Патент на изобретение №2169146
|
||||||||||||||||||||||||||
(54) БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ПОДАВЛЕНИЯ СЛИПАНИЯ ТРОМБОЦИТОВ И КОМПОЗИЦИЯ ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ
(57) Реферат: Описываются новые бициклические соединения, имеющие ядро, сформированное из двух конденсированных шестичленных колец, А и В, представленное формулой I, либо фармацевтически приемлемая соль, сольват или пролекарственная производная этого соединения, в котором бициклическое ядро из колец А и В выбирается из группы, состоящей из формул (17)-(21), R3 является кислотной группой, содержащей кислотный радикал, выбранный из формул группы (С), n = 0-6; R0 является одинаковым или разным и выбирается независимо из водорода, алкила, связующая группа -(L)- является связью или выбирается из формул -C(=O)-N(H)- и -С(Н2)-O-, Q является основной группой и выбирается из амидиногруппы и пиперидина. Новые бициклические соединения полезны в качестве препаратов, нейтрализующих действие гликопротеина IIв/IIIа, для предотвращения тромбоза. Описывается также способ подавления слипания тромбоцитов и композиция для его осуществления. 13 с. и 12 з.п. ф-лы, 1 табл. 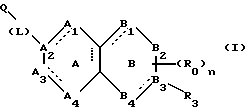 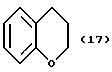 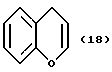 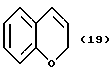 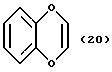 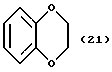 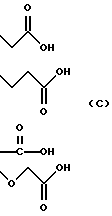
Изобретение относится к бициклическим соединениям, полезным в качестве препаратов, нейтрализующих действие гликопротеина IIb/IIIa, для предотвращения тромбоза. Наиболее частые случаи сосудистых заболеваний связаны с зависящим от тромбоцитов сужением кровеносных сосудов, – такие как атеросклероз и артериосклероз, острый инфаркт миокарда, хроническая постоянная стенокардия, непостоянная стенокардия, приступы ишемической болезни, периферические сосудистые заболевания, артериальный тромбоз, преэклампсия, эмболия, рестеноз или внезапная закупорка с последующей ангиопластикой, каротидная эндартеректома, анастомоз сосудистых тканей и т.д. Эти условия представляют собой различные нарушения, которые, как полагают, инициируются активацией тромбоцитов на стенках сосудов. Слипание и агрегация тромбоцитов предположительно являются важной частью формирования тромба. Эта деятельность происходит при посредничестве нескольких склеивающих тромбоциты гликопротеинов. Активные центры связывания для фибриногена, фибронектина и других факторов свертывания размещаются на тромбоцитном мембранном гликопротеиновом комплексе IIb/IIIa. Когда тромбоцит активизирован агонистом, таким как тромбин, активный центр связи гликопротеина IIb/IIIa становится доступным для фибриногена, что может привести к агрегации тромбоцитов и формированию сгустка. До сих пор предлагалось блокировать эти центры образования тромба путем использования различных терапевтических средств. Патент США 1 5064814 предлагает в качестве антитромбозных агентов циклические аминокислотные производные N-амидинопипериндинкарбоксила. Патент США 1 5039805 предлагает различные производные бензойной кислоты и фенилуксусной кислоты для подавления связывания фибриногена с фибриногенным рецептором, гликопротеином IIb/IIIa. В международной патентной заявке PCT WO 93/00095 предлагается использовать содержащие семичленные кольца бициклические соединения. Заявка ЕР 1 456835 описывает бициклические соединения, имеющие слившиеся шестичленные кольца (производные хиназолин-3-алканойной кислоты), которые, как сообщается, имеют подавляющее воздействие на агрегацию тромбоцитов. Международная патентная заявка PCT WO 93/08174 описывает непептидиловые интегриновые подавители, которые являются бициклическими системами с 6- и 7-членными слившимися кольцами, которые имеют терапевтические приложения при заболеваниях, для которых показано блокирование агрегации тромбоцитов. Патентная заявка WO 94/12478 описывает приготовление 6,5-бициклических соединений, для которых установлено, что они эффективны при подавлении агрегации тромбоцитов. Патентная заявка WO 94/08962 описывает приготовление 6,5-бициклических соединений, для которых установлено, что они эффективны при подавлении агрегации тромбоцитов. Патентная заявка Великобритании GB 2276384 описывает новые производные оксохиназолина, для которых установлено, что они оказывают нейтрализующее воздействие на фибриногенный рецептор. Статья Роберта С. Макдауэлла и др. (Robert S. McDowell, et al.) “From Peptide to Non-Peptide. 1. The Elucidation of a Bioactive Conformation of the arginine-glycine-aspartic Acid Recognition Sequence”, в Журнале американского химического общества (J. Am. Chem. Soc.), 1994, 116, с. 5069-5076, описывает строение непептидных средств, подавляющих связывание фибриногена-гликопротеина IIb/IIIa. Публикация Брента К. Блэкберна и Томаса Р. Гадека (Brent К. Blackburn and Thomas R. Gadek) “Chapter 9. Glycoprotein IIb/IIIa Antagonists” в Годовых отчетах по медицинской химии (Annual Reports in Medical Chemistry) – 28, часть II – “Cardiolvascular and Pulmonary Agents”, с. 79-88, 1993, опубликованная Academic Press, Inc., описывает непептиды в качестве нейтрализаторов взаимодействия гликопротеина IIb/IIIa и фибриногена. Статья Роберта С. Макдауэлла и др. (Robert S. McDowell, et al.) “From Peptide to Non-Peptide. 2. The de Novo Design of Potent, Non-Peptidal Inhibitors of Platelet Aggregation Based on a Benzodiazepinedione Scaffold” в Журнале американского химического общества (J. Am. Chem. Soc.), 1994, 116, с. 5077-5083, описывает бензодиазапинедион, который является средством, подавляющим агрегацию тромбоцитов. Соединения хинолина упоминаются в патентной литературе для различных использований в медицине. К примеру, европейская патентная заявка 1 0315399; патент США 1 5041453; патентная заявка PCT WO 89/04303 и патентная заявка PCT WO 89/4304 описывают производные хинолина, полезные в качестве подавителей липоксигеназы и/или нейтрализаторов лейкотриена, обладающих антивоспалительными и антиаллергическими свойствами. Эти соединения должны содержать три ароматических кольца, каждое разорвано кислородом или серой и, возможно, другими группами. В сфере сердечно-сосудистой и церебрально-сосудистой терапии существует необходимость альтернативных средств, которые могут быть использованы в профилактике и лечении тромбов. В настоящем изобретении открыто, что определенные новые бициклические соединения блокируют фибриногенный рецептор гликопротеина IIb/IIIa, тем самым подавляя агрегацию тромбоцитов и последующее образование тромба. Фармацевтические составы, содержащие бициклические соединения по настоящему изобретению, подавляют агрегацию и полезны для профилактики и лечения тромбогенных заболеваний, таких как инфаркт миокарда, стенокардия, удары, периферические артериальные заболевания, распространяющееся внутрисосудистое свертывание и венозный тромбоз. Настоящее изобретение состоит в новом бициклическом соединении с ядром, сформированным из двух слившихся шестичленных колец А и В, представленных формулой (I), как определяется ниже, и всех его фармацевтически приемлемых солей, сольватов и пролекарственных производных 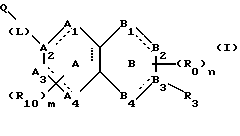 Другим аспектом настоящего изобретения является фармацевтический состав, содержащий новое бициклическое соединение по настоящему изобретению. Еще одним аспектом настоящего изобретения является способ подавления агрегации тромбоцитов, подавления фибриногенного связывания или предотвращения тромбоза путем введения млекопитающему бициклических соединений по настоящему изобретению. Еще одним аспектом настоящего изобретения является способ лечения человека для облегчения патологического воздействия атеросклероза и артериосклероза, острого инфаркта миокарда, хронической постоянной стенокардии, непостоянной стенокардии, приступов ишемической болезни, периферических сосудистых заболеваний, артериального тромбоза, преэклампсии, эмболии, рестеноза с последующей ангиопластикой, каротидной эндартеректомы и анастомоза сосудистых тканей; способ содержит введение упомянутому человеку нового бициклического соединения по настоящему изобретению. Подробное описание изобретения. Термин “алкил”, используемый здесь, относится к одновалентному прямому или разветвленному цепочечному радикалу из одного-десяти атомов углерода, в том числе метил, этил, n-пропил, изопропил, n-бутил, изобутил, трет-бутил, n-гексил и тому подобное, но не ограничивается ими. Термин “галогенозамещенный алкил”, используемый здесь, обозначает только что определенную алкильную группу, замещенную одним, двумя или тремя галогенными атомами, выбранными из хлора, фтора, брома и йода. Примеры таких групп содержат хлорметил, бромэтил, трифторметил и тому подобное. Термин “арил”, используемый отдельно, означает гомоциклический ароматический радикал, связанный или несвязанный. Предпочтительными арильными группами являются фенил, нафтил, бифенил, фенантренил, нафтаценил и тому подобное. Термин “замещенный арил” обозначает арильную группу, замещенную одним, двумя или тремя заместителями, выбранными из галогенов, гидроксилов, защищенных гидроксилов, циановых групп, нитрогрупп, C1-C10-алкила, C1-С10-алкоксогрупп, трифторметила, аминогрупп, аминометила и тому подобного. Примерами таких групп являются 4-хлорфенил, 2-метилфенил, 3-метил-4-гидроксифенил и 3-этоксофенил. Термин “арилалкил” означает одну, две или три арильные группы с несколькими атомами углерода, присоединенными к алкильному радикалу с несколькими атомами углерода. Типичной арилалкильной группой является бензильная группа. Термин “алкенил”, используемый здесь, обозначает одновалентный прямой или разветвленный цепочечный радикал, из двух-шести углеродных атомов, содержащих двойную углеродную связь, в том числе 1-пропенил, 2-пропенил, 2-метил-1-пропенил, 1-бутенил, 2-бутенил, и тому подобное, но не ограниченный ими. Термин “алкилен”, используемый здесь, обозначает двухвалентную прямую или разветвленную цепочечную группу из одного-десяти углеродных атомов, в том числе (но не ограничиваясь), -CH2-, -(CH2)2-, -(CH2)3, -CH(CH3)-, -CH(C2H5)-, -CH(CH3)CH2– и тому подобное. Термин “алкенилен”, используемый здесь, обозначает двухвалентную прямую или разветвленную цепочечную группу из двух-десяти углеродных атомов, содержащую двойную связь углерод-углерод, в том числе (но не ограничиваясь) -CH= CH-, -C(CH3)= CH-, -CH=CH-CH2-, -CH=C(CH3)-CH2-, -CH2CH(CH=CH2)CH2– и тому подобное. Термин “алкинилен”, используемый здесь, обозначает двухвалентную прямую или разветвленную цепочечную группу из двух-десяти углеродных атомов, содержащую тройную связь углерод-углерод, в том числе (но не ограничиваясь) 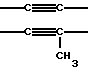 и тому подобное. Термин “амидиногруппа” обозначает радикал, имеющий структурную формулу 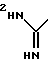 Термин “основной радикал” обозначает органический радикал, являющийся протонным акцептором. Примерами основных радикалов являются амидино-, гуанидино-, амино- и пиперидильные группы. Термин “основная группа” обозначает органическую группу, содержащую один или более основных радикалов. Основная группа может состоять только из одного основного радикала. Термин “неинтерферирующий органический радикал” обозначает любой органический заместитель, присутствующий в бициклическом соединении с формулой (I), который не является вредным для эффективности этого соединения как нейтрализатора гликопротеина IIb/IIIa. Термин “кислотный радикал” обозначает органический радикал, являющийся протонным донором. Примерами кислотных радикалов являются 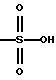 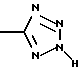 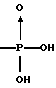 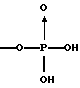 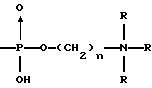 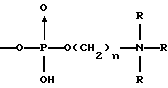 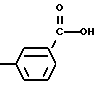 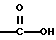 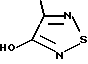 Термин “кислотная группа” обозначает органическую группу, содержащую один (или более) кислотный радикал. Кислотная группа может состоять только из одного кислотного радикала. Соединения по изобретению. Соединения по настоящему изобретению имеют общую формулу (I), приведенную ниже 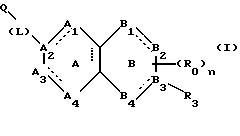 а также все фармацевтически приемлемые соли, сольваты и пролекарственные производные этого соединения. Бициклическое ядро формулы (I) формируется слиянием двух шестичленных колец А и В, имеющих углеродные атомы, соединенные мостиковой связью. Пунктирные линии в структурной формуле (I) обозначают возможное наличие дополнительной связи, то есть ненасыщенности, которая придаст кольцевой структуре ароматический характер. Следует понять, что соединенные мостиковой связью углеродные атомы будут либо незамещены, либо замещены (водородом) в зависимости от степени ненасыщенности в бициклической кольцевой системе. Атомы A1, A2, А3 и А4 кольца А и атомы B1, B2, В3, В4 кольца В формулы (I) выбираются независимо из углерода, кислорода, серы и азота с условием, что как минимум два атома из B1, В2, В3, В4 являются атомами углерода. Более точно, A1, А3 и А4 выбираются независимо из углерода, кислорода, серы и азота, а А2 выбирается независимо из углерода или азота при условии, что A2 имеет ненасыщенную связь, если А2 – азот, и при условии, что как минимум два атома из A1, А2, А3 и А4 являются атомами углерода. Соответственно B1, В2 и В4 выбираются независимо из углерода, кислорода, серы и азота, а B3 выбирается независимо из углерода или азота при условии, что В3 имеет ненасыщенную связь, если В3 является азотом, и при условии, что как минимум два атома из B1, B2, В3 и В4 являются атомами углерода. Бициклические ядра соединений по настоящему изобретению могут формироваться из кольцевых систем, состоящих из любых ядер (1-16), показанных ниже (но не ограничиваются ими) 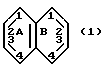    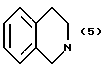 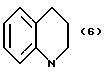 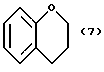 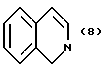 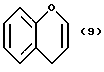 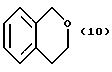 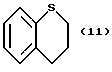 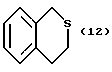 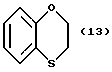 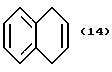 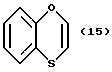 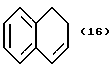 Ядра, показанные формулами от (1) до (16) выше и от (17) до (30) ниже, имеют нумерацию атомов в кольцах А и В и соответствующие положения заместителей, как показано выше в (1). К примеру, ядра (Imm) и (Ipp) 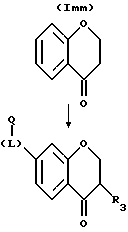 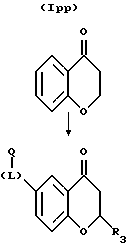 дали бы различные продукты в рамках формулы (I). Соединения по настоящему изобретению, соответствующие формуле (I) с ядрами от (1) до (19), представлены ниже формулами от (Ia) до (Ie): 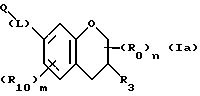 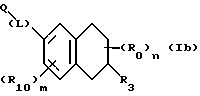 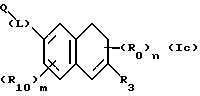 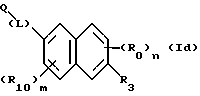 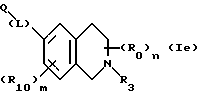 Другие бициклические ядра, подходящие для формирования соединений по формуле (I), представлены ниже формулами от (17) до (21): 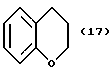 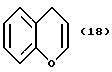 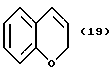 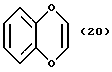 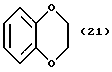 Соединения по настоящему изобретению, соответствующие формуле (I) с ядрами (17)-(21), представлены ниже при помощи формул от (If) до (Ii): 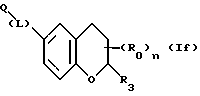 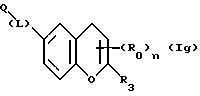 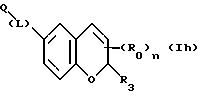 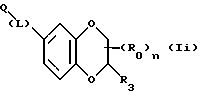 Бициклические ядерные кольца, замещенные при помощи =O, подходящие для формирования соединений по формуле (I), представлены ниже формулами от (22) до (27): 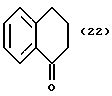 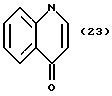 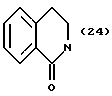 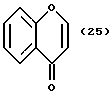 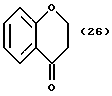 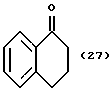 Соединения по настоящему изобретению, соответствующие формуле (I) с кислородозамещенными ядрами (22)-(27), представлены ниже с помощью формул от (Ik) до (Im): 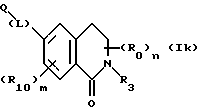 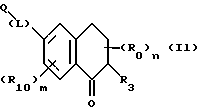 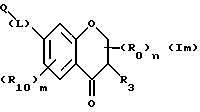 Другие бициклические ядерные кольца, с замещением при помощи =O, подходящие для формирования соединений по формуле (I), представлены ниже при помощи формул от (28) до (30): 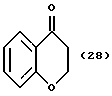 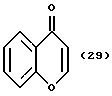 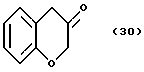 Соединения по настоящему изобретению, соответствующие формуле (I) с ядрами (28)-(30), представлены ниже с помощью формул от (In) до (Ip): 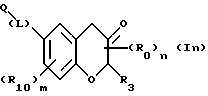 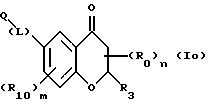 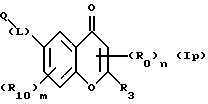 Наиболее предпочтительными ядрами для соединений по настоящему изобретению являются изохинолин, изохинолон, нафталин, тетрагидронафталин, тетралон, дигидронафталин и бензопиран. Заместитель R3 является кислотной группой либо ее фармацевтически приемлемой солью или сольватом (или пролекарственным производным упомянутой оксидной группы) и предпочтительно является кислотной группой, содержащей карбоксильную функциональность. Группа R3 может быть единственным заместителем атома В3 кольца. Альтернативно, когда атом В3 может принять две связи, эти связи могут быть заняты двойной связью с группой R3 (при двойной связи R3, приложенной прямо к кольцу В по формуле (I)) или второй R3-группой, или второй группой, выбранной из водорода, C1-C10-алкила, C1-С10-галогенозамещенного алкила, С2-С10-алкенила, C2-C10-алкинила С3-С10-циклоалкила, арила, C7-C12-аралкила, гидроксильной группы, C1-C10-алкоксигруппы, C1-C10-аралкоксигруппы, карбоксильной группы, ацила, циано-, галогено-, нитро- и сульфогруппы. R3, кислотная группа, предпочтительно выбирается из групп, имеющих члены, представленные следующими формулами: 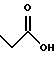 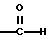 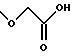 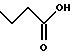 Заместители R0 являются неинтерферирующими органическими радикалами и являются одинаковыми или разными на каждом атоме B1, B2 и B4 и одинаковыми или разными между атомами B1, B2 и B4, и независимо выбираются из водорода, C1-C10-алкила, C1-C10-галогенозамещенного алкила, C2-C10-алкенила, С2-С10-алкинила, С3-С10-циклоалкила, арила, C6-C12-арилалкила, гидроксильной группы, C1-C10-алкоксильной группы, C6-C12-арилалкоксильной группы, аминогруппы, замещенной аминогруппы, карбамойловой группы, карбоксильной группы, ацила, циано-, галогено-, нитро-, сульфогрупп при условии, что только один атом из B1, B2 и В4 может быть также замещен на =O или =S. Количество n заместителей R0 у атомов B1, B2 и В4 кольца В является целым числом от 0 до 6 и зависит от суммы количеств незанятых связей, наличествующих у отдельных атомов B1, В2 и В4. Обычно n будет в диапазоне от 2 до 6 для большинства соединений по настоящему изобретению. Так, к примеру, когда кольцо В предельно, B2 – кислород, a B1 и В4 – углерод, то на атоме В2 не будет ни одного заместителя R0, как показано на структурной формуле (Iq) ниже: 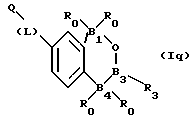 Для ненасыщенных колец В количество незанятых связей, имеющихся у отдельных атомов B1, В2 и В4, уменьшается, а количество требуемых заместителей R0 соответственно меньше. Так, к примеру, когда кольцо В ненасыщенное, B2 – азот, а B1 и В4 – углерод, то на B2 не будет ни одного заместителя R0, как показано на структурной формуле (Ir) ниже: 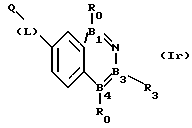 Атомы A1, A2, А3 и А4 кольца А выбираются независимо из углерода, водорода, серы и азота при условии, что как минимум два атома из A1, A2, А3 и А4 являются атомами углерода. Заместители R10 являются одинаковыми или разными на каждом из атомов A1, А3 и А4 и одинаковыми или разными между атомами A1, А3 и А4 и независимо выбираются из водорода, C1-C10-алкила, C1-C10-галогенозамещенного алкила, C1-C10-алкенила, C2-C10-алкинила, С3-С10-циклоалкила, арила, С6-С12-арилалкила, гидроксильной группы, алкоксильной группы, С6-С12-арилалкоксильной группы, карбоксильной группы, ацила, циано-, галогено-, нитро- и сульфогрупп; при условии, что только один атом из A1, А3 и А4 может также быть замещен на =O или =S, когда на одном атоме доступны две позиции для замещения (т.е. когда одна или более из пунктирных линий в кольце А по формуле I отсутствует, а один из атомов А является атомом углерода). Количество m заместителей R10 у атомов A1, А3 и А4 кольца А является целым числом от 0 до 6 и зависит от суммы количеств незанятых связей, наличествующих у отдельных атомов A1, А3 и А4 аналогично замещению группами R0 на кольце В, как описано выше. Обычно n будет в диапазоне от 2 до 6 для большинства соединений по настоящему изобретению. Атом А2 кольца А замещается только связующей группой -(L)-, когда А2 имеет только одну незанятую связь, однако, когда А2 имеет две незанятых связи, вторая связь может быть занята группой, выбранной из водорода, алкила, галогенозамещенного 1-C10-алкила, C2-C10-алкенила, C2-C10-алкинила, С3-С10-циклоалкила, арила, C7-C12-арилалкила, гидроксильной группы, C1-C10-алкоксильной группы, C7-C12-арилалкоксильной группы, ацила, циано-, галогено-, нитро-, сульфогрупп и основной группы. Связующая группа -(L)- присоединена к атому А2 кольца А и является (i) связью или (ii) двухвалентной замещенной или незамещенной цепочкой из 1-10 атомов (т. е. существует от 1 до 10 атомов в цепи между связующими двухвалентными связями при том, что все остальные атомы находятся на ответвлениях от этих атомов цепочки). К примеру, когда -(L)- является связью, соединение по изобретению может иметь структурную формулу (Is) следующего вида: 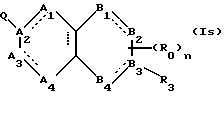 Альтернативно, когда -(L)- является связующей группой  Соединение по настоящему изобретению может иметь структурную формулу (It) следующего вида: 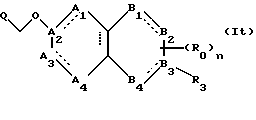 Алкиленовые, алкениленовые и алкиниленовые группы являются подходящими для связующих групп. Предпочтительные связующие группы имеют от 1 до 4 атомов цепи и соответствуют общим формулам:     где Z1, Z2, Z3 и Z4 являются атомами, выбранными из группы, состоящей из углерода, азота, серы и кислорода. Связующие группы, содержащие три атома цепи, такие как 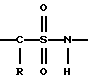 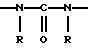 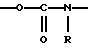 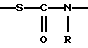 где R является водородом или алкилом, также могут использоваться. В частности, предпочтительными являются связующие группы, содержащие два атома цепи, такие как: 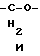 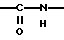 Связующая группа 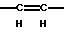 имеет цис- и трансформы, и обе эти формы и их смеси во всех пропорциях находятся в рамках данного изобретения. Асимметрические связыватели, например, связыватели  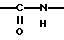 могут быть перевернуты в своей точке присоединения между кольцом А ядра и основной группой Q, как показано формулами (Iu) и (Iv) ниже: 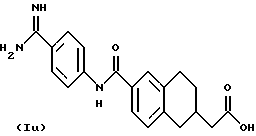 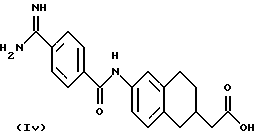 Заместителем Q в формуле (I) является основная группа. Основная группа содержит один или более основных радикалов 1. Подходящие основные радикалы содержат один или более атомов азота и содержат амино-, имино-, амидиногруппы, N-алкиламидины, N,N’-диалкиамидины, N-ариламидины, аминометиленаминогруппу, иминометиламиногруппу, гуанидиногруппу, аминогуанидиногруппу, алкиламиногруппу, диалкиламиногруппу, триалкиламиногруппу, алкилиденаминогруппу, пирролиловую, имидазолиловую, пиразолиловую, пиридиловую, пиразиниловую, пиримидиниловую, индолизиниловую, изоиндолиловую, 3H-индолиловую, индолиловую, 1H-индазолиловую, пуриниловую, 4Н-хинолизиноловую, изохинолиловую, хинолиловую, фталазиниловую, нафтиридиниловую, хиноксалиниловую, хиназолиниловую, циннолиниловую, амидную, тиоамидную, бензамидиногруппу, птеридиниловую, 4аН-карбозолиловую, карбозолиловую, бета-карболиниловую, фенантридиниловую, акридиниловую, пиримидиниловую, фенантролиниловую, феназиниловую, фенарсазиниловую, фенотиазиниловую, пирролиниловую, имидазолидиниловую, имидазолиниловую, пиразолидиниловую, пиразолиниловую, пиперидиловую, пиперазиниловую, индолиниловую, изоиндолиниловую, хинуклидиниловую, морфолиниловую группы, или любую из предыдущих, замещенную амино-, имино-, амидино-, аминометиленамино-, иминометиламино-, гуанидино-, алкиламино-, диалкиламино-, триалкиламиногруппами, тетрагидроизохинолином, дигидрозиоиндолом, алкилиденаминогруппами или группой, представленной формулой 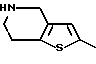 Предпочтительные основные радикалы выбираются из амино-, пиперидиловых, гуанидино-, и амидиногрупп. Наиболее предпочтительные основные радикалы являются радикалами амидино- и пиперидиловых групп, представленные формулами 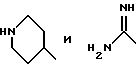 Основная группа Q может иметь форму основного радикала (такого, как Q1 в формуле 1w ниже), ответвляющегося от циклического кольца. Таким образом, основная группа Q может содержать две части, а именно (i) один или более основных радикалов Q1 и (ii) циклическую группу D, имеющую от 5 до 8 атомов кольца. Кольцо D присоединено к атому A2 кольца А бициклического ядра через связующую группу -(L)-, как показано выше в формуле (I). Кольцо D может также иметь заместители R20, которые выбираются из хлора, фтора или неинтерферирующих органических радикалов. Заместители R20 могут быть в количестве t, где t является целым числом от нуля до количества незанятых связей в кольце D. Основной радикал Q1 присоединяется к кольцу D способом, показанным формулой (Iw) ниже 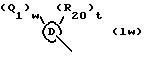 (присоединяется к атому A2 через связующую группу). Подходящие кольца D формируются из ядра, выбранного из группы, состоящей из бензола, циклогептадиена, циклогептатриена, циклогептана, циклогексана, циклогексена, циклогексадиена, циклогептена, циклооктадиена, циклооктана, циклооктатетраена, циклооктена, циклопентана, циклопентена, имидазола, изооксазола, морфолина, оксазола, пиперазина, пиперидина, пиразина, пиразола, пиридина, пиримидина, пиррола, пирролидина, пирролина, тетрагидропиридина, тетрагидропиримидина, 1H-тетразола, тиазолидина, тиазола, тиопирана, 1,3,5-триазина, 1,2,3-триазола, 1,2,4-триазола, дигидрофурана, дигидропирана, диоксана, диоксепина, диоксолана, фурана, оксакана, тетрагидрофурана, тетрагидропирана, тиофена и тетрагидротиофена. Общая формула (Ix) для предпочтительных соединений по настоящему изобретению, имеющих основной радикал, присоединенный к циклическому кольцу из 5-8 атомов, показана ниже 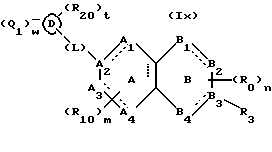 в которой A1, А3 и A4 независимо выбираются из углерода, кислорода, серы и азота; А2 выбирается независимо из углерода или азота при условии, что А2 имеет незанятую связь, если A2 является азотом, и при условии, что как минимум два атома из A1, A2, А3 и А4 являются атомами углерода; B1, B2, В4 выбираются независимо из углерода, кислорода, серы и азота; B3 выбирается независимо из углерода или азота при условии, что B3 имеет незанятую связь, если B3 является азотом, и при условии, что как минимум два атома из B1, B2, B3 и B4 являются атомами углерода; R3 является кислотной группой, содержащей один или более кислотных радикалов; n – число от 0 до 6; R0 являются одинаковыми или разными и выбираются независимо из водорода, алкила, галогенозамещенного алкила, алкенила, алкинила, циклоалкила, арила, арилалкила, гидроксильной, алкоксильной, аралкоксильной, амино-, замещенной амино-, карбамойловой, карбоксильной, ациловой, циано-, галогено-, нитро-, сульфогрупп, = O или =S; при условии, что, если R0 является =O или =S, то только один атом из B1, В2, В3 и В4 может быть атомом азота; m – число от 0 до 6; R10 являются одинаковыми или разными и выбираются независимо из водорода, алкила, галогенозамещенного алкила, алкенила, алкинила, циклоалкила, арила, арилалкила, гидроксильной, алкоксильной, аралкоксильной, карбоксильной, ациловой, циано-, галогено-, нитро-, сульфогрупп, =O и =S; при условии, что только один R10 может быть =O или =S; t – число от 0 до 3; R20 являются одинаковыми или разными и выбираются независимо из водорода, галогенов, алкила, галогенозамещенного алкила, алкенила, алкинила, циклоалкила, арила, арилалкила, гидроксильной, алкоксильной, аралкоксильной, карбоксильной, ациловой, циано-, галогено-, нитро-, сульфогрупп; связующая группа -(L)- является связью или двухвалентной замещенной или незамещенной цепочкой из 1-10 атомов, выбранных из группы, состоящей из углерода, азота, серы, и кислорода; и D является кольцом, сформированным из 5-8 атомов кольца, и упомянутые атомы кольца выбираются независимо из углерода, азота, кислорода или серы, при условии, что как минимум два атома кольца D являются атомами углерода; w – число от 1 до 3; Q1 – основной радикал. Соединения по настоящему изобретению, имеющие кольца А, B и D, представлены следующими формулами (Iy)-(Iah) ниже 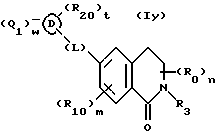 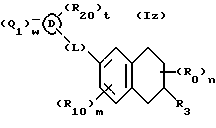 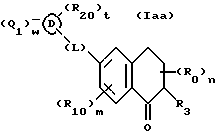 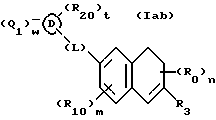 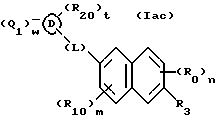 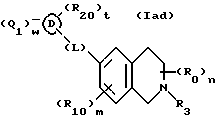 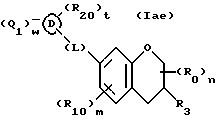 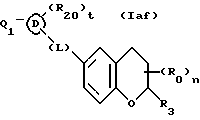 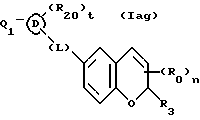 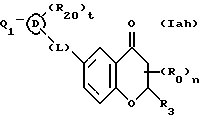 Предпочтительная основная группа Q имеет шестичленное кольцо D, как показано ниже формулой (Iai): 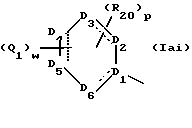 где p является целым числом от 0 до 8, как, к примеру, в специфических группах Q: 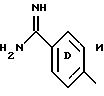 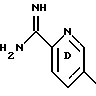 Предпочтительный вариант выполнения соединения по настоящему изобретению представлен формулой II ниже: 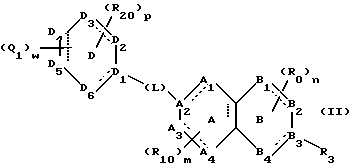 В формуле II основная группа на атоме А2 ядра имеет две части, а именно (i) шестичленное кольцо D, которое присоединяется к связующей группе -(L)-, и (ii) основной радикал (основные радикалы) Q1 (где w – целое число от 1 до 3), присоединенные к кольцу D. Основные радикалы являются такими, как определено ранее. Атомы D2, D3, D4, D5 и D6 выбираются независимо из углерода, азота, кислорода или серы, а атом D1 выбирается из углерода или азота; при условии, что как минимум два из атомов D2, D3, D4, D5 и D6 являются атомами углерода. Q1 является основным радикалом, определенным ранее. Предпочтительными кольцевыми структурами с ответвлением Q1 являются те, в которых атомы D1, D2, D3, D4, D5 и D6 формируют циклическое кольцо, выбираемое из группы, состоящей из бензола, пиридина, пиперидина, 1,2-пиперазина, 1,3-пиперазина, 1,4-пиперазина, пирана, тиопирана, тиабензола, циклогексена и циклогексана, наиболее предпочтителен бензол. Предпочтительным основным радикалом Q1 является амидиновый радикал. Заместители R20 являются одинаковыми или разными на каждом атоме D2, D3, D5 и D6 и одинаковыми или разными между атомами D2, D3, D5 и D6 и являются неинтерферирующими органическими радикалами, независимо выбираемыми из водорода, алкила, галогенозамещенного алкила, алкенила, алкинила, циклоалкила, арила, арилалкила, гидроксильной, алкоксильной, аралкоксильной, амино-, замещенной амино-, карбамойловой, карбоксильной-, ациловой, циано-, галогено-, нитро- и сульфогрупп. Число p заместителей R20 является целым от 0 до 8 в зависимости от суммы количества незанятых связей, наличествующих на отдельных атомах D2, D3, D5 и D6. Предпочтительными соединениями по настоящему изобретению являются базирующиеся на ядрах замещенного бензамидином изохинолина, изохинолина, нафталина, тетрагидронафталина, дигидронафталина, бензопирана и тетралона, как частично показано формулами (III)-(IIIe) ниже: 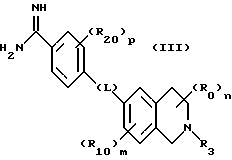 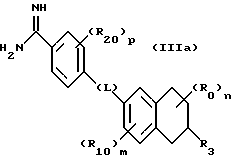 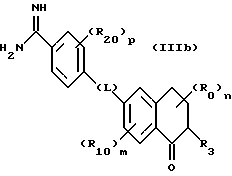 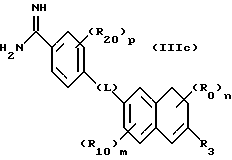 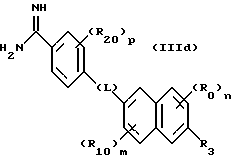 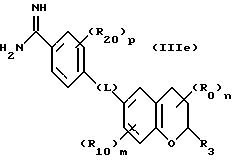 где -(L)-, n, m, p, R0, R3, R10 и R20 являются такими, как определено ранее. Наиболее предпочтительны соединения, в которых R10 и R20 являются водородом, а -(L)- имеет два атома углерода. В другом предпочтительном аспекте настоящего изобретения кольцо D содержит один или более (предпочтительно 1 или 2) заместителей, независимо выбираемых из хлора или фтора. Хлоровые и фторовые заместители могут добавляться к любому кольцу D, которое, как описано выше, имеет от 5 до 8 членов. Иллюстративные соединения по настоящему изобретению с замещением шестичленных колец D показаны формулами (IV)-(IVb) ниже: 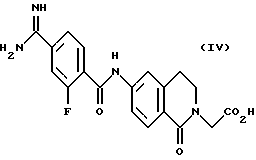 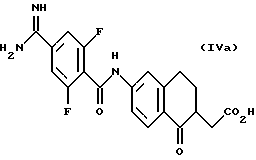 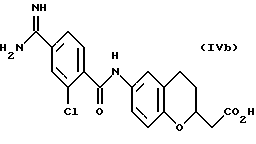 Без привязки к какой-либо теории действия считается, что отнимающие электрон группы, такие как фтор, уменьшают основность основной группы и увеличивают оральную биополезность соединений по настоящему изобретению. Специфические соединения по настоящему изобретению изохинолинового типа, которые в высшей степени предпочтительны, представлены следующими структурными формулами (V)-(Vv) или фармацевтически приемлемыми солями, сольватами или пролекарственными производными этих соединений: 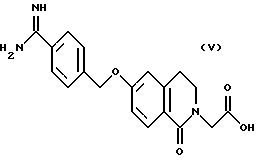 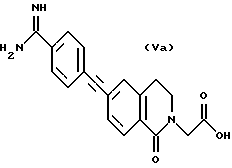 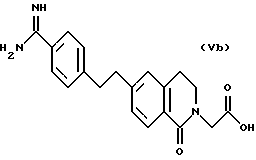 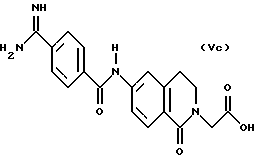 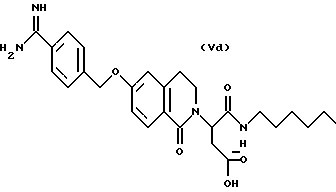 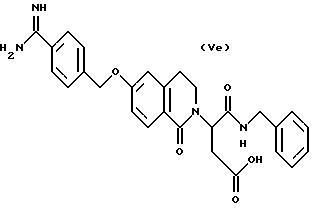 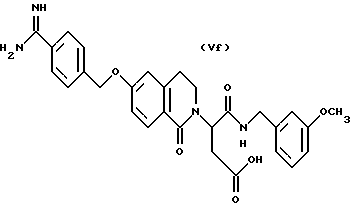 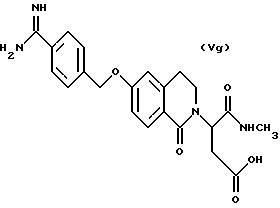 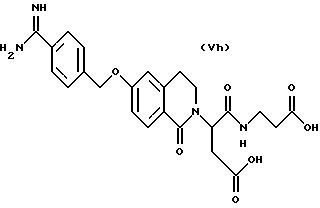 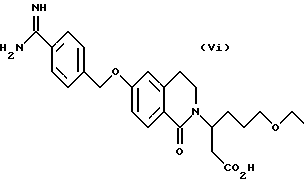 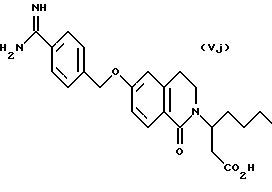 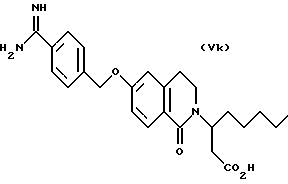 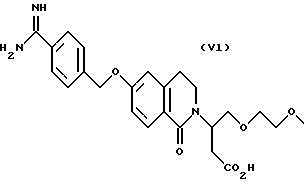 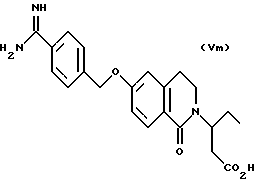 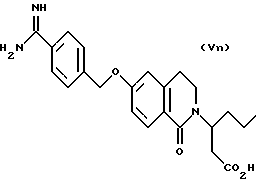 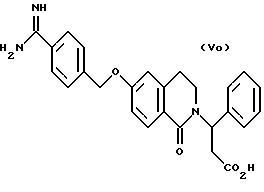 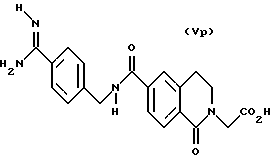 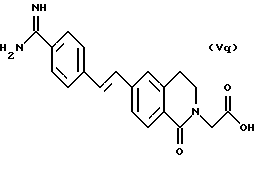 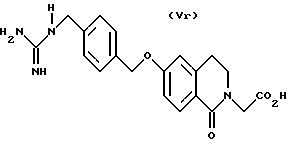 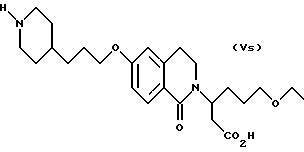 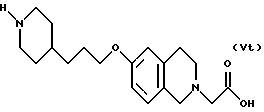 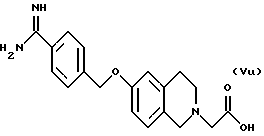 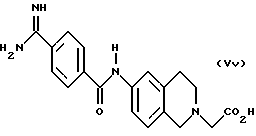 и смеси соединений (V)-(Vv). Другие специфические соединения по настоящему изобретению нафталинового/тетралинового типа, которые в высшей степени предпочтительны, представлены следующими структурными формулами (VI)-(VIp) или фармацевтически приемлемыми солями, сольватами или пролекарственными производными этих соединений: 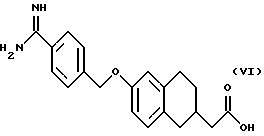 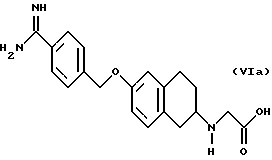 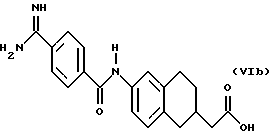 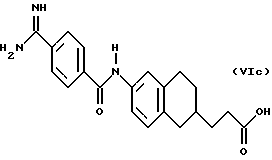 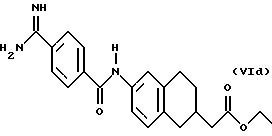 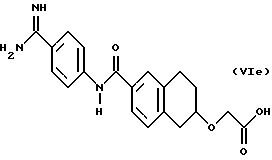 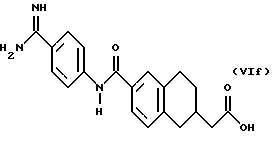 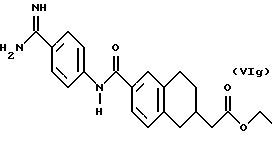 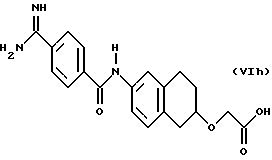 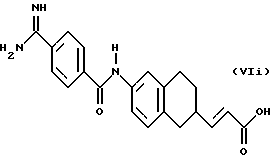 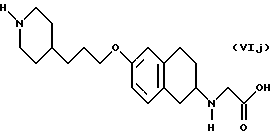 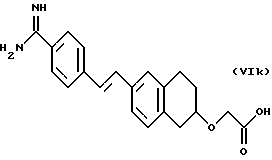 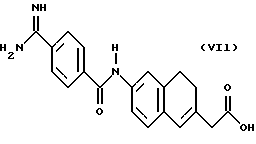 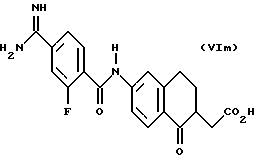 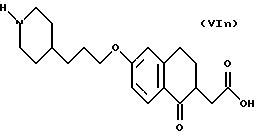 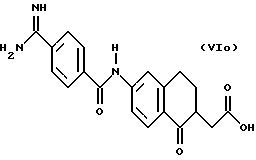 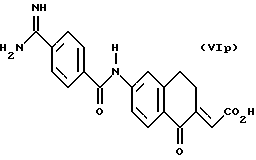 и смесями соединений (VI)-(VIp). Другие предпочтительные специфические соединения по настоящему изобретению представлены следующими структурными формулами (L)-(LXIII) и всеми фармацевтически приемлемыми солями, сольватами и пролекарственными производными этих соединений: 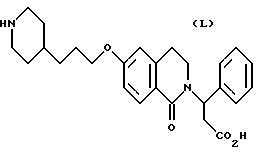 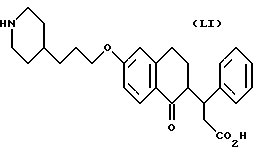 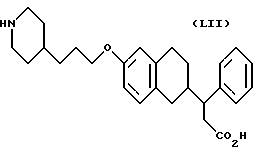 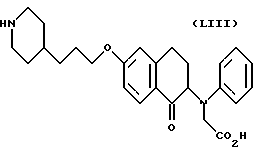 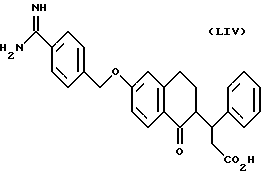 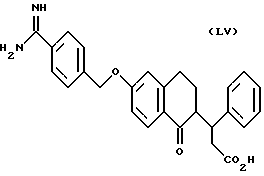 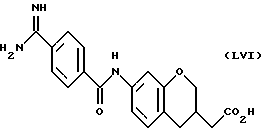 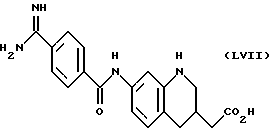 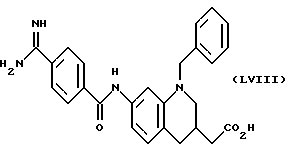 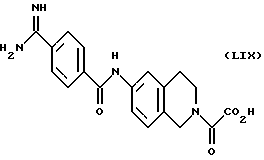 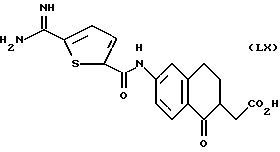 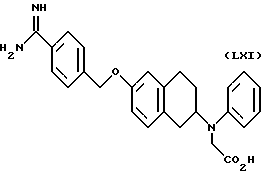 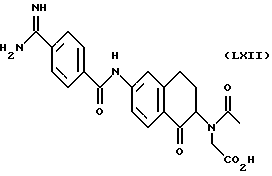 