Патент на изобретение №2168995
|
||||||||||||||||||||||||||
(54) ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОЕДИНЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ
(57) Реферат: Описывается новая фармацевтическая композиция для лечения опухоли у животного-хозяина, содержащая эффективное количество  -L- энантиомера формулы (I), где R1 и R2 выбраны из группы, содержащей водород, ацил и C1-C18 алкил, f представляет собой Н или F, которое по меньшей мере на 95% свободно от соответствующего -D-энантиомера, или его фармацевтически приемлемой соли в фармацевтически приемлемом носителе. Композиция может найти применение для лечения опухолей, включая рак, и других состояний с аномальной или нежелательной пролиферацией клеток у животных, включая людей. 8 с. и 41 з.п.ф-лы, 5 ил., 3 табл. -L- энантиомера формулы (I), где R1 и R2 выбраны из группы, содержащей водород, ацил и C1-C18 алкил, f представляет собой Н или F, которое по меньшей мере на 95% свободно от соответствующего -D-энантиомера, или его фармацевтически приемлемой соли в фармацевтически приемлемом носителе. Композиция может найти применение для лечения опухолей, включая рак, и других состояний с аномальной или нежелательной пролиферацией клеток у животных, включая людей. 8 с. и 41 з.п.ф-лы, 5 ил., 3 табл.
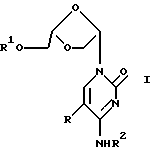
Данное изобретение относится к области медицинской химии и, в частности, касается (-)-(2S, 4S)-1-(2-гидроксиметил-1,3-диоксолан- 4-ил)цитозина (на который также ссылаются как на (-)-OddC) или его производного и их применения для лечения рака у животных, включая людей. Предпосылки изобретения. Опухоль представляет собой состояние с нерегулируемой, дезорганизованной пролиферацией клеток. Опухоль является злокачественной, или раковой, если она обладает свойствами инвазивности и метастазирования. Понятие инвазивности относится к склонности ткани проникать в окружающую ткань, прорастая базальные мембраны, которые определяют границы тканей, таким образом, зачастую проникая в циркуляторную систему организма. Понятие метастазирования относится к склонности опухоли мигрировать в другие участки тела и образовывать зоны пролиферации на удалении от места первоначального возникновения. Сейчас рак является второй по частоте ведущей причиной смертности в Соединенных Штатах. Рак диагносцирован у более чем 8000000 человек в Соединенных Штатах, причем ожидается, что в 1994 году будет поставлено 1208000 новых диагнозов. Ежегодно в данной стране от данного заболевания умирает более чем 500000 человек. Природа рака на молекулярном уровне не полностью ясна. Известно, что воздействие на клетки канцерогена, такого как некоторые вирусы, определенные химические вещества или радиация, ведет к изменению в ДНК, которые инактивируют “супрессирующий” ген или активируют “онкоген”. Супрессирующие гены представляют собой гены, регулирующие рост, которые при возникновении мутации больше не могут сдерживать клеточный рост. Онкогены представляют собой первоначально нормальные гены (так называемые проонкогены), которые при возникновении мутации или при изменении условий экспрессии становятся трансформирующими генами. Продукты трансформирующих генов вызывают ненормальный клеточный рост. Более чем двадцать различных нормальных клеточных генов могут стать онкогенами при возникновении генетических изменений. Трансформированные клетки отличаются от нормальных клеток по многим показателям, включая клеточную морфологию, межклеточные взаимодействия, содержание мембранных структур, структуру клеточного скелета, секрецию белка, экспрессию генов и смертность (трансформированные клетки могут расти неограниченно). Все разнообразные типы клеток организма могут трансформироваться в клетки доброкачественных или злокачественных опухолей. Наиболее частым местом возникновения опухоли является легкое, далее следуют ободочнопрямокишечная область, молочная железа, предстательная железа, мочевой пузырь, поджелудочная железа и затем яичник. Другие широко распространенные типы рака включают лейкоз, злокачественные опухоли центральной нервной системы, включая злокачественную опухоль головного мозга, меланому, лимфому, эритромиелоз, рак матки и рак тканей головы и шеи. Сейчас рак лечат с помощью одного или комбинации трех типов лечения: хирургии, лучевой терапии и химиотерапии. Хирургическое вмешательство включает в себя объемное иссечение пораженной ткани. Тогда как хирургическое лечение иногда является эффективным в удалении опухолей, локализованных в определенных участках тела, например, в молочной железе, толстом кишечнике и коже, оно не применимо при лечении опухолей, локализованных в других участках, таких как позвоночник, а также при лечении диссеминированных неопластических состояний, таких как лейкоз. Химиотерапия включает в себя прерывание репликации клеток или клеточного метаболизма. Ее чаще применяют как при лечении лейкозов, так и рака молочной железы, легкого и яичка. Существует пять основных классов химиотерапевтических средств, применяемых в настоящее время для лечения рака: природные продукты и их производные; антрациклины; алкилирующие агенты; антипролиферативные средства (также называемые антиметаболитами) и гормональные средства. На химиотерапевтические средства часто ссылаются как на антинеопластические средства. Считается, что алкилирующие агенты действуют посредством алкилирования и сшивания гуанина и, возможно, других оснований в ДНК, прекращая клеточное деление. Типичные алкилирующие агенты включают этилениминовые соединения, алкилсульфаты, цисплатин и различные нитрозомочевины. Недостатком данных соединений является то, что они воздействуют не только на злокачественные клетки, но и на другие клетки, деление которых естественно, такие как клетки костного мозга, кожи, слизистой оболочки желудочно- кишечного тракта и тканей плода. Антиметаболиты обычно являются обратимыми или необратимыми ингибиторами ферментов или соединениями, которые вмешиваются в процесс репликации, трансляции или транскрипции нуклеиновых кислот иным образом. Было найдено, что несколько синтетических нуклеозидов проявляют противораковую активность. Хорошо известным нуклеозидным производным с сильной противораковой активностью является 5-фторурацил. 5-Фторурацил использовали в клинике для лечения злокачественных опухолей, включая, например, карциномы, саркомы, рак кожи, рак органов пищеварения и рак молочной железы. Однако, 5-фторурацил вызывает серьезные побочные реакции, такие как тошнота, алопеция, диаррея, стоматит, лейкоцитарная тромбоцитопения, анорексия, пигментация и отеки. Производные 5-фторурацила с противораковой активностью описаны в Патенте США N 4336381 и в Японских патентных изданиях NN 50-50383, 50-50384, 50-64281, 51-146482 и 53-84981. В Патенте США N 4000137 описано, что продукт перекисного окисления инозина, аденозина или цитидина с метанолом или этанолом обладает активностью против лимфолейкоза. Цнтозинарабинозид (на который также ссылаются как на Цитарабин, аrаС и Цитозар) является нуклеозидным аналогом дезоксицитидина, который был впервые синтезирован в 1950 году и введен в клиническую медицину в 1963 году. В настоящее время он является важным лекарством при лечении острого миелолейкоза. Он также обладает активностью против острого лимфолейкоза и, в меньшей степени, применим в лечении хронического миелолейкоза и не-Ходжкинской лимфомы. Основным действием аrаС является ингибирование синтеза ДНК в ядре. Handschumacher, R. и Cheng, Y., “Purine and Pyrimidine Antimetabolites”, Cancer Medicine, Глава XV-1, 3-е издание, под редакцией J. Holland, et al., Lea and Febigol, издатели. 5-Азацитидин является аналогом цитидина, который применяют, в первую очередь, для лечения острого миелолейкоза и миелодиспластического синдрома. 