Патент на изобретение №2169149
|
||||||||||||||||||||||||||
(54) СОЕДИНЕНИЯ СО СТРУКТУРОЙ КАМПТОТЕЦИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
(57) Реферат: Изобретение относится к новым алкалоидам формулы I 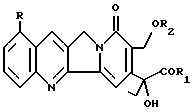 присутствующим в различных частях Mappia foetida, а также к их фармацевтическому использованию и использованию их в качестве новых синтонов для получения соединений с противоопухолевой и антивирусной активностью, те же самые продукты являются новыми синтонами для получения новых аналогов камптотецина и фоетидинов. Описывается также способ получения соединений I. Особая водорастворимость этих новых соединений исключительно важна, так как позволяет вводить их парентерально без необходимости превращения в производные. 5 с. и 2 з.п. ф-лы, 1 табл. Изобретение относится к алкалоидам со структурой камптотецина, выделенным из Mappia foetida или полученным полусинтезом из указанных алкалоидов. Известно, что Mappia foetida, растение, растущее в индейском субконтиненте, содержит в своих различных частях, главным образом, в семенах, камптотецин, маппицин и фоетидин I и II (ЕР А-685481). В “Journal of Medicinal Chemistry”, 1979, vol. 22, N 3, описываются производные камптотецина и их получение. Алкалоиды изобретения имеют следующую общую формулу I 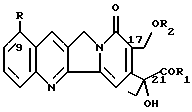 в которой R представляет атом водорода или метоксигруппу; R1 представляет гидрокси, ОМ-группу, где М представляет катион щелочного металла, предпочтительно натрия или калия, 1-C6-алкоксигруппу, необязательно замещенную феноксигруппу, амино, C1-C6-моноалкиламино или C2-C12-диалкиламиногруппу, в которой алкильная часть необязательно замещена аминогруппами, ариламиногруппу; R2 представляет C2-C6-алкильную группу или группу формулы COR3, где R3 представляет алкил C1-C6 или необязательно замещенный фенил или бензил. Фенокси, фенильные или бензильные группы могут быть замещены атомами галогена; C1-С6-алкильными, C1-С6-алкокси, нитро, циано, C1-C3-галогеналкильными группами. Соединения формулы I, в которой R представляет водород или метокси, R1 представляет гидрокси или ОМ-группу (М = натрий или калий) и R2 представляет ацетил, могут быть выделены из Mappia foetida экстракцией искусственно высушенной растительной биомассы при температурах не выше, чем, 50oC, предпочтительно при 35oC, сначала алифатическими кетонами или алифатическими сложными эфирами и затем алифатическими спиртами. В этих рабочих условиях 17-ацетилпроизводные камптотециновой и 9-метоксикамптотециновой кислот можно экстрагировать с высокими выходами. Несмотря на то, что Mappia foetida был широко изучен в качестве селективного источника камптотецина, указанные алкалоиды не были идентифицированы, вследствие, вероятно, их разложения до камптотецина во время экстракции с использованием неподходящих растворителей. В присутствии алифатических спиртов эти алкалоиды легко превращаются в камптотецин даже при природном pH экстракции. Эту же группу соединений можно получить селективным ацетилированием C17-гидроксила камптотецина в щелочной среде. Получаемые соединения можно, в свою очередь, использовать в качестве исходных материалов для получения других соединений формулы I, в которой R2 отличается от ацетила и/или R1 представляет алкокси, фенокси или аминогруппу, как определено выше, или для получения фоетидинов I и II. Для этих целей можно использовать общепринятые способы получения сложных эфиров или амидов, например, реакцию соединений I, в которых R1 представляет ОМ-группу, с алкилгалогенидами, такими как этил- или бензилбромацетат, для получения сложных эфиров, или реакцию соединений I, в которых R1 представляет ОН, с амином и дициклогексилкарбодиимидом для получения амидов. Соединения I обладают цитотоксической активностью против клеточных линий опухолей. Например, в таблице 1 представлена цитотоксическая активность против линии карциномы толстой кишки (НСТ116) и против той же самой линии, устойчивой к наиболее обычной химиотерапии (HCT116/VM46). Результаты доказали, насколько соединение изобретения является более активным, чем камптотецин. Соединения I можно, следовательно, использовать в качестве действующего начала в противоопухолевых фармацевтических композициях в смеси с подходящими носителями, например, инъецируемыми физиологическими растворами. Дозы могут изменяться в широких пределах (от 5 до 500 мг/день), но в принципе они будут составлять примерно 10 мг алкалоида в день. Следующие примеры дополнительно иллюстрируют изобретение. Пример 1 Выделение 17-ацетилкамптотециновой и 17-ацетил-9-метокси-камптотециновой кислот 3 кг семян Mappia foetida экстрагируют три раза сухим ацетоном (3 х 3 л) при комнатной температуре. Объединенные экстракты концентрируют досуха для получения 580 г воскообразной массы, содержащей камптотецин, 9-метоксикамптотецин и небольшое количество 17-ацетилкамптотециновой кислоты. Растительный материал от экстракции ацетоном снова неоднократно экстрагируют метанолом (3 х 3 л) при 10oC; после концентрирования экстрактов при низкой температуре получают 200 г сухого остатка, который суспендируют в 1 л воды и экстрагируют три раза 500 мл н-бутанола; объединенные бутанольные экстракты концентрируют досуха в вакууме при температуре не выше, чем 30oC. Получают 28,9 г алкалоидной фракции, обогащенной смесью 17-ацетилкамптотециновой и 9-метокси-17-ацетилкамптотециновой кислот, и хроматографируют в обращенной фазе через колонку RP18, элюируя смесью метанол/вода и метанолом с получением трех фракций, состоящих соответственно из кумароилагматина и камптотециновых кислот. Эту фракцию очищают дополнительно на силикагеле, получая 3,8 г 17-ацетилкамптотециновой кислоты, имеющей следующие спектроскопические и химико-физические характеристики: т. пл. 258oC,  D = +63,4 (с = 0,05, H2O); D = +63,4 (с = 0,05, H2O);1H-NMR (DMSO-d6)  : 0.85 (t, 3H, H-18), 1.95 (m+s, 5H, Н-19+COCH3), 5.20 (s, 2H, H-17), 5.40,60 (q, JAB = 10.6 Hz, H-5), 7.65-8.65 (m, 6H, arom).
