)ацетофенон (170 мг, 0,69 ммоля), tпл 181-182oC. Стадия (ж) Смесь 5′-хлор-2′,3′-этилендиокси-4′-(метилкарбониламино)ацетофенона (152 мг, 0,56 ммоля) и гидроксида натрия (4 н., 1,4 мл, 5,6 ммоля) в 5 мл метанола нагревали с обратным холодильником в течение 3,5 часов. Смесь концентрировали с помощью роторного испарителя и остаток перемешивали с водой. Остаток выделяли путем фильтрации и промывали водой. Путем сушки получали 4′-амино-5′-хлор-2′, 3′- этилендиоксиацетофенон (95 мг, 0,41 ммоля), tпл 133-136oC. ПРИМЕР 7 1-(4-Амино-5-хлор-2-метоксифенил)-3-(пиперидин-4-ил) пропан-1-он Ниже описано получение соединения формулы I, где R1 обозначает хлор, R3 обозначает метокси, R4 обозначает группу формулы (b), где p равно 0, q равно 2 и R7 обозначает водород. Стадия (a) 4-Амино-5-хлор-2-метоксиацетофенон (12,75 г, 64 ммоля), полученный в соответствии с примером 5, суспендировали в ТГФ (125 мл). Суспензию добавляли к охлажденному до -50oC раствору диизопропиламида лития (16,1 г, 150 ммолей) в ТГФ (200 мл) и смесь перемешивали при 0oC в течение 15 минут. Добавляли пиридин-4-карбоксальдегид (8,0 г, 75 ммолей) и смеси давали нагреться до 5oC. Добавляли водный аммонийхлорид и смесь экстрагировали трижды этилацетатом. Объединенные экстракты промывали водой, сушили над сульфатом натрия и концентрировали в вакууме с получением полутвердого остатка. Добавляли этилацетат-гексан и путем последующей фильтрации получали 1-(4-амино-5-хлор- 2-метоксифенил)-3-гидрокси-3-(пиридин-4-ил)пропан-1-он (12,1 г, 40 ммолей), tпл 181-183oC. Стадия (б) 1-(4-Амино-5-хлор-2- метоксифенил)-3-гидрокси-3-(пиридин-4-ил)пропан-1-он (8,6 г, 28 ммолей), полученный в соответствии со стадией (а) примера 6, растворяли в концентрированной серной кислоте (40 мл) и раствор перемешивали при комнатной температуре в течение 15 минут. Раствор сливали на лед, повышали основность с помощью гидроксида аммония и затем фильтровали, собирая желтое твердое вещество. Путем сушки получали 1-(4-амино-5-хлор-2-метоксифенил)-3-(пиридин-4-ил)-2- пропен-1-он (7,6 г, 26 ммолей), tпл 209-211oC. Стадия (в) 1-(4-Амино-5-хлор-2-метоксифенил)-3-(пиридин-4-ил)-2-пропен-1-он (0,5 г, 1,7 ммоля), полученный в соответствии со стадией (б) примера 6, растворяли в уксусной кислоте (25 мл) и раствор гидрогенизировали над катализатором 5% родия на окисноалюминиевом носителе (0,2 г) при давлении 50 фунт/кв.дюйм в течение 24 часов. Раствор фильтровали, разбавляли водой, повышали основность с помощью гидроксида аммония и трижды экстрагировали метиленхлоридом. Объединенные экстракты сушили над сульфатом натрия. Путем выпаривания получали 1-(4-амино-5-хлор-2-метоксифенил)-3- (пиперидин-4-ил) пропан-1-он (0,35 г, 1,2 ммоля). ПРИМЕР 8 1-(4-Амино-5-хлор-2-метоксифенил)-3-(пиперидин-4- ил)пропен-1-он Ниже описан способ, альтернативный способу примера 7, стадиям (а)-(б). Гидроксид калия (5,0 г, 89,3 ммоля) растворяли в воде (25,0 мл). К раствору добавляли этанол (100 мл) и смесь перемешивали. 4-Амино-5-хлор-2-метоксиацетофенон (10,0 г, 50,5 ммоля), полученный в соответствии с примером 5, добавляли к перемешиваемому раствору и через 5 минут добавляли пиридин-4- карбоксальдегид (6,4 г, 60,0 ммолей) и смесь перемешивали в течение приблизительно 12 часов. Затем смесь разбавляли водой и фильтровали, собирая желтое твердое вещество. Путем сушки получали 1-(4-амино-5-хлор-2-метоксифенил)-3-(пиридин-4-ил)-2-пропен-1-он (12,85 г, 44,5 ммоля). Тем же способом, что и в примере 8, но заменяя 4-амино-5-хлор-2-метоксиацетофенон на 4-амино-5-хлор- 2,3-этилендиоксиацетофенон, получали 1-(4-амино-5-хлор-2,3- этилендиоксифенил)-3-(пиридин-4-ил)пропен-1-он, tпл 209-211oC. Тем же способом, что и в примере 8, но заменяя 4-амино-5-хлор-2- метоксиацетофенон на 4-амино-5-хлор-2,3-диметоксиацетофенон, получали 1-(4-амино-5-хлор-2,3-диметоксифенил)-3-(пиридин-4- ил)пропен-1-он, tпл 220-223oC. ПРИМЕР 9 1-(4-Амино-5-хлор-2,3-этилендиоксифенил)-3-(пиперидин-4-ил)-2-пропан- 1-он Ниже описан способ, альтернативный способу примера 7, стадии (в). Стадия (а) Раствор 1-(4-амино-5-хлор-2,3-этилендиоксифенил)-3- (пиридин-4-ил)-2-пропен-1-она (1,55 г, 5 ммолей) в 50 мл ТГФ гидрогенизировали над 5% палладием на угле (0,5 г) в течение приблизительно 2 часов. Смесь фильтровали и концентрировали с помощью роторного испарителя. Путем очистки остатка с помощью хроматографии на силикагеле (2% CH3OH-CH2Cl2 + 0,1% NH4OH) получали 1-(4 -амино-5-хлор-2,3-этилендиоксифенил)-3-(пиридин-4-ил)пропан-1-он (1,23 г, 3,8 ммоля). Стадия (б) Раствор 1-(4-амино-5-хлор-2,3-этилендиоксифенил)- 3-(пиридин-4-ил)пропан-1-она (1,23 г, 3,8 ммоля) в 15 мл ледяной уксусной кислоты гидрогенизировали над катализатором 5% родия на окисноалюминиевом носителе (1,0 г) при давлении 50 фунт/кв.дюйм в течение приблизительно 20 часов. Смесь фильтровали и фильтр промывали несколько раз этанолом. Объединенные фильтраты концентрировали в вакууме. Путем очистки остатка с помощью хроматографии на силикагеле (20% CH3OH-CH2Cl2 + 0,2% NH4OH) получали 1-(4-амино-5-хлор-2,3-этилендиоксифенил)-3-(пиперидин-4- ил)пропан-1-он (0,22 г, 0,68 ммоля). ПРИМЕР 10 1-(4-Амино-5-хлор-2-метоксифенил)-3-[1-(проп-1-ил)пиперидин-4-ил] пропан-1-он Ниже описано получение соединения формулы I, где R1 обозначает хлор, R3 обозначает метокси, R4 обозначает группу формулы (b), где p равно 0, q равно 2 и R7 обозначает проп-1-ил. 1-(4-Амино-5-хлор-2-метоксифенил)-3-(пиперидин-4-ил) пропан-1-он (0,2 г, 0,67 ммоля), полученный в соответствии с примером 3 или примером 7, триэтиламин (0,2 мл) и 1-бромпропан (0,065 мл, 0,75 ммоля) растворяли в ДМФ (4 мл) и раствор перемешивали при комнатной температуре в течение 12 часов. Раствор разделяли между водным гидроксидом аммония и этилацетатом. Этилацетатный слой отделяли, промывали водой и затем соляным раствором, сушили над сульфатом натрия и выпаривали. Путем очистки остатка с помощью хроматографии на силикагеле (10% CH3OH- CH2Cl2) получали 1-(4-амино-5-хлор-2-метоксифенил)-3-[1-(проп- 1-ил)пиперидин-4-ил] пропан-1-он, который затем растворяли в этанольной соляной кислоте. Путем кристаллизации с простым эфиром получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифе-нил)-3-[1- (проп-1-ил)пиперидин-4-ил]пропан-1-она (0,15 г, 0,4 ммоля), пл 200-201oC. Тем же способом, что и в примере 10, но заменяя 1-бромпропан на йодметан, получали гидрохлорид 1-(4-амино-5-хлор-2- метоксифенил)-3-(1-метилпиперидин-4-ил)пропан-1-она, tпл 179-180oC. Тем же способом, что и в примере 10, но заменяя 1-бромпропан на йодэтан, получали гидрохлорид 1-(4-амино-5-хлор-2- метоксифенил)-3-(1-этилпиперидин-4-ил)пропан-1-она, tпл 128-130oC. Тем же способом, что и в примере 10, но заменяя 1-бромпропан на йодбутан, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)- 3-[1-(бут-1-ил)пиперидин-4-ил]пропан-1-она, tпл 195-196oC. Тем же способом, что и в примере 10, но заменяя 1-бромпропан на 1-бром-2-метилпропан, получали гидрохлорид 1-(4-амино-5-хлор-2- метоксифенил)-3-[1-(2-метилпроп-1-ил)пиперидин-4-ил]пропан-1 -она, tпл 198-199oC. Тем же способом, что и в примере 10, но заменяя 1-бромпропан на 1-бром-3-метилбутан, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-3-[1-(3-метилбут-1- ил)пиперидин-4-ил]пропан-1-она, tпл 178-179oC. Тем же способом, что и в примере 10, но заменяя 