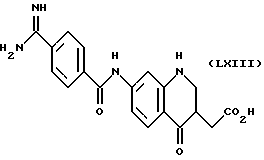 и смесями любых соединений (L)-(LXIII). Другие специфические соединения по настоящему изобретению бензопиранового типа, которые являются в высшей степени предпочтительными, представлены следующими структурными формулами (VIII)-(VIIIi) или фармацевтически приемлемыми солями, сольватами или пролекарственными производными этих соединений: 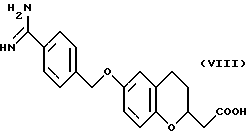 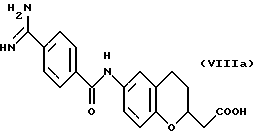 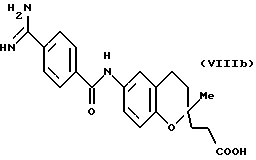 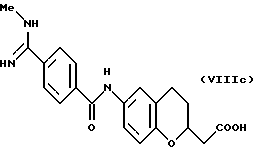 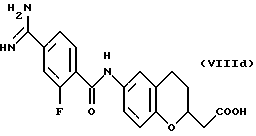 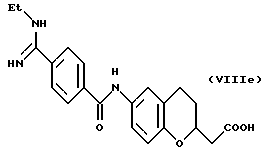 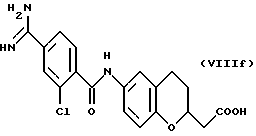 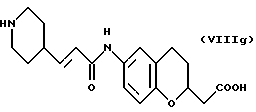 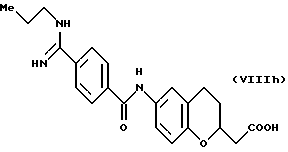 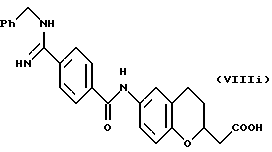 Другие соединения по настоящему изобретению, имеющие бициклическое ядро с атомом кислорода в кольце А, представлены следующими формулами (IX)-(IXl) ниже: 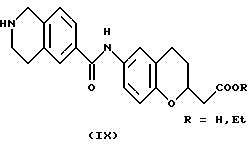 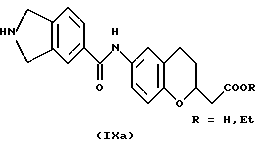 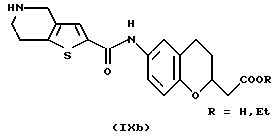 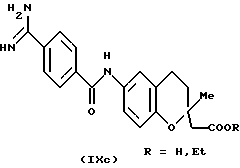 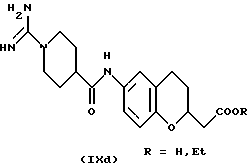 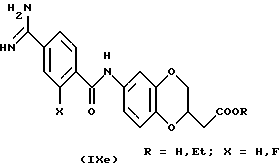 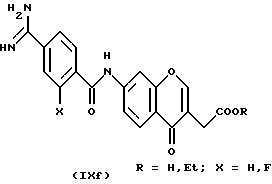 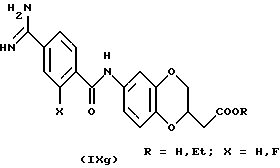 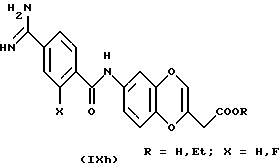 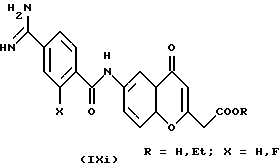 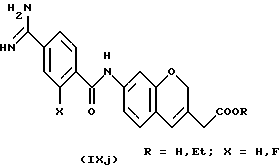 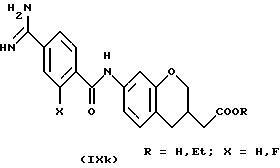 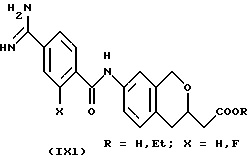 Соединения по настоящему изобретению обладают как минимум одним кислотным функциональным заместителем (R3 в формуле I) и поэтому способны образовывать соли. Примеры фармацевтически приемлемых солей включают в себя (но не ограничиваются ими) соли щелочных и щелочноземельных металлов, таких как литий, натрий, калий, кальций, магний, алюминий и подобные. Соли традиционно приготавливаются из свободной кислоты путем взаимодействия кислоты в растворе с основанием либо путем экспозиции кислоты на анионообменной смоле в солевом цикле. Попадающие под определение фармацевтически приемлемых соли являются относительно нетоксичными неорганическими и органическими основными дополнительными солями соединений по настоящему изобретению, к примеру, аммония, четвертичного аммония и аминных реакций, производные от азотных оснований значительной основности для формирования солей соединениями по настоящему изобретению (см. , например, S.M.Berge et. al., “Pharmaceutical Salts” в J. Phar. Sci., 66: 1-19 (1977)). Основная часть соединений по настоящему изобретению (группа Q формулы I и группа Q1 формулы II) может реагировать с подходящими органическими или неорганическими кислотами для формирования солей по изобретению. Примеры солей могут быть выбраны из группы, содержащей ацетат, бензолосульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, камсилат, карбонат, хлорид, клавуланат, дигидрохлорид, эдетат, эдисилат, эстолат, эсилат, фумарат, глюцептат, глюконат, глютамат, гликоллиларсанлат, гексилресорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, йодид, изотионат, лактат, лактобионат, лаурат, малат, мальзеат, манделат, мезилат, метилбромид, метилнитрат, метилсульфат, мукат, напсилат, нитрат, олеат, оксалат, пальмитат, пантотенат, фосфат, полигалактуронат, салицилат, стеарат, субацетат, сукцинат, таннат, тартрат, тосилат, трифторацетат, трифторметансульфонат и валерат. Соединения по формуле (I) могут также быть в виде амфотерных ионов, так как они содержат и кислотную, и основную функциональность и способны к самопротонированию. Определенные соединения по настоящему изобретению обладают одним или более хиральными центрами и, таким образом, могут существовать в оптически активных формах. Подобным образом, когда соединения содержат алкениловую или алкениленовую группу, существует возможность цис- и трансизомерных форм этих соединений. R- и S- изомеры и их смеси, в том числе рацемические смеси, а также смеси цис- и трансизомеров, подразумеваются настоящим изобретением. Дополнительные асимметричные атомы углерода могут присутствовать в замещающей группе, такой, например, как алкиловая группа. Все такие изомеры, а также их смеси следует рассматривать в рамках настоящего изобретения. Если желателен отдельный стереоизомер, он может быть приготовлен хорошо известными из уровня техники способами путем использования стереоспецифических реакций с исходными материалами, содержащими асимметричные центры, и уже растворенными или альтернативно при помощи способов, которые дают смеси стереоизомеров и последующее растворение с помощью известных способов. Пролекарственные производные соединений по настоящему изобретению. Пролекарства являются производными соединений по настоящему изобретению, которые имеют метаболически расщепляемые группы и становятся путем сольволиза или при физиологических условиях соединениями по настоящему изобретению, которые фармацевтически активны в естественных условиях. К примеру, сложноэфирные производные соединений по настоящему изобретению часто активны в естественных условиях, но неактивны в искусственных. Другие производные соединений по настоящему изобретению имеют активность как в форме кислоты, так и в форме кислотной производной, но форма кислотной производной часто дает преимущества растворимости, совместимости с тканями или задержанного высвобождения в организме млекопитающего (см. Bundgard, Н. Design of Prodrugs, p. 7-9, 21-24, Elsevier, Amsterdam, 1985). Пролекарства включают в себя кислотные производные, хорошо известные специалистам-практикам, например, такие как сложные эфир, полученные реакцией исходной кислоты с подходящим спиртом, или амиды, полученные реакцией соединения исходной кислоты с амином. Простые алифатические или ароматические сложные эфиры, полученные из кислотных групп, ответвляющихся от соединений по настоящему изобретению, являются предпочтительными пролекарствами. В некоторых случаях желательно получать пролекарства типа двойного сложного эфира, такие как (алкоксил)алкильные сложные эфиры или ((алкоксикарбонил)окси)алкильные сложные эфиры. Предпочтительными являются C1-C8-алкиловые, C2-C8-алкениловые, ариловые, C7-C12-замещенные ариловые и C7-C12-арилалкиловые сложные эфиры соединений по настоящему изобретению (по формуле I). В частности, предпочтительны C1-C4-алкиловые сложные эфиры, к примеру, в которых кислотная группа R3 была превращена в сложный эфир, сформировав группу, представленную одной из следующих формул: 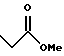 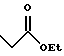 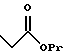 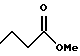 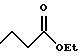 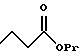 Другие специфические пролекарственные производные, которые являются соединениями по настоящему изобретению, представлены формулами (Xa)-(Xb), показанными ниже: 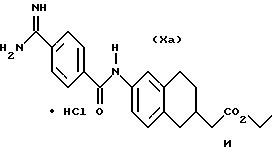 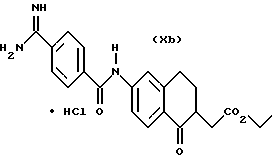 Ацилированные основные радикалы, которые являются частью основной группы на соединениях по настоящему изобретению, как было обнаружено, значительно повышают биополезность. Без привязки к какой-либо теории действия предполагается, что понижение основности основной группы (Q) делает соединения по настоящему изобретению менее подверженными “пищевому эффекту”, то есть они имеют хорошую пригодность для терапевтической подачи животному без голодания. Соединения по настоящему изобретению выгодным образом могут являться двойными пролекарственными производными. К примеру, кислотная группа (R3) может вступать в реакцию, формируя сложный эфир, а основная группа Q (или основной радикал Q1) может дополнительно вступать в реакцию, формируя ацилированное основное производное. Более того, пролекарственные производные соединений по настоящему изобретению могут комбинироваться с другими характеристиками, описанными здесь, для увеличения биополезности, к примеру замещением атомами фтора на кольце D соединений по формуле (II). Эти комбинированные характеристики дают в результате такое соединение, как представленное формулой (Xc): 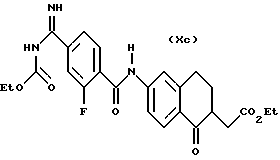 Другим в высшей степени предпочтительным классом пролекарств по настоящему изобретению являются те, которые сформированы ацилированием основных радикалов (например, Q1), имеющихся у соединений по настоящему изобретению. Ациловая часть ацилированного основного радикала имеет общую формулу: 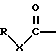 где R является C1-C8-алкилом, C2-C8-алкенилом, арилом, C7-C12-замещенным арилом и C7-C12-арилалкилом; а X является связью, С, О, S или N. Предпочтительно R является C1-C4-алкилом, а X – кислородом. К примеру, пролекарства ацилированных основных радикалов по изобретению получаются, как показано пунктами А, В, С и D ниже: А) ацилирование амидина дает группу пролекарственного производного: 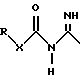 В) ацилирование циклического амина, например, пиперидина, дает группу пролекарственного производного: 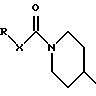 С) ацилирование гуанидина дает группу пролекарственного производного: 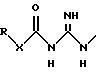 D) ацилирование первичного амина дает группу пролекарственного производного: 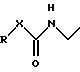 где, для вышеупомянутых А, В, С и D, R является таким же, как определено выше для ацилированной части основной группы. Терапевтические соединения по настоящему изобретению включают в себя пролекарственные производные бициклических соединений, имеющих ядро, сформированное двумя слившимися шестичленными кольцами, А и В, представленных формулой (Xd): 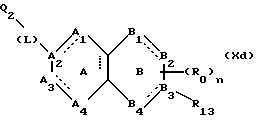 в которой A1, А3, А4 выбираются независимо из углерода, кислорода, серы и азота; А2 выбирается независимо из углерода или азота при условии, что A2 имеет незанятую связь, если А2 является азотом, и при условии, что как минимум два атома из A1, А2, А3 и А4 являются атомами углерода; B1, В2, В4 выбираются независимо из углерода, кислорода, серы и азота; В3 выбирается независимо из углерода или азота при условии, что В3 имеет незанятую связь, если В3 является азотом, и при условии, что как минимум два атома из B1, В2, В3, В4 являются атомами углерода; n – число от 2 до 6; R0 являются одинаковыми или различными и выбираются независимо из водорода, алкила, галогенозамещенного алкила, алкенила, алкинила, циклоалкила, арила, арилалкила, гидроксильной, алкоксильной, аралкоксильной, амино-, замещенной амино-, карбамойловой, карбоксильной, ациловой, циано-, галогено-, нитро-, сульфогрупп, =O или =S при условии, что, если R0 является =O или =S, то только один из атомов B1, В2, В3 и В4 может быть атомом азота; m – число от 2 до 6; R10 являются одинаковыми или различными и выбираются независимо из водорода, алкила, галогенозамещенного алкила, алкенила, алкинила, циклоалкила, арила, арилалкильной, гидроксильной, алкоксильной, аралкоксильной, карбоксильной, ациловой, циано-, галогено-, нитро-, сульфогрупп, =O и =S; при условии, что только один R10 может являться =O или =S; связующая группа -(L)- является связью или двухвалентной замещенной или незамещенной цепочкой из 1-10 атомов, выбираемых из группы, состоящей из углерода, азота, серы и кислорода; и Q2 выбирается из (i) основных групп, либо (ii) основных групп, содержащих ацилированный основной радикал; R13 выбирается из (i) кислотных групп, содержащих кислотный радикал, либо (ii) кислотных групп, содержащих сложноэфирную производную кислотного радикала; при условии, что Q2 является основной группой, содержащей ацилированный основной радикал, или R13 является кислотной группой, содержащей сложноэфирную производную кислотного радикала. Предпочтительной формой пролекарственной производной является соединение с формулой (Xd), имеющее двойную пролекарственную функциональность, то есть в котором Q2 является основной группой, содержащей ацилированный основной радикал, a R13 является кислотной группой, содержащей сложноэфирную производную кислотного радикала. Другой предпочтительной формой пролекарства является соединение с формулой (Xd), в котором ацилированная часть ацилированного основного радикала имеет общую формулу: 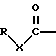 где R является C1-C8-алкилом, C2-C8-алкенилом, арилом, C7-C12-замещенным арилом и C7-C12-арилалкилом, а X является связью, С, О, S или N. Предпочтительно R является C1-C4-алкилом, а X – кислородом. Группа Q2 может содержать две части, а именно (i) один или более радикалов, выбираемых из основных радикалов или ацилированных основных радикалов, каждый из которых обозначен Q3, и (ii) циклической группы D (как ранее определено формулой Iw). Общая формула для пролекарственных производных по настоящему изобретению описывает бициклическое соединение, имеющее ядро, сформированное из двух слившихся шестичленных колец, А и В, представленное формулой (Xe), или фармацевтически приемлемые соли, сольваты или пролекарственные производные этого соединения: 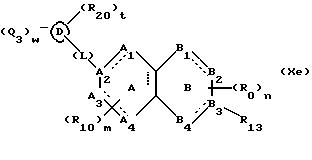 где A1, А3, А4 выбираются независимо из углерода, кислорода, серы и азота; А2 выбирается независимо из углерода или азота при условии, что А2 имеет незанятую связь, если А2 является азотом, и при условии, что как минимум два атома из A1, А2, А3 и А4 являются атомами углерода; B1, B2, В4 независимо выбираются из углерода, кислорода, серы и азота; B3 выбирается независимо из углерода или азота при условии, что В3 имеет незанятую связь, если B3 является азотом, и при условии, что как минимум два атома из B1, В2, В3, В4 являются атомами углерода; n – число от 0 до 6; R0 являются одинаковыми или различными и независимо выбираются из водорода, алкила, галогенозамещенного алкила, алкенила, алкинила, циклоалкила, арила, арилалкильной, гидроксильной, алкоксильной, аралкоксильной, амино-, замещенной амино-, карбомойловой, карбоксильной, ациловой, циано-, галогено-, нитро-, сульфогрупп, =O или =S, при условии, что, если R0 является =O или =S, то только один атом из B1, B2, В3 и B4 может быть атомом азота; m – число от 0 до 6; R10 являются одинаковыми или различными и независимо выбираются из водорода, алкила, галогенозамещенного алкила, алкенила, алкинила, циклоалкила, арила, арилалкильной, гидроксильной, алкоксильной, аралкоксильной, карбоксильной, ациловой, циано-, галогено-, нитро-, сульфогрупп, =O и =S, при условии, что только один R10 может являться =O или =S; t – число от 0 до 3; R20 являются одинаковыми или различными и независимо выбираются из водорода, галогенов, алкила, галогенозамещенного алкила, алкенила, алкинила, циклоалкила, арила, арилалкильной, гидроксильной, алкоксильной, аралкоксильной, карбоксильной, ациловой, циано-, галогено-, нитро-, сульфогрупп; связующая группа -(L)- является связью или двухвалентной замещенной или незамещенной цепочкой из 1-10 атомов, выбираемых из группы, состоящей из углерода, азота, серы и кислорода; и D является кольцом из 5-8 атомов кольца, и упомянутые атомы кольца независимо выбираются из углерода, азота, кислорода или серы с условием, что как минимум два атома кольца D являются атомами углерода; w – целое число от 1 до 3; Q3 выбирается из (i) основных радикалов, или (ii) ацилированных основных радикалов; R13 выбирается из (i) кислотной группы, содержащей кислотный радикал, либо (ii) кислотной группы, содержащей сложноэфирную производную кислотного радикала; при условии, что Q3 является ацилированным основным радикалом или R13 является кислотной группой, содержащей сложноэфирную производную кислотного радикала. Целое число w предпочтительно равно 1. В формуле (Xe) предпочтительно, чтобы R20 были хлором и/или фтором, a t равнялось 1 или 2. Также предпочтительными являются соединения по формуле (Xe), в которых Q3 является ацилированным основным радикалом, а R13 является кислотной группой, содержащей сложноэфирную производную кислотного радикала. Наиболее предпочтительными ацилированными основными группами являются основные радикалы карбаминокислотных сложных эфиров амидиновых, пиперидиновых или гуанидиновых основных радикалов. Наиболее предпочтительны радикалы карбаминокислотных C1-C4 алкильных сложных эфиров амидина. Карбаминокислотные сложноэфирные пролекарственные производные по изобретению могут быть получены таким способом, как показанный на схеме 27. Предпочтительные пролекарственные производные соединений по настоящему изобретению, имеющие различные характеристики, обсуждаемые в этом разделе, представлены формулами (Xf)-(Xr) ниже: 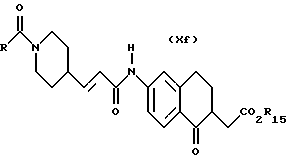 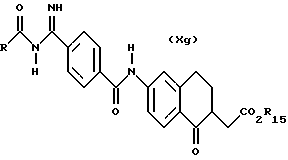 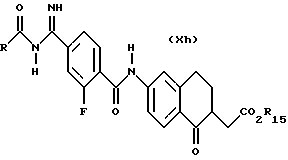 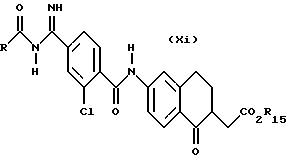 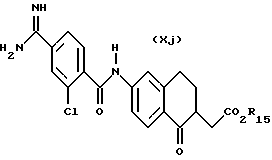 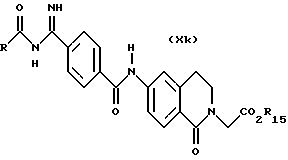 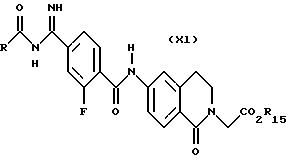 где R = -OMe, -OEt, -OPr, C1-C4алкил; R15=Me, Et, Pr 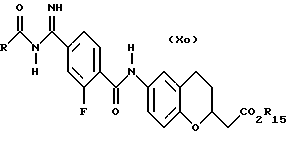 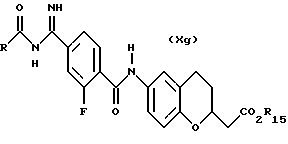 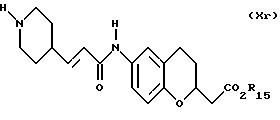 где R = -H, -OMe, -OEt, -OPr, X = -Cl, -F, -H, R15 = Me, Et, Pr. Способ получения соединений по изобретению Общие схемы 1-33 синтеза, приведенные в конце описания, используются для получения соединений по настоящему изобретению. Во всех схемах и примерах используются следующие сокращения: TBAF – фторид тетрабутиламмония Tf – (трифлат)-сульфонат трифторметана Boc – третичный бутоксикарбонил Bn – бензил But – третичный бутил DMF – формамид диметила Cbz – бензилоксикарбонил EDCI – 1-(3-диметиламинопропил)-3-этилкарбодиимид DMAP – диметиламинопиридин LHMDS – гексаметилдисилазан лития THF – тетрагидрофуран DIBAH – диизобутилгидрид алюминия Boc 20 – ди-трет-бутилбикарбонат HMDS – гексаметилдисилазан TsOH – p-толуолсульфокислота MCPBA – мета-хлоропероксибензойная кислота NMO – 4-метилморфолин-N-оксид TFAA – трифторацетатный ангидрид TBSCL – трет-бутилдиметилсилилхлорид TMEDA – N,N,N’,N’-тетраметилэтилендиамин LDA – диизопропиламид лития. Общие комментарии. Реакции, описываемые схемами реакций, выполняются с использованием стандартных химических методологий, описанных и указанных в стандартной литературе. Исходные материалы являются коммерчески доступными реагентами, а реакции выполняются в стандартной лабораторной посуде при нормальных температуре и давлении, за исключением тех случаев, когда специально оговариваются другие условия. Схема 1 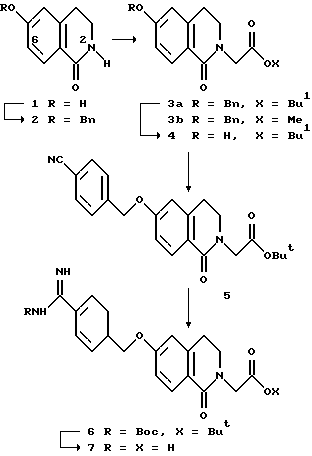 Схема 1 описывает способ получения 2,6-двузамещенных изохинолонов, имеющих эфирно-связанный аргининовый изостер у С6 и радикал уксусной кислоты в позиции 2. На первом шаге схемы 1 изохинолин (1) реагирует с бензилбромидом в присутствии карбоната калия в орошающем ацетоне для получения бензилозащищенного фенола (2). Это соединение реагирует с гидридом натрия и затем алкилируется на азоте либо при помощи альфа-бромо-трет-бутил ацетата, либо альфа-бромометилацетата для получения 2-замещенного изохинолона (3а) (6-бензилокси-3,4-дигидро-1-оксо-2(1H)изохинолон уксусной кислоты-1,-диметилэтил сложный эфир) или (3b). Бензильная группа С6 затем удаляется с помощью водорода и палладия, а последующее алкилирование 6-гидроксильной группы выполняется при помощи K2CO3 и алкилбромида для получения двузамещенного изохинолона (5). Соединение (5) затем преобразуется в Boc-защищенный амидин (6) с помощью нескольких реакций, а именно: (i) реагирования нитрила с H2S, (ii) алкилирования промежуточного тиоамида метилйодидом, (iii) реагирования промежуточного тиоимидата с ацетатом аммония, и (iv) затем Boc-протектирования сформированного амидина для получения соединения (6). Соединение (6) депротектируется с помощью неразбавленной TFA, дающей (7) в виде соли TFA. Схема 2 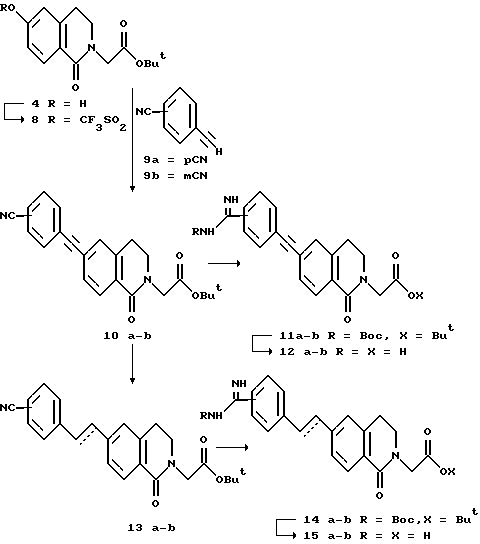 —- возможная ненасыщенность Схема 2 описывает способ синтеза, пригодный для получения углеродного замещения в позиции С6 бициклического ядра. В этой схеме соединение (4) (6-гидрокси-3,4-дигидро-1-оксо-2(1H)изохинолин уксусной кислоты-1,1-диметил сложный эфир) по схеме 1 преобразуется в трифлат (8) с помощью трифторуксусного ангидрида и пиридина. Затем это соединение реагирует с ацетиленовым соединением (9a) или (9b) в присутствии палладия для получения ацетиленово-связанного бензонитрила (10a) или (10b). Соединение (10a) или (10b) снова преобразуется при помощи такого же набора процедур, какой используется для преобразования соединения (5) (6-{(4-цианофенил)метокси}-3,4-дигидро-1-оксо-2(1H) изохинолон уксусной кислоты, -1,1-диметилэтил сложный эфир) в соединение (6) (6-{{4-(1,1-диметилэтоксикарбонил аминоиминометил)фенил}метокси}-3,4-дигидро-1-оксо-2(1H)изохинолон уксусной кислоты-1,1-диметилэтил сложный эфир) для получения амидинового продукта (11a) или (11b). Соединения (11a) или (11b) также могут снова быть депротектированы с помощью TFA для получения соединения (12a) или (12b). Альтернативно промежуточное вещество (10a) или (10b) может быть либо частично, либо полностью гидрогенизировано, как показано на схеме, давая алкиленово- или алкениленовосвязанное соединения (13a) или (13b). Соединение (13a) или (13b) снова преобразуется с помощью преобразования нитрила в амидин, описанного ранее (схема 1, шаги 5 > 6), давая соединение (14a) или (14b), которое затем депротектируется при помощи TFA для получения соединения (15a) или (15b). Схема 3 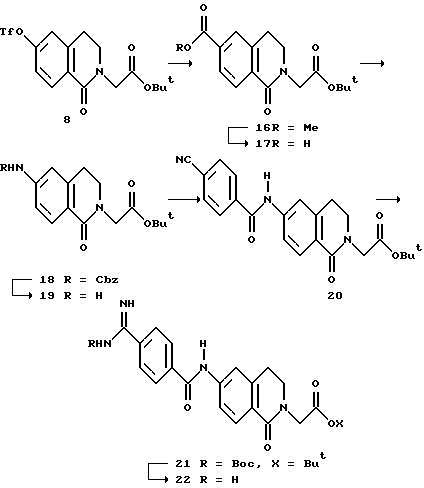 Схема 3 описывает получение изохинолонов, содержащих азотное замещение в позиции С6. Эта схема начинается с трифлата (8), получение которого описано ранее на схеме 2. Трифлат преобразуется в сложный ариловый эфир (16) путем использования палладия, монооксида углерода и метанола. Сложный эфир (16) затем омыливается гидроксидом лития в водном растворе TFA. Свободная кислота (7) затем подвергается перегруппировке Кертиуса (т.е. образованию изоцианата путем температурного разложения ацилазидов). Требуемый ацилазид формируется с помощью трифенилфосфорилазида, и затем пиролизуется на месте для получения изоцианата, который затем поглощается бензиловым спиртом, давая Cbz-защищенный анилин (18). Cbz-анилин (18) затем преобразуется в свободный амин (19) с помощью каталитической гидрогенизации. Амин (19) затем ацилируется парацианбензойной кислотой в присутствии EDCI и DMAP, давая амидосвязанное соединение (20). Соединение (20) затем преобразуется в Boc-защищенный амидин (21) при условиях по схеме 1, а это вещество затем депротектируется с помощью TFA для получения соединения (22). Схема 4 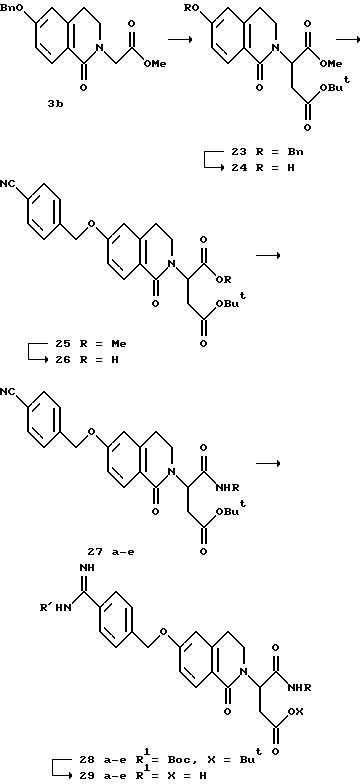 Амины (а)-(e): (a) гексиламин (b) бензиламин (с) p-метоксифенэтиламин (d) метиламин (e) бета-амино-1-бутилаланин Схема 4 описывает получение 2,6-двузамещенных изохинолонов, в которых позиция 2 замещена долей аспарагиновой кислоты. Схема 4 начинается с соединения (3b), получение которого описывается схемой 1. Соединение (3b) отдает протон LHMDS, а результирующий анион затем нейтрализуется при помощи альфа-бромо-t-бутилацетата для получения соединения (23). 6-бензиловая группа соединения (23) удаляется с помощью палладия и водорода для получения свободного фенола (24). Соединение (24) затем алкилируется, как описано для получения соединения (5) в схеме 1. Метиловый сложный эфир (25) затем смыливается гидроксидом лития в THF для получения свободного карбоксилата (26). Затем свободный карбоксилат соединяется с различными аминами в присутствии EDCI и DMAP для получения полуамидных сложных эфиров (27a)-(27e). Полуамидные сложные эфиры (27a)-(27e) затем снова преобразуются с помощью ранее описанного в схеме 1 протокола (шаги 5-6) для получения Boc-защищенных амидинов (28a)-(28e). Boc-защищенный амидин затем депротектируется с помощью TFA для получения соединений (29a)-(29e). Схема 5 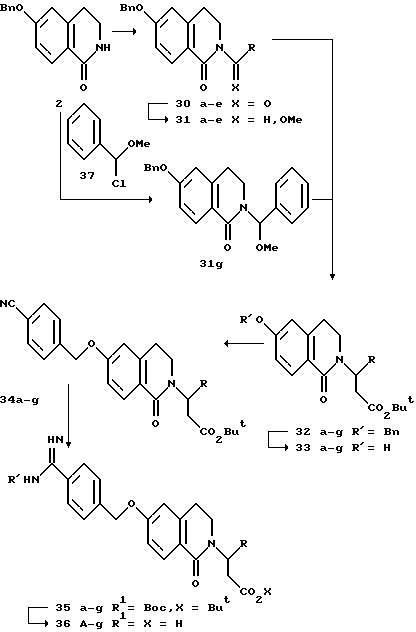 Типы кислот: (а) 4-этоксибутановая (b) пентановая (с) гексановая (d) 2-(2-метоксиэтокси)уксусная (e) пропионовая (f) бутановая Схема 5 описывает получение 2,6-двузамещенных изохинолонов, в которых позиция 2 замещена изостером аспарагиновой кислоты. Соединения схемы 5 отличаются от соединений, получаемых по схеме 4, в том, что группа R соединения (36) по схеме 5 не содержит амидинового связывания, как у соединений (29a)-(29e) по схеме 4. Соединение (2), исходный материал, получается по способу схемы 1, затем ацилируется разными активированными кислотами (кислотными галогенидами и ангидридами) для получения соответствующих имидов (30a)-(30e). Затем имид выборочно восстанавливается до своего экзоциклического карбонила при помощи DIBAH и затем улавливается кислым метанолом для получения альфа-метоксиамидов (31a)-(31e). Альтернативно альфа-метоксиамиды (31) могут быть получены путем реакции соли натрия из (2) с подходящим альфа-хлорэфиром (37). Все альфа-метоксиамиды (31a)-(31g) реагируют с эфиром трифторида бора в присутствии кетенацетали для получения бета, бета-двузамещенных пропионатов (32a)-(32g). После этого бензильная группа удаляется из позиции 6 с помощью каталитической гидрогенизации, а фенолы могут быть снова алкилированы способом, показанным в схеме 1 (шаги 4-5) для получения эфирно-связанных нитрилов (34a)-(34g). Этот нитрил затем может преобразовываться в Boc-защищенный амидин (35a)-(35g), как показано на схеме 1 (шаги 5-6). Затем депротекция дает окончательные соединения (36a)-(36g). Схема 6 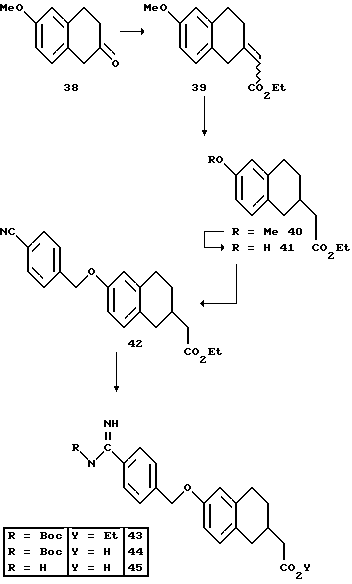 Схема 6 описывает получение соединений по настоящему изобретению, имеющих тетралиновое ядро. 6-метокси-2-тетралон (38) реагирует с трет-бутил диэтилфосфоноацетатом для получения ненасыщеного сложного эфира (39). Последующая гидрогенизация удаляет ненасыщенность для получения соединения (40). Соединение (40) обрабатывается трибромидом бора, а неочищенный продукт переэстерифицируется с помощью HCl и этанола для получения (41). Фенол (41) затем алкилируется, как показано на схеме 1 (шаги 4-5), давая (42). Нитрил затем может преобразовываться в Boc-защищенный амидин (43), как показано на схеме 1 (шаги 5-6). Амидиновый сложный эфир (43) затем омыливается с помощью гидроксида натрия для получения соединения (44), которое затем депротектируется с помощью TFA и анизола для получения окончательного продукта (45). Схема 7 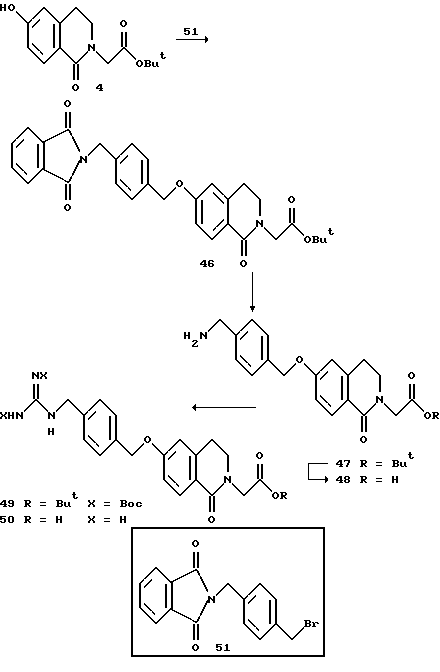 Схема 7 описывает получение соединений по настоящему изобретению, имеющих гуанидиновую группу в качестве основной функциональности. Фенол (4), полученный по схеме 1, алкилируется бромидом (51) (полученным из дибромида и фталимида калия), давая аддукт (46). Это соединение депротектируется водным гидрозином, давая амин (47). Соединение (47) преобразуется в защищенный гуанидин (49) при помощи N,N’-бис(трет-бутоксикарбонил)-S-метил-изотиомочевины. Соединение (49) депротектируется при помощи TFA, давая продукт (50) в виде трифторацетатной соли. Схема 8 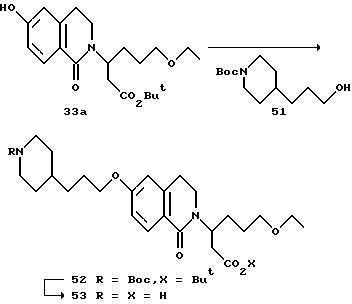 Схема 8 описывает получение соединений по настоящему изобретению, имеющих аминовую группу в качестве основной функциональности. Соединение (33a), полученное по схеме 5, соединяется со спиртом (51) (полученным из 3-(4-пиридил)-пропанола с помощью стандартных протоколов) при помощи трифенилфосфена и диэтилазодикарбоксилата, давая соединение (52). Соединение (52) депротектируется с помощью чистой TFA, давая продукт (53) в виде соли TFA. Схема 9 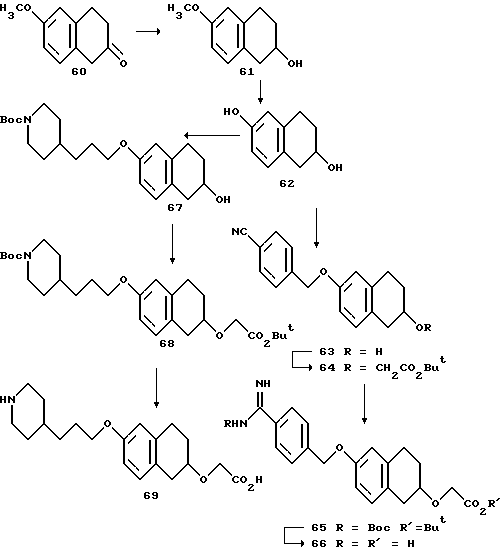 Схема 9 описывает получение 2-6 двузамещенных тетралинов, в которых позиция 2 занята радикалом a-алкоксиуксусной кислоты, а позиция 6 содержит либо эфирно-связанный бензамидин, либо составляющую эфирно-связанного 4-алкилпиперидина. Схема начинается с 6-метокси-2-тетралона (60), который последовательно обрабатывается NaBH4, а затем DIBAH, давая дигидроксильное соединение 62. Фенольный гидроксил может быть выборочно алкилирован с помощью либо альфа-бромо-p-толунитрила, либо подходящего 4-алкилпиперидина, давая соединения 63 и 67 соответственно. Оба соединения затем алкилируются при помощи трет-бутилбромацетата в условиях фазового переноса, давая соединения 64 и 68. Нитрил 64 преобразовывается в Boc-защищенный амидин 65, а затем в продукт 66 при помощи последовательности реакций, описанных в схеме 1. Соединение 68 преобразуется в полностью депротектированное соединение 69 путем обработки TFA. Схема 10 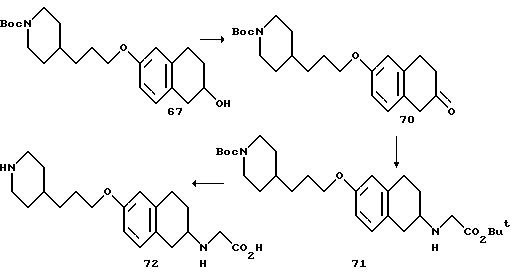 Схема 10 описывает получение 2,6-двузамещенных тетралинов, в которых позицию 2 занимает составляющая альфа-аминоуксусной кислоты, а позицию 6 – эфирно-связанный 4-алкилпиперидин. Спирт 67, полученный по схеме 9, оксидируется с помощью DMSO и TFAA при условиях Сверна, давая кетон (70), который аминируется с восстановлением при помощи глицин трет-бутил сложного эфира, давая соединение 71. Этот материал затем депротектируется с помощью TFA, давая соединение 72. Схема 11 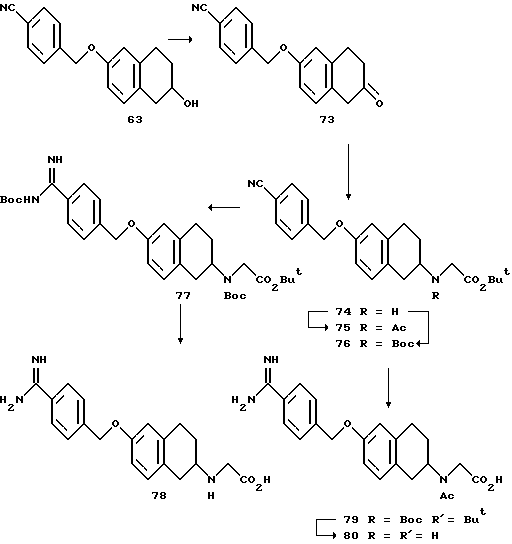 Схема 11 обрисовывает получение 2,6-двузамещенных тетралинов, в которых позиция 2 содержит радикал альфа-аминоуксусной кислоты, а позиция 6 занята эфирно-связанным бензамидином. Синтез начинается со спирта 63 (схема 9), который оксидируется с помощью TFAA и DMSO (способ Сверна), давая кетон 73. Этот материал затем аминируется с восстановлением с помощью глицин трет-бутил сложного эфира, давая 74. Вторичный азот затем либо Boc-защищается (76), либо ацилируется (75). Затем Boc-производная преобразуется в защищенный амидин 77 с помощью последовательности реакций, выведенных в схеме 1. Затем этот материал полностью депротектируется с помощью TFA, давая соединение 78. Подобным образом ацетиловая производная 75 преобразуется в соединение 80. Схема 12 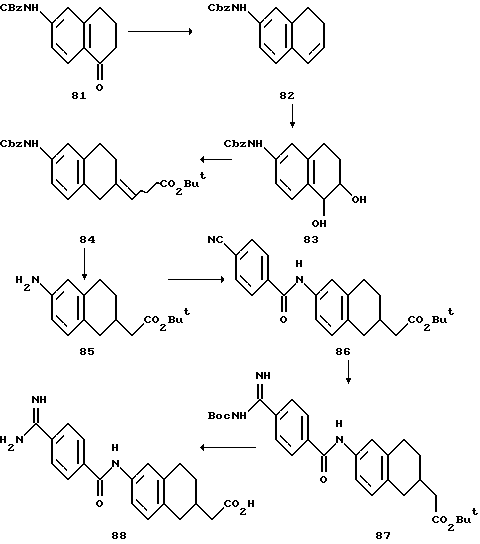 Схема 12 обрисовывает получение тетралинов, имеющих радикал уксусной кислоты у C2 и амидосвязанный бензамидин у C6. На первом шаге тетралон 81 восстанавливается с помощью NaBH4, а получающийся в результате нестабильный спирт дегидрируется с помощью TsOH в бензоле, давая дигидронафталин 82. Осимиляция соединения 82 дает диол 83, который затем подвергается действию TsOH в орошающем бензоле. Таким образом сформированный нестабильный 2-тетралон не изолируется, а допускается в реакцию с натриевой солью трет-бутилдиэтилфосфоацетата, давая ненасыщенный сложный эфир 84 в виде смеси олефиновых изомеров. Этот материал подвергается гидрогенизации на палладии, что приводит к предельности олефина и удалению Cbz-группы, обеспечивая анилин 85. Ацилирование соединения 85 при помощи 4-цианобензойной кислоты выполняется с помощью EDCI, а получающийся в результате амид 86 преобразуется в Boc-защищенный амидин 87 в условиях, ранее описанных в схеме 1. Удаление составляющей Вос и расщепление трет-бутилового сложного эфира выполняется при помощи TFA, давая соединение 88. Схема 13 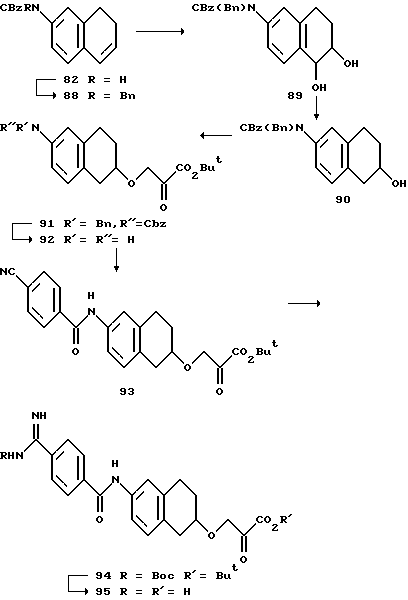 Схема 13 описывает получение производных тетралина, в которых позиция 2 замещена составляющей a-алкоксиуксусной кислоты, а позиция 6 замещена амидосвязанным бензамидином. В этой схеме соединение 82 из схемы 12 вводится в реакцию с NaH и бензилбромидом, давая третичный карбамат 88. Этот материал затем подвергается осимиляции и дегидрации так же, как описано для соединения 83 в схеме 12. Сформированный нестабильный 2-тетралон немедленно восстанавливается до спирта 90 при помощи NaBH4. Этот материал алкилируется с помощью трет-бутилбромацетата в условиях фазового переноса, что дает в результате эфир 91. Каталитическая гидрогенизация высвобождает 6-амино составляющую (92), которая ацилируется с помощью 4-цианобензойной кислоты в присутствии EDCI, давая 93. Нитрил 93 преобразуется в Boc-защищенный амидин 94 с помощью нескольких преобразований, описанных в схеме 1. Одновременная депротекция этого амидина и кислотных составляющих выполняется с помощью TFA, давая окончательный продукт 95. Схема 14 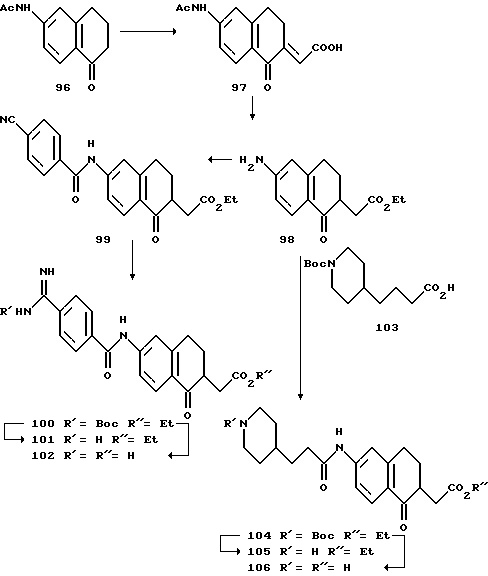 Схема 14 обрисовывает синтез тетралонов, имеющих составляющую уксусной кислоты в позиции 2 и либо амидосвязанный бензамидин, либо амидосвязанный 4-алкилпиперидин в позиции 6. Схема начинается с тетралона 96, который вводится в реакцию с глиоксиловой кислотой в присутствии NaOH, давая продукт 97 конденсации. Ненасыщенный сложный эфир 97 восстанавливается с помощью Zn в HOAc, а получающееся в результате соединение преобразуется в анилин 98 сначала путем удаления ацетата с помощью 6N HCl, а затем путем превращения в сложный эфир кислотной составляющей с помощью этаноловой HCl. Затем этот материал ацилируется с помощью 4-цианобензойной кислоты при посредничестве EDCI, давая 99. Нитрильная составляющая соединения 99 преобразуется в Boc-защищенный амидин 100 с помощью последовательности реакций, описанных в схеме 1. Омыливание сложноэфирной составляющей с помощью NaOH, сопровождаемое обработкой TFA, дает соединение 102. Соединения, содержащие амидосвязанный 4-алкилпиперидин, могут быть получены путем ацилирования анилина 98 с помощью соединения 103, что дает аналог 104. Омыливание сложного эфира 104, сопровождаемое TFA-депротекцией пиперидина, дает соединение 106. Схема 15 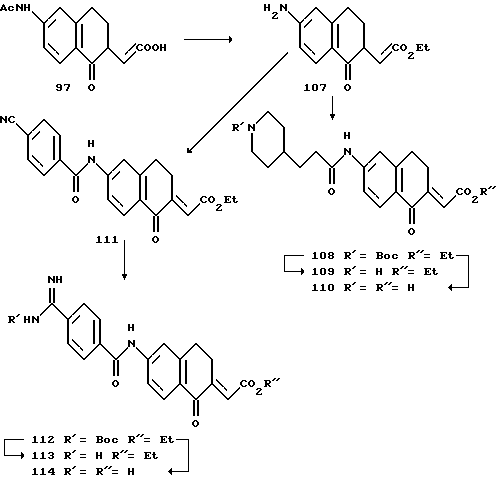 Схема 15 описывает способ получения производных тетралона, в которых позиция 2 занята ненасыщенной кислотой, а позиция 6 замещена либо амидосвязанным бензамидином, либо 4-алкилпиперидином. На первом шаге соединение 97 (схема 14) может преобразовываться в анилин 107 путем удаления ацетата с помощью 6N HCl и последующего превращения в сложный эфир с помощью этанольной HCl. Затем этот материал может быть ацилирован с помощью либо 4-цианбензойной кислоты, либо подходящего 4-алкилпиперидина (103). В первом случае нитрил 111 может преобразовываться в амидин 112 с помощью последовательности реакций, описанных в схеме 1. Омыливание соединения 112, сопровождаемое обработкой с помощью TFA, должно давать 114. Пиперидиновый замыкатель 108 может быть подвергнут омыливанию и TFA-депротекции, что подобным образом дает соединение 110. Схема 16 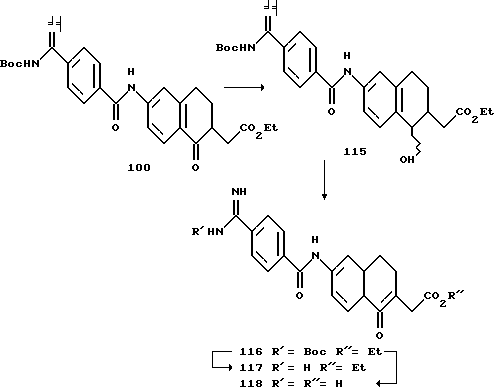 Схема 16 описывает получение производных дигидронафталина, содержащих в позиции 2 составляющую уксусной кислоты, а в позиции 6 – амидосвязанный бензамидин. Тетралон 100 (схема 14) вводится в реакцию с помощью NaBH4 в этаноле, давая нестабильный спирт 115. Этот материал обрабатывается с помощью TsOH в THF, давая дегидрированный продукт 116. Сложноэфирное омыливание, сопровождаемое деблокированием амидина с помощью TFA, дает желаемый продукт 118. Схема 17 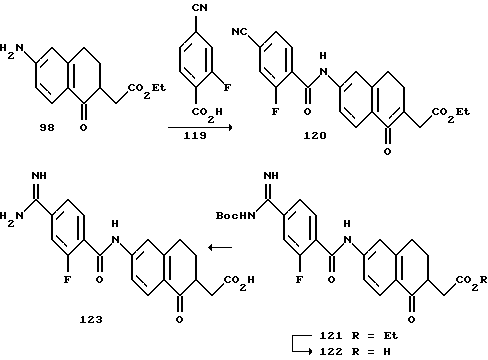 Схема 17 обрисовывает общее получение 2,6-двузамещенных тетралонов, в которых позиция 2 замещена радикалом уксусной кислоты, а позиция 6 содержит амидосвязанный галогенозамещенный бензамидин. Анилин 98 (полученный по схеме 14) вводится в реакцию с бензойной кислотой 119 (получаемой стандартными способами из 4-амино-2-фтортолуола) в присутствии EDCI и DMAP. Получающийся амид (120) преобразуется в Boc-защищенный амидин 121 с помощью процедур, обозначенных в схеме 1. Затем сложноэфирная составляющая гидролизуется, давая кислоту 122, а последующая обработка TFA обеспечивает соединение 123. Схема 18 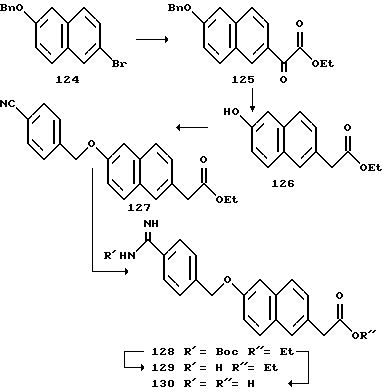 Схема 18 описывает способ получения 2,6-двузамещенных нафталинов, имеющих радикал уксусной кислоты в позиции 2 и эфирно-связанный аргининовый изостер в позиции 6. На первом шаге схемы 18 бромонафталин 124 подвергается переметаллированию с помощью t-BuLi, а результирующий анион нейтрализуется этилоксалатом. Получающийся аддукт 125 затем восстанавливается с помощью NaBH4, а получившийся спирт ацилируется с помощью уксусного ангидрида. Каталитическая гидрогенизация удаляет бензильный ацетат и высвобождает 6-гидроксильную составляющую, давая соединение 126. Затем свободный фенол алкилируется с помощью альфа-бромо-p-толунитрила в присутствии К2СО3, давая двузамещенный нафталин 127. Нитрильная составляющая затем преобразуется в Boc-защищенный амидин 128 с помощью последовательности реакций, ранее описанных в схеме 1. Омыливание сложного эфира в соединении 128, сопровождаемое удалением Boc-группы с помощью TFA, дает окончательное соединение 130. Схема 19 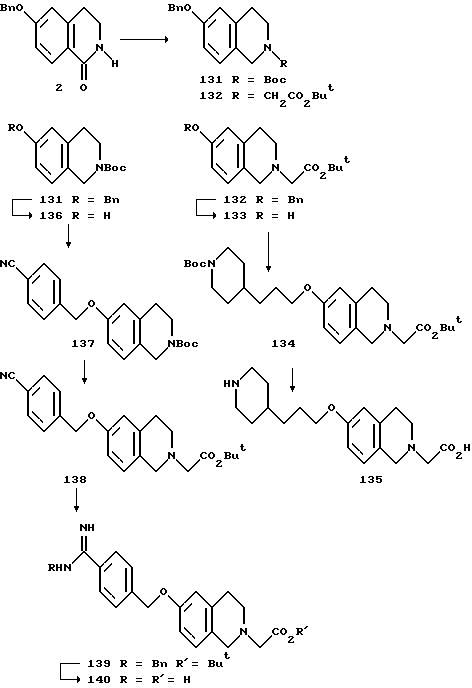 Схема 19 описывает получение двузамещенных производных тетрагидроизохинолина, имеющих составляющую уксусной кислоты в позиции 2 и либо эфирно-связанный бензамидин, либо 4-алкил пиперидиновую составляющую в позиции 6. Начальное изохинолиновое ядро получается путем восстановления бензилзащищенного изохинолона 2 (схема 1) с помощью LiAlH4. Этот материал подвергается либо Boc-протектированию), что дает соединение 131, либо алкилированию с помощью трет-бутилбромацетата, что