Количество 9-метокси-17-ацетилкамптотециновой кислоты составляет одну пятую часть предшествующего количества и она имеет следующие физико-химические характеристики: т. пл. 208oC, D = +56,4 (с = 0,05, H2O).
Пример 2 : 0.85 (t, 3H, H-18), 1.95 (m+s, 5H, Н-19+COCH3), 5.20 (s, 2H, H-17), 5.40,60 (q, JAB = 10.6 Hz, H-5), 7.65-8.65 (m, 6H, arom).
Количество 9-метокси-17-ацетилкамптотециновой кислоты составляет одну пятую часть предшествующего количества и она имеет следующие физико-химические характеристики: т. пл. 208oC, D = +56,4 (с = 0,05, H2O).
Пример 2Получение 17-ацетилкамптотециновой кислоты из камптотецина 1 г камптотецина суспендируют в 30 мл воды, добавляют 340 мг NaOH и выдерживают при перемешивании при 40oC в течение двух часов или, в любом случае, до полного растворения; воду удаляют в вакууме и остаток растворяют в 20 мл ДМФ при сильной реакции; к раствору постепенно добавляют 600 мг уксусного ангидрида и всю массу выдерживают при перемешивании в течение примерно 2 часов. Растворитель удаляют в вакууме и остаток распределяют в смеси хлороформ/метанол/вода, 5: 6:4. Фазу метанола концентрируют досуха и остаток кристаллизуют, получая 17-ацетилкамптотециновую кислоту, имеющую такие же характеристики, как характеристики, описанные в примере 1. Пример 3 17-ацетилкамптотецин-21-метиловый эфир 17-Ацетилкамптотецин (100 мг, 0,25 ммоль) растворяют в сухом ДМФ (8 мл) и к нему добавляют сухой карбонат калия (68 мг, 0,49 ммоль) и иодметан (69 мг, 0,49 ммоль), перемешивают при комнатной температуре в течение 20 часов. Реакционную смесь фильтруют и промывают хлороформом (5 мл). Фильтраты разбавляют хлороформом (10 мл) и промывают водой (5 мл х 3). Органическую фазу сушат над сухим сульфатом натрия. После фильтрования растворитель удаляют в вакууме и остаток (170 мг) подвергают флэш-хроматографии (CHCl3:CH3ОН, 9:1). Указанное в заголовке соединение получают (45 мг, выход: 45%) в виде твердого вещества. 1H NMR (CDCl3) : 1.02 (t, J=7 Hz, 3H, H-18), 2.09 (s, 3H, OCOCH3), 2.26-2.45 (m, 2H, H-19), 3.82 (s, 3H, OCH3), 5.38 (s, 2H, H-5), 5.52 (s, 2H, H-17), 7.51-8.42 (m, 6H, arom). MS (EI) M+ 422. Т. пл. (разложение) 234-235oC.
Следуя тому же способу, но используя этилбромацетат или трет-бутилбромацетат вместо иодметана, получают соответствующий этиловый (а) или трет-бутиловый (b) эфиры.
(а) 1H NMR (CDCl3) : 1.10 (t, J=7.5 Hz, 3H, H-18), 1.30 (t, J=7.5 Hz, 3H, CH3), 2.10 (s, 3H, OCOCH3), 2.30-2.55 (m, 2H, H-19), 4.25 (q, J=7.5 Hz, 2H, CH2), 4.70 (q, JAB= 15 Hz, 2H, OCOCH2CO), 5.32 (s, 2H, H-5), 5.52 (s, 2H, H-17), 7.6 (m, 5H, arom).
(b) 1H NMR (CDCl3) : 1.10 (t, J= 7.5 Hz, 3H, H-18), 1.46 (s, 9Н, С(CH3)3), 2.10 (s, 3H, OCOCH3), 2.35-2.52 (m, 2H, H-19), 4.60 (q, JAB= 15 Hz, 2H, OCOCH2CO), 5.30 (s, 2H, H-5), 5.52 (s, 2H, H-17), 7.58-8.38 (m, 6H, arom).