2-Фтораденозин-5′-фосфат (Fludara, на который также ссылаются как на FaraA) является одним из наиболее активных средств для лечения хронического миелолейкоза. Соединение действует путем ингибирования синтеза ДНК. Обработка клеток F-araA сопряжена с сосредоточением клеток на границе G1/S-фаз и в S-фазе; таким образом, он является лекарством, специфичным для S-фазы клеточного цикла. Включение активного метаболита, F-araАТФ, тормозит элонгацию цепи ДНК. F-araA также является мощным ингибитором рибонуклеотидредуктазы, ключевого фермента, ответственного за образование дАТФ. 2-Хлордезоксиаденозин применяют для лечения B- клеточных новообразований с низкой степенью дифференцировки, таких как лимфолейкоз, не-Ходжкинская лимфома и волосато-клеточный лейкоз. Спектр действия схож с таковым Fludara. Данное соединение подавляет синтез ДНК в растущих клетках и подавляет репарацию ДНК в оставшихся клетках. Хотя ряд химиотерапевтических средств обнаружен и используется в настоящее время для лечения рака, ведется поиск новых средств, которые были бы эффективны и которые проявляли бы низкую токсичность по отношению к здоровым клеткам. Таким образом, целью настоящего изобретения является разработка соединений, которые обладают противоопухолевой и, в частности, противораковой активностью. Другой целью настоящего изобретения являются фармацевтические композиции для лечения рака. Дальнейшей целью настоящего изобретения является способ лечения рака. Сущность изобретения. Описываются способ и композиция для лечения опухолей и, в частности, рака у людей и других животных- хозяев, который заключается во введении эффективного количества (-) -(2S, 4S)-1-(2-гидроксиметил-1,3-диоксолан-4-ил) цитозина (на который также ссылаются как на (-)-OddC, L-OddC или (-)-L-OddC), его фармацевтически приемлемого производного, включая 5′- или N4-алкилированное или ацилированное производное, или его фармацевтически приемлемой соли, необязательно в фармацевтически приемлемом носителе. В альтернативном осуществлении описанные здесь соединения могут применяться при лечении особенно таких состояний, которые отличны от опухолей и рака и заключаются в аномальной или нежелательной пролиферации клеток. Примеры включают кожные заболевания, такие как гиперкератоз (включая ихтиоз, кератодермию, красный плоский лишай и псориаз), бородавки, включая остроконечные кондиломы, и буллезные поражения, а также любые состояния с аномальной или нежелательной клеточной пролиферацией, которые могут излечиваться с помощью метотрексата. Активные соединения, описанные здесь, могут также применяться для вызывания или способствования выкидышу. При предпочтительном осуществлении (-)-(2S, 4S)-1-(2-гидроксиметил-1,3-диоксолан-4-ил)цитозин предоставлен в виде указанного энантиомера (L-энантиомера) и, по существу, в отсутствие соответствующего ему энантиомера (т.е. в энантиомерно обогащенной, включая энантиомерно чистую, форме). Считается, что (-)-(2S, 4S)-1-(2-гидроксиметил-1, 3-диоксолан-4- ил)цитозин является первым примером “L”- нуклеозида, который проявляет противоопухолевую активность. (-)- (2S, 4S) -1- (2-гидроксиметил-1, 3-диоксолан-4-ил) цитозин имеет структуру, представленную формулой I 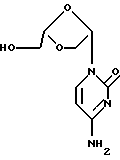 Было обнаружено, что (-)-(2S,4S)-1-(2-гидроксиметил-1,3- диоксолан-4-ил)цитозин проявляет значительную активность против раковых клеток и имеет низкую токсичность по отношению к здоровым клеткам хозяина. Неограничивающие примеры злокачественных опухолей, которые можно лечить с помощью данного соединения, включают рак легкого, ободочнопрямокишечной области, молочной железы, предстательной железы, мочевого пузыря, носоглотки, поджелудочной железы, яичника, лейкозы, лимфомы, рак тканей головы и шеи, злокачественные опухоли центральной нервной системы (включая злокачественные опухоли головного мозга), карциному шейки матки, меланому и печеночно-клеточный рак. В альтернативном осуществлении описаны способ и композиция для лечения опухолей и, в частности, рака или иных аномальных или нежелательных пролиферативных состояний у людей и других животных-хозяев, который включает в себя введение эффективного количества производного L-OddC формулы: 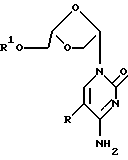 где R является F, Cl, -CH3, -С(Н)=CH2, -Br, -NO2, -C  CH или -CN, и R1 представляет собой водород, алкил, ацил, монофосфат, дифосфат или трифосфат, или его фармацевтически приемлемого производного необязательно в фармацевтически приемлемом носителе, предпочтительно, в энантиомерно обогащенной форме.
Хотя предпочтительным осуществлением данного изобретения является применение активных соединений или их производных или их солей в не встречающейся в природе конфигурации (L-конфигурация), соединения, описанные здесь, или их производные или соли могут альтернативно вводиться в виде естественной конфигурации (D-конфигурация) или в виде рацемической смеси.
Любое соединение, описанное здесь для применения в лечении опухолей, может вводиться в сочетании или в чередовании с другими противоопухолевыми фармацевтическими средствами в целях повышения эффективности лечения. Примеры включают природные продукты и их производные; антрациклины; алкилирующие агенты; антипролиферативные средства (также называемые антиметаболитами) и гормональные средства. Конкретно, средства включают, но не ограничиваются nitrogen mustards (соединения с хлоралкильной группой на конце азотной группы), этилениминовые соединения, алкилсульфаты, цисплатин, нитрозомочевины, 5-фторурацил, цитозинарабинозид, 5-азацитидин, 2-фтораденозин-5′-фосфат, 2-хлордезоксиаденозин, тамоксифен, актиномицин, амсакрин, блеомицин, карбоплатин, кармустин, циклофосфамид, циклоспорин, даунорубицин, доксирубицин, интерлейкин, ломустин, меркаптопурин, метотрексат, митомицин, тиогуанин, винбластин, факторы роста, включая Г-КСФ, ГМ-КСФ и фактор роста тромбоцитов; адреамицин, WP-16, гидроксимочевину, этопозид; CH или -CN, и R1 представляет собой водород, алкил, ацил, монофосфат, дифосфат или трифосфат, или его фармацевтически приемлемого производного необязательно в фармацевтически приемлемом носителе, предпочтительно, в энантиомерно обогащенной форме.
Хотя предпочтительным осуществлением данного изобретения является применение активных соединений или их производных или их солей в не встречающейся в природе конфигурации (L-конфигурация), соединения, описанные здесь, или их производные или соли могут альтернативно вводиться в виде естественной конфигурации (D-конфигурация) или в виде рацемической смеси.
Любое соединение, описанное здесь для применения в лечении опухолей, может вводиться в сочетании или в чередовании с другими противоопухолевыми фармацевтическими средствами в целях повышения эффективности лечения. Примеры включают природные продукты и их производные; антрациклины; алкилирующие агенты; антипролиферативные средства (также называемые антиметаболитами) и гормональные средства. Конкретно, средства включают, но не ограничиваются nitrogen mustards (соединения с хлоралкильной группой на конце азотной группы), этилениминовые соединения, алкилсульфаты, цисплатин, нитрозомочевины, 5-фторурацил, цитозинарабинозид, 5-азацитидин, 2-фтораденозин-5′-фосфат, 2-хлордезоксиаденозин, тамоксифен, актиномицин, амсакрин, блеомицин, карбоплатин, кармустин, циклофосфамид, циклоспорин, даунорубицин, доксирубицин, интерлейкин, ломустин, меркаптопурин, метотрексат, митомицин, тиогуанин, винбластин, факторы роста, включая Г-КСФ, ГМ-КСФ и фактор роста тромбоцитов; адреамицин, WP-16, гидроксимочевину, этопозид;  -, – и -, – и  -интерфероны и винкристин. Способы введения эффективных количеств данных средств легко определяются или описаны, например, в The Physician’s Desk Reference, последнее издание, изданное Medical Economics Data Production Company, и в Martindale, The Extra Pharmacopoeia, последнее издание, изданное The Pharmaceutical Press. Данные способы могут быть модифицированы в установленном порядке в целях оптимизации эффективности сочетанной или чередующейся терапии.
Краткое описание чертежей.
Фиг. 1 показывает ИД50 (-)- OddC и сочетание (-)-OddC + ТГУ (тетрагидроуридин, ингибитор цитидиндезаминазы) для раковых клеток из толстого кишечника. График отображает ингибирование роста в процентном отношении к контрольному росту от концентрации (мкМ). На графике данные отдельно по (-)-OddC представлены -интерфероны и винкристин. Способы введения эффективных количеств данных средств легко определяются или описаны, например, в The Physician’s Desk Reference, последнее издание, изданное Medical Economics Data Production Company, и в Martindale, The Extra Pharmacopoeia, последнее издание, изданное The Pharmaceutical Press. Данные способы могут быть модифицированы в установленном порядке в целях оптимизации эффективности сочетанной или чередующейся терапии.
Краткое описание чертежей.
Фиг. 1 показывает ИД50 (-)- OddC и сочетание (-)-OddC + ТГУ (тетрагидроуридин, ингибитор цитидиндезаминазы) для раковых клеток из толстого кишечника. График отображает ингибирование роста в процентном отношении к контрольному росту от концентрации (мкМ). На графике данные отдельно по (-)-OddC представлены  ), а данные по (-)-OddC + ТГУ представлены ), а данные по (-)-OddC + ТГУ представлены  Фиг. 2 представляет собой график роста массы опухоли для мышиной карциномы (Colon 38), обрабатываемой дважды в день (-)- OddC, дозами по 25 мг/кг массы тела. График отображает рост опухоли в процентном отношении к исходной массе опухоли от количества дней. Лечение мышей проводили в 1, 2, 3, 4 и 5 дни. На графике данные по контролю (без введения (-)-OddC представлены ( ), а данные по (-)-OddC представлены  Фиг. 3 показывает уровень выживаемости лейкозных мышей P388, которых лечили (-)-OddC. График отображает процент выживаемости от дней лечения. Лечение мышей проводили на 1, 2, 3, 4 и 5 день. На графике отображены уровень выживаемости контроля (без введения (- )-OddC) представлен ), уровень выживаемости мышей, которым вводили (-)-OddC по 25 мг/кг массы тела дважды в день, представлен (– –), а уровень выживаемости мышей, которым вводили (-)-OddC по 50 мг/кг массы тела дважды в день, представлен (О).