дает образование соединения 132. Boc-защищенный материал подвергается гидрогенизации, которая высвобождает С6-фенол, который затем алкилируется с помощью альфа-бромотолунитрила, давая 137. Boc-группа этого соединения расщепляется с помощью TFA, а затем результирующий амин алкилируется с помощью трет-бутилбромацетата, давая соединение 138. Это соединение преобразуется в Boc-защищенный амидин 139, а затем в депротектированный вариант 140 с помощью процедур, обозначенных в схеме 1. N-алкилированное соединение 132 подобным образом подвергается гидрогенизации, а получающийся фенол алкилируется с помощью подходящего 4-алкилпиперидина, давая соединение 134. Этот материал депротектируется с помощью TFA, давая соединение 135. Схема 20 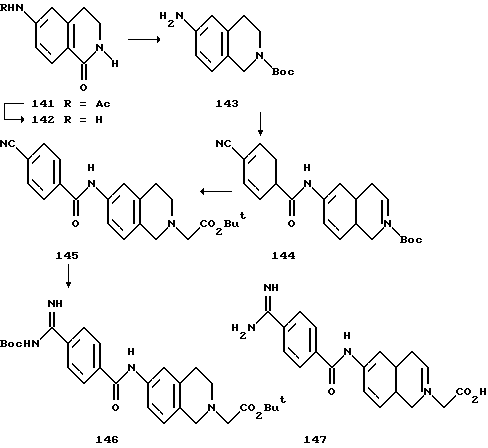 Схема 20 описывает получение 2,6-двузамещенных производных тетрагидроизохинолина, содержащих радикал уксусной кислоты в позиции 2 и амидосвязанный бензамидин в позиции 6. Синтез начинается с кислотного гидролиза 6-ацетамидогруппы изохинолона 141, давая анилин 142. Затем сырье подвергается действию бензилбромида и К2СО3 в CH3CN, что дает смесь моно- и дибензилзащищенных изохинолонов. Эта смесь подвергается восстановлению с помощью LiAlH4, формируя тетрагидроизохинолин, который сразу обрабатывается ди-трет-бутилбикарбонатом. Сформированный Boc-защищенный материал затем гидрогенизируется на палладии, давая анилин 143. Этот материал ацилируется с помощью p-цианбензойной кислоты, давая соединение 144. Обработка этого материала с помощью TFA дает вторичный амин, который алкилируется с помощью трет-бутилбромацетата, давая соединение 145. Преобразование соединения 145 в Boc-защищенный амидин 146, а затем в его депротектированный гомолог 147 выполняется с помощью процедур, описанных в схеме 1. Схема 21 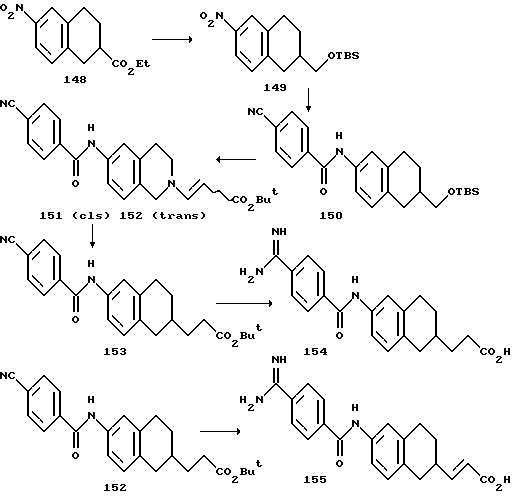 Схема 21 описывает способ синтеза, пригодный для получения 2,6-двузамещенных тетралинов, содержащих пропионатную или пропеноатную составляющую в позиции 2 и амидосвязанный бензамидин в позиции 6. На первом шаге сложный нитроэфир 148 восстанавливается с помощью LiBH4, а получающийся спирт протектируется в виде своего TBS-эфира. Соединение 149 затем подвергается гидрогенизации, а сформированный анилин сразу обрабатывается с помощью EDCI и p-цианбензойной кислоты, давая амид 150. Силиловая группа соединения 150 удаляется, а полученный спирт подвергается оксидированию с помощью DMSO и оксалилхлорида (способ Сверна). Таким образом полученный альдегид не очищается, а вводится в реакцию с натриевой солью t-бутил диэтилфосфоноацетата, которая дает разделяемую смесь олефиновых изомеров 151 (цис) и 152 (транс). Транс-изомер 152 преобразуется в Boc-защищенный амидин, а затем в депротектированное соединение 155 с помощью последовательности действий, описанной в схеме 1. Цис изомер подвергается гидрогенизации на палладии, дает предельный аналог 153. Этот материал также преобразуется в Boc-защищенный амидин, а затем в его депротектированный гомолог 154, как описано в схеме 1. Схема 22 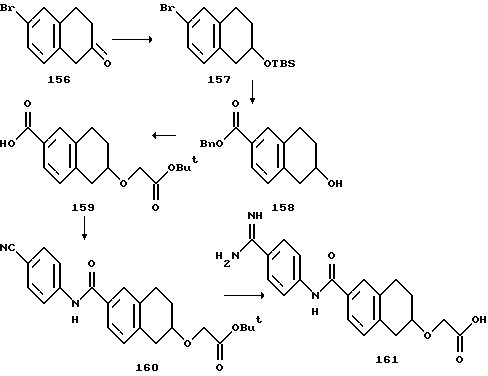 Схема 22 описывает способ синтеза для двузамещенных тетралинов, имеющих радикал альфа-алкоксиуксусной кислоты у 2 и карбоксилсвязанный бензамидин у С6. Эта схема начинается с 6-бромо-2-тетралона (156), который восстанавливается с помощью NaBH4, а получающийся спирт протектируется в виде своего трет-бутилдиметилсилил (TBS) эфира, давая соединение 157. Обработка этого соединения с помощью t-BuLi вызывает металлогалогеновый обмен, а формирующийся анион нейтрализуется с помощью CO2. Получающийся карбоксилат сразу преобразуется в бензиловый сложный эфир при помощи бензилового спирта и EDCI. TBS-группа удаляется в процессе обработки при помощи TBAF, что приводит к получению спирта 158. Свободная вторичная гидроксильная группа алкилируется при помощи трет-бутилбромацетата с использованием условий фазового переноса, а 6-карбоксилат высвобождается путем каталитической гидрогенизации, что дает соединение 159. Амид 160 является результатом ввода соединения 159 в реакцию с 4-циананилином в присутствии EDCI и DMAP. Нитрил 160 преобразуется в Boc-защищенный амидин, а затем – в полностью депротектированное соединение 161 при условиях, описанных в схеме 1. Схема 23 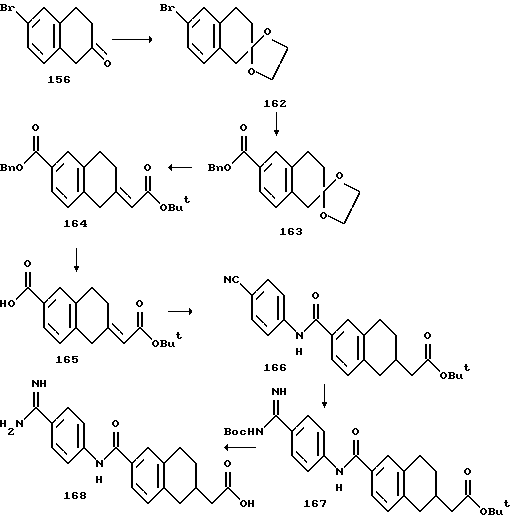 Схема 23 обрисовывает получение тетралинов, имеющих радикал уксусной кислоты у С2 и карбоксилсвязанный бензамидин у С6. На первом шаге бромтетралон 156 обрабатывается этиленгликолем и TsOH в условиях дегидрации, что дает кетал 162. Этот материал обрабатывается с помощью t-BuLi, а результирующий анион нейтрализуется с помощью CO2. Сформированная кислота сразу превращается в сложный эфир с помощью бензилового спирта и EDCI, что дает соединение 163. Спирокетал, содержащийся в 163, расщепляется с помощью водной HCl в ацетоне, а полученный кетон вводится в реакцию с натриевой солью трет-бутилдиэтилфосфоноацетата, давая соединение 164 в виде смеси олефиновых изомеров. Каталитическая гидрогенизация на палладии удаляет ненасыщенность и высвобождает С6 карбоксилат, давая кислоту 165. Конденсация этого соединения с помощью 4-аминобензонитрила дает амид 166. Преобразование соединения 166 в Boc-защищенный амидин 167, а затем в окончательное соединение 168 выполняется с использованием последовательности действий, описанной в схеме 1. Схема 24 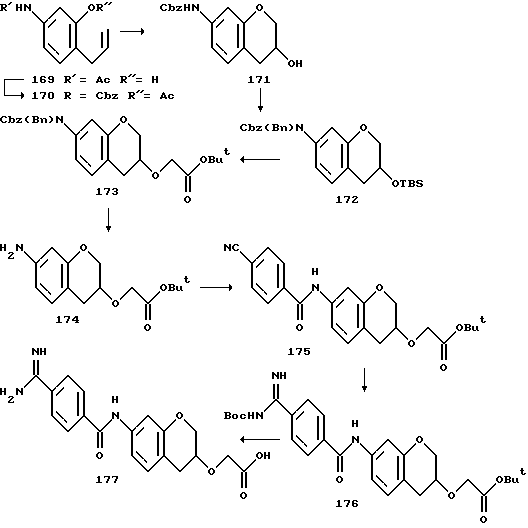 Схема 24 описывает получение 3,7-двузамещенных бензопиранов, в которых позиция 3 замещена составляющей альфа-алкоксиуксусной кислоты, а позиция 7 замещена амидосвязанным бензамидином. Синтез начинается с аллилозамещенного ароматического углеводорода 169. Ацетамидный гидролиз выполняется с помощью NaOH в EtOH (щелочь Клейзена), а получающийся анилин заново протектируется в виде своего CBz-варианта. Свободный фенол затем ацилируется уксусным ангидридом, давая соединение 170. Олефин реагирует с MCPBA, давая соответствующий эпоксидный полимер, который перегруппировывается в присутствии NaI, давая смесь 3-гидрокси- и 3-ацетоксибензопиранов. Эта смесь обрабатывается с помощью LiOH, что дает спирт 171. Спиртовая составляющая соединения 171 затем преобразуется в свой TBS-эфир, а получающееся соединение адкилируется на азоте для получения полностью защищенного соединения 172. Высвобождение гидроксирадикала у С3 при помощи TBAF, сопровождаемое алкилированием при помощи трет-бутилбромацетата при условиях фазового переноса, дает соединение 173. Каталитическая гидрогенизация обеспечивает анилин 174, который ацилируется с помощью 4-цианбензойной кислоты, давая амид 175. Этот материал сначала преобразуется в соответствующий защищенный бензамидин 176, а затем в его деблокированный гомолог 177 при помощи последовательности действий, описанной в схеме 1. Схема 25 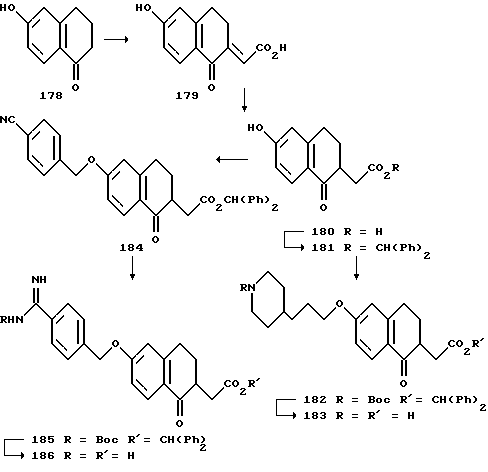 Схема 25 обрисовывает получение 2,6-двузамещенных тетралонов, в которых позиция 2 замещена уксуснокислотной составляющей, а позиция 6 замещена либо алкоксисвязанным бензамидином, либо алкоксисвязанным 4-алкилпиперидином. На первом шаге тетралон 178 обрабатывается NaOH и глиоксиловой кислотой, давая аддукт 179. Этот материал восстанавливается с помощью Zn в уксусной кислоте, а получающаяся кислота (180) реагирует с дифенилдиазометаном, давая бензгидриловый сложный эфир 181. Затем свободный фенол может алкилироваться с помощью альфа-бромо-p-толунитрила, давая соединение 184, либо с помощью подходящего 4-алкилпиперидина, давая соединение 182. Нитрил 184 затем преобразуется в соответствующий Boc-защищенный амидин 185, а затем в полностью депротектированное соединение 186 при помощи последовательности реакций, описанной в схеме 1. Соединение 182 депротектируется с помощью TFA, давая соединение 183. Схема 26 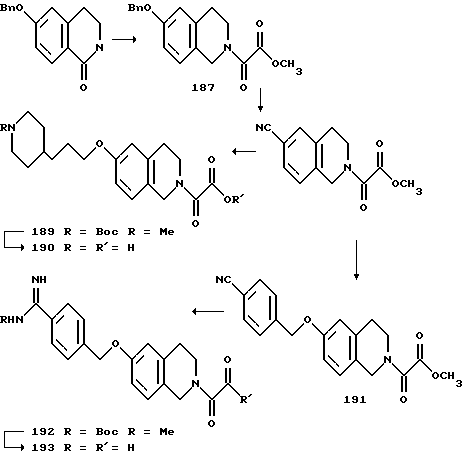 Схема 26 описывает способ получения тетрагидроизохинолинов, в которых позиция 2 замещена радикалом оксаминовой кислоты, а позиция 6 содержит эфирно-связанный бензамидин. На первом шаге изохинолон 2 обрабатывается LiAlH4, а получающийся продукт восстановления ацилируется метилоксалилхлоридом, давая соединение 187. Этот материал подвергается гидрогенизации, а получающийся фенол алкилируется либо с помощью альфа-бромотолунитрила, либо с помощью подходящего 4-алкилпиперидина, давая соединения 191 и 189 соответственно. Нитрильная составляющая соединения 191 преобразуется в Boc-защищенный амидин 192 при помощи процедур, описанных в схеме 1. Этот материал затем oмыливается с помощью NaOH, а получающаяся кислота обрабатывается с помощью TFA, давая соединение 193. Соединение 190 получается с помощью подобной последовательности омыливания и депротекции. Схема 27 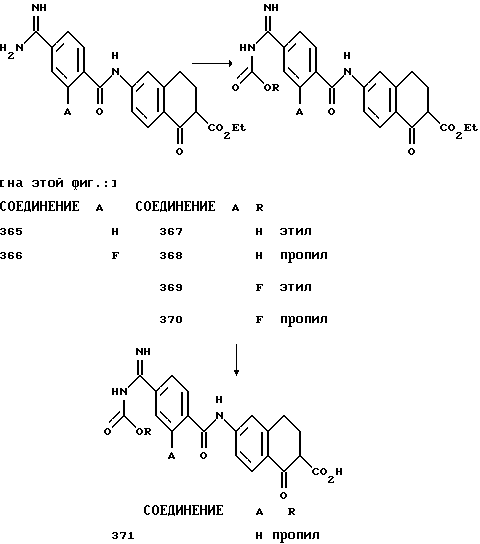 Схема 27 описывает ацилирование амидиновой составляющей, содержащейся в 2,6-двузамещенных тетралонах 365 и 366, которые получаются из соединений 102 и 123 схем 14 и 17 (соответственно) путем превращения в сложный эфир с помощью этанола. Ацилирование выполняется посредством реагирования соединения, содержащего бензамидин, с алкилхлорформиатом в присутствии водного основания, таким образом получаются производные 367 и 370. Эти материалы затем могут подвергаться омыливанию с помощью этанольного NaOH, что дает свободную кислоту (см. 371 в схеме 27). Схема 28 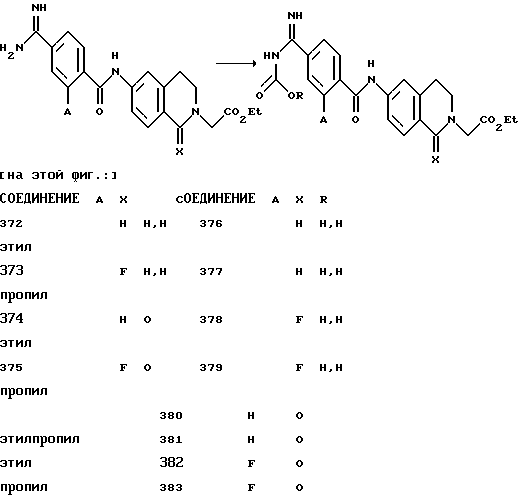 Процедура по схеме 27 является общей и также была применена к соединениям, содержащим изохинолоновое ядро, как показано на схеме 28. Подобным образом, можно также получить N-ацилированные производные бензамидина, содержащие тетрагидроизохинолины или бензопираны. Схема 29 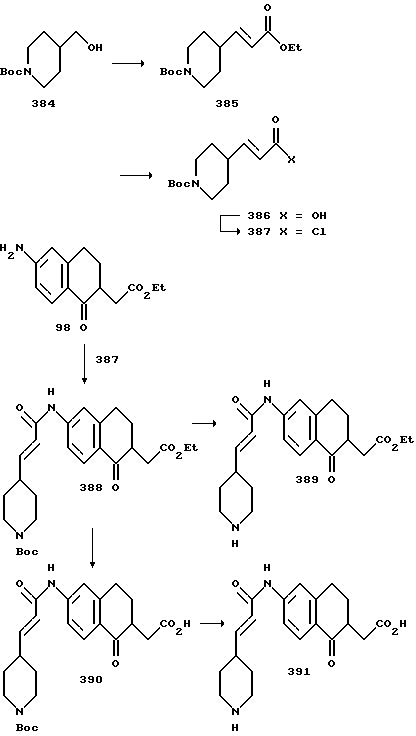 Схема 29 описывает получение 2,6-двузамещенных тетралонов, в которых позиция 2 занята радикалом уксусной кислоты, а позиция 6 содержит амидосвязанную 4-пропенойлпиперидиновую составляющую. На первом шаге спирт 384 (полученный из 4-пиридилкарбинола путем гидрогенизации и протектирования) оксидируется с помощью оксалилхлорида и DMSO, давая соответствующий альдегид. Этот материал не отделяется, а реагирует в неочищенном виде с натриевой солью триэтилфосфоноацетата, что дает желаемый ненасыщенный сложный эфир 385. Этот материал oмыливается с помощью LiOH, а получающаяся кислота 386 активируется оксалилхлоридом, давая соединение 387. Анилин 98 реагирует с кислым хлоридом 387, давая аддукт 388. Это соединение N-депротектируется с помощью TFA, давая сложный эфир 389 после солевого обмена с помощью HCl. Альтернативно омыливание с помощью LiOH сначала дает свободную кислоту 390, которая затем может N-депротектироваться с помощью TFA, давая соединение 391. Схема 30 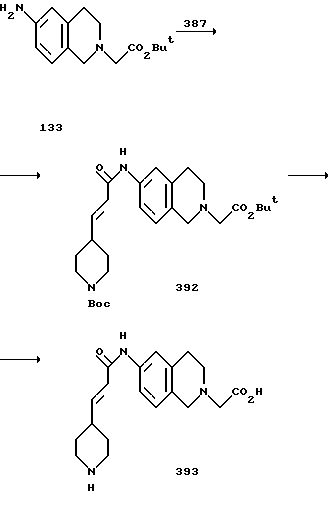 Схема 30 описывает получение 2,6-двузамещенных тетрагидроизохинолинов, имеющих радикал уксусной кислоты в позиции 2, и амидосвязанный 4-пропенойлпиперидин в позиции 6. На первом шаге 6-аминосоставляющая соединения 133 ацилируется кислым хлоридом 387 (схема 29), давая аддукт 392. Этот материал может полностью депротектироваться с помощью TFA, обеспечивая соединение 393. Схема 31 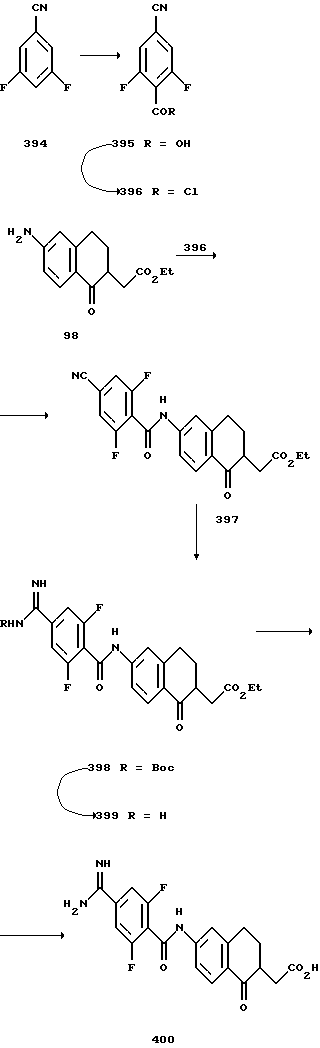 Схема 31 обрисовывает получение 2,6-двузамещенных тетралонов, в которых позиция 2 занята уксусно-кислотной составляющей, а позиция 6 поддерживает амидосвязанный дифторбензамидин. На первом шаге к дифторбензонитрилу 394 добавляется литий с помощью n-бутиллития, а получающийся анион нейтрализуется с помощью CO2, что дает кислоту 395. Это соединение затем обрабатывается оксалилхлоридом, а получающийся кислый хлорид 396 реагирует с анилином 98, давая аддукт 397. Нитрильная составляющая в соединении 397 преобразуется в Boc-защищенный амидин 398 с помощью последовательности реакций, используемых для преобразования 5 в 6, как описано в схеме 1. Соединение 398 может депротектироваться с помощью TFA, давая соединение 399. Альтернативно соединение 398 может быть полностью депротектировано сначала путем расщепления сложно-эфирной составляющей с помощью NaOH, а затем путем депротектирования амидина с помощью TFA, что дает соединение 400. Схема 32 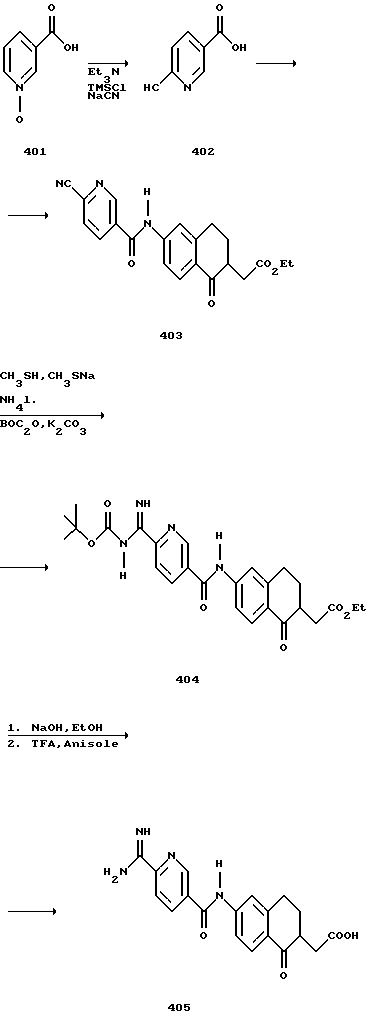 Схема 32 описывает получение 2,6-двузамещенных тетралонов, имеющих уксусно-кислотную составляющую в позиции 2 и амидосвязанный амидинопиридин в позиции 6. На первом шаге пиридин 401 реагирует с Et3N, TMSCl и NaCN, давая кислоту 402. Этот материал соединяется с анилином 98, давая аддукт 403. Нитрил в соединении 403 затем реагирует с натриевой солью метантиола, давая метилтиоимидат. Это промежуточное соединение реагирует с йодидом аммония, давая амидин, который Boc-протектируется, давая соединение 404. Соединение 404 сначала реагирует с этанольным NaOH для расщепления сложного эфира, а затем – с TFA для депротектирования амидина, обеспечивая полностью депротектированный гомолог 405. Схема 33 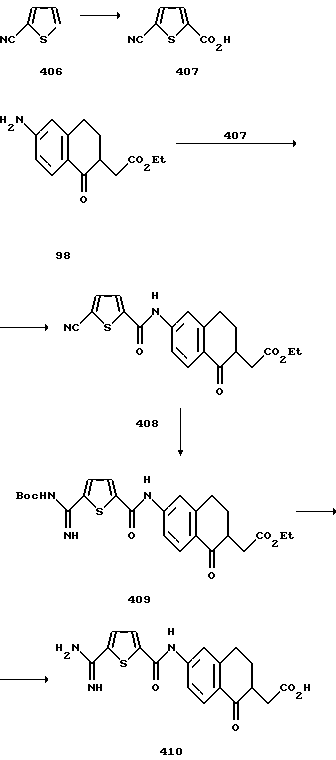 Схема 33 описывает получение 2,6-двузамещенных тетралонов, в которых амидосвязанный амидин содержит тиофеновое ядро. На первом шаге тиофен 406 металлируется с помощью LDA, а получающийся анион нейтрализуется с помощью CO2, давая кислоту 407. Эта кислота реагирует с соединением 98 в присутствии EDCI, давая амид 408. Нитрильная составляющая в соединении 408 преобразуется в Boc-защищенный амидин 409 при помощи последовательности реакций, используемой для формирования соединения 6 в схеме 1. Получающееся соединение сначала oмыливается при помощи этанолового NaOH, а затем N-депротектируется с помощью TFA, давая соединение 410 в виде соли TFA. Схема 34 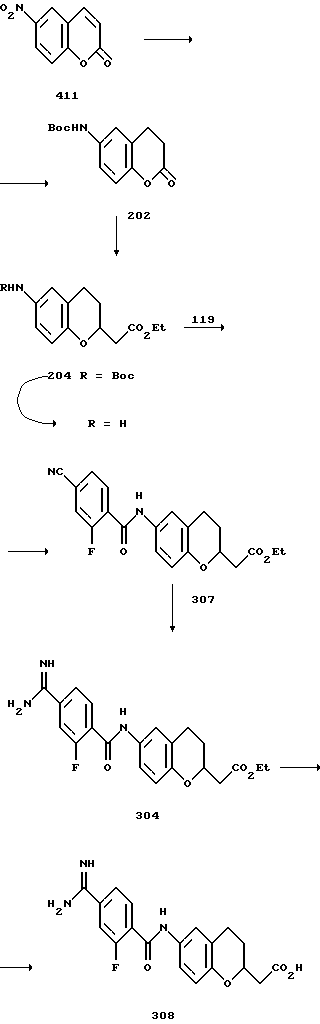 Схема 34 описывает получение 2,6-двузамещенных бензопиранов, в которых позиция 2 содержит уксуснокислотную составляющую, а позиция 6 содержит амидосвязанный фторозамещенный бензамидин. При первом преобразовании нитрогруппа восстанавливается формиатом аммония и палладием, а получающийся анилин Boc-протектируется. Затем при помощи Pd/C устраняется ненасыщенность кольца В, что дает лактон 202. Этот материал восстанавливается с помощью DIBAH, давая промежуточный лактол, который реагирует с этоксикарбонилметилентрифенилфосфораном, давая бензопиран 204. Этот материал затем N-депротектируется с помощью TFA, а получающийся анилин реагирует с кислым хлоридом, полученным из фторокислоты 119, давая аддукт 307. Этот материал затем подвергается действию HCl в этаноле, что дает промежуточный иминоэфир, который не отделяется, а вместо этого реагирует с аммиаком, что приводит к образованию соединения 304. Этот материал затем гидролизуется с помощью NaOH в этаноле, давая желаемую свободную кислоту 308 после нейтрализации. Примеры Следующие примеры даются, чтобы позволить специалисту пользоваться настоящим изобретением. Эти примеры, однако, не должны считаться ограничением рамок изобретения, как определено в формуле изобретения. Номера ссылок, используемые в примерах, относятся к соответствующим соединениям, показанным в предшествующих схемах 1-26 реакций. Пример 1 Получение 6-{ { 4-(аминоиминометил)фенол}метокси}-3,4- дигидро-1-оксо-2(1H)-изохинолинуксусного кислого трифрорацетата, соединение имеет формулу (7): 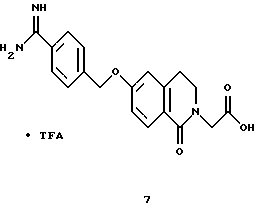 Часть A: Смесь фенола (1) (6-гидрокси-3,4-дигидро-1-оксо- 2(1H)-изохинолина) (1,0 г, 6,14 ммоль), бензилбромида (1,0 г, 6,14 ммоль), К2СО3 (0,93 г, 6,74 ммоль) и ацетона (15 мл) поддерживалась при орошении в течение 12 ч, а затем охлаждалась до комнатной температуры. Затем смесь разбавлялась EtOAc и промывалась H2O. Органический материал обезвоживался (MgSO4) и концентрировался. Неочищенный остаток перекристаллизовывался из EtOAc/гексана, давая 1,53 г (98%) соединения (2) (6-бензилокси- 3,4-дигидро-1-оксо-2(1H)-изохинолина) в виде белого сухого остатка. Часть B: В раствор лактама (2) (0,1 г, 0,39 ммоль) в THF (4 мл) был добавлен гидрид натрия (0,017 г 60%-ной дисперсии в минеральном масле, 0,43 ммоль). Полученная смесь поддерживалась при орошении в течение 1 ч, а затем охлаждалась до комнатной температуры. Затем смесь обрабатывалась трет-бутилбромацетатом (0,07 г, 0,43 ммоль). По истечении 1 ч реакция нейтрализовалась добавлением H2O (10 мл), и полученная смесь экстрагировалась с помощью EtOAc. Объединенные экстракты обезвоживались (с помощью MgSO4) и концентрировались. Неочищенный материал очищался путем хроматографии (силикагель, 2: 1 гексан:EtOAc), давая 0,14 г (99%) соединения (3a) в виде белого сухого остатка. Часть C: Смесь соединения (3а) (0,13 г, 0,37 ммоль), Pd/C (0,14 г, 10% на углероде), и EtOAc (5 мл) перемешивалась в атмосфере водорода (стеклянная пробирка) в течение 1,5 ч, а затем фильтровалась. Затем фильтрат концентрировался, давая 0,13 г (100%) вещества (4) в виде в основном чистого белого сухого остатка. Часть D: Смесь соединения (4) (1,00 г, 3,60 ммоль), альфа-бромо-p-толунитрила (0,71 г, 3,60 ммоль), К2СО3 (0,50 г, 3,60 ммоль) и ацетона (35 мл) поддерживалась при орошении в течение 4 ч, а затем охлаждалась до комнатной температуры. Полученная смесь концентрировалась, а остаток хроматографировался на двуокиси кремния (1:1 гексан/EtOAc), давая 1,38 г (98%) соединения (5) в виде прозрачного масла. Часть E: Смесь соединения (5) (0,385 г, 0,982 ммоль), пиридина (5,5 мл), и Et3N (0,55 мл) насыщалась H2S и оставлялась на 2 дня. Этот раствор затем разбавлялся H2O, а полученная смесь экстрагировалась с помощью EtOAc, и экстракты концентрировались. Неочищенное выделенное вещество вводилось в смесь ацетона (5 мл) и CH3I (2,5 мл), и поддерживалось при орошении в течение 1 ч. Эта смесь охлаждалась до комнатной температуры и концентрировалась. Неочищенное выделенное вещество вводилось в MeOH (5 мл) и обрабатывалось с помощью NH4OAc. Полученный раствор поддерживался в течение 2 ч при 60oC, а затем концентрировался. Неочищенное выделенное вещество вводилось в раствор THF/H2O (1:1, 6 мл) и обрабатывалось с помощью К2СО3 (0,179 г, 1,30 ммоль) и Boc2O (0,202 г, 0,95 ммоль), а полученная смесь перемешивалась при комнатной температуре в течение 2 ч. Затем реакционная смесь разбавлялась EtOAc и промывалась водой. Затем органический материал концентрировался, и неочищенное выделенное вещество очищалось путем хроматографии (силикагель 200-400 меш, 30: 1 CHCl/MeOH), давая 0,311 г (62%) соединения (6) в виде прозрачного масла. Часть F: Смесь соединения (6) (0,311 г, 0,612 ммоль) и TFA (5 мл) поддерживалась при комнатной температуре в течение 1 ч, а затем концентрировалась. Остаток добавлялся в H2O, и смесь промывалась Et2O. Оставшийся водный материал лиофолизировался, давая 0,31 г соединения (7) в виде белого сухого остатка. 1H NMR (300 MHz, CD3OD) 3,03 (t, J=6,5 Hz, 2H), 3,68 (t, J=6,5 Hz, 2H), 4,29 (s, 2H), 5,30 (s, 2H), 6,94 (d, J=1,9 Hz, 1H), 7,0 (dd, J=1,9, 8,6 Hz, 2H), 7,69 (d, J=8,5 Hz, 2H), 7,84 (d, J=8,5 Hz, 2H), 7,87 (d, J=8,6 Hz, 1H), IR (CHCl3) 2928, 1695, 1435, 1286 см-1; MS (FAB) m/e 354,1451 (354,1454 рассчитано для С19H20N3O4). Пример 2 Получение 6-{ { 4-(аминоиминометил) фенил} этил}-3,4- дигидро-1-оксо-2(1H)изохинолинуксусного кислого трифторацетата, соединение представлено формулой (12a): 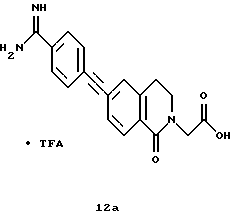 Часть А: В раствор соединения (4) (9,5 г, 34,2 ммоль) и свежедистиллированного пиридина (250 мл) был добавлен трифторметансульфоангидрид (5,8 мл, 34,2 ммоль) при 0oC. Полученный раствор был оставлен нагреваться до комнатной температуры, а затем нейтрализовался добавлением H2O (125 мл). Смесь экстрагировалась с помощью EtOAc, экстракт обезвоживался (MgSO4) и концентрировался. Неочищенный материал очищался путем хроматографии (силикагель, 4:1 гексан:этилацетат), давая 11,54 г (82,4%) соединения (8) (6-{{ (трифторметил)-сульфонил} окси} -3,4- дигидро-1-оксо-2(1H)изохинолинуксусной кислоты-1,1-сложный диметилэфир) в виде белого сухого остатка. Часть B: Смесь соединения (8) (0,325 г, 0,79 ммоль), соединения (9a) (0,141 г, 1,11 ммоль), бис(трифенилфосфин)-палладий (II) хлорида (0,014 г, 0,02 ммоль), безводного DMF (2,5 мл) и свежедистиллированного Et3N (0,5 мл) перемешивалась при 90oC в течение 1 ч. В это время добавлялась H20 (25 мл), и смесь экстрагировалась при помощи EtOAc (2 х 75 мл). Экстракты обезвоживались на MgSO4 и концентрировались. Неочищенный материал очищался путем колоночной хроматографии (силикагель, 5:2 гексан:EtOAc), давая 0,173 г (57%) соединения (10a) в виде оранжевого сухого остатка. Часть С: Следуя общей процедуре, используемой для получения соединения (6), (пример 1, часть Е) соединение (11a) получалось с выходом 53% при исходных 0,13 г соединения (10a). Часть D: Следуя общей процедуре, используемой для получения соединения (7), (пример 1, часть F) соединение (12a) (6-{{4-(аминоиминометил)фенил}этинил} -3,4-дигидро-1-оксо-2(1H)- изохинолинуксусный кислый трифторацетат) получалось с выходом 76% при исходных 0,089 г соединения (11a). 1H NMR (300 MHz, CD3OD) 3,11 (t, J=6,6 Hz, 2H), 3,73 (t, J=7,8 Hz, 1H), 4,34 (s, 2H), 7,51 (s, 1H), 7,55 (d, J=7,8 Hz, 1H), 7,78 (d, J=8,7 Hz, 2H), 7,83 (d, J=7,4 Hz, 2H), 7,97 (d, J=8,0 Hz, 1H); IR (KBr) 3355, 3085, 1709, 1610, 1183 см-1; MS (FAB) m/e 348,1332 (348,1348 рассчитано для С20H18N3О3). Пример 3 Получение 6-{ 2-{ 4-(аминоиминометил)фенил} этил}-3,4- дигидро-1-оксо-2(1H)-изохинолинуксусного кислого трифторацетата, соединение представлено формулой (15a): 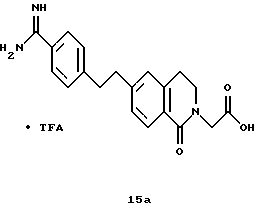 Часть А: Смесь соединения (10a) (0,10 г, 0,26 ммоль), Pd/C (0,10 г 10% на углеродe) и EtOAc (15 мл) перемешивалась в атмосфере водорода (стеклянная колба) в течение 1,5 ч, а затем фильтровалась и концентрировалась, давая 0,10 г (100%) соединения (13a) в виде нечистого белого сухого остатка. Часть B: Следуя общей процедуре, используемой при получении соединения (6) (пример 1, часть Е), соединение (14a) получается с выходом 78% при исходных 0,095 г соединения (13a). Часть C: Следуя общей процедуре, используемой для получения соединения (7) (пример 1, часть F), соединение (15a) получается с выходом 60% при исходных 0,09 г соединения (14a). 1H NMR (300 MHz, CD3OD), 3,01 (m, 6H), 3,64 (t, J=6,6 Hz, 2H), 4,28 (s, 2H), 7,10 (m, 3HO), 7,39 (d, J=8,2 Hz, 2H), 7,67 (d, 8,2 Hz, 2H), 7,77 (d, J= 8,4 Hz, 1H); IR (KBr) 3337, 3112, 1641, 1210, 1188 см-1; MS (FAB) m/e 352,1655 (352,1661 рассчитано для C20H22N3O3). Пример 4 Получение 6-{ { 4-(аминоиминометил)бензойл}амино}-3,4- дигидро-1-оксо-2(1H)-изохинолинуксусного кислого трифторацетата, соединение представлено формулой (22): 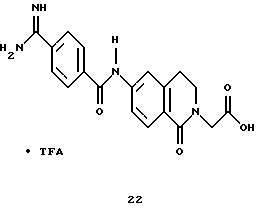 Часть А: Раствор из соединения (8) (пример 2, часть A) (5,0 г, 12,2 ммоль), DMF (25 мл), ацетата палладия (II) (0,082 г, 0,37 ммоль), трифенилфосфина (0,19 г, 0,73 ммоль), свежедистиллированного Et3N (3,4 мл, 24,4 ммоль) и безводного MeOH (9,9 мл, 244 ммоль) перемешивался в атмосфере СО (стеклянная пробирка) при температуре 65oC в течение 15 ч. Затем реакционная смесь остывала и разбавлялась H2O. Полученная смесь экстрагировалась с помощью EtOAc (2 х 100 мл). Объединенные экстракты обезвоживались (MgSO4) и концентрировались. Неочищенный материал очищался путем колоночной хроматографии (силикагель, 3: 1 гексан:EtOAc) для получения 2,80 г (72%) соединения (16) (6-(метоксикарбонил)-3,4-дигидро-1-оксо-2(1H)-изохинолонуксусной кислоты-1,1-сложный диметилэфир) в виде нечистого белого сухого остатка. Часть B: Раствор из соединения (16) (2,8 г, 8,7 ммоль) и THF (87 мл) обрабатывался водным LiOH (87 мл 0,1 N раствора, 8,7 ммоль), а полученный раствор выдерживался при комнатной температуре в течение 1 ч. Затем реакционная смесь концентрировалась до 1/2 объема и экстрагировалась с помощью EtOAc. Часть водного материала затем подкислялась (pH = 5) с помощью 1N HCl, и эта смесь затем экстрагировалась с помощью EtOAc. Объединенные экстракты затем обезвоживались (MgSO4) и концентрировались, давая 0,37 г соединения (17) в виде вязкого масла. Оставшийся водный материал лиофолизировался, давая 2,06 г соединения (17) в виде соли лития. Часть C: Раствор из соединения (17) (0,200 г, 0,66 ммоль) и безводного толуола (50 мл) обрабатывается дифенилфосфорилазидом (282,3 мл, 1,31 ммоль) и свежедистиллированным EtaN (0,18 мл, 1,31 ммоль), а полученный раствор поддерживался при температуре 85oC в течение 2 ч. Затем реагирующее соединения остывало до комнатной температуры, после чего обрабатывалось бензиловым спиртом (0,14 мл, 1,31 ммоль) и перемешивалось в течение еще одного часа. Затем реакционная смесь концентрировалась, и неочищенное выделенное вещество очищалось путем колоночной хроматографии (силикагель, 1:1 гексан:EtOAc), что давало 0,21 г (79%) соединения (18) (6-{(бензилоксикарбонил)амино}-3,4-дигидро-1-оксо-2(1H)-изохинолонуксусной кислоты-1,1-сложный диметилэфир) в виде белого сухого остатка. Часть D: Смесь соединения (18) (0,20 г, 0,49 ммоль), EtOH (20 мл), EtOAc (20 мл) и Pd/C (0,2 г 10% на C) перемешивалась в атмосфере водорода (стеклянная пробирка) в течение 1 ч, а затем фильтровалась и концентрировалась, давая 0,138 г (100%) соединения (19) (6-амино-3,4-дигидро-1-оксо-2(1H)- изохинолинуксусной кислоты-1,1-сложный диметилэтилэфир) в виде белого сухого остатка. Часть E: Раствор из соединения (19) (0,125 г, 0,45 ммоль), безводного дихлорметана (2,5 мл), парацианобензойной кислоты (0,066 г, 0,45 ммоль), 1-(3-диметиламинопропил-3-этилкарбоимид гидрохлорида (EDCI) (0,095 г, 0,50 ммоль) и 4-диметиламинопиридина (DMAP) (10,0 мг) выдерживался при комнатной температуре в течение 2 ч, а затем концентрировался. Неочищенное выделенное вещество очищалось путем колоночной хроматографии (силикагель, 2:1 EtOAc:гексан), давая 0,176 г (96%) соединения (20) в виде белого сухого остатка. Часть F: Следуя общей процедуре, используемой для синтеза соединения (6) (пример 1, часть Е), соединение (21) получалось с выходом 36% при исходных 0,17 г соединения (20). Часть G: Следуя общей процедуре, используемой для синтеза соединения (7) (пример 1, часть F), соединение (22) получалось с выходом 76% при исходных 0,07 г соединения (21). 1H NMR (300 MHz, CD3OD) 3,09 (t, J=6,6 Hz, 2H), 3,72 (t, J=6,6 Hz, 2H), 4,32 (s, 2H), 7,67 (d, 1H), 7,80 (br s, 1H), 7,94 (d, J=8,3 Hz, 3H), 8,16 (d, J=8,2 Hz, 2H); IR (CHCl3) 3354, 3007, 1634, 1538, 1196 см-1; MS (FD) m/e 367. Аналитически рассчитано для C21H19F3N4O6: С 52,50; H 3,99; N 11,66. Найдено: С 52,62; H 4,2; N 11,41. Пример 5 Получение (+-)-6-{ {4-(аминоиминометил)фенил}метокси}-3,4-дигидро-1-оксо-бета{ гексиламинокарбонил] -2(1H)-изохинолонуксусного кислого трифторацетата, соединение представлено формулой (29a): 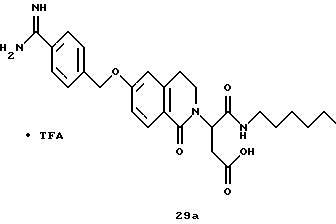 Часть А: Следуя процедуре, описанной для получения соединения (3a) (пример 1, часть B), соединение (3b) было получено с выходом 60%, при исходном лактаме (2) и метилбромацетате. Часть B: Раствор соединения (3b) (1,95 г, 6,0 ммоль) и THF (10 мл) добавлялся к раствору LHMDS (полученному из n-BuLi и HMDS по стандартным протоколам, 6,6 ммоль) и THF (10 мл) при -78oC. Через 1 ч раствор обрабатывался трет-бутилбромацетатом (1,1 мл, 6,6 ммоль) и оставлялся нагреваться до комнатной температуры. Смесь разбавлялась с помощью EtOAc (100 мл) и промывалась H2O. Органический материал обезвоживался (MgSO4) и концентрировался. Хроматография (силикагель, 200-400 меш, 2:1 гексан/EtOAc) давала 2,17 г (82%) соединения (23) в виде прозрачного масла. Часть C: Следуя процедуре, используемой для получения соединения (4) (пример 1, часть C), соединение (24) было получено с выходом 94% при исходных 2,17 г соединения (23). Часть D: Смесь из соединения (24) (1,79 г, 5,12 ммоль), альфа-бромо-p-толунитрила (1,11 г, 5,64 ммоль), К2СО3 (0,78 г, 5,64 ммоль), Bu4NI (кат.) и DMF (10 мл) перемешивалась при 80oC в течение 3 ч, а затем остывала до комнатной температуры. Затем смесь разбавлялась с помощью EtOAc (100 мл) и промывалась H2О. Органический материал концентрировался, и неочищенное выделенное вещество очищалось путем хроматографии (силикагель, 200-400 меш, 1,5:1 гексан/EtOAc), давая 2,32 г (98%) соединения (25) в виде прозрачного масла. Часть E: Смесь из соединения (25) (0,46 г, 1,0 ммоль), водного LiOH (11 мл, 0,1 ммоль) и THF (11 мл) перемешивалась при комнатной температуре в течение 3 ч, а затем концентрировалась до 1/2 объема. Оставшийся водный материал сначала промывался Et2O, а затем подкислялся до pH = 3 с помощью 1N HCl. Эта смесь экстрагировалась с помощью EtOAc, а объединенные экстракты концентрировались. Неочищенный остаток вводился в CH2Cl2 (5 мл) и обрабатывался гексиламином (0,15 мл, 1,1 ммоль), EDCI (0,28, 1,5 ммоль), и DMAP (кат.). Полученная смесь выдерживалась при комнатной температуре в течение 4 ч, а затем разбавлялась EtOAc и промывалась H2O. Органический материал концентрировался, и неочищенный остаток очищался путем хроматографии (силикагель 200-400 меш, 1:1 гексан/EtOAc), что дает 0,52 г (92%) соединения (27a) в виде прозрачного масла. Часть F: Следуя процедуре, используемой для получения соединения (6) (пример 1, часть Е), соединение (28a) было получено с выходом 75% при исходных 0,52 г соединения (27a). Часть G: Следуя процедуре для получения соединения (7) (пример 1, часть F), соединение (29a) было получено с выходом 82% при исходных 0,47 г соединения (28a). 1H NMR (300 MHz, CD3OD), 0,83 (m, 3H), 1,27 (m, 6H), 1,45 (m, 2H), 2,71 (dd, J=8,0, 15,9 Hz, 1H), 3,1 (m, 5H), 3,59 (m, 2H), 5,28 (s, 2H), 5,48 (t, J= 7,7 Hz, 1H), 6,90 (d, J=2,0 Hz, 1H), 6,98 (dd, J=2,0, 8,7 Hz, 1H), 7,70 (d, J=8,2 Hz, 2H), 7,83 (d, J=8,2 Hz, 2H), 7,90 (d, J=8,7 Hz, 1H); IR (KBr) 3331, 1668, 1605, 1278, 1188 см-1; MS (FAB) m/e 495,2612 (495,2607 рассчитано для C27H35N4O5). Пример 6 Получение (+-)-6-{ { 4-(аминоиминометил)фенил}метокси}-3,4-ди-гидро-1-оксо-бета-{{(фенилметил)амино}карбонил}-2(1H)-изохинолон-пропанового кислого трифторацетата, соединение представлено формулой (29b). 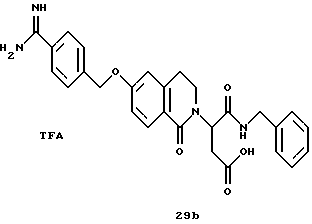 Часть А: Следуя процедуре, используемой для получения соединения (27a) (пример 5, часть E) соединение (27b) было получено с выходом 84% при исходных 0,46 г соединения (26) (пример 5, часть Е) и 0,12 г бензиламина. Часть B: Следуя процедуре, используемой для получения соединения (6) (пример 1, часть Е), соединение (28b) было получено с выходом 76% при исходных 0,45 г соединения (27b). Часть C: Следуя процедуре, используемой для получения соединения (7) (пример 1, часть F), соединение (29b) было получено с выходом 72% при исходных 0,41 г соединения (28b). 1H NMR (300 MHz, СО3OD), 2,70 (dd, J=7,2, 16,1 Hz 1H), 2,90 (br t, J=6,4 Hz, 2H), 3,08 (dd, J=7,9, 15,8 Hz, 1H), 3,60 (m, 2H), 4,3 (dd, J=5,7, 14,9 Hz, 1H), 4,43 (dd, J=6,3, 14,8 Hz, 1H), 5,28 (s, 2H), 5,50 (t, J=7,5 Hz, 1H), 6,87 (m, 1H), 6,97 (dd, J=2,0, 8,6 Hz, 1H), 7,25 (m, 5H), 7,71 (d, J= 8,3 Hz, 2H), 7,87 (d, J=8,3 Hz, 2H), 7,90 (d, J=8,5 Hz, 1H); IR (KBr) 3333, 3092, 1668, 1604, 1278, 1185 см-1; MS (FAB) m/e 501,2151 (501,2138 рассчитано для C28H29N4O5). Пример 7 Получение (+-)-6-{ { 4-(аминоиминометил)фенил}метокси}-3,4- дигидро-1-оксо-бета-{ {(фенилметил)амино}кaрбонил}-2(1H)-изохинолонпропанового кислого трифторацетата, соединение представлено формулой (29c). 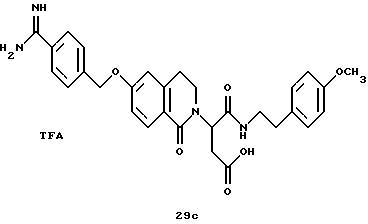 Часть А: Следуя общей процедуре, используемой для получения соединения (27a) (пример 5, часть Е) соединение (27c) было получено с выходом 76% при исходных 0,46 г соединения (26) и 0,17 г p-метоксифенэтиламина. Часть B: Следуя процедуре, используемой для получения соединения (6) (пример 1, часть Е), соединение (28c) было получено с выходом 85% при исходных 0,44 г соединения (27c). Часть C: Следуя процедуре, используемой для получения соединения (7) (пример 1, часть F), соединение (29c) было получено с выходом 80% при исходных 0,45 г соединения (28c). 1H NMR (300 MHz, CD3OD), 2,75 (m, 5H), 3,05 (dd, J=7,4, 15,8 Hz, 1H), 3,30 (m, 2H), 3,50 (m, 2H), 3,66 (s, 3H), 5,30 (s, 2H), 5,47 (t, J-7,7 Hz, 1H), 6,65 (d, J= 8,4 Hz, 2H), 6,90 (m, 1H), 6,98 (dd, J=2,2, 8,5 Hz, 1H), 7,05 (d, J=8,4 Hz, 1H), 7,71 (d, J=8,4 Hz, 2H), 7,81 (d, J=8,4 Hz, 2H), 7,90 (d, J=8,5 Hz, 1H). Пример 8 Получение (+-)-6-{{4-(аминоиминометил)фенил}метокси}-3,4-дигидро-бета- { (метиламино)карбонил} -1-оксо-2(1H)-изохинолинпропанового кислого трифторацетата, соединение представлено формулой (29d). 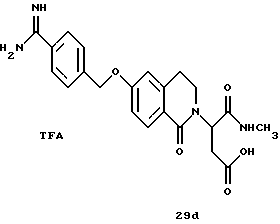 Часть А: Следуя общей процедуре, используемой для получения соединения (27a), соединение (27d) было получено с выходом 80% при исходных 0,46 г соединения (26), 0,07 г метиламингидрохлорида и 0,15 мл Et3N. Часть B: Следуя процедуре, используемой для получения соединения (6) (пример 1, часть Е), соединение (28d) было получено с выходом 63% при исходных 0,37 г соединения (27d). Часть C: Следуя процедуре, используемой для получения соединения (7) (пример 1, часть F), соединение (29d) было получено с выходом 76% при исходных 0,30 г соединения (28d). 1H NMR (300 MHz, CD3OD), 2,75 (m, 4H), 3,0 (m, 2H), 3,10 (dd, J=7,4, 15,9 Hz, 1H), 3,60 (m, 2H), 5,29 (s, 2H), 5,44 (t, J=7,6 Hz, 1H), 6,90 (d, J= 2,2 Hz, 1H), 6,98 (dd, J=2,2, 8,4 Hz, 1H), 7,69 (d, J=8,2 Hz, 2H), 7,82 (d, J=8,2 Hz, 2H), 7,89 (d, J=8,4 Hz, 1H); IR (KBr) 3335, 3105, 1668, 1605, 1480, 1278, 1185 см-1; MS (FAB) m/e 425,1819 (425,1825 рассчитано для C22H25N4O5). Пример 9 Получение (+-)-6-{ { 4-(аминоиминометил)фенил} метокси}-бета-{{(2- карбоксиэтил)амино} карбонил} -3,4-дигидро-1-оксо-2(1H)-изохинолинпропанового кислого трифторацетата, соединение представлено формулой (29e). 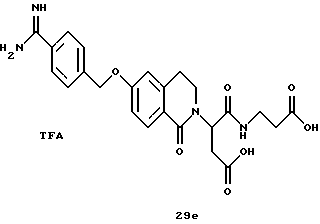 Часть А: Следуя общей процедуре, используемой для получения соединения (27a) (пример 5, часть Е), соединение (27e) было получено с выходом 74% при исходных 0,46 г соединения (26), 0,2 г бета-амино-1-бутилаланина гидрохлорида и 0,15 мл Et3N. Часть B: Следуя процедуре, используемой для получения соединения (6) (пример 1, часть Е), соединение (28e) было получено с выходом 65% при исходных 0,42 г соединения (27e). Часть C: Следуя процедуре, используемой для получения соединения (7) (пример 1, часть F), соединение (29e) было получено с выходом 89% при исходных 0,45 г соединения (28e). 1H NMR (300 MHz, CD3OD), 2,75 (t, J=6,2 Hz, 2H), 2,65 (dd, J=8,2, 1589 Hz, 1H), 3,05 (m, 3H), 3,35 (m, 2H), 3,50 (m, 2H), 5,28 (s, 2H), 5,49 (t, J= 7,7 Hz, 1H), 6,89 (m, 1H), 6,95 (dd, J=2,2, 8,4 Hz, 1H), 7,68 (d, J=8,5 Hz, 2H), 7,84 (d, J=8,5 Hz, 2H), 7,90 (d, J=8,4 Hz, 1H); IR (KBr) 3338, 3108, 1669, 1604, 1278, 1187 см-1; MS (FAB) m/e 483. Аналитически рассчитано для C26H27N4O9F3: С 52,35; H 4,56; N 9,39. Найдено: С 52,43; H 4,82; N 9,13. Пример 10 Получение (+-)-6-{ { 4-(аминомимнометил)фенил}метокси}-b- (3-этоксипропил)-3,4-дигидро-1-оксо-2(1H)-изохинолинпропанового кислого трифторацетата, соединение представлено формулой (36a): 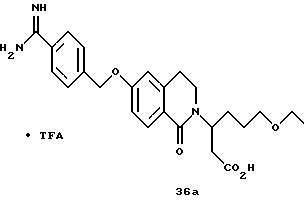 Часть A: Раствор соединения (2) (пример 1, часть А) (6,53 г, 25,8 ммоль) и THF (100 мл) обрабатывался NaH (1,13 г 60%-ной дисперсии в масле, 28,3 ммоль), а полученная смесь выдерживалась при орошении в течение 1 ч. Смесь оставлялась остывать до комнатной температуры, а затем обрабатывалась с помощью 4-этокси-бутанойлхлорида (28,4 ммоль, полученного из кислоты с помощью стандартных протоколов) и DMAP (кат.). Полученная смесь перемешивалась при комнатной температуре в течение 16 ч, а затем разбавлялась EtOAc. Органическая смесь промывалась H2О и концентрировалась. Неочищенный материал очищался путем хроматографии (силикагель, 200-400 меш, гексан/EtOAc, 4:1), что давало 6,12 г (65%) соединения (30a) в виде прозрачного масла. Часть B: Раствор соединения (30a) (6,12 г, 16,7 ммоль) в THF (10 мл) обрабатывался DIBAH (3,9 мл, 21,68 ммоль) при температуре -78oC. Через 1 ч реакция нейтрализовалась путем добавления метанольной HCl (79 мл 1,1 М раствора). Смесь затем разбавлялась EtOAc и промывалась водой и насыщенным водным NaHCO3. Органический материал концентрировался, а неочищенный остаток очищался путем хроматографии (силикагель, 200-400 меш, гекcaн/EtOAc/Et3N, 3:1:0,01), давая 4,09 г (64%) соединения (31a) в виде прозрачного масла. Часть C: Смесь из соединения (31a) (3,25 г, 8,48 ммоль), диметил-t-бутилсилокси-1-1-бутоксиэтена (9,24 г, 42,4 ммоль) и CH2Cl2 (30 мл) обрабатывалась с помощью BF3  Et2O (1,1 мл, 8,48 ммоль) при -78oC. Полученный раствор оставлялся нагреваться до комнатной температуры спустя 2 ч, а затем нейтрализовался путем добавления насыщенного водного NaHCO3 (20 мл). Полученная смесь экстрагировалась с помощью EtOAc, а экстракты концентрировались. Неочищенный продукт очищался путем хроматографии (силикагель, 200-400 меш, гексан/EtOAc 4:1), давая 3,1 г (78%) соединения (32a) в виде прозрачного масла.