Пример 417-деацетилкамптотециновая кислота, 21-эфир Соединение b (60 мг, 0,11 ммоль) растворяют в сухом хлороформе (2 мл). При 0oC в атмосфере азота добавляют иодтриметилсилан (33 мг, 0,17 ммоль), перемешивая при 0oC в течение 1 часа и при комнатной температуре в течение 1 часа. Реакционную смесь выливают в 5% раствор NaHCO3 (5 мл). Водную фазу промывают неоднократно хлороформом до тех пор, пока фаза хлороформа не стала бесцветной. Водную фазу нейтрализуют 2,5% раствором HCl при 0oC до pH 7 и экстрагируют бутанолом (5 мл х 6). Фазы бутанола объединяют и выпаривают в вакууме, получая остаток (51 мг), который подвергают флэш-хроматографии через силикагель, элюируя смесью хлороформ-метанол, получая указанное в заголовке соединение (11 мг). 1H NMR (DMSO-d6) : (ppm) 0.82 (t, J=7Hz, 3H, Н-18), 2.12 (s+m, 5H, Н-19 and H-17), 4.29 (f, JAB = 15 Hz, 2H, 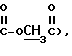 5.22 (s, 2H, H-5), 6.62 (s, 1H, OH), 7.50-8.62 (m, 6H, arom).
13H NMR (DMSO-d6) :: (ppm) 7.7 (t, C-19), 13.7 (t, C-17), 30.01 (f, C-18), 50.2 (f, C-5), 63.9 (f, C-5), 77.5 (s, C-20), 99.1 (d, C-14), 125.9 (s, C-16), 127.3 (d, C-10), 127.8 (s, C-8), 128.6 (d, C-9), 128.9 (d, C-12), 129.7 (s, C-6), 130.3 (d, C-11), 131.4 (d, C-7), 141.3 (s, C-3), 148.0 (s, C-13), 150.7 (s, C-15), 153.9 (s, C-2), 160.8 (s, 16a), 171.0 (s, 5.22 (s, 2H, H-5), 6.62 (s, 1H, OH), 7.50-8.62 (m, 6H, arom).
13H NMR (DMSO-d6) :: (ppm) 7.7 (t, C-19), 13.7 (t, C-17), 30.01 (f, C-18), 50.2 (f, C-5), 63.9 (f, C-5), 77.5 (s, C-20), 99.1 (d, C-14), 125.9 (s, C-16), 127.3 (d, C-10), 127.8 (s, C-8), 128.6 (d, C-9), 128.9 (d, C-12), 129.7 (s, C-6), 130.3 (d, C-11), 131.4 (d, C-7), 141.3 (s, C-3), 148.0 (s, C-13), 150.7 (s, C-15), 153.9 (s, C-2), 160.8 (s, 16a), 171.0 (s, 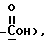 172.8 (s, C-21).
Основные методы исследования. Спектры 1H-NMR и 13C-NMR получали на спектрометре Bruker АС 200 либо Bruker АС 300; все значения представлены в м.д. (). ВЭЖХ анализ осуществляли на хроматографе Merk Hitachi LC.6200А. Данные ИК-спектров получали на FT/IR JASCO-300E. В случаях, когда требовалось проведение исследований в безводной среде, использовали атмосферу азота и растворители, свежеперегнанные из CaH2 или Na.
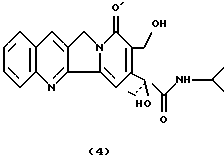
Камптотецин 21-изопропиламид (4). Камптотецин (400 мг, 1,04 ммоль) суспендировали в изопропиламине (80 мл) в атмосфере азота и нагревали в течение 2 дней при температуре кипения с обратным холодильником с получением прозрачного раствора оранжевого цвета. После упаривания растворителя сырой продукт очищали флэш-хроматографией (силикагель, CHCl3: CH3OH 95:5) с получением соединения 4 в виде твердого вещества желтого цвета (457 мг, 98%).
1H-NMR(Py-d5) : 1.2-1.4 (9H), 2.55(dq, 1H), 2.75 (dq, 1H), 4.45 (m, 1H), 5.1 (s, 2H), 5.5 (d, 1H, J=8), 5.8 (d, 1H, J=8), 7.6 (t, 1H, J=5.3), 7,8 (t, 1H, J=5.3), 7.95 (d, 1H, J=5.3), 8.05 (s, 1H), 8.20 (d+s, 2H), 8.35 (d, 1H, J=5.3).
13C-NMR (Py-d5) : 10.111 (q), 23.927 (q), 24.186(q), 34.469 (t), 43.425 (d), 52.326 (t), 58.693 (t), 81.450 (s), 103.435 (d), 129.625 (d), 130.154 (s), 130.605 (d), 130.951 (d), 131.189 (s), 132.039 (s), 132.463 (d), 133.609 (d), 145.170 (s), 150.262 (s), 154.638 (s), 157.126 (s), 164.053 (s), 175.149 (s).
ИК (см-1): 1667, 1650, 1511, 1458, 1405, 1240.
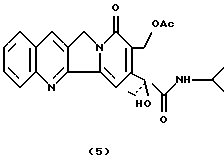
Камптотецин 17-ацетил-21-изопропиламид (5).
