Патент на изобретение №2169732
|
||||||||||||||||||||||||||
(54) ПРОИЗВОДНЫЕ 5-ОКСО-5Н-[1]-БЕНЗОПИРАНО-[5,6-B]4-ОКСО-4Н-[1,2]-ПИРИМИДО-1,4,5,6-ТЕТРАГИД РО-1,3-ТИАЗИНА
(57) Реферат: Описываются новые производные 5-оксо-5Н-/1/-бензопирано-/5,6-в/-4-оксо-/1,2/-пиримидо-1,4,5,6-тетрагидро-1,3-тиазина формулы 1, где R1 – H или галоген; R2 – H или галоген, или нитрогруппа, или гидроксигруппа, или метоксигруппа, обладающие антимикробной активностью. Они могут быть использованы в медицине для лечения туберкулеза и других вирусных заболеваний, а также заболеваний, сопровождающихся иммунодефицитом. 10 з.п. ф-лы, 11 табл. 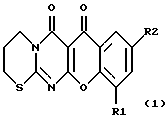
Изобретение относится к медицине, точнее к фармакологии, ветеринарии, косметологии, конкретно к синтетическим биологически активным соединениям гетероциклического ряда, обладающим антимикробной, противовирусной, антихламидийной и иммуностимулирующей активностями: производным 5-оксо-5Н-[1]бензопирано-[5,6-b] -4-оксо- 4Н-[1,2]-пиримидо-1,4,5,6-тетрагидро-1,3-тиазина. Указанные производные имеют высокую антимикробную активность по отношению к различным микобактериям, значительную противовирусную активность по отношению к вирусам простого герпеса, высокую антихламидийную активность, а также активность как индукторы интерферона. Соединения предназначены в основном для использования в медицинской практике для лечения туберкулеза, микобактериозов, вирусных заболеваний, инфекций, вызванных хламидиями, а также заболеваний, сопровождающихся иммунодефицитом, в частности злокачественных новообразований. Кроме того, указанные соединения могут быть использованы для тех же целей в ветеринарии и косметологии. Уровень техники Как известно, в современной химиотерапии инфекционных заболеваний существует много нерешенных задач. Среди бактериальных инфекций одну из серьезнейших проблем представляет туберкулез, лечение которого особенно затруднено из-за высокой устойчивости туберкулезных микобактерий к воздействию химиотерапевтических агентов (Grosset J., Current problems with tuberculosis treatment, Res. Microbiology, 1996, Vol. 147, N 10-16). Кроме того, имеющимся препаратам присущ существенный недостаток – широкое распространение устойчивости к ним у циркулирующих микобактерий. Исторически первыми противотуберкулезными препаратами были производные фенолов, бензойных кислот (Дайсон Г., Мей П. Химия синтетических лекарственных средств. – Пер. с англ., М.: Мир, 1964 г.), меркаптобензимидазолы (Bockmuhl, Persch, патент США N 2323445. ), п-аминосалициловая кислота (Bernhiem H. , J. of Bacteriology, 1941, Vol. 41, стр.387.). Наиболее активными противотуберкулезными агентами оказались открытые в конце 40-х годов нашего столетия производные изоникотиновой кислоты, такие как изониазид и его аналоги, применяемые по настоящее время, этионамид, этамбутол, аминогликозиды, рифампицин, фторхинолоны (Grosset J., Current problems with tuberculosis treatment, Res. Microbiology. 1996, Vol. 147, N 10-16). При терапии туберкулеза и микобактериозов обычно назначают одновременно 2-3 препарата, что замедляет появление резистентных форм микобактерий, но не решает проблемы привыкания и резистентности полностью. Поэтому поиск новых активных противотуберкулезных агентов, в том числе действующих на резистентные к изониазиду формы микобактерий, остается актуальным. Не менее серьезную проблему для современной медицины представляют вирусные заболевания. Как правило, вирусные инфекции, например, вызываемые вирусами группы герпеса, крайне плохо поддаются лечению, что связано как с недостаточной эффективностью существующих препаратов, так и быстрой изменчивостью вирусов, мутации которых приводят к появлению устойчивых форм (Pharmaceutical microbiology, Ed. by W. B. Hugo and A.D.Russel Blackwell Scientific Publications, Oxford, 1987; Benson C. A. Treatment of disseminated Mycobacterium avium complex disease: a clinician’s perspective, Res. Microbiology, 1996, Vol. 147, P. 16-24; Saltzman R. , Jurewicz R., Boon B, Safety of famciclovir in patients with herpes zoster and genital gerpes, Aritimicrobiaf agents and Chemotheraphy, 1994, Vol. 38, N 10, P. 2454-2457). Из синтетических препаратов для лечения заболеваний, вызванных вирусами группы герпеса, применяют ацикловир, ганцикловир, ретровир (Pharmaceutical microbiology, Ed. by W.B.Hugo and A.D.Russel Blackwell Scientific Publications, Oxford, 1987) и другие, эффективность которых не всегда достаточна. Много проблем существует при лечении заболеваний, вызванных хламидиями, включая широко распространенные урогенитальные инфекции (Fenelon L.E., Mumtaz G., Ridgway G.L. The in-vitro susceptibility of Chlamydia pneumoniae, J. of Antimicrobial Chemotherahy, 1990, Vol. 26, P. 763-767). Хламидии, являясь внутриклеточными