1-бромпропан на 1-бромпентан, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-3-[1-(пент- 1-ил)пиперидин-4-ил]пропан-1-она, tпл 196-197oC. Тем же способом, что и в примере 10, но заменяя 1-бромпропан на 1-бромгексан, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-3-[1-(гекс- 1-ил)пиперидин-4-ил]пропан-1-она, tпл 212-213oC. Тем же способом, что и в примере 10, но заменяя 1-бромпропан на 3-бромпропен, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-3-[1-(2- пропенил)пиперидин-4-ил]пропан-1-она, tпл 162-163oC. Тем же способом, что и в примере 10, но заменяя 1-бромпропан на 1-хлор-2- метоксиэтан, получали гидрохлорид 1-(4-амино-5-хлор-2- метоксифенил)-3-[1-(2-метоксиэтил)пиперидин-4-ил]пропан-1-она. Тем же способом, что и в примере 10, но заменяя 1-бромпропан на 2-бром-1-[(диметиламиносульфонил)амино] этил, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-3-{ 1-[2- (диметиламиносульфонил)аминоэтил]пиперидин-4-ил}пропан-1-она, tпл 195-196oC. Тем же способом, что и в примере 10, но заменяя 1-бромпропан на 2-бром-1-[(диметиламинокарбонил)амино] этил, получали гидрохлорид 1-(4- амино-5-хлор-2-метоксифенил)-3-{1-[2-(диметиламинокарбонил)аминоэтил] пиперидин-4-ил}пропан-1-она, tпл 167-171oC. Тем же способом, что и в примере 10, но заменяя 1-бромпропан на 2-бром-1-[(метоксикарбонил)амино] этил, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-3-{ 1-[2- (метоксикарбонил)аминоэтил] пиперидин-4-ил} пропан-1-она, tпл 239-240oC. Тем же способом, что и в примере 10, но заменяя 1-бромпропан на 2-бром-1-[(трифторметилсульфонил)амино] этил, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-3-{ 1-[2- (трифторметилсульфонил)аминоэтил]пиперидин-4-ил}пропан-1-она, tпл 235-238oC. Тем же способом, что и в примере 10, но заменяя 1-бромпропан на 3-бром-1-[(метилсульфонил)амино] пропил, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-3-{1-[3- (метилсульфонил)аминопроп-1-ил]пиперидин-4-ил} пропан-1-она, tпл 194-195oC. Тем же способом, что и в примере 10, но заменяя 1-бромпропан на йодбутан и 1-(4-амино-5-хлор-2-метоксифенил)-3- (пиперидин-4-ил)пропан-1-он на 1-(4-амино-5-хлор-2,3- этилендиоксифенил)-3-(пиперидин-4-ил)пропан-1-он, получали гидрохлорид 1-(4-амино-5-хлор-2,3-этилендиоксифенил)-3-[1-(бут-1- ил)пиперидин-4-ил]пропан-1-она, tпл 265-267oC. Тем же способом, что и в примере 10, но заменяя 1-бромпропан на йодбутан и 1-(4-амино- 5-хлор-2-метоксифенил)-3-(пиперидин-4-ил) пропан-1-он на 1-(4-амино-5-хлор-2,3-диметоксифенил)-3-(пиперидин-4-ил) пропан-1-он, получали гидрохлорид 1-(4-амино-5-хлор-2,3-диметоксифенил)-3-[1- (бут-1-ил)пиперидин-4-ил]пропан-1-она, tпл 175-176oC. ПРИМЕР 11 1-(4-Амино-5-хлор-2-метоксифенил)-3-{1-[2-(метилсульфонил)аминоэтил] пиперидин-4-ил}пропан-1-он Ниже описано получение соединения формулы I, где R1 обозначает хлор, R3 обозначает метокси, R4 обозначает группу формулы (b), где p равно 0, q равно 2 и R7 обозначает 2-[(метилсульфонил)амино]этил. Этиленамин (40 мг, 0,9 ммоля) растворяли в толуоле (5 мл) и раствор охлаждали в ледяной бане. Медленно добавляли раствор метансульфонилхлорида в толоуоле (1 мл, 0,1 мг, 1 ммоль) и смесь перемешивали в течение 20 минут с образованием раствора 1-[2-(метилсульфонил)этил]этиленимина. 1-(4-Амино-5-хлор-2- метоксифенил)-3-(пиперидин-4-ил)пропан-1-он (240 мг, 0,8 ммоля), полученный в соответствии с примером 3 или примером 7, растворяли в ТГФ (20 мл) и через фильтр добавляли раствор 1-[2- (метилсульфонил)этил]этиленимина. Смесь нагревали с обратным холодильником в течение 1 часа и затем концентрировали в вакууме. Остаток растворяли в этилацетате и затем этот раствор трижды промывали водой, сушили над сульфатом натрия и выпаривали. Путем очистки с помощью хроматографии на силикагеле (15% CH3OH- CH2Cl2) получали 1-(4-амино-5-хлор-2-метоксифенил)-3-{ 1-[2- (метилсульфонил)аминоэтил] пиперидин-4-ил} пропан-1-он. Путем кристаллизации из этанольной соляной кислоты получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-3-{ 1-[2- (метилсульфонил)аминоэтил]пиперидин-4-ил}пропан-1-она, tпл 123-126oC. ПРИМЕР 12 1-[4-Амино-5-хлор-2-(3,5-диметоксибензилокси)фенил]-3-{1-[2- (метилсульфонил)аминоэтил]пиперидин-4-ил}пропан-1-он Ниже описано получение соединения формулы I, где R1 обозначает хлор, R3 обозначает 3,5-диметоксибензилокси, R4 обозначает группу формулы (b), где p равно 0, q равно 2 и R7 обозначает 2-(метилсульфонил)аминоэтил. Стадия (а) 1-[4-Амино-5-хлор-2-метоксифенил] -3-{ 1-[2- (метилсульфонил)аминоэтил] пиперидин-4-ил} пропан-1-он (1,19 г, 2,8 ммоля), полученный в соответствии с примером 11, растворяли в дихлорэтане (50 мл). Добавляли трехбромистый бор в метиленхлориде (4 мл, 1,0 г, 4,0 ммоля) и смесь перемешивали приблизительно в течение 12 часов. Затем смесь сливали на лед и добавляли метиленхлорид (150 мл) и гидроксид аммония. Эту двухслойную смесь перемешивали до образования раствора в слое метиленхлорида. Водный слой удаляли и смешивали со свежим метиленхлоридом (100 мл) и эту двухслойную смесь перемешивали. Метиленхлоридные слои объединяли и сушили над карбонатом калия. Путем фильтрации и концентрации получали 1- (4-амино-5-хлор-2-гидроксифенил)-3-{ 1-[2- (метилсульфонил)аминоэтил]пиперидин-4-ил}пропан-1-он (0,8 г, 2,0 ммоля). Стадия (б) 1-(4-Амино-5-хлор-2-гидроксифенил)-3-{ 1-[2- (метилсульфонил)аминоэтил] пиперидин-4-ил} пропан-1-он (0,78 г, 1,93 ммоля), 3,5-диметоксибензилхлорид (0,396 г, 2,12 ммоля) и карбонат калия (2,6 г, 18,8 ммоля) смешивали в ДМФ (  2 мл) и смесь перемешивали при температуре окружающей среды в течение 48 часов. Дополнительно добавляли 3,5-диметоксибензилхлорид (0,05 г, 0,13 ммоля) и смесь перемешивали в течение 24 часов. Растворитель удаляли при пониженном давлении и к оставшимся твердым веществам добавляли метиленхлорид и водный бикарбонат натрия и эту двухслойную смесь перемешивали. Метиленхлоридный слой отделяли, дважды промывали водой и сушили над карбонатом калия. Путем фильтрации и концентрации получали неочищенный продукт (1,2 г).
Путем очистки с помощью хроматографии на силикагеле (3% CH3OH-CH2Cl2) получали 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил] -3-{ 1-[2- (метилсульфонил)аминоэтил] пиперидин-4-ил} пропан-1-он (0,53 г, 0,98 ммоля). Путем кристаллизации из этанольной соляной кислоты получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил]-3- {1-[2-(метилсульфонил)аминоэтил] пиперидин-4-ил}пропан-1-она (0,54 г, 0,93 ммоля), tпл 123-126oC.
Тем же способом, что и в примере 12, стадия (а), но заменяя 1-[4-амино-5-хлор-2-метоксифенил]-3-{1- [2-(метилсульфонил)аминоэтил]пиперидин-4-ил} пропан-1-он на 1-(4- амино-5-хлор-2-метоксифенил)-3-[1-(бут-1-ил)пиперидин-4-ил]пропан- 1-он, и затем в соответствии с примером 12, стадия (б), получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил]- 3-[1-(бут-1-ил)пиперидин-4-ил]пропан-1-она, tпл 179-182oC.
Тем же способом, что и в примере 12, стадия (а), но заменяя 1-[4-амино-5-хлор-2-метоксифенил]-3-{1-[2-(метилсульфонил)аминоэтил] пиперидин-4-ил} пропан-1-он на 1-(4-амино-5-хлор-2-метоксифенил)-3- [1-(пент-1-ил)пиперидин-4-ил] пропан-1-он, и затем в соответствии с примером 12, стадия (б), получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5- диметоксибензилокси)фенил]-3-[1-(пент-1-ил)пиперидин-4-ил] пропан-1-она, tпл 174-176oC.