К раствору соединения 1 (95 мг, 0,23 ммоль) в CH2Cl2 (5 мл) добавляли безводный пиридин (0,74 мл) и Ас2О (0,73 мл). Реакционную смесь оставляли на ночь в условиях перемешивания, комнатной температуры и атмосферы азота. Смесь промывали водой, а затем 5% HCl и экстрагировали CHCl3. После упаривания растворителя неочищенный продукт очищали флэш-хроматографией (силикагель, CHCl3: CH3ОН 96: 4) с получением соединения 5 в виде твердого вещества желтого цвета (95 мг, 92%).
1H-NMR (CDCl3) : 1.10-120 (9H), 2.15 (s, 3H), 2.30 (dq, 1H), 2.60 (dq, 1H), 4.04 (m, 1H), 4.85 (d, 1H, J=18.6), 5.10 (d, 1H, J=18.6), 5.30 (s, 1H), 5.40 (d, 1H, J=13.3), 5.55 (d, 1H, J=13.3), 6.9 (d, 1H), 7.20-7.90 (5H).
13С-NMR (CDCl3) : 7.816, 20.921, 22.273, 22.679, 33.125, 41.659, 50.236, 59.199, 78.681, 100.654, 125.088, 125.100, 127.528, 127.675, 127.824, 128.188, 129.319, 130.087, 130.474, 144.297, 148.457, 151.921, 161.567, 171.298 (x2).
N-Ди-трет-бутоксикарбонил-1,4-диаминобутан (6).
К раствору 1,4-диаминобутана (2 г, 0,023 моль) и Et3N (2,27 г, 0,023 моль) в ТГФ (35 мл) по каплям добавляют раствор BOC2О (2,47 г, 0,0113 моль) в ТГФ (10 мл). Реакционную смесь перемешивают в течение 1 дня при комнатной температуре. Белый осадок отфильтровывают и твердое вещество несколько раз промывают ТГФ. Объединенные фильтраты упаривают в вакууме и неочищенный продукт очищают хроматографией (силикагель, CHCl3:CH3OH 9:1 и CHCl3:CH3ОН: изопропиламин 9:1:0,5) с получением соединения 6 в виде масла (1,03 г).
1H-NMR (CDCl3) : 1.4 (9H), 1.5(m, 4H), 2.66 (t,2H, J=6), 3.09 (2H).
Камптотецин 21-N-ди-трет-бутоксикарбонил-4-аминобутанамид(7) 172.8 (s, C-21).
Основные методы исследования. Спектры 1H-NMR и 13C-NMR получали на спектрометре Bruker АС 200 либо Bruker АС 300; все значения представлены в м.д. (). ВЭЖХ анализ осуществляли на хроматографе Merk Hitachi LC.6200А. Данные ИК-спектров получали на FT/IR JASCO-300E. В случаях, когда требовалось проведение исследований в безводной среде, использовали атмосферу азота и растворители, свежеперегнанные из CaH2 или Na.
Камптотецин 21-изопропиламид (4). Камптотецин (400 мг, 1,04 ммоль) суспендировали в изопропиламине (80 мл) в атмосфере азота и нагревали в течение 2 дней при температуре кипения с обратным холодильником с получением прозрачного раствора оранжевого цвета. После упаривания растворителя сырой продукт очищали флэш-хроматографией (силикагель, CHCl3: CH3OH 95:5) с получением соединения 4 в виде твердого вещества желтого цвета (457 мг, 98%).
1H-NMR(Py-d5) : 1.2-1.4 (9H), 2.55(dq, 1H), 2.75 (dq, 1H), 4.45 (m, 1H), 5.1 (s, 2H), 5.5 (d, 1H, J=8), 5.8 (d, 1H, J=8), 7.6 (t, 1H, J=5.3), 7,8 (t, 1H, J=5.3), 7.95 (d, 1H, J=5.3), 8.05 (s, 1H), 8.20 (d+s, 2H), 8.35 (d, 1H, J=5.3).
13C-NMR (Py-d5) : 10.111 (q), 23.927 (q), 24.186(q), 34.469 (t), 43.425 (d), 52.326 (t), 58.693 (t), 81.450 (s), 103.435 (d), 129.625 (d), 130.154 (s), 130.605 (d), 130.951 (d), 131.189 (s), 132.039 (s), 132.463 (d), 133.609 (d), 145.170 (s), 150.262 (s), 154.638 (s), 157.126 (s), 164.053 (s), 175.149 (s).
ИК (см-1): 1667, 1650, 1511, 1458, 1405, 1240.
Камптотецин 17-ацетил-21-изопропиламид (5).
К раствору соединения 1 (95 мг, 0,23 ммоль) в CH2Cl2 (5 мл) добавляли безводный пиридин (0,74 мл) и Ас2О (0,73 мл). Реакционную смесь оставляли на ночь в условиях перемешивания, комнатной температуры и атмосферы азота. Смесь промывали водой, а затем 5% HCl и экстрагировали CHCl3. После упаривания растворителя неочищенный продукт очищали флэш-хроматографией (силикагель, CHCl3: CH3ОН 96: 4) с получением соединения 5 в виде твердого вещества желтого цвета (95 мг, 92%).