Фиг. 4 представляет собой график относительной чувствительности определенных линий раковых клеток к (-)-OddC на основе ИР50. Столбцы, вытянутые направо, представляют чувствительность клеточной линии к (-)-OddC, превышающую среднюю чувствительность всех исследованных клеточных линий. Поскольку масштаб столбцов является логарифмическим, столбец в 2 единицы вправо означает, что соединение достигает ИР50 на клеточной линии в концентрации, равной одной сотой от средней концентрации, требуемой на других клеточных линиях, и, таким образом, клеточная линия необычно чувствительна к (-)-OddC. Столбцы, вытянутые налево, соответственно означают чувствительность меньшую, чем среднюю.
Фиг. 5 представляет собой график ингибирования роста человеческой опухоли (-)-OddC. Трех- шестинедельным мышам NCr nude подкожно прививали 2106 клеток HepG2 или DU-145 на каждый бок. Лечение начинали, когда опухоли находились в прогрессивной стадии роста. Лекарства вводили дважды в день в от 0 до 4 включительно дни и размеры опухолей измеряли в указанные дни. Кривые А и В показывают лекарственные эффекты на опухоли HepG2 и опухоли DU-145 соответственно (-O-Контроль; –-Ara C 25 мг/кг, внутрибрюшинно; – –), а уровень выживаемости мышей, которым вводили (-)-OddC по 50 мг/кг массы тела дважды в день, представлен (О).
Фиг. 4 представляет собой график относительной чувствительности определенных линий раковых клеток к (-)-OddC на основе ИР50. Столбцы, вытянутые направо, представляют чувствительность клеточной линии к (-)-OddC, превышающую среднюю чувствительность всех исследованных клеточных линий. Поскольку масштаб столбцов является логарифмическим, столбец в 2 единицы вправо означает, что соединение достигает ИР50 на клеточной линии в концентрации, равной одной сотой от средней концентрации, требуемой на других клеточных линиях, и, таким образом, клеточная линия необычно чувствительна к (-)-OddC. Столбцы, вытянутые налево, соответственно означают чувствительность меньшую, чем среднюю.
Фиг. 5 представляет собой график ингибирования роста человеческой опухоли (-)-OddC. Трех- шестинедельным мышам NCr nude подкожно прививали 2106 клеток HepG2 или DU-145 на каждый бок. Лечение начинали, когда опухоли находились в прогрессивной стадии роста. Лекарства вводили дважды в день в от 0 до 4 включительно дни и размеры опухолей измеряли в указанные дни. Кривые А и В показывают лекарственные эффекты на опухоли HepG2 и опухоли DU-145 соответственно (-O-Контроль; –-Ara C 25 мг/кг, внутрибрюшинно; – – (-)-OddC, 25 мг/кг, перорально; – – (-)-OddC, 25 мг/кг, перорально; – – (-)-OddC, 25 мг/кг, внутрибрюшинно). Каждая точка данных представляет собой среднее – (-)-OddC, 25 мг/кг, внутрибрюшинно). Каждая точка данных представляет собой среднее  СО по 10 опухолям на графике А и шести опухолям на графике В.
Подробное описание изобретения.
Изобретение, как описано здесь, представляет собой способ и композицию для лечения опухолей и, в частности, рака у людей или других животных-хозяев, который заключается во введении эффективного количества (-)- (2S, 4S) -1- (2-гидроксиметил-1, 3-диоксолан-4-ил) цитозина, производного соединения, указанного здесь далее, включая 5-замещенное или 5′- или N4-алкилированное или ацилированное производное или его физиологически приемлемой соли, необязательно, в фармацевтически приемлемом носителе.
На (-) – (2S, 4S) -1- (2-гидроксиметил-1, 3- диоксолан-4-ил)цитозин ссылаются как на “L”-нуклеозид. Поскольку 2 и 5 углероды диоксоланового кольца являются хиральными, их неводородные заместители (CH2OH или цитозиновое основание, соответственно) могут быть в конфигурации как цис (на той же стороне), так и транс (на противоположной стороне) по отношению к системе диоксоланового кольца. Таким образом, следующими конфигурациями представлены четыре оптических изомера (когда диоксолановый радикал ориентирован в горизонтальной плоскости, так что кислород в 3-положении находится впереди): цис (когда обе группы “наверху”, что соответствует конфигурации природных нуклеозидов, на которые ссылаются как на “D”-нуклеозид), цис (когда обе группы “внизу”, что является не встречающейся в природе конфигурацией, на которую ссылаются как на “L”-нуклеозид), транс (когда заместитель при C2 “наверху”, а заместитель при C5 “внизу”) и транс (когда заместитель при C2 “внизу”, а заместитель при C5 “наверху”). Считается, что (-)-(2S, 4S)-1-(2- гидроксиметил-1, 3-диоксолан-4-ил) цитозин или его производное представляет собой первый пример “L”-нуклеозида, который проявляет противоопухолевую активность. Это является неожиданным в свете того факта, что данная конфигурация “L”-нуклеозида не существует в природе.
Как используется здесь, термин “энантиомерно обогащенный” относится к нуклеозидному составу, который включает в себя, по крайней мере, около 95% и предпочтительно около 97%, 98%, 99% или 100% отдельного энантиомера данного нуклеозида. При предпочтительном осуществлении (-)-(2S, 4S)-1-(2- гидроксиметил-1, 3-диоксолан-4-ил) цитозин или его производное или соль представлены в нуклеозидном составе, который, по существу, состоит из одного энантиомера, т.е. в виде указанного энантиомера (L-энантиомера) и, по существу, в отсутствие соответствующего ему D-энантиомера (т.е. в энантиомерно обогащенной, включая энантиомерно чистую, форме).
Активное соединение может быть введено, таким образом, как и любое производное, так чтобы при введении реципиенту оно было способно прямо или косвенно давать исходное соединение (-)-L-OddC или 5-замещенное производное, как иначе определяется здесь, или которое само по себе проявляет активность. Неограничивающими примерами являются фармацевтически приемлемые соли (на которые альтернативно ссылаются как на “физиологически приемлемые соли”) (-)- OddC, 5-производные, как показаны выше, и 5′- и N4– ацилированные или алкилированные производные активного соединения (на которые альтернативно ссылаются как на “физиологически активные производные”). В одном осуществлении ацильной группой является эфир карбоновой кислоты (-C(О)R), в которой некарбонильный радикал эфирной группы выбран из неразветвленного, разветвленного или циклического алкила (обычно от C1 до C18 и более обычно от C1 до C5), алкарила, аралкила, алкоксиалкила, включая метоксиметил, аралкила, включая бензил, алкил или алкокси от C1 до C4; сульфоновых эфиров, таких как алкил- или аралкилсульфонил, включая метансульфонил, моно-, ди- или трифосфорного эфира, тритила или монометокситритила, замещенного бензила, триалкилсилила (например, диметил-трет-бутилсилила) или дифенилметилсилила. Арильные группы сложных эфиров оптимально содержат фенильную группу.
Конкретные примеры фармацевтически приемлемых производных L-O-ddC включают, но не ограничиваются: СО по 10 опухолям на графике А и шести опухолям на графике В.
Подробное описание изобретения.
Изобретение, как описано здесь, представляет собой способ и композицию для лечения опухолей и, в частности, рака у людей или других животных-хозяев, который заключается во введении эффективного количества (-)- (2S, 4S) -1- (2-гидроксиметил-1, 3-диоксолан-4-ил) цитозина, производного соединения, указанного здесь далее, включая 5-замещенное или 5′- или N4-алкилированное или ацилированное производное или его физиологически приемлемой соли, необязательно, в фармацевтически приемлемом носителе.
На (-) – (2S, 4S) -1- (2-гидроксиметил-1, 3- диоксолан-4-ил)цитозин ссылаются как на “L”-нуклеозид. Поскольку 2 и 5 углероды диоксоланового кольца являются хиральными, их неводородные заместители (CH2OH или цитозиновое основание, соответственно) могут быть в конфигурации как цис (на той же стороне), так и транс (на противоположной стороне) по отношению к системе диоксоланового кольца. Таким образом, следующими конфигурациями представлены четыре оптических изомера (когда диоксолановый радикал ориентирован в горизонтальной плоскости, так что кислород в 3-положении находится впереди): цис (когда обе группы “наверху”, что соответствует конфигурации природных нуклеозидов, на которые ссылаются как на “D”-нуклеозид), цис (когда обе группы “внизу”, что является не встречающейся в природе конфигурацией, на которую ссылаются как на “L”-нуклеозид), транс (когда заместитель при C2 “наверху”, а заместитель при C5 “внизу”) и транс (когда заместитель при C2 “внизу”, а заместитель при C5 “наверху”). Считается, что (-)-(2S, 4S)-1-(2- гидроксиметил-1, 3-диоксолан-4-ил) цитозин или его производное представляет собой первый пример “L”-нуклеозида, который проявляет противоопухолевую активность. Это является неожиданным в свете того факта, что данная конфигурация “L”-нуклеозида не существует в природе.