паразитами, мало доступны для большинства существующих препаратов и, кроме того, распространившаяся устойчивость к последним привела к появлению большого числа хронических форм заболевания. Для лечения хламидийных инфекций используют обычно препараты тетрациклинового ряда, макролиды и фторхинолоны, например ципрофлоксацин (там же). Часто заболевания вирусной и бактериальной природы протекают на фоне снижения активности иммунной системы организма, поэтому проблема создания эффективных препаратов для лечения иммунодефицитных состояний различного происхождения также является одной из актуальнейших. Кроме того стимуляция активности иммунной системы способствует в борьбе организма против опухолевых процессов, при которых также наблюдается множественная устойчивость к существующим лечебным препаратам (Boyd M.R. The Future of new drug development. Current therapy in oncology 1992, P. 11-22). Необходимо отметить, что в настоящее время проводится интенсивный поиск новых биологически активных соединений среди производных пиримидинового ряда. В частности, в ряду пиримидина известно немало производных, используемых в качестве лекарственных препаратов (Negwer M., Organic-chemical drugs and their synonyms, Akademic-Verlag, Berlin, 1987.). Моноциклические производные пиримидина, в частности барбитуровые кислоты, обладают универсальным антибактериальным действием за счет антагонизма с пиримдиновыми основаниями ДНК (Biswas C. Dissert. Abstracts 1964, Vol. 24, N 9, P. 3501). Активные бактерицидные препараты обнаружены среди гидразиновых производных пиримидина (Langley B.W. , патент Великобритании N 845378 от 24.08.60; Спасов А.В., Райков З.Д., авт. св. Болгарии N 17122, Мкл C 07 D 51/22, заявл. 5.06.71), тиопиримидинов (Davies G. D. , Robins P.K., Cheng C.C., J. of American Chemical Society., 1962, Vol. 82, N 9, P. 1724-1729), алкокси- и аминопиримидинов (Minchle H., Milzher K., Keisser F. Заявка Германии N 2412854, Мкл C 07 D от 26.09. 1974. ). Эфиры пиримидокарбоновых кислот проявляют противоопухо левую активность (Krepelka J. , Kotva R. , Pijman V., Semonsky M., патенты Чехословакии NN 215554; 215593; 215580, Мкл C 07 D 239/54 от 15.04. 1984). Многие производные барбитуровых кислот обладают значительной противовирусной активностью (Спасов А. В., Райков З.Д., авт.св. Болгарии N 17122, Мкл C 07 D 51/22, заявл. 5.06.71., 17-Ueda T., Kato S., патент Японии 11832 (’62), Aug. 23, заявлен 10.05.1958). Производные [4,2-b]пиримидопирана обладают противовоспалительными свойствами (Sigao S. , Hirosi I., J. of Pharmaceutical Society of Japan, 1969, Vol. 89, N 2, P. 266-271.). Это же действие обнаружено у многих N-арил и N-циклогексил-барбитуровых кислот (Senda S., Fujimura H., Izumi I., патенты Японии NN 7912; 7913; 7914; 7915; 7916; April 22, заявлены в 5.10.1961). Все приведенные выше примеры объединяет общность химической структуры, т.к. они являются моноциклическими производными пиримидина (содержат один пиримидиновый гетероцикл, не сопряженный с другими циклическими системами). Известен пример обнаружения биологических свойств у трициклических производных пиримидинового ряда, однако их действие ограничено антиаллергической активностью (Blythin D. J. , Coldwell N. J., патент США N 4272535 опубл. 09.071981, заявлен 27.07.1979), не распространяется на область антимикробного и антивирусного действия. Наиболее близкими по химическому строению заявляемым соединениям являются производные 2,4-[1Н, 3Н, 5Н]-[1]бензопирано[2,3-d] пиримидина, запатентованные в качестве анти-аллергических агентов (там же). В качестве прототипа выбран гидразид изоникотиновой кислоты (изониазид) (Boyd M. R. The Future of new drug development. Current therapy in oncology 1992, P. 11-22). Выбор основан на его совпадении с заявляемым веществом по наиболее важному назначению – противотуберкулезному действию. Изониазид является одним из наиболее эффективных противотуберкулезных препаратов, может использоваться в любых лекарственных формах (таблетки, инъекции, ингаляции), при этом по своему химическому строению объект изобретения и прототип принадлежат к классу азотистых гетероциклов. Задача изобретения Задачей изобретенья является получение новых химических соединений, обладающих высокой противомикробной активностью (особенно в отношении штаммов микобактерий, резистентных к прототипу – изониазиду), одновременно обладающих противовирусной активностью (по отношению к вирусам простого герпеса), антихламидийной активностью, а также стимулирующих выработку эндогенных интерферонов в организме. Другими словами, задача изобретения сводится к химическому синтезу биологически активных веществ, превосходящих прототип не только по широте воздействия на штаммы туберкулезных и других микобактерий, но обладающих противовирусным, антихламидийным