1H-NMR (CDCl3) : 1.10-120 (9H), 2.15 (s, 3H), 2.30 (dq, 1H), 2.60 (dq, 1H), 4.04 (m, 1H), 4.85 (d, 1H, J=18.6), 5.10 (d, 1H, J=18.6), 5.30 (s, 1H), 5.40 (d, 1H, J=13.3), 5.55 (d, 1H, J=13.3), 6.9 (d, 1H), 7.20-7.90 (5H).
13С-NMR (CDCl3) : 7.816, 20.921, 22.273, 22.679, 33.125, 41.659, 50.236, 59.199, 78.681, 100.654, 125.088, 125.100, 127.528, 127.675, 127.824, 128.188, 129.319, 130.087, 130.474, 144.297, 148.457, 151.921, 161.567, 171.298 (x2).
N-Ди-трет-бутоксикарбонил-1,4-диаминобутан (6).
К раствору 1,4-диаминобутана (2 г, 0,023 моль) и Et3N (2,27 г, 0,023 моль) в ТГФ (35 мл) по каплям добавляют раствор BOC2О (2,47 г, 0,0113 моль) в ТГФ (10 мл). Реакционную смесь перемешивают в течение 1 дня при комнатной температуре. Белый осадок отфильтровывают и твердое вещество несколько раз промывают ТГФ. Объединенные фильтраты упаривают в вакууме и неочищенный продукт очищают хроматографией (силикагель, CHCl3:CH3OH 9:1 и CHCl3:CH3ОН: изопропиламин 9:1:0,5) с получением соединения 6 в виде масла (1,03 г).
1H-NMR (CDCl3) : 1.4 (9H), 1.5(m, 4H), 2.66 (t,2H, J=6), 3.09 (2H).
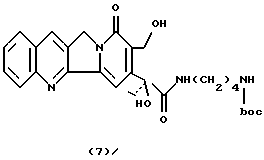
Камптотецин 21-N-ди-трет-бутоксикарбонил-4-аминобутанамид(7)К суспензии камптотецина (250 мг, 0,72 ммоль) в сухом пиридине (15 мл), добавляют соединение 6 (541 мг, 2,88 ммоль) в атмосфере азота. Реакционную смесь нагревают до 80oC с получением прозрачного раствора желтого цвета. После выдерживания в течение трех дней при температуре 80oC охлажденную смесь промывают 5% HCl, экстрагируют CHCl3 и сушат над Na2SO4. После упаривания растворителя неочищенный продукт очищают флэш-хроматографией (силикагель, CHCl3: CH3ОН 95: 5) с получением соединения 7 в виде твердого вещества желтого цвета (224 мг, 58%). 1H-NMR (CDCl3+D2O) : 1.05 (t, 3H, J =7.4), 1.40 (9H), 1.59 (m, 4H), 2.20 (dq, 1H), 2.41 (dq, 1H), 3.15 (m, 2H), 3.35 (m, 2H), 4.85 (d, 1H, J= 12), 4.94 (d+s, 3H, J=12), 7.30 (t, 1H), 7.41 (t+s, 2H), 7.48 (d, 1H, J= 8.4), 7.61 (t, 1H, J=8.4), 7.82 (s. 1H), 7.95 (d, 1H, J=8.4).
13С-NMR (CDCl3) : 7.769, 26.543, 27.311, 28.211. 32.409, 38.962, 39.972, 49.817, 57.223, 78.771, 78.800 100.985, 127.278, 127.400, 127.892, 128.927, 129.049, 129.969, 130.191, 130.300, 142.961, 148.142, 151.689, 154.332, 155.966. 161.632, 173.011.
ВЭЖХ (RP8, 75%H2O, 25%CH3CN): 9,44 мин.
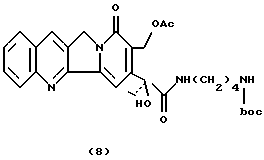
Камптотецин 17 ацeтил-21-N-ди-тpeт-бутoкcикapбoнил-14-аминобутанамид (8)Используя ту же процедуру, что и при получении соединения 5, соединение 7 (94 мг, 0,17 ммоль) подвергают ацетилированию с использованием Ас2О (0,33 мл) и безводного пиридина (0,34 мл) с получением соединения 8 в виде твердого вещества желтого цвета (81 мг. 82%). 1H-NMR(CDCl3) : 1.10 (t, 3H, J=7.4), 1.40 (9H), 1.60 (m, 4H), 2.01 (s, 3H), 2.30 (dq, 1H), 2.50 (dq, 1H), 3.15 (t, 2H), 3.30-3.40 (m, 2H), 4.82 (d, 1H, J= 18.6). 5.05 (d, 1H, J=18.6), 5.40 (d+d, 2H, J=13.3), 7.20 (t, 1H), 7.32 (d, 1H, J=8), 7.40 (2H), 7.55 (t, 1H, J=8), 7.9 (2H).
13C-NMR (CDCl3) : 7.891, 20.885, 26.675, 27.499, 28.339, 32.964, 39.059, 40.151, 50.227, 58.800, 78.506, 79.031, 100.612, 124.778, 127.265, 127.691 (x2), 127.800, 128.988, 130.039, 130.406, 144.179, 148.146, 151.425, 155.950, 157.120, 161.530, 171.127, 172.126.
ВЭЖХ (RP8, 75%H2O, 25%CH3CN): 10,48 мин.
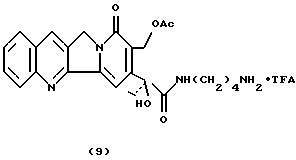
Камптотецин 17 ацетил-21-4-аминобутанамид трифторацетат (9)К раствору соединения 8 (30 мг, 0,052 ммоль) в безводном CH2Cl2 (2 мл) в атмосфере азота при температуре 0oC по каплям добавляют TFA (0,5 мл). Реакционную смесь перемешивают в атмосфере азота при температуре 0oC в течение 1,5 час, затем растворитель упаривают в вакууме и остаток растворяют в CH3ОН (с целью удаления оставшегося ТFА). После упаривания растворителя в результате повторного осаждения из CHCl3/CH3OH-петролейного эфира получают соединение 9 в виде твердого вещества желтого цвета (30,7 мг, 100%). 1H-NMR (DMSO-d6) : 0.9 (t, 3H), 1.5 (4H), 1.95 (s, 3H), 2.15 (m. 2H), 2.77 (2H), 3.1 (m, 2H). 5.3 (2H), 5.4 (2H), 7.4-8.7 (6H).
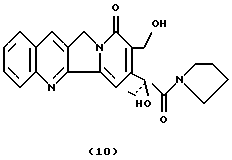
Камптотецин 21-пирролидинамид (10)Камптотецин (200 мг, 0,57 ммоль) растворяют в пирролидине (30 мл) и нагревают в течение 2 дней при температуре 70-80oC. После упаривания растворителя остаток повторно осаждают из CHCl3-петролейного эфира и неочищенный продукт очищают флэш-хроматографией (силикагель, CHCl3: CH3ОН 97:3) с получением соединения 10 в виде твердого вещества (124 мг, 52%). 1H-NMR (CDCl3) : 1.05 (t, 3H, J=7.4), 1.80 (m, 4H), 2.35 (q, 2H, J= 7.4),3.05 (m, 1H), 3.19 (m, 1H), 3.60 (m, 1H), 3.65 (m, 1H), 3.86 (t, 1H, J= 5.3), 4.74 (d, 2H, J=5.3), 5.30 (s, 1H), 5.35 (s, 2H), 7.64 (t+s, 2H), 7.82 (t, 1H, J=8), 7.92 (d, 1H, J=8), 8.24 (d, 1H, J=8), 8.40 (s, 1H).
13C-NMR (CDCl3) : 7.656, 23.209, 26.470, 30.731, 47.323, 47.829, 49.924, 57.650, 77.183, 100.048, 125.500, 127.757, 128.068, 128.530, 129.487, 130.451, 130.600, 130.997, 143.644, 151.549, 162.068, 170.856.
ВЭЖХ (RP8, 75%H20, 25%CH3CN): 4,23 мин.
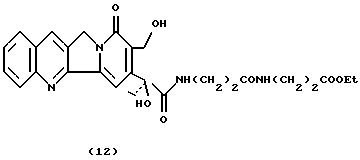
Камптотецин 21- -этиловый эфир-аланинамид (11) и (12) -этиловый эфир-аланинамид (11) и (12)К раствору гидрохлорида- -аланин этилового эфира (1,86 г, 12 ммоль) в CH2Cl2 (7 мл) при комнатной температуре в атмосфере азота добавляют Et3N (6,7 мл, 48 ммоль), и реакционную смесь перемешивают в течение 2 часов. Белый осадок отфильтровывают и фильтрат упаривают в вакууме (Т<30oC) с получением раствора -аланин этилового эфира в Et3N.
К суспензии камптотецина (300 мг, 0,86 ммоль) в безводном пиридине (16 мл) добавляют в атмосфере азота раствор -аланин этилового эфира в Et3N. Реакционную смесь нагревают до 80oC с получением прозрачного раствора желтого цвета. После трех дней выдерживания при 80oC охлажденную реакционную смесь промывают 5% HCl, экстрагируют CHCl3 и сушат над Na2SO4. После упаривания растворителя неочищенный продукт очищают флэш-хроматографией (силикагель, CHCl3: CH3ОН сначала 97:3, затем 95:5) с получением соединения 11 (252 мг, 63%) и соединения 12 (42 мг, 9,1%) в виде твердых веществ бледно-желтого цвета.
11: 1H NMR (CDCl3) : 1.05 (t, 3H, J=7.4), 1.28 (t, 3H, J=8), 2.30 (dq, 1H), 2.40 (dq, 1H), 2.64 (t, 2H, J=5.3), 3.60 (dt, 2H, J=5.3), 4.18 (q, 2H, J=8) 4.90 (d, 2H, J=3.2), 4.98 (s, 2H), 7.5 (m, 3H), 7.70 (t, 1H, J=8), 7.85 (s, 1H), 8.04 (d, 1H, J=8).
* двойные пики из-за наличия енольной формы амида.
12: 1H-NMR (CDCl3) : 1.05 (t, 3H, J=7.4), 1.25 (t, 3H, J=8), 2.35 (dq, 1H), 2.45 (dq, 1H), 2.50 (m, 4H), 3.45 (m, 2H), 3.60 (m, 2H), 4.10 (q, 2H, J= 8), 4.30 (d, 1H, J=10.6), 4.95 (d, 1H, J=10.6), 5.10 (s, 2H), 6.72 (t, 1H), 7.40 (t, 1H), 7.55 (2H), 7.70 (2H), 8.05 (d, 1H, J=8), 8.15 (s, 1H).