Как используется здесь, термин “энантиомерно обогащенный” относится к нуклеозидному составу, который включает в себя, по крайней мере, около 95% и предпочтительно около 97%, 98%, 99% или 100% отдельного энантиомера данного нуклеозида. При предпочтительном осуществлении (-)-(2S, 4S)-1-(2- гидроксиметил-1, 3-диоксолан-4-ил) цитозин или его производное или соль представлены в нуклеозидном составе, который, по существу, состоит из одного энантиомера, т.е. в виде указанного энантиомера (L-энантиомера) и, по существу, в отсутствие соответствующего ему D-энантиомера (т.е. в энантиомерно обогащенной, включая энантиомерно чистую, форме).
Активное соединение может быть введено, таким образом, как и любое производное, так чтобы при введении реципиенту оно было способно прямо или косвенно давать исходное соединение (-)-L-OddC или 5-замещенное производное, как иначе определяется здесь, или которое само по себе проявляет активность. Неограничивающими примерами являются фармацевтически приемлемые соли (на которые альтернативно ссылаются как на “физиологически приемлемые соли”) (-)- OddC, 5-производные, как показаны выше, и 5′- и N4– ацилированные или алкилированные производные активного соединения (на которые альтернативно ссылаются как на “физиологически активные производные”). В одном осуществлении ацильной группой является эфир карбоновой кислоты (-C(О)R), в которой некарбонильный радикал эфирной группы выбран из неразветвленного, разветвленного или циклического алкила (обычно от C1 до C18 и более обычно от C1 до C5), алкарила, аралкила, алкоксиалкила, включая метоксиметил, аралкила, включая бензил, алкил или алкокси от C1 до C4; сульфоновых эфиров, таких как алкил- или аралкилсульфонил, включая метансульфонил, моно-, ди- или трифосфорного эфира, тритила или монометокситритила, замещенного бензила, триалкилсилила (например, диметил-трет-бутилсилила) или дифенилметилсилила. Арильные группы сложных эфиров оптимально содержат фенильную группу.
Конкретные примеры фармацевтически приемлемых производных L-O-ddC включают, но не ограничиваются: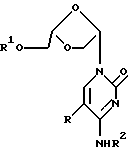 где R является F, Cl, -CH3, -C(H)=CH2, -C CH или -CN, -Br, -NO2, a R1 и R2 независимо выбраны из группы, содержащей водород, алкил и ацил, конкретно включая, но не ограничиваясь ими, метил, этил, пропил, бутил, пентил, гексил, изопропил, изобутил, втор-бутил, трет-бутил, изопентил, амил, трет-пентил, 3-метилбутирил, гидросукцинат, 3-хлорбензоат, циклопентил, циклогексил, бензоил, ацетил, пивалоил, мезилат, пропионил, бутирил, валерил, капроновую, каприловую, каприновую, лауриновую, миристиновую, пальмитиновую, стеариновую, олеиновую кислоты и аминокислоты, включая, но не ограничиваясь, аланил, валинил, лейцинил, изолейцинил, пролинил, фенилаланинил, триптофанил, метионинил, глицинил, серинил, треонинил, цистеинил, тирозинил, аспарагинил, глютаминил, аспартоил, глютаоил, лизинил, аргининил и гистидинил. При предпочтительном осуществлении производное представлено в виде L-энантиомера и, по существу, в отсутствие соответствующего ему энантиомера (т.е. в энантиомерно обогащенной, включая энантиомерно чистую, форме).
L-OddC или его производное может быть предоставлено в виде фармацевтически приемлемых солей. Как используется здесь, термин “фармацевтически приемлемые соли или комплексы” относится к солям или комплексам L-OddC или его производных, которые сохраняют желательную биологическую активность исходного соединения и, если и проявляют, то минимальные токсические эффекты. Неограничивающими примерами таких солей являются (а) соли добавления кислот, образованные неорганическими кислотами (например, хлорводородная кислота, бромводородная кислота, серная кислота, фосфорная кислота, азотная кислота и тому подобное), и соли, образованные органическими кислотами, такими как уксусная кислота, винная кислота, янтарная кислота, яблочная кислота, аскорбиновая кислота, бензойная кислота, дубильная кислота, памоевая кислота, альгиновая кислота, полиглютаминовая кислота, нафталинсульфокислоты, нафталиндисульфокислоты и полигалактуроновая кислота; (б) соли добавления оснований, образованные поливалентными катионами металлов, таких как цинк, кальций, висмут, барий, магний, алюминий, медь, кобальт, никель, кадмий и тому подобное, или органическим катионом, образовавшимся из N,N- дибензилэтилендиамина, аммония или этилендиамина; или (в) сочетание (а) и (б); например, соль танната цинка и тому подобное.
Модификации активного соединения, особенно по N4– и 5′-0-положениям, могут сильно влиять на растворимость, биодоступность и скорость метаболизма активных продуктов, таким образом, предоставляя возможность контролировать транспорт активных продуктов. Далее, модификации могут влиять на противораковую активность соединения, в некоторых случаях увеличивая активность по сравнению с исходным соединением. Это может быть легко оценено путем получения производного и исследования его противораковой активности методами, описанными здесь, или другим методом, известным специалистам в данной области.
Таким образом, настоящее изобретение включает в себя следующие объекты изобретения:(а) (-)-(2S, 4S)-1-(2-гидроксиметил-1, 3-диоксолан-4-ил) цитозин и его производные и соли; (б) (+)-(2S, 4S)-1-(2-гидроксиметил-1, 3-диоксолан-4-ил) цитозин и его производные и соли; (в) (-/+)-(2S, 4S)-1-(2-гидроксиметил-1, 3-диоксолан-4-ил) цитозин и его производные и соли; (г) (-)-(2S, 4S) -1- (2-гидроксиметил-1, 3-диоксолан-4-ил) цитозин и его производные и соли или его (+)-энантиомер или их рацемическая смесь и их фармацевтически приемлемые производные и соли для применения в медицинском лечении, например, для лечения или профилактики опухоли, включая раковую опухоль; (д) применение (-)-(2S, 4S)-1-(2-гидроксиметил-1, 3-диоксолан-4-ил)цитозина и его фармацевтически приемлемых производных и солей или его (+) -энантиомера или их рацемической смеси и их фармацевтически приемлемых производных и солей в производстве лекарства для лечения опухоли, включая раковую опухоль; (е) фармацевтические составы, содержащие (-)-(2S, 4S)-1-(2-гидроксиметил-1, 3-диоксолан-4-ил) цитозин и его фармацевтически приемлемые производные и соли или его (+)-энантиомер или их рацемическую смесь или их фармацевтически приемлемые производные и соли совместно с фармацевтически приемлемым носителем; (ж) способ получения (-)-(2S, 4S)-1-(2- гидроксиметил-1,3-диоксолан-4-ил)цитозина, который заключается: (1) во взаимодействии необязательно защищенного цитозина с 1,3- диоксоланом формулы А 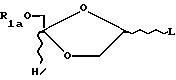 где R1a является водородной или гидроксильной защитной группой, включая ацильную группу, a L является уходящей группой; и необязательное удаление любой гидроксильной защитной группы. (2) во взаимодействии соединения формулы В 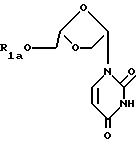 (где R1a указан выше) с агентом, служащим для превращения оксогруппы в 4-положении урацильного кольца в аминогруппу; причем удаляют все оставшиеся защитные группы, что дает желательный продукт; (з) способ получения (-)- или (+)- энантиомера (2S, 4S)-1-(2-гидроксиметил-1, 3-диоксолан-4-ил) цитозина, который заключается в том, что соединение или его производное (например, 5′-сложного эфира) в виде смеси (-) и (+)- энантиомеров подвергают условиям или проводят взаимодействию с реагентами (например, с подходящим ферментом), служащим для разделения энантиомеров и, если необходимо, превращают полученное производное в исходное соединение. Альтернативно, смесь может быть пропущена через колонку для хиральной жидкостной хроматографии, которая разделяет энантиомеры данного типа. (и) способ получения (2S, 4S)-1-(2-гидроксиметил-1,3-диоксолан-4-ил) цитозина, который включает взаимодействие защищенного 1,3-диоксолана формулы: 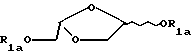 с защищенным