Тем же способом, что и в примере 12, но заменяя 1-[4-амино-5-хлор-2-метоксифенил] -3-{1-[2- (метилсульфонил)аминоэтил]пиперидин-4-ил}пропан-1-он на 1-(4- амино-5-хлор-2-метоксифенил)-3-[1-(2-метоксиэтил) пиперидин-4-ил] пропан-1-он, и затем в соответствии с примером 12, стадия (б), получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил] – 3-[1-(2-метоксиэтил)пиперидин-4-ил]пропан-1-она, tпл 183-184oC.
Тем же способом, что и в примере 12, стадия (а), но заменяя 1-[4-амино-5-хлор-2-метоксифенил]-3-{1-[2- (метилсульфонил)аминоэтил]пиперидин-4-ил} пропан-1-он на 1-[4-амино-5-хлор-2-метоксифенил]-3-{1-[2- (диметиламиносульфонил)аминоэтил] пиперидин-4-ил}пропан-1-он, и затем в соответствии с примером 12, стадия (б), получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил] – 3-{1-[2-(диметиламиносульфонил)аминоэтил] пиперидин-4-ил} пропан-1-она, tпл 173-176oC.
Тем же способом, что и в примере 12, стадия (б), но заменяя 3,5-диметоксибензилхлорид на бензилхлорид, получали гидрохлорид 1-(4-амино-5-хлор-2-бензилоксифенил)-3-{ 1- [2-(метилсульфонил)аминоэтил] пиперидин-4-ил}пропан-1-она, пл 193-194oC.
Тем же способом, что и в примере 12, стадия (б), но заменяя 3,5-диметоксибензилхлорид на 3-метоксибензилхлорид, получали гидрохлорид 1-[4-амино-5-хлор-2-(3- метоксибензилокси)фенил]-3-{1-[2-(метилсульфонил)аминоэтил] пиперидин-4-ил}пропан-1-она, tпл 174-176oC.
Тем же способом, что и в примере 12, стадия (а), но заменяя 1-[4-амино-5-хлор-2- метоксифенил] -3-{ 1-[2-(метилсульфонил)аминоэтил]пиперидин-4- ил} пропан-1-он на 1-[4-амино-5-хлор-2-метоксифенил] -5- (пиперидин-1-ил)пентан-1-он, и затем в соответствии с примером 12, стадия (б), но заменяя 3,5-диметоксибензилхлорид на йодэтан, получали гидрохлорид 1-[4-амино-5-хлор-2-этоксифенил] -5- (пиперидин-1-ил)пентан-1-она, tпл 230-231oC.
Тем же способом, что и в примере 12, стадия (а), но заменяя 1-(4-амино-5-хлор-2- метоксифенил)-3-{ 1-[2-(метилсульфонил)аминоэтил] пиперидин-4- ил} пропан-1-он на 1-[4-амино-5-хлор-2-метоксифенил] -5- (пиперидин-1-ил)пентан-1-он, и затем в соответствии с примером 12, стадия (б), но заменяя 3,5-диметоксибензилхлорид на 1-бромпропан, получали гидрохлорид 1-[4-амино-5-хлор-2-(проп-1-илокси)фенил] -5- (пиперидин-1-ил)пентан-1-она, tпл 232-233oC.
Тем же способом, что и в примере 12, стадия (а), но заменяя 1-(4-амино-5-хлор-2- метоксифенил)-3-{1-[2-(метилсульфонил)аминоэтил]пиперидин-4-ил} пропан-1-он на 1-[4-амино-5-хлор-2-метоксифенил]-5-(пиперидин-1- ил)пентан-1-он, и затем в соответствии с примером 12, стадия (б), но заменяя 3,5-диметоксибензилхлорид на 3,5-диметилбензилхлорид, получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5- диметилбензилокси)фенил] -5-(пиперидин-1-ил)пентан-1-она, tпл 218-233oC.
Тем же способом, что и в примере 12, стадия (а), но заменяя 1-[4-амино-5-хлор-2-метоксифенил]-3-{1-[2- (метилсульфонил)аминоэтил]пиперидин-4-ил} пропан-1-он на 1-(4-амино-5-хлор-2-метоксифенил)-5-(4- аминокарбонилпиперидин-1- ил)пентан-1-он, и затем в соответствии с примером 12, стадия (б), но заменяя 3,5-диметоксибензилхлорид на 3-метоксибензилхлорид, получали гидрохлорид 1-[4-амино-5-хлор-2-(5- метоксибензилокси)фенил] -5-(4-аминокарбонилпиперидин-1-ил)пентан- 1-она, tпл 204-205oC.
ПРИМЕР 13 2 мл) и смесь перемешивали при температуре окружающей среды в течение 48 часов. Дополнительно добавляли 3,5-диметоксибензилхлорид (0,05 г, 0,13 ммоля) и смесь перемешивали в течение 24 часов. Растворитель удаляли при пониженном давлении и к оставшимся твердым веществам добавляли метиленхлорид и водный бикарбонат натрия и эту двухслойную смесь перемешивали. Метиленхлоридный слой отделяли, дважды промывали водой и сушили над карбонатом калия. Путем фильтрации и концентрации получали неочищенный продукт (1,2 г).
Путем очистки с помощью хроматографии на силикагеле (3% CH3OH-CH2Cl2) получали 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил] -3-{ 1-[2- (метилсульфонил)аминоэтил] пиперидин-4-ил} пропан-1-он (0,53 г, 0,98 ммоля). Путем кристаллизации из этанольной соляной кислоты получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил]-3- {1-[2-(метилсульфонил)аминоэтил] пиперидин-4-ил}пропан-1-она (0,54 г, 0,93 ммоля), tпл 123-126oC.
Тем же способом, что и в примере 12, стадия (а), но заменяя 1-[4-амино-5-хлор-2-метоксифенил]-3-{1- [2-(метилсульфонил)аминоэтил]пиперидин-4-ил} пропан-1-он на 1-(4- амино-5-хлор-2-метоксифенил)-3-[1-(бут-1-ил)пиперидин-4-ил]пропан- 1-он, и затем в соответствии с примером 12, стадия (б), получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил]- 3-[1-(бут-1-ил)пиперидин-4-ил]пропан-1-она, tпл 179-182oC.
Тем же способом, что и в примере 12, стадия (а), но заменяя 1-[4-амино-5-хлор-2-метоксифенил]-3-{1-[2-(метилсульфонил)аминоэтил] пиперидин-4-ил} пропан-1-он на 1-(4-амино-5-хлор-2-метоксифенил)-3- [1-(пент-1-ил)пиперидин-4-ил] пропан-1-он, и затем в соответствии с примером 12, стадия (б), получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5- диметоксибензилокси)фенил]-3-[1-(пент-1-ил)пиперидин-4-ил] пропан-1-она, tпл 174-176oC.
Тем же способом, что и в примере 12, но заменяя 1-[4-амино-5-хлор-2-метоксифенил] -3-{1-[2- (метилсульфонил)аминоэтил]пиперидин-4-ил}пропан-1-он на 1-(4- амино-5-хлор-2-метоксифенил)-3-[1-(2-метоксиэтил) пиперидин-4-ил] пропан-1-он, и затем в соответствии с примером 12, стадия (б), получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил] – 3-[1-(2-метоксиэтил)пиперидин-4-ил]пропан-1-она, tпл 183-184oC.
Тем же способом, что и в примере 12, стадия (а), но заменяя 1-[4-амино-5-хлор-2-метоксифенил]-3-{1-[2- (метилсульфонил)аминоэтил]пиперидин-4-ил} пропан-1-он на 1-[4-амино-5-хлор-2-метоксифенил]-3-{1-[2- (диметиламиносульфонил)аминоэтил] пиперидин-4-ил}пропан-1-он, и затем в соответствии с примером 12, стадия (б), получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5-диметоксибензилокси)фенил] – 3-{1-[2-(диметиламиносульфонил)аминоэтил] пиперидин-4-ил} пропан-1-она, tпл 173-176oC.
Тем же способом, что и в примере 12, стадия (б), но заменяя 3,5-диметоксибензилхлорид на бензилхлорид, получали гидрохлорид 1-(4-амино-5-хлор-2-бензилоксифенил)-3-{ 1- [2-(метилсульфонил)аминоэтил] пиперидин-4-ил}пропан-1-она, пл 193-194oC.
Тем же способом, что и в примере 12, стадия (б), но заменяя 3,5-диметоксибензилхлорид на 3-метоксибензилхлорид, получали гидрохлорид 1-[4-амино-5-хлор-2-(3- метоксибензилокси)фенил]-3-{1-[2-(метилсульфонил)аминоэтил] пиперидин-4-ил}пропан-1-она, tпл 174-176oC.
Тем же способом, что и в примере 12, стадия (а), но заменяя 1-[4-амино-5-хлор-2- метоксифенил] -3-{ 1-[2-(метилсульфонил)аминоэтил]пиперидин-4- ил} пропан-1-он на 1-[4-амино-5-хлор-2-метоксифенил] -5- (пиперидин-1-ил)пентан-1-он, и затем в соответствии с примером 12, стадия (б), но заменяя 3,5-диметоксибензилхлорид на йодэтан, получали гидрохлорид 1-[4-амино-5-хлор-2-этоксифенил] -5- (пиперидин-1-ил)пентан-1-она, tпл 230-231oC.