ВЭЖХ (RP8, 75%H2O, 25%CH3CH): 2,80 мин.
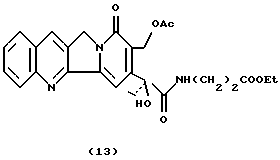
Камптотецин 17-ацетил-21--этиловый эфир-аланинамид (13)С использованием той же процедуры, что и для получения соединения 5, соединение 11 (80 мг, 0,18 ммоль) подвергают ацетилированию с использованием Ас2О (0,32 мл) и безводного пиридина (0,36 мл) с получением соединения 13 в виде твердого вещества желтого цвета (76 мг, 85%). 1H-NMR (CDCl3+D2O) : 1.10 (t, 3H, J=7.4), 1.30 (t, 3H, J=8), 2.10 (s, 3H), 2.35 (m, 2H), 2.50 (m, 2H), 2.55 (t, 2H, J=5.3), 3.55 (m, 2H), 4.15 (q, 2H, J=8), 5.10 (d, 1H, J=18.6), 5.20 (d, 1H, J=18.6), 5.42 (d, 1H, J=10.6), 5.55 (d, 1H, J=10.6), 7.4-8.1 (6H).
ВЭЖХ (RP8, 75%H2O, 25%CH3CN): 4,83 мин.
N-(2-цианоэтил)-1,4-диаминобутан (14)К 1,4-диаминобутану (26,4 г, 0,3 моль) в атмосфере азота при температуре 0oC добавляют по каплям ацетонитрил (15,9 г, 0,3 моль). Реакционную смесь перемешивают в течение 20-30 мин при температуре 4-5oC, в течение 30 мин при температуре 20-40oC и в течение 2 часов при температуре 100oC. Продукт очищают вакуумной перегонкой с получением соединения 14 в виде масла (т.кип. = 96-97oC, P = 6,6  10-2 мбар, 17,5 г, 41%).
1H-NMR (CDCl3) : 1.1 (s, 2H), 15 (m, 4H), 2.5 (t, 2H, J=7.2), 2.6-2.7 (4H, J=7.2), 2.9 (t, 2H, J=7.2).
N,N-Ди-трет-бутоксикарбонил-N-(2-цианоэтил)-1,4-диаминобутан (15) 10-2 мбар, 17,5 г, 41%).
1H-NMR (CDCl3) : 1.1 (s, 2H), 15 (m, 4H), 2.5 (t, 2H, J=7.2), 2.6-2.7 (4H, J=7.2), 2.9 (t, 2H, J=7.2).
N,N-Ди-трет-бутоксикарбонил-N-(2-цианоэтил)-1,4-диаминобутан (15)К раствору соединения 14 (5,16 г, 36,5 ммоль) и Et3N (12,2 г, 120,6 ммоль) в ТГФ (30 мл) добавляют по каплям раствор BOC2О (16,7 г, 76,6 ммоль) в ТГФ (20 мл). Реакционную смесь перемешивают при комнатной температуре в течение дня. Белый осадок отфильтровывают и твердое вещество промывают несколько раз ТГФ. Объединенные фильтраты упаривают в вакууме с получением соединения 15 в виде масла (12,45 г, 100%). 1H-NMR (CDCl3) : 1.5 (18H+4H), 2.55 (2H), 3.1 (dd, 2H, J=7.3), 3.25 (t, 2H, J=7.3), 3.45 (t, 2H, J=7.3).
N2,N3-Ди-трет-бутоксикарбонилспермидин (16)К безводному эфиру (170 мл) при температуре 0oC осторожно добавляют LiAlH4 (1,4 г, 36,3 ммоль), затем к суспензии LiAlH4 при температуре 0oC добавляют по каплям раствор соединения 15 (3,1 г, 9.08 ммоль) в безводном эфире (60 мл). Реакционную смесь перемешивают в течение 3 часов при температуре 0oC, затем гасят путем последовательного добавления 4/5 мл 1 н. NaOH и 15 мл воды при температуре 0oC. Реакционную смесь фильтруют через стеклянную воронку. Объединенные фильтраты промывают насыщенным солевым раствором, водный слой экстрагируют CHCl3 и сушат над Na2SO4. Растворитель удаляют в вакууме, получая соединение 16 в виде масла (2,8 г, 98%). 1H-NMR (CDCl3) : 1.4 (18H+4H), 1.55 (2H), 2.6 (t, 2H), 3.0-3.2 (6H).
13C-NMR (CDCl3) : 25.49 (t), 27.20 (t), 6х28.25 (q), 31.63 (t), 33.81 (t), 39.85 (t), 2х46.31 (t), 78.80 (s), 79.16 (s), 155.50 (s), 155.86 (s).