цитозиновым основанием, которое необязательно замещено по 5-положению, применяя кислоту Льюиса, которая не рацемизует продукт, такую как триметилсилилтрифлат. Что касается способа ж) (1), гидроксизащитная группа включает в себя защитные группы, подробно описанные ниже, включая ацил (например, ацетил), арилацил (например, бензоил или замещенный бензоил), тритил или монометокситритил, бензил или замещенный бензил, тризамещенный силил, включая триалкилсилил (например, диметил-трет-бутилсилил) или дифенилметилсилил. Цитозиновое соединение может быть необязательно защищено тризамещенными силильными группами. Защитные группы могут быть удалены традиционным способом. Уходящая группа L является одной из уходящих групп, известных в области химии нуклеозидов, например, галогеном, таким как хлор, фтор или бром, тозилом, мезилом, трифлатом, алкокси, такие как метокси и этокси; или ацилом, таким как ацетил или бензоил. Взаимодействие по способу ж) (1) может быть проведено в органическом растворителе (например, в 1,2-дихлорэтане или ацетонитриле) в присутствии кислоты Льюиса, такой как SnCl4, хлорид титана или триметилсилилтрифлат. Соединения формулы А (где L представляет собой ацильную группу, например, ацетильную группу) могут быть получены путем взаимодействия соединения формулы С. 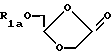 (где R1a определен выше) с восстановителем, например, с литийалюминийгидридом, с последующей обработкой подходящим традиционным реагентом с получением желательного промежуточного продукта, например, для ацилирования ангидридом карбоновой кислоты, в частности, ацетангидридом, для ацилирования хлорирующими или бромирующими реагентами для галогенирования, или алкилирующими реагентами. Соединение формулы С может быть получено путем взаимодействия соединения формулы D, где R1a является H, 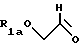 D или E с HOCH2СО2H при повышенной температуре. Соединение формулы E, где R1a обозначает R, может быть получено путем озонолиза аллилового простого эфира или сложного эфира, имеющего формулу CH2= CH-CH2-OR, или простого диэфира или сложного диэфира 2-бутен-1,3-диола, имеющего формулу ROCH2-CH=CH-CH2OR, в которой R является защитной группой, такой как алкильная, силильная или ацильная группа. Что касается способа ж) 2), соединение формулы С может быть обработано 1,2,4-триазолом совместно с 4-хлорфенилдихлорфосфатом с образованием соответствующего 4-(1,2,4-триазоилильного) соединения, которое затем превращают в желательное 4-амино(цитидиновое) соединение путем взаимодействия, например, с метанолом. Исходные продукты формул В и С могут быть получены, например, путем взаимодействия подходящего (необязательно защищенного) основания с соединением формулы А способом, аналогичным описанному в способе ж) 1). Урацил и цитозин могут быть поставлены Aldrich Chemical Co., Milwaukee, W1 53233, USA. Li-OddC или его производное могут быть преобразованы в фармацевтически приемлемый сложный эфир путем взаимодействия с подходящим этерифицирующим агентом, например, с галогенангидридом или ангидридом карбоновой кислоты. L-OddC или его фармацевтически приемлемое производное может быть преображено в его фармацевтически приемлемую соль обычным способом, например, путем обработки подходящим основанием. Сложный эфир или соль могут быть превращены в исходное соединение, например, путем гидролиза. При альтернативном осуществлении соединения, описанные здесь, могут быть применены для лечения состояний, специфически отличающихся от опухолей или рака, которые включают в себя аномальную или нежелательную пролиферацию клеток. Примеры включают кожные заболевания, такие как гиперкератоз (включая ихтиоз, кератодермию, плоский красный лишай и псориаз), бородавки, включая остроконечные кондиломы, и буллезные поражения, а также любое состояние аномальной клеточной пролиферации, которые могут излечиваться метотрексатом. Активные соединения, описанные здесь, также могут применяться для вызывания или способствования выкидышу. Таким образом, изобретение также включает (-)-(2S, 4S)-1-(2- гидроксиметил-1,3-диоксолан-4-ил)цитозин и его производные и соли или его (+)-энантиомер или их рацемическую смесь и их фармацевтически приемлемые производные и соли для применения в медикаментозной терапии, например, для лечения или профилактики состояний с аномальной или нежелательной пролиферацией клеток; а также применение (-)-(2S, 4S)-1-(2-гидроксиметил-1, 3- диоксолан-4-ил)цитозина и его производных и солей или его (+)- энантиомера или их рацемической смеси и их фармацевтически приемлемых производных и солей при получении лекарственного средства для лечения состояний аномальной или нежелательной пролиферации клеток. II. Получение активных соединений. (-)-L-OddC и его производные могут быть получены, как описано выше, по методу, подробно описанному в Международной Публикации PCT N WO 92/18517, опубликованной 29 октября 1992 года, по методу, описанному на Схеме 1 (см. в конце описания) и в рабочих примерах 1-7, приведенных ниже, или по любому другому методу, известному специалистам в данной области. Данные методы или другие известные методы могут быть приспособлены для получения производных L-OddC, приведенных в качестве примера. Пример 1. Получение 6-ангидро-L-гулозы. 6-Ангидро-L-гулозу получали в одну стадию из L-гулозы путем обработки L-гулозы кислотой, например, 0,5 н. HCl, с выходом 60% (Evans, M.E., et al., Carbohydr. Res. (1973), 28, 359). (2) прямо преобразовывали без избирательной защиты, как делалось до этого (Jeong, L.S. et al., Tetrahedron Lett. (1992), 33, 595 и Beach, J.W. et al., J.Я Org. Chem. (1992, в печати)), в диоксолантриол (3) путем окисления с помощью NaIO4 с последующим восстановлением с помощью NaBH4, который без выделения превращали в изопропилиденовое производное (4). Бензоилирование до (5), удаление защиты до (6) и окисление диола (6) давали кислоту (7). Окислительное декарбоксилирование (7) с помощью Pb(OAc)4 в сухом ТГФ давало ацетат (8), ключевой промежуточный продукт, с хорошим выходом. Ацетат конденсировали с желаемыми пиримидинами (например, силилированный тимин и N-ацетилцитозин) в присутствии TMCOTf, что позволяло получить ,-смесь, которую разделяли на колонке с силикагелем, что давало индивидуальные изомеры (9 и 10). Дебензоилирование метанольным аммиаком давало желательный (-)-OddC (11).
Пример 2: Получение (-)-1,6-ангидро- –-L- -гулопиранозы(2).
Смесь L-гулозы (1) (33 г, 0,127 моль) и 0,5 н. HCl (330 мл, 0,165 моль) нагревали с обратным холодильником в течение 20 часов. Смесь охлаждали и нейтрализовали до pH 6 с помощью смолы (Dovex-2, HCO3-форма) при барботировании воздуха. Смолу регенерировали промыванием 10% HCl, водой, метанолом, водой и насыщенным раствором NaHCO3. Реакционную смесь фильтровали и смолу промывали водой (500 мл). Объединенный фильтрат концентрировали досуха и сушили в вакууме в течение ночи. Остаток очищали с помощью колонки (высота 5 см, силикагель, пористый, CHCl3-CH3ОН, 10: 1), что давало бледно-желтое твердое вещество, которое перекристаллизовывали из абсолютного спирта, что давало бесцветное твердое вещество (2) [Rf = 0,43 (CHCl3-CH3ОН, 5:1), 7,3 г, 35,52%] . Полученную L-гулозу (Rf = 0,07, 11 г) вновь рециклизовывали, что давало (2) (5 г, общий выход 60%): т.пл. 142,5-145oC; 1H ЯМР (DMCO-d6)  3,22-3,68 (м, 4H, Н-2, -3, -4 и -6a), 3,83 (Д, J6b,6a = 7,25 Гц, 1H, Hb-6), 4,22 (псевдо т, J5,6a = 4,61 и 4,18 Гц, H, Н-5), 4,46 (д, J2-OH,2 = 6,59 Гц, 1H, 2- ОН, способный к обмену с D2O), 4,62 (д, J3-OH,3 = 5,28 Гц, 1H, 3-OH, способный к обмену с D2O), 5,07 (д, J4-OH,4 = 4,84 Гц, 1H, 4-ОН, способный к обмену с D2O), 5,20 (д, J1,2 = 2,19 Гц, 1H, Н-1). []2D5 -50,011 (с, 1,61, CH3ОН).
Пример 3: Получение (-)-(1’S,2S,4S)-4-(1,2-дигидроксиэтил- 1,2-О-изопропилиден)-2-гидроксиметил)диоксолана (4).