Часть D: Et2O (1,1 мл, 8,48 ммоль) при -78oC. Полученный раствор оставлялся нагреваться до комнатной температуры спустя 2 ч, а затем нейтрализовался путем добавления насыщенного водного NaHCO3 (20 мл). Полученная смесь экстрагировалась с помощью EtOAc, а экстракты концентрировались. Неочищенный продукт очищался путем хроматографии (силикагель, 200-400 меш, гексан/EtOAc 4:1), давая 3,1 г (78%) соединения (32a) в виде прозрачного масла.
Часть D:Следуя процедуре, используемой для получения соединения (4) (пример 1, часть С), соединение (33a) было получено с выходом 88% при исходных 3,1 г соединения (32a). Часть E: Следуя процедуре, используемой для получения соединения (25) (пример 5, часть D), соединение (34a) было получено с выходом 95% при исходных 0,53 г соединения (33a). Часть F: Следуя процедуре, используемой для получения соединения (6) (пример 1, часть E), соединение (35a) было получено с выходом 40% при исходных 0,71 г соединения (34a). Часть G: Следуя процедуре, используемой для получения соединения (7) (пример 1, часть F), соединение (36a) было получено с выходом 85% при исходных 0,32 г соединения (35a). 1H NMR (300 MHz, CD3OD) 1,15 (t, J=6,9 Hz, 3H), 1,40-1,80 (m, 4H), 2,60 (m, 2H), 2,95 (m, 2H), 3,49 (m, 6H), 5,10 (m, 1H), 5,29 (s, 2H), 6,94 (d, J= 2,3 Hz, 1H), 6,97 (dd, J=2,2, 8,7 Hz, 1H), 7,70 (d, J=8,3 Hz, 2H), 7,82 (d, J= 8,5 Hz, 2H), 7,85 (d, J=8,8 Hz); IR (KBr) 3334, 3105, 1668, 1604, 1134 см-1; MS (FAB) m/e 454,2380 (454,2342 рассчитано для C25H32N3O5). Пример 11 Получение (+-)-6-{ { 4-(аминоиминометил)фенил}метокси}-6-бутил-3,4- дигидро-1-оксо-2(1H)-изохинолинпропанового кислого трифторацетата, соединение представлено формулой (36b): 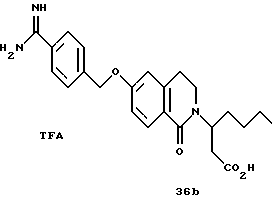 Часть A: Следуя процедуре, используемой для получения соединения (30a) (пример 10, часть А), соединение (30b) было получено с выходом 90% при исходных соединении (2) (0,3 г) и пентановом ангидриде (0,24 г). Часть B: Следуя процедуре, используемой для получения соединения (31a) (пример 10, часть В), соединение (31b) было получено с выходом 83% при исходных 0,39 г соединения (30b). Часть C: Следуя процедуре, используемой для получения соединения (32a) (пример 10, часть С), соединение (32b) было получено с выходом 52% при исходных 0,33 г соединения (31a). Часть D: Следуя процедуре, используемой для получения соединения (4) (пример 1, часть С), соединение (33b) было получено с выходом 98% при исходных 0,22 г соединения (32b). Часть E: Следуя процедуре, используемой для получения соединения (25) (пример 5, часть D), соединение (34b) было получено с выходом 95% при исходных 0,17 г соединения (33b). Часть F: Следуя процедуре, используемой для получения соединения (6) (пример 1, часть Е), соединение (33b) было получено с выходом 56% при исходных 0,23 г соединения (34b). Часть G: Следуя процедуре, используемой для получения соединения (7) (пример 1, часть F), соединение (36b) было получено с выходом 89% при исходных 0,14 г соединения (33b). 1H NMR (300 MHz, CD3OD) 0,89 (t, J=7,15 Hz, 3H), 1,35 (m, 4H), 1,65 (m, 2Н), 2,60 (m, 2H), 2,95 (m, 2H), 3,50 (m, 2H), 5,05 (m, 1H), 5,29 (s, 2H), 6,95 (m, 2H), 7,70 (d, J=8,4 Hz, 2H), 7,84 (app t, J=8,2 Hz, 3H); IR (KBr) 3333, 3107, 1667, 1604, 1138 см-1; MS (FAB) m/e 424. Аналитически рассчитано для C26H30N3O6: С 58,10; H 5,12; N 7,82. Найдено: С 57,85; H 5,56; N 7,56. Пример 12 Получение (+-)-6-{{4-(аминоиминометил)фенил}метокси}-3,4-дигидро-1- оксо-b-пентил-2(1H)-изохинолинпропанового кислого трифторацетата, соединение представлено формулой (36c): 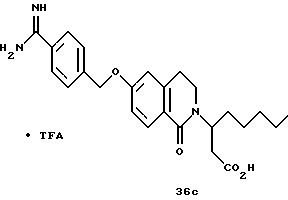 Часть А: Следуя процедуре, используемой для получения соединения (30a) (пример 10, часть А), соединение (30c) было получено с выходом 95% при исходных соединении (2) (0,75 г) и гексанохлориде (0,43 г). Часть B: Следуя процедуре, используемой для получения соединения (31a) (пример 10, часть В), соединение (31c) было получено с выходом 64% при исходных 1,1 г соединения (30c). Часть C: Следуя процедуре, используемой для получения соединения (32a) (пример 10, часть C), соединение (32c) было получено с выходом 70% при исходных 0,80 г соединения (31c). Часть D: Следуя процедуре, используемой для получения соединения (4) (пример 1, часть C), соединение (33c) было получено с выходом 87% при исходных 0,69 г соединения (32c). Часть E: Следуя процедуре, используемой для получения соединения (25) (пример 5, часть D), соединение (34c) было получено с выходом 88% при исходных 0,13 г соединения (33c). Часть F: Следуя процедуре, используемой для получения соединения (6) (пример 1, часть E), соединение (35c) было получено с выходом 65% при исходных 0,18 г соединения (34c). Часть G: Следуя процедуре, используемой для получения соединения (7) (пример 1, часть F), соединение (36c) было получено с выходом 80% при исходном соединении (35b). 1H NMR (300 MHz, CD3OD) 0,90 (m, 3H), 1,30 (m, 6H), 1,60 (m, 2H), 1,26 (m, 2H), 2,97 (m, 2H), 3,45 (m, 2H), 5,05 (m, 1H), 5,30 (2, 2H), 6,88 (m, 1H), 6,94 (m, 1H), 7,70 (d, J=8,3 Hz, 2H), 7,83 (d, J=8,4 Hz, 2H), 7,85 (d, J= 9 Hz, 1H); IR (KBr) 3335, 3115, 1668, 1481, 1188 см-1; MS (FAB) m/e 438,2366 (438,2393 рассчитано для C25H32N3O4). Пример 13 Получение (+-)-6-{ {4-(аминоиминометил)фенил}метокси}-3,4-дигидро-1-оксо-бета-(1,4-диоксигексил)-2(1H)-изохинолинпропанового кислого трифторацетата, соединение представлено формулой (36d): 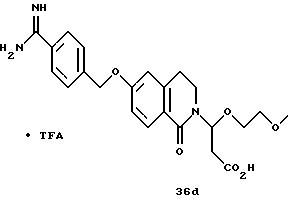 Часть A: Следуя процедуре, используемой для получения соединения (30a) (пример 10, часть А), соединение (30d) было получено с выходом 81% при исходных соединении (2) (2,0 г) и 2-метоксиэтоксиацетилхлориде (2,35 г). Часть B: Следуя процедуре, используемой для получения соединения (31a) (пример 10, часть В), соединение (31d) было получено с выходом 52% при исходных 2,35 г соединения (30d). Часть C: Следуя процедуре, используемой для получения соединения (32a) (пример 10, часть С), соединение (32d) было получено с выходом 42% при исходных 0,57 г соединения (31d). Часть D: Следуя процедуре, используемой для получения соединения (4) (пример 1, часть С), соединение (33d) было получено с выходом 96% при исходных 0,30 г соединения (32d). Часть E: Следуя процедуре, используемой для получения соединения (25) (пример 5, часть D), соединение (34d) было получено с выходом 91% при исходных 0,23 г соединения (33d). Часть F: Следуя процедуре, используемой для получения соединения (6) (пример 1, часть Е), соединение (35d) было получено с выходом 15% при исходных 0,27 г соединения (34d). Часть G: Следуя процедуре, используемой для получения соединения (7) (пример 1, часть F), соединение (36d) было получено с выходом 98% при исходных 0,05 г соединения (35d). 1H NMR (300 MHz, CD3OD) 2,70 (t, J=6,2 Hz, 2H), 2,93 (t, J=6,2 Hz, 2H), 3,30 (s, 3H), 3,47-3,78 (m, 8H), 5,09 (br s, 1H), 5,29 (s, 2H), 6,88 (d, J= 2,2 Hz, 1H), 6,95 (dd, J=2,2, 8,7 Hz, 1H), 7,82 (d, J=8,0 Hz, 2H), 7,84-7,86 (m, 3H); IR (KBr) 3350, 3114, 1669, 1604, 1482, 1385, 1279, 1186, 1029, 842 см-1; MS (FAB) m/e 456,3. Аналитически рассчитано для C26H30N3O8: С 54,84; H 5,31; N, 7,38. Найдено: С 54,61; H 5,26; N 7,37. Пример 14 Получение (+-)-6-{ {4-(аминоиминометил)фенил}метокси}-b-этил-3,4-дигидро-1-оксо-2(1H)-изохинолинпропанового кислого трифторацетата, соединение представлено формулой (36e): 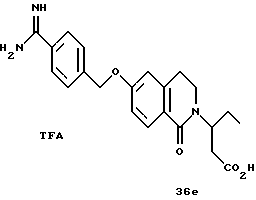 Часть А: Следуя процедуре, используемой для получения соединения (30a) (пример 10, часть А), соединение (30e) было получено с выходом 69% при исходных соединении (2) (1,5 г) и пропанойлхлориде (1,26 г). Часть B: Следуя процедуре, используемой для получения соединения (31a) (пример 10, часть B), соединение (31e) было получено с выходом 73% при исходных 1,2 г соединения (30e). Часть C: Следуя процедуре, используемой для получения соединения (32a) (пример 10, часть C), соединение (32e) было получено с выходом 49% при исходных 0,92 г соединения (32e). Часть D: Следуя процедуре, используемой для получения соединения (4) (пример 1, часть C), соединение (33e) было получено с выходом 89% при исходных 0,55 г соединения (32e). Часть E: Следуя процедуре, используемой для получения соединения (25) (пример 5, часть D), соединение (34e) было получено с выходом 86% при исходных 0,36 г соединения (33e). Часть F: Следуя процедуре, используемой для получения соединения (6) (пример 1, часть Е), соединение (35e) было получено с выходом 36% при исходных 0,38 г соединения (34e). Часть G: Следуя процедуре, используемой для получения соединения (7) (пример 1, часть F), соединение (36e) было получено с выходом 92% при исходных 0,22 г соединения (33e). 1H NMR (300 MHz, CD3OD) 0,91 (t, J=7,3 Hz, 3H), 1,62-1,69 (m, 2H), 2,55-2,62 (m, 2H), 2,92-2,97 (m, 2H), 3,42-3,53 (m, 2H), 4,94 (m, 1H), 5,29 (s, 2H), 6,89 (d, J=2,5Hz, 1H), 6,95 (dd, J=2,5, 8,6 Hz, 1H), 7,70 (d, J=8,4 Hz, 2H), 7,84-7,87 (m, 3H). IR (KBr) 3330, 3109, 2973 1670, 1604, 1481, 1344, 1256, 1041, 835 см-1; MS (FAB) m/e 396,1923 (396,1923 рассчитано для C22H26N3O4). Пример 15 Получение (+-)-6-{ { 4-(аминоиминометил)фенил} метокси} -3,4-дигидро-1- оксо-b-пропил-2(1H)-изохинолинпропанового кислого трифторацетата, соединение представлено формулой (36f): 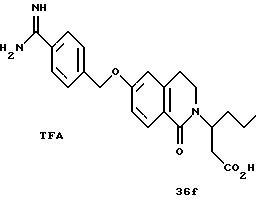 Часть A: Следуя процедуре, используемой для получения соединения (30a) (пример 10, часть A), соединение (30f) было получено с выходом 77% при исходных соединении (2) (пример 1, часть A) и бутанойлхлориде (0,98 г). Часть B: Следуя процедуре, используемой для получения соединения (31a) (пример 10, часть B), соединение (31f) было получено с выходом 73% при исходных 0,6 г соединения (30f). Часть C: Следуя процедуре, используемой для получения соединения (32a) (пример 10, часть C), соединение (32f) было получено с выходом 46% при исходных 0,44 г соединения (31f). Часть D: Следуя процедуре, используемой для получения соединения (4) (пример 1, часть C), соединение (33f) было получено с выходом 90% при исходных 0,24 г соединения (32f). Часть E: Следуя процедуре, используемой для получения соединения (25) (пример 5, часть D), соединение (34f) было получено с выходом 88% при исходных 0,16 г соединения (33f). Часть F: Следуя процедуре, используемой для получения соединения (6) (пример 1, часть E), соединение (35f) было получено с выходом 44% при исходных 0,19 г соединения (34f). Часть G: Следуя процедуре, используемой для получения соединения (7) (пример 1, часть F), соединение (36f) было получено с выходом 66% при исходных 0,085 г соединения (35f). 1H NMR (300 MHz, CD3OD) 0,95 (t, J=7,3 Hz, 3H), 1,29-1,36 (m, 2H), 1,54-1,71 (m, 2H), 2,56-2,62 (m, 2H), 2,91-2,96 (m, 2H), 3,43-3,53 (m, 2H), 5,09 (br s, 1H), 5,29 (s, 2H), 6,88 (d, J=2,1 Hz, 1H), 6,96 (dd, J=2,1, 8,5 Hz, 1H), 7,70 (d, J=8,2 Hz, 2H), 7,82 (d, J=8,2 Hz, 2H), 7,85 (d, J=8,5 Hz, 1H); IR (KBr) 3327, 3106, 2963, 2874, 1670, 1628, 1604, 1480, 1278, 1136 см-1; MS (FAB) m/e 410,2077 (410,2079 рассчитано для C23H28N3O4). Пример 16 Получение (+-)-6-({ 4-(аминоиминометил)фенил} метокси} -3,4-дигидро-1- оксо-b-фенил-(1H)-изохинолинпропанового кислого трифторацетата, соединение представлено формулой (36g): 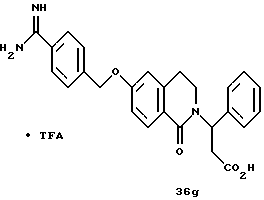 Часть A: Изохинолон (2) (1,0 г, 3,95 ммоль) и 60 вес.% NaH, суспендированные в минеральном масле (0,174 г, 4,35 ммоль), орошались в THF (40 мл) в течение 1 ч. Смесь остывала до комнатной температуры, и добавлялась одна порция альфа-метоксиметилхлорида (0,683 г, 4,35 ммоль) (см. Leibigs, в Ann. Chem., 191, 1932). Реакционная смесь перемешивалась в течение ночи при комнатной температуре. Смесь разбавлялась водой (100 мл) и экстрагировалась с помощью EtOAc (2 х 50 мл). Объединенные экстракты обезвоживались на безводном сульфате натрия и концентрировались. Остаток хроматографировался на силикагеле, элюированном гексаном/EtOAc в пропорции 2:1. Получено 1,02 г соединения (31g) в виде прозрачного масла (68% от теоретического). Часть B: Следуя процедуре, используемой для получения соединения (32a) (пример 10, часть C), соединение (32g) было получено с выходом 36% при исходных 2,29 г соединения (31g). Часть C: Следуя процедуре, используемой для получения соединения (4) (пример 1, часть C), соединение (33g) было получено с выходом 83% при исходных 1,02 г соединения (32g). Часть D: Следуя процедуре, используемой для получения соединения (25) (пример 5, часть D), соединение (34g) было получено с выходом 91% при исходных 0,675 г соединения (33g). Часть E: Следуя процедуре, используемой для получения соединения (6) (пример 1, часть E), соединение (35g) было получено с выходом 50% при исходных 0,80 г соединения (34g). Часть F: Следуя процедуре, используемой для получения соединения (7) (пример 1, часть F), соединение (36g) было получено с выходом 79% при исходных 0,43 г соединения (35g). 1H NMR (300 MHz, СО3OD) 2,76-3,30 (m, 5H), 3,47-3,54 (m, 1H), 5,27 (s, 2H), 6,38 (t, J=7,4 Hz, 1H), 6,84 (d, J=2,3 Hz, 1H), 6,96 (dd, J=2,3, 8,7 Hz, 1H), 7,28-7,40 (m, 5H), 7,68 (d, J=8,2 Hz, 2H), 7,81 (d, J=8,2 Hz, 2H), 7,91 (d, J=8,7 Hz, 1H); IR (KBr) 3328, 3107, 1671, 1604, 1421, 1278, 1189, 1134, 1020 см-1; MS (FAB) m/e 444,1931 (444,1923 рассчитано для C26H26N3O4). Пример 17 Получение 6-{ { 3-(аминоиминометил)фенил} этинил} -3,4-дигидро-1-оксо- 2(1H)-изохинолинуксусного кислого трифторацетата, соединение представлено формулой (12b): 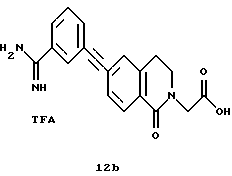 Часть А: Следуя процедуре, используемой для получения соединения (10a) (пример 2, часть B), соединение (10b) было получено с выходом 54% при исходных соединении (8) (пример 2, часть A) и 0,9 г соединения (9b). Часть B: Следуя процедуре, используемой для получения соединения (6) (пример 1, часть E), соединение (11b) было получено с выходом 10% при исходных 0,1 г соединения (9b). Часть C: Следуя процедуре, используемой для получения соединения (7) (пример 1, часть F), соединение (12b) было получено с выходом 87% при исходных 0,01 г соединения (11b). 1H NMR (300 MHz, СО3OD) 3,07 (t, J=6,5 Hz, 2H), 3,70 (t, J=6,6 Hz, 2H), 4,31 (s, 2H), 7,46 (s, 1H), 7,50 (d, J=8,2 Hz, 1H), 7,63 (t, J=7,8 Hz, 1H), 7,78 (d, J=7,6 Hz, 1H), 7,88 (d, J=7,7 Hz, 1H), 7,92 (s, 1H), 7,96 (d, J=4,8 Hz, 1H); IR (CHCl3) 3010, 1647, 1607, 1277, 1156 см-1; MS (FAB) m/e 348,1338 (348,1348 рассчитано для C20H18N3O3). Пример 18 Получение 6-{ 2-{ 3-(аминоиминометил)фенил} этинил}-3,4-дигидро-1-оксо- 2(1H)-изохинолинуксусного кислого трифторацетата, соединение представлено формулой (15b): 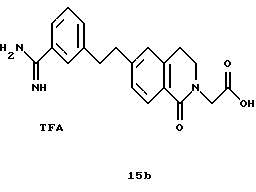 Часть A: Следуя процедуре, используемой для получения соединения (13a) (пример 2, часть A), соединение (13b) было получено с выходом 98% при исходных 0,13 г соединения (10b). Часть B: Следуя процедуре, используемой для получения соединения (6) (пример 1, часть E), соединение (14b) было получено с выходом 64% при исходных 0,09 г соединения (13b). Часть C: Следуя процедуре, используемой для получения соединения (7) (пример 1, часть F), соединение (15b) было получено с выходом 86% при исходных 0,09 г соединения (14b). 1H NMR (300 MHz, СО3OD) 3,00 (m, 6H), 3,65 (t, J=6,6 Hz, 2H), 4,28 (s, 2H), 7,09 (s, 1H), 7,13 (d, J=8,2 Hz, 1H), 7,49 (m, 2H), 7,59 (m, 2H), 7,79 (d, J= 7,9 Hz, 1H); IR (KBr) 1716, 1679, 1639, 1195, 1134 см-1; MS (FD) m/e 352. Аналитически рассчитано для C22H22F3N3O5: С 56,77; H 4,76; N 9,03. Найдено: С 56,65; H 4,71; N 8,73. Пример 19 Получение 6-{ {4-(аминоиминометил)фенил}метиламинокарбонил}-3,4- дигидро-1-оксо-2(1H)-изохинолинуксусного кислого трифторацетата, соединение представлено формулой 50: 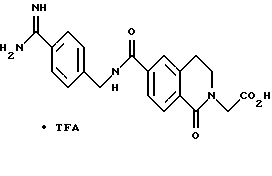 Часть A: Раствор соединения (17) (6-карбокси-3,4-дигидро-1-оксо-2(1H)- изохинолинуксусной кислоты-1,1-сложный диметилэтилэфир) (0,20 г, 0,66 ммоль), p-цианобензиламина (0,10 г, 0,66 ммоль), EDCI (0,15 г, 0,8 ммоль), и DMAP (0,18 г, 1,4 ммоль) в CH2Cl2 (7,0 мл) выдерживался при комнатной температуре в течение 18 ч и затем концентрировался. Остаток очищался путем хроматографии (силикагель 200-400 меш, 25:1 CHCl3/MeOH), давая 0,98 г (37%) 6-{{ (4-цианофенил)метиламино} карбонил} -3,4-дигидро-1-оксо-2(1H)-изохинолин уксусной кислоты-1,1-сложного диметилэтилэфира в виде белого сухого остатка. Часть B: Следуя процедуре, используемой для получения соединения (6) (пример 1, часть E), [[4-(1,1-диметилэтоксикарбонил аминоиминометил)-фенил]метиламинокарбонил]-3,4-дигидро-1-оксо- 2(1H)изохинолин уксусной кислоты-1,1-сложный диметилэтилэфир был получен с выходом 38% при исходном 6-{{(4-цианофенил)метиламино} карбонил}-3,4-дигидро-1-оксо-2(1H)-изохинолин уксусной кислоты-1,1-сложном диметилэтилэфире. Часть C: Следуя процедуре используемой для получения соединения (7) (пример 1, часть F), 6-{{4-(аминоиминометил)фенил}метиламинокарбонил}-3,4- дигидро-1-оксо-2(1H)-изохинолинуксусного кислого трифторацетата, было получено с выходом 83%, при исходных 0,05 г [[4-(1,1- диметилэтоксикарбониламиноиминометил)фенил]метиламинокарбонил]-3,4-дигидро- 1-оксо-2(1H)-изохинолинуксусной кислоты-1,1-сложного диметилэтилэфира. 1H NMR (300 MHz, CD3OD) 3,14 (t, J=6,4 Hz, 2H), 3,73 (t, J=6,7 Hz, 2H), 4,34 (br s, 2H), 4,68 (d, J=5,9 Hz, 2H), 7,6 (d, J=8,4 Hz, 2H), 7,79 (m, 4H), 8,03 (d, J=8,0 Hz, 1H); IR (KBr) 3327, 3109, 1670, 1639, 1190 см-1; MS (FD) m/e 381. Пример 20 Получение 40 (+-)-6-[[4-(аминоимидометил)фенил]метокси]-1,2,3,4- тетрагидронафтилен-2-уксусного кислого трифторацетата, соединение представлено формулой (45): 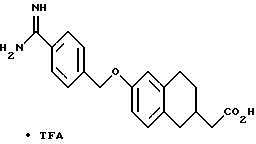 Часть A: Имеющая температуру 0oC суспензия из 650 мг (16,3 ммоль, 60%-ная дисперсия в минеральном масле) NaOH в THF обрабатывалась 2,70 мл (3,0 г, 13,6 ммоль) триэтилфосфоноацетата. После перемешивания при 0oC в течение 0,25 ч раствор 2,0 (11,3 ммоль) 6-метокси-2-тетралона (38) (см. схему 6) в 10 мл THF добавлялся по каплям. Охлаждающая ванна удалялась, а реакция шла при перемешивании и комнатной температуре в течение 16 ч. Реакция нейтрализовалась путем добавления 50 мл рассола. Были разделены два слоя, и органическая фаза обезвоживалась на Na2SO4. Выпаривание дало 3,50 г коричневого масла. Очищение путем вспышечной хроматографии (SiO2, 20% EtOAc в гексане) давало 2,10 г (8,52 ммоль, 75%) соединения (39) в виде светло-желтого масла. Часть B: В раствор 1,00 г (4,06 ммоль) соединения (39) в 20 мл EtOH добавлялась суспензия 0,2 г 10% Pd/C в 10 мл EtOH. Смесь гидрогенизировалась при давлении 50 фунтов/дюйм2 в течение 3 ч при комнатной температуре. Катализатор отфильтровывался, а над реакцией производится выпаривание в вакууме, дающее 1,10 г масла. Очищение путем радиальной хроматографии (SiO2, 5% EtOAc в гексане) дало 910 мг (3,66 ммоль, 90%) соединения (40) в виде прозрачного масла. Часть C: Имеющий температуру -78oC раствор 100 мг (0,40 ммоль) соединения (40) в 4 мл CH2Cl2 обрабатывался BBr3 (1,0 мл 1М раствора в CH2Cl2). Через 4 ч реакция достигала комнатной температуры, и перемешивалась при комнатной температуре в течение 18 ч. Реакция охлаждалась до -78oC и обрабатывалась 5 мл EtOH. Смесь нагревалась и перемешивалась при комнатной температуре в течение 3 ч. Летучие вещества выпаривались в вакууме, а остаток растворялся в 5 мл EtOH, и смесь перемешивалась в течение 2 ч. Выпаривание EtOH давало коричневое масло, которое снова растворялось в 20 мл EtOH, а раствор обрабатывался струей HCl (г) в течение 10 мин. Реакция завершалась и перемешивалась при комнатной температуре в течение 16 ч. Концентрация в вакууме давала 61 мг фенола (41). Материал вводился в 2 мл DMF и обрабатывался 41 мг (0,30 ммоль) K2CO3, 8 мг (0,05 ммоль) NaI и 57 мг (0,29 ммоль) альфа-бромо-p-толунитрила. Реакция перемешивалась при комнатной температуре в течение 16 ч, и DMF удалялась в вакууме. Остаток разделялся между 10 мл H2O и 10 мл EtOAc. Органический слой отделялся, промывался 10 мл H2O и обезвоживался на Na2SO4. Выпаривание в вакууме давало 91 мг сухого остатка. Очищение сухого остатка путем радиальной хроматографии (SiO2, 25% EtOAc в гексане) давало 82 мг (0,24 ммоль, 60% от соединения (40)) соединения (42) в виде белого сухого остатка. Часть D: Следуя процедуре, используемой для получения соединения (6) (пример 1, часть E), соединение (43) было получено с выходом 50% при исходных 0,429 г соединения (42). Часть E: Раствор 250 мг (0,54 ммоль) соединения (43) в 5 мл EtOH обрабатывался 0,5 мл 5N водного NaOH (2,5 ммоль). Реакция перемешивалась при комнатной температуре в течение 6 ч, в это время добавлялось 3,0 мл 1N водной лимонной кислоты (3,0 ммоль). EtOH выпаривался в вакууме. Белый сухой остаток фильтровался, промывался H2O и сушился в вакууме для получения 130 мг кислоты (44) в виде белого порошка. Сухой остаток суспендировался в 1 мл анизола, и смесь обрабатывалась 10 мл трифторуксусной кислоты. Реакция перемешивалась при комнатной температуре в течение 3 ч и выпаривалась в вакууме. Остаток суспендировался в 10 мл H2O, и раствор экстрагировался с помощью гексана (5 х 5 мл). Водный слой лиофилизировался для получения 96 мг (0,26 ммоль, 48% от соединения (43)) трифторацетатной соли соединения (45) в виде белого сухого остатка. MS (FD), m/e 339 (M+1, 100). IR (KBr) 3301, 3145, 2915, 1711, 1664, 1503, 1437, 1196, 1143, 1057 см-1. Аналитически рассчитано для C27H34N2O5 1,5 H2O: С 55,11; H 5,47; N 5,84. Hайдено С 55,46; H 5,15; N 5,45.
Пример 21Получение 6-[[4-(гуанидинометил)фенил] метокси] -3,4-дигидро-1-оксо- 2(1H)-изохинолинуксусного кислого трифторацетата, соединение представлено формулой: 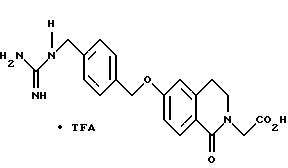 Часть А: Смесь соединений (4) и (51) (полученных из дибромида и фталамида калия с помощью стандартных протоколов), K2CO3, и DMF выдерживалась при 80oC в течение 4 ч, а затем оставлялась остывать до комнатной температуры. Реакционная смесь разбавлялась EtOAc и промывалась H2O. Органический материал концентрировался, и неочищенное выделенное вещество очищалось на двуокиси кремния, давая соединение (46) в виде прозрачного масла. Часть B: Смесь гидрата гидразина (0,079 мл 85%-ного раствора в H2O, 1,4 ммоль), соединения (46) (0,075 г, 0,14 ммоль) и EtOH (3 мл) выдерживалась при 60oC в течение 1 ч, а затем оставлялась остывать до комнатной температуры. Реакционная смесь разбавлялась EtOAc и промывалась водным NaHCO3. Органический материал концентрировался, давая 0,055 г (100%) соединения (47) в виде прозрачного масла. Часть C: Смесь соединения (47) (0,049 г, 0,12 ммоль) N,N’-бис(трет-бутоксикарбонил)-S-метилизотиомочевины (0,043 г, 0,15 ммоль) и THF (1 мл) выдерживалась при комнатной температуре в течение 60 ч, а затем концентрировалась. Хроматография (2: 1 гексан/EtOAc) дала 0,073 г (90%) соединения (49) в виде прозрачного масла. Часть D: Следуя процедуре, используемой для получения соединения (7) (пример 1, часть F), соединение (50) было получено с выходом 78% при исходных 0,07 г соединения (49). 1H NMR (300 MHz, CD3OD) 3,05 (bt, 2H), 3,65 (bt, 2H), 4,28 (s, 2H), 5,20 (s, 2H), 6,90 (m, 2H), 7,35 (d, 2H), 7,50 (d, 2H), 7,85 (d, 2H); IR (KBr) 3364, 3199, 1736, 1687, 1633, 1609, 1179 см-1; MS (FAB) m/e 383,1732 (383,1717 рассчитано для C20H23N4O2). Пример 22 Получение 6[4-(пиперидин-4-ил)пропилокси]-3,4-дигидро-1-оксо-В- (3-этоксипропил)-1-оксо-2(1H)-изохинолинпропанового кислого трифторацетата, соединение представлено формулой: 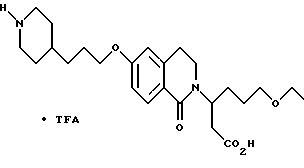 Часть А: Раствор соединения (33a) (0,053 г, 0,14 ммоль) и спирта (51) (полученного из 3-(4-пиридил)-пропанола при помощи стандартных протоколов), трифенилфосфина (0,046 г, 0,17 ммоль), диэтилазодикарбоксилата (0,028 мл, 0,17 ммоль) в THF (1,3 мл) выдерживался при комнатной температуре в течение 1 ч, а затем концентрировался. Неочищенный остаток очищался путем хроматографии (1:1 гексан/EtOAc), давая 0,047 г (61%) соединения 52 в виде прозрачного масла. Часть B: Следуя процедуре, используемой для получения соединения (7) (пример 1, часть F), соединение (53) было получено с выходом 95% при исходных 0,042 г соединения (52). 1H NMR (300 MHz, CD3OD) 1,13 (t, J=7,0 Hz, 3H), 1,27-1,98 (m, 15H), 2,58 (m, 2H), 2,96 (m, 4H), 3,28-3,51 (m, 6H), 4,02 (t, J=6,1 Hz, 2H), 5,05 (m, 1H), 6,75 (br s, 1H), 6,83 (d, J=8,7 Hz, 1H), 7,82 (d, J=8,6 Hz, 1H); MS (FAB) m/e 447. Аналитически рассчитано для C27H39N2O7: С 57,85; H 7,01; N 5,00. Найдено: С 58,13; H 7,18; N 5,28. Пример 23 Получение соединения, представленного формулой 66: 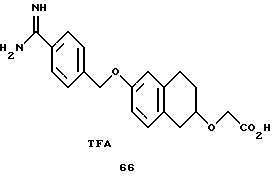 Часть A: Раствор DIBAH в толуоле (100 мл 1,5 М раствора, 150 ммоль) и 6-метокси-2-тетралона (60) (5,19 г, 28 ммоль) выдерживался при орошении в течение 17 ч, а затем остужался до 0oC. Этот раствор нейтрализовался путем медленного добавления насыщенного водного NH4Cl (25 мл), а затем 1N HCl (25 мл) и медленным нагреванием до комнатной температуры с перемешиванием. Полученная студенистая смесь фильтровалась через Селит, а бесцветный водный фильтрат экстрагировался с помощью EtOAc. Объединенные экстракты промывались 1N HCl, H2O и рассолом, обезвоживались (MgSO4) и концентрировались в вакууме. Неочищенный материал очищался путем хроматографии (силикагель 230-400 меш, толуол: EtOAc градиент) для получения 1,75 г (38%) соединения 62 в виде рыжевато-коричневого сухого остатка. Часть B: К раствору соединения 62 (1,64 г, 10 ммоль) в DMF (40 мл) при -5oC медленно добавлялся гидроксид бензилтриметиламмония (Тритон В, 4,5 мл, 10 ммоль). После 0,75 ч перемешивания добавлялся альфа-бромо-p-толунитрил (1,98 г, 10 ммоль) в сухом виде, и раствор постепенно в течение ночи нагревался до комнатной температуры. Смесь разбавлялась EtOAc, промывалась H2O, 1N HCl, насыщенным NaHCO3 и рассолом, обезвоживалась (MgSO4) и концентрировалась в вакууме. Неочищенный материал очищался путем хроматографии (силикагель 230-400 меш, толуол: EtOAc градиент) для получения 2,05 г (73%) соединения 63 в виде белого сухого остатка. Часть C: К энергично перемешиваемой смеси соединения 63 (2,0 г, 7,16 ммоль), KOH (50% w/v водный, 20 мл) и кислого сульфата тетрабутиламмония (1,25 г, 3,58 ммоль) в бензоле (30 мл) добавлялся по каплям чистый трет-бутилбромацетат (3,51 мл, 21,72 ммоль). Смесь перемешивалась при комнатной температуре в течение 3 ч, затем разбавлялась EtOAc и промывалась 1N HCl, насыщенным NaHCO3, H2O и рассолом, обезвоживалась (MgSO4) и концентрировалась в вакууме. Неочищенный материал очищался путем хроматографии (силикагель 230-400 меш, толуол:EtOAc градиент) для получения 2,38 г (85%) соединения 64 в виде белого сухого остатка. Часть D: Следуя общей процедуре, описанной для получения соединения 6 (пример 1, часть E), соединение 65 было получено с выходом 63% при исходных 2,33 г соединения 64. Часть E: Следуя общей процедуре, описанной для получения соединения 7 (пример 1, часть F), соединение 66 было получено с выходом 98% при исходных 1,78 г соединения 65. MS (FD) m/e 355. Пример 24 Получение соединения, представленного формулой 69: 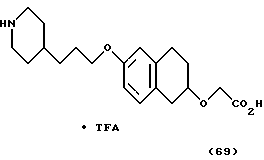 Часть А: К раствору соединения 62 (0,64 г, 3,9 ммоль) в DMF (25 мл) при -5oC медленно добавлялся гидроксид бензилтриметиламмония (Тритон В, 1,77 мл, 3,9 ммоль). После перемешивания в течение 0,5 ч добавлялся чистый 1-tBOC-4-(3-бромпропил)пиперидин (1,19 г, 3,9 ммоль), и раствор оставлялся нагреваться до комнатной температуры постепенно в течение ночи. Смесь разбавлялась EtOAc, промывалась H2O, 1N HCl, насыщенным NaHCO3 и рассолом, обезвоживалась (MgSO4) и концентрировалась в вакууме. Неочищенный материал очищался путем хроматографии (силикагель 230-400 меш, толуол:EtOAc градиент) для получения 1,37 г (90%) соединения 67 в виде бесцветной смолы. Часть B: К энергично перемешиваемой смеси соединения 67 (1,32 г, 3,4 ммоль), КОН (50% w/v водного, 10 мл) и кислого сульфата тетрабутиламмония (0,6 г, 1,7 ммоль) в бензоле (15 мл) был добавлен по каплям чистый трет-бутилбромацетат (0,61 мл, 3,74 ммоль). Смесь перемешивалась при комнатной температуре в течение 3 ч, затем разбавлялась EtOAc и промывалась 1N HCl, H2O и рассолом, обезвоживалась (MgSO4) и концентрировалась в вакууме. Неочищенный материал очищался путем хроматографии (силикагель 230-400 меш, толуол:EtOAc градиент) для получения 1,56 г (91%) соединения 68 в виде бледно-желтого масла. Часть C: Смесь соединения 68 (1,51 г, 3 ммоль) и TFA (15 мл) перемешивалась при комнатной температуре в течение 2 ч, а затем концентрировалась в вакууме. К полученному маслу добавлялось Et2O/гексан, и после разрушения ультразвуком получался сухой остаток. Материал фильтровался, промывался Et2O и обезвоживался для получения 1 г (77%) соединения 69 в виде рыжевато-коричневого сухого остатка. MS (FD) m/e 348. Пример 25 Получение соединения, представленного формулой 72: 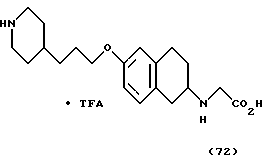 Часть A: К раствору DMSO (0,26 мл, 3,6 ммоль) в CH2Cl2 (13 мл), охлажденному до -78oC, добавлялся по каплям чистый ангидрид трифторацетата (0,51 мл, 3,6 ммоль). Бесцветный раствор перемешивался в течение 0,25 ч при -78oC, затем соединение 67 (0,7 г, 1,8 моль) в CH2Cl2 (12 мл) добавлялось по каплям в течение 5 мин. Раствор перемешивался 1 ч при -78oC, затем оставлялся нагреваться до комнатной температуры и снова перемешивался в течение 1,5 ч. В чистом виде добавлялся диизопропилэтиламин (0,72 мл, 4,14 ммоль), и перемешивание при комнатной температуре продолжалось в течение 1,5 ч. Раствор разбавлялся CH2Cl2 (50 мл) и промывался 1N HCl, насыщенным NaHCO3, H2O и рассолом, обезвоживался (MgSO4) и концентрировался для получения 0,7 г (> 99%) соединения 70 в виде бесцветного масла, которое сразу использовалось на следующем шаге без дополнительного очищения. Часть B: Смесь соединения 70 (0,70 г, 1,8 ммоль), NaBH3CN (0,12 г, 1,8 ммоль), глицин-t-бутил сложного эфира (0,47 г, 3,6 ммоль), ледяного HOAc (0,1 мл, 1,8 ммоль) и порошковых молекулярных фильтров 3А (0,4 г) в абсолютном EtOH (20 мл) перемешивалась при комнатной температуре в течение 17 ч. Смесь фильтровалась, фильтрат концентрировался, а полученное масло заново растворялось в EtOAc/H2O и с помощью 1N NaOH pH доводилась до 7,4. Слои разделялись, и водный слой экстрагировался с помощью EtOAc. Экстракты EtOAc объединялись и промывались насыщенным NaHCO3, H2O и рассолом, обезвоживались (Na2SO4) и концентрировались. Неочищенное выделенное вещество очищалось путем хроматографии (силикагель 230-400 меш, толуол:EtOAc градиент) для получения 0,17 г (19%) соединения 71 в виде бесцветной смолы. Часть C: Смесь соединения 71 (0,2 г, 0,4 ммоль) и TFA (10 мл) перемешивалась при комнатной температуре в течение 3 ч, а затем концентрировалась в вакууме. К полученному маслу медленно добавлялся Et2O, и после разрушения ультразвуком получался сухой остаток. Материал фильтровался, промывался Et2O и обезвоживался для получения 0,2 г (87%) соединения 72 в виде рыжевато-коричневого сухого остатка. MS (FD) m/e 347. Пример 26 Получение соединения, представленного формулой 78: 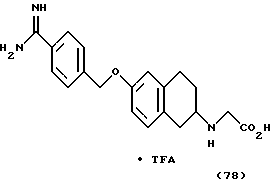 Часть A: К раствору DMSO (0,28 мл, 4 ммоль) в CH2Cl2 (13 мл), охлажденному до -78oC, был добавлен по каплям чистый трифторацетатный ангидрид (0,56 мл, 4 ммоль). Мутный белый раствор перемешивался в течение 0,25 ч при -78oC, а затем в течение 5 мин добавлялось по каплям соединение 63 (0,558 г, 2 ммоль) в CH2Cl2 (12 мл). Раствор перемешивался в течение 1 ч при -78oC, затем оставлялся нагреваться до комнатной температуры и снова перемешивался в течение 1,5 ч. Добавлялся чистый диизопропилэтиламин (0,8 мл, 4,6 ммоль), и перемешивание при комнатной температуре продолжалось в течение 1 ч. Раствор разбавлялся CH2Cl2 (50 мл), промывался 1N HCl, насыщенным NaHCO3, H2O и рассолом, обезвоживался (MgSO4) и концентрировался для получения 0,55 г (> 99%) соединения 73 в виде светло-желтого сухого остатка, который сразу использовался на следующем шаге без дополнительного очищения. Часть B: Смесь соединения 73 (0,55 г, 2 ммоль), NaBH3CN (0,13 г, 2 ммоль), глицин t-бутил сложного эфира (0,52 г, 4 ммоль), ледяного HOAc (0,11 мл, 2 ммоль) и порошковых молекулярных фильтров 3A (0,4 г) в абсолютном EtOH (25 мл) перемешивалась при комнатной температуре в течение 17 ч. Смесь фильтровалась, фильтрат концентрировался, а полученная смола заново растворялась в EtOAc/H2O и с помощью 1N NaOH pH доводилaсь до 7,5. Слои разделялись, и водный слой экстрагировался с помощью EtOAc. Объединенные экстракты EtOAc промывались насыщенным NaHCO3, H2O и рассолом, обезвоживались (Na2SO4) и концентрировались, давая 0,8 г (99%) соединения 74 в виде бесцветной смолы, без дополнительной очистки. Часть C: Смесь соединения 74 (0,784 г, 2 ммоль), K2CO3 (0,829 г, 6 ммоль) и BOC2O (0,873 г, 4 ммоль) в THF/H2O (1:1, 20 мл) перемешивалась при комнатной температуре в течение 5 ч. THF выпаривалась в вакууме, а водный остаток разбавлялся рассолом (50 мл) и экстрагировался с помощью EtOAc. Объединенные экстракты промывались рассолом, обезвоживались (MgSO4) и концентрировались. Неочищенный материал очищался путем хроматографии (силикагель 230-400 меш, толуол: EtOAc градиент) для получения 0,74 г (75%) соединения 76 в виде бледно-желтого сухого остатка. Часть D: Следуя общей процедуре, используемой для получения соединения 6 (пример 1, часть E), соединение 77 было получено с выходом 31% при исходных 0,66 г соединения 76. Часть E: Следуя общей процедуре, используемой для получения соединения 7 (пример 1, часть F), соединение 78 было получено с выходом 81% при исходных 0,22 г соединения 77. MS (FD) m/e 354. Пример 27 Получение соединения, представленного формулой 80: 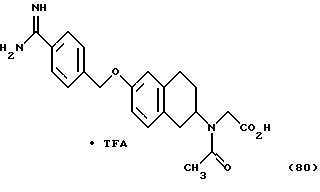 Часть A: Соединение 74 было растворено (1,96 г, 5 ммоль) в CH2Cl2 (20 мл), был добавлен пиридин (2 мл, 26 ммоль), после чего по каплям был добавлен чистый уксусный ангидрид (0,47 мл, 5 ммоль). Золотистый раствор перемешивался при комнатной температуре в течение 6 ч, затем промывался 1N HCl, H2O и рассолом, обезвоживался (MgSO4) и концентрировался. Неочищенный материал очищался путем хроматографии (силикагель 230-400 меш, толуол:EtOAc градиент), давая 0,86 г (39%) соединения 75 в виде белого сухого остатка. Часть B: Следуя общей процедуре, используемой для получения соединения 6 (пример 1, часть E), соединение 79 было получено с выходом 81% при исходных 1,19 г соединения 75. Часть C: Следуя общей процедуре, используемой для получения соединения 7 (пример 1, часть F), соединение 80 было получено с выходом 92% при исходных 0,96 г соединения 79. MS (FD) m/e 396. Пример 28 Получение соединения, представленного формулой 88: 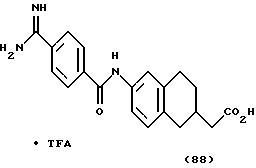 Часть A: Смесь соединения 81 (3,9 г, 13,3 ммоль) и EtOH (20 мл) обрабатывалась NaBH4 (1,0 г, 26,6 ммоль). Смесь выдерживалась при орошении в течение 1 ч, а затем остывала. Реакционная смесь затем разбавлялась EtOAc и промывалась H2O. Органический материал концентрировался, и остаток, полученный таким образом, подвергался дегидрации с помощью TsOH (кат.) в орошающем бензоле. Неочищенная дегидрационная смесь разбавлялась EtOAc и промывалась H2О. Органический материал концентрировался, и неочищенный остаток очищался путем хроматографии (5:1 гексан/EtOAc), давая 2,6 г соединения 82. Часть B: Смесь соединения 82 (2,6 г, 9,5 ммоль), NMO (1,53 г, 11,3 ммоль), tBuOH (8 мл), H2O (8 мл) и ацетона (8 мл) обрабатывалась с помощью OsO4 (0,1 мл 1 мг/мл раствора в CCl4), и полученная смесь перемешивалась при комнатной температуре в течение ночи. Затем смесь разбавлялась EtOAc и промывалась H2О и насыщенным водным NaHCO3. Органический материал затем концентрировался. Неочищенный остаток перекристаллизовывался из EtOAc/гексана, давая 2,8 г соединения 83 в виде белого сухого остатка. Часть C: Диол 83 (2,8 г) суспендировался в бензоле и добавлялся (0,1 г) TsOH. Затем смесь поддерживалась при орошении в течение 15 мин. Затем раствор разбавлялся EtOAc и промывался 0,1 N водным NaOH. Органический материал затем концентрировался. Неочищенный остаток вводился в THF (25 мл), и получающийся раствор добавлялся к смеси NaH (0,5 г 60%-ной дисперсии в масле, 14,7 ммоль), триэтилфосфоноацетата (3,3 г, 14,7 ммоль) и THF (25 мл) при oC. Полученная смесь нагревалась до комнатной температуры и спустя 3 ч разбавлялась EtOAc и промывалась H2O. Органический материал концентрировался, а неочищенное выделенное вещество очищалось на двуокиси кремния (3: 1 гексан/EtOAc), давая 2,52 г соединения 84 в виде прозрачного масла. Часть D: Смесь соединение 84 (2,51 г, 6,87 ммоль), Pd/C (10% на углероде, 2,5 г) и EtOH (20 мл) выдерживалась в атмосфере H2 (стеклянная колба) в течение 2 ч, а затем фильтровалась и концентрировалась. Остаток растворялся в CH2Cl2 (5 мл) и обрабатывался p-цианобензойной кислотой (1,21 г, 8,3 ммоль), EDCI (1,6 г, 8,3 ммоль) и DMAP (кат.). Полученный раствор перемешивался в течение 4 ч, а затем разбавлялся EtOAc и промывался H2O. Органический материал концентрировался, а полученный сухой материал кристаллизовывался из (EtOAc/гексана), давая 1,35 г (54%) соединения 86 в виде белого сухого остатка. Часть E: Следуя общей процедуре, описанной для получения соединения (6) (пример 1, часть E), соединение 87 было получено с выходом 80% при исходных 1,35 г соединения 86. Часть F: Следуя общей процедуре, описанной для получения соединения 7 (пример 1, часть F), соединение 88 получалось с выходом 70% при исходных 0,2 г соединения 87. 1H NMR (300 MHz, CD3OD) 1,5 (m, 1H), 2,0 (m, 1H), 2,2 (m, 1H), 2,4 (m, 2H), 2,45 (dd, J=10,2, 16,2 Hz, 1H), 2,91 (m, 3H), 7,05 (d, J=8,2 Hz, 1H), 7,40 (m, 2H), 7,92 (d, J=8,4 Hz, 2H), 8,12 (d, J=8,4 Hz, 2H); IR (KBr) 3322, 3104, 1712, 1667, 1207 см-1; MS (FAB) m/e 352,1661 (352,1654 рассчитано для C20H22N3O3). Пример 29 Получение соединения, представленного формулой 95: 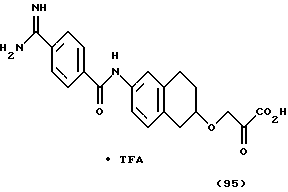 Часть A: Смесь соединения 82 и NaH в THF обрабатывалась бензилбромидом и Bu4NI (кат. ), а полученный раствор оставлялся при комнатной температуре на 2 ч. Затем раствор разбавлялся EtOAc и промывался H2O. Органический материал концентрировался, давая практически чистое соединение 88 в виде желтого масла. Часть B: Смесь соединения 88 (1,0 г, 2,71 ммоль), NMO (0,40 г, 3,0 ммоль), t-BuOH (2,0 мл), ацетона (2,0 мл) и H2O (2 мл) обрабатывалась с помощью OsO4 (0,1 мл 1 мг/мл раствора CCl4), и полученное соединение оставлялось на ночь. Смесь затем разбавлялась EtOAc и промывалась насыщенным водным NaHCO3 и H2O. Органический материал концентрировался, и неочищенный остаток вводился в бензол (25 мл) и обрабатывался TsOH (кат.). Полученная смесь выдерживалась при орошении в течение 15 мин, а затем концентрировалась. Неочищенное выделенное вещество вводилось в EtOH и обрабатывалось NaBH4 (0,25 г) и оставлялось на 1 ч. Эта смесь разбавлялась EtOAc и промывалась H2O. Органический материал концентрировался, а неочищенное выделенное вещество очищалось путем хроматографии (1:1 гексан/EtOAc), давая 0,19 г соединения 90 в виде прозрачного масла. Часть C: Смесь соединения 90 (0,18 г, 0,64 ммоль) и t-бутилбромацетата (0,18 г, 0,95 ммоль), бензола (5 мл), 50% NaOH (5 мл) и Bu4NHSO4 (кат.) энергично перемешивалась при комнатной температуре в течение 12 ч. Эта смесь затем разбавлялась EtOAc и промывалась H2O. Органический материал концентрировался, и неочищенное выделенное вещество очищалось путем хроматографии (5:1 гексан/EtOAc), что давало 0,09 г (35%) соединения 91 в виде прозрачного масла. Часть D: Смесь соединения 91 (0,31 г) и 10% Pd/C (0,3 г) в EtOAc выдерживалась в атмосфере H2 (стеклянная пробирка) в течение 4 ч, а затем фильтровалась и фильтрат концентрировался. Неочищенный остаток вводился в CH2Cl2 (5 мл) и обрабатывался p-цианобензойной кислотой (0,12 г, 0,70 ммоль), EDCI (0,23 г, 0,79 ммоль) и DMAP (кат.). Полученный раствор выдерживался при комнатной температуре в течение 2 ч, а затем разбавлялся EtOAc и промывался H2O. Органический материал концентрировался, и неочищенный остаток очищался на двуокиси кремния (3: 1 гексан/EtOAc), что давало 0,24 г соединения 93 в виде прозрачного масла. Часть E: Следуя общей процедуре, используемой для получения соединения 6 (пример 1, часть E), соединение 94 было получено с выходом 56% при исходных 0,23 г соединения 93. Часть F: Следуя общей процедуре, используемой для получения соединения 7 (пример 1, часть F), соединение 95 было получено с выходом 63% при исходных 0,16 г соединения 94. 