Камптотецин 21-N2, N3-ди-трет-бутоксикарбонилспермидинамид (17)К суспензии камптотецина (700 мг, 2,01 ммоль) в безводном пиридине (50 мл) добавляют в атмосфере азота соединение 16 (2,8 г, 8,1 ммоль). Реакционную смесь нагревают до 80oC с получением прозрачного раствора желтого цвета. После выдерживания при температуре 80oC в течение 3 дней охлажденную реакционную смесь промывают 5% HCl, экстрагируют CHCl3 и сушат над Na2SO4. После упаривания растворителя неочищенный продукт очищают флэш-хроматографией (силикагель, CHCl3: CH3ОН 96: 4) с получением соединения 17 в виде твердого вещества бледно-желтого цвета (730 мг, 52%). 1H-NMR(CDCl3) : 1.15 (t, 3H), 1.25 (4H), 1.45 (18H), 1.65 (2H), 2.39 (m, 2H), 3.1 (4H), 3.25 (m, 4H), 4.35 (d, 1H, J=13.2), 5.0 (d, 1H, J=13.2), 5.15 (2H), 7.5-8.2 (6H).
Камптотецин 17-ацетил-21-N2, N3-ди-трет-бутоксикарбонил спермидинамид (18)С использованием той же процедуры, что и для получения соединения 5, соединение 17 (570 мг, 0,82 ммоль) подвергают ацетилированию с использованием Ас2О (1,54 мл) и безводного пиридина (1,65 мл) с получением соединения 18 в виде твердого вещества желтого цвета (488 мг, 81%). 1H-NMR (CDC3) : 1.15 (t, 3H, J=6.9), 1.2 (6H), 1.45 (18H), 2.05 (s, 3H), 2.35 (m, 2H), 3.05 (4H), 3.2 (4H), 5.25 (s, 2H), 5.4 (2H), 7.5-8.4 (6H).
13C-NMR (CDCl3) : 7.87 (q), 20.97 (q), 25.66 (t), 2х27.39 (t), 6х28.37 (q), 32.93 (t), 40.15 (t), 3х46.58 (t), 50.2 (t), 58.98 (t), 78.74 (s), 79.64 (s), 100.6 (d), 125.0 (s), 127.72 (d), 127.89 (d), 2х128.35 (s), 129.32 (d), 130.19 (d), 130.61 (d), 2х144.47 (s), 2х148.45 (s), 2х155.97 (s), 161.58 (s), 2х171.15 (s).
ВЭЖХ (RP8; 75%H2O, 25%CH3CN): 18,47 мин.
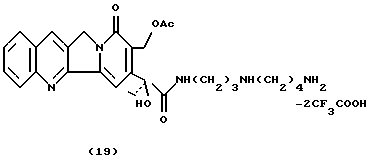
Камптотецин 17-ацетил-21-спермидинамид-ди-трифторацетат (19)С использованием той же процедуры, что и для получения соединения 9, снимают защиту соединения 18 (488 мг, 0,664 ммоль) при помощи TFA (6,8 мл) с получением соединения 19 в виде твердого желтого вещества (505 мг, 99,7%). 1H- NMR (CDCl3+CD3OD) : 0.8 (t, 3H, J=6.5), 1.56 (4H), 1.73 (2H), 1.86 (s, 3H), 2.14 (m, 2H), 2.78 (m, 4H), 3.15 (2H), 5.03 (2H), 5.22 (2H), 7.4-8.3 (6H).
13C-NMR (CDCl3+CD3OD) : 7.15 (q), 20.19 (q), 22.52 (t), 23.74 (t), 25.82 (t), 31.73 (t), 35.53 (t), 38.31 (t), 44.93 (t); 46.73 (t), 50.0 (t), 58.85 (t), 78.71 (s), 100.83 (d), 123.90 (s), 127.77 (d), 127.99 (d), 2х128.39 (s), 128.54 (d), 130.53 (d), 131.54 (d), 144.20 (s), 148.04 (s), 151.89 (s), 156.25 (s), 161.68 (s), 171.74 (s), 174.89 (s).
Формула изобретения
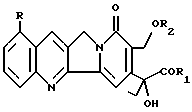 в которой R представляет атом водорода или метоксигруппу; R1 представляет гидрокси, С1 – С6 алкоксигруппу, амино, С1 – С6 моноалкиламино или С2 – С12 диалкиламиногруппу, в которой алкильная часть необязательно замещена аминогруппами; R2 представляет С1 – С6 алкильную группу или группу формулы COR3, где R3 представляет С1 – С6 алкил, при условиии, что R, R1 и R2 не могут быть одновременно H, NHCH(CH3)2, COCH3/COC5H11 соответственно. 2. Соединения по п. 1, где R представляет водород или метокси, R1 представляет гидрокси, R2 представляет ацетил. 3. Соединения по п.2, в качестве промежуточных продуктов. 4. Способ получения соединений по п.2, отличающийся тем, что проводят экстракцию предварительно высушенной растительной биомассы Mappia foetida при температуре ниже, чем 50oC, алифатическими кетонами или алифатическими сложными эфирами и затем алифатическими спиртами. 5. Способ получения соединений формулы I по п.2, отличающийся тем, что проводят селективное ацетилирование С17-гидроксила камптотецина в щелочной среде. 6. Способ получения соединений формулы I по п.1, отличающийся тем, что проводят этерификацию соединений по п.3. 7. Фармацевтические композиции, обладающие противоопухолевой активностью, содержащие в качестве активного ингредиента соединение по пп.1 – 3 в смеси с подходящими носителями. РИСУНКИ
|
||||||||||||||||||||||||||