и иммуностимулирующим действием. Сущность изобретения Поставленная задача решается путем синтеза нового класса гетероциклических соединений – производных 5-оксо-5Н-[1]-бензопирано-[5,6-b]-4-оксо-4Н-[1,2]-пиримидо-1,4,5,6- тетрагидро-1,3-тиазина (I) общей формулы (1) 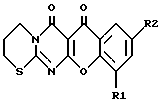 (I-X) Где: R1 – H или галоген; R2 – H, или галоген, или нитрогруппа, или гидроксигруппа, или метоксигруппа. Поставленная задача может быть решена при R1 = R2 = H (I); R1 = H и R2 = Cl (II); R1 = R2 = Cl (III); R1 = H и R2 = Br (IV); R1 = R2 = Br (V): R1 = H и R2 = NO2 (VI); R1 = Cl и R2 = NO2 (VII); R1 = Br и R2 = NO2 (VIII); R1 = H и R2 = OCH3 (IX); R1 = H и R2 = OH (X). Заявляемое вещество является новым, поскольку оно не известно из доступных источников информации. Заявляемое решение является неочевидным. Как известно, практически невозможно заранее предсказать биологическую активность новых конденсированных гетероциклических систем. Поэтому получение группы заявляемых соединений, которые являются сложными тетрациклическими системами, включающими 4 гетероатома (2 атома азота, 1 кислород и 1 сера) и обнаружение у них антимикробных, иммуностимулирующих и противовирусных свойств никак не вытекает очевидным образом из современного уровня техники. Раскрытие изобретения Предлагаемый нами синтез заявляемых веществ состоит из двух основных этапов: 1) синтезируют 4-оксо-4Н-6-окси-[1,2-d]-пиримидо-1,4,5,6- тетрагидро-1,3-тиазин (XI) из 2-тиобарбитуровой кислоты (XII) и 1,3-дигалогенпропана (XIII). 2) получают целевое соединение (I-X) из полученного на первом этапе промежуточного вещества (XI) и соответствующего производного салицилового альдегида (XIV). Способ является общим для всех членов группы. Сущность изобретения поясняется приведенными ниже двумя примерами синтеза промежуточного вещества и тремя примерами синтеза заявленных веществ, двумя таблицами выходов промежуточного и целевых продуктов, двумя таблицами характеристик целевых продуктов и данными семи серий экспериментов по определению их биологических свойств где: примеры 1 и 2 – конкретные варианты выполнения 1-го этапа синтеза заявляемого вещества (получение промежуточного вещества (XI), т.е. 4-оксо-4Н-6-окси-[1,2-d] -пиримидо-1,4,5,6-тетрагидро-1,3-тиазина, причем пример 2 содержит обобщенный множественный вариант; примеры 3, 4, и 5 – конкретные варианты выполнения второго этапа синтеза заявляемых веществ (получение целевых продуктов (I-X), т.е. производных 5-оксо-5Н-[1] -бензопирано-[5,6-b] -4-оксо-4Н-[1,2] -пиримидо- 1,4,5,6-тетрагидро-1,3-тиазина); таблица 1 – выход промежуточного вещества (XI), т.е. 4-оксо-4Н-6-окси[1,2-d]-пиримидо-1,4,5,6-тетрагидро-1,3-тиазина. таблица 2 – методы синтеза и выход целевых продуктов (I-X), т.е. производных 5-оксо-5Н-[1] -бензопирано-[5,6-b] -4-оксо-4Н-[1,2] -пиримидо- 1,4,5,6-тетрагидро-1,3-тиазина; таблица 3 – температуры плавления и данные спектров ПМР целевых продуктов (I-X), т.е. производных 5-оксо-5Н-[1]-бензопирано-[5,6-b]-4- оксо-4Н-[1,2]-пиримидо-1,4,5,6-тетрагидро-1,3-тиазина; таблица 4 – данные элементного анализа целевых продуктов (I-X), т.е. производных 5-оксо-5Н-[1] -бензопирано-[5,6-b] -4-оксо-4Н-[1,2]- пиримидо-1,4,5,6-тетрагидро-1,3-тиазина. Данные семи серий экспериментов по определению биологической активности заявляемых соединений содержат: эксперимент 1 – определение антимикробного действия; эксперимент 2 – определение действия заявляемых соединений на патогенные микобактерии; эксперимент 3 – определение острой токсичности; эксперимент 4 – терапевтическая эффективность при экспериментальном туберкулезе; эксперимент 5 – определение действия на вирус герпеса; эксперимент 6 – определение интерферониндуцирующей активности; эксперимент 7 – определение действия на C.trachomatis. Пример 1. Вариант выполнения 1-го этапа синтеза заявляемых веществ (получение промежуточного вещества, т.е. 4-оксо-4Н-6-окси-[1,2-d]-пиримидо-1,4,5,6-тетрагидро-1,3-тиазина) (XI) 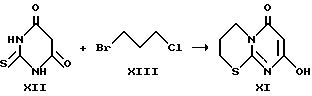 14,4 г (0,1 моль) 2-тиобарбитуровой кислоты (XII) прибавляют к 100-150 мл водно-спиртового раствора 0,2 моль щелочи и перемешивают. Затем прибавляют 0,1 моль 1-бром-3-хлорпропана (XIII) и перемешивают смесь при нагревании. В процессе реакции pH раствора понижается. После завершения реакции охлаждают раствор, выпавший осадок отделяют и промывают. Для очистки продукт растворяют в водной щелочи и высаживают соляной кислотой. Выход 4-оксо-4Н-6-окси-[1,2-d] -пиримидо-1,4,5,6-тетрагидро-1,3-тиазина 9,7 г (43,8%), Т пл. 250-260oC (с разл.) Найдено, %: C 45,02; H 5,27; N 14,09; S 17,11; C7H8N2O2S Вычислено, %: C 45,07; H 5,21; N 14,15; S 17,02 Спектр ПМР, ДМСО-d6,  , м.д.: 2,18, м (2H, CH2); 3,19 т (2H, SCH2); 3,90 т (2H, NCH2); 5,08 с (1H, CH); 11,06 с (1H, OH).
Пример 2. Вариант выполнения 1-го этапа синтеза заявляемых веществ (получение промежуточного вещества, т.е. 4-оксо-4Н-6-окси-[1,2-d]-пиримидо-1,4,5,6-тетрагидро-1,3-тиазина) (XI): , м.д.: 2,18, м (2H, CH2); 3,19 т (2H, SCH2); 3,90 т (2H, NCH2); 5,08 с (1H, CH); 11,06 с (1H, OH).
Пример 2. Вариант выполнения 1-го этапа синтеза заявляемых веществ (получение промежуточного вещества, т.е. 4-оксо-4Н-6-окси-[1,2-d]-пиримидо-1,4,5,6-тетрагидро-1,3-тиазина) (XI):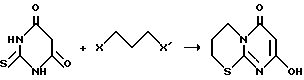 Производят алкилирование 2-тиобарбитуровой кислоты (XII) 1,3-дигалогенпроизводными пропана общей формулы X-CH2-CH2-CH2-X’ (XIII) (где X, X’ – Cl, Br или J). Процесс ведут аналогично примеру 1. Выходы продукта приведены в табл.1. Пример 3. Вариант выполнения второго этапа синтеза заявляемого вещества (получение целевого продукта, а именно 5-оксо-5Н-[1]-бензопирано-[5,6-b]-4-оксо-4Н-[1,2]-пиримидо-1,4,5,6- тетрагидро-1,3-тиазина (1): 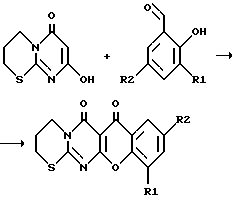 Смесь 1,84 г (0,01 моль) 4-оксо-4Н-6-окси-[1,2-d]-пиримидо-1,4,5,6-тетрагидро-1,3-тиазина (XI) и 1,83 г (0,015 моль) салицилового альдегида (XV), (R1 = R2 = H) нагревают до прекращения выделения паров образующейся в ходе реакции воды. Затем тщательно промывают образовавшуюся реакционную массу спиртом для удаления исходных и побочных продуктов, в остаток перекристаллизовывают из уксусной кислоты, получая целевой продукт – 5-оксо-5Н-[1] -бензопирано-[5,6-b] -4-оксо-4Н-[1,2] – пиримидо-1,4,5,6-тетрагидро-1,3-тиазин. Выход приведен в табл. 2, характеристики – в табл. 3, данные элементного анализа – в табл. 4. Пример 4. Вариант выполнения второго этапа синтеза заявляемого вещества (получение целевого продукта, а именно 3-хлор-5-оксо-6Н-[1]-бензопирано-[5,6-b]-4-оксо-4Н-[1,2]-пиримидо- 1,4,5,6-тетрагидро-1,3-тиазина (11): 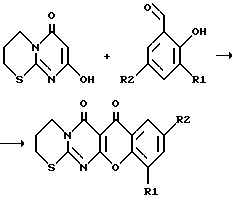 (R1 = H, R2 = Cl) (R1 = H, R2 = Cl). Смесь 1,84 г (0,01 моль) 4-оксо-4Н-6-окси-] [1,2-d] -пиримидо-1,4,5,6-тетрагидро-1,3-тиазина (XI) и 2,35 г (0,015 моль) 5-хлорсалицилового альдегида (XV), (R1 = H, R2 = Cl) в высококипящем инертном растворителе (хлорбензол, анизол, диглим, этиленгликоль) нагревают до исчезновения исходного пиримидотиазина. После этого отделяют осадок, тщательно промывают и сушат. Получают целевой продукт – 3-хлор-5-оксо-5Н-[1] -бензопирано-[5,6-b] -4-оксо-4Н-[1,2] -пиримидо- 1,4,5,6- тетрагидро-1,3-тиазин. Выход приведен в табл. 2, характеристики – в табл. 3, данные элементного анализа – в табл. 4. Пример 5. Вариант выполнения второго этапа синтеза заявляемого вещества (получение целевого продукта, а именно 1,3-дихлор-5-оксо-5Н-[1]-бензопирано-[5,6-b]-4-оксо-4Н-[1,2]-пиримидо- 1,4,5,6-тетрагидро-1,3-тиазина (III). Смесь 1,84 г (0,01 моль) 4-оксо-4Н-6-окси-[1,2-d]-пиримидо-1,4,5,6-тетрагидро-1,3-тиазина (XI) и 1,93 г (0,01 моль) 3,5-дихлорсалицилового альдегида (XV), (R1 = R2 = Cl) кипятят в этаноле 6-8 часов. Образовавшийся осадок отфильтровывают, промывают горячим этанолом и сушат. Получают целевой продукт – 1,3-дихлор-5-оксо-5Н-[1]-бензопирано-[5,6-b]-4-оксо- 4Н-[1,2]-пиримидо-1,4,5,6-тетрагидро-1,3-тиазин. Выход приведен в табл. 2, характеристики – в табл. 3, данные элементного анализа – в табл. 4. Остальные целевые соединения – производные 5-оксо-5Н-[1]-бензопирано-[5,6-b] -4-оксо-4Н-[1,2] -пиримидо-1,4,5,6- тетрагидро-1,3-тиазина (IV-X), где: 3-бром-5-оксо-5Н-[1] -бензопирано-[5,6-b] -4-оксо-4Н-[1,2] – пиримидо-1,4,5,6-тетрагидро-1,3-тиазин (IV), R1 = H, R2 = Br; 1,3-дибром-5-оксо-5Н-[1] -бензопирано-[5,6-b] -4-оксо-4Н-[1,2] – пиримидо-1,4,5,6-тетрагидро-1,3-тиазин (V), R1 = R2 = Br; 3-нитро-5-оксо-5Н-[1] -бензопирано-[5,6-b] -4-оксо-4Н-[1,2] – пиримидо-1,4,5,6-тетрагидро-1,3-тиазин (VI), R1 = H, R2 = NO2; 1-хлор-3-нитро-5-оксо-5Н-[1] -бензопирано-[5,6-b]-4-оксо-4Н-[1,2]- пиримидо-1,4,5,6-тетрагидро-1,3-тиазин (VII), R1 = Cl, R2 = NO2; 1-бром-3-нитро-5-оксо-5Н-[1] -бензопирано-[5,6-b]-4-оксо-4Н-[1,2]- пиримидо-1,4,5,6-тетрагидро-1,3-тиазин (VIII), R1 = Br,R2 = NO2; 3-метокси-5-оксо-5Н-[1] -бензопирано-[5,6-b]-4-оксо-4Н-[1,2]- пиримидо-1,4,5,6-тетрагидро-1,3-тиазин (IX), R1 = H, R2 = OCH3; 3-окси-5-оксо-5Н-[1] -бензопирано-[5,6-b] -4-оксо-4Н-[1,2] – пиримидо-1,4,5,6-тетрагидро-1,3-тиазин (X), R1 = H, R2 = OH – получают из 4-оксо-4Н-6-окси-[1,2-d] -пиримидо-1,4,5,6-тетрагидро-1,3-тиазина (XI) и соответствующих салициловых альдегидов (XV) по приведенным выше примерам 3, 4 или 5. Методы синтеза и выходы продуктов приведены в табл. 2, характеристики – в табл. 3, данные элементного анализа – в табл. 4. Экспериментальная проверка биологической активности заявляемых соединений Эксперимент 1 Определение антимикробного действия соединений Для определения антимикробной активности были использованы стандартные штаммы Mycobacteium smegmatis ATCC607 и Mycobacterium tuberculosis H37RV, чувствительные ко всем антимикробным препаратам. Оценку антимикобактериального действия проводили методом серийных разведений (Pharmaceutical microbiology, Ed. by W.B.Hugo and A.D.Russel Blackwell Scientific Publications, Oxford, 1987, 511 p.) M.smegmatis ATCC607 для засева выращивали на жидкой синтетической среде N-1. Соединения растворяли в диметил-сульфокисиде (ДМСО) и титровали в среде N-1 так, что данный препарат содержался в отдельных пробирках со средой в концентрациях от 200 до 0,025 мг/л. Концентрация препарата в среде соседних пробирок отличалась в два раза. В контроле использовали ДМСО, который титровали так же, как и препарат. Тест-штамм бактерий добавляли в количестве 1-2  106 мл. Результат учитывали после 72-часового культивирования пробирок при 37oC.
Для M. tuberculosis H37Rv условия постановки опыта были идентичными, за исключением того, что бактерии выращивали на среде Сотона, содержащей 10% лошадиной сыворотки, и плотность микробной суспензии при засеве составляла 50106.
В качестве контроля в обоих случаях были использованы известные туберкулостатические препараты. Результаты, полученные для использованных штаммов, суммированы в таблице 5.
Приведенные в табл. 5 данные показывают, что заявляемые соединения обладают антимикробной активностью по отношению к использованным штаммам микобактерий в концентрациях от 100,0 до 0,1 мг/л. Активность действия на микобактерии ряда заявляемых соединений in vitro не уступает, а порой и превосходит таковую у известных противотуберкулезных препаратов.
Эксперимент 2 106 мл. Результат учитывали после 72-часового культивирования пробирок при 37oC.
Для M. tuberculosis H37Rv условия постановки опыта были идентичными, за исключением того, что бактерии выращивали на среде Сотона, содержащей 10% лошадиной сыворотки, и плотность микробной суспензии при засеве составляла 50106.
В качестве контроля в обоих случаях были использованы известные туберкулостатические препараты. Результаты, полученные для использованных штаммов, суммированы в таблице 5.
Приведенные в табл. 5 данные показывают, что заявляемые соединения обладают антимикробной активностью по отношению к использованным штаммам микобактерий в концентрациях от 100,0 до 0,1 мг/л. Активность действия на микобактерии ряда заявляемых соединений in vitro не уступает, а порой и превосходит таковую у известных противотуберкулезных препаратов.
Эксперимент 2Определение действия соединений на патогенные микобактерии В работе использованы штаммы, выделенные от больных в клиниках С.Петербурга (Россия) в 1996 году; M. tuberculosis 61(S) – чувствительный к действию известных антимикробных препаратов (аминогликозидов, рифампицина, изониазида, этамбутола); M. tuberculosis 16(R) – устойчивый к основным антимикобактериальным препаратам; Mycobacterium avium 84 – микобактерия, вызывающая заболевания и гибель лиц, инфицированных вирусом иммунодефицита человека. Исследование проводили на среде Левенштейна-Йенсена. Препараты, растворенные в (ДМСО), добавляли в отдельные пробирки со средой в конечной концентрации 10 мг/л. После засева тест-штаммов на поверхность среды пробирки инкубировали в течение 30 суток. В контроле использовали ДМСО в соответствующих концентрациях и стандартные препараты, используемые для лечения туберкулеза и микобактериозов (табл. 6). Полученные данные свидетельствуют, что указанные в таблице 6 заявляемые соединения угнетают рост всех использованных в опыте видов и штаммов микобактерий. Таким образом, in vitro они действуют и на штамм микобактерий, выделенный от больных, устойчивый к основным препаратам, используемым для лечения туберкулеза. Кроме того эти соединения в данных условиях угнетают рост M.avium. Остальные заявляемые соединения обладают в аналогичных условиях меньшей антимикобактериальной активностью. Эксперимент 3 Определение острой токсичности Определение острой токсичности проведено на беспородных белых мышах массой 18-20 г. Эмульсии заявляемых соединений вводили в различных концентрациях: 1500, 700, 500, 100, 20 и 5 мг/кг. Для исследования каждой концентрации соединения использовали по 5 животных. Препарат вводили один раз в сутки через рот или внутрибрюшинно. Период наблюдения составил 14 дней. На 1, 8 и 15 день проводили взвешивание животных в каждой группе. Для контроля использовали животных, которым вводили эмульсию, приготовленную без испытуемых соединений. Для макроскопического исследования внутренних органов проводили вскрытие всех животных, умерших в ходе опыта и выживших к концу эксперимента. В ходе проведенных исследований не наблюдалось потери веса, изменения в поведении и внешнем виде, а также гибели животных. По результатам вскрытия макроскопической патологии со стороны внутренних органов животных как в опытной так и в контрольной группах обнаружено не было (табл. 7). Полученные результаты свидетельствуют, что при приеме через рот или внутрибрюшинно заявляемые соединения в концентрации 1500 мг/кг не обладают острой токсичностью для мышей. Эксперимент 4 Терапевтическая эффективность действия заявляемых соединений при экспериментальном туберкулезном процессе у мышей Исследование проведено на беспородных мышах-самцах массой 18-20 г. Туберкулезный процесс вызывали у мышей введением в хвостовую вену M. bovis bovinus 8 в дозе 0,1 мг/мышь в виде бактериальной суспензии (0,2 мл). Испытуемые соединения использовали в трех дозах (10, 20 и 30 мг/кг). В качестве препарата сравнения использовали стрептомицин. Испытуемые соединения вводились животным один раз в сутки внутрь, через рот, стрептомицин – внутримышечно. Выведение животных из опыта (путем декапитации) производили на 42 день после инфицирования, когда в группе контроля заражения (нелеченные мыши) погибло 45% животных. Эффективность терапии оценивали по выживаемости животных, относительной массе легких, индексам поражения легких и высеваемости микобактерий из селезенки. В последнем случае гомогенат органа засевали на плотную яичную среду Левенштейна-Йенсена. Подсчет колоний проводили через 25-30 дней культивирования. Рассчитывали среднюю высеваемость микобактерий на одну пробу, что выражали как число колониеобразующих единиц (КОЕ) на 100 мг (табл. 8). Проведенные исследования показали, что у экспериментальных животных с генерализованным туберкулезным процессом заявляемые соединения показывают терапевтический эффект, по проявленной активности не уступающий таковому у стрептомицина. Эксперимент 5 Определение действия заявляемых соединений на вирус герпеса Антивирусная активность изучалась по отношению к вирусу герпеса I типа (ВПГ-I /Ленинград/248/88) по общепринятому методу (Gentry G.A., Lawrency N., Lushbaugh N. Isolation and differentiation of Herpes simplex virus and Trichomonas vaginalis in cell culture, J. of Clinical Microbiology 1985, Vol. 22, N 2, P. 199-204). Вирусы выращивали на перевиваемой культуре клеток Vero, полученной из банка клеточных культур Института цитологии РАН. Схема постановки опыта К клеткам, выращенным на среде RPMI-1640 с 10% телячьей сыворотки и помещенным в лунки 96 луночного плато, добавляли вирус в конечной концентрации 10 частиц/мл и заявляемые соединения, растворенные в ДМСО, в конечной концентрации 100, 10 и 1 мг/л. Для каждой испытанной концентрации препарата использовали 5 независимых лунок. Плато инкубировали в течение 60 мин при 38oC в CO2-инкубаторе. После инкубации вирус удаляли и снова вносили свежую среду, содержащую заявляемые соединения в использованных концентрациях. Результаты оценивали по наличию цитопатогенного действия вируса на клетки через 36 часов инкубации при 38oC в CO2-инкубаторе. В опыте были использованы следующие контроли: 1. Контроль культуры клеток (способность к нормальному росту) 2. Контроль вируса (оценка способности к репродукции) 3. Контроль антивирусной активности противовирусного препарата – ацикловира 4. Контроль соединений (токсичность соединений) 5. Контроль растворителя (ДМСО) на токсичность. Для оценки цитопатического действия вируса подсчитывали число неизмененных клеток в 100 полях, образованных специальной сеткой окуляра инвертированного микроскопа. Полученные результаты представлены в таблице 9. Полученные результаты указывают, что приведенные в таблице 9 заявляемые соединения обладают антегерпетической активностью, сравнимой с таковой у стандартного препарата ацикловира. Остальные заявляемые соединения имели менее выраженную активность в процессе подавления репродукции вируса герпеса в выбранных условиях эксперимента. Эксперимент 6 Определение интерферониндуцирующей активности заявляемых соединений Индукцию синтеза интерферонов заявляемыми препаратами проводили на первичной культуре человеческих лимфоцитов (именно данные клетки в организме человека являются основными продуцентами интерферонов). Для получения культуры лимфоцитов использовали свежую (12 часов после забора) кровь здоровых доноров (не второй группы). Для выделения лимфоцитов гепаринизированная кровь, полученная от здорового донора, подвергалась центрифугированию в градиенте плотности фиколл-верографин 1.71 г/см3 для выделения фракции иммунокомпетентных клеток. Указанная фракция отбиралась и разводилась питательной средой RPMI-1640, содержащей 5% эмбриональной сыворотки крупного рогатого скота, 0.3 мг/мл L-глутамина, 100 ед/мл пенициллина, 50 мг/мл стрептомицина. Концентрацию лимфоцитов учитывали после окрашивания метиленовым синим и подсчета количества клеток в камере Горяева. Исходные растворы заявляемых веществ разводили питательной средой RPMI-1640 так, чтобы конечные концентрации веществ составляли ряд: 100, 10, 1 мг/л после внесения суспензии лимфоцитов. Конечная концентрация лимфоцитов в индукционной смеси составила 3 106 клеток/мл. Параллельно с опытными пробами проставлялись следующие контроли:1) контроль спонтанной продукции интерферонов (ИФН) лимфоцитами; 2) контроль протекания процесса при воздействии стандартизированного индуктора ИФН N-метил-N-(a, D-глюкопиранозил)аммоний-10-метиленкарбоксилат акридона (циклоферон). 3) контроль протекания процесса при воздействии стандартизированного индуктора ИФН – Неовира (натрия 10-метиленкарбоксилат-9-акридон) с соответствующим содержанием DMCO в опытных пробах. 4) контроль спонтанной продукции интерферонов в присутствии DMCO, в количестве, соответствующем испытуемым образцам. Контрольные и опытные образцы инкубировали 24 часа при 37oC. После инкубации пробы центрифугировались при 2000 g для осаждения клеточных элементов и из проб отбирался ИФН-содержащий супернатант, который анализировали на количественное содержание ИФН. Осадок клеток ресуспендировали в прежнем объеме питательной среды, окрашивали витальным красителем – трипановым синим – и подсчитывали число клеток в камере Горяева (как описано выше) для определения цитотоксического действия препаратов. Количественное определение содержания ИФН в контрольных и опытных образцах производили с использованием иммуноферментной тест-системы на ИФН-а производства ТОО “Протеиновый контур” ProCon IF2 plus. Для определения количества интерферона в пробе использовали твердофазный иммуноферметный метод с использованием пероксидазы хрена в качестве индикаторного фермента. Активность связанной пероксидазы измеряли с использованием автоматического фотометра для микропланшетов с микропроцессором при длине волны – 450 нм. Для подсчета результатов параллельно определяли активность ИФН у стандартных растворов ИФН, содержащих известное количество препарата. На основании полученных результатов строилась калибровочная кривая, позволяющая при использовании микропроцессора автоматического фотометра получать данные, выраженные в Международных Единицах активности (МЕ). Результаты анализа выражаются в МЕ активности ИФН на мл в данной индукционной системе, содержащей 3 106 лимфоцитов/мл. Каждая опытная и контрольная точка исследовалась в 4 параллелях.
Контроли иммуноферментной реакции1. Контроль DMCO с питательной средой. 2. Контроль компонентов системы (согласно инструкции). 3. Все результаты учитывались только при соответствии контролей паспортным данным системы. Полученные результаты подвергались статистическому анализу по критерию и расчетом доверительного интервала при p = 0.05. Произведен анализ сходимости результатов в параллельных опытах. В результате проведенных исследований установлено, что среди заявляемых соединений имеются пробы, обладающие способностью индуцировать синтез ИФН (таблица 10). Эксперимент 7 Определение действия заявляемых соединений на Chlamydia trachomatis Антимикробную активность заявляемых соединений изучали по отношению к C. trachomatis D323 – стандартному штамму из коллекции кафедры микробиологии С. Петербургского Государственного университета им. ак. И.П.Павлова. Данный штамм был выделен от больного с хламидийным уретритом, имеет морфологию и физиологическую активность, характерную для представителей данного вида, чувствителен к действию препаратов, используемых для лечения хламидийной инфекции. В работе использованы клеточные культуры McCoy и L929, полученные из института Цитологии РАН. Схема постановки опыта Клетки выращивали во флаконах из нейтрального стекла в среде RPMI-1640 с добавлением 10% эмбриональной телячьей сыворотки. Опыт ставили в стеклянных (лишенных токсичности) плоскодонных флаконах с покровными стеклами. Клетки вносили в среду в конечной концентрации 1 10 кл/мл. После получения монослоя в пробирки вносили стандартные заражающие дозы хламидий, хранящиеся в замороженном состоянии при -70oC. Одновременно к клеткам добавляли испытуемые соединения в конечной концентрации 100 мг/л. Пробу центрифугировали при 2400g в течении 60 минут при комнатной температуре и инкубировали при 37oC в течение 2 часов. После этого меняли питательную среду на новую, содержащую 5% эмбриональной телячьей сыворотки и циклогексимид (2 мкг/мл) с повторным внесением заявляемых соединений в той же концентрации. Параллельно дублировали пробы, используя среду без циклогексемида, чтобы исключить его влияние на изучаемые субстанции. Пробы инкубировали в течение 48 часов в CO2-инкубаторе.