1H NMR (300 MHz, CD3OD) 1,90 (m, 1H), 2,05 (m, 1H), 2,7-3,3 (m, 4H), 3,90 (m, 1H), 4,20 (s, 2H), 7,10 (d, J=8,0 Hz, 1H), 7,40 (m, 2H), 7,90 (d, J=8,3 Hz, 2H), 8,15 (d, J=8,3 Hz, 2H); IR (KBr) 3326, 2936, 1664, 1598 см-1; MS (FAB) m/e 368. Аналитически рассчитано для C22H22N3O6F3: С 54,89; H 4,61; N, 8,73. Найдено: С 54,90; H 4,67; N 8,50. Пример 30 Получение соединения, представленного формулой 102: 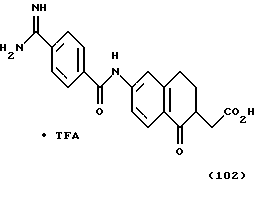 Часть A: Смесь тетралона 96 (5,0 г, 24,6 ммоль), моногидрата гликоксикислоты (8,4 г, 93,6 ммоль), NaOH (4,35 г, 168,9 ммоль), метанола (50 мл) и H2O (50 мл) выдерживалась при орошении в течение 1,25 ч, а затем охлаждалась до 0oC. Затем реакция подкислялась (при перемешивании) концентрированной HCl. Полученный осадок (97) (5,8 г) собирался путем фильтрации. Часть B: Смесь соединения 97 (20,0 г, 77,2 ммоль) и Zn (14,1 г, 216 ммоль) в HOAc (160 мл) и H2O (60 мл) выдерживалась при орошении в течение 1,25 ч, а затем фильтровалась. Фильтрат разбавлялся H2O, и полученная смесь экстрагировалась с помощью EtOAc. Объединенные экстракты концентрировались. Неочищенное выделенное вещество вводилось в концентрированную HCl (100 мл) и выдерживалось при орошении в течение 0,5 часов. Затем смесь разбавлялась H2O (300 мл) и охлаждалась до 5oC. Смесь осторожно нейтрализовывалась до pH = 4 путем добавления кристаллического NaCO3. Сформированный осадок собирался путем фильтрования и обезвоживался в вакууме. Затем этот материал суспендировался в EtOH, и полученный раствор насыщался HCl (г). Затем смесь концентрировалась. Таким образом сформированный материал суспендировался в H2O, а pH полученного раствора доводился до 10 с помощью кристаллического NaOH. Этот материал экстрагировался с помощью EtOAc, а экстракты концентрировались. Неочищенный продукт перекристаллизовывался из EtOAc/гексанов, давая 12,1 г чистого соединения 98 в виде рыжевато-коричневого сухого остатка. Часть C: Смесь соединения 98 (6,8 г, 27,5 ммоль), p-цианбензойной кислоты (4,4 г, 30,2 ммоль), EDCI (7,86 г, 41,2 ммоль), DMAP (0,1 г) и CH2Cl2 (10 мл) перемешивалась при комнатной температуре в течение 4 ч. Эта смесь затем разбавлялась EtOAc и промывалась H2O. Затем органический материал концентрировался, давая неочищенное соединение 99 в виде рыжевато-коричневого сухого остатка. Перекристаллизация из EtOAc/гексанов дает 7,86 г чистого соединения 99. Часть D: Следуя общей процедуре, описанной для получения соединения 6 (пример 1, часть E), соединение 100 было получено с выходом 74% при исходных 7,85 г соединения 99. Часть E: Следуя общей процедуре, описанной для получения соединения 7 (пример 1, часть F), соединение 101 было получено с выходом 90% при исходных 5,0 г соединения 100. Часть F: Смесь соединения 100 (2,0 г, 4,1 ммоль) и EtOAc (5 мл) обрабатывалась NaOH (0,49 г, 12,1 ммоль), и полученный раствор выдерживался при комнатной температуре в течение 2 ч. Затем раствор концентрировался, а полученный остаток вводился в H2О. Водный материал сначала промывался EtOAc, а затем осторожно подкислялся (pH = 4) с помощью KHSO4. Сформированный осадок собирался путем фильтрации и обезвоживался в вакууме. Этот материал затем обрабатывался TFA (10 мл) в течение 1 ч, а затем концентрировался. Неочищенный материал вводился в горячую H2O, фильтровался, и затем лиофолизировался, давая чистое соединение 102 в виде белого порошка. 1H NMR (300 MHz, CD3OD) 2,0 (m, 1H), 2,25 (m, 1H), 2,50 (dd, J=6,4, 16,4 Hz, 1H), 2,90 (dd, J=4,2, 16,5 Hz, 1H), 2,90-3,2 (m, 3H), 7,6 (dd, J=1,9, 8,6 Hz, 1H), 7,80 (s, 1H), 7,95 (m, 3H), 8,14 (d, J=8,3 Hz, 2H); IR (KBr) 3330, 3108, 1712, 1669, 1538 см-1; MS (FAB) 366. Аналитически рассчитано для C22H20N3O6AF3: С 55,12; H 4,20; N 8,76. Найдено: С 54,88; H 4,31; N 8,46. Пример 31 Получение соединения, представленного формулой 118: 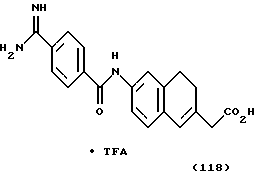 Часть A: Смесь соединения 100 (0,2 г, 0,4 ммоль) и EtOH (10 мл) обрабатывалась NaBH4 (0,025 г, 0,4 ммоль) и оставлялась при комнатной температуре на 1 ч. Эта смесь затем концентрировалась, а остаток растворялся в EtOAc. Эта смесь промывалась H2O и концентрировалась. Неочищенный остаток вводился в THF (15 мл) и обрабатывался TsOH (кат.). Полученный раствор выдерживался при орошении в течение 1,5 ч. Эта смесь концентрировалась, и остаток вводился в EtOAc, полученный раствор промывался 0,1N NaOH, а затем концентрировался. Хроматография (1:1 гексан/EtOAc) давала 0,08 г чистого соединения 116 в виде белого сухого остатка. Часть B: Следуя общей процедуре, описанной для получения соединения 102 (пример 30, часть F), соединение 118 было получено с выходом 80% при исходных 0,08 г соединения 116. 1H NMR (300 MHz, CD3OD) 2,34 (br t, J=8,0 Hz, 2H), 2,83 (br t, J=8,0 Hz, 2H), 3,28 (s, 2H), 6,40 (s, 1H), 7,0 (d, J=8,7 Hz, 1H), 7,5 (m, 2H), 7,92 (d, J= 8,3 Hz, 2H), 8,10 (d, J= 2H); IR (KBr) 3385, 3089, 1716, 1672, 1194 см-1; MS (FAB) m/e 350,1505 (350,1505 рассчитано для C20H20N3O3). Пример 32 Получение соединения, представленного формулой 123: 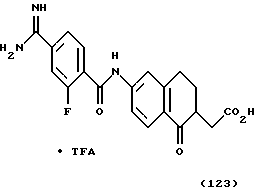 Часть A: Смесь соединения 98 (0,14 г, 0,58 ммоль), кислоты 119 (0,095 г, 0,58 ммоль), EDCI (0,16 г, 0,86 ммоль), DMAP (кат.) и CH2Cl2 (3 мл) выдерживалась при комнатной температуре в течение ночи. Затем смесь разбавлялась EtOAc и промывалась H2O. Органический материал концентрировался, и неочищенный остаток очищался на двуокиси кремния (гексан/EtOAc 2:1), давая 0,095 г (40%) соединения 120. Часть B: Следуя общей процедуре, описанной для получения соединения 6 (пример 1, часть E), соединение 121 было получено с выходом 37% при исходных 0,95 г соединения 120. Часть C: Смесь соединения 121 (0,04 г, 0,08 ммоль), NaOH (0,003 г, 0,08 ммоль) и EtOH (5 мл) выдерживалась при комнатной температуре в течение 6 ч, а затем концентрировалась. Остаток растворялся в H2O и подкислялся до pH = 4 с помощью KHSO4. Полученная смесь экстрагировалась с помощью EtOAc, и экстракты концентрировались. Хроматография (EtOAc) давала 0,014 г соединения 122. Обработка этого материала TFA (5 мл) в течение 1 ч с последующей концентрацией давала 0,014 г соединения 123. 1H NMR (300 MHz, CD3OD) 2,0 (ddd, J=4,5, 13,0, 25,8 Hz, 1H), 2,30 (m, 1H), 2,45 (dd, J=6,4, 16,5 Hz, 1H), 2,90 (dd, J=5,7, 16,5 Hz, 1H), 2,9-3,2 (m, 3H), 7,6 (m, 1H), 7,75 (m, 3H), 7,95 (m, 2H); IR (KBr) 3341, 3118, 1664, 1205 см-1; MS (FAB) m/e 384. Пример 33 Получение соединения, представленного формулой 130: 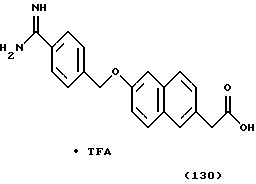 Часть A: Смесь 2-бромо-6-бензилоксинафтилена (124) (1,0 г, 3,2 ммоль) и THF (25 мл) обрабатывалась t-BuLi (4,2 мл, 1,7М раствора в пентане, 7,0 ммоль) при -78oC. Через 1 ч добавлялся диэтилоксалат (0,5 мл, 3,5 ммоль), и полученная смесь оставлялась нагреваться до комнатной температуры. Реакционная смесь затем разбавлялась EtOAc и промывалась H2О. Органический слой концентрировался. Неочищенный материал очищался путем хроматографии (3:1 гексан/EtOAc), что давало 0,52 г чистого соединения 125. Часть B: Смесь соединения 125 (7,0 г, 6,0 ммоль) и EtOH (50 мл) обрабатывалась NaBH4 (0,12 г, 6,0 ммоль) и перемешивалась в течение 1 ч. Затем смесь разбавлялась EtOAc и промывалась 1N HCl. Органический материал затем концентрировался. Неочищенный материал вводился в пиридин (10 мл) и обрабатывался Ас2O (10 мл). Через 1 ч раствор концентрировался до безводного состояния, и остаток пропускался через пробку из двуокиси кремния (4:1 гексан/EtOAc). Материал, полученный таким образом, подвергался каталитической гидрогенизации, использующей 10% Pd/C (стеклянная пробирка). После удаления катализатора путем фильтрации и концентрации получается 0,48 г (35%) желательного соединения 126. Часть C: Смесь соединения 126 (0,48 г, 2,1 ммоль), альфа-бромо-p-толунитрила (0,45 г, 2,3 ммоль), K2CO3 (0,32 г, 2,3 ммоль), Bu4NI (кат.) и DMF (5 мл) выдерживалась при 80oC в течение 4 ч, а затем оставлялась остывать до комнатной температуры. Этот раствор разбавлялся EtOAc, и полученный раствор промывался H2O. Органический материал затем концентрировался. Неочищенный остаток перекристаллизовывался из EtOAc/гексана, давая 0,33 г (45%) соединения 127 в виде рыжевато-коричневого остатка. Часть D: Следуя общей процедуре, описанной для получения соединения 6 (пример 1, часть E), соединение 128 было получено с выходом 50% при исходных 0,33 г соединения 127. Часть E: Смесь соединения 128 (0,10 г, 0,22 моль), EtOH (5 мл), и водного NaOH (0,22 мл 2N раствора, 0,44 ммоль) перемешивалась при комнатной температуре в течение 5 ч, а затем концентрировалась. Остаток вводился в H2О, и полученный раствор экстрагировался с помощью EtOAc. Затем pH водного материала доводилась до 4 с помощью HCl (1N), и полученная смесь экстрагировалась с помощью EtOAc. Экстракты концентрировались, и неочищенный материал обрабатывался TFA (10 мл) в течение 1 ч при комнатной температуре. Реакционная смесь затем концентрировалась до безводного состояния, давая 0,07 г соединения 130 в виде белого сухого остатка. 1H NMR (300 MHz, CD3OD) 3,85 (s, 2H), 5,2 (s, 2H), 7,2-7,4 (m, 3H), 7,6-7,9 (m, 7H); IR (KBr) 3334, 3106, 1695, 1669, 1130 см-1; MS (FAB) m/e 335. Аналитически рассчитано для C22H19N2O5F3: С 58,93; H 4,27; N 6,25. Найдено: С 58,70; H 4,46; N 5,97. Пример 34 Получение соединения, представленного формулой: 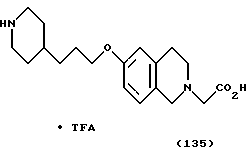 Часть A: Смесь соединения 2 (0,5 г, 2,0 ммоль) и THF (10 мл) обрабатывалась LiAlH4 (0,15 г, 4,0 ммоль), а затем выдерживалась при орошении в течение 2 ч. Смесь охлаждалась до комнатной температуры, а затем гасилась H2O и 15% NaOH. Полученная смесь фильтровалась и концентрировалась. Эта процедура позволяла выделение 0,45 г материала, чистота которого являлась существенной для следующего преобразования. Часть этого материала (0,25 г, 1,1 ммоль), К2СО3 (0,16 г, 1,17 ммоль), трет-бутилбромацетата (0,25 г, 1,17 ммоль) и CH3CN (5 мл) перемешивались при комнатной температуре в течение 15 ч. Смесь затем разбавлялась EtOAc и промывалась 2O. Органический материал концентрировался, и неочищенный остаток очищался на двуокиси кремния (2,5:1 гексан/EtOAc), давая 0,34 г (90%) соединения 132. Часть C: Смесь соединения 132 (0,1 г, 0,28 ммоль) Pd/C (10% на углероде 0,1 г) и EtOAc выдерживалась в атмосфере H2 в течение 12 ч, а затем фильтровалась и концентрировалась. Хроматография (1,5:1 гексан/EtOAc) давала 0,039 г (52%) соединения 133. Часть D: Смесь соединения 133 (0,073 г, 0,28 ммоль), NaH (0,012 г 60%-ной дисперсии в масле, 0,31 ммоль) в THF (10 мл) перемешивалась при комнатной температуре в течение 1/2 ч, а затем обрабатывалась раствором 1-tВОС-4-(3-бромпропил)пиперидина (0,093 г, 0,31 ммоль) в THF (1 мл). Полученный раствор выдерживался при орошении в течение 2 ч, а затем остывал до комнатной температуры. Реакционная смесь разбавлялась EtOAc и промывалась H2O. Органический материал концентрировался, и полученный материал хроматографировался на двуокиси кремния (3:1 гексан/EtOAc), давая 0,086 г алкилированного продукта. Этот материал (0,076 г) растворялся в TFA (5 мл) и выдерживался при комнатной температуре в течение 1 ч. Этот материал затем концентрировался. Неочищенный остаток вводился в 10% HCl (5 мл) и лиофолизировался, давая 0,51 г соединения 135 в виде белого порошка. 1H NMR (300 MHz, CD3OD) 1,30-1,58 (m, 4H), 1,60-1,75 (m, 1H), 1,85 (m, 2H), 1,95 (m, 2H), 3,0 (m, 2H), 3,2 (m, 2H), 3,4 (m, 2H), 3,65 (br s, 2H), 4,0 (t, J=6,2 Hz, 2H), 4,18 (s, 2H), 4,45 (s, 2H), 6,82 (m, 2H), 7,15 (d, J= 8,4 Hz, 1H); IR (KBr) 3406, 2946, 1741, 1614 см-1; MS (FAB) m/e 333,2182 (333,2178 рассчитано для C19H29N2O3). Пример 35 Получение соединения, представленного формулой 140: 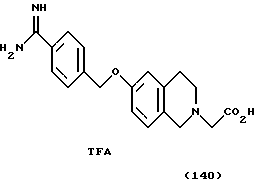 Часть A: Смесь соединения 2 (0,5 г, 2,0 ммоль) и THF (10 мл) обрабатывалась LiAlH4 (0,15 г, 4,0 ммоль), и полученная смесь выдерживалась при орошении в течение 16 ч. Смесь остывала до комнатной температуры, а затем гасилась с помощью H2O и 15% NaOH. Полученная смесь фильтровалась и концентрировалась. Неочищенный продукт восстановления вводился в THF/H2O (1:1, 10 мл) и обрабатывался Boc2O (0,64 г, 2,9 ммоль) и К2СО3 (0,41 г, 2,9 ммоль). Полученная смесь перемешивалась при комнатной температуре в течение 2 ч, а затем разбавлялась EtOAc. Органический материал промывался H2O и концентрировался. Неочищенное выделенное вещество хроматографировалось на двуокиси кремния (1: 1 гексан/EtOAc), давая 0,58 г чистого соединения 131. Часть B: Смесь соединения 131 (0,58 г), Pd/C (10% на углероде, 0,58 г) и EtOAc (30 мл) выдерживалась в атмосфере H2 (стеклянная пробирка) в течение 1 ч, а затем фильтровалась и концентрировалась. Получалось 0,46 г практически чистого соединения 136. Часть C: Смесь соединения 136 (0,46 г, 1,95 ммоль), К2СО3 (0,3 г, 2,1 ммоль), aльфа-бромо-p-толунитрила (0,42 г, 2,1 ммоль), Bu4NI (кат.) и ацетона выдерживалась при орошении в течение 6 ч. Реакционная смесь затем разбавлялась EtOAc и промывалась 2O. Органический материал концентрировался, и неочищенный остаток очищался путем хроматографии (1:1 гексан/EtOAc), давая 0,34 г соединения 137. Часть D: Смесь соединения 137 (0,34 г, 0,94 ммоль) и TFA (10 мл) выдерживалась при комнатной температуре в течение 1 ч, а затем концентрировалась. Остаток вводился в насыщенный водный NaHCO3, а полученная смесь экстрагировалась с помощью EtOAc. Экстракты объединялись и концентрировались. Неочищенный остаток вводился в CH3CN (10 мл), и полученный раствор обрабатывался К2СО3 (0,14 г, 1,0 ммоль) и трет-бутилбромацетата (0,20 г, 1,0 ммоль). Полученная смесь перемешивалась при 60oC в течение 2,5 ч, а затем разбавлялась EtOAc. Органический материал промывался H2O и концентрировался. Неочищенный остаток очищался на двуокиси кремния (2,5:1 гексан/EtOAc), давая 0,18 г соединения 138. Часть E: Следуя процедуре, описанной для получения соединения 6 (пример 1, часть E), соединение 139 было получено с выходом 33% при исходных 0,18 г соединения 138. Часть F: Следуя процедуре, описанной для получения соединения 7 (пример 1, часть F), соединение 140 было получено с выходом 66% при исходных 0,075 г соединения 139. 1H NMR (300 MHz, CD3OD) 3,19 (m, 2H), 3,62 (m, 2H), 4,05 (s, 2H), 4,21 (s, 6H), 6,92 (m, 2H), 7,11 (d, J=8,3 Hz, 1H), 7,65 (d, J=8,4 Hz, 2H), 7,82 (d, J=8,4 Hz, 2H); IR (KBr) 3333, 3104, 1668, 1617, 1191 см-1; MS (FAB) m/e 340,1. Пример 36 Получение соединения, представленного формулой 146: 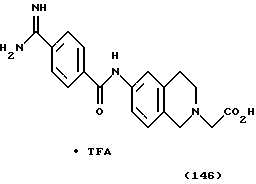 Часть A: Смесь соединения 141 (12,3 г, 60,2 ммоль) и 5N HCI (75 мл) выдерживалась при орошении в течение 12 ч, а затем концентрировалась до безводного состояния. Остаток вводился в насыщенный водный NaHCO3, и эта смесь экстрагировалась с помощью EtOAc. Затем экстракты обезвоживались на Na2SO4 и концентрировались. Неочищенный продукт очищался на двуокиси кремния (15:85 MeOH/CH2Cl2), давая 5,0 г соединения 142 в виде рыжевато-коричневого сухого остатка. Часть B: Смесь соединения 142 (2,6 г, 16,0 ммоль), бензил бромида (5,5 г, 32,0 ммоль), K2CO3 (4,43 г, 32,0 ммоль), CH3CN (30 мл) и Bu4NI (кат.) выдерживалась при орошении в течение 3,5 ч, а затем разбавлялась EtOAc и промывалась H2O. Органический материал обезвоживался и концентрировался. Хроматография (15:85 MeOH/CH2Cl2) позволяла выделить фракцию, содержащую и моно-, и дибензилированный материал. Это соединение растворялось в THF, и полученный раствор обрабатывался LiAlH4 (1,52 г, 40 ммоль). Смесь орошалась 4 ч, а затем гасилась водой и 15% NaOH. Полученная смесь фильтровалась и концентрировалась. Неочищенный продукт, выделенный таким образом, сразу вводился в THF/H2O и обрабатывался Boc2O (3,84 г, 17,6 ммоль) и K2CO3 (6,6 г, 48,0 ммоль). Через 1 ч смесь разбавлялась EtOAc и промывалась H2O и рассолом. Органический материал концентрировался, и неочищенное выделенное вещество очищалось на двуокиси кремния, давая 6,25 г смеси монобензила и дибензилированных тетрагидроизохинолинов. Эта смесь подвергалась каталитической гидрогенизации (Pd/C) в EtOH, давая 1,92 г чистого соединения 143 после хроматографии (1:3 MeOH/CH2Cl2) на двуокиси кремния. Часть C: Смесь соединения 143 (1,92 г, 8,2 ммоль) p-цианбензойной кислоты (1,2 г, 8,2 ммоль), EDCI (1,7 г, 9,0 ммоль) и DMAP (кат.) в CH2Cl2 (20 мл) выдерживалась при комнатной температуре в течение 2 ч. Смесь затем разбавлялась EtOAc и промывалась водой. Органический материал затем концентрировался, давая неочищенное соединение 144, чистота которого было значима для следующей реакции. Неочищенное вещество 144 растворялось в TFA и оставлялось при комнатной температуре в течение 1 ч, а затем концентрировалось. Остаток вводился в насыщенный водный NaHCO3, и полученная смесь экстаргировалась с помощью EtOAc. Органические экстракты концентрировались, давая желаемый амин. Хроматография (двуокись кремния, 10% TFA в MeOH) давала 1,23 г материала, чистота которого была значима на следующем шаге. Смесь этого материала (1,2 г, 4,7 ммоль), t-бутилбромацетата (0,99 г, 5,1 ммоль), K2CO3 (0,70 г, 5,1 ммоль), Bu4NI (кат.) и CH3CN перемешивалась при комнатной температуре в течение 3 ч. Смесь затем разбавлялась EtOAc и промывалась водой. Органический материал обезвоживался с помощью Na2SO4 и концентрировался. Хроматография (1:9 MeOH/CHCl3) давала 0,62 г соединения 145 в виде желтого масла. Часть E: Следуя общей процедуре, используемой для получения соединения 6 (пример 1, часть E), соединение 146 было получено с выходом 26% при исходных 0,1 г соединения 145. Часть F: Слeдуя общей процедуре, используемой для получения соединения 7 (пример 1, часть F), соединение 147 было получено с выходом 80% при исходных 0,034 г соединения 146. 1H NMR (300 MHz, CD3OD) 3,23 (m, 2H), 3,62 (m, 2H), 4,10 (s, 2H), 4,51 (m, 2H), 7,2 (d, J=8,2 Hz, 1H), 7,6 (m, 1H), 7,75 (s, 1H), 7,92 (d, J=8,4 Hz, 2H), 8,16 (d, J=8,4 Hz, 2H). Пример 37 Получение соединения, представленного формулой 155: 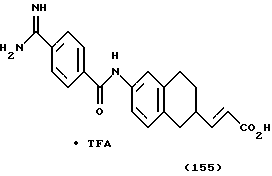 Часть A: Раствор сложного эфира 148 (0,81 г, 3,23 ммоль) и THF (7 мл) обрабатывался LiBH4 (0,14 г, 6,5 ммоль) и оставлялся при комнатной температуре на 6 ч. Смесь затем разбавлялась EtOAc и промывалась H2O. Органический материал концентрировался, давая 0,65 г материала, чистота которого являлась значимой для следующего шага. Смесь этого материала (0,65 г, 3,1 ммоль), TBSCl (0,51 г, 3,5 ммоль), имидазола (0,24 г, 3,47 ммоль) и DMF (5 мл) выдерживалась при комнатной температуре в течение 1 ч. Затем смесь разбавлялась EtOAc и промывалась H2O. Органический материал концентрировался, и неочищенный остаток очищался на двуокиси кремния (5:1 гексан/EtOAc), давая 0,96 г чистого соединения 149. Часть B: Смесь соединения 149 (0,96 г) и Pd/C (10% на углероде, 0,96 г) в EtOAc выдерживалась в атмосфере H2 (стеклянная пробирка) в течение 1 ч, а затем фильтровалась и концентрировалась. Неочищенное выделенное вещество вводилось в CH2Cl2 (5 мл) и обрабатывалось p-цианбензойной кислотой (0,45 г, 3,1 ммоль), EDCI (0,64 г, 3,34 ммоль) и DMAP (кат.). Полученный раствор выдерживался при комнатной температуре в течение 2 ч, а затем разбавлялся EtOAc. Органический материал промывался H2O, а затем концентрировался. Хроматография (1:1 гексан/EtOAc) давала 1,09 г чистого соединения 150. Часть C: Смесь соединения 150 (1,09 г, 2,59 ммоль) и TBAF (5,2 мл 1М раствора в THF, 5,2 ммоль) выдерживалась при комнатной температуре в течение 1 ч. Эта смесь разбавлялась EtOAc, промывалась H2O, а затем концентрировалась, давая 0,71 г практически чистого первичного спирта. Этот материал (0,65 г, 2,11 ммоль) оксидировался с помощью DMSO, оксалилхлорида и TFA (способ Сверна). Неочищенное выделенное вещество, полученное таким образом, вводилось в THF (5 мл) и добавлялось к смеси t-бутилдиэтилфосфоноацетата (0,71 г, 3,2 ммоль), NaH (0,13 г 60%-ной дисперсии в масле, 3,2 ммоль) и THF (10 мл). Через 1 ч смесь разбавлялась EtOAc и промывалась H2О. Органический материал концентрировался, и неочищенный остаток фракционировался на двуокиси кремния (5: 1 гексан/EtOAc), давая 0,27 г соединения 151, 0,197 г соединения 152 и 0,47 г заново полученного исходного спирта. Часть D: Следуя процедуре, описанной для получения соединения 7 (пример 1, части E и F), соединение 155 было получено с выходом 54% при исходных 0,27 г соединения 152. 1H NMR (300 MHz, CD3OD) 1,65 (m, 1H), 2,05 (m, 1H), 2,60-2,95 (m, 5H), 5,85 (d, J=15,5 Hz, 1H), 7,05 (dd, J=9,6, 15,8 Hz, 1H), 7,10 (d, J=8,3 Hz, 1H), 7,4 (m, 2H), 7,91 (d, J=8,4 Hz, 2H), 8,17 (d, J=8,4 Hz, 2H); IR (KBr) 3313, 3102, 1670, 1203 см-1; MS (FAB) m/e 364. Аналитически рассчитано для C23H22N3O5F3: C 57,86; H 4,65; N 8,80. Найдено: C 57,59; H 4,84; N 8,78. Пример 38 Получение соединения, представленного формулой 154: 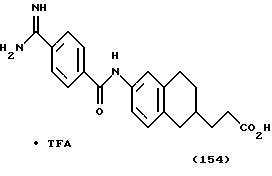 Часть A: Смесь соединения 151 (0,18 г, 0,43 ммоль) и Pd/C (10% на углероде, 0,18 г) в EtOH выдерживалась в атмосфере H2 (стеклянная пробирка) в течение 30 мин, а затем фильтровалась и концентрировалась. Хроматография (3:1 гексан/EtOAc) давала 0,09 г соединения 153 в виде прозрачного масла. Часть B: Следуя общей процедуре, используемой для получения соединения 7 (пример 1, части E и F), соединение 154 было получено с выходом 51% при исходных 0,09 г соединения 153. 1H NMR (300 MHz, CD3OD) 1,4 (m, 1H), 1,7 (m, 3H), 1,97 (m, 1H), 2,4 (m, 3H), 2,85 (m, 3H), 7,08 (d, J=8,3 Hz, 1H), 7,40 (m, 2H), 7,90 (d, J=8,4 Hz, 2H), 8,17 (d, J= 8,4 Hz, 2H); IR (KBr) 3317, 3102, 2926, 1708, 1666, 1142 см-1; MS (FRB) m/e 366,1815 (366,1818 рассчитано для C21H24N3O3). Пример 39 Получение соединения, представленного формулой 161: 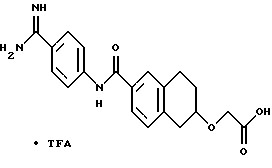 Часть A: Смесь 6-бромтетралона 156 (1,0 г, 4,4 ммоль) и EtOH (10 мл) обрабатывалась NaBH4 (1 г) при комнатной температуре. Через 1 ч смесь разбавлялась EtOAc и промывалась H2O. Органический материал концентрировался до безводного состояния, и неочищенное выделенное вещество растворялось в безводном DMF (10 мл) и обрабатывалось TBSCl (1,0 г, 6,6 ммоль) и имидозолом (0,45 г, 6,6 ммоль). Полученный раствор оставлялся на ночь при комнатной температуре. Эта смесь затем разбавлялась EtOAc и промывалась H2O и концентрировалась. Неочищенное выделенное вещество очищалось на двуокиси кремния (гексан), давая 0,8 г соединения 157 (52%) в виде прозрачного масла. Часть B: Смесь соединения 157 (1,93 г, 5,7 ммоль) и THF (25 мл) обрабатывалась t-BuLi (8,4 мл 1,7M раствора в пентане) при -78oC. Через 30 мин струя сухого CO2 прокачивалась через раствор, и реакция оставлялась нагреваться до комнатной температуры. Полученная THF-смесь разбавлялась H2O, подкислялась 1N HCl и экстрагировалась с помощью EtOAc. Экстракты концентрировались, давая 1,59 г неочищенной кислоты. Часть (0,5 г, 1,63 ммоль) этого материала растворялась в CH2Cl2 (2,0 мл), и полученный раствор обрабатывался бензиловым спиртом (0,19 г, 1,8 ммоль), EDCI (0,34 г, 1,8 ммоль) и DMAP (кат.). Эта смесь оставлялась на 2 ч, а затем разводилась EtOAc и промывалась H2O. Органический материал концентрировался, и неочищенный остаток обрабатывался TBAF (1,8 мл 1М раствора в THF, 1,8 ммоль). Через 25 мин смесь разбавлялась EtOAc и промывалась 2O. Органический материал концентрировался, давая 0,45 г соединения 158 в виде практически чистого масла. Часть C: Смесь соединения 158 (0,45 г, 1,59 ммоль), t-бутилбромацетата (0,96 г, 4,9 ммоль), бензола (5 мл), 50% водного NaOH (5 мл) и Bu4NHSO4 (кат.) энергично перемешивалась при комнатной температуре в течение 5 ч. Смесь затем разбавлялась EtOAc и промывалась 2O. Органический материал концентрировался, и неочищенный остаток очищался на двуокиси кремния (5:1 гексан/EtOAc), давая 0,44 г (69%) желаемого алкилированного продукта в виде прозрачного масла. Смесь этого материала (0,44 г, 1,1 ммоль), Pd/C (10% на углероде, 0,44 г) и EtOAc (10 мл) перемешивалась в атмосфере H2 (стеклянная пробирка) в течение 2 ч. Материал затем фильтровался и концентрировался, давая 0,29 г практически чистого соединения 159. Часть D: Смесь соединения 159 (0,29 г, 0,94 ммоль) EDCI (0,2 г, 1,0 ммоль), 4-аминобензонитрила (0,12 г, 1,0 ммоль), DMAP (кат.) и CH2Cl2 (5 мл) выдерживалась при комнатной температуре в течение 4 ч. Эта смесь затем разбавлялась EtOAc и промывалась H2O. Органический материал затем концентрировался. Хроматография (2,5: 1 гексан/EtOAc) давала фракцию (0,28 г), содержащую желаемый амид 160 и то, что по предположению является симметричным ангидридом 159. Этот материал подавался на следующий шаг. Часть E: Материал, полученный на предыдущем шаге (0,28 г), вводился в пиридин (20 мл) и TEA (2 мл), и полученный раствор насыщался H2S. Эта смесь оставлялась при комнатной температуре на 12 ч, а затем разбавлялась EtOAc и промывалась H2O. Органический материал концентрировался, и неочищенная смесь хроматографировалась на двуокиси кремния (EtOAc), давая 0,13 г чистого промежуточного тиоамида. Этот материал затем обрабатывался так же, как описано в примере 1 части E, давая 0,07 г чистого Boc-защищенного материала. Этот материал вводился в TFA и перемешивался при комнатной температуре в течение 1 ч, а затем концентрировался, что давало 0,056 г соединения 161. 1H NMR (300 MHz, CD3OD) 1,92-2,17 (m, 2H), 2,80-3,22 (m, 4H), 4,05 (m, 1H), 4,22 (s, 2H), 7,27 (d, J=8,3 Hz, 1H), 7,72 (m, 2H), 7,82 (d, J=8,4 Hz, 2H), 8,02 (d, J=8,4 Hz, 2H); IR (KBr) 3318, 3147, 1739, 1656, 1137 см-1; MS (FAB) m/e 368. Пример 40 Получение соединения, представленного формулой 168: 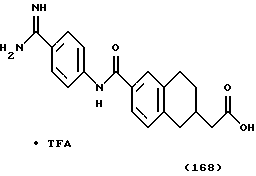 Часть A: Смесь соединения 156 (1,25 г, 5,5 ммоль), этиленгликоля (3,4 г, 55 ммоль), TsOH (кат) и бензола (25 мл) выдерживалась при орошении с удалением H2O в течение 3 ч. Затем смесь разбавлялась EtOAc, и полученный раствор промывался 1N NaOH. Органический материал затем концентрировался, и неочищенный остаток очищался путем хроматографии (5:1 гексан/EtOAc), что давало 1,15 г (77%) соединения 162 в виде прозрачного масла. Часть B: Раствор соединения 162 (1,15 г, 4,3 ммоль) и THF (15 мл) обрабатывается t-BuLi (6,3 мл 1М раствора в пентане, 10,7 ммоль) при -78oC в течение 30 мин, а затем гасился добавлением CO2 (г). Реакционная смесь оставлялась нагреваться до комнатной температуры, а затем разбавлялась H2O. Полученная смесь подкислялась концентрированной HCl и экстрагировалась с помощью EtOAc. Органические экстракты концентрировались, и неочищенное выделенное вещество вводилось в CH2Cl2 (10 мл) и обрабатывалось бензиловым спиртом (0,58 г, 5,4 ммоль), EDCI (1,02 г, 5,4 ммоль) и DMAP (кат.). Полученный раствор выдерживался в течение ночи при комнатной температуре, а затем разбавлялся EtOAc и промывался H2O. Органический материал концентрировался, и неочищенный остаток хроматографировался на двуокиси кремния, давая одну фракцию (1,06 г), содержащую желаемый продукт 163 и бензиловый спирт в соотношении 1:1. Этот материал пригоден для использования в следующей реакции. Часть C: Вышеописанная смесь растворялась в ацетоне (20 мл) и обрабатывалась 1N HCl (2 мл) и выдерживалась при орошении в течение 1 ч. Затем смесь разбавлялась EtOAc и промывалась насыщенным водным NaHCO3. Органический материал затем концентрировался. Неочищенное выделенное вещество вводилось в THF и добавлялось к смеси t-бутилдиэтилфосфоноацетата (1,1 г, 4,93 ммоль), NaH (0,1 г, 60% дисперсии в масле, 4,93 ммоль) и THF (25 мл) при -78oC. Полученный раствор оставлялся нагреваться до комнатной температуры и затем выдерживался при орошении в течение 1 ч. Затем смесь разбавлялась EtOAc и промывалась H2O. Органический материал концентрировался, и неочищенное выделенное вещество очищалось на двуокиси кремния (2,5:1 гексан/EtOAc), давая 0,47 г соединения 164 в виде смеси олефиновых изомеров. Часть D: Смесь соединения 164 (0,47 г) и Pd/C (10% на углероде, 0,47 г) в EtOH выдерживалась в атмосфере H2 (стеклянная пробирка) в течение 2 ч, затем фильтровалась и концентрировалась, давая 0,29 г практически чистого соединения 165. Часть E: Смесь соединения 165 (0,29 г, 1,0 ммоль), EDCI (0,28 г, 1,5 ммоль), p-цианбензойной кислоты (0,12 г, 1,0 ммоль), DMAP (кат.) и CH2Cl2 (5 мл) выдерживалась при 100oC в запечатанной трубке в течение 2 ч, а затем разбавлялась EtOAc и промывалась H2O. Органический материал концентрировался, и остаток хроматографировался на двуокиси кремния (80:1 CHCl3/THF), давая 0,28 г (69%) соединения 166. Часть F: Следуя процедуре, описанной для получения соединения 6 (пример 1, часть E), соединение 67 было получено с выходом 56% при исходных 0,28 г соединения 166. Часть G: Следуя процедуре, описанной для получения соединения 7 (пример 1, часть F), соединение 168 было получено с выходом 91% при исходных 0,22 г соединения 167. 1H NMR (300 MHz, CD3OD) 1,5 (m, 1H), 2,0 (m, 1H), 2,2 (m, 1H), 2,35-2,55 (m, 3H), 2,95 (m, 3H), 7,05 (d, J=8,25 Hz, 1H), 7,4 (m, 2H), 7,93 (d, J=8,4 Hz, 2H), 8,15 (d, J=8,4 Hz, 2H); IR (KBr) 3322, 3104, 1712, 1667 см-1; MS (FAB) m/e 352,1654 (352,1661 рассчитано для C20H22N3O3). Пример 41 Получение соединения, представленного формулой (177): 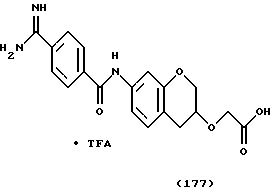 Часть A: Смесь соединения 169 (3,5 г) и щелочи Клейзена (NaOH в EtOH) (75 мл) выдерживалась при орошении в течение 6 ч, а затем охлаждалась. Смесь концентрировалась до 1/2 объема, и оставшийся водный материал нейтрализовывался до pH=7 концентрированной HCl. Смесь затем экстрагировалась с помощью EtOAc, а объединенные экстракты концентрировались. Остаток вводился в THF/H2O (1:1, 20 мл) и обрабатывался K2CO3 (3,2 г, 23 ммоль) и CBz-хлоридом (3,92 г, 23 ммоль). Смесь энергично перемешивалась в течение 1 ч, а затем разбавлялась EtOAc и промывалась водой. Органический материал концентрировался, и неочищенный остаток подвергался ацилированию с помощью Ac2O (5 мл) в пиридине (10 мл). Через 2 ч смесь концентрировалась до безводного состояния, и остаток хроматографировался (3: 1 гексан/EtOAc), давая 6,42 г чистого соединения 170. Часть B: Смесь соединения 170 (6,42 г, 19,75 ммоль), MCPBA (4,27 г, 24,69 ммоль) и CH2Cl2 (40 мл) выдерживалась при комнатной температуре в течение 15 ч. В это время смесь разбавлялась EtOAc и промывалась насыщенным водным NaHCO3 и H2O. Затем органический материал концентрировался. Неочищенный материал вводился в ацетон (450 мл) и обрабатывался NaI (4 г). Полученный раствор выдерживался при орошении в течение 4 ч, а затем остывал. Смесь концентрировалась, растворялась в EtOAc, промывалась H2O и концентрировалась заново. Этот материал затем обрабатывался 0,1N LiOH (290 мл) в THF (290 мл) в течение 12 ч. Смесь разбавлялась EtOAc и промывалась водой, а оставшийся органический материал концентрировался. Хроматография (2:1 гексан/EtOAc) давала 3,58 г соединения 171. Часть C: Смесь соединения 171 (6,8 г, 2,6 ммоль), TBSCl (0,43 г, 2,9 ммоль), имидазола (0,21 г, 3,2 ммоль) и DMF (5 мл) перемешивалась при комнатной температуре в течение 16 ч. Этот материал затем разбавлялся EtOAc и промывался H2O. Органический материал концентрировался, давая практически чистый TBS-эфир. Смесь этого материала (0,98 г, 2,4 ммоль) и THF (10 мл) обрабатывалась NaH (0,07 г 60%-ной дисперсии в масле, 2,6 ммоль) и оставлялась на 1 ч. Смесь затем обрабатывалась бензилбромидом (0,45 г, 2,6 ммоль) и Bu4NI (кат.) и оставлялась на 5 ч. Затем смесь разбавлялась EtOAc и промывалась H2O. Органический материал затем концентрировался. Неочищенный остаток вводился в THF и обрабатывался TBAF (2,9 мл 1М раствора в THF, 2,9 ммоль). Через 1 ч нахождения при комнатной температуре смесь разбавлялась EtOAc и промывалась H2O. Органический материал концентрировался, и неочищенный материал хроматографировался на двуокиси кремния (гексан/EtOAc 1:1), давая 0,94 г (95%) соединения 172. Часть D: Следуя процедуре, используемой для получения соединения 68 (пример 24, часть B), соединение 173 получалось с выходом 80% при исходных 0,43 г соединения 172. Часть E: Смесь соединения 173 (0,65 г, 1,16 ммоль) и Pd/C (10% на углероде, 0,65 г) в EtOH (10 мл) выдерживалась в атмосфере 2 (стеклянная пробирка) в течение 2,5 ч, а затем фильтровалась, а фильтрат концентрировался. Неочищенный материал затем вводился в CH2Cl2 (5 мл) и обрабатывался EDCI (0,23 г, 1,2 ммоль), p-цианбензойной кислотой (0,18 г, 1,2 ммоль) и DMAP (кат.). Полученный раствор выдерживался при комнатной температуре в течение 1 ч, а затем разбавлялся EtOAc. Полученная смесь промывалась H2O и затем концентрировалась. Неочищенный остаток хроматографировался на двуокиси кремния (1:2 гексан/EtOAc), давая 0,32 г (71%) соединения 175. Часть F: Следуя процедуре, используемой для получения соединения 6 (пример 1, часть E), соединение 176 было получено с выходом 59% при исходных 0,31 г соединения 175. Часть G: Следуя процедуре, используемой для получения соединения 7 (пример 1, часть F), соединение 177 было получено с выходом 70% при исходных 0,23 г соединения 176. 1H NMR (300 MHz, CD3OD) 2,85 (dd, J=5,4, 16,4 Hz, 1H), 3,06 (dd, J=4,4, 16,5 Hz, 1H), 4,0-4,2 (m, 5H), 7,05 (d, J=8,25 Hz, 1H), 7,18 (m, 1H), 7,22 (s, 1H), 7,92 (d, J= 8,4 Hz, 2H), 8,10 (d, J=8,4 Hz, 2H); IR (KBr) 3340, 1667, 1603, 1201 см-1; MS (FAB) m/e 370. Аналитически рассчитано для C21H20N3O7F3: C 52,18; H 4,17; N 8,69. Найдено: C 52,15; H 4,02; N 8,54. Пример 42 Получение соединения, представленного формулой 186: 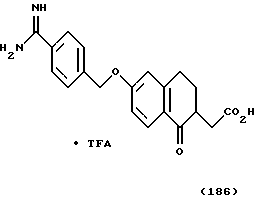 Часть A: К смеси соединения 178 (2,17 г, 13,4 ммоль) и глиоксилата натрия (4,25 г, 37,4 ммоль) добавлялся 1N NaOH (50 мл, 50 ммоль). Раствор перемешивался в течение 4 ч при комнатной температуре, доводился до pH=1 с помощью концентрированной HCl, добавлялась 5N HCl (200 мл), и в течение 24 ч поддерживалось орошение. Смесь охлаждалась, и собирался полученный осадок. Фильтрат экстрагировался с помощью EtOAc, объединенные экстракты промывались рассолом, обезвоживались (MgSO4) и концентрировались в вакууме, давая сухой остаток, который объединялся с вышеупомянутым осадком, что давало 3,02 г (99%) соединения 179 в виде коричневого сухого остатка без дальнейшей очистки. Часть B: К перемешиваемому раствору соединения 179 (0,95 г, 4,36 ммоль) в ледяном HOAc/H2O (2: 1, 15 мл) добавлялась цинковая пыль (1,0 г, 15,3 ммоль). Смесь орошалась в течение 2 ч, охлаждалась до комнатной температуры, разбавлялась EtOAc и промывалась 1N HCl, H2O и рассолом. Органический материал обезвоживался (MgSO4) и концентрировался, что давало 0,89 г (93%) соединения 180 в виде коричневого сухого остатка без дальнейшей очистки. Часть C: Соединение 180 (0,88 г, 4,0 ммоль) растворялось в THF/EtOAc (1:4, 25 мл), добавлялся дифенилдиазометан (0,97 г, 5,0 ммоль) в сухом виде, и красный раствор перемешивался 5 дней при комнатной температуре, после чего орошался в течение 4 ч. Смесь разбавлялась EtOAc, промывалась 1N HCl, насыщенным NaHCO3, H2O и рассолом, обезвоживалась (MgSO4) и концентрировалась в вакууме. Неочищенное выделенное вещество очищалось путем хроматографии (силикагель 230-400 меш, толуол:EtOAc градиент) для получения 0,77 г (50%) соединения 181 в виде рыжевато-коричневого сухого остатка. Часть D: К раствору соединения 181 (0,77 г, 2 ммоль) в DMF (20 мл) добавлялся K2CO3 (0,276 г, 2 ммоль) в сухом виде. После 0,5 ч перемешивания при комнатной температуре добавлялся альфа-бромо-p-толунитрил (0,40 г, 2,0 ммоль) в сухом виде, и раствор перемешивался при комнатной температуре в течение 4 ч. Смесь разбавлялась EtOAc, промывалась H2O, 1N HCl, насыщенным NaHCO3 и рассолом, обезвоживалась (MgSO4) и концентрировалась в вакууме. Неочищенный материал очищался путем хроматографии (силикагелевая препарационная пластина, 8: 2 толуол:EtOAc), давая 0,83 г (83%) соединения 184 в виде светло-желтого сухого остатка. Часть E: Следуя общей процедуре, описанной для получения соединения 6 (пример 1, часть E), соединение 185 было получено с выходом 41% при исходных 0,8 г соединения 184. Часть F: Следуя общей процедуре, описанной для получения соединения 6 (пример 1, часть F), соединение 186 было получено с выходом 41%) при исходных 0,8 г соединения 185; MS (FD) m/e 355. Пример 43 Получение соединения, представленного формулой 190: 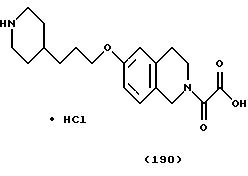 Часть A: Смесь соединения 2 (1,0 г, 3,95 ммоль) и THF (20 мл) обрабатывалась LiAlH4 (0,30 г, 7,9 ммоль) и выдерживалась при орошении в течение 2 ч. Смесь охлаждалась до комнатной температуры, а затем гасилась водой и 15% NaOH. Полученная смесь фильтровалась и концентрировалась. Неочищенный материал, полученный таким образом, растворялся в пиридине (10 мл) и обрабатывался метилоксалилхлоридом (0,38 мл, 4,3 ммоль). Полученная смесь выдерживалась при комнатной температуре в течение 1 ч, а затем разбавлялась EtOAc и промывалась водой. Органический материал концентрировался, и неочищенный остаток очищался на двуокиси кремния (3:1 гексан/EtOAc), давая 0,65 г соединения 187. Часть B: Смесь соединения 187 (0,65 г) и Pd/C (10%, на углероде, 0,65 г) и EtOH (10 мл) выдерживалась в атмосфере H2 (стеклянная пробирка) в течение 2 ч, а затем фильтровалась, и фильтрат концентрировался. Этот процесс давал 0,45 г практически чистого соединения 188. Часть C: Смесь соединения 188 (0,098 г, 0,42 ммоль), NaH (0,018 г 60%-ной дисперсии в масле, 0,46 ммоль) и THF (2 мл) перемешивалась при орошении в течение 0,5 ч, а затем обрабатывалась 1-tBOC-4-(3-бромпропил) пиперидином (0,141 г, 0,42 ммоль). Полученная смесь выдерживалась при орошении в течение 8 ч, а затем разбавлялась EtOAc, промывалась H2O и рассолом. Органический материал концентрировался, и неочищенное выделенное вещество очищалось на двуокиси кремния (1,5:1 гексан/EtOAc), давая 0,11 г соединения 189. Часть D: Смесь соединения 189 (0,11 г, 0,25 ммоль), NaOH (0,02 г, 0,5 ммоль) и EtOH (5 мл) выдерживалась при комнатной температуре в течение 1 ч, а затем концентрировалась. Остаток вводился в H2O, и смесь подкислялась до pH=4 с помощью KHSO4. Эта смесь экстрагировалась с помощью EtOAc, а экстракты концентрировались. Неочищенный остаток обрабатывался TFA (5 мл) в течение 1 ч, а затем концентрировался. Остаток вводился в 0,1 N HCl и лиофолизировался, давая 0,051 г соединения 190. 1H NMR (300 MHz, CD3OD) 1,2-1,7 (m, 6H), 1,8 (m, 2H), 1,95 (m, 2H), 2,9 (m, 4H), 3,35 (m, 2H), 3,75 (m, 2H), 3,95 (m, 2H), 4,6 (m, 2H), 6,25 (m, 2H), 7,1 (m, 1H); IR (KBr) 2940, 1735, 1653, 1187 см-1; MS (FAB) m/e 347. Пример 44 Получение соединения, представленного формулой 193: 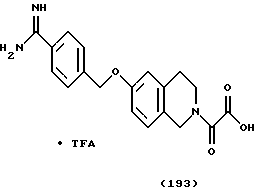 Часть A: Смесь соединения 188 (0,19 г, 0,81 ммоль), NaH (0,021 г, 60%-ной дисперсии в масле, 0,89 ммоль) и THF (5 мл) перемешивалась при комнатной температуре в течение 0,5 ч, а затем обрабатывалась альфа-бромо-p-толунитрилом (0,17 г, 0,89 ммоль). Полученная смесь выдерживалась при орошении в течение 8 ч, а затем разбавлялась EtOAc и промывалась H2O. Органический материал концентрировался, и неочищенный остаток очищался на двуокиси кремния (1:1 гексан/EtOAc), давая 0,22 г соединения 191. Часть B: Следуя процедуре, описанной для получения соединения 6 (пример 1, часть E), соединение 192 было получено с выходом 44% при исходных 0,22 г соединения 191. Часть C: Смесь соединения 192 (0,12 г, 0,27 ммоль), NaOH (0,22 г, 0,55 ммоль), EtOH (5 мл) выдерживалась при комнатной температуре в течение 1 ч, а затем концентрировалась. Остаток растворялся в H2O, и полученный раствор подкислялся до pH=4 с помощью KHSO4. Этот раствор затем лиофолизировался. Неочищенный остаток, полученный таким образом, экстрагировался с помощью MeOH, и объединенные экстракты фильтровались и концентрировались. Выделенный материал обрабатывался TFA (5 мл) в течение 1 ч, а затем концентрировался. Таким образом, выделяли 0,05 г соединения 193. 1H NMR (300 MHz, CD3OD) 2,91 (m, 2H), 3,72 (m, 2H), 4,6 (m, 2H), 5,25 (s, 2H), 6,8 (m, 2H), 7,0 (m, 1H), 7,6 (d, J=8,3 Hz, 2H), 7,8 (d, J=8,3 Hz, 2H); IR (KBr) 3336, 3114, 1668, 1506 см-1; MS (FAB) m/e 354. Пример 45 Получение этил-рац-(6-(4-(аминоиминометил)фенилметокси)-3,4-дигидро-2H-1-бензопиран-2-ил)ацетата гидрохлорида, соединение представлено формулой 194: 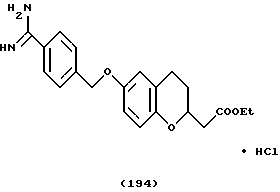 Шаг A: Получение этил-рац-(6-(4-(цианфенил)метокси)-3,4-дигидро-2H-1-бензопиран-2-ил)ацетата, промежуточного соединения, представленного формулой (195) 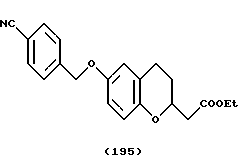 6,0 г (25,4 ммоль) этил-рац-(3,4-дигидро-6-гидрокси-2H-1- бензопиран-2-ил)ацетата (полученного по eвропейской патентной заявке EP 129906, которая включена сюда посредством ссылки) и 4,9 г (25,0 ммоль) 4-цианбензилбромида растворялись в 36 мл обезвоженного ацетона, и было добавлено 3,5 г (25,3 ммоль) карбоната калия. После перемешивания в течение ночи при 50oC добавлялось еще 0,3 г (1,3 ммоль) бензопирана, и реакция продолжалась еще столько же времени. Неорганический сухой остаток удалялся фильтрацией, фильтрат концентрировался в вакууме, и из остатка путем хроматографии на силикагеле с гексаном/ацетоном 40:5 получался чистый нитрил. Выход: 6,7 г (76%) бледно-желтого сухого остатка, tпл 75-76oC. Шаг B: Получение этил-рац-(3,4-дигидро-6-(4-этоксикарбонимидойл) фенилметокси)-2H-1-бензопиран-2-ил)ацетата гидрохлорида, промежуточного соединения, представленного формулой (196): 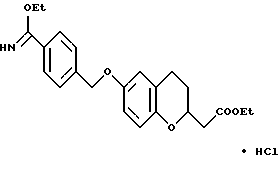 7,54 г (21,2 ммоль) нитрила из шага (A) суспендировалось в 340 мл безводного этанола. Суспензия охлаждалась в ледяной ванне и насыщалась газообразным хлороводородом (примерно 5 ч). За ночь отстаивания был сформирован прозрачный раствор. Раствор выпаривался до безводного состояния в вакууме, и соединение (196) перемешивалось с гексаном, фильтровалось с помощью всасывания и обезвоживалось в вакууме. Выход: 6,43 г (70%) бледно-желтого порошка, tпл 118-119oC. Шаг C: Получение соединения (194), этил-рац-(6-(4-(аминоиминометил)фенилметокси)-3,4-дигидро-2H-1- бензопиран-2-ил)ацетата гидрохлорида. 385 мл насыщенного раствора аммиака в этаноле охлаждалось льдом и добавлялось 6,43 г (14,8 ммоль) промежуточного соединения из шага B. Промежуточное соединение из шага B перемешивалось в течение ночи при комнатной температуре, и растворитель удалялся в вакууме. Оставшееся сухое объявленное соединение перемешивалось с гексаном, фильтровалось с помощью всасывания и обезвоживалось в вакууме при 40oC. Выход: 5,32 г (89%) белого порошка, tпл 123-125oC. Пример 46 Получение рац-(6-(4-(аминоиминометил)фенилметокси)-3,4- дигидро-2H-1-бензопиран-2-ил)уксусного кислого трифторацетата, соединение представлено формулой (197) 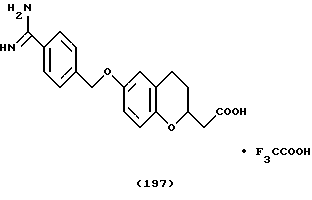 Шаг A: Получение этил-рац-(6-(4-(N-трет-бутоксикарбонил (аминоиминометил))фенилметокси)-3,4-дигидро-2H-1-бензопиран-2-ил)ацетата, промежуточного соединения, представленного формулой (198): 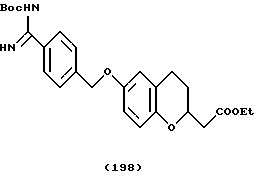 3,7 г (9,1 ммоль) амидина из примера 45 растворялось в 55 мл смеси из THF/H2O 1: 1. После добавления 1,65 г (11,9 ммоль) карбоната калия, 1,99 г (9,1 ммоль) Boc2O добавлялось по каплям, и смесь перемешивалась в течение ночи при комнатной температуре. Затем смесь разбавлялась с помощью 100 мл этилацетата. Органический слой отделялся, промывался водой, обезвоживался на сульфате натрия и концентрировался в вакууме, давая чистый защищенный амидин. Выход: 4,3 (100%) масла. Шаг B: Получение рац-(6-(4-(N-трет-бутоксикарбонил(аминоиминометил) фенилметокси)-3,4-дигидро-2H-1-бензопиран-2-ил)уксусной кислоты, промежуточного соединения, представленного формулой (199): 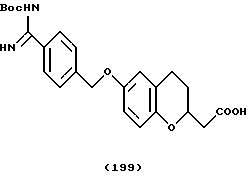 5,2 г (11,1 ммоль) сложного эфира из шага A растворялось в 75 мл этанола. После добавления 36 мл 2N водного гидроксида натрия смесь нагревалась до 60oC на несколько минут. Затем смесь доводилась до pH=6 с помощью уксусной кислоты. Этанол удалялся в вакууме, и добавлялся этилацетат. Органический слой отделялся, обезвоживался на сульфате натрия, концентрировался в вакууме, и из остатка путем хроматографии на силикагеле с дихлорметаном/этанолом 40: 5 получалась объявленная карбоновая кислота. Выход: 2,0 г (41%) белого порошка, tпл 220-222oC (dec). Шаг C: Получение рац-(6-(4-(аминоиминометил)фенилметокси)-3,4- дигидро-2H-1-бензопиран-2-ил)уксусного кислого трифторацетата. 0,15 г (0,34 ммоль) защищенного амидина из шага B и 2,8 мл трифторуксусной кислоты смешивались и перемешивались при комнатной температуре в течение 1,5 ч. Растворитель удалялся в вакууме, и объявленный амидин получался после добавления 10 мл воды. Продукт фильтровался с помощью всасывания, перемешивался снова с 10 мл воды, фильтровался и обезвоживался в вакууме. Выход: 0,09 г (58%) белого порошка, tпл 210-212oC. Пример 47 Получение этил-рац-(6-(N-(4-(аминоиминометил)бензойл)амино)-3,4- дигидро-2H-1-бензопиран-2-ил)ацетата гидрохлорида, соединение представлено формулой (200): 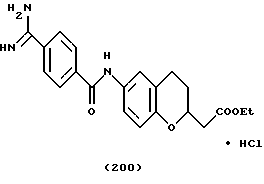 Шаг A: Получение трет-бутил(2-оксо-2H-1-бензопиран-6-ил)карбомата, промежуточного соединения, представленного формулой (201): 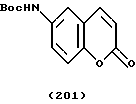 100 г (523 ммоль) 6-нитрокумарина растворялось в 600 мл безводного этанола, и раствор держался в атмосфере аргона. 86 г (1,364 моль) формиата аммония и 6 г 10% Pd/C добавлялись при перемешивании, в то время как температура росла до 45oC, наблюдалось выделение газа. Реакционная смесь орошалась в течение 3 ч и разбавлялась еще 200 мл этанола. Горячая смесь фильтровалась сквозь Селит и промывалась 200 мл горячего этанола. Незащищенный амин получался после охлаждения, фильтрации с помощью всасывания, промывания гексаном и обезвоживания в вакууме. Еще одна порция амина получалась путем концентрации фильтрата. Общий выход промежуточного амина составил 74 г. Неочищенный амин растворялся в 300 мл смеси THF/H2O 1:1 и выдерживался в атмосфере аргона. Добавлялись 110 г (504 ммоль) Boc2O и 95 г (687 ммоль) безводного карбоната калия. Реакционная смесь перемешивалась в течение ночи при комнатной температуре, разбавлялась 750 мл воды и экстрагировалась с помощью этилацетата (3 х 1 л). Объединенные органические слои обезвоживались на сульфате натрия и концентрировались в вакууме. Остаток растворялся в 2 л дихлорметана и перемешивался в присутствии 1 кг силикагеля, который фильтровался с помощью всасывания и промывался дихлорметаном. Фильтрат концентрировался в вакууме, давая объявленное соединение. Аналитический образец очищался путем хроматографии на силикагеле с дихлорметаном. Выход: 112 г (82%) бледно-желтых кристаллов, пл 142-143oC. Шаг B: Получение трет-бутил(3,4-дигидро-2-оксо-2H-1-бензопиран-6-ил)карбoмата, промежуточного соединения, представленного формулой (202): 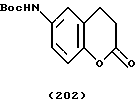 85 г (325 ммоль) кумарина из шага A растворялись в смеси 1150 мл этанола и 115 мл уксусной кислоты, и раствор заливался в гидрогенизационный автоклав. 6 г 10% Pd/C добавлялось, и все это гидрогенизировалось при комнатной температуре и давлении водорода 20 атм. Через два дня добавлялось еще 3 г Pd/C, и реакция продолжалась еще два дня. Катализатор удалялся путем фильтрации, а фильтрат концентрировался в вакууме. Остаток растворялся в 1 л этилацетата, промывался 500 мл насыщенного водного бикарбоната натрия, обезвоживался на сульфате натрия, а растворитель удалялся в вакууме. Объявленный лактон очищался путем хроматографии на силикагеле с дихлорметаном, содержащим 1% этанола. Выход: 21 г (25%) бесцветных кристаллов, tпл 158-159oC. Шаг C: Получение трет-бутил-рац-(3,4-дигидро-2-гидрокси-2H-1-бензопиран-6-ил)карбомата, промежуточного соединения, представленного формулой (203): 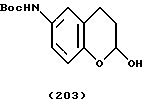 21 г (79,8 ммоль) лактона из предыдущего шага растворялись в 340 мл безводного дихлорметана и выдерживались в атмосфере аргона. Раствор охлаждался до -70oC и выдерживался при этой температуре, в то время как в течение 45 мин по каплям добавлялось 67 мл 25%-ного раствора гидрида диизобутилалюминия (DIBAl-H) в толуоле. После еще 1 ч перемешивания при этой температуре медленно добавлялось 20 мл метанола, и смесь заливалась в 1 л насыщенного водного раствора хлорида аммония. Твердые составляющие удалялись путем фильтрации сквозь Селит и промывались дихлорметаном. Фильтрат обезвоживался на сульфате натрия, концентрировался в вакууме, давая чистое объявленное соединение, как фиксировалось его 1H-NMR. Выход: 16,5 г (78%) желтого масла. Шаг D: Получение этил-рац-(6-(N-трет-бутоксикарбониламино)-3,4-дигидро-2H-1-бензопиран-2-ил)ацетата, промежуточного соединения, представленного формулой (204): 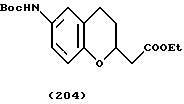 11,2 г (42,2 ммоль) соединения из шага C и 14,7 г (42,2 ммоль) этоксикарбонилметиленатрифенилфосфорана растворялись в 130 мл безводного толуола и орошались в течение 22 ч. Реакционная смесь охлаждалась до комнатной температуры, и добавлялись 300 мг гидрида натрия. Еще через 5 ч нагревания смесь заливалась в 1 л ледяной воды. Смесь трижды экстрагировалась с помощью этилацетата, и объединенные органические слои промывались 300 мл воды, обезвоживались на сульфате натрия и концентрировались в вакууме. Бензопиран (204) получался из остатка путем хроматографии на силикагеле с дихлорметаном. Выход: 5,5 г (39%) бесцветного аморфного сухого остатка, tпл 67-69oC. Шаг E: Получение этил-рац-(6-(-(4-цианбензойл)амино)-3,4- дигидро-2H-1-бензопиран-2-ил)ацетата, промежуточного соединения, представленного формулой (205): 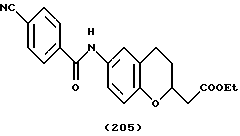 2,0 г (6,0 ммоль) защищенного амина из предыдущего шага обрабатывалось 6 мл трифторуксусной кислоты и перемешивалось в течение 2 ч при комнатной температуре. Смесь нейтрализовывалась насыщенным водным раствором бикарбоната натрия и экстрагировалась с помощью этилацетата. Органический слой обезвоживался на сульфате натрия и концентрировался при пониженном давлении, оставалось темное масло из незащищенного этил-рац-(6-амино-3,4-дигидро-2H-1-бензопиран-2-ил)ацетата. Оно растворялось в 40 мл безводной THF, обрабатывалось 4 мл безводного пиридина и 1,0 г (6,0 ммоль) 4-цианбензойлхлорида, и перемешивалось в течение ночи при комнатной температуре. Смесь заливалась в ледяной водный раствор бикарбонатнатрия и экстрагировалась с помощью этилацетата. Органический слой последовательно промывался водным раствором сульфата меди (II) и рассолом, обезвоживался на сульфате натрия и концентрировался в вакууме. Нитрил (205) получался из остатка путем хроматографии на силикагеле с дихлорметаном/этанолом 96:4. Выход: 1,4 г (64%) бледно-желтых кристаллов, tпл 146-148oC. Шаг F: Получение этил-рац-(6-(N-(4-аминоиминометил)бензойл)амино)- 3,4-дигидро-2H-1-бензопиран-2-ил)ацетата гидрохлорида. 1,4 г (3,8 ммоль) нитрила из предыдущего шага растворялось в 50 мл безводного этанола. Раствор охлаждался льдом и насыщался газообразным хлороводородом. После перемешивания в течение ночи при комнатной температуре растворитель удалялся при пониженном давлении, а остаток обрабатывался насыщенным раствором аммиака в этаноле. Реакционная смесь перемешивалась в течение трех дней, выпаривалась в вакууме, и объявленное соединение получалось в виде желтого масла путем хроматографии на силикагеле с дихлорметаном/этанолом 65:35 с 5%-ным содержанием аммиака в этаноле. Кристаллический образец для аналитических и биологических тестов получался путем перемешивания со смесью этанола/эфирного хлороводорода/эфира. Выход: 0,9 г (56%) желтых кристаллов, tпл 253oC (dec). Пример 48 Получение рац-(6-(N-(4-аминоиминометил)бензойл)амино)-3,4- дигидро-2H-1-бензопиран-2-ил)уксусной кислоты, соединение представлено формулой (206): 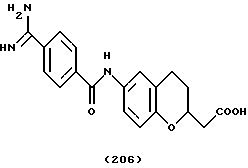 0,2 г (0,48 ммоль) сложного эфира из примера 47 добавлялось к смеси 4 мл этанола и 0,5 мл 2N водного гидроксида натрия. Смесь разбавлялась водой до тех пор, пока не превращалась в прозрачный раствор. После легкого нагревания он перемешивался при комнатной температуре в течение 3 ч и подкислялся с помощью 2N уксусной кислоты, в это время образовывался осадок, который фильтровался, промывался водой и обезвоживался в вакууме. Очень плохо растворимое соединение (206) исследовалась с помощью элементного анализа и масс-спектрографии. Выход: 0,16 г (95%) бесцветного аморфного сухого остатка tпл 290-291oC (dec). Пример 49 Получение рац-(6-(N-(4-карбомойлбензойл)амино)-3,4-дигидро-2H-1- бензопиран-2-ил)уксусной кислоты, соединение представлено формулой (207): 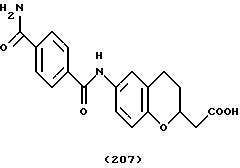 Способ A: 0,47 г (1,12 ммоль) сложного эфира из примера 47 добавлялось к смеси 5 мл этанола и 5 мл 2N водного гидроксида натрия. Смесь нагревалась в паровой ванне в течение 15 мин, охлаждалась до комнатной температуры, и доводилась до pH= 4 с помощью 2N водной соляной кислоты, формировался осадок. Осадок фильтровался с помощью всасывания, и амид (207) очищался путем суспендирования в небольшом количестве горячего этанола. Выход: 60 мг (15%) бежевого аморфного сухого остатка, tпл 273-274oC. Способ B: 0,7 (1,92 ммоль) нитрила из примера 47, шаг E, растворялись в 15 мл 98%-ной муравьиной кислоте, и в течение 4 ч через смесь пропускалась струя газообразного хлороводорода. Реакционная смесь перемешивалась в течение ночи при комнатной температуре, и растворитель удалялся в вакууме. Оставшиеся сухие остатки перемешивались с водой, фильтровались с помощью всасывания, и последовательно промывались этанолом и эфиром. Амид суспенидровался в горячем этаноле, фильтровался, и обезвоживался в вакууме. Выход: 0,45 г (66%) серых кристаллов, tпл 264-265oC. Пример 50 Получение этил-рац-(6-(N-(4-аминоиминометил)бензойл)амино)-3,4- дигидро-2H-1-бензопиран-2-ил)пропаноата гидрохлорида, соединение представлено формулой (208): 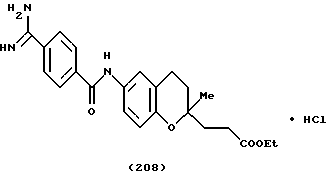 Шаг A: Получение этил-рац-3-(3,4-дигидро-2-метил-6-нитро-4- оксо-2H-1-бензопиран-2-ил)пропаноата, промежуточного соединения, представленного формулой (209): 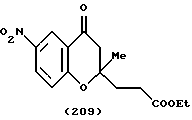 5,4 г (29,8 ммоль) 2-гидрокси-5-нитроацетофенона (полученного способом из Journal of American Chemical Society, 1954, 76, 4993, рассмотрение в котором включено сюда посредством ссылки), 5,8 г (40,2 ммоль) этил-4-оксопентаноата, и 1,7 мл пирролидина растворялись в 50 мл толуола, и смесь нагревалась в течение 6 ч с азеотропным удалением воды. Смесь концентрировалось в этилацетате. Раствор последовательно промывался 1N водной соляной кислотой и рассолом, обезвоживался на сульфате натрия, а растворитель удалялся при пониженном давлении. Хроманон (209) кристаллизовался из полученного остатка. Выход: 5,3 г (58%) бледно-желтого аморфного сухого остатка, пл 88-90oC. Шаг B: Получение этил-3-(3,4-дигидро-4-гидрокси-2-метил-6-нитро- 2H-1-бензопиран-2-ил)пропаноата, промежуточного соединения, представленного формулой (210): 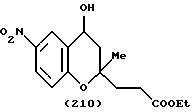 3,2 г (10,4 ммоль) хроманона из шага A растворялось в 100 мл этанола. Малыми порциями добавлялось 0,76 г (20,0 ммоль) борного гидрида натрия, и смесь перемешивалась при комнатной температуре в течение 5 ч. Реакционная смесь концентрировалась в вакууме, подкислялась с помощью 1N водной соляной кислоты и экстрагировалась с помощью этилацетата. Органический слой обезвоживался на сульфате натрия, и растворитель удалялся при пониженном давлении. Бензопиран (210) получался путем хроматографии на силикагеле с дихлорметаном, содержащим 41% этанола. Выход: 2,0 г (62%) масла. Шаг C: Получение этил-рац-3-(2-метил-6-нитро-2H-1-бензопиран-2-ил)пропаноата, промежуточного соединения, представленного формулой (211): 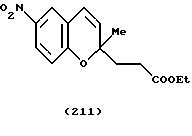 2,0 г (6,5 ммоль) спирта из предыдущего шага растворялось в 75 мл толуола, добавлялось каталитическое количество 4-толуолсульфокислоты. Смесь орошалась в течение 7 ч, в то время как вода азеотропно удалялась. Реакционная смесь промывалась водным раствором бикарбоната натрия, органический слой обезвоживался на сульфате натрия, и концентрировался в вакууме. Объявленный хромен получался из остатка путем хроматографии на силикагеле с этилацетатом/гексаном 1:3. Выход: 0,85 г (45%) масла. Шаг D: Получение этил-рац-3-(6-амино-3,4-дигидро-2-метил-2H-1- бензопиран-2-ил)пропаноата, промежуточного соединения, представленного формулой (212): 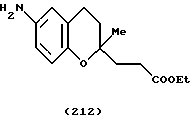 1,6 г (5,5 ммоль) 6-нитрохромена из предыдущего шага растворялось в смеси 20 мл этанола и 10 мл уксусной кислоты. Добавлялось 400 мг Pd/C, и смесь заливалась в автоклав. Она перемешивалась в течение ночи при комнатной температуре в атмосфере 20 бар водорода до завершения восстановления. Катализатор удалялся путем фильтрации, а этанол дистиллировался в вакууме. Реакционная смесь разбавлялась этилацетатом, промывалась концентрированным водным раствором бикарбоната натрия, обезвоживалась на сульфате натрия и концентрировалась при понижении давления, давая неочищенный объявленный амин, который был чистым, как фиксировалось 1H-NMR. Выход: 1,6 г масла, темнеющего со временем. Шаг E: Получение этил-рац-(6-(N-(4-цианбензойл)амино)-3,4- дигидро-2-метил-2H-1-бензопиран-2-ил) пропаноата, соединение представлено формулой (213): 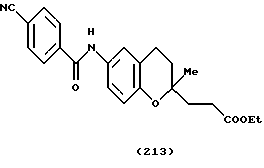 1,6 г (6,1 ммоль) неочищенного амина из шага D растворялось в 40 мл безводной THF. Последовательно добавлялись безводный пиридин и 1,0 г (6,0 ммоль) 4-цианбензойл хлорида. Смесь перемешивалась в течение ночи при комнатной температуре и заливалась в ледяной раствор водного бикарбоната натрия. Она экстрагировалась с помощью этилацетата, и органический слой промывался водным сульфатом меди (II) и рассолом, обезвоживался на сульфате натрия и концентрировался в вакууме. Чистый нитрил получался путем хроматографии на силикагеле с дихлорметаном/этанолом 97:3. Выход: 1,5 г (63%) темного вязкого масла. Шаг F: Получение этил-рац-(6-(N-(4-аминоиминометил)бензойл)амино)- 3,4-дигидро-2-метил-2H-1-бензопиран-2-ил)пропаноата гидрохлорида. 1,5 г (3,8 ммоль) нитрила из шага E растворялось в 50 мл безводного этанола. Раствор охлаждался в ледяной ванне, насыщался газообразным хлороводородом и перемешивался в течение ночи при комнатной температуре. Реакционная смесь концентрировалась при пониженном давлении, и остаток обрабатывался насыщенным раствором аммиака в этаноле. Он перемешивался еще 24 ч при комнатной температуре концентрировался в вакууме и хроматографировался на силикагеле с дихлорметаном/этанолом 65:35 с 5%-ным содержанием аммиака в этаноле, давая объявленный амидин в виде масла. Кристаллический образец для аналитических и биологических тестов получался перемешиванием в смеси этанола/эфирного хлороводорода/эфира. Выход: 0,94 г (55%) желтых кристаллов, tпл 118-120oC. Пример 51 Получение рац-(6-(N-(4-карбомойлбензойл)амино)-3,4-дигидро-2- метил-2H-1-бензопиран-2-ил)пропановой кислоты, соединение представлено формулой (214): 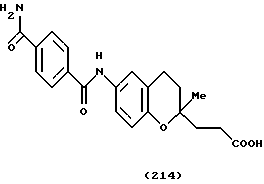 0,5 г (1,12 ммоль) сложного эфира из примера 50 добавлялось к смеси 5 мл 2N водного гидроксида натрия и 5 мл этанола. Реакционная смесь перемешивалась при нагреве в паровой бане в течение 20 мин, в это время смесь превращается в прозрачный раствор, а затем доводилась до pH=4 с помощью 2N водной соляной кислоты и экстрагировалась с помощью этилацетата. Органический слой обезвоживался на сульфате натрия и концентрировался в вакууме. Реакционная смесь очищалась путем хроматографии на силикагеле с хлороформом/этанолом 1: 1. Оставшееся масло, полученное из чистых фракций, обрабатывалось 2 мл трифторуксусной кислоты и перемешивалось в течение 2 ч при комнатной температуре. Растворитель удалялся при пониженном давлении, и объявленный амид кристаллизовывался из остатка путем обработки этанолом. Выход: 70 мг (16%) бежевого аморфного сухого остатка, tпл 237-238oC. Пример 52 Получение этил-рац-3-(6-(4-аминоиминометил)фенилметокси)-2- метил-2H-1-бензопиран-2-ил)пропаноата гидрохлорида, соединение представлено формулой (215): 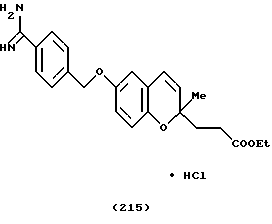 Шаг A: Получение 4-((3-ацетил-4-гидроксифенокси)метил)бензонитрила, промежуточного соединения, представленного формулой (216): 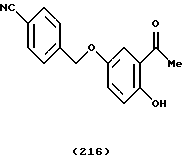 45 г (296 ммоль) 2,5 дигидроксиацетофенона и 58,4 г (298 ммоль) 4-цианбензилбромида растворялись в 650 мл безводного ацетона, добавлялись 45 г (326 ммоль) карбоната калия и 4,5 г йодида калия. После орошения в течение 6,5 ч неорганические твердые частицы удалялись путем фильтрации и промывались ацетоном. Объединенные фильтраты концентрировались в вакууме, и остаток перемешивался с 600 мл горячего метанола. Метаноловый раствор охлаждался до комнатной температуры, и соединение (216) фильтровалось с помощью всасывания, последовательно промывалось метанолом и гексаном и обезвоживалось в вакууме при 40oC. Выход: 69,5 г (88%) бежевых кристаллов, tпл 123-127oC. Шаг B: Получение этил-рац-3-(6-(4-цианфенилметокси-3,4-дигидро-2- метил-4-оксо-2H-1-бензопиран-2-ил)пропаноата, промежуточного соединения, представленного формулой (217): 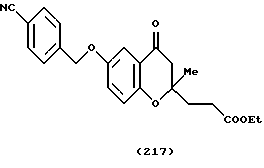 30 г (112,2 ммоль) ацетофенола из предыдущего шага, 20,4 г (141,5 ммоль) этил-4-оксопентаноата и 9,6 мл пирролидина растворялись в 500 мл безводного толуола. Смесь перемешивалась 20 ч при комнатной температуре, после чего орошалась 5,5 ч с азеотропным удалением воды. Смесь концентрировалась при пониженном давлении, а оставшееся масло перемешивалось в течение 30 мин в 200 мл водной 2N соляной кислоты. Оно экстрагировалось с помощью дихлорметана, и органический слой последовательно промывался 2N соляной кислотой, водой и рассолом. Затем оно обезвоживалось на решетке с ячейками 0,4 нм и концентрировалось в вакууме, оставляя коричневое масло, которое хроматографировалось на силикагеле с дихлорметаном. Отвердение маслянистого бензопирана (217) происходило путем перемешивания с водной 2N соляной кислотой. Соединение фильтровалось с помощью всасывания, последовательно промывалось водой и гексаном, и обезвоживалось в вакууме. Выход: 12,27 г (28%) желтых кристаллов, tпл 88-92oC. Шаг C: Получение этил-3-(6-(4-цианфенилметокси-3,4-дигидро-4- гидрокси-2-метил-2H-1-бензопиран-2-ил)пропаноата, промежуточного соединения, представленного формулой (218): 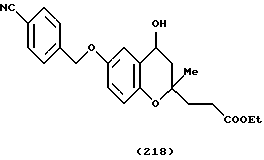 0,18 г (2,0 ммоль) хроманона из шага B растворялось в 10 мл безводного этанола и добавлялось 40 мг (1,06 ммоль) боргидрида натрия. После перемешивания в течение ночи при комнатной температуре добавлялось еще 40 мг гидрида, а перемешивание продолжалось еще 4 ч до тех пор, пока не завершалось восстановление. Растворитель удалялся в вакууме, а остаток обрабатывался смесью воды и дихлорметана. Водный слой экстрагировался с помощью дихлорметана, и объединенные органические слои обезвоживались на решетке с ячейками 0,4 нм. После концентрации при пониженном давлении бензопиран получался путем хроматографии на силикагеле с дихлорметаном, содержащим до 2% этанола. Выход: 0,46 г (57%) масла. Шаг D: Получение этил-рац-3-(6-(4-цианфенилметокси)-2-метил-2H-1- бензопиран-2-ил)пропаноата, промежуточного соединения, представленного формулой (219): 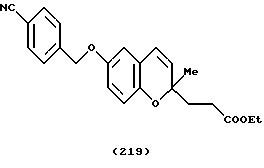 380 мг (0,96 ммоль) соединения из предыдущего шага растворялись в 25 мл толуола. После добавления каталитического количества 4-толуолсульфокислоты смесь нагревалась в течение 1 ч с азеотропным удалением воды вплоть до завершения реакции. Смесь дважды промывалась насыщенным водным бикарбонатом натрия, обезвоживалась на решетке с ячейками 0,4 нм и концентрировалась в вакууме. Неочищенный хромин очищался путем хроматографии на силикагеле с дихлорметаном. Выход: 230 мг (63%) масла. Шаг E: Получение этил-рац-3-(6-(4-аминоиминометил)фенилметокси)-2- метил-2H-1-бензопиран-2-ил)пропаноата гидрохлорида. 1,5 г (4,0 ммоль) нитрила из предыдущего шага растворялось в 100 мл безводного этанола. Раствор охлаждался до 5-10oC, насыщался хлороводородом, перемешивался при комнатной температуре в течение ночи и концентрировался в вакууме. Оставшееся коричневое масло обрабатывалось 100 мл насыщенного раствора аммиака в этаноле и перемешивалось в течение 2 дней. Растворитель удалялся в вакууме, и объявленный амидин получался из остатка путем хроматографии на силикагеле с дихлорметаном/этанолом 95:5 и уменьшения соотношения до 80: 20. Выход: 1,19 г (69%) желтого аморфного сухого остатка, tпл <50oC. Пример 53 Получение рац-3-(6-(4-(аминоиминометил) фенилметокси)-2-метил- 2H-1-бензопиран-2-ил)пропанового кислого гидрохлорида, соединения, представленного формулой (220): 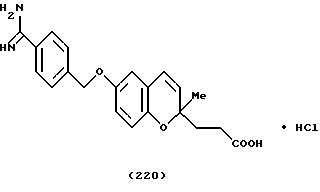 200 мг (0,464 ммоль) сложного эфира из примера 52 растворялось в 5 мл этанола. Добавлялись две капли воды и 0,64 мл 0,9 этанолового раствора этилата натрия, и смесь перемешивалась в течение 3 ч при 50oC. После добавления еще одного такого же количество этилата натрия реакция продолжалась 2 ч при 50oC и 3 дня при комнатной температуре. Формировался осадок, который фильтровался с помощью всасывания, повторно промывался этанолом и гексаном. Неочищенный сухой остаток нагревался в смеси 4 мл воды и 1 мл водной 2N соляной кислоты в течение нескольких минут и перемешивался 2 ч при комнатной температуре. Смесь выпаривалась до безводного состояния, и объявленный гидрохлорид суспендировался три раза в 3 мл горячего изопропанола. Горячие растворы сцеживались, объединялись и концентрировались до безводного состояния. Остаток промывался два раза эфиром и обезвоживался в вакууме. Выход: 68 мг (36%) бежевого аморфного сухого остатка, tпл 56oC. Пример 54 Получение рац-(3,4-дигидро-6-(4-(пиперидин-4-ил)бутокси)-2H-1- бензопиран-2-ил)уксусного кислого трифторацетата, соединения, представленного формулой (225): 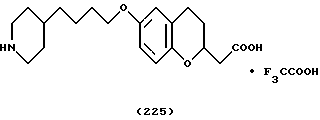 Шаг A: Получение этил-рац-(6-(4-(1-(трет-бутоксикарбонил)пиперидин- 4-ил)бутокси)-3,4-дигидро-2H-1-бензопиран-2-ил)ацетата, промежуточного соединения, представленного формулой (226): 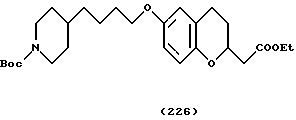 0,92 г (3,9 ммоль) этил-рац-(3,4-дигидро-6-гидрокси-2H-1-бензопиран-2-ил)ацетата (полученного в европейской патентной заявке EP 129906, которая включена сюда посредством ссылки) растворялось в 25 мл безводной DMF. Раствор охлаждался до -5oC, и по каплям добавлялось 1,8 мл 40%-ного гидроксида бензилтриметиламмония (тритон В) в метаноле. Через 40 мин при этой температуре добавлялось 1,25 г (3,9 ммоль) 4-(4-бромбутил)-1-(трет-бутоксикарбонил)пиперидина (полученного по европейской патентной заявке EP 478328, которая включена сюда посредством ссылки). Смесь перемешивалась при -5oС еще 3 ч, нагревалась до комнатной температуры, перемешивалась в течение ночи и заливалась в 150 мл этилацетата. Затем она последовательно промывалась водой, 1N водно-соляной кислотой, водой, насыщенным водным бикарбонатом натрия, водой и рассолом. Органический слой обезвоживался на сульфате натрия и концентрировался в вакууме. Объявленное соединение получалось путем хроматографии на силикагеле с гексаном/этилацетатом 4:1. Выход: 0,64 г (35%) бесцветного масла. Шаг B: Получение рац-(6-(4-(1-(трет-бутоксикарбонил)пиперидин-4- ил)бутокси)-3,4-дигидро-2H-1-бензопиран-2-ил)уксусной килоты, промежуточного соединения, представленного формулой (227): 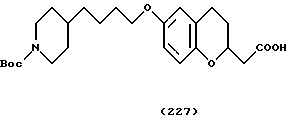 0,64 г (1,35 ммоль) сложного эфира из шага A растворялось в 10 мл этанола и добавлялось 5,6 мл 1N этанолового раствора этилата натрия. Реакционная смесь перемешивалась при комнатной температуре в течение семи дней и концентрировалась до безводного состояния при пониженном давлении. Остаток обрабатывался водой и нейтрализовывался 10%-ным водным раствором KHSO4. Он экстрагировался с помощью этилацетата. Органический слой обезвоживался на сульфате натрия, а растворитель удалялся в вакууме, что давало чистую карбоновную кислоту. Выход: 0,58 г (96%) бледно-желтого масла, которое медленно отвердевало со временем. Шаг C: Получение рац-(3,4-дигидро-6-(4-(пиперидин-4-ил)бутокси)- 2H-1-бензопиран-2-ил)уксусного кислого трифторацетата. 0,4 г (0,9 ммоль) защищенного пиперидина из предыдущего шага обрабатывалось 6 мл трифторуксусной кислоты. Смесь перемешивалась в течение 2 ч при комнатной температуре и выпаривалась в вакууме. После добавления воды смесь экстрагировалась с помощью эфира, и органический слой обезвоживался на сульфате натрия и концентрировался при пониженном давлении. Объявленное соединение очищалось путем хроматографии на силикагеле с дихлорметаном/этанолом 96: 4. Выход: 120 мг (29%) бежевого масла, которое со временем частично отвердевало. Пример 55 Получение рац-(6-(5-(аминоиминометил)пентокси)-3,4-дигидро-2H-1- бензопиран-2-ил)уксусного кислого трифторацетата, промежуточного соединения, представленного формулой (253): 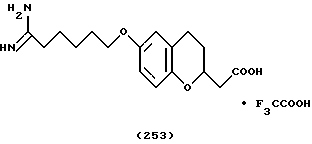 Шаг A: Получение этил-рац-(6-(5-цианпентокси)-3,4-дигидро-2H-1- бензопиран-2-ил)ацетата, промежуточного соединения, представленного формулой (254): 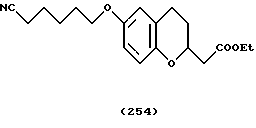 2,109 г (9,27 ммоль) этил-рац-(3,4-дигидро-6-гидрокси-2H-1-бензопиран-2-ил)ацетата (полученного по европейской патентной заявке EP 129906, которая включена сюда посредством ссылки) и 2,0 г (11,4 ммоль) 6-бромкапронитрила растворялись в 30 мл безводного ацетона. Добавлялись 2,0 г (14,5 ммоль) карбоната калия, 250 мг йодида калия, 100 мг хлорида триэтилбензиламмония, и смесь орошалась в течение 10 ч, а затем перемешивалась при комнатной температуре в течение 2 дней. Неорганический твердый остаток удалялся путем фильтрации и промывался ацетоном, а объединенные фильтраты концентрировались в вакууме. Нитрил (254) получался из остатка путем хроматографии на силикагеле с дихлорметаном, содержащим до 4% этанола. Выход: 1,32 г (43%) масла. Шаг B: Получение этил-рац-(6-(5-(аминоиминометил)пентокси)-3,4- дигидро-2H-1-бензопиран-2-ил)ацетата гидрохлорида, промежуточного соединения, представленного формулой (255): 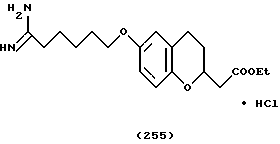 1,25 г (3,77 ммоль) нитрила из шага A растворялось в 50 мл безводного этанола. Раствор охлаждался до 0oC и насыщался хлороводородом. После перемешивания в течение ночи он концентрировался в вакууме. Остаток обрабатывался смесью 10 мл жидкого аммиака и 50 мл безводного этанола, и перемешивался в течение ночи при комнатной температуре. Растворитель удалялся при пониженном давлении, а оставшийся материал последовательно перемешивался с этанолом и дихлорметаном. Твердые частицы удалялись путем фильтрации после каждой процедуры, а фильтраты концентрировались при пониженном давлении. Неочищенное объявленное соединение, полученное из последнего фильтрата, очищалось путем хроматографии на силикагеле с дихлорметаном/этанолом 9:1, а затем 8:2. Выход: 0,97 г (67%) белого порошка, tпл 82-84oC. Шаг C: Получение этил-рац-(6-(5-(N-трет- бутоксикарбониламиноиминометил)пентокси)-3,4-дигидро-2H-1-бензопиран- 2-ил)ацетата, промежуточного соединения, представленного формулой (256): 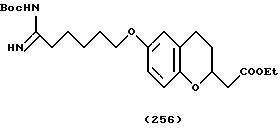 0,7 г (1,82 ммоль) амидина из шага B растворялось в 11 мл THF/H2O 1:1. После добавления 335 г (2,42 ммоль) карбоната калия и 0,4 г (1,83 ммоль) Boc2O смесь перемешивалась в течение ночи при комнатной температуре. Она разбавлялась 25 мл этилацетата. Водный слой отделялся и экстрагировался с помощью этилацетата. Объединенные органические слои промывались водой, обезвоживались на сульфате натрия и концентрировались в вакууме до безводного состояния, давая неочищенный защищенный амидин, используемый на следующем шаге. Выход: 0,87 г желтого масла. Шаг D: Получение рац-(6-(5-(N-трет-бутоксикарбониламиноиминометил) пентокси)-3,4-дигидро-2H-1-бензопиран-2-ил)уксусной кислоты, промежуточного соединения, представленного формулой (257): 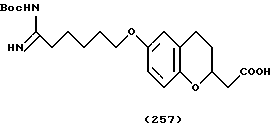 Смесь 13 мл этанола и 6 мл 2N водного гидроксида натрия добавлялась к 0,81 г (1,81 ммоль) сложного эфира из предыдущего шага. Смесь перемешивалась при комнатной температуре в течение 4 ч и нейтрализовалась с помощью разбавленной уксусной кислоты. После выпаривания в вакууме остаток перемешивался со смесью дихлорметана/метанола 1:1. Твердые частицы удалялись путем фильтрации и промывались, а объединенные фильтраты концентрировались при пониженном давлении. Карбоновая кислота (257) получалась путем хроматографии на силикагеле с дихлорметаном и увеличения полярности добавлением 3% этанола. Выход: 215 мг (28%) масла. Шаг E: Получение рац-(6-(5-(аминоиминометил)пентокси)-3,4- дигидро-2H-1-бензопиран-2-ил)уксусного кислого трифторацетата. 112 мг (0,266 ммоль) защищенного амидина из Шага D обрабатывалось при комнатной температуре в течение 1 ч. Растворитель удалялся в вакууме, а остаток перемешивался с 10 мл воды, в это время объявленное соединение выпадало в осадок. Продукт фильтровался с помощью всасывания, промывался последовательно водой и эфиром и обезвоживался в вакууме. Другая порция получалась из объединенных фильтратов, которые дважды промывались эфиром и концентрировались при пониженном давлении. Общий выход: 91 мг (79%) бежевого порошка, tпл 132-134oC. Пример 56 Получение рац-3-(6-(N-(4-(аминоиминометил)бензойл)амино)-3,4- дигидро-2-метил-2H-1-бензопиран-2-ил)пропановой кислоты, соединение представлено формулой (258): 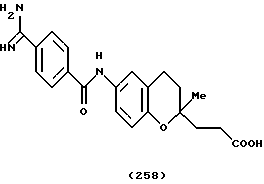 0,4 г (0,9 ммоль) сложного эфира из примера 50 добавлялось к смеси 8 мл этанола и 0,5 мл 2N водного гидроксида натрия. Все это перемешивалось в течение ночи при комнатной температуре, разбавлялось 10 мл воды, и доводилось до pH=4 с помощью уксусной кислоты. Объявленное соединение выпадало в осадок из раствора. Оно фильтровалось, промывалось водой и обезвоживалось в вакууме. Выход: 280 мг (82%) бесцветного аморфного порошка tпл 278-280oC (dec). Пример 57 Получение этил-рац-(3,4-дигидро-6-(N-(4-((метиламино)иминометил) бензойл)амино)-2H-1-бензопиран-2-ил)ацетата гидрохлорида, вещества представленного формулой (260): 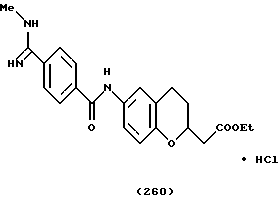 Шаг A: Получение этил-рац-(3,4-дигидро-6-(N-(4-((метиламино) иминометил)бензойл)амино)-2H-1-бензопиран-2-ил)ацетата гидрохлорида, промежуточного соединения, представленного формулой (261): 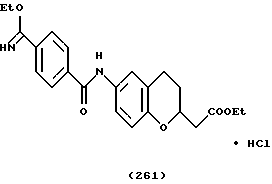 2,0 г (5,5 ммоль) нитрила из примера 47, шаг E, растворялось в 65 мл безводного этанола. Раствор охлаждался льдом и насыщался газообразным хлороводородом. Он перемешивался в течение ночи при комнатной температуре и концентрировался при пониженном давлении, давая промежуточное соединение в виде кристаллического сухого остатка, который использовался в следующем шаге. Выход: 2,4 г (98%). Шаг B: Получение этил-рац-(3,4-дигидро-6-(N-(4-((метиламино) иминометил)бензойл)амино)-2H-1-бензопиран-2-ил)ацетата гидрохлорида. 1,35 г (3,0 ммоль) неочищенного промежуточного соединения из предыдущего шага в 50 мл безводного этанола охлаждалось льдом. Раствор нейтрализовался с помощью 30%-ного этанолового раствора метиламина с последующими 2 ч перемешивания. Добавлялось еще 5 мл раствора метиламина, и перемешивание продолжалось еще 6 ч, в то время как температура поддерживалась ниже 5oC. Получался прозрачный раствор, который концентрировался при пониженном давлении. Объявленное соединение кристаллизовывалось после обработки остатка этанолом и очищалось путем нагрева суспензии в этаноле. Выход: 0,9 г (69%) желтого аморфного сухого остатка, tпл 278-279oC. Пример 58 Получение рац-(3,4-дигидро-6-(N-(4-((метиламино)иминометил) бензойл)амино)-2H-1-бензопиран-2-ил)уксусной кислоты, соединение представлено формулой (262): 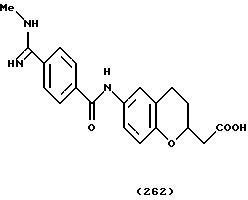 К 207 мг (0,48 ммоль) сложного эфира из примера 57 добавлялись 4 мл этанола, 0,5 мл 2N водного гидроксида натрия и три капли воды, и смесь перемешивалась в течение ночи при комнатной температуре. Формировался осадок. Он доводился до pH=5 с помощью 2N уксусной кислоты, и объявленное соединение фильтровалось с помощью всасывания, промывалось последовательно водой и этанолом, и обезвоживалось в вакууме. Выход: 0,12 г (68%) бледно-желтого порошка, tпл 275-276oC (dec). Пример 59 Получение этил-рац-(6-(N-(4-((бензойл)амино)иминометил)бензойл)амино)- 3,4-дигидро-2H-1-бензопиран-2-ил)ацетата, соединение представлено формулой (263): 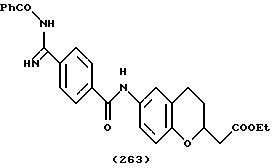 Раствор 0,42 г (1,0 ммоль) сложного эфира из примера 47, 0,22 г триэтиламина и 20 мг 4-диметиламинопиридина в 20 мл безводного дихлорметана охлаждался до -20oC, и по каплям при этой температуре добавлялся раствор 0,15 г (1,1 ммоль) бензойлхлорида в 2 мл дихлорметана. Смесь становилась прозрачной при медленном нагревании до комнатной температуры и перемешивалась в течение 3 ч. После добавления воды смесь экстрагировалась с помощью этилацетата. Органический слой обезвоживался на сульфате натрия и концентрировался при пониженном давлении. Объявленный бензоат получался путем хроматографии на силикагеле с дихлорметаном/этанолом 96:4 в виде масла. Кристаллический образец для аналитических и биологических тестов получался путем перемешивания с эфиром. Выход: 0,21 г (43%) бежевого порошка, tпл 149-150oC (dec). Пример 60 Получение рац-9-(6-аминоиминометил-2-метил-2H-1-бензопиран-2- ил)нонановой кислоты, соединение представлено формулой (264) 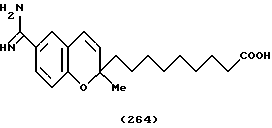 Шаг A: Получение этил-рац-9-(6-циано-3,4-дигидро-2-метил-4- оксо-2H-1-бензопиран-2-ил)нонаноата, промежуточного соединения, представленного формулой (265): 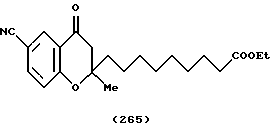 5-циано-2-гидроксиацетофенон получался путем переработки 4-ацетоксибензонитрила (Arch. Pharm. 1977, 310, 119, рассмотрение в которой включено сюда посредством ссылки), а этил-10-оксоундеканоата – путем PdCl2-катализаторного оксидирования этил-10-ундекеноата (J.Organomet. Chem. 1987, 334, C5, рассмотрение в которой включено сюда посредством ссылки). 32,4 г (201 ммоль) ацетофенона, 35,0 г (153,3 ммоль) сложного эфира и 7 мл пирролидина растворялись в 160 мл толуола. После нахождения в течение 1 ч при комнатной температуре раствор нагревался с азеотропным удалением воды в течение 8 ч. Растворитель удалялся в вакууме, а остаток растворялся в дихлорметане. Он промывался водой, и водный слой экстрагировался с помощью дихлорметана. Объединенные органические слои обезвоживались на сульфате натрия и концентрировались при пониженном давлении. Объявленный хроманон получался путем хроматографии на силикагеле с дихлорметаном. Выход: 12,6 г (22%) масла. Шаг B: Получение этил-9-(6-циано-3,4-дигидро-4-гидрокси-2-метил- 2H-1-бензопиран-2-ил)нонаноата, промежуточного соединения, представленного формулой (266): 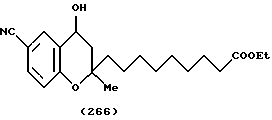 12,6 г (33,9 ммоль) хроманона из шага A растворялось в 300 мл безводного этанола. 2,6 г (68,7 ммоль) борного гидрата натрия добавлялись маленькими порциями, а температура поддерживалась ниже 25oC. Раствор перемешивался в течение ночи, концентрировался в вакууме, гидролизировался с помощью льда и водного хлороводорода, и экстрагировался три раза с помощью дихлорметана. Объединенные органические слои обезвоживались на сульфате натрия и концентрировались при пониженном давлении. Объявленный бензопиран получался путем хроматографии на силикагеле с дихлорметаном. Выход: 11,2 г (88%) масла. Шаг C: Получение этил-рац-9-(6-циано-2-метил-2H-1-бензопиран-2-ил)нонаноата, промежуточного соединения, представленного формулой (267): 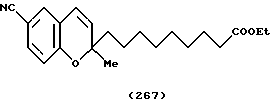 11,2 г (30,0 ммоль) соединения из шага B растворялось в 200 мл толуола. Добавлялось каталитическое количество p-толуолсульфокислоты, и раствор нагревался с азеотропным удалением воды в течение 7 ч. Смесь промывалась насыщенным водным раствором бикарбоната натрия, обезвоживалась на сульфате натрия и концентрировалась при пониженном давлении. Объявленный бензопиран очищался путем хроматографии на силикагеле с дихлорметаном. Выход: 3,0 г (28%) масла. Шаг D: Получение этил-рац-9-(6-аминоиминометил-2-метил-2H-1- бензопиран-2-ил)нонаноата гидрохлорида, промежуточного соединения, представленного формулой (268): 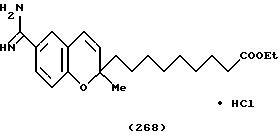 1,5 г (4,2 ммоль) хромина из предыдущего шага растворялось в 50 мл безводного этанола. Раствор насыщался хлороводородом и перемешивался в течение ночи при комнатной температуре. Растворитель удалялся в вакууме, а остаток обрабатывался насыщенным раствором аммиака в этаноле. Он перемешивался в течение ночи и концентрировался до безводного состояния при пониженном давлении, давая чистый объявленный амидин. Выход: 1,4 г (81%) масла. Шаг E: Получение рац-9-(6-аминоиминометил-2-метил-2H-1-бензопиран-2- ил)нонановой кислоты. 7 мл 2N водного гидроксида натрия и 5 мл ацетонитрила были добавлены к 0,6 г (1,47 ммоль) сложного эфира из предыдущего шага, и смесь нагревалась в течение 30 мин в паровой бане. Смесь доводилась до pH=7 с помощью 2N уксусной кислоты, образовывался осадок. Он фильтровался с помощью всасывания, последовательно промывался ледяной водой и ацетоном и обезвоживался в вакууме. Выход: 0,4 г (79%) белого порошка, tпл 223-225oC (dec). Пример 61 Получение рац-(3,4-дигидро-6-(2-(пиперидин-4-ил)этокси)-2H-1- бензопиран-2-ил)уксусного кислого трифторацетата, соединение представлено формулой (269): 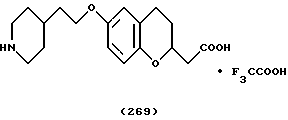 Шаг A: Получение этил-рац-(6-(2-(1-(трет-бутоксикарбонил)пиперидин-4- ил)этокси)-3,4-дигидро-2H-1-бензопиран-2-ил)ацетата, промежуточного соединения, представленного формулой (270): 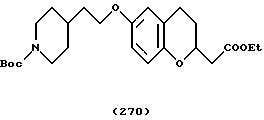 К раствору 2,2 г (7,5 ммоль) 4-(2-бромэтил)-1-(трет-бутоксикарбонил)пиперидина (полученного бромированием 2-(1-(трет-бутоксикарбонил)пиперидин-4-ил)этанола по европейской патентной заявке EP 478328) в 50 мл безводного ацетонитрила добавлялись 2,84 г (8,72 ммоль) безводного карбоната цезия. После добавления по каплям раствора 3,05 г (12,9 ммоль) этил-рац-(3,4-дигидро-6-гидрокси-2H-1-бензопиран-2-ил) ацетата (полученного по европейской патентной заявке EP 129906) в 20 мл безводного ацетонитрила раствор перемешивался в течение ночи при комнатной температуре. Добавлялся еще 1 г карбоната цезия, и перемешивание продолжалось 2 ч при 70oC и в течение ночи при комнатной температуре. Смесь фильтровалась сквозь Селит, который промывался ацетонитрилом и ацетоном. Фильтрат концентрировался при пониженном давлении, и остаток последовательно перемешивался с гексаном/этилацетатом 4:1 и дважды с этилацетатом. Объединенные растворы, отделенные от нерастворимого материала, концентрировались в вакууме, и соединение (270) получалось путем хроматографии на силикагеле с гексаном, затем гексаном/этилцетатом 4: 1. Выход: 1,9 г (86%) желтого масла, которое со временем отвердевало. Шаг B: Получение рац-(6-(2-(1-(трет-бутоксикарбонил)пиперидин-4- ил)этокси)-3,4-дигидро-2H-1-бензопиран-2-ил)уксусной кислоты, промежуточного соединения, представленное формулой (271): 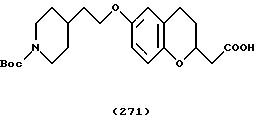 К раствору 1,5 г (3,35 ммоль) сложного эфира из предыдущего шага в 100 мл этанола добавлялось 6,8 мл 2N водного гидроксида натрия, и смесь перемешивалась в течение ночи при комнатной температуре. Смесь доводилась до pH= 5 с помощью разбавленной уксусной кислоты и концентрировалась при пониженном давлении. Объявленная кислота получалась путем хроматографии на силикагеле с дихлорметаном с последующим добавление до 3% этанола. Выход: 0,54 г (38%) бежевого масла. Шаг C: Получение рац-(3,4-дигидро-6-(2-(пиперидин-4-ил)этокси)- 2H-1-бензопиран-2-ил)уксусного кислого трифторацетата. 117 мг (0,28 ммоль) защищенного амидина из шага B перемешивалось в 2,3 мл трифторуксусной кислоты при комнатной температуре в течение 30 мин, и смесь концентрировалась до безводного состояния в вакууме, оставляя чистое объявленное соединение. Выход: 115 мг (95%) коричневой смолы. Пример 62 Получение этил-рац-(6-(N-(4-аминоиминометил-2-хлорбензойл)амино)- 1-оксо-1,2,3,4-тетрагидронафталин-2-ил)ацетата гидрохлорида, соединение представлено формулой (278): 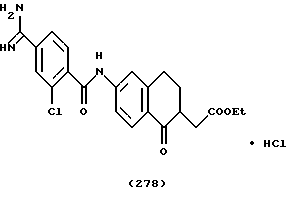 Шаг A: Получение этил-рац-(6-(N-(2-хлор-4-цианбензойл)-1- оксо-1,2,3,4-тетрагидронафталин-2-ил)ацетата гидрохлорида, соединение представлено формулой (280): 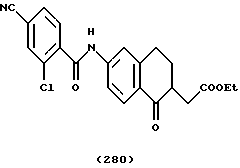 5,7 г (31,4 ммоль) 2-хлор-4-цианбензойной кислоты, которая получалась путем оксидирования 3-хлор-4-метил-бензонитрила (по Chem. Ber. 1936, 69, 537, рассмотрение в которой включено сюда посредством ссылки), растворялось в 300 мл толуола, и добавлялось 10,25 г (86,2 ммоль) хлористого тионила. После 4 ч орошения добавлялось такое же количество хлористого тионила, и нагревание продолжалось в течение ночи. Растворитель удалялся при пониженном давлении, остаток растворялся в 100 мл толуола и снова концентрировался в вакууме, оставляя неочищенный кислый хлорид в виде желтого масла. Выход: 4,1 г (65%). Раствор 0,98 г (3,96 ммоль) тетралона 98 и 3,3 мл безводного пиридина в 33 мл безводной THF охлаждался льдом, и по каплям добавлялся раствор 1,0 г (5,0 ммоль) неочищенного 2-хлор-4-цианбензойл хлорида в 33 мл безводной THF. Смесь медленно нагревалась до комнатной температуры, перемешивалась в течение ночи, и заливалась в ледяную воду. Смесь экстрагировалась с помощью этилацетата, и органический слой промывался насыщенным водным бикарбонатом натрия, а затем рассолом, и обезвоживался на сульфате натрия. Продукт концентрировался при пониженном давлении, а остаток перемешивался с гексаном, давая кристаллический нитрил (280), который фильтровался и обезвоживался в вакууме при 50oС. Выход: 1,5 г (92%) бежевого кристаллического сухого остатка, tпл 173-175oC. Шаг B: Получение этил-рац-(6-(N-(2-хлор-4-(этоксикарбонимидойл) бензойл)амино)-1-оксо-1,2,3,4-тетрагидронафталин-2-ил)ацетата гидрохлорида, промежуточного соединения, представленного формулой (281): 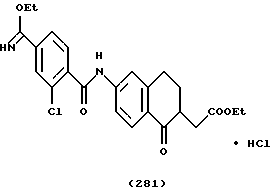 1,35 (3,3 ммоль) нитрила из шага A суспендировалось в 50 мл безводного этанола, а температура смеси поддерживалась ниже 5oC, в то время как смесь перемешивалась и насыщалась хлороводородом (примерно 6 ч). После ночи отстаивания еще в течение 3 ч пропускался поток при пониженном давлении, и остаток перемешивался с гексаном, давая кристаллическое объявленное соединение, которое фильтровалось и обезвоживалось в вакууме. Выход: 1,3 г (80%) бежевого кристаллического сухого остатка, tпл 234-235oC. Шаг C: Получение этил-рац-(6-(N-(4-аминоиминометил-2-хлорбензойл)амино)-1-оксо-1,2,3,4- тетрагидронафталин-2-ил)ацетата гидрохлорида. 1,2 г (2,43 ммоль) соединения из предыдущего шага вводилось в 50 мл охлажденного насыщенного раствора аммиака в этаноле. После перемешивания в течение ночи при комнатной температуре добавлялось еще 20 мл этанолового раствора, и перемешивание продолжалось еще столько же времени. Твердые частицы удалялись путем фильтрации, а фильтрат концентрировался при пониженном давлении. Остаток перемешивался с гексаном, давая неочищенное кристаллическое объявленное соединение, которое очищалось путем хроматографии на силикагеле с хлороформом/метанолом 8:1. Выход: 0,2 г (18%) бежевого кристаллического порошка, tпл 225-226oC. Пример 63 Получение рац-(6-(N-(4-аминоиминометил-2-хлорбензойл)амино)-1- оксо-1,2,3,4-тетрагидронафталин-2-ил)уксусной кислоты, соединение представлено формулой (282): 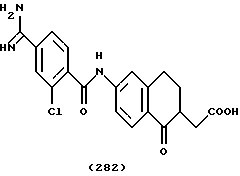 0,2 г (0,43 ммоль) сложного эфира из примера 62 в смеси с 5 мл этанола и 0,7 мл 2N водного гидроксида натрия перемешивались при комнатной температуре в течение ночи. Реакционная смесь доводилась до pH=4 с помощью 2N уксусной кислоты, осадок фильтровался с помощью всасывания, промывался последовательно водой и ацетоном и обезвоживался в вакууме. Выход: 0,13 г (75%) белого порошка, tпл 240-242oC. Пример 64 Получение рац-(6-(N-(4-аминоиминометил)амино)бензойл)амино)-3,4- дигидро-2H-1-бензопиран-2-ил)ацетата, соединение представлено формулой (283): 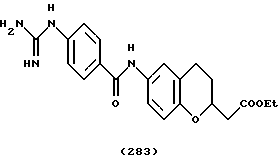 Гидрохлорид 4-гуанидинбензойной кислоты получался из 4-аминобензойной кислоты по процедуре, описанной в литературе (Recl. Trav. Chim. Pays-Bas, 1953, 72, 643, рассмотрение в которой включено сюда посредством ссылки). Он нагревался в хлористом тиониле в течение 1 ч, а затем концентрировался до безводного состояния, давая неочищенный гидрохлорид бензойлхлорида. 0,62 г (1,85 ммоль) соединения из примера 47, шаг D, перемешивалось в течение 1 ч при комнатной температуре в 2 мл трифторуксусной кислоты, и смесь концентрировалась в вакууме до безводного состояния. Смесь обрабатывалась насыщенным водным раствором бикарбоната натрия и экстрагировалась с помощью этилацетата. Органический слой обезвоживался на сульфате натрия и концентрировался при пониженной температуре, и маслянистый остаток растворялся в 20 мл безводного пиридина, что сопровождалось добавлением 0,44 г (1,88 ммоль) неочищенного гидрохлорида 4-гуанидинбензойлхлорида. После перемешивания в течение ночи при комнатной температуре смесь заливалась в 100 мл воды, формировался осадок, который собирался путем фильтрации, промывался водой и обезвоживался в вакууме. Объявленное соединение очищалось путем хроматографии на силикагеле с этанолом/концентрированным водным аммиакам 85:15. Выход: 0,15 г (20%) коричневого аморфного сухого остатка. Пример 65 Получение рац-(6-(N-(4-аминоиминометил)амино)бензойл)амино)-3,4- дигидро-2H-1-бензопиран-2-ил)уксусной кислоты, соединение представлено формулой (284): 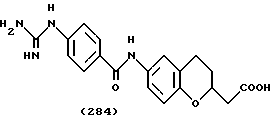 0,08 г (0,2 ммоль) сложного эфира из примера 64 растворялось в 30 мл этанола, после чего добавлялось 0,4 мл 2N водного раствора гидроксида натрия. Смесь перемешивалась при комнатной температуре в течение двух дней, и концентрировалась до безводного состояния при пониженном давлении. Остаток растворялся в воде, и раствор нейтрализовался уксусной кислотой, в это время объявленная кислота образовывалась в виде осадка. Он фильтровался с помощью всасывания, промывался водой и обезвоживался в вакууме. Выход: 0,045 г (61%) бежевого кристаллического сухого остатка, tпл 258-260oC. Пример 66 {BL-43} Этил-рац-3-(6-(4-аминоиминометил)фенилметокси)-3,4-дигидро-2- метил-4-оксо-2H-1-бензопиран-2-ил)пропаноата гидрохлорид, соединение представлено формулой (299): 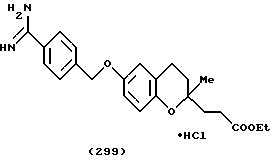 Пример 67 Рац-3-(6-(4-аминоиминометил)фенилметокси)-3,4-дигидро-2-метил-4- оксо-2H-1-бензопиран-2-ил)пропановой кислоты гидрохлорид, соединение представлено формулой (300): 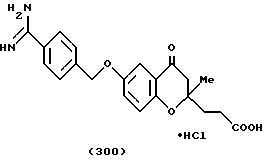 Пример 68 Получение этил-рац-(3,4-дигидро-6-(N-(4-((этоксикарбониламино) иминометил)бензойл)амино)-2H-1-бензопиран-2-ил)ацетата, соединение представлено формулой (303): 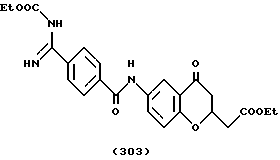 418 мг (1 ммоль) бензопирана из примера 47, 0,3 мл триэтиламина и 20 мг 4-диметиламинопиридина растворялись в 20 мл безводного дихлорметана, и раствор охлаждался до -20oC. При этой температуре по каплям добавлялось 119 мг (1,1 ммоль) этилхлорформиата в 2 мл безводного дихлорметана, и по прошествии 30 мин раствор нагревался до комнатной температуры и перемешивался еще в течение 2 ч. Смесь заливалась в холодную воду, и органический слой обезвоживался на сульфате натрия и концентрировался при пониженном давлении. Оставшееся кристаллическое объявленное соединение перемешивалось с эфиром, фильтровалось и обезвоживалось в вакууме. Выход: 360 мг (79%) бледно-желтого сухого остатка, tпл 163-165oC. Пример 69 Получение этил-рац-(6-(N-(4-аминоиминометил)-2-фторбензойл)амино)- 3,4-дигидро-2H-1-бензопиран-2-ил)ацетата гидрохлорида, соединение представлено формулой (304): 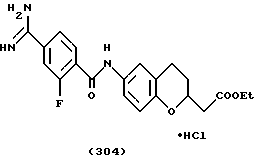 Шаг A: Получение этил-рац-(6-(N-(4-циано-2-фторбензойл)амино-3,4- дигидро-2H-1-бензопиран-2-ил)ацетата, промежуточного соединения, представленного формулой (307): 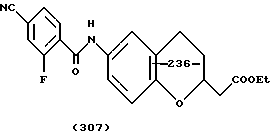 5,0 г (30,3 ммоль) кислоты 119 нагревалось в течение 1 ч при орошении в 50 мл хлористого тионила, содержащего одну каплю DMF. Реакционная смесь концентрировалась при пониженном давлении, и неочищенный кислый хлорид растворялся в 130 мл безводной THF. Этот раствор добавлялся по каплям при 0oC к неочищенному этил(6-амино-3,4-дигидро-2H-1-бензопиран-2-ил)ацетату в 200 мл безводной THF и 20 мл безводного пиридина, который был получен в соответствии с примером 47, шаг E, из 10,0 г (29,8 ммоль) защищенного производного при помощи 20 мл трифторуксусной кислоты. После перемешивания в течение ночи при комнатной температуре смесь заливалась в ледяную воду, содержащую бикарбонат натрия. Осадок объявленного нитрила фильтровался с помощью всасывания, нагревался несколько минут в этаноле, снова фильтровался после охлаждения, и кристаллы промывались гексаном и обезвоживались в вакууме. Выход: 8,6 г (75%) бежевого аморфного сухого остатка, tпл 154-155oC. Шаг B: Получение этил-рац-(6-(N-(4-аминоиминометил)-2- фторбензойл)амино)-3,4-дигидро-2H-1-бензопиран-2-ил)ацетата гидрохлорида. 4,5 г (11,8 ммоль) нитрила из предыдущего шага в 150 мл безводного этанола охлаждалось до 0oС и насыщалось газообразным хлороводородом. После перемешивания в течение ночи при комнатной температуре смесь концентрировалась при пониженном давлении. Кристаллический остаток обрабатывался 150 мл насыщенного этанолового раствора аммиака и снова перемешивался в течение ночи. Неочищенное объявленное вещество получалось после концентрации в вакууме, рекристаллизации из этанола, сопровождаемой кристаллизацией из смеси этанола/воды/эфира. Выход: 3,0 г (58%) бледно-желтого порошка, tпл 198-200oC. Пример 70 Получение рац-(6-(N-(4-аминоиминометил)-2-фторбензойл)амино)-3,4- дигидро-2H-1-бензопиран-2-ил)уксусной кислоты, соединение представлено формулой (308): 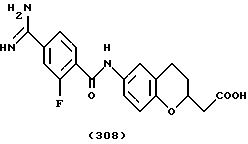 1,2 г (2,75 ммоль) сложного эфира из примера 69 перемешивалось в течение ночи при комнатной температуре в смеси 20 мл этанола и 5 мл 2N водного раствора гидроксида натрия. Смесь доводилась до pH=4 с помощью 2N уксусной кислоты, и осадок объявленного соединения фильтровался при помощи всасывания, последовательно промывался водой и ацетоном, и обезвоживался в вакууме при 50oC. Выход: 0,84 г (82%) бледно-желтого порошка, tпл 250oC. Пример 71 Получение этил-рац-(3,4-дигидро-6-(N-(4-этиламино)иминометил) бензойл)амино)-2H-1-бензопиран-2-ил)ацетата гидрохлорида, соединение представлено формулой (309): 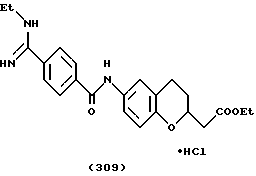 1,8 г (4,0 ммоль) неочищенного промежуточного соединения из примера 57, шаг A, растворялось в 50 мл этанола. Раствор охлаждался льдом, и добавлялось 2 мл 50%-ного этанолового раствора этиламина. Раствор перемешивался в течение ночи при комнатной температуре, а растворитель удалялся при пониженном давлении. Остаток перемешивался с небольшим количеством этанола, давая чистое кристаллическое объявленное соединение, которое фильтровалось и обезвоживалось в вакууме. Выход: 1,6 г (89%) бледно-желтого порошка, tпл 290-291oC. Пример 72 Получение рац-(3,4-дигидро-6-(N-(4-этиламино)иминометил)бензойл) амино)-2H-1-бензопиран-2-ил)уксусной кислоты, соединение представлено формулой (310): 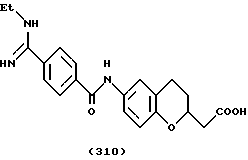 1,8 г (4,0 ммоль) неочищенного соединения из примера 57, шаг A, растворялось в 50 мл этанола. Раствор охлаждался льдом, добавлялось 2 мл 50%-ного этанолового раствора диметиламина. Раствор перемешивался в течение ночи при комнатной температуре, растворитель удалялся при пониженном давлении. Остаток перемешивался с небольшим количеством этанола, давая чистое кристаллическое объявленное соединение, которое фильтровалось и обезвоживалось в вакууме. Выход: 1,35 г (75%) белого порошка, tпл 248oC. Пример 73 Получение этил-рац-(3,4-дигидро-6-(N-(4-((диметиламино)иминометил) бензойл)амино)-2H-1-бензопиран-2-ил)ацетата гидрохлорида, соединение представлено формулой (311): 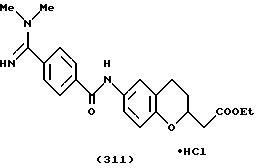 1,8 г (4,0 ммоль) неочищенного соединения из примера 57, шаг A, растворялось в 50 мл этанола. Раствор охлаждался льдом, добавлялось 2 мл 50%-ного этанолового раствора диметиламина. Раствор перемешивался в течение ночи при комнатной температуре, растворитель удалялся при пониженном давлении. Остаток перемешивался с небольшим количеством этанола, давая чистое кристаллическое объявленное соединение, которое фильтровалось и обезвоживалось в вакууме. Выход: 1,35 г (75%) белого порошка, tпл 248oC. Пример 74 Получение этил-рац-(3,4-дигидро-6-(N-(4-((метоксикарбониламина) иминометил)бензойл)амино)-2H-1-бензопиран-2-ил)ацетата, соединение представлено формулой (312): 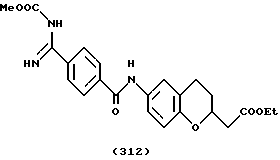 К 417 мг (1,0 ммоль) амидина из примера 47, 0,3 мл триэтиламина и 20 мг 4-диметиламинопиридина в 40 мл дихлорметана было добавлено при 0oC 104 мг (1,1 ммоль) метилхлорформиата, и смесь перемешивалась в течение ночи при комнатной температуре. Образовывался белый осадок, который собирался путем фильтрации и хроматографировался на силикагеле с дихлорметаном/этанолом 9:1, давая чистый объявленный карбомат. Выход: 0,21 г (48%) белого порошка, tпл 204-206oC. Пример 75 Получение этил-рац-(6-(N-(4-(аминоиминометил)-2-хлорбензойл)амино)- 3,4-дигидро-2H-1-бензопиран-2-ил)ацетата гидрохлорида, соединение представлено формулой (313): 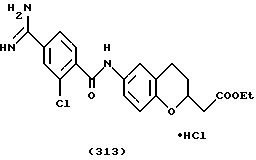 Шаг A: Получение этил-рац-(6-(N-(2-хлор-4-цианбензойл)амино)-3,4- дигидро-2H-1-бензопиран-2-ил)ацетата, промежуточного соединения, представленного формулой (314): 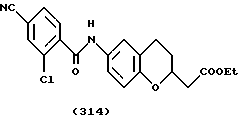 3,0 г (8,94 ммоль) бензопирана из примера 47, шаг D, перемешивалось с 6 мл трифторуксусной кислоты в течение 2 ч при комнатной температуре. Смесь обрабатывалась насыщенным водным раствором бикарбоната натрия и экстрагировалась этилацетатом. Органический слой обезвоживался на сульфате натрия, а растворитель удалялся в вакууме, давая неочищенный незащищенный аминобензопиран, который растворялся в смеси 50 мл безводной THF и 8 мл безводного пиридина. 50 мл раствора неочищенного 2-хлор-4-цианбензойлхлорида в безводной THF, который получался из 1,1 г (6,1 ммоль) 2-хлор-4-цианбензойной кислоты (Chem. Ber. 1936, 69, 537, рассмотрение в которой включено сюда посредством ссылки) в соответствии с примером 62, шаг A, добавлялось по каплям при 0oC. После перемешивания в течение ночи при комнатной температуре смесь заливалась в ледяную воду, содержащую бикарбонат натрия. Смесь экстрагировалась с помощью этилацетата, и органический слой последовательно промывался водным раствором сульфата меди (II) и рассолом, обезвоживался на сульфате натрия и концентрировался при пониженном давлении. Нитрил (314) получался из остатка путем хроматографии на силикагеле с хлороформом/метанолом 96:4. Он кристаллизовался из чистых фракций после перемешивания с небольшим количеством этанола. Выход: 1,1 г (46%) бежевых кристаллов, tпл 152-154oC. Шаг B: Получение этил-рац-(6-(N-(4-(аминоиминометил)-2- хлорбензойл)амино)-3,4-дигидро-2H-1-бензопиран-2-ил)ацетата гидрохлорида. 1,1 г (2,76 ммоль) нитрила из предыдущего шага растворялось в 25 мл этанола, и раствор охлаждался до 0oC и насыщался газообразным хлороводородом. Он перемешивался в течение ночи при комнатной температуре с последующей концентрацией при пониженном давлении. Остаток обрабатывался 20 мл насыщенного раствора аммиака в этаноле и снова перемешивался в течение ночи. После удаления растворителя в вакууме оставшееся объявленное соединение перекристаллизовывалось из этанола/воды/эфира. Выход: 0,9 г (72%) бледно-желтой пудры, tпл 225-226oC. Пример 76 Получение рац-(6-(N-(4-(аминоиминометил)-2-хлорбензойл)амино)-3,4- дигидро-2H-1-бензопиран-2-ил)уксусной кислоты, соединение представлено формулой (314): 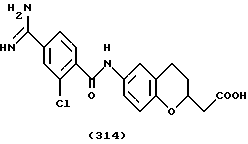 0,3 г (0,66 ммоль) сложного эфира из примера 75 перемешивалось в течение ночи при комнатной температуре со смесью 5 мл этанола и 0,5 мл 2N водного раствора гидроксида натрия. Смесь доводилась до pH=5 с помощью 2N уксусной кислоты. Осадок из объявленного соединения фильтровался с помощью всасывания, последовательно промывался водой и ацетоном и обезвоживался в вакууме. Выход: 0,19 г (74%) белого порошка, tпл 269-270oC (dec). Пример 77 Получение этил-рац-(3,4-дигидро-6-(N-(3-пиперидин-4- ил)пропенойл)амино)-2H-1-бензопиран-2-ил)ацетата трифторацетата, соединение представлено формулой (327): 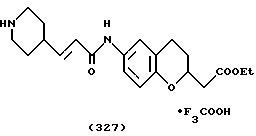 Шаг A: Получение этил-рац-(6-(N-(3-(1-(трет-бутоксикарбонил)пиперидин- 4-ил)пропенойл)амино)-3,4-дигидро-2H-1-бензопиран-2-ил)ацетата, промежуточного соединения, представленного формулой (329): 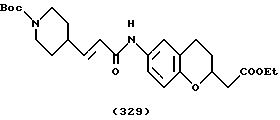 0,85 г (3,33 ммоль) кислоты 386 растворялось в смеси 23 мл безводного дихлорметана и 0,26 мл безводной DMF. Раствор выдерживался при температуре между -10oC и 0oC, в это время медленно добавлялось 0,46 г (3,6 ммоль) оксалилхлорида. После перемешивания в течение 40 мин при этой температуре смесь по каплям добавлялась к раствору 0,5 мл триэтиламина и неочищенного этил(6-амино-3,4-дигидро-2H-1-бензопиран-2-ил)ацетата (полученного с помощью трифторуксусной кислоты из 1,5 г (4,5 ммоль) Boc-защищенного амидина, как описано в примере 47) в 35 мл безводного дихлорметана. После перемешивания в течение 90 мин при этой температуре смесь заливалась в ледяную воду, и водный слой экстрагировался с помощью дихлорметана. Объединенные органические слои промывались водой, обезвоживались на сульфате натрия и концентрировались при пониженном давлении. Объявленное соединение очищалось путем хроматографии на силикагеле с дихлорметаном/этанолом 80:1. Выход: 0,81 г (51%) масла. Шаг B: Получение этил-рац-(3,4-дигидро-6-(N-(3-(пиперидин-4- ил)пропенойл)амино)-2H-1-бензопиран-2-ил)ацетата трифторацетата. 0,81 г (1,71 ммоль) защищенного пиперидина из предыдущего шага перемешивалось в течение 2 ч при комнатной температуре в 6 мл трифтороуксусной кислоты. Смесь заливалась в ледяную воду, доводилась до pH=7 с помощью бикарбоната натрия и экстрагировалась с помощью дихлорметана. Органический слой обезвоживался на сульфате натрия и концентрировался при пониженном давлении, а остаток перемешивался со смесью гексана и эфира, что давало кристаллическое объявленное вещество, которое собиралось путем фильтрации и обезвоживалось при 50oC в вакууме. Выход: 0,18 г (22%) красноватых кристаллов, tпл 72-73oC. Пример 78 Получение рац-(3,4-дигидро-6-(N-(3-(пиперидин-4-ил)пропенойл)амино)- 2H-1-бензопиран-2-ил)уксусной кислоты, соединение представлено формулой (330): 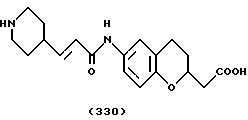 0,1 г (0,2 ммоль) сложного эфира из примера 77 перемешивалось в течение ночи при комнатной температуре в смеси 2,5 мл этанола и 0,35 мл 2N водного раствора гидроксида натрия. Реакционная смесь охлаждалась льдом и доводилась до pH= 5,5 с помощью 2N уксусной кислоты. Осадок из объявленной кислоты фильтровался с помощью всасывания, промывался небольшим количеством холодной воды и обезвоживался при 50oC в вакууме. Выход: 42 мг (59%) бежевых кристаллов, tпл 175-178oC. Пример 79 Получение этил-рац-(6-(N-(4-(аминоиминометил)бензойл)амино)-3,4- дигидро-4, -диметил-2H-1-бензопиран-2-ил)ацетата гидрохлорида, соединение представлено формулой (331): 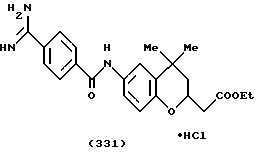 Шаг A: Получение трет-бутил(3,4-дигидро-4,4-диметил-2-оксо-2H-1- бензопиран-6-ил)карбомата, промежуточного вещества, представленного формулой (332): 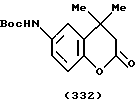 К суспензии 1,5 г 10% Pd/C в 300 мл этанола добавлялись в атмосфере аргона 29,2 г (132,0 ммоль) 3,4-дигидро-4,4-диметил-6-нитро-2-оксо-2H-1-бензопирана (полученного путем нитрования 3,4-дигидро-4,4-диметил-2-оксо-2H-1-бензопирана в соответствии с J. Am. Chem. Soc., 1970, 92, 4377, рассмотрение в которой включено сюда посредством ссылки) и 21,4 г (339,4 ммоль) формиата аммония. В течение приблизительно 1 ч температура повышалась до 50oC, что сопровождалось выделением газа, и смесь нагревалась еще 3 ч при 80oC. Катализатор фильтровался через Селит, который промывался 500 мл горячего этанола. Объединенные фильтраты концентрировались при пониженном давлении до объема в 100 мл, а сформировавшийся осадок фильтровался с помощью всасывания. Вторая порция неочищенного 6-амино-3,4-дигидро-4,4-диметил-2-оксо-2H-1-бензопирана получалась после дальнейшей концентрации. Обе порции объединялись и растворялись в 80 мл THF, после чего добавлялось 80 мл воды, 25,3 г (183 ммоль) карбоната калия и 29,9 г (133,8 ммоль) Boc2O и 2,5 г карбоната калия, а перемешивание продолжалось в течение ночи. Смесь заливалась в 300 мл воды и экстрагировалась с помощью этилацетата, и органический слой обезвоживался на сульфате натрия и концентрировался при пониженном давлении. Первая порция объявленного соединения кристаллизовывалась из маслянистого остатка с помощью диизопропилового эфира. Дополнительное количество получалось из концентрированной исходной жидкости путем хроматографии на силикагеле с толуолом и кристаллизации из маслянистых чистых фракций с помощью диизопропилового эфира. Общий выход: 31,6 г (82%) бесцветных кристаллов, tпл 106-108oC. Шаг B: Получение трет-бутил рац-(3,4-дигидро-4,4-диметил-2- гидрокси-2H-1-бензопиран-6-ил)карбoмата, промежуточного соединения, представленного формулой (333): 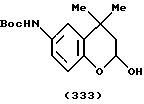 31,6 г (108,5 ммоль) бензопирана из предыдущего шага растворялось в 450 мл безводной THF и охлаждалось до -70oC в атмосфере аргона. При этой температуре в течение 1 ч по каплям добавлялось 133 мл 25%-ного раствора DIBAH в толуоле, и смесь перемешивалась еще 2 ч. Смесь осторожно гасилась 35 мл метанола и нагревалась до комнатной температуры, затем заливалась в 1000 мл насыщенного водного раствора хлорида аммония. После энергичного перемешивания верхний органический слой отвердевал до желеобразного состояния, желе отделялось и перемешивалось с 1000 мл этилацетата. Затем смесь фильтровалась через Селит и промывалась 500 мл этилацетата. Объединенные фильтраты обезвоживались на сульфате натрия и концентрировались в вакууме, давая коричневую смолу, из которой получался чистый объявленный ацетал путем хроматографии на силикагеле с толуолом/ацетоном 95:5. Выход: 25,9 г (81%) желтой смолы. Шаг C: Получение этил-рац-(6-(N-трет-бутоксикарбониламино)-3,4- дигидро-4,4-диметил-2H-1-бензопиран-2-ил)ацетата, промежуточного соединения, представленного формулой (334): 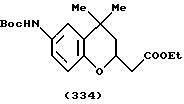 К раствору 25,9 г (88,3 ммоль) соединения из шага B в 150 мл толуола добавлялось 32,4 г (93,0 ммоль) этоксикарбонилметиленатрифенилфосфорана, а затем 0,6 г (15 ммоль) 60% гидрида натрия маленькими порциями. Смесь нагревалась до 120oC в течение 3 ч, охлаждалась до комнатной температуры, и твердые частицы удалялись путем фильтрации. Фильтрат концентрировался при пониженном давлении, и соединение (334) получалось путем хроматографии на силикагеле с гексаном/этилацетатом 4:1 до 1:1. Выход: 6,9 г (22%) бесцветных кристаллов, tпл 116-119oC. Шаг D: Получение этил-рац-(6-(N-(4-цианбензойл)амино)-3,4- дигидро-4,4-диметил-2H-1-бензопиран-2-ил)ацетата, промежуточного соединения, представленного формулой (335): 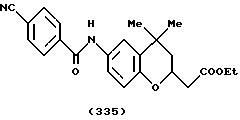 2,18 г (6,0 ммоль) соединения из предыдущего шага депротектировалось путем перемешивания в 10 мл трифторуксусной кислоты, как описано в примере 47, шаг E, и коричневое масло из неочищенного 6-аминобензопирана растворялось в 40 мл безводного пиридина. После добавления 0,99 г (6,0 ммоль) 4-цианбензойлхлорида смесь перемешивалась при комнатной температуре в течение ночи. Она концентрировалась в вакууме до безводного состояния и трижды растворялась в толуоле с последующей концентрацией при пониженном давлении для удаления оставшегося пиридина. Объявленный нитрил кристаллизовывался из коричневого остатка после перемешивания с небольшим количеством этанола. Он фильтровался с помощью всасывания, промывался холодным этанолом и обезвоживался в вакууме. Выход: 1,7 г (72%) белого порошка, tпл 163-165oC. Шаг E: Получение этил-рац-(6-(N-(4-аминоиминометил)бензойл)амино)- 3,4-дигидро-4,4-диметил-2H-1-бензопиран-2-ил)ацетата гидрохлорида. Суспензия 1,7 г (4,33 ммоль) нитрила из шага D в 100 мл безводного этанола охлаждалась льдом и насыщалась газообразным хлороводородом. После отстаивания в течение ночи при комнатной температуре она концентрировалась до безводного состояния при пониженном давлении, и остаток растворялся в 50 мл 10%-ного этанолового раствора аммиака. Смесь перемешивалась при комнатной температуре в течение 3 дней до завершения реакции. Растворитель удалялся в вакууме, а смолистый остаток растворялся в толуоле/ацетоне 7:3. Первая порция объявленного амидина кристаллизовывалась из раствора, а другая порция получалась путем хроматографии на силикагеле с помощью той же самой смеси растворителей. Общий выход: 1,6 г (83%) желтого кристаллического сухого остатка, tпл 122-124oC. Пример 80 Получение рац-(6-(N-(4-аминоиминометил)бензойл)амино)-3,4- дигидро-4,4-диметил-2H-1-бензопиран-2-ил)уксусной кислоты, соединение представлено формулой (336): 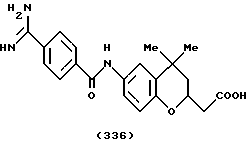 0,3 г (0,67 ммоль) сложного эфира из примера 79 суспендировалось в 100 мл этанола с последующим добавлением 1 мл водного 2N гидроксида натрия. Смесь перемешивалась при комнатной температуре в течение 4 ч. Смесь фильтровалась, фильтрат концентрировался до безводного состояния при пониженном давлении, и остаток растворялся в воде. Раствор затем нейтрализовывался разбавленной уксусной кислотой, и осадок соединения (336) фильтровался с помощью всасывания, промывался тщательно водой и обезвоживался в вакууме при 50oC. Выход: 0,2 г (78%) желтого кристаллического сухого остатка, tпл 248-250oC. Пример 81 Получение этил-рац-(3,4-дигидро-6-(N-(4-((пропиламино)иминометил) бензойл)амино)-2H-1-бензопиран-2-ил)ацетат гидрохлорида, соединение представлено формулой (337): 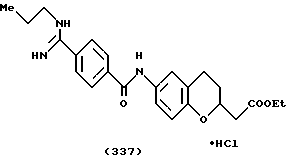 К раствору 0,8 г (1,8 ммоль) неочищенного промежуточного соединения из примера 57, шаг A, в 20 мл этанола добавлялось при 0oC 0,3 мл n-пропиламина. Смесь перемешивалась в течение ночи при комнатной температуре и концентрировалась при пониженном давлении. Остаток обрабатывался этанолом и эфиром, давая чистое кристаллическое объявленное соединение, которое собиралось путем фильтрации и обезвоживалось в вакууме. Выход: 0,63 г (76%) белого порошка, tпл 271-273oC. Пример 82 Получение рац-(3,4-дигидро-6-(N-(4-((пропиламино)иминометил) бензойл)амино)-2H-1-бензопиран-2-ил)уксусной кислоты, соединение представлено формулой (338): 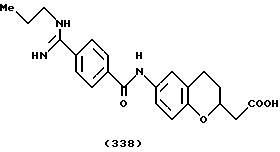 0,25 г (0,54 ммоль) сложного эфира из примера 81 перемешивалось при комнатной температуре в течение ночи с 5 мл этанола и 0,7 мл 2N водного гидроксида натрия. Смесь доводилась до pH=4 с помощью 2N уксусной кислоты, осадок кислоты (338) фильтровался с помощью всасывания, последовательно промывался водой и ацетоном и обезвоживался в вакууме. Выход: 0,16 г (74%) бледно-желтого порошка, tпл 241-242oC (dec). Пример 83 Получение рац-(6-(N-(4-(бутиламино)иминометил)бензойл)амино)-3,4- дигидро-2H-1-бензопиран-2-ил)ацетата гидрохлорида, соединение представлено формулой (339): 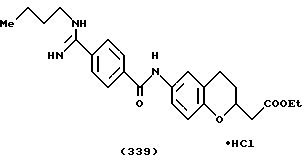 Соединение получалось из 0,8 г (1,8 ммоль) неочищенного промежуточного соединения из примера 57, шаг A, и 0,3 мл n-бутиламина, как описано в примере 81. Выход: 0,6 г (71%) бледно-желтого порошка, tпл 265-267oC. Пример 84 Получение рац-(6-(N-(4-(бутиламино)иминометил)бензойл)амино)-3,4- дигидро-2H-1-бензопиран-2-ил)уксусной кислоты, соединение представлено формулой (340): 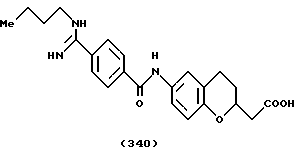 0,25 г (0,53 ммоль) сложного эфира из примера 83 гидролизовалось в объявленную кислоту, как описано в примере 82. Выход 0,13 г (60%) белого порошка, tпл 240-241oC (dec). Пример 85 Получение этил-рац-(3,4-дигидро-6-(N-(4-(пропоксикарбониламино) иминометил)бензойл)амино)-2H-1-бензопиран-2-ил)ацетата, соединение представлено формулой (341): 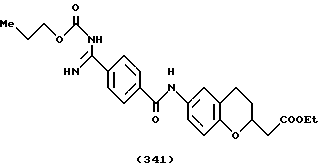 417 мг (1,0 ммоль) соединения из примера 47, 0,3 мл триэтиламина, 20 мг 4-диэтиламинопиридина и 135 мг (1,1 ммоль) пропилхлороформиата растворялись в 20 мл дихлорметана при 0oС с последующим перемешиванием в течение ночи при комнатной температуре. После добавления такого же объема воды смесь экстрагировалась с помощью этилацетата. Органический слой обезвоживался на сульфате натрия и концентрировался при пониженном давлении, давая неочищенное объявленное соединение, которое рекристаллизовывалось из дихлорметана/гексана. Выход: 0,27 г (58%) бледно-желтого аморфного сухого остатка, tпл 183-184oC. Пример 86 Получение пропил-рац-(6-(N-(4-(аминоиминометил)бензойл)амино)-3,4- дигидро-2H-1-бензопиран-2-ил)ацетата гидрохлорида, соединение представлено формулой (342): 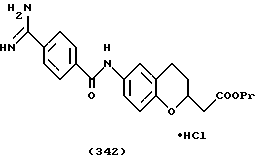 Раствор 0,5 г (1,37 ммоль) нитрила из примера 47, шаг E, в 50 мл n-пропанола охлаждался льдом и насыщался газообразным хлороводородом. После перемешивания в течение ночи при комнатной температуре смесь концентрировалась при пониженном давлении, и остаток обрабатывался 50 мл насыщенного раствора аммиака в n-пропаноле. Смесь снова перемешивалась в течение ночи при комнатной температуре, растворитель удалялся в вакууме, а оставшийся объявленный амидин перекристаллизовывался из n-пропанола. Выход: 0,36 г (61%) желтого порошка, tпл 238-240oC (dec). Пример 87 Получение метил-рац-(6-(N-(4-(аминоиминометил)бензойл)амино)-3,4- дигидро-2H-1-бензопиран-2-ил) ацетата гидрохлорида, соединение представлено формулой (343): 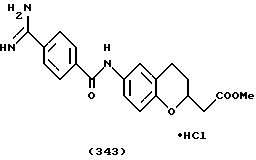 Суспензия 0,35 г (1,0 ммоль) кислоты из примера 48 в 20 мл метанола охлаждалась до 0oC и насыщалась газообразным хлороводородом. Через 2 ч перемешивания она концентрировалась при пониженном давлении, и оставшееся соединение (343) перекристаллизовывалось из метанола/эфира. Выход: 0,29 г (72%) желтых кристаллов, tпл 235-237oC. Пример 88 Получение этил-рац-(3,4-дигидро-6-(N-(2-фтор-4-((метоксикарбониламино)иминометил)бензойл)амино)-2H-1-бензопиран-2-ил)ацетата, соединение представлено формулой (349): 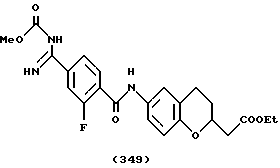 436 мг (1,0 ммоль) амидина из примера 69, 0,3 мл триэтиламина и 20 мг 4-диметиламинпиридина растворялись в 29 мл дихлорметана с последующим добавлением 104 мг (1,1 ммоль) метилхлорформиата при 0oC. После перемешивания в течение ночи при комнатной температуре сформировался белый осадок из объявленного вещества. Он фильтровался с помощью всасывания и последовательно промывался водой и эфиром. Выход: 0,35 г (77%) бледно-желтого порошка, tпл 202-204oC. Пример 89 Получение этил-рац-(3,4-дигидро-6-(N-(4-(этоксикарбониламино) иминометил)-2-фторбензойл)амино)-2H-1-бензопиран-2-ил)ацетата, соединение представлено формулой (350): 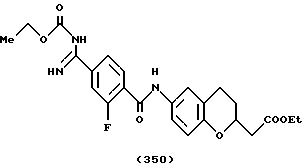 Объявленный карбомат получался, как описано в примере 88, из 435 мг (1,0 ммоль) амидина из примера 69 и 119 мг (1,1 ммоль) этилхлорформиата. Соединение перекристаллизовывалось из дихлорметана/гексана. Выход: 0,35 г (74%) белого порошка, tпл 168-169oC. Пример 90 Получение этил-рац-(3,4-дигидро-6-(N-(2-фтор-4- ((пропоксикарбониламино)иминометил)бензойл)амино)-2H-1-бензопиран-2- ил)ацетата, соединение представлено формулой (351): 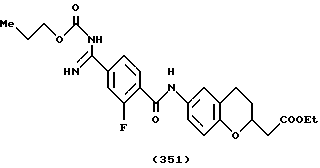 Объявленное соединение получалось, как описано в примере 85, из 436 мг (1,0 ммоль) амидина из примера 69 и 135 мг (1,1 ммоль) пропилхлорформиата. Выход: 0,29 г (60%) белого кристаллического сухого остатка, tпл 157-159oC. Пример 91 Предлагаемый способ для получения рац-4-аминоиминометил-N-(3,4- дигидро-2-(1H-тетразол-5-ил)метил-2H-1-бензопиран-6-ил)бензамида, соединение представлено формулой (352): 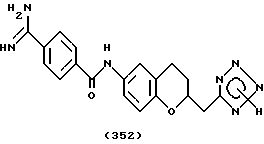 Шаг A: Получение трет-бутил-рац-(2-цианметил-3,4-дигидро-2H-1- бензопиран-6-ил)карбомата, промежуточного соединения, представленного формулой (353): 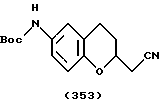 Шаг B: Получение трет-бутил-рац-(3,4-дигидро-2-(1H-тетразол-5- ил)метил-2H-1-бензопиран-6-ил)карбомата, промежуточного соединения, представленного формулой (354): 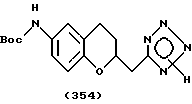 Шаг C: Получение рац-4-циано-N-(3,4-дигидро-2-(1H-тетразол-5- ил)метил-2H-1-бензопиран-6-ил)бензамида, соединения, представленного формулой (355): 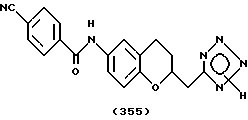 Пример 92 Получение этил-рац-(3,4-дигидро-6-(N-(4-(N-фенилметиламино) иминометил)бензойл)амино)-2H-1-бензопиран-2-ил)ацетата гидрохлорида, соединение представлено формулой (356): 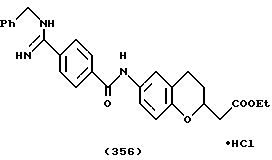 Смесь из 0,49 г (1,1 ммоль) промежуточного соединения из примера 57, шаг A, и 0,5 мл бензойламина в 15 мл безводного этанола перемешивалась в течение 5 ч при комнатной температуре. Растворитель удалялся при пониженном давлении, а оставшееся объявленное соединение кристаллизовывалось из этанола/эфира. Кристаллы собирались путем фильтрации, промывались эфиром и обезвоживались в вакууме. Выход: 0,49 г (88%) бледно-желтых кристаллов, tпл 254-256oC. Пример 93 Получение рац-(3,4-дигидро-6-(N-(4-(N’-фенилметиламино) иминометил)бензойл)амино)-2H-1-бензопиран-2-ил)уксусной кислоты, соединение представлено формулой (357): 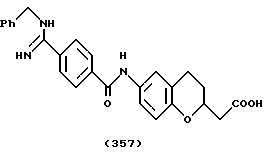 0,2 г (0,39 ммоль) сложного эфира из примера 92 добавлялось к смеси 10 мл этанола и 0,5 мл 2N водного раствора гидроксида натрия. После легкого нагревания смесь перемешивалась в течение ночи при комнатной температуре. Смесь доводилась до pH=4 с помощью 2N уксусной кислоты, и осадок из объявленной кислоты фильтровался с помощью всасывания, последовательно промывался водой и ацетоном и обезвоживался в вакууме. Выход: 0,12 г (69%) бледно-желтого порошка, tпл 220oC (dec). Пример 94 Получение этил-рац-(3,4-дигидро-6-(N-(4-(пентиламино)иминометил) бензойл)амино)-2H-1-бензопиран-2-ил)ацетата гидрохлорида, соединение представлено формулой (363): 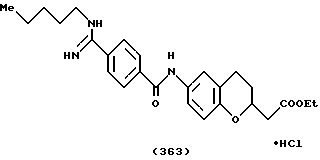 К суспензии 447 мг (1,0 ммоль) промежуточного соединения из примера 57, шаг А, в 15 мл безводного этанола при 0oC добавлялось 0,5 мл n-пентиламина. Смесь превращалась в прозрачный раствор после перемешивания в течение ночи при комнатной температуре. Раствор концентрировался при пониженном давлении, и кристаллический остаток объявленного соединения перекристаллизовывался из этанола/эфира. Выход: 0,33 г (68%) бледно-желтых кристаллов, tпл 267-269oC. Пример 95 Получение рац-(3,4-дигидро-6-(N-(4-((пентиламино)иминометил) бензойл)амино)-2H-1-бензопиран-2-ил)уксусной кислоты, соединение представлено формулой (364): 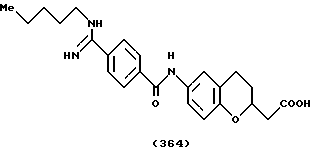 0,15 г (0,31 ммоль) сложного эфира из примера 94 перемешивалось в течение ночи при комнатной температуре в смеси 10 мл этанола и 0,7 мл 2N водного раствора гидроксида натрия. Смесь доводилась до pH=4 с помощью 2N уксусной кислоты, осадок из объявленной кислоты фильтровался, последовательно промывался водой и ацетоном и обезвоживался в вакууме. Выход: 98 мг (75%) бесцветных кристаллов, tпл 223-225oC. Ссылочные номера в следующих примерах находятся в реакционных схемах 27-33 выше. Пример 96 Получение соединения 368 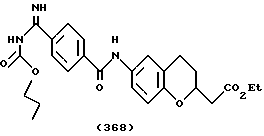 Смесь соединения 365 (1,25 г, 2,0 ммоль), H2O (5 мл) и THF (5 мл) обрабатывалась пропилхлорформиатом (0,34 г, 2,7 ммоль) и K2CO3 (0,19 г, 13,5 ммоль) при комнатной температуре. Через 1 ч смесь разбавлялась EtOAc и промывалась H2O. Органический материал концентрировался, и неочищенный остаток перекристаллизовывался из EtOAc/гексана, давая 0,8 г соединение 368 в виде белого сухого остатка. 1H NMR (300 MHz, DMSO) 10,63 (s, 1H), 9,17 (br s, 2H), 8,05 (dd, J=8,3, 19,6 Hz, 4H), 7,85 (m, 2H), 7,73 (d, J=8,5 Hz, 1H), 4,06 (q, J=7,0 Hz, 2H), 3,99 (t, J=6,5 Hz, 2H), 3,1 (m, 1H), 2,93 (m, 2H), 2,73 (dd, J=6,2, 16,3 Hz, 1H), 2,42 (m, 1H), 2,11 (m, 1H), 1,98 (m, 1H), 1,60 (m, 2H), 1,16 (t, J=7,0 Hz, 3H), 0,91 (t, J=7,3 Hz, 3H); IR (KBr) 1737, 1661, 1603, 1525, 1256 см-1; MS (FAB) m/e 480. Пример 97 Получение соединения 367 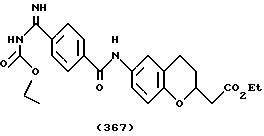 Следуя процедуре, используемой для получения соединения 368, соединение 267 было получено с выходом 91% при исходных 0,50 г соединения 365 и 0,10 г этилхлорформиата. 1H NMR (300 MHz, CDCl3) 8,55 (s, 1H), 7,80 (m, 5H), 7,48 (d, J=8,8 Hz, 1H), 4,26 (m, 4H), 3,05 (m,4H), 2,52 (m, 1H), 2,28 (m, 1H), 2.0 (m, 1H), 1,39 (t, J=7,1 Hz, 3H), 1,32 (t, J=7,2 Hz, 3H); IR (KBr) 1727, 1686, 1661, 1602, 1256 см-1; MS (FAB) m/e 466. Анализ для C22H27N3O6. Расчет: C 64,51; H 5,85; N 9,03. Найдено: C 64,77; H 5,87; N 8,82. Пример 98 Получение соединения 369: 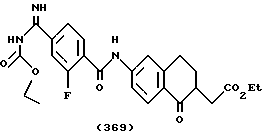 Следуя процедуре, используемой для получения соединения 368, соединение 369 было получено с выходом 87% при исходных 0,26 г соединения 366 и 0,07 г этилхлорформиата. 1H NMR (300 MHz, CDCl3) 8,64 (app d, J=15,0 Hz, 1H), 8,20 (t, J=7,9 Hz, 1H), 8,01 (d, J=8,5 Hz, 1H), 7,84 (d, J=12,7 Hz, 1H), 7,79 (s, 1H), 7,72 (s, J= 8,3 Hz, 1H), 7,42 (d, J=8,7 Hz, 1H), 4,24 (m, 4H), 3,13 (m, 4H), 2,44 (m, 1H), 2,26 (m, 1H), 1,99 (m, 1H), 1,37 (t, J=7,0 Hz, 3H), 1,28 (t, J=7,3 Hz, 3H); IR (KBr) 1731, 1673, 1520, 1256 см-1. Пример 99 Получение соединения 370: 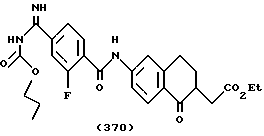 Следуя процедуре, используемой для получения соединения 368, соединение 370 получалось с выходом 86% при исходных 0,26 г соединения 366 и 0,08 г пропилхлорформиата. 1H NMR (300 MHz, CDCl3) 8,65 (app d, J=15,1 Hz, 1H), 8,20 (t, J=8,1 Hz, 1H), 8,0 (d, J=8,5 Hz, 1H), 7,84 (d, J=12,8 Hz, 1H), 7,78 (s, 1H), 7,72 (d, J= 8,2 Hz, 1H), 7,41 (d, J=8,4 Hz, 1H), 4,17 (m, 4H), 3,0 (m, 4H), 2,42 (m, 1H), 2,25 (m, 1H), 2,0 (m, 1H), 1,78 (m, 2H), 1,28 (t, J=7,3 Hz, 3H), 1,00 (t, J=7,5 Hz, 3H); IR (KBr) 1733, 1662, 1520, 1251 см-1; MS (FAB) m/e 498. Пример 100 Получение соединения 371: 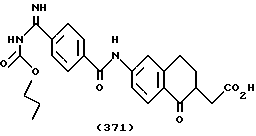 Смесь соединения 368 (0,11 г, 0,23 ммоль) и EtOH (5 мл) обрабатывалась NaOH (1,1 мл 2N раствора, 2,3 ммоль), и полученный раствор перемешивался в течение 5 ч при комнатной температуре. Реакционная смесь затем концентрировалась, и остаток вводился в H2O. Этот материал экстрагировался с помощью EtOAc, и экстракты удалялись. Оставшийся водный материал подкислялся до pH=5 с помощью 1N HCl, а полученный материал экстрагировался с помощью EtOAc. Экстракты обезвоживались (MgSO4) и концентрировались, давая 0,06 г (57%) желаемой кислоты 371 в виде белого сухого остатка. 1H NMR (300 MHz, CD3OD) 8,0-7,9 (m, 5H), 7,79 (br s, 1H), 7,64 (dd, J= 2,1, 8,6 Hz, 1H), 4,11 (t, J=6,7 Hz, 2H), 3,15-2,95 (m, 3H), 2,89 (dd, J= 5,6, 16,4 Hz, 1H), 2,48 (dd, J=6,6, 16,5 Hz, 1H), 2,27 (m, 1H), 2,06 (m, 1H), 1,71 (m, 2H), 1,00 (t, J=7,4 Hz, 3H); MS (FD) m/e 452. Пример 101 Получение соединения 381: 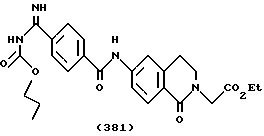 Следуя процедуре, используемой для получения соединения 368, соединение 381 получалось с выходом 47% при исходных 0,082 г соединения 374 и 0,13 г пропилхлорформиата. 1H NMR (300 MHz, CDCl3) 8,60 (br s, 1H), 7,89 (d, J=8,5 Hz, 1H), 7,85 (s, 5H), 7,40 (d, J=10 Hz, 1H), 4,31 (s, 2H), 4,21 (q, J=7,1 Hz, 2H), 4,15 (t, J= 6,9 Hz, 2H), 3,64 (t, J=6,6 Hz, 2H), 3,0 (t, J=6,5 Hz, 2H), 1,8 (m, 2H), 1,28 (t, J=7,1 Hz, 3H), 0,99 (t, J=7,4 Hz, 3H); IR (KBr) 3390, 3286, 1732, 1655, 1617, 1274 см-1; MS (FAB) m/e 481. Анализ для C25H28N4O6. Расчeт: C 62,49; H 5,82; N 11,66. Найдено: C 62,65; H 5,87; N 11,43. Пример 102 Получение соединения 391: 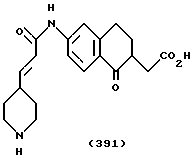 Шаг A: Смесь спирта 334 (5,0 г, 23,5 ммоль, полученного из 4-пиридилкарбинола путем гидрогенизации и протектирования) и CH2Cl2 (25 мл) добавлялась к раствору оксалилхлорида (2,8 мл, 32,5 ммоль), DMSO (2,5 мл, 34,9 ммоль) и CH2Cl2 (25 мл) при -78oC. Через 1 ч реакция обрабатывалась Et3N (6,5 мл, 46,5 ммоль) и оставлялась нагреваться до комнатной температуры. Эта смесь разбавлялась EtOAc и промывалась H2O. Органический материал обезвоживался (MgSO4) и концентрировался. Неочищенный остаток вводился в THF (25 мл) и добавлялся к смеси триэтилфосфоноацетата (6,23 г, 28,2 ммоль), NaH (1,12 г 60%-ной дисперсии в масле, 28,2 ммоль) и THF (25 мл) с температурой -78oC. Эта смесь оставлялась нагреваться до комнатной температуры. Через 1 ч смесь разбавлялась EtOAc и промывалась H2O. Органическая фаза концентрировалась, и неочищенный материал очищался путем хроматографии (3:1 гексан/EtOAc), давая 4,7 г (71%) соединения 1016 в виде прозрачного масла. Шаг B: Смесь соединения 385 (1,0 г, 3,53 ммоль), LiOH (71 мл 0,1N раствора в H2O, 7,1 ммоль) и THF (70 мл) перемешивалась при комнатной температуре в течение ночи. Затем смесь концентрировалась до половины объема и подкислялась до pH=5 с помощью 1N HCl. Полученная смесь экстрагировалась с помощью EtOAc, и экстракты концентрировались, давая 0,72 г (80%) желаемой кислоты 385 в виде практически чистого белого сухого остатка. Шаг C: Оксалилхлорид (0,08 мл, 1,0 ммоль) добавлялся к смеси соединения 386 и бензола (5 мл). Добавлялась одна капля DMF, и смесь перемешивалась при комнатной температуре в течение 1 ч. Полученный раствор концентрировался, давая неочищенный кислый хлорид 387 в виде масла. Этот материал растворялся в CH2Cl2 (5 мл) и добавлялся к раствору анилина 98 (0,17 г, 0,69 ммоль), пиридина (5 мл) и CH2Cl2 (5 мл). Полученная смесь перемешивалась при комнатной температуре в течение 1 ч, а затем разбавлялась EtOAc и промывалась H2O. Органический материал концентрировался, и неочищенный материал очищался путем хроматографии на двуокиси кремния (1,5:1 гексан/EtOAc), давая 0,25 г (90%) соединения 388 в виде белого сухого остатка. Шаг D: Раствор соединения 388 (0,24 г, 0,49 ммоль) и THF (10 мл) обрабатывался LiOH (10 мл 0,1 N раствора в H2O, 1,0 ммоль) и перемешивался при комнатной температуре в течение ночи. Эта смесь затем концентрировалась до половины объема и осторожно подкислялась с помощью 1N HCl (pH=5). Водная смесь затем экстрагировалась с помощью EtOAc, и экстракты концентрировались. Неочищенная кислота 390 растворялась в TFA (10 мл), отстаивалась в течение 1 ч при комнатной температуре, а затем концентрировалась. Таким образом сформированный материал растворялся в 1N HCl, а полученный раствор лиофолизировался, давая 0,15 г (80%) соединения 391 в виде белого порошка. 1H NMR (300 MHz, CDCl3) 7,89 (d, J=8,5 Hz, 1H), 7,68 (s, 1H), 7,51 (d, J= 8,5 Hz, 1H), 6,87 (dd, J=6,5, 15,4 Hz, 1H), 6,20 (d, J=15,4 Hz, 1H), 3,44 (br d, J=12,8 Hz, 2H), 3,0 (m, 6H), 2,6 (m, 1H), 2,4 (m, 1H), 2,25 (m, 1H), 2,05 (m, 3H), 1,70 (m, 2H); IR (KBr) 3384, 2953, 1726, 1672, 1533 см-1; MS (FAB) m/e 357. Анализ для C20H25N2O4Cl. Расчeт: C 61,14; H 6,41; N 7,13. Найдено: C 61,40; H 6,60; N 7,18. Пример 103 Получение соединения 389: 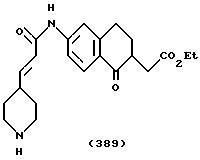 Раствор соединения 388 (0,60 г, 1,2 ммоль) и TFA (10 мл) выдерживался при комнатной температуре в течение 1 ч, а затем концентрировался. Остаток растворялся в H2O (10 мл), обрабатывался HCl (2,5 мл 1N раствора, 2,5 ммоль), и полученный раствор лиофолизировался, давая 0,49 г (95%) соединения 389 в виде белого порошка. 1H NMR (300 MHz, CDCl3) 7,92 (d, J=8,6 Hz, 1H), 7,72 (s, 1H), 7,54 (d, J= 8,7 Hz, 1H), 6,93 (dd, J=6,5, 15,6 Hz, 1H), 6,24 (d, J=15,5 Hz, 1H), 4,18 (q, J= 7,1 Hz, 2H), 3,49 (m, 2H), 3,10 (m, 5H), 2,82 (dd, J=6,3, 16,4 Hz, 1H), 2,65 (m, 1H), 2,51 (dd, J=6,2, 16,4 Hz, 1H), 2,3 (m, 1H), 2,1 (m, 3H), 1,68 (m, 2H), 1,22 (t, J=7,0 Hz, 3H); IR (KBr) 3346, 3286, 2935, 1734, 1676, 1584 см-1; MS (FAB) m/e 385. Анализ для C22H29N2O4. Расчeт: C 62,78; H 6,94; N 6,66. Найдено: C 63,01; H 6,90; N 6,75. Пример 104 Получение соединения 400: 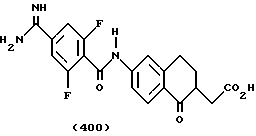 Шаг A: Смесь 3,5-дифторбензонитрила 394 (0,5 г, 3,59 ммоль), TMEDA (0,77 г, 7,3 ммоль) и THF (15 мл) обрабатывалась n-BuLi (2,6 мл 1,4 М раствора в гексане, 3,6 ммоль) при -78oC. Реакционная смесь перемешивалась при -78oC в течение 15 мин, а затем гасилась CO2 (г). Смесь оставлялась нагреваться до комнатной температуры и концентрировалась. Остаток вводился в H2O и промывался Et2O. Оставшийся водный материал подкислялся до pH=2 с помощью 1N HCl, а затем экстрагировался с помощью EtOAc. Затем экстракты концентрировались, давая 0,53 г соединения 395 в виде белого сухого остатка. Шаг B: Смесь соединения 395 (0,28 г, 1,51 ммоль), DMF (1 капля) и бензола (10 мл) обрабатывалась оксалилхлоридом при комнатной температуре. Эта смесь перемешивалась в течение 30 мин и затем концентрировалась. Неочищенный осадок вводился в CH2Cl2 (5 мл) и добавлялся в раствор соединения 98 (0,38 г, 1,51 ммоль), пиридина (3 мл) и CH2Cl2 (3 мл). Эта смесь перемешивалась при комнатной температуре в течение 0,5 ч, а затем разбавлялась EtOAc и промывалась водой. Органический материал концентрировался, и остаток хроматографировался на двуокиси кремния (1:1 гексан/EtOAc), давая 0,56 г соединения 397. Шаг C: Нитрил преобразовывался в Boc-защищенный амидин 398 с помощью общей процедуры, описанной для получения соединения 6 (пример 1, часть E). Шаг D: Амидин 398 преобразовывался в полностью депротектированное соединение 400 с помощью общей процедуры, описанной для получения соединения 130 (пример 33, часть E). 1H NMR (300 MHz, CD3OD) 7,98 (d, J=8,6 Hz, 1H), 7,74 (s, 1H), 7,68 (m, 2H), 7,64 (d, J= 8,6 Hz, 1H), 3,0 (m, 3H), 2,88 (dd, J=6,4, 16,6 Hz, 1H), 2,46 (dd, J= 6, 4, 16,6 Hz, 1H), 2,25 (m, 1H), 2,0 (m, 1H); IR (KBr) 3306, 1660, 1603, 1426, 1039 см-1; MS (FAB) m/e 402. Пример 105 Получение соединения 405: 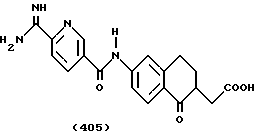 Шаг A: Получение промежуточного соединения (402): 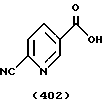 К смеси N-оксида никотиновой кислоты (8,8 г, 63 ммоль) в 100 мл безводной DMF добавлялся Et3N (32 г, 316 ммоль), что давало гомогенный раствор. Смесь обрабатывалась TMSCl (34 г, 316 ммоль) и перемешивалась в течение 30 мин при комнатной температуре перед добавлением NaCN (12,4 г, 253 ммоль). Реакция нагревалась до 100oC в течение 16 ч, затем охлаждалась до комнатной температуры и фильтровалась. Фильтрат концентрировался до безводного состояния в вакууме, обрабатывался 2N водной HCl (150 мл) и экстрагировался несколько раз с помощью CH2Cl2. Экстракты обезвоживались на MgSO4, концентрировались до безводного состояния и очищались путем хроматографии (SiO2, 1% v/v MeOH-CHCl3), давая 4,01 г сухого остатка. Шаг B: Получение промежуточного соединения (408): 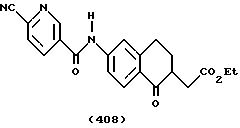 Смесь 6-циан-3-пиридин-карбоновой кислоты (1,2 г, 8,1 ммоль), 4-диметиламинопиридина (1,0 г, 8,1 ммоль), диизопропилметиламина (1,1 г, 8,1 ммоль), соединения 98 (2,0 г, 8,1 ммоль) в 25 мл CH2Cl2 обрабатывалась 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлоридом (2,4 г, 12,2 ммоль). Реакция перемешивалась при комнатной температуре в течение 60 ч. Смесь наносилась на подложку из силикагеля и вымывалась 0,5% v/v MeOH-CHCl3, давая 2,76 г (90%) соединения 408 в виде белого сухого остатка. Шаг C: Получение промежуточного соединения (409): 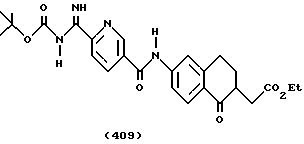 Раствор нитрила 403 (0,214 г, 0,57 ммоль) в 50 мл 1M раствора CH3SH в EtOH обрабатывался 75 г (1,1 ммоль) CH3SNa, и смесь перемешивалась при комнатной температуре в течение 2 ч. Добавлялся сухой NH4I (0,7 г), и смесь нагревалась при орошении, пока реакция не заканчивалась, что показывала тонкослойная хроматография. Раствор концентрировался до безводного состояния, и остаток вводился в 5 мл THF. Раствор обрабатывался K2CO3 (0,75 г, 5,4 ммоль), Boc2O (1,2 г, 5,4 ммоль), THF (5 мл) и H2O (5 мл), и смесь перемешивалась в течение ночи. Реакционная смесь разбавлялась EtOAc и промывалась H2O. Органический слой обезвоживался на MgSO4, концентрировался до безводного состояния и очищался путем хроматографии (SiO2, 40% v/v EtOAc/гексан), давая 183 мг соединения 409 в виде сухого остатка. Шаг D: Защищенный амидин (70 мг, 0,14 ммоль) 404 смешивался с EtOH (2 мг) и 1N водным NaOH (0,57 мл). Реакция перемешивалась при комнатной температуре в течение ночи (16 ч) и подкислялась HOAc (1 мл). Этот продукт концентрировался до безводного состояния, затем очищался путем хроматографии (SiO2, 5: 1: 94 v/v MeOH/HOAc/CHCl3). Полученная пена обрабатывалась трифторуксусной кислотой (5 мл) и анизолом (5 мл) в течение 60 ч. Затем реакция концентрировалась до безводного состояния, давая 25 мг соединения 405 в виде сухого остатка. Пример 106 Получение соединения 410: 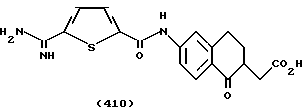 Шаг A: В высушенную над пламенем колбу добавлялись диизопропиламин (3,85 мл, 27,45 ммоль) и безводная THF (20 мл, дистиллированной из CaH). Смесь перемешивалась в атмосфере азота и охлаждалась до -78oC. К этому раствору добавлялся n-BuLi так, чтобы температура раствора не поднималась выше -70oC. В отдельную высушенную над пламенем колбу добавлялись 2-тиофенкарбонитрил (2,0 г, 18,3 ммоль) и безводная THF (20 мл). Раствор охлаждался до -78oC и перемешивался в атмосфере азота в течение 15 мин. Раствор 2-тиофенкарбонитрила затем переносился с помощью полой иглы в раствор LDA, и реакционная смесь перемешивалась еще 1 ч при -78oC. Когда раствор достигал комнатной температуры, в течение 20 мин добавлялся углекислый газ. После концентрации в вакууме полученный остаток вводился в KOH (100 мл, 0,01 М) и промывался EtOAC (2 х 75 мл). Водный слой затем подкислялся до pH=2 с помощью концентрированной HCl и насыщался NaCl. Продукт экстрагировался с помощью EtOAc (4х100 мл), и объединенные органические экстракты концентрировались в вакууме. Полученный осадок очищался путем колоночной хроматографии на силикагеле 60 (230-400 меш) с вымыванием продукта с помощью 20% MeOH в CHCl3 (2% v/v уксусной кислоты). Подходящие фракции объединялись и концентрировались в вакууме, давая 760 мг (27%) соединения 407 в виде рыжевато-коричневого сухого остатка. Шаг B: К соединению 407 (260 мг, 1,7 ммоль) добавлялись соединение 98 (420 мг, 1,7 ммоль), EDCI (520 мг, 2,72 ммоль), 4-DMAP (каталитическое количество) и CH2Cl2 (6 мл). Смесь перемешивалась при комнатной температуре в течение 12 ч, а затем разбавлялась EtOAc (40 мл), и органическая часть промывалась HCl (40 мл, 0,1 М), NaOH (40 мл, 0,1 М) и H2O (40 мл). Затем органический слой концентрировался в вакууме, и полученный осадок очищался путем колоночной хроматографии на силикагеле 60 (230-400 меш) с вымыванием продукта с помощью 10% THF в CHCl3. Подходящие фракции объединялись и концентрировались в вакууме, давая 200 мг (31%) соединения 408 в виде рыжевато-коричневого сухого остатка. Шаг C: Нитрил 408 преобразовывался в соединение 409 с помощью общей процедуры, описанной для получения соединения 6 (пример 1, часть E). Шаг D: Амидин 409 преобразовывался в полностью депротектированное соединение 410 с помощью общей процедуры, описанной для получения соединения 130 (пример 33, часть E). 1H NMR (300 MHz, CD3OD) 2,02 (m, 1H), 2,26 (m, 1H), 2,48 (dd, J=6,4, 10,1 Hz, 1H), 2,86 (dd, J=5,7, 10,7 Hz, 1H), 3,11 (m, 3H), 7,63 (dd, J=2, 6,6 Hz, 1H), 7,76 (bs, 1H), 7,95 (m, 2H), 8,03 (d, J=4,1 Hz, 1H, 2H); IR (KBr) 3322, 3096, 1667, 1585, 1537, 1502, 1425, 1384, 1342, 1284, 1267 см-1; MS (FAB) m/e 372,2; tпл 205-208 (dec). Анализ для C20H18N3O6F3S. Расчeт: C 49,48; H 3,74; N 8,66. Найдено: C 49,45; H 3,76; N 8,44. Способы испытаний. Идентификация соединений, являющихся активными ингибиторами тромбоцитного слипания (ИТС), возможна с помощью наблюдения того, что соединения, блокирующие связывание фибриногена с гликопротеиновым комплексом IIb-IIIa в лабораторных условиях, также способны подавлять инициируемое тромбином или ADP слипание человеческих тромбоцитов и формирование тромбов в естественных условиях. Это наблюдение дает базу для получения эффективных ИТС путем оценки способности тестовых материалов нарушать взаимодействие фибриноген-гликопротеин IIb-IIIa. Следующие способы испытаний использовались для оценки соединений по настоящему изобретению, охватывающихся соединениями, представленными формулами (I), (II), (IX), (Xd) и (Xe), как было описано ранее. 1 1 – Испытание ELISA IIb-IIIa: В нижеописанном испытании гликопротеин IIb-IIIa приготавливается в очищенном виде с помощью способа, такого как, например, описанный в Fitzgerald L.A. et al. Anal. Biochem. (1985) 151:169-177 (рассмотрение в которой включено сюда посредством ссылки). Гликопротеин IIb-IIIa наносится на микротитровую пластинку. Покрытый носитель затем контактирует с фибриногеном и с тестовым материалом (например, соединениями по формуле I) и выдерживается достаточное время для того, чтобы произошло максимальное связывание фибриногена с иммобилизированным гликопротеином IIb-IIIa. Фибриноген обычно обеспечивается с концентрацией приблизительно 5-50 нМ, а тестовый материал может по желанию добавляться с разбавлением. Обычное время выдерживания составляет от 2 до 4 ч при 25oC, время и температура взаимозависимы. После выдерживания раствор, содержащий фибриноген и тестовый материал, удаляется, а уровень связывания фибриногена измеряется путем подсчета связанного с гликопротеином IIb-IIIa фибриногена. Могут использоваться любые подходящие средства фиксирования, однако традиционно используется маркированный фибриноген, к примеру, с использованием биотинилированных меток. Такие способы хорошо известны из уровня техники. A. Описание исследований – пластиночные исследования Очищенный рецептор тромбоцитного гликопротеина IIb-IIIa приготавливался так, как описано в Fitzgerald L.A. et al., Anal. Biochem. (1985) 151:169-177 (1985). Витронектиновый рецептор приготавливался так, как описано в Smith J. W. J. Biol. Chem (1988) 263:18726-18731. После очистки рецепторы помещались в 0,1%-ный Тритон X-100 при концентрации 0,1-1,0 мг/мл. Рецепторы наносились на ячейки 96-ячеечных пластин ELISA с плоским дном (микротитровая пластина Linbro EIA-Plus, Flow Laboratories) после разбавления 1: 2000 раствором 20 мМ Трис-HCl, 150 мМ NaCl, 1 мМ CaCl2, pH=7,4 для уменьшения концентрации Тритон X-100 до уровня ниже его критической мицеллярной концентрации и добавления составляющей 100 ul к каждой ячейке. Затем ячейки выдерживались в течение ночи при 4oC, а затем высасывались до сухого состояния. Дополнительные активные центры блокировались путем добавления альбумина бычьей сыворотки (АБС) при концентрации 35 мг/мл в буфер на 2 ч при 30oC для предотвращения неспецифического связывания. Затем ячейки промывались вместе с буфером связывания (50 нМ Трис-HCl, 100 мМ NaCl, 2 мМ CaCl2, 1 мг/мл АБС). Соответствующие лиганды (фибриноген, фактор фон Виллебранда, или витронектин) соединялись с биотином с помощью коммерчески доступных реагентов и стандартных протоколов. Маркированные лиганды добавлялись в покрытые рецепторами ячейки с конечной концентрацией 10 нМ (100 ul/ячейка) и выдерживались в течение 3 ч при 25oC в присутствии или в отсутствии тестовых образцов. После выдерживания ячейки высасывались до сухого состояния, а связанный лиганд подсчитывался. Связанный протеин фиксировался путем добавления антибиотинного антитела, соединенного с щелочной фосфатазой, с последующим добавлением субстрата (p-нитрофенил-фосфат) и определением оптической плотности каждой ячейки при 405 нМ. Уменьшенная цветность наблюдалась в ячейках, выдерживавшихся с тестовыми образцами, которые подавляют связывание лиганда с рецептором. 1 2 – Исследование слипания тромбоцитов. В дополнение к ранее описанному исследованию ELISA IIb-IIIa, исследование слипания человеческих PRP/ADP полезно при оценке терапевтических соединений. Богатая тромбоцитами плазма приготавливалась из крови здоровых доноров-людей для использования при определении подавления соединениями слипания тромбоцитов. Кровь собиралась посредством 21-мерной перекрываемой полой иглы с помощью метода двойного впрыскивания в 1/10 объема 3,8%-ного тринатриевого цитрата. Богатая тромбоцитами плазма приготавливалась при комнатной температуре путем центрифугирования цитрированной цельной крови при 100 х g в течение 12 мин. Богатая тромбоцитами плазма содержит приблизительно 200-400000 тромбоцитов/мкл. Бедная тромбоцитами плазма приготавливалась путем центрифугирования цитрированной цельной крови при 12000 х g в течение 2 мин. Слипание тромбоцитов исследовалось в 4-канальном приборе слипания тромбоцитов (РАР-и, Biodata, Hatboro, PA) в соответствии с инструкциями производителя. Подавление слипания тромбоцитов изучалось путем добавления различных количеств дифосфата аденозина (ADP) к перемешиваемой человеческой богатой тромбоцитами плазме. Человеческая богатая тромбоцитами плазма выдерживалась с тестируемым соединением в течение 1 мин при 37oC перед добавлением различных агрегирующих агентов, чаще всего ADP 5 мкМ, но также и 1 мкг/мл коллагена, 1 мкМ U46619 и 0,3 мкМ фактора активации тромбоцита. Таблица результатов исследования Пример 1 ELISA IIb-IIIa Слипание: человеческие PRP/ADP [см. таблицу в конце описания] Примечание: NT = не тестировалось. Фармацевтические составы Фармацевтические составы, содержащие соединения по настоящему изобретению, могут применяться перорально в виде таблеток, капсул, растворов, эмульсий или суспензий, ингаляционных жидких или твердых частиц, в виде спрея, через кожу путем таких приспособлений, как чрескожный пластырь (как, к примеру, описано в патентах США 1 5296222 и 5271940, рассмотрение в которых включено сюда посредством ссылки) или ректально, например в виде свечей. Липофильные пролекарственные производные по настоящему изобретению (к примеру, формулы Xd, Xe) частично хорошо подходят для применения путем кожной абсорбции и для систем доставки. Прием также может быть парентеральным, к примеру, в виде инъекционных растворов. Таблетки приготавливаются путем смешивания активного ингредиента (“активным ингредиентом” является одно или более соединений по настоящему изобретению из тех, которые соответствуют формулам I, II, Xd или Xe) с фармацевтически инертными, неорганическими или органическими носителями, разбавителями и/или наполнителями. Примерами таких наполнителей, которые могут использоваться для таблеток, являются лактоза, кукурузный крахмал или его производные, тальк, стеариновая кислота или ее соли. Примерами подходящих наполнителей для мягких желатиновых капсул являются растительные масла, воски, жиры, полутвердые или жидкие полиолы. Подходящими наполнителями для приготовления растворов и сиропов являются вода, полиолы, сахароза, инвертированный сахар и глюкоза. Подходящими наполнителями для инъекционных растворов являются вода, спирты, полиолы, глицерол и растительные масла. Эти фармацевтические продукты могут дополнительно содержать предохранители, растворители, стабилизаторы, увлажняющие агенты, эмульгаторы, подсластители, красители, ароматические вещества, буферы, покрывающие агенты и антиоксиданты. Фармацевтические составы по данному изобретению для парентеральных инъекций содержат фармацевтически приемлемые стерильные водные или неводные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки для получения стерильных инъекционных растворов или дисперсий непосредственно перед использованием. Активный ингредиент также может быть в виде микрокапсул. Примеры составов с активным ингредиентом описаны ниже: Состав 1 Жесткие желатиновые капсулы приготавливаются с помощью следующих ингредиентов: – (мг/капсула) Активный ингредиент – 250,0 Крахмал – 305,0 Стеарат магния – 5,0 Вышеописанные ингредиенты смешиваются, и ими заполняются жесткие желатиновые капсулы по 560 мг. Состав 2 Таблетки приготавливаются с помощью нижеуказанных ингредиентов: – (мг/таблетка) Активный ингредиент – 250,0 Целлюлоза микрокристаллическая – 400,0 Коллоидная двуокись кремния – 10,0 Стеариновая кислота – 5,0 Компоненты смешиваются и спрессовываются в таблетки каждая весом 665 мг. Состав 3 Сухой ингаляционный порошок приготавливается из следующих компонентов: – вес.% Активный ингредиент – 5 Лактоза – 95 Активная смесь смешивается с лактозой, и смесь добавляется в порошковое ингаляционное приспособление. Состав 4 Таблетки, содержащие каждая 60 мг активного ингредиента, приготавливаются следующим образом: – (мг) Активный ингредиент – 60,0 Крахмал – 45,0 Микрокристаллическая целлюлоза – 35,0 Поливинилпирролидон (в виде 10%-ного раствора в воде) – 4,0 Натрий-карбоксиметилкрахмал – 4,5 Стеарат магния – 0,5 Тальк – 1,0 Итого – 150,0 Активный ингредиент, крахмал и целлюлоза пропускались через сетчатый фильтр 1 20 меш США и тщательно перемешивались. Раствор поливинилпирролидона перемешивался с полученными порошками, которые затем пропускались через сетчатый фильтр 16 меш США. Образованные гранулы обезвоживались при 50-60oC и пропускались через сетчатый фильтр 16 меш США. Натрий-карбоксиметилкрахмал, стеарат магния и тальк, предварительно пропущенные через сетчатый фильтр 30 меш США, затем добавлялись к гранулам, которые после перемешивания спрессовывались в таблетирующей машине в таблетки каждая весом 150 мг. Состав 5 Капсулы, содержащие каждая 80 мг медикамента, приготавливались следующим образом: – (мг) Активный ингредиент – 80,0 Крахмал – 109,0 Стеарат магния – 1,0 Итого – 190,0 Активный ингредиент, целлюлоза, крахмал и стеарат магния смешиваются, пропускаются через сетчатый фильтр 20 меш США, и смесью заполняются жесткие желатиновые капсулы по 190 мг. Состав 6 Свечи, содержащие каждая 225 мг активного ингредиента, приготавливаются следующим образом: Активный ингредиент – 225 мг Насыщенные глицериды жирной кислоты – До 2000 мг Активный ингредиент пропускается сквозь сетчатый фильтр 60 меш США и суспендируется в насыщенных глицеридах жирной кислоты, предварительно растопленных минимально необходимым количеством тепла. Затем смесь заливается в свечевую форму номинального объема 2,0 г и охлаждается. Состав 7 Суспензии, содержащие каждая 50 мг медикамента на дозу в 5,0 мл, приготавливаются следующим образом: Активный ингредиент – 50,0 Ксантановая смола – 4,0 Натрий-карбоксиметилцеллюлоза – (11%) Микрокристаллическая целлюлоза (89%) – 50,0 мг Сахароза – 1,75 г Бензоат натрия – 10,0 мг Ароматизатор – q.v. Краситель – q.v. Очищенная вода – До 5,0 мл Медикамент, сахароза и ксантановая смола смешиваются, пропускаются сквозь сетчатый фильтр 10 меш США, а затем смешиваются с предварительно приготовленным раствором микрокристаллической целлюлозы и натрий-карбоксиметилцеллюлозы в воде. Бензоат натрия, ароматизатор и краситель разбавляются небольшим количеством воды и добавляются при перемешивании. Затем добавляется значительное количество воды для получения требуемого объема. Состав 8 Капсулы, содержащие каждая 150 мг медикамента, приготавливаются следующим образом: – (мг) Активный ингредиент – 150,0 Крахмал – 407,0 Стеарат магния – 3,0 Итого – 560,0 Активный ингредиент, целлюлоза, крахмал и стеарат магния смешиваются, пропускаются сквозь сетчатый фильтр 20 меш США, и смесью заполняются жесткие желатиновые капсулы объемом 560 мг. Способ применения Настоящее изобретение обеспечивает способ предотвращения или лечения тромбоза у млекопитающих, в особенности у людей. Cпособ содержит введение человеку или млекопитающему терапевтически эффективного количества соединений по настоящему изобретению. Ингибиторы слипания тромбоцитов по изобретению терапевтически полезны для предотвращения образования тромба. Такое лечение показано, в частности (но не ограничивается данным списком), при атеросклерозе и артериосклерозе, остром инфаркте миокарда, хронической нестабильной стенокардии, кратковременных ишемических ударах, периферийных сосудистых заболеваниях, артериальных тромбозах, преэклампсии, эмболии, рестенозе и/или тромбозе с последующей ангиопластикой, каротидной эндартеректоме, анастомозе сосудистых тканей и хронических сердечно-сосудистых устройствах (например, вживленных катетерах или шунтах “внеорганизменных устройств циркуляции”). Эти синдромы представляют различные связанные с сужением и закупоркой сосудистые нарушения, которые предположительно инициируются путем активации тромбоцитов на стенках сосудов. ИТС могут использоваться для предотвращения или прекращения формирования артериального тромба при нестабильной стенокардии и артериальной эмболизации или тромбозе, а также при лечении или предотвращении инфаркта миокарда (ИМ) и образования пристеночного тромба после ИМ. При нарушениях, связанных с мозгом, возможно лечение или предотвращение кратковременных ишемических ударов и лечение тромбовых приступов или развивающихся приступов. ИТС могут также использоваться для предотвращения слипания тромбоцитов, эмболизации или увядания при внеорганизменных циркуляциях, в том числе улучшения почечного диализа, сердечно-легочных обводов, гемоперфузий и плазмофареза. ИТС предотвращают слипание тромбоцитов, эмболизацию или потери, связанные с внутрисосудистыми устройствами, а также их прием дает в результате улучшенное использование интрааортальных баллонных насосов, желудочковых вспомогательных устройств и артериальных катетеров. ИТС также будут полезны при лечении или предотвращении венного тромбоза в таких случаях, как глубокий венный тромбоз, тромбоз нижней полой вены, тромбоз почечной вены или воротной вены и тромбоз легочной вены. Также поддаются лечению различные нарушения, вызванные увяданием тромбоцитов, такие как thrombotic thrombocytopenic purpura. Вдобавок, ИТС по настоящему изобретению могут использоваться в многочисленных нетерапевтических применениях, где желательно подавление слипания тромбоцитов. К примеру, может быть достигнуто усовершенствованное хранение тромбоцитов и цельной крови путем добавления значительных количеств соединений, количество которых будет варьировать в зависимости от предполагаемого времени хранения, условий хранения, конечного использования хранимого материала и т.д. Предпочтительно соединения по настоящему изобретению подаются в виде фармацевтических составов. Так, соединения по настоящему изобретению могут вводится перорально, парентерально, топикально, ректально и т.д., в желаемых единицах дозировки. Использующийся здесь термин “парентеральный” подразумевает без ограничения подкожный, внутривенный, внутриартериальный ввод путем инъекции или вливания. Термин “топикально” касается ввода ректально и путем ингаляционного спрея, а также более общих путей ввода через кожу и слизистые оболочки рта и носа. Конкретные уровни дозировок активных ингредиентов в фармацевтических составах по настоящему изобретению могут варьировать так, чтобы ввести такое количество активного соединения (соединений), которое является эффективным для достижения желаемой терапевтической реакции у конкретного пациента. Выбранный уровень дозировки будет зависеть от активности конкретного соединения, путей ввода, серьезности излечиваемых болезней, а также состояния и истории болезни лечащегося пациента. Однако из уровня техники известно, что начальные дозы соединения должны быть ниже, чем требуется для достижения желаемого терапевтического эффекта, а затем дозы постепенно увеличиваются до тех пор, пока желаемый эффект не достигнут. По желанию эффективная дневная доза может делиться на несколько доз в целях простоты приема, например, от двух до четырех отдельных доз в день. Понятно, однако, что конкретный уровень дозировки для любого конкретного пациента будет зависеть от множества факторов, в том числе веса тела, общего состояния здоровья, диеты, времени и способа ввода, сочетания с другими лекарствами и серьезностью излечиваемой болезни. Диапазон терапевтических дозировок от примерно 0,01 до примерно 10000 мг/день, предпочтительно от 1 до 300 мг/день. Многие модификации и изменения настоящего изобретения могут быть сделаны, не выходя за рамки изобретения, что очевидно для специалиста. Описанные здесь конкретные варианты выполнения предложены только в качестве примеров, а изобретение ограничивается только терминами прилагаемой формулы изобретения. Формула изобретения
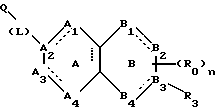 где бициклическое ядро из колец А и В выбирается из группы, состоящей из формул (17) – (21) 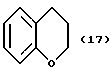 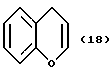 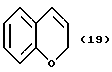 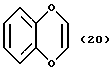 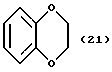 R3 является кислотной группой, содержащей кислотный радикал, выбранный из формул 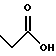 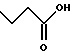 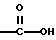 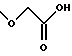 n = 0 – 6; R0 является одинаковым или разным и выбирается независимо из водорода, алкила, связующая группа -(L)- является связью или выбирается из формул 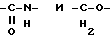 Q является основной группой и выбирается из амидиногруппы и пиперидина. 2. Соединение по п.1, представленное формулой I f 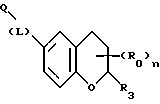 3. Соединение по п.1, представленное формулой Ig 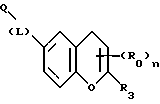 4. Соединение по п.1, представленное формулой Ih 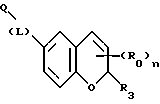 5. Соединение по п.1, представленное формулой Ii: 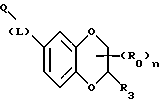 6. Соединение по п.1, в котором кислотный радикал кислотной группы R3, представляет группу 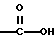 7. Соединение по п. 1, в котором кислотный радикал в R3 выбирается из группы: 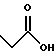 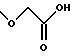 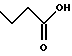 8. Соединение по п.1, в котором Q содержит основной радикал, выбираемый из амидиногруппы и пиперидила. 9. Соединение по п.1, в котором связующая группа – (L)- выбирается из групп, представленных формулами 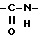 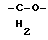 10. Бициклическое соединение или его фармацевтически приемлемая соль, сольват или пролекарственная производная, имеющие ядро, сформированное из двух конденсированных шестичленных колец, выбранное из формул Iaf, Iag и Iah 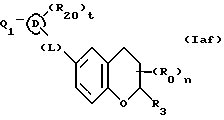 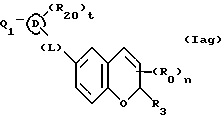 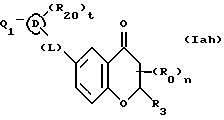 где R3 является кислотной группой, содержащей кислотный радикал, выбранный из формул 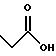 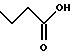 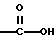 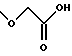 n = 0 – 6; R0 – является одинаковым или разным и выбирается независимо из водорода, алкила, t = 0 – 3; R20 – является одинаковым или разным и выбирается независимо из водорода, галогенов; связующая группа -(L) – является связью или выбирается из формул 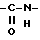 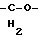 D – фенил или тиофен; Q1 – основной радикал и выбирается из амидино- и гуанидиногрупп. 11. Соединение по п.10, в котором кольцо D выбирается из фенила и тиофена. 12. Соединение по п.10, в котором Q1 выбирается из амидино- и гуанидиногрупп. 13. Соединение по п.10, в котором R20 является хлором или фтором, а t = 1 или 2. 14. Соединение по п.10, в котором ядро выбирается из бензопирена, как показано структурной формулой 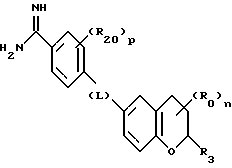 где R20, R0, R3, р, n и L как определены в п.10. 15. Соединение, выбираемое из группы, состоящей из соединений, представленных следующими формулами VIII – VIIIi либо фармацевтически приемлемые соль, сольват или пролекарственная производная этого соединения: 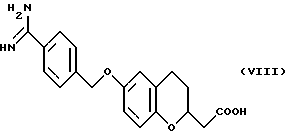 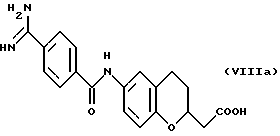 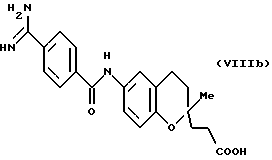 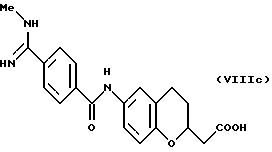 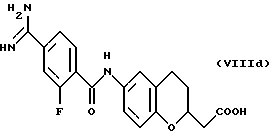 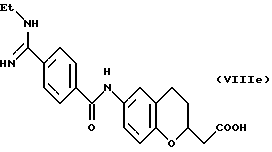 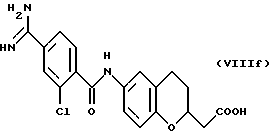 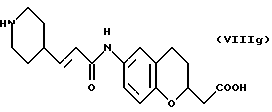 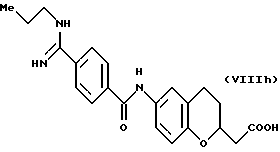 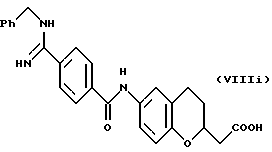 или смеси любых из соединений (VIII – VIIIi). 16. Фармацевтический состав, подавляющий слипание тромбоцитов, содержащий бициклические соединения по пп.1 – 15 в эффективном количестве, носитель или разбавитель. 17. Бициклическое соединение по пп.1 – 15 или его фармацевтически приемлемая соль, используемое при атеросклерозе и артериосклерозе, остром инфаркте миокарда, хронической стабильной стенокардии, нестабильной стенокардии, кратковременных ишемических ударах, периферических заболеваний сосудов, артериальном тромбозе, преэклампсии, эмболии, рестенозе с последующей ангиопластикой, каротидной эндартеректоме и анастомозе сосудистых тканей. 18. Способ подавления связывания фибриногена на активных центрах гликопротеина IIb-IIIa у млекопитающего в том числе человека, путем введения, как минимум, одного соединения по п.1 в эффективном количестве. 19. Пролекарственная производная бициклического соединения, имеющего ядро, сформированное из двух конденсированных шестичленных колец, А и В, представленного формулой Xd 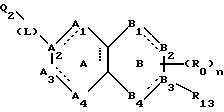 где А1, А2, А3, А4 являются атомами углерода; B1, B2, B4 выбираются независимо из атомов углерода, кислорода; В3 независимо выбирается из атомов углерода или азота, при условии, что В3 имеет незаполненную связью, если В3 является азотом, и при условии, что как минимум два из атомов В1, В2, В3 В4 являются атомами углерода; n = 0 – 6; R0 является одинаковым или разным и выбирается независимо из водорода, алкила, связующая группа -(L)-является связью или выбирается из формул 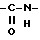 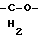 Q2 основная группа и выбирается из амидиногруппы и пиперидила, R13 кислотная группа, содержащая либо кислотный радикал, выбранный из формул 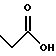 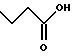 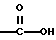 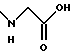 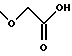 либо С1-С4 сложный эфир кислотного радикала. 20. Пролекарственное производное бициклического соединения, выбираемое из группы, состоящей из соединений, представленных формулами Xg, Xh, Xi, Xk, Xl 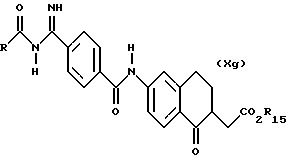 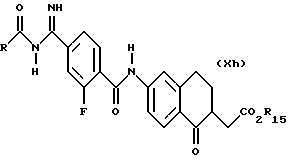 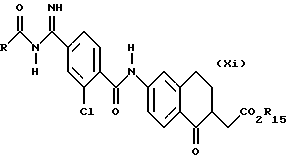 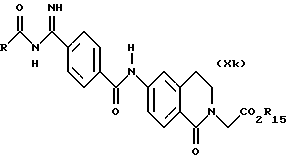 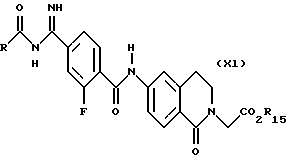 где R = -OMe, -OEt, -OPr; R15 = Me, Et, Pr. 21. Пролекарственное производное бициклического соединения, выбираемое из группы, состоящей из соединений, представленных формулами Xo, Xg, Xr 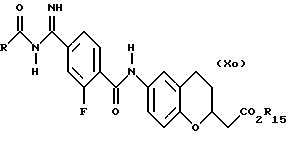 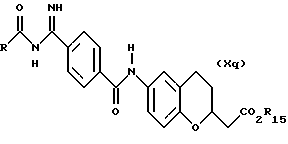 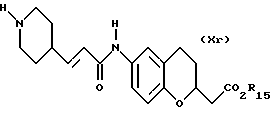 где R = -OMe, -OEt, -OPr; R15 = -Me, Et, Pr. 22. Фармацевтический состав, подавляющий слипание тромбоцитов, содержащий бициклическое соединение по п.19 в эффективном количестве, носитель или разбавитель. 23. Способ подавления слипания тромбоцитов у млекопитающего путем введения бициклического соединения по п.1 или 19 в эффективном количестве. 24. Способ подавления связывания фибриногена путем контактирования активных центров гликопротеина IIb-IIIa с соединением по п.1 или 19. 25. Способ подавления связывания фибриногена на активных центрах гликопротеина IIb-IIIa у млекопитающего, в том числе человека, путем введения, как минимум, одного соединения по п.19 в эффективном количестве при атеросклерозе и артериосклерозе, остром инфаркте миокарда, хронической стабильной стенокардии, кратковременных ишемических ударах, периферических заболеваний сосудов, артериальном тромбозе, преэклампсии, эмболии, рестенозе с последующей ангиопластикой, каротидной эндартеректоме и анастомозе сосудистых тканей. РИСУНКИ
MM4A Досрочное прекращение действия патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 19.01.2004
Извещение опубликовано: 20.10.2005 БИ: 29/2005
|
||||||||||||||||||||||||||