Тем же способом, что и в примере 12, стадия (а), но заменяя 1-(4-амино-5-хлор-2- метоксифенил)-3-{ 1-[2-(метилсульфонил)аминоэтил] пиперидин-4- ил} пропан-1-он на 1-[4-амино-5-хлор-2-метоксифенил] -5- (пиперидин-1-ил)пентан-1-он, и затем в соответствии с примером 12, стадия (б), но заменяя 3,5-диметоксибензилхлорид на 1-бромпропан, получали гидрохлорид 1-[4-амино-5-хлор-2-(проп-1-илокси)фенил] -5- (пиперидин-1-ил)пентан-1-она, tпл 232-233oC.
Тем же способом, что и в примере 12, стадия (а), но заменяя 1-(4-амино-5-хлор-2- метоксифенил)-3-{1-[2-(метилсульфонил)аминоэтил]пиперидин-4-ил} пропан-1-он на 1-[4-амино-5-хлор-2-метоксифенил]-5-(пиперидин-1- ил)пентан-1-он, и затем в соответствии с примером 12, стадия (б), но заменяя 3,5-диметоксибензилхлорид на 3,5-диметилбензилхлорид, получали гидрохлорид 1-[4-амино-5-хлор-2-(3,5- диметилбензилокси)фенил] -5-(пиперидин-1-ил)пентан-1-она, tпл 218-233oC.
Тем же способом, что и в примере 12, стадия (а), но заменяя 1-[4-амино-5-хлор-2-метоксифенил]-3-{1-[2- (метилсульфонил)аминоэтил]пиперидин-4-ил} пропан-1-он на 1-(4-амино-5-хлор-2-метоксифенил)-5-(4- аминокарбонилпиперидин-1- ил)пентан-1-он, и затем в соответствии с примером 12, стадия (б), но заменяя 3,5-диметоксибензилхлорид на 3-метоксибензилхлорид, получали гидрохлорид 1-[4-амино-5-хлор-2-(5- метоксибензилокси)фенил] -5-(4-аминокарбонилпиперидин-1-ил)пентан- 1-она, tпл 204-205oC.
ПРИМЕР 131-(4-Амино-5-хлор-2-метоксифенил)-3-{ 1-[3-(4-метоксифенил)проп-1-ил] пиперидин-4-ил}пропан-1-он Ниже описано получение соединения формулы I, где R1 обозначает хлор, R2 обозначает водород, R3 обозначает метокси и R4 обозначает группу формулы (b), где p равно 0, q равно 2 и R7 обозначает 3-(4-метоксифенил)проп-1-ил. Стадия (а) Смесь 1-(4-амино-5-хлор-2- метоксифенил)-3-пиридин-4-илпропан-1-она (1,5 г, 5,16 ммоля), полученного согласно примеру 3 или 7, 3-(4-метоксифенил)-1- йодпропана (1,64 г, 5,93 ммоля) в 13 мл ацетонитрила нагревали в атмосфере азота с обратным холодильником в течение приблизительно 4 часов. Смесь концентрировали в вакууме и путем очистки остатка с помощью хроматографии на силикагеле (5% CH3OH-CH2Cl2 + 0,1% NH4OH) получали гидройодид 1-(4-амино-5-хлор-2-метоксифенил- 3-{1-[3-(4-метоксифенил)проп-1-ил]пиридинил-4-ил}пропан-1-она (1,82 г, 3,28 ммоля). Стадия (б) Раствор гидройодида 1-(4-амино-5- хлор-2-метоксифенил)-3-{1-[3-(4-метоксифенил)проп-1- ил]пиридинил-4-ил}пропан-1-она (1,82 г, 3,28 ммоля) в 30 мл ДМФ гидрогенизировали над катализатором из оксида платины (IV) (350 мг) при давлении 50 фунт/кв.дюйм. Реакционную смесь фильтровали и концентрировали при высоком вакууме. Остаток растворяли в 150 мл метиленхлорида и раствор разбавляли раствором гидроксида аммония. Метиленхлоридный слой отделяли и промывали ледяной водой (3 раза). Метиленхлоридый слой сушили над сульфатом натрия и концентрировали в вакууме. Путем кристаллизации остатка из этанольной соляной кислоты получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-3- {1-[3-(4-метоксифенил)проп-1-ил]пиперидин-4-ил}пропан-1-она (1,33 г, 3,09 ммоля), tпл 188-190oC. Тем же способом, что и в примере 13, стадия (а), но заменяя 3-(4-метоксифенил)-1-йодпропан на 3-(3,5-диметоксифенил)-1-йодпропан, и затем в соответствии с примером 13, стадия (б), получали гидрохлорид 1-(4-амино-5-хлор-2- метоксифенил)-3-{1-[3-(3,5-диметоксифенил)проп-1-ил]пиперидин- 4-ил} пропан-1-она, tпл 160-163oC. Тем же способом, что и в примере 13, стадия (а), но заменяя 3-(4-метоксифенил)-1-йодпропан на 3-(3,4-диметоксифенил)-1-йодпропан, и затем в соответствии с примером 13, стадия (б), получали гидрохлорид 1-(4-амино-5-хлор-2- метоксифенил)-3-{1-[3-(3,4-диметоксифенил)проп-1-ил]пиперидин-4- ил} пропан-1-она, tпл 177-179oC. Тем же способом, что и в примере 13, стадия (а), но заменяя 3-(4-метоксифенил)-1-йодпропан на 3-(3,4-этилендиоксифенил)-1-йодпропан, и затем в соответствии с примером 13, стадия (б), получали гидрохлорид 1-(4-амино-5-хлор-2- метоксифенил)-3-{1-[3-(3,4-этилендиоксифенил)проп-1- ил]пиперидин-4-ил}пропан-1-она, tпл 168-170oC. Тем же способом, что и в примере 13, стадия (а), но заменяя 3-(4-метоксифенил)-1-йодпропан на 3-(3,4-метилендиоксифенил)-1- йодпропан, и затем в соответствии с примером 13, стадия (б), получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-3-{ 1-[3- (3,4-метилендиоксифенил)проп-1-ил]пиперидин-4-ил}пропан-1-она, tпл 200-202oC. Тем же способом что и в примере 13, стадия (а), но заменяя 3-(4-метоксифенил)-1-йодпропан на 3-(3,4,5- триметоксифенил)-1-йодпропан, и затем в соответствии с примером 13, стадия (б), получали гидрохлорид 1-(4-амино-5-хлор-2- метоксифенил)-3-{1-[3-(3,4,5-триметоксифенил)проп-1- ил]пиперидин-4-ил}пропан-1-она, tпл 180-182oC. Тем же способом,что и в примере 13, стадия (а), но заменяя 1-(4-амино-5-хлор-2- метоксифенил)-3-пиридин-4-илпропан-1-он на 1-(4-амино-5-хлор-2,3- этилендиоксифенил)-3-пиридин-4-илпропан-1-он, и затем в соответствии с примером 13, стадия (б), получали гидрохлорид 1-(4- амино-5-хлор-2,3-этилендиоксифенил)-3-{ 1-[3-(4- метоксифенил)проп-1-ил]пиперидин-4-ил}пропан-1-она, tпл 188-189oC. ПРИМЕР 14 1-(4-Амино-5-хлор-2-метоксифенил)-5-пиперидин-1-илпентан-1-о Ниже описано получение соединения формулы I, где R1 обозначает хлор, R2 обозначает водород, R3 обозначает метокси и R4 обозначает группу формулы (а), где p равно 0, n равно 4 и R5 и R6 вместе обозначают пиперидин-1-ил. Стадия (а) Магний (236 г, 9,71 моля) суспендировали в 3,5 л ТГФ в атмосфере азота и добавляли 1-бром-4-хлорбутан (1,11 л, 8,68 М, 9,63 моля) с такой скоростью, чтобы температура смеси оставалась ниже 25oC. Раствор N-метокси-N- метил-4-амино-5-хлор-2-метоксибензамида (400 г, 1,64 моля), полученного в соответствии с примером 2, охлаждали до -20oC в атмосфере азота в 4 л ТГФ и добавляли 410 мл хлортриметилсилана. Смеси давали нагреться до температуры между -12 и 30oC и затем в течение приблизительно 20 минут добавляли 2,5 л раствора, содержащего магний и 1-бром-2-хлорбутан. Смесь охлаждали до 5oC и затем разбавляли 420 мл концентрированной соляной кислоты в 5 л воды. Смесь нагревали до 32oC, перемешивали в течение 15 минут и затем разбавляли приблизительно 2 л этилацетата. Органический слой отделяли и промывали последовательно 2 л воды и 2 л смеси 1:1 воды/насыщенного раствора хлорида натрия и насыщенным раствором хлорида натрия. Все водные слои объединяли и экстрагировали этилацетатом. Все органические слои объединяли, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток смешивали с горячим гексаном и оставляли стоять при комнатной температуре в течение приблизительно 12 часов, получая осадок. Осадок выделяли с помощью фильтрации и промывали гексаном. Путем сушки в вакуумной печи при температуре между 50 и 55oC при продувании газообразным азотом получали 1-(4-амино-5-хлор-2- метоксифенил)-5-хлорпентан-1-он (365 г, 1,33 моля). Стадия (б) Смесь 1-(4-амино-5-хлор-2-метоксифенил)-5-хлорпентан-1-она (365 г, 1,33 моля), пиперидина (620 г, 7 молей) и йодистого натрия (30 г, 0,2 моля) в 1 л ДМФ выдерживали между 78 и 82oC в течение 4 часов. Реакционной смеси давали охладиться до 50oC, затем перемешивали в течение приблизительно 12 часов и добавляли 5 л воды. Смесь перемешивали при 25-30oC в течение 1 часа, получая осадок, и осадок выделяли с помощью фильтрации. Осадок промывали 4 л воды и сушили с отсасыванием в течение 1 часа. Осадок растворяли приблизительно в 2 л этилацетата при нагревании до приблизительно 50oC и затем добавляли 130 мл раствора концентрированной соляной кислоты в общем объеме 4 л воды. Смесь перемешивали в течение 30 минут и затем перемешивали в течение 3 часов в водно-ледяной бане, получая осадок. Осадок выделяли с помощью фильтрации и промывали 1 л холодной смеси 1:1 воды/этилацетата и затем 1 л этилацетата. Осадок сушили в вакуумной печи при температуре между 45 и 50oC при продувании газообразным азотом в течение 24 часов, при 50-55oC в течение 24 часов и затем при комнатной температуре в течение 48 часов. Сухой осадок растворяли приблизительно в 9 л кипящего метанола. Раствор фильтровали и затем перегоняли в вакууме до объема приблизительно 4 л. Добавляли реактивный спирт и смесь перегоняли при атмосферном давлении до точки кипения между 72 и 74oC и до общего объема приблизительно 5 л. Оставшуюся смесь перемешивали при комнатной температуре в течение приблизительно 12 часов и затем в ледяной бане в течение 4 часов, получая осадок. Осадок выделяли с помощью фильтрации и промывали приблизительно 1,5 л спирта. Выделенный осадок сушили с отсасыванием в течение 2 часов и затем в вакуумной печи при температуре между 55 и 60oC при продувании газообразным азотом в течение приблизительно 12 часов и сухой осадок пропускали через клиническое сито 10 размера. Путем дальнейшей сушки в вакуумной печи при температуре между 55 и 60oC при продувании газообразным азотом в течение приблизительно 48 часов получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-5-пиперидин-1-илпентан-1-она (357 г, 0,99 моля), tпл 220-222oC. Тем же способом, что и в примере 14, но заменяя пиперидин на 4-метилпиперидин, получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-5-(4-метилпиперидин-1-ил)пентан- 1-она, tпл 197-198oC. ПРИМЕР 15 1-(4-Амино-5-хлор-2-метоксифенил)-5-пиперидин-1-илпентан-1-о Ниже описано получение соединения формулы I, где R1 обозначает хлор, R2 обозначает водород, R3 обозначает метокси и R4 обозначает группу формулы (а), где p равно 0, n равно 4 и R5 и R6 вместе обозначают пиперидин-1-ил. Суспензию гидрохлорида 5-(пиперидин-1-ил)валериановой кислоты (6,6 г, 30 молей) в 100 мл ТГФ охлаждали в ледяной бане и добавляли 100 мл 1 н. литийбис(триметилсилил)амида в ТГФ. Ледяную баню удаляли и получившийся раствор перемешивали при комнатной температуре в течение 1 часа. Смесь метил-2-метокси-4-амино-5-хлорбензоата (2,15 г, 10 ммолей) и хлортриметилсилана (2,5 мл, 20 ммолей) в 25 мл ТГФ охлаждали в ледяной бане и добавляли 20 мл 1 н. литийбис(триметилсилил)амида в ТГФ. Раствор, содержащий 5-(пиперидин-1-ил)валериановую кислоту и 1 н. литийбис(триметилсилил)амид, охлаждали в ледяной бане и в течение 5 минут добавляли смесь, содержащую метил-2-метокси-4-амино-5- хлорбензоат. Реакционную смесь удаляли из ледяной бани и перемешивали при 50-55oC в течение 2 часов и затем добавляли раствор 25 мл концентрированной соляной кислоты в 175 мл воды. Смесь перемешивали при 50-55oC в слабом потоке азота и затем добавляли 120 мл этилацетата. Смесь перемешивали в ледяной бане в течение 1 часа, получая осадок. Осадок выделяли путем фильтрации и промывали этилацетатом. Осадок перемешивали приблизительно в 40 мл кипящего изопропанола в течение 30 минут и затем в течение 1 часа в ледяной бане. Осадок выделяли путем фильтрации и промывали изопропанолом. Путем сушки в вакуумной печи при температуре между 55 и 60oC при продувании газообразным азотом в течение 18 часов получали гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-5- пиперидин-1-илпентан-1-она (2,9 г, 8 ммолей). ПРИМЕР 16 Ниже представлены характерные фармацевтические композиции, содержащие соединение формулы I. КОМПОЗИЦИЯ ДЛЯ ОРАЛЬНОГО ВВЕДЕНИЯ Характерный раствор для орального введения включает: Соединение формулы I – 100-1000 мг Моногидрат лимонной кислоты – 105 мг Гидроксид натрия – 18 мг Корригент – Вода – д.к. до 100 мл КОМПОЗИЦИЯ ДЛЯ ВНУТРИВЕННОГО ВВЕДЕНИЯ Характерный раствор для внутривенного введения включает: Соединение формулы I – 10-100 мг Моногидрат декстрозы – д.к. до изотоничности Моногидрат лимонной кислоты – 1,05 мг Гидроксид натрия – 0,18 мг Вода для инъекции – д.к. до 1,0 мл КОМПОЗИЦИЯ ДЛЯ ТАБЛЕТКИ Характерная таблетированная форма соединения формулы I может содержать: Соединение формулы I – 1% Микрокристаллическая целлюлоза – 73% Стеариновая кислота – 25% Коллоидный кремний – 1% ПРИМЕР 17 Исследование 5-НТ4-рецептора торакальной части пищевода Ниже описан метод изучения in vitro с использованием выделенной мышечной слизистой оболочки пищевода крысы для идентификации испытываемых соединений, которые являются лигандами 5-НТ4-рецептора. Торакальные части пищеводов выделяли из самцов крыс линии Sprague-Dawley и помещали в раствор Тироде. Внешнюю поперечно-полосатую мышцу удаляют для обнажения мышечной слизистой оболочки. Каждую слизистую оболочку подвешивали вертикально в 10-миллилитровой тканевой ванне, содержащей метисергид (1 мкМ), кокаин (30 мкМ) и кортикостерон (30 мкМ) в растворе Тироде, поддерживая температуру 37oC и постоянно продувая газовой смесью 95% O2 и 5% CO2. К каждому образцу ткани прилагают постоянное растягивающее натяжение в 1 г и после этого через 15-минутные интервалы повторно прикладывают по 0,5 г. Под действием карбахола (3 мкМ) достигают стационарно сокращенного состояния и затем ткань обрабатывают 5-НТ в условиях кумулирования концентрации, которая повышается до достижения максимальной или близкой к максимальной релаксации. 5-НТ дает зависящую от концентрации релаксацию мышечной ткани слизистой оболочки, где функцию медиатора выполняет 5-НТ4. Ткань подвергают действию раствора Тироде без лекарства в течение 30 минут, а затем вновь сокращают с помощью карбахола. Затем ткань подвергают действию испытываемого соединения. Если само испытываемое соединение не вызывает релаксацию мышечной ткани слизистой оболочки пищевода, ткань подвергают действию 5-НТ в присутствии испытываемого соединения. Соединения, которые в действительности вызывают релаксацию, характеризуются как агонисты 5-НТ4-рецептора. Соединения, которые ингибируют реакцию релаксации, вызванную 5-НТ, характеризуются как антагонисты 5-НТ4-рецептора. С помощью метода, описанного в примере 17, получили следующие результаты для соединений по настоящему изобретению. Соединение – Константа сродства (-log) А – 7,4 Б – 7,5 В – 8,4 Г – 8,2 Д – 8,8 Е – 10,6 А: гидрохлорид 1-(4-амино-5- хлор-2-метоксифенил)-5-(пиперидинил-1-ил)пентан-1-она Б: гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-5-(4-метилпиперидинил-1- ил)пентан-1-она В: гидрохлорид 1-(4-амино-5-хлор-2- метоксифенил)-3-{2-[(метилсульфонил)амино]этил}пиперидин-4- ил)пропан-1-она Г: гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)- 3-[1-(н-бутил)пиперидин-4-ил]пропан-1-она Д: гидрохлорид 1-(4- амино-5-хлор-2-метоксифенил)-3-{ 1-[3-(3,4-диметоксифенил)проп-1-ил]пиперидин-4-ил}пропан-1-она E: 3-(4-метоксифенил)проп-1-ил, а именно гидрохлорид 1-(4-амино-5- хлор-2,3-этилендиоксифенил)-3- {1-[3-(4-метоксифенил)-проп-1-ил]пиперидин-4-ил}пропан-1-она ПРИМЕР 18 Исследование прокинетической активности Ниже описан метод исследования in vivo, позволяющий определить прокинетическую активность путем измерения степени воздействия лекарства на скорость опорожнения желудка крыс от пробного завтрака. Этот метод описан Droppleman и др., упомянутым выше. Пробный завтрак приготавливают, медленно добавляя 20 г целлюлозной смолы (Hercules Inc. , Вилмингтон, Делавэр) к 200 мл холодной дистиллированной воды, смешивая в гомогенизаторе Уоринга при приблизительно 20000 об/мин. Смешивание продолжают до тех пор, пока не произойдет полное диспергирование и гидратация целлюлозной смолы (приблизительно через 5 минут). Три мясных бульонных кубика растворяют в 100 мл теплой воды и затем смешивают с целлюлозным раствором, после чего добавляют 16 г очищенного казеина (Sigma Chemical Co. , Сент-Луис, МО), 8 г кондитерской сахарной пудры, 8 г кукурузного крахмала и 1 г угольного порошка. Каждый ингредиент добавляют медленно и тщательно смешивают, получая в результате приблизительно 325 мл гомогенной пасты темно-серого или черного цвета. Эту пищу затем выдерживают в холодильнике в течение ночи, и за это время поглощенный воздух улетучивается. Перед экспериментом пищу вынимают из холодильника и дают нагреться до комнатной температуры. Самцов крыс линии Sprague-Dawley (весом 170-204 г), достигших полного развития, лишают пищи в течение 24 часов, позволяя по желанию пить воду. Утром в день эксперимента каждое животное взвешивают и распределяют случайным образом по предназначенным для обработки группам по десять животных в каждой. Каждая крыса получает путем внутрибрюшинной инъекции либо наполнитель, либо пробный завтрак, либо в качестве стандарта метоклопрамид. Через 0,5 часа после инъекции с помощью 5,0-миллилитрового одноразового шприца каждой крысе оральным путем вводят 3,0 мл пробного завтрака. Пять образцов пробного завтрака взвешивают на аналитических весах, и эти веса усредняют для определения среднего веса пробного завтрака. Через 1,5 часа после инъекции каждую крысу умерщвляют путем асфикции двуокисью углерода и извлекают желудок путем вскрытия брюшного отдела, тщательно зажимая и отсекая пищевод точно ниже пилорического сфинктера. Приняв меры предосторожности, чтобы избежать любой потери его содержимого, каждый желудок помещают на маленькую, предварительно взвешенную и соответственно помеченную 7 мл ванночку для взвешивания и сразу взвешивают на аналитических весах. Каждый желудок рассекают вдоль по линии меньшей кривизны, промывают водопроводной водой, аккуратно промокают досуха для удаления избытка влаги и взвешивают. Оставшееся в желудке количество пробного завтрака определяют как разницу между весом полного желудка и весом опорожненного желудка. Разница между оставшимся количеством пробного завтрака и средним весом пробного завтрака представляет собой количество пробного завтрака, которое вышло в течение 1,5 часов после инъекции. Результаты представляют в виде граммов вышедшей пищи или процентов изменения по отношению к контролю. Средние величины и стандартные отклонения в испытываемых группах сравнивают с таковыми в контрольных группах. Значимость определяют с помощью t-теста Дуннета (Statistical Association Journal, December 1955, 1096-112). С помощью метода, описанного в примере 18, установили, что соединения по настоящему изобретению обладают прокинетической активностью. ПРИМЕР 19 Исследование антифобического поведения Ниже описан метод определения in vivo антифобической активности путем измерения степени влияния лекарства на естественный страх мышей, помещенных в новое, ярко освещенное окружение. Незараженных самцов мышей линии C5BI/6J, весом 18-20 г, держали группами по 10 мышей в клетках с контролируемыми шумом, температурой и влажностью. Корм и вода были доступны по желанию. Мышей содержали при следующем световом цикле: 12 часов света и 12 часов темноты, причем свет включают в 6.00 часов утра, а выключают в 6.00 часов вечера. Все эксперименты начинают по меньшей мере через 7 дней после доставки на место. Автоматическая аппаратура для определения изменений исследовательской активности была приобретена у фирмы Omni-Tech Electronics Columbus Ohio, она подобна той, которая использовалась Crawley и Goodwin (1980), как описано у упомянутых выше Kilfoil и др. Вкратце, камера состоит из плексигласового ящика (44 х 21 х 21 см), разделенного на две камеры с помощью темной плексигласовой перегородки. В перегородке, разделяющей две камеры, имеется отверстие размером 13 х 5 см, через которое мышь может легко проходить. Темная камера имеет прозрачные стенки и белый пол. Единственное освещение дает флуоресцентная лампа (40 Вт), помещенная над камерами. Система текущего контроля активности животных Digiscan Animal Activity Monitor System RXYZCM16 (фирмы Omni-Tech Electronics) регистрирует исследовательскую активность мышей внутри тест-камер. Перед началом опыта мышам позволяли в течение 60 минут акклиматизироваться к лабораторным условиям. После того, как мыши получали путем внутрибрюшинной инъекции либо испытываемое соединение, либо наполнитель, ее возвращали в ее исходный садок на 15 мин после обработки. Затем мышь помещали в центр освещенной камеры и контролировали в течение 10 минут. Антифобия проявляется как общее увеличение исследовательской активности в освещенной области. Увеличение исследовательской активности проявляется в увеличении латентного состояния (промежутка времени, требуемого для того, чтобы мышь начала двигаться в темную камеру, когда ее впервые помещают в центр освещенной области), увеличении челночной активности, увеличении или не изменении локомоторной активности (количества пересеченных линий разметки) и уменьшении времени, проведенном в темном отделении. ПРИМЕР 20 Исследование страха при синдроме отмены Ниже описана методика определения in vivo уменьшения интенсивности симптомов, вызванных отменой наркотических веществ, путем измерения степени воздействия лекарства на страх, который возникает у мышей после хронического введения наркотического вещества и затем резкого прекращения этих обработок. Незараженных самцов мышей линии BKW (25-30 г) содержали группами по 10 особей в клетках с контролируемыми шумом, температурой и влажностью. Корм и вода доступны по желанию. Мышей содержали при следующем световом цикле: 12 часов света и 12 часов темноты, причем свет включали в 6.00 часов утра, а выключали в 6.00 часов вечера. Все эксперименты начинали по меньшей мере через 7 дней после доставки на место. Уровни страха определяли с помощью двухкамерной исследовательской модели Crawley и Goodwin (см. пример 19). Антифобия проявляется как общее увеличение исследовательской активности в освещенной области. Увеличение исследовательской активности проявляется в увеличении латентного состояния (промежутка времени, требуемого для того, чтобы мышь начала двигаться в темную камеру, когда ее впервые помещают в центр освещенной области), увеличении или не изменении локомоторной активности (количества пересеченных линий разметки), увеличении числа подъема на задние лапы и уменьшении времени, проведенном в темном отделении. Увеличение исследовательской активности в освещенной области вызывается путем введения мышам в течение 14 дней этанола (8,0% в отношении веса к объему в питьевой воде), никотина (0,1 мг/кг, внутрибрюшинно, дважды в день) или кокаина (1,0 мг/кг, внутрибрюшинно, дважды в день). Антифобию оценивали через 1, 3, 7 и 14 дней после начала применения наркотика. Применение резко прекращали и через 8, 24 и 48 часов после этого определяли исследовательскую активность в освещенной области. Наполнитель или испытываемые соединения вводили во время фазы отмены путем внутрибрюшинной инъекции. Ответные реакции оценивали по ингибированию снижения антифобного поведения после прекращения введения этанола, кокаина или никотина. ПРИМЕР 21 Исследование увеличения познавательной способности Ниже описана модель для определения активности увеличения познавательных способностей путем измерения степени, в какой испытываемое соединение способно скомпенсировать дефицит познавательной способности, вызванный атропином (30 мг/кг, внутрибрюшинно) с использованием водного лабиринта Морриса. Крыс линии Sprague Dawley (240-260 г) держали в лаборатории в течение ночи перед испытанием и оставляли там же в течение всего эксперимента. Водный лабиринт Морриса состоял из круглого бассейна, выполненного из черного плексигласа (диаметром 122 см, высотой 46 см, с 15-сантиметровым ободом), наполненного непрозрачной водой на высоту 35 см. Скрытую платформу, выполненную из черного плексигласа, помещали на 1-2 см ниже поверхности воды. Бассейн разделяли на четыре квадранта, условно соответствующих северу, югу, востоку и западу. Платформу помещали в южном квадранте, приблизительно на рассеянии 24 см от края. В комнате помещали предметы с большой контрастностью, служащие в качестве пространственных ориентиров. Телевизионная камера отслеживала пути, по которым плавали крысы, и получаемые таким образом данные анализировали для определения времени в секундах, необходимого крысам для того, чтобы найти платформу (латентность избегания опасности). Тесты начинали, помещая крысу в один из четырех квадрантов мордочкой к стене. Испытания состояли из блока из шести опытов (первый начинался в северном квадранте, затем в восточном, южном, западном, северном и, наконец, снова в восточном) в каждый из двух последовательных дней. В течение каждого опыта крысе давали 90 секунд, чтобы найти платформу. Если крыса успешно находила платформу, ей давали 30 секунд для “изучения” пространственных ориентиров. Если крысе не удавалось найти платформу в течение 90 секунд, ей начисляли счет в 90 секунд и помещали на платформу на 30 секунд. Использовали следующие группы по 8 крыс в каждой: 1) контрольные, обработанные наполнителем; 2) контрольные, обработанные атропином; 3) обработанные атропином плюс испытываемым лекарством. Таким образом, данные исследования предназначались для определения, могло ли испытываемое лекарство скомпенсировать дефицит познавательной способности, вызванный атропином (30 мг/кг, внутрибрюшинно). Статистические методы анализа применяли для изучения гетерогенности кривых обучаемости и разделения кривых обучаемости. Следуя процедуре, изложенной в примере 21, выявляли соединения по настоящему изобретению, обладающие свойствами повышения познавательной способности. Например, гидрохлорид 1-(4- амино-5-хлор-2-метоксифенил)-5-(пиперидинил-1-ил)пентан-1-она и гидрохлорид 1-(4-амино-5-хлор-2-метоксифенил)-5-(4- метилпиперидинил-1-ил)пентан-1-она оказались эффективными после внутрибрюшинного введения (10 нг/кг-1 мг/кг) в данном исследовании. ПРИМЕР 22 Токсичность Крысам вводили орально дозы 5, 15 и 50 мг/кг/день гидрохлорида 1-(4-амино-5-хлор-2- метоксифенил)-5-(пиперидинил-1-ил) пентан-1-она в течение одного месяца. Не было выявлено никаких патологических изменений, связанных с обработкой любой из указанных доз. Хотя настоящее изобретение описано в отношении конкретных примеров выполнения изобретения, специалистам в данной области техники понятно, что могут быть внесены различные изменения и могут быть заменены эквиваленты без отступления от истинной сущности и объема изобретения. Подразумевается, что все такие модификации подпадают под объем приложенной формулы изобретения. Пример 23 1-(4-Амино-5-хлор-2-метоксифенил)-5-пиперидин-1-илпентан-1-о Раствор N-оксида 1-(4-амино-5-хлор-2-метоксифенил)-5- пиперидин-1-илпентан-1-она (3,38 г, 10 ммоль) и комплекса триметиламин-диоксид серы (3,02 г, 20 ммоль) в 25 мл диоксана нагревали при кипячении с обратным холодильником в течение 2,5 часа. Смесь охлаждали до комнатной температуры, разбавляли водой и подщелачивали карбонатом калия. Смесь экстрагировали этилацетатом и экстракт промывали водой, сушили над сульфатом натрия и упаривали до получения сырого свободного основания. Превращение в гидрохлорид при помощи HCl в изопропаноле приводил к получению гидрохлорида 1-(4-амино-5-хлор-2-метоксифенил)-5-пиперидин-1- илпентан-1-она (2,40 г, 6,7 ммоля), tпл 220-222oC. Формула изобретения
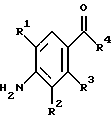 где R1 обозначает галоген; R2 обозначает водород или С1-С4алкокси; R3 обозначает С1-С4алкокси или фенилС1-С4алкокси (где фенил необязательно замещен одним-тремя заместителями, независимо выбранными из С1-С4алкила, С1-С4алкилокси, 3,4-метилендиокси), или R2 и R3 вместе обозначают метилендиокси или этилендиокси; R4 обозначает группу формулы (а) или (b) 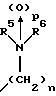 или 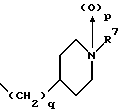 где n равно 3, 4 или 5; p равно 0; q равно 1 или 2; R5 и R6 каждый обозначает С1-С4алкил или вместе образуют -(СН2)4-, -(СН2)6-, -(СН2)2О(СН2)2– или -CHR8CH2CR9R10CHR11CH2-, где R8 и R11 каждый обозначает водород или вместе обозначают -(СН2)t-, где t равно 1, R9 обозначает водород, гидроксил, С1-С8алкил, или С1-С4алкилокси; R10 обозначает водород, С1-С8алкил или фенил, или -(CH2)xR12, где х равно 0, 1, 2 или 3 и R12 обозначает гидроксил, С1-С4алкилокси, -C(O)NR13R14, -NR13C(O)OR14, -SO2NR13R14, -NR13SO2R14, -NR13SO2NR14R15 или -NR13C(O)NR14R15, где R13, R14 и R15 независимо обозначают водород, С1-С4алкил, трифторметил; R7 обозначает водород, С1-С8алкил, С3-С8алкенил, или фенилС1-С4алкил (где фенил необязательно замещен одним-тремя заместителями, независимо выбранными из С1-С4алкилокси, метилендиокси, этилендиокси), или -(CH2)zR12, где z равно 2 или 3 и R12 имеет значения, указанные выше, и его фармацевтически приемлемые соли, отдельные изомеры и смеси изомеров. 2. Соединение по п.1, имеющее следующую формулу I(a): 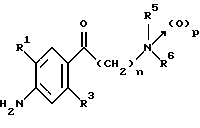 где n равно 3, 4 или 5; p равно 0; R1 обозначает галоген; R3 обозначает С1-С4алкилокси; R5 и R6 каждый обозначает С1-С4алкил или вместе образуют -(СН2)4-, -(СН2)6-, -(CH2)2O(CH2)2– или -CHR8CH2CR9R10CHR11CH2-, где R8 и R11 каждый обозначает водород или вместе обозначают -(СН2)t-, где t равно 1, R9 обозначает водород, гидроксил, С1-С8алкил или C1-C4-алкилокси и R10 обозначает водород, С1-С8алкил или фенил, или -(СН2)xR12, где х равно 0, 1, 2 или 3 и R12 обозначает гидроксил, С1-С4алкилокси, -C(O)NR13R14, -NR13C(O)OR14, -SO2NR13R14, -NR13SO2R14, -NR13SO2NR14R15 или -NR13C(O)NR14R15, где R13, R14 и R15 независимо обозначают водород, C1-С4алкил или трифторметил, и его фармацевтически приемлемые соли, отдельные изомеры и смеси изомеров. 3. Соединение по п. 2, где p равно 0, a R5 и R6 вместе образуют -CHR8CH2CR9R10CHR11СН2-. 4. Соединение по п.3, где n равно 4. 5. Соединение по п.4, где R1 обозначает хлор, R3 обозначает метокси и каждый из R8, R9 и R11 обозначает водород. 6. Соединение по п.5, где R10 обозначает водород, а именно 1-(4-амино-5-хлор-2-метоксифенил)-5-(пиперидинил-1-ил)пентан-1-он и его фармацевтически приемлемые соли. 7. Соединение по п. 6, которое является гидрохлоридом 1-(4-амино-5-хлор-2-метоксифенил)-5-(пиперидинил-1-ил)пентан-1-она. 8. Соединение по п.5, где R10 обозначает метил, а именно, 1-(4-амино-5-хлор-2-метоксифенил)-5-(4-метилпиперидинил-1-ил)пентан-1-он и его фармацевтически приемлемые соли. 9. Соединение по п. 8, которое является гидрохлоридом 1-(4-амино-5-хлор-2-метоксифенил)-5-(4-метилпиперидинил-1-ил)пентан-1-она. 10. Соединение по п.1, имеющее следующую формулу I(b): 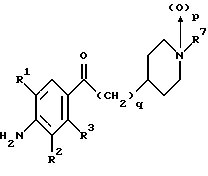 где p равно 0; q – равно 1 или 2; R1 обозначает галоген; R2 обозначает водород или С1-С4алкилокси, и R3 обозначает С1-С4алкилокси или R2 и R3 вместе обозначают метилендиокси или этилендиокси; R7 обозначает водород, С1-С8алкил, С3-С8алкенил, или фенилС1-С4алкил, где фенил необязательно замещен одним-тремя заместителями, независимо выбранными из С1-С4алкилокси, метилендиокси, этилендиокси, или -(CH2)zR12, где z равно 2 или 3 и R12 обозначает гидроксил, С1-С4алкилокси, -C(O)NR13R14, -NR13C(O)OR14, -SO2NR13R14, -NR13SO2R14, -NR13SO2NR14R15 или -NR13C(O)NR14R15, где R13, R14 и R15 независимо обозначают водород, C1-С4алкил, трифторметил, и его фармацевтически приемлемые соли, отдельные изомеры и смеси изомеров. 11. Соединение по п.10, где q равно 2. 12. Соединение по п.11, где R1 обозначает хлор, R2 обозначает водород и R3 обозначает метокси. 13. Соединение по п.12, где p равно 0 и R7 обозначает н-бутил, а именно 1-(4-амино-5-хлор-2-метоксифенил)-3-[1-(н-бутил)пиперидин-4-ил]пропан-1-он и его фармацевтически приемлемые соли. 14. Соединение по п.13, которое является гидрохлоридом 1-(4-амино-5-хлор-2-метоксифенил)-3-[1-(н-бутил)пиперидин-4-ил]пропан-1-она. 15. Соединение по п.12, где p равно 0 и R7 обозначает 2-[(метилсульфонил)амино] этил, а именно 1-(4-амино-5-хлор-2-метоксифенил)-3-{ 2-[(метилсульфонил)амино] этил} пиперидин-4-ил]пропан-1-он и его фармацевтически приемлемые соли. 16. Соединение по п.15, которое является гидрохлоридом 1-(4-амино-5-хлор-2-метоксифенил)-3-{2-[(метилсульфонил)амино]этил}пиперидин-4-ил]пропан-1-она. 17. Соединение по п.12, где p равно 0 и R7 обозначает 3-(3,4-диметоксифенил)проп-1-ил, а именно 1-(4-амино-5-хлор-2-метоксифенил)-3-{ 1-[3-(3,4-диметоксифенил)проп-1-ил]пиперидин-4-ил}пропан-1-он и его фармацевтически приемлемые соли. 18. Соединение по п.17, которое является гидрохлоридом 1-(4-амино-5-хлор-2-метоксифенил)-3-{ 1-[3-(3,4-диметоксифенил)проп-1-ил] пиперидин-4-ил} пропан-1-она. 19. Соединение по п.11, где R1 обозначает хлор и R2 и R3 вместе обозначают этилендиокси. 20. Соединение по п.19, где p равно 0 и R7 обозначает 3-(4-метоксифенил)проп-1-ил, а именно 1-(4-амино-5-хлор-2,3-этилендиоксифенил)-3-{1-[3-(4-метоксифенил)проп-1-ил] пиперидин-4-ил} пропан-1-он и его фармацевтически приемлемые соли. 21. Соединение по п.20, в котором R7 является 3-(4-метоксифенил)проп-1-илом, а именно, гидрохлорид 1-(4-амино-5-хлор-2,3-этилендиоксифенил)-3-{ 1-[3-(4-метоксифенил)проп-1-ил]пиперидин-4-ил}пропан-1-она. 22. Соединение по п.1, имеющее следующую формулу I(с) 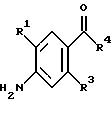 где R1 обозначает галоген; R3 обозначает фенилC1-C4алкокси, где фенил необязательно замещен одним-тремя заместителями, независимо выбранными из C1-C4алкила, C1-C4алкилокси, 3,4-метилендиокси; R4 обозначает группу формулы (а) или (b) 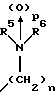 или 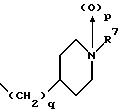 где n равно 3, 4 или 5; p равно 0; q равно 1 или 2; R5 и R6 каждый обозначает C1-C4алкил или вместе образуют -(СН2)4-, -(СН2)6-, -(CH2)2O(CH2)2– или -CHR8CH2CR9R10CHR11CH2-, где R8 и R11 каждый обозначает водород или вместе обозначают -(СН2)t-, где t равно 1, R9 обозначает водород, гидроксил, C1-C8алкил или C1-C4алкилокси и R10 обозначает водород, C1-C8алкил или фенил, или -(CH2)xR12, где х равно 0, 1, 2 или 3 и R12 обозначает гидроксил, C1-C4алкилокси, -C(O)NR13R14, -NR13C(O)OR14, -SO2NR13R14, -NR13SO2R14, -NR13SO2NR14R15 или -NR13C(O)NR14R15, где R13, R14 и R15 независимо обозначают водород, C1-C4алкил или трифторметил; R7 обозначает водород, C1-C8алкил, или C3-C8алкенил, или фенилC1-C4алкил, где фенил необязательно замещен одним-тремя заместителями, независимо выбранными из C1-C4алкилокси, метилендиокси, этилендиокси, или -(СН2)zR12, где z равно 2 или 3 и R12 имеет значения, указанные выше, и его фармацевтически приемлемые соли, отдельные изомеры и смеси изомеров. 23. Соединение по п.22, где R3 обозначает необязательно замещенный фенилС1-С4алкилокси и R4 обозначает группу формулы (а). 24. Соединение по п.23, где R5 и R6 вместе образуют -CHR8CH2CR9R10CHR11СН2-. 25. Соединение по п.24, где n равно 4. 26. Соединение по п.22, где R3 обозначает необязательно замещенный фенилС1-С4алкилокси и R4 обозначает группу формулы (b). 27. Соединение по п.26, где q равно 2. 28. Производные фенилалканонов формулы I по пп. 1 – 27, обладающие сродством к 5-НТ4-рецепторам. 29. Фармацевтическая композиция для лечения состояний, которые могут быть облегчены воздействием на 5-НТ4 рецепторы, включающая активный ингредиент, отличающаяся тем, что в качестве последнего используют соединение формулы I по пп.l – 27 в терапевтически эффективном количестве. 30. Способ лечения состояний, которые могут быть облегчены введением средства, взаимодействующего с 5-НТ4-рецепторами животного, отличающийся тем, что животному вводят соединения формулы I по пп.1 – 27 в терапевтически эффективном количестве. 31. Способ по п. 29, в котором состояние выбрано из расстройства ЦНС, расстройства желудочно-кишечного тракта, расстройства сердечно-сосудистой системы и расстройства мочевых путей. 32. Способ получения соединения формулы I 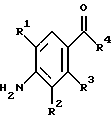 где R1 обозначает галоген; R2 обозначает водород или C1-С4алкилокси и R3 обозначает C1-С4алкилокси или фенилC1-С4алкилокси, где фенил необязательно замещен одним-тремя заместителями, независимо выбранными из C1-С4алкила, C1-С4алкилокси, 3,4-метилендиокси, или R2 и R3 вместе обозначают метилендиокси или этилендиокси; R4 обозначает группу формулы (а) или (b) 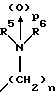 или 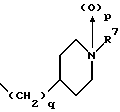 где n равно 3, 4 или 5; p равно 0; q равно 1 или 2; R5 и R6 каждый обозначает C1-С4алкил или вместе образуют -(СН2)4-, -(СН2)6-, -(СН2)2O(СН2)2– или -CHR8CH2CR9R10CHR11CH2-, где R8 и R11 каждый обозначает водород или вместе обозначают -(СН2)t-, где t равно 1, R9 обозначает водород, гидроксил, C1-С8алкил, или C1-С4алкилокси и R10 обозначает водород, С1-С8алкил или фенил, или -(CH2)xR12, где х равно 0, 1, 2 или 3, и R12 обозначает гидроксил, C1-С4алкилокси, -C(O)NR13R14, -NR13C(O)OR14, -SO2NR13R14, -NR13SO2R14, -NR13SO2NR14R15 или -NR13C(O)NR14R15, где R13, R14 и R15 независимо обозначают водород, C1-С4алкил, трифторметил и R7 обозначает водород, C1-С8алкил, или C3-С8алкенил, или фенилC1-С4алкил, (где фенил необязательно замещен одним-тремя заместителями, независимо выбранными из C1-С4алкилокси, метилендиокси, этилендиокси) или -(CH2)zR12, где z равно 2 или 3 и R12 имеет значения, указанные выше, и его фармацевтически приемлемых солей, отдельных изомеров и смесей изомеров, отличающийся тем, что соединение формулы II 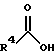 или его защищенное производное, где R4 имеет значения, указанные выше, подвергают взаимодействию с соединением формулы III 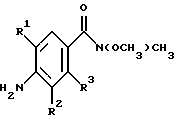 где каждый из R1, R2 и R3 имеет значения, указанные в выше, с последующим последовательным подкислением, декарбоксилированием, в случае необходимости, удалением защитных групп. 33. Способ получения соединений формулы I по п.1, в которой R4 обозначает группу формулы (b), где p равно 0 и q равно 2, отличающийся тем, что соединение формулы VIII 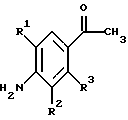 где R1, R2 и R3 имеют значения, указанные в п.1, подвергают взаимодействию с соединением формулы IX 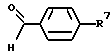 где R7 имеет значения, указанные в п.1, с последующим проведением дегидратации и гидрогенизации. 34. Способ получения соединений формулы I по п.1, где R4 обозначает группу формулы (b), где p равно 0 , q равно 1 или 2; R7 имеет значения по п. 1, кроме водород, отличающийся тем, что соединение формулы I, в котором R4, p, q приведены выше, a R7 означает водород, подвергают алкилированию соединением L-R20 или L-(CH2)zR12, где L означает уходящую группу, R20 означает C1-C8алкил, C3-C8алкенил или фенилC1-C4aлкил, где фенил необязательно замещен одним или тремя заместителями, независимо выбранными из C1-C4алкилокси, метилендиокси, тилендиокси; R12 имеет значения, приведенные в п.1; z равно 2 или 3. 35. Способ получения соединений формулы I по п.1, где R4 обозначает группу формулы (b), p равной 0 и q равно 1 или 2, a R7 означает – CH2CH2NHC(O)R14, -CH2CH2NHSO2R14, -CH2CH2NHSO2NR14R15 или CH2CH2NHC(O)NR14R15, где значения R14 и R15 приведены в п.1, отличающийся тем, что соединение формулы I, где значения R4, р, q приведены выше, а R7 означает водород, подвергают алкилированию соединением формулы X-R21, где Х означает ацациклопроп-1-ил, R21 означает -C(O)R14, -SO2R14, -SO2NR14R15, -C(O)NR14R15, где значения R14 и R15 приведены выше. 36. Способ получения соединения формулы I по п.1, где R1, R2, R3 и R4 имеют значения, указанные в п.1, а р означает 0, восстановлением соединения формулы I, где R1, R2, R3 и R4 указаны выше, a p означает 1. 37. Способ получения соединений формулы I по п.1 в виде их солей путем взаимодействия соответствующей несолевой формы с фармацевтически приемлемой неорганической или органической кислотой или основанием. 38. Способ получения соединений формулы I по п.1 в свободном виде путем взаимодействия соответствующей кислотно-аддитивной соли или соли присоединения основания соединения формулы I с пригодными основаниями или кислотой соответственно. Приоритет по признакам: 26.05.1993 кроме R2 – C1-C4алкокси, R2 и R3 вместе – метилендиокси или этилендиокси; R12 – NR13C(O)OR14; R7 – фенил C1-C4алкил, где фенил необязательно замещен одним или тремя заместителями, независимо выбранными из C1-C4алкилокси, метилендиокси, этилендиокси; 26.04.1994, где R2 – C1-C4алкокси; R2 и R3 вместе – метилендиокси или этилендиокси; R12 -NR13C(O)OR14; R7 – фенил C1-C4алкил, где фенил необязательно замещен одним или тремя заместителями, независимо выбранными из C1-C4алкилокси, метилендиокси, этилендиокси. РИСУНКИ
|
||||||||||||||||||||||||||