Раствор NaIO4 (22,36 г, 0,1 моль) в воде (300 мл) по каплям добавляли к раствору (2) (11,3 г, 0,07 моль) в метаноле (350 мл), охлажденному до 0oC, в течение 10 минут. Смесь механически перемешивали в течение 15 минут. К данной смеси добавляли NaBH4 (7,91 г, 0,21 моль) и реакционную смесь перемешивали в течение 10 минут при 0oC. Белое твердое вещество отфильтровывали и твердое вещество промывали метанолом (300 мл). Объединенный фильтрат нейтрализовали с помощью 0,5 н. HCl (~200 мл) и концентрировали досуха. Остаток сушили в вакууме в течение ночи. Сиропообразный остаток растирали с метанолом-ацетоном (1:5, 1200 мл), применяя механическую мешалку (5 часов), и белое твердое вещество (первое) отфильтровывали. Фильтрат концентрировали досуха и остаток растворяли в ацетоне (500 мл) и далее в п-толуолсульфокислоте (6,63 г, 0,035 моль). После перемешивания в течение 6 часов смесь нейтрализовали триэтиламином, твердое вещество (второе) отфильтровывали и фильтрат концентрировали досуха. Остаток растворяли в этилацетате (350 мл) и промывали водой (50 мл х 2), сушили (MgSO4), фильтровали и выпаривали, с получением сырого (4) (3,6 г) в виде желтоватого сиропа. Водный слой концентрировали досуха и сушили в вакууме. Полученное твердое вещество (первое и второе) объединяли с высушенным веществом из водного слоя и рециклизовали путем перемешивания в течение 1 часа в 10% метаноле-ацетоне (900 мл) и п-толуолсульфоновой кислоте (16 г, 0,084 моль) и получали сырой (4) (5,6 г). Сырой (4) очищали с помощью сухой колонки с силикагелем CH3OH-CHCl3, 1-5%) с получением (4) [Rf = 0,82 (CHCl3-CH3ОН, 10:1), 8,8 г, 61,84%] в виде бесцветного масла. 1H ЯМР (DMCO-d6) 1,26 и 1,32 (2 х с, 2 х 3H, изопропилиден), 3,41 (дд, 3,22-3,68 (м, 4H, Н-2, -3, -4 и -6a), 3,83 (Д, J6b,6a = 7,25 Гц, 1H, Hb-6), 4,22 (псевдо т, J5,6a = 4,61 и 4,18 Гц, H, Н-5), 4,46 (д, J2-OH,2 = 6,59 Гц, 1H, 2- ОН, способный к обмену с D2O), 4,62 (д, J3-OH,3 = 5,28 Гц, 1H, 3-OH, способный к обмену с D2O), 5,07 (д, J4-OH,4 = 4,84 Гц, 1H, 4-ОН, способный к обмену с D2O), 5,20 (д, J1,2 = 2,19 Гц, 1H, Н-1). []2D5 -50,011 (с, 1,61, CH3ОН).
Пример 3: Получение (-)-(1’S,2S,4S)-4-(1,2-дигидроксиэтил- 1,2-О-изопропилиден)-2-гидроксиметил)диоксолана (4).
Раствор NaIO4 (22,36 г, 0,1 моль) в воде (300 мл) по каплям добавляли к раствору (2) (11,3 г, 0,07 моль) в метаноле (350 мл), охлажденному до 0oC, в течение 10 минут. Смесь механически перемешивали в течение 15 минут. К данной смеси добавляли NaBH4 (7,91 г, 0,21 моль) и реакционную смесь перемешивали в течение 10 минут при 0oC. Белое твердое вещество отфильтровывали и твердое вещество промывали метанолом (300 мл). Объединенный фильтрат нейтрализовали с помощью 0,5 н. HCl (~200 мл) и концентрировали досуха. Остаток сушили в вакууме в течение ночи. Сиропообразный остаток растирали с метанолом-ацетоном (1:5, 1200 мл), применяя механическую мешалку (5 часов), и белое твердое вещество (первое) отфильтровывали. Фильтрат концентрировали досуха и остаток растворяли в ацетоне (500 мл) и далее в п-толуолсульфокислоте (6,63 г, 0,035 моль). После перемешивания в течение 6 часов смесь нейтрализовали триэтиламином, твердое вещество (второе) отфильтровывали и фильтрат концентрировали досуха. Остаток растворяли в этилацетате (350 мл) и промывали водой (50 мл х 2), сушили (MgSO4), фильтровали и выпаривали, с получением сырого (4) (3,6 г) в виде желтоватого сиропа. Водный слой концентрировали досуха и сушили в вакууме. Полученное твердое вещество (первое и второе) объединяли с высушенным веществом из водного слоя и рециклизовали путем перемешивания в течение 1 часа в 10% метаноле-ацетоне (900 мл) и п-толуолсульфоновой кислоте (16 г, 0,084 моль) и получали сырой (4) (5,6 г). Сырой (4) очищали с помощью сухой колонки с силикагелем CH3OH-CHCl3, 1-5%) с получением (4) [Rf = 0,82 (CHCl3-CH3ОН, 10:1), 8,8 г, 61,84%] в виде бесцветного масла. 1H ЯМР (DMCO-d6) 1,26 и 1,32 (2 х с, 2 х 3H, изопропилиден), 3,41 (дд, 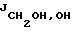 = 3,96 Гц, = 3,96 Гц, 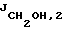 = 3,96 Гц, 2H, CH2OH), 3,56-4,16 (м, 6H, Н-4, -5, -1′ и -2′), 4,82 (т, = 3,96 Гц, 2H, CH2OH), 3,56-4,16 (м, 6H, Н-4, -5, -1′ и -2′), 4,82 (т, 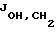 = 6,0 Гц, 1H, CH2OH, способный к обмену с D2O), 4,85 (т, = 6,0 Гц, 1H, CH2OH, способный к обмену с D2O), 4,85 (т, 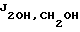 = 3.96 Гц, 1H, Н-2). []2D5 – 12,48 (с, 1,11, CHCl3), Анал. расч. для C9H16O5: С 52,93; H 7,90. Получ.: С 52,95; H 7,86.
Пример 4; Получение (+)-(1’S, 2S,4S)-4-(1,2-дигидроксиметил- 1,2-О-изопропилиден)-2-(О-бензоилоксиметил) диоксолана (5).
К раствору (4) (8,5 г, 0,042 моль) в пиридине-CH2Cl2 (1:2, 120 мл) при 0oC по каплям добавляли бензоилхлорид (6,5 мл, 0,056 моль) и температуру повышали до комнатной температуры. После перемешивания в течение 2 часов реакцию гасили добавлением метанола (10 мл) и смесь концентрировали досуха в вакууме. Остаток растворяли в CH2Cl2 (300 мл) и промывали водой (100 мл х 2), солевым раствором, сушили (MgSO4), фильтровали и выпаривали, с получением желтоватого сиропа, который очищали методом хроматографии на колонке с силикагелем (EtOAc-гексан 4-30%), и получали (5) [Rа=0,45 (гексан-EtOAc, 3: 1), 10,7 г, 83,4%] в виде бесцветного масла. 1H ЯМР (CDCl3) 1,35 и 1,44 (2 х с, 2 х 3H, изопропилиден), 3,3-4,35 (м, 6H, Н-4, -5, -1′ и -2′), 4,44 (д, J= 3,96 Гц, 2H, CH2-OBz), 5,29 (т, J=3,74 Гц, 1H, Н-2), 7,3-7,64, 8,02-8,18 (м, 3H, 2H, -OBz). []2D5 + 10,73 (с, 1,75, CH3ОН). Анал. расч. для C16H20O6: С 62,33; H 6,54. Получ.: С 62,39; H 6,54.
Пример 5: Получение (+)-(1’S,2S,4S)-4-(1,2-диrидpoкcиэтил-2-(О-бензоилоксиметил)диоксолана (6).
Смесь (5) (5,7 г, 0,018 моль) и п-толуолсульфокислоты (1,05 г, 0,0055 моль) в метаноле (70 мл) перемешивали при комнатной температуре в течение 2 часов. Не завершая реакцию, растворитель выпаривали до половины исходного объема и добавляли еще метанол (50 мл) и п-толуолсульфоновую кислоту (0,7 г, 3,68 ммоль). После перемешивания в течение еще одного часа реакционную смесь нейтрализовали триэтиламином и растворитель выпаривали досуха.
Остаток очищали методом хроматографии на колонке с силикагелем (EtOAc-гексан 10- 33%), с получением (6) [Rf=0,15 (гексан-EtOAc, 1:1), 4,92 г, 99,2%] в виде бесцветного сиропа. 1H ЯМР (ДМСО-d6) 3,43 (м, 2H, Н-2′), 3,67-4,1 (м, 4H, Н-4, -5 и -1′), 4,32 (д, J=3,73 Гц, 2H, CH2-OBz), 4,60 (т, J= 5,72 Гц, 2′-ОН, способный к обмену с D2O), 5,23 (т, J=3,96 Гц, 1H, Н-2), 7,45-7,7, 7,93-8,04 (м, 3H, 2H, -OBz), []2D5 + 9,16 (c, 1,01, CHCl3), Анал. расч. для C13H16O6: С 58,20; H 6,01. Получ.: С 58,02; H 6,04.
Пример 6: Получение (-)-(2S,4S)- и (2S,4R)-4-ацетокси-2-(O- бензоилоксиметил)диоксолана (8).
К раствору (6) (3,04 г, 0,011 моль) в CCl4:CH3CN (1:1, 160 мл) добавляли раствор NaIO4 (10,18 г, 0,048 моль) в воде (120 мл) с последующим добавлением гидрата RuO2 (0,02 г). После того, как реакционную смесь перемешивали в течение 5 часов, твердое вещество удаляли путем фильтрования через Цилит и фильтрат выпаривали до 1/3 объема. Остаток растворяли в CH2CI2 (100 мл) и водный слой экстрагировали добавлением CH2Cl2 (100 мл х 2). Объединенный органический слой промывали солевым раствором (50 мл), сушили (MgSO4), фильтровали, выпаривали досуха в вакууме в течение 16 часов, с получением сырого (7) (2,6 г, 91%).
К раствору сырого (7) (2,6 г, 0,01 моль) в сухом ТГФ (60 мл) добавляли Pb(ОАс)4 (5,48 г, 0,0124 моль) и пиридин (0,83 мл, 0,103 моль) в атмосфере N2. Смесь перемешивали в течение 45 минут в атмосфере N2 и твердое вещество удаляли путем фильтрования. Твердое вещество промывали этилацетатом (60 мл) и объединенный органический слой выпаривали досуха. Остаток очищали методом хроматографии на колонке с силикагелем (гексан-EtOAc, 2:1) и получали (8) [Rf= 0,73 и 0,79 (гексан-EtOAc, 2: 1), 1,9 г, 69,34%] в виде бесцветного масла. 1H ЯМР (CDCl3) 1,998, 2,11 (2 х с, 3H, -ОАс), 3,93-4,33 (м, 2H, Н-5), 4,43, 4,48 (2 х д, J=3,73, 3,74 Гц, 2H, CH2OBz), 5,46, 5,55 (2 х т, J= 4,18, 3,63 Гц, IH, H-2), 6,42 (м, 1H, H-4), 7,33-7,59, 8,00-8,15 (м, 3H, 2H, -OBz). []2D5 – 12,53 (с, 1,11, CHCl3). Анал. расч. для C13H14O6: С 58,64; H 5,30. Получ.: C 58,78; H 5,34.
Пример 7: Получение (-)-(2S,4S)-1-[2-(бензоил)-1,3- диоксолан-4-ил]цитозина (9) и (+)-(2S, 4S)-1-[2-(бензилокси)-1,3- диoкcoлaн-4-ил]lцитoзинa (10).
Смесь N4-ацетилцитозина (1,24 г, 7,52 ммоль) в сухом дихлорэтане (20 мл), гексаметилдисилазана (15 мл) и сульфата аммония (каталитическое количество) нагревали с обратным холодильником в течение 4 часов в атмосфере азота. Полученный прозрачный раствор охлаждали до комнатной температуры. К данному силилированному ацетилцитозину добавляли раствор (8) (1,0 г, 3,76 ммоль) в сухом дихлорэтане (10 мл) и ТМСОТf (1,46 мл, 7,55 ммоль). Смесь перемешивали в течение 6 часов. Добавляли насыщенный NaHCO3 (10 мл) и смесь перемешивали еще в течение 15 минут и фильтровали через пад из Цилита. Фильтрат выпаривали и твердое вещество растворяли в EtOAc и промывали водой и солевым раствором, сушили, фильтровали и выпаривали, с получением сырого продукта. Данный сырой продукт очищали на колонке с силикагелем (5% CH3ОН/CHCl3) и получали чистой ,-смеси (9) и (10) (0,40 г, 30%) и ,-смеси (13) и (14) (0,48 г, 40%). Смесь (14) вновь ацилировали для разделения, объединенную ,-смесь разделяли на длинной колонке с силикагелем (3% CH3ОН/CHCl3) и получали (9) (0,414 г, 30,7%) и (10) (0,481 г, 35,6%) в виде пены. Эти пены растирали с CH3ОН и получали белые твердые вещества. 9: УФ (CH3ОН) = 3.96 Гц, 1H, Н-2). []2D5 – 12,48 (с, 1,11, CHCl3), Анал. расч. для C9H16O5: С 52,93; H 7,90. Получ.: С 52,95; H 7,86.
Пример 4; Получение (+)-(1’S, 2S,4S)-4-(1,2-дигидроксиметил- 1,2-О-изопропилиден)-2-(О-бензоилоксиметил) диоксолана (5).
К раствору (4) (8,5 г, 0,042 моль) в пиридине-CH2Cl2 (1:2, 120 мл) при 0oC по каплям добавляли бензоилхлорид (6,5 мл, 0,056 моль) и температуру повышали до комнатной температуры. После перемешивания в течение 2 часов реакцию гасили добавлением метанола (10 мл) и смесь концентрировали досуха в вакууме. Остаток растворяли в CH2Cl2 (300 мл) и промывали водой (100 мл х 2), солевым раствором, сушили (MgSO4), фильтровали и выпаривали, с получением желтоватого сиропа, который очищали методом хроматографии на колонке с силикагелем (EtOAc-гексан 4-30%), и получали (5) [Rа=0,45 (гексан-EtOAc, 3: 1), 10,7 г, 83,4%] в виде бесцветного масла. 1H ЯМР (CDCl3) 1,35 и 1,44 (2 х с, 2 х 3H, изопропилиден), 3,3-4,35 (м, 6H, Н-4, -5, -1′ и -2′), 4,44 (д, J= 3,96 Гц, 2H, CH2-OBz), 5,29 (т, J=3,74 Гц, 1H, Н-2), 7,3-7,64, 8,02-8,18 (м, 3H, 2H, -OBz). []2D5 + 10,73 (с, 1,75, CH3ОН). Анал. расч. для C16H20O6: С 62,33; H 6,54. Получ.: С 62,39; H 6,54.
Пример 5: Получение (+)-(1’S,2S,4S)-4-(1,2-диrидpoкcиэтил-2-(О-бензоилоксиметил)диоксолана (6).
Смесь (5) (5,7 г, 0,018 моль) и п-толуолсульфокислоты (1,05 г, 0,0055 моль) в метаноле (70 мл) перемешивали при комнатной температуре в течение 2 часов. Не завершая реакцию, растворитель выпаривали до половины исходного объема и добавляли еще метанол (50 мл) и п-толуолсульфоновую кислоту (0,7 г, 3,68 ммоль). После перемешивания в течение еще одного часа реакционную смесь нейтрализовали триэтиламином и растворитель выпаривали досуха.
Остаток очищали методом хроматографии на колонке с силикагелем (EtOAc-гексан 10- 33%), с получением (6) [Rf=0,15 (гексан-EtOAc, 1:1), 4,92 г, 99,2%] в виде бесцветного сиропа. 1H ЯМР (ДМСО-d6) 3,43 (м, 2H, Н-2′), 3,67-4,1 (м, 4H, Н-4, -5 и -1′), 4,32 (д, J=3,73 Гц, 2H, CH2-OBz), 4,60 (т, J= 5,72 Гц, 2′-ОН, способный к обмену с D2O), 5,23 (т, J=3,96 Гц, 1H, Н-2), 7,45-7,7, 7,93-8,04 (м, 3H, 2H, -OBz), []2D5 + 9,16 (c, 1,01, CHCl3), Анал. расч. для C13H16O6: С 58,20; H 6,01. Получ.: С 58,02; H 6,04.
Пример 6: Получение (-)-(2S,4S)- и (2S,4R)-4-ацетокси-2-(O- бензоилоксиметил)диоксолана (8).
К раствору (6) (3,04 г, 0,011 моль) в CCl4:CH3CN (1:1, 160 мл) добавляли раствор NaIO4 (10,18 г, 0,048 моль) в воде (120 мл) с последующим добавлением гидрата RuO2 (0,02 г). После того, как реакционную смесь перемешивали в течение 5 часов, твердое вещество удаляли путем фильтрования через Цилит и фильтрат выпаривали до 1/3 объема. Остаток растворяли в CH2CI2 (100 мл) и водный слой экстрагировали добавлением CH2Cl2 (100 мл х 2). Объединенный органический слой промывали солевым раствором (50 мл), сушили (MgSO4), фильтровали, выпаривали досуха в вакууме в течение 16 часов, с получением сырого (7) (2,6 г, 91%).
К раствору сырого (7) (2,6 г, 0,01 моль) в сухом ТГФ (60 мл) добавляли Pb(ОАс)4 (5,48 г, 0,0124 моль) и пиридин (0,83 мл, 0,103 моль) в атмосфере N2. Смесь перемешивали в течение 45 минут в атмосфере N2 и твердое вещество удаляли путем фильтрования. Твердое вещество промывали этилацетатом (60 мл) и объединенный органический слой выпаривали досуха. Остаток очищали методом хроматографии на колонке с силикагелем (гексан-EtOAc, 2:1) и получали (8) [Rf= 0,73 и 0,79 (гексан-EtOAc, 2: 1), 1,9 г, 69,34%] в виде бесцветного масла. 1H ЯМР (CDCl3) 1,998, 2,11 (2 х с, 3H, -ОАс), 3,93-4,33 (м, 2H, Н-5), 4,43, 4,48 (2 х д, J=3,73, 3,74 Гц, 2H, CH2OBz), 5,46, 5,55 (2 х т, J= 4,18, 3,63 Гц, IH, H-2), 6,42 (м, 1H, H-4), 7,33-7,59, 8,00-8,15 (м, 3H, 2H, -OBz). []2D5 – 12,53 (с, 1,11, CHCl3). Анал. расч. для C13H14O6: С 58,64; H 5,30. Получ.: C 58,78; H 5,34.
Пример 7: Получение (-)-(2S,4S)-1-[2-(бензоил)-1,3- диоксолан-4-ил]цитозина (9) и (+)-(2S, 4S)-1-[2-(бензилокси)-1,3- диoкcoлaн-4-ил]lцитoзинa (10).
Смесь N4-ацетилцитозина (1,24 г, 7,52 ммоль) в сухом дихлорэтане (20 мл), гексаметилдисилазана (15 мл) и сульфата аммония (каталитическое количество) нагревали с обратным холодильником в течение 4 часов в атмосфере азота. Полученный прозрачный раствор охлаждали до комнатной температуры. К данному силилированному ацетилцитозину добавляли раствор (8) (1,0 г, 3,76 ммоль) в сухом дихлорэтане (10 мл) и ТМСОТf (1,46 мл, 7,55 ммоль). Смесь перемешивали в течение 6 часов. Добавляли насыщенный NaHCO3 (10 мл) и смесь перемешивали еще в течение 15 минут и фильтровали через пад из Цилита. Фильтрат выпаривали и твердое вещество растворяли в EtOAc и промывали водой и солевым раствором, сушили, фильтровали и выпаривали, с получением сырого продукта. Данный сырой продукт очищали на колонке с силикагелем (5% CH3ОН/CHCl3) и получали чистой ,-смеси (9) и (10) (0,40 г, 30%) и ,-смеси (13) и (14) (0,48 г, 40%). Смесь (14) вновь ацилировали для разделения, объединенную ,-смесь разделяли на длинной колонке с силикагелем (3% CH3ОН/CHCl3) и получали (9) (0,414 г, 30,7%) и (10) (0,481 г, 35,6%) в виде пены. Эти пены растирали с CH3ОН и получали белые твердые вещества. 9: УФ (CH3ОН)  max 298 нм; Анал. (C17H17N3О8) С, H, N. 10: УФ (CH3ОН) max 298 нм.
Пример 8: Получение (-)-(2S,4S)-1-(2-гидроксиметил-1,3- диoкcoлaн-4-ил)цитoзинa (11).
Раствор (9) (0,29 г, 0,827 ммоль) в CH3ОН/NH3 (50 мл, насыщенном при 0oC) перемешивали при комнатной температуре в течение 10 часов. Растворитель выпаривали и неочищенный (11) очищали на препаративных силикагелевых пластинках (20% CH3ОН/CHCl3) с получением масла. Его кристаллизовали из CH2Cl2/гексана и получали (11) (0,136 г, 77,7%) в виде белого твердого вещества. УФ max 278,0 нм ( max 298 нм; Анал. (C17H17N3О8) С, H, N. 10: УФ (CH3ОН) max 298 нм.
Пример 8: Получение (-)-(2S,4S)-1-(2-гидроксиметил-1,3- диoкcoлaн-4-ил)цитoзинa (11).
Раствор (9) (0,29 г, 0,827 ммоль) в CH3ОН/NH3 (50 мл, насыщенном при 0oC) перемешивали при комнатной температуре в течение 10 часов. Растворитель выпаривали и неочищенный (11) очищали на препаративных силикагелевых пластинках (20% CH3ОН/CHCl3) с получением масла. Его кристаллизовали из CH2Cl2/гексана и получали (11) (0,136 г, 77,7%) в виде белого твердого вещества. УФ max 278,0 нм ( 11967) (pH 2), 270,0 нм ( 774) (pH 7), 269,0,0 нм ( 8379) (pH 11; Анал. (С8H11N3О4) С, H, N).
II. Фармацевгические композиции.
Люди, лошади, собаки, коровы и другие животные, в частности, млекопитающие, страдающие опухолями и, в частности, раком, могут подвергаться лечению путем введения пациенту эффективного количества (-)-OddC или его производного или его фармацевтически приемлемой соли, необязательно, в фармацевтически приемлемом носителе или разбавителе либо самого по себе или в сочетании с другими известными противораковыми фармацевтическими средствами. Данное лечение также может проводиться в сочетании с другими традиционными способами лечения рака, такими как лучевая терапия или хирургическое лечение.
Данные соединения могут вводиться любым подходящим путем, например, перорально, парентерально, внутривенно, внутрикожно, подкожно или местно в виде жидкости, мази, геля или твердого вещества или в форме аэрозоля. Активное соединение входит в состав с фармацевтически приемлемым носителем или разбавителем в количестве, достаточном для обеспечения пациента терапевтически эффективным количеством по желательному показанию, не вызывая у пациента, подвергающегося лечению, серьезных токсических эффектов. Предпочтительная доза соединения для лечения всех упомянутых здесь состояний колеблется в пределах от около 10 нг/кг до 300 мг/кг, предпочтительно от 0,1 до 100 мг/кг в день, более обычно от 0,5 до около 25 мг на килограмм массы тела реципиента в день. Обычная доза при местном применении будет колебаться от 0,01 до 3% по массе в подходящем носителе.
Соединение удобно вводить в любой подходящей стандартной дозированной форме, включая, но не ограничиваясь, стандартную дозированную форму, содержащую от 1 до 3000 мг, предпочтительно от 5 до 500 мг активного ингредиента в стандартной дозированной форме. При пероральном применении обычно удобной является доза в 25-250 мг.
Активный ингредиент предпочтительно вводится с тем, чтобы получить пиковые концентрации активного соединения в плазме около 0,00001-30 мМ, предпочтительно, около 0,1-30 мкМ. Этого можно достигнуть, например, путем внутривенной инъекции раствора или состава активного ингредиента необязательно в физиологическом растворе или в водной среде или введения в виде болюса с активным ингредиентом.
Концентрация активного соединения в лекарственной композиции будет зависеть от скоростей всасывания, распределения, инактивации и экскреции лекарства, а также от других факторов, известных специалистам в данной области. Также следует отметить, что уровни дозировок будут также варьировать в зависимости от тяжести состояния, подлежащего облегчению. Далее следует учесть, что для каждого отдельного субъекта особая схема введения лекарственного вещества должна устанавливаться по прошествии некоторого времени в соответствии с индивидуальной потребностью и профессиональным суждением лица, вводящего или наблюдающего за введением композиций, и что интервалы концентраций, изложенные здесь, приведены лишь в качестве примера и не направлены на ограничение сферы или применения на практике заявляемой композиции. Активный ингредиент может вводиться сразу или может быть разделен на ряд меньших доз, подлежащих введению через варьирующие интервалы времени.
Композиции для перорального применения будут, как правило, включать в себя инертный разбавитель или съедобный носитель. Они могут быть заключены в желатиновые капсулы или спрессованы в таблетки. В целях терапевтического перорального введения активное соединение или его пролекарственное производное может быть смешано с наполнителями и применено в форме таблеток, пастилок или капсул. Фармацевтически совместимые связывающие агенты и/или адъювантные твердые материалы могут входить в состав в качестве части композиции.
Таблетки, пилюли, капсулы, пастилки и тому подобное могут содержать любые из следующих ингредиентов или соединения сходной природы: связывающее вещество, такое как микрокристаллическая целлюлоза, смола трагаканта или желатин; наполнитель, такой как крахмал или лактоза, диспергирующее средство, такое как альгиновая кислота, Primogel или кукурузный крахмал; смазывающее вещество, такое как стеарат магния или Sterotes; скользящее вещество, такое как коллоидный диоксид кремния; подсластитель, такой как сахароза или сахарин; или корригент, такой как перечная мята, метилсалицилат или корригент со вкусом апельсина. Когда стандартной дозированной формой является капсула, она может содержать в дополнение к материалам выше перечисленного типа жидкий носитель, такой как нелетучее масло. Кроме этого, стандартные дозированные формы могут содержать другие разнообразные материалы, которые меняют физическую форму дозированной единицы, например, оболочки из сахара, шеллака или энтеросолюбильных веществ.
Активное соