Патент на изобретение №2169727
|
||||||||||||||||||||||||||
(54) ЗАМЕЩЕННЫЕ 1,2,3,4-ТЕТРАГИДРО-2-НАФТАЛИНАМИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ
(57) Реферат: Изобретение относится к новым замещенным 1,2,3,4-тетрагидро-2-нафталинамина формулы I, где R1 и R2 независимо представляют собой водород, С1-4алкил, С1-4алкоксил, галоген, трифторметил; R3 представляет собой водород, гидроксил, С1-4алкоксил, циaнoгpуппу, карбамоил; R4 и R5 независимо представляют собой водород, С1-4алкил, гидрокси С2-4алкил, или образуют вместе с атомом азота, к которому они присоединены, пиперидиногруппу; в виде свободного основания или соли, полученной присоединением кислоты. Способ получения соединения формулы I путем взаимодействия соединения формулы II с соединением формулы III, и полученное соединение выделяют в виде свободного основания или соли, полученной присоединением кислоты. Фармацевтическая композиция, обладающая антиконвульсивной активностью, активностью при ишемии, вызванной нейрональными повреждениями и активностью в условиях, включающих высвобождение глютамата, содержащая соединение формулы I в форме свободного основания или фармацевтически приемлемой соли, в терапевтически эффективном количестве. 3 c. и 5 з.п. ф-лы, 1 табл. 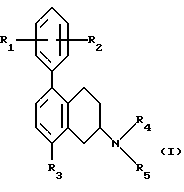 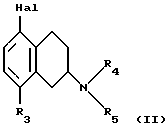 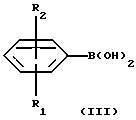
Настоящее изобретение относится к новым амино-тетралинам (тетрагидро-нафталинаминам), их получению, их использованию в качестве фармацевтических средств, и к содержащим их фармацевтическим композициям. В первом аспекте в соответствии с изобретением предлагаются 1,2,3,4-тетрагидро-2-нафталинамины, несущие фенильный заместитель в ароматическом кольце, и их соли, полученные присоединением кислоты. Фенильный заместитель предпочтительно находится в положении 5 2-нафталинамина. Фенильный заместитель может нести дополнительные заместители, например, как в случае формулы I, приведенной ниже. Дополнительные заместители могут быть также представлены удобным образом, но не исключительно на ароматическом кольце, например, в положении 8 2-нафталинамина, когда фенильная группа находится в положении 5. В частности, в настоящем изобретении предлагается соединение формулы I 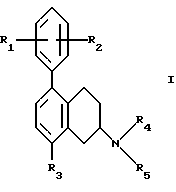 где R1 и R2 независимо представляют собой водород, (C1-4)алкил, (C1-4)алкоксил, (C1-4)алкилтиогруппу, галоген, трифторметил, трифторометоксил, цианогруппу, (C2-5)алканоил, (C1-4)алкилсульфонил или сульфамоил, R3 представляет собой водород, гидроксил, (C1-4)алкил, (C1-4)алкоксил, галоген, цианогруппу, (C2-5)алканоил, карбамоил, (C1-4)алкилсульфонилоксил или трифторметилсульфонилоксил, и R4 и R5 независимо представляют собой водород, (C1-4)алкил, гидрокси(C2-4)алкил или фенил(C1-4)алкил, или образуют, вместе с атомом азота, к которому они присоединены, пирролидинил, пиперидиногруппу или пиперазинил, в форме свободного основания или соли, полученной присоединением кислоты. Галоген представляет собой фтор, хлор, бром, или иод, предпочтительно фтор или хлор. Любые алкильные, алкоксильные и алкилтио-радикалы предпочтительно являются радикалами с прямой цепью. Они предпочтительно имеют от 1 до 3-х атомов углерода, более предпочтительно они являются метилом, метоксилом и метилтиогруппой. Предпочтительны следующие значения и их комбинации: R1 и R2 независимо представляют собой водород, (C1-4)алкил, (C1-4)алкоксил, галоген или трифторметил, R3 представляет собой водород, гидроксил, (C1-4)алкоксил, цианогруппу или карбамоил, R4 и R5 независимо представляют собой водород или (C1-4)алкил, или образуют вместе с азотом, к которому они присоеденены, пиперидиногруппу. В частной группе соединений формулы I, R1 и R2 независимо представляют собой водород, (C1-4)алкил, (C1-4)алкоксил, (C1-4)алкилтиогруппу, галоген, трифторметил, цианогруппу или (C2-5)алканоил, R3 – такой, как описано выше, и R4 и R5 независимо представляют собой водород, (C1-4)алкил или фенил(C1-4)алкил, или образуют, вместе с атомом азота, к которому они присоединены, пирролидинил, пиперидиногруппу или пиперазинил. Соединения по изобретению имеют асимметричный углеродный атом в положении 2. Поэтому они могут существовать в оптически активной форме или в форме смеси оптических изомеров, например в форме рацемических смесей. Все оптические изомеры и их смеси, включая рацемические смеси, являются частью настоящего изобретения. Если соединение по изобретению находится в оптически активной форме, то предпочтительной является R конфигурация. В еще одном аспекте изобретения предлагается способ получения соединений по изобретению, при котором 1,2,3,4-тетрагидро-2-нафталинамин, несущий галоген в ароматическом кольце, подвергают взаимодействию с возможно замещенной фенилбороновой кислотой и полученное соединение выделяют в форме свободного основания или соли, полученной присоединением кислоты. В частности, в изобретении предлагается способ получения соединений по изобретению, при котором соединение формулы II 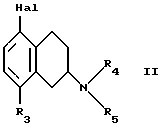 где R3, R4, и R5 такие, как определено выше, и Hal представляет собой галоген, подвергают взаимодействию с соединением формулы III 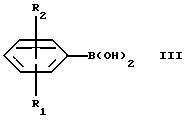 где R1 и R2 такие, как определено выше, и полученное соединение выделяют в форме свободного основания или соли, полученной присоединением кислоты. Реакция может быть осуществлена известным способом, предпочтительно путем осуществления катализируемого металлом переходного арил-арил сочетания, например, как описано в примере 1. В качестве галогена предпочтителен бром или иод, особенно бром. Обработку реакционных смесей, полученных согласно вышеописанному способу, и очистку полученных таким образом соединений можно выполнять в соответствии с известными процедурами. Соли, полученные присоединением кислоты, могут быть произведены известным способом из форм свободного основания и наоборот. Подходящие фармацевтически приемлемые соли, полученные присоединением кислоты, для использования в соответствии с настоящим изобретением включают в себя, например, гидрохлорид, гидромалеат, гидрофумарат и гидромалонат. Рацемические соединения по изобретению могут быть получены из рацемических исходных материалов. Оптически активные изомеры могут быть получены из оптически активных исходных материалов или из рацемата. Энантиомеры могут быть получены из рацемата известными методами, например фракционной кристаллизацией диастериоизомерных солей, например их солей с (+)-ди-O,O’-п-толуоил-D-винной кислотой или (-)-ди-O,O’-p-толуоил-L-винной кислотой. Исходные материалы формулы II могут быть получены путем галогенизирования соединений формулы IV 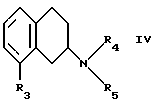 где R3, R4 и R5 такие, как определено выше, в соответствии с известными процедурами, например как описано в примере 1. Исходные материалы формул III и IV известны или могут быть произведены аналогичным образом известными способами. Соединения по изобретению, например соединения формулы I и их фармацевтически приемлемые соли, полученные присоединением кислоты, в дальнейшем именуемые как агенты по изобретению, проявляют фармакологическую активность, поэтому успешно применимы как фармацевтические препараты. Агенты по изобретению обеспечивают продолжительную защиту против максимального судорог у мышей, вызываемых электрошоком, в дозах примерно от 1 до 100 мг/кг перорально и примерно от 0,32 до 32 мг/кг внутрибрюшинно [cf. E. A. Swinyard, J. Am. Pharm. Assoc. Scient. Ed. 38, 201 (1949); J. Pharmacol. Exptl. Therap. 106, 319 (1952)]. Агенты по изобретению поэтому пригодны для лечении эпилепсии и других судорожных состояний, таких как судорожная форма гипертонического криза. Более того, агенты по изобретению уменьшают неврологическое повреждение, вызываемое ишемией, и сопутствующие симптомы в модели окклюзии средней мозговой артерии на крысах в дозе от 1 до 30 мг/кг внутрибрюшинно, внутривенно и перорально [cf. A. Tamura et al., J. Cereb. Blood Flow Metabol. 1, 53-60 (1981)), A. Sauter, M. Rudin, Stroke IZ, 1228-1234 (1986)). Поэтому агенты по изобретению пригодны для лечения любых клинических состояний, включая компонент церебральной аноксии, гипоксии и/или ишемии, например при ишемическом повреждении серого и белого вещества, ударе, субарахноидальном кровоизлиянии, повреждениях и травме головного и спинного мозга, высоком внутричерепном давлении, при деменции вследствие множественных инфарктов и сосудистой деменции, при любых хирургических вмешательствах, потенциально связанных с церебральной аноксией, гипоксией и/или ишемией (например при искусственном кровообращении, операциях на внемозговых сосудах). Агенты по изобретению проявляют свойство связываться с чувствительным к вератридину натриевым каналом с ИК50, составляющими от примерно 0,1 до примерно 100 мкМ. Процедура связывания известна (J.B. Braun, Journal of Neuroscience 6, 2064-2070 (1986). Они блокируют вератридин-индуцированное высвобождение глютамата в препаратах срезов крысиного гипокампа в концентрациях примерно 0,1 – 1 мкМ. Эксперимент проводят согласно известной модификации [M. J. Leach et al. In Epilepsia 27, 490-497 (1986) and Stroke 24, 1063-1067 (1993)], используя экзогенный глютамат. Как результат, агенты по изобретнию показаны для лечения любой патологии, нарушения или клинического состояния, этиология которых включает в себя высвобождение глютамата, включая психические расстройства (такие как шизофрения, депрессия, чувство тревоги, приступы паники, дефицит внимания и расстройство познавательной способности, социальное отчуждение); гормональные нарушения (избыток секреции гормона роста (GH) [например, при лечении диабета, ангиопатии и акромегалии] или избыток секреции лютеинизирующего гормона (LH) [гипертрофия предстательной железы, климактерический синдром], избыток секреции кортикостерона при стрессе); метаболически индуцированное поражение головного мозга (гипогликемия, некетотиновая гиперглицинемия [глициновая энцефалопатия] , дефицит сульфитоксидазы, печеночная энцефалопатия, связанная с несостоятельностью печени), рвоту, спазмы, шум в ушах, боль (онкологическая, при артритах) и злоупотребление лекарственными средствами (этанол, опиаты [включая синтетические с опиатоподобным действием, например петидин, метадон], кокаин, амфетамин, барбитураты и другие седативные средства; бензодиазепины) и абстиненцию. Кроме того, агенты по изобретению показаны при лечении любых патологий, включающих в себя неврологическое повреждение, например нейродегенеративных расстройств (таких как болезни Альцгеймера (Aizheimer’s), Хантингтона (Huntington’s), Паркинсона (Parkinson’s)), нейродегенерации, индуцированной вирусами (включая ВИЧ), амиотрофического бокового склероза (ALS), надъядерного паралича, оливомостомозжечковую атрофию (OPCA), воздействия окружающей среды, экзогенных нейротоксинов. Для вышеуказанных показаний подходящая доза будет, конечно, варьировать в зависимости от, например, используемого соединения, реципиента, способа введения и природы и тяжести состояния, подлежащего лечению. Однако в общем удовлетворительные результаты на животных получают при суточной дозе от примерно 0,1 до примерно 100, предпочтительно от примерно 0,5 до примерно 100 мг/кг массы тела животного. Для крупных млекопитающих, например человека, указанная суточная доза лежит в пределах от примерно 1 до примерно 500, предпочтительно от примерно 1 до примерно 300 мг агента по изобретению, при соответствующем введении, например в виде разделенных доз до 4-х раз в день или в форме поддерживаемого высвобождения. Для всех этих показаний предпочтительным соединением является (R)-1,2,3,4-тетрагидро-8-метокси-N, N-диметил-5-[4-трифторометил)фенил] -2- нафталинамин, который является соединением примера 21. Например, было установлено, что в вышеупомянутой модели электрошока это соединение обеспечивает защиту против максимальных индуцируемых электрошоком судорог в пороговой дозе 10 мг/кг перорально на период до 8 часов после введения. В модели MCA окклюзии было установлено, что соединение, введенное внутрибрюшинно немедленно после окклюзии, дозозависимым образом уменьшает размер инфаркта при 3,2, 10 и 32 мг/кг (19, 43 и 53% соответственно). В тесте с вератридин-индуцированным высвобождением глютамата было обнаружено, что соединение блокирует высвобождение с ИК50, составляющей 0,5 мкМ, что согласуется с его сродством к сайту связывания вератридина (ИК50 = 125 нМ). Соединение примера 21, например, превосходит стандарт лифразин в модели MCA окклюзии (снижение размеров инфаркта на 43% против 25% после дозы 10 мг/кг, введенной внутрибрюшинно). В тесте с вератридин-индуцированным высвобождением глютамата было обнаружено, что оно примерно равно по силе действия лифразину, но превосходит стандарты рилюзола и ламотригин (ИК50 = 0.5 мкМ против 5 мкМ и 20 мкМ соответственно). Предпочтительными показаниями являются эпилепсия, удар, травма головного мозга и спинальная травма. Агент по изобретению может быть введен любым традиционным способом, в частности энтерально, предпочтительно перорально, например в форме таблеток или капсул, или парентерально, например в форме инъекционных растворов или суспензий. Согласно вышеупомянутому, в настоящем изобретении предлагается также агент по изобретению для использования в качестве фармацевтического средства, например, для лечения эпилепсии, удара, травмы головного мозга и спинальной травмы. В настоящем изобретении дополнительно предлагается фармацевтическая композиция, содержащая агент по изобретению в сочетании с по меньшей мере одним фармацевтическим наполнителем или разбавителем. Такие композиции могут быть изготовлены традиционным способом. Формы стандартной дозы содержат, например, от примерно 0,25 до примерно 150, предпочтительно от 0,25 до примерно 25 мг соединения по изобретению. Кроме того, в настоящем изобретении предлагается использование агента по изобретению для изготовления лекарственного средства для лечения любого из вышеуказанных состояний, например эпилепсии, удара, травмы головного мозга и спинальной травмы. В еще одном аспекте настоящего изобретения предлагается способ лечения любого из вышеуказанных состояний, например эпилепсии, удара, травмы головного мозга и спинальной травмы, у субъекта в случае необходимости в таком лечении, при котором на указанного субъекта воздействуют терапевтически эффективным количеством агента по изобретению. Следующие примеры иллюстрируют изобретение. Температуры даны в градусах Цельсия и нескорректированы. Пример 1: (+/-)-1,2,3,4-тетрагидро-5-(4-хлорфенил)-8-метокси-N, N- диметил-2-нафталинамин 0,57 г (2 ммоль) (+/-)-1,2,3,4-тетрагидро-5-бром-8-метокси-N,N- диметил-2-нафталинамина растворяют в 8 мл толуола. Добавляют 0,55 г (3,54 ммоль) 4-хлорфенилбороновой кислоты, 0,07 г (0,23 ммоль) три(ортотолил)фосфина, 3 мл 2N водного карбоната натрия и 0,7 мл метилового спирта. После дегазации и заполнения системы аргоном добавляют 0,031 г (0,14 ммоль) ацетата палладия (II) и смесь перемешивают в течение ночи при температуре 80 градусов. Водную фазу отделяет и экстрагируют этилацетатом. Объединенные органические фазы экстрагируют 2N уксусной кислотой, кислые экстракты подщелачивают водным раствором аммиака и снова экстрагируют этилацетатом. После высушивания с сульфатом натрия, фильтрации и испарения органической фазы полученное светло-коричневое масло обрабатывают фумаровой кислотой в метил-трет.-бутилэфире. Полученную соль перекристаллизовывают от изопропанола, получая белые кристаллы гидрофумарата соединения, указанного в заголовке, с температурой плавления 213-216o. Исходный материал может быть получен следующим образом: 4,1 г (20 ммоль) (+/-)-1,2,3,4-тетрагидро-8-метокси-N,N-диметил-2- нафталинамин растворяют в 50 мл уксусной кислоты и добавляют 1,8 г (22 ммоль) ацетата натрия. Раствор 1,02 мл (20 ммоль) брома в 5 мл уксусной кислоты добавляют капля за каплей при комнатной температуре приблизительно в течение 30 минут. Формируется бесцветный осадок. После перемешивания в течение ночи растворитель отгоняют в условиях вакуума, остаток собирают водой и экстрагируют этилацетатом. Оставшуюся водную фазу подщелачивают водным раствором аммиака и экстрагируют этилацетатом. Органическую фазу сушат сульфатом натрия, фильтруют и выпаривают до сухого остатка. Оставшееся масло перегоняют в аппарате для перегонки “колба в колбу”, получая светло-желтое масло с температурой кипения 170-180o при 0,04 мбар. Следующие соединения получают аналогично примеру (см. таблицу). Желатиновые капсулы могут быть получены обычным способом и пригодны для использования при лечении эпилепсии и мозговых ударов. Желатиновые капсулы Соединение формулы I, например, пример 21 в виде свободного основания – 10,0 мг Лактоза – 165,5 мг Диоксид кремния (Аэросил) – 1,5 мг Кукурузный крахмал – 120,0 мг Стеарат магния – 3,0 мг Всего – 300,0 мг Пустая капс. – 77,0 мг Результ. вес – 377,0а Формула изобретения
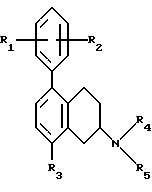 R1 и R2 независимо представляют собой водород, C1-4алкил, C1-4алкоксил, галоген, трифторметил; R3 представляет собой водород, гидроксил, C1-4алкоксил, цианогруппу, карбамоил; R4 и R5 независимо представляют собой водород, C1-4алкил, гидрокси-C2-4алкил, или образуют вместе с атомом азота, к которому они присоединены, пиперидино-группу, в виде свободного основания или соли, полученной присоединением кислоты. 2. Соединение по п.1, отличающееся тем, что R1 и R2 независимо представляют собой водород, C1-4алкил, C1-4алкоксил, галоген, трифторметил; R3 представляет собой водород, гидроксил, C1-4алкоксил, цианогруппу, карбамоил R4 и R5 независимо представляют собой водород, C1-4алкил, или образуют вместе с атомом азота, к которому они присоединены, пиперидино-группу, в виде свободного основания или соли, полученной присоединением кислоты. 3. Соединение по п.1, отличающееся тем, что представляет собой соединение формулы I, в оптически активной или рацемической форме, где R1=4-Cl, R2=H, R3=OMe, R4=Me, R5=Me, R1=H, R2=H, R3=OMe, R4=Me, R5=Me, R1=2-Cl, R2=H, R3=OMe, R4=Me, R5=Me, R1=2-Me, R2=H, R3=OMe, R4=Me, R5=Me, R1=2-OMe, R2=H, R3=OMe, R4=Me, R5=Me, R1=H, R2=H, R3=OH, R4=Me, R5=Me, R1=H, R2=H, R3=H, R4=Me, R5=Me, R1=4-Me, R2=H, R3=OMe, R4=Me, R5-Me, R1=4-CF3, R2=H, R3=OMe, R4=Me, R5=Me, R1=2-Cl, R2=4-Cl, R3=OMe, R4=Me, R5=Me, R1=2-F, R2H, R3=OMe, R4=Me, R5=Me, R1=3-Cl, R2=H, R3=OMe, R4=Me, R5=Me, R1=2-Cl, R2=H, R3=OMe, R4=Me, R5=H, R1=H, R2=H, R3=OMe, R4+R5=пиперидино, R1=2-Cl, R2=H, R3=OMe, R4+R5=пиперидино, R1=H, R2=H, R3=OMe, R4=n-Pr, R5=n-Pr, R1=4-CF3, R2=H, R3=OH, R4=Me, R5=Me, R1=2-Et, R2=H, R3=OMe, R4=Me, R5=Me, R1=2-Cl, R2=H, R3=CN, R4=Me, R5=Me, R1=2-Cl, R2=4-F, R3=OMe, R4=Me, R5=Me, R1=2-F, R2=4-F, R3=OMe, R4=Me, R5=Me, R1=2-OMe, R2=4-OMe, R3=OMe, R4=Me, R5=Me, R1=2-F, R2=3-F, R3=OMe, R4=Me, R5=Me, R1=2-OMe, R2=3-OMe, R3=OMe, R4=Me, R5=Me, R1=2-Me, R2=5-Me, R3=OMe, R4=Me, R5=Me, R1=2-Cl, R2=3-Cl, R3=OMe, R4=Me, R5=Me, R1=2-F, R2=5-F, R3=OMe, R4=Me, R5=Me, R1=2-Me, R2=4-Me, R3=OMe, R4=Me, R5=Me, R1=2-Cl, R2=H, R3=CONH2, R4=Me, R5=Me, R1=4-CF3, R2-H, R3=OMe, R4=Me, R5=H, R1=4-OMe, R2=H, R3=OMe, R4=Me, R5=Me, R1=4-F, R2=H, R3=OMe, R4=Me, R5=Me, R1=4-CF3, R2=H, R3=OMe, R4=Et-OH, R5=Me, и R1=4-CF3, R2=H, R3=H, R4=Me, R5=Me Me -метил, Et – этил и Pr – пропил, в виде свободного основания или соли, полученной присоединением кислоты. 4. Соединение по п.1, которое представляет собой (R)-1,2,3,4-тетрагидро-8-метокси-N, N-диметил-5-[4-(трифторметил)фенил] -2-нафталинамин в виде свободного основания или соли, полученной присоединением кислоты. 5. Соединение по п. 1 в виде свободного основания или фармацевтически приемлемой соли, полученной присоединением кислоты, для использования в фармацевтическом средстве. 6. Соединение по п. 1 в виде свободного основания или фармацевтически приемлемой соли, полученной присоединением кислоты, обладающее антиконвульсивной активностью, активностью при нейрональных повреждениях, вызванных ишемией, и активностью в условиях, включающих высвобождение глютамата. 7. Способ получения замещенных 1,2,3,4-тетрагидронафтиламинов общей формулы I по п.1 в виде свободного основания или соли, полученной присоединением кислоты, отличающийся тем, что соединение формулы II, 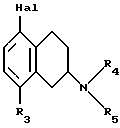 где R3, R4 и R5 такие, как определено в п.1; Hal представляет собой галоген, подвергают взаимодействию с соединением формулы III 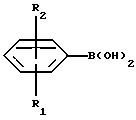 где R1 и R2 такие, как определено в п.1, и полученное соединение выделяют в виде свободного основания или соли, полученной присоединением кислоты. 8. Фармацевтическая композиция, обладающая антиконвульсивной активностью, активностью при ишемии, вызванной нейрональными повреждениями и активностью в условиях, включающих высвобождение глютамата, содержащая активный агент в сочетании с фармацевтическим наполнителем или разбавителем, отличающаяся тем, что в качестве активного агента она содержит соединение I по п. 1 формулы в форме свободного основания или фармацевтически приемлемой соли, полученной присоединением кислоты, в терапевтически эффективном количестве. РИСУНКИ
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 04.05.2005
Извещение опубликовано: 20.02.2007 БИ: 05/2007
|
||||||||||||||||||||||||||