Контроли включили: контроли культур клеток, контроль действия растворителей, контроль действия хламидий в отсутствии каких бы-то ни было препаратов, контроль чувствительности хламидий к стандартному антимикробному препарату – ципрофлоксацину (19), контроль испытуемых соединений на токсичность по отношению к культурам клеток.
Оценку результатов проводили путем выявления хламидийных цитоплазматических включений с помощью метода иммунофлюоресценции (MicroTrac Chlamydia trachomatis Direct Specimen Test) и хламидийных антигенов с помощью CylaMonoScreen (Russian-British Joint Venture 66 Regent’s Parc Road London NW1 7SX) (Wang S-P., Grayston J.T. Serotyping of Clamydia trachomatis by inderect fluorescent-antibody staining of inclusions in cell culture with monoclonal antibodies. J. of Clinical Microbiology, 1991, Vol. 29, N 7, P. 1295-1298. и Judson B. A. , Lambert P.P. Improved Syva MicroTrac Clamydia trachomatis direct test method, Journal of Clinical Microbiology, 1988, Vol. 26, N 12, P. 2657-2658). Эффект действия препарата определяли, анализируя состояние монослоя и число клеток с ЦПВ по сравнению с контролем (культура клеток, зараженная C.trachomatis D323), при этом учитывали число неизмененных клеток в 100 полях зрения, полученных при использовании специальной сетки окуляра микроскопа.
Результаты контрольных проб, удовлетворяющие требованиям эксперимента: контроль культуры клеток – морфология клеток и состояние монослоя соответствуют данному типу клеток, контроль роста хламидий в культуре клеток – наличие ЦПВ в монослое, контроль действия стандартного антимикробного препарата – уменьшение числа ЦПВ в монослое по сравнению с предыдущим контролем, контроль токсичности заявляемых соединении – токсичность отсутствует, контроль действия растворителей – токсическое действие на клетки отсутствует. Результаты проведенных испытаний представлены в таблице 11.
Полученные данные свидетельствуют, что заявляемые соединения, приведенные в таблице 11, обладают выраженной активностью против хламидий, превосходящей таковую у стандартного препарата – ципрофлоксацина.
Остальные заявляемые соединения обладают менее выраженной активностью по защите клеток от хламидий в выбранных условиях эксперимента.
Промышленная применимостьПримеры 1-5 и результаты практического синтеза и анализа заявляемых соединений, приведенные в таблицах 1-4, подтверждают возможность лабораторного и промышленного синтеза всех десяти заявляемых соединений средствами, освоенными современной фармацевтической промышленностью, а также их четкую идентификацию общепринятыми методами контроля. Серия экспериментов по определению биологической активности, приведенная в семи представленных отчетах, показала, что заявляемые соединения обладают биологической активностью по отношению к различным микроорганизмам, включая микобактерии (чувствительные и устойчивые к существующим препаратам), хламидии, вирус простого герпеса, а также интерферониндуцирующей активностью. Последнее указывает на возможность их использования в лечении некоторых раковых заболеваний. Приведенные факты доказывают достижение задач, поставленных изобретением: синтезирован новый класс гетероциклических соединений, обладающих высокой и широкой биологической активностью, в частности антимикобактериальной (возбудители туберкулеза и микобактериозов), иммуностимулирующей, противохламидийной и противовирусной. Таким образом, по нашему мнению, заявляемые средства (вещества) (удовлетворяют всем требованиям, предъявляемым к изобретению: они новы, неочевидны и промышленно применимы. Формула изобретения
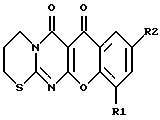 где R1 – H или галоген; R2 – H, или галоген, или нитрогруппа, или гидроксигруппа, или метоксигруппа. 2. Соединение формулы 1 по п.1, где R1=R2-H (I). 3. Соединение формулы 1 по п.1, где R1=R2-H и R2-Cl (II). 4. Соединение формулы 1 по п.1, где R1=R2-Cl (III). 5. Соединение формулы 1 по п.1, где R1=H и R2=Br (IV). 6. Соединение формулы 1 по п.1, где R1=R2=Br (V). 7. Соединение формулы 1 по п.1, где R1=H и R2=NO2 (VI). 8. Соединение формулы 1 по п.1, где R1=Cl и R2=NO2 (VII). 9. Соединение формулы 1 по п.1, где R1=Br и R2=NO2 (VIII). 10. Соединение формулы 1 по п.1, где R1=H и R2=OCH3 (IX). 11. Соединение формулы 1 по п.1, где R1=H и R2=OH (X). РИСУНКИ
|
||||||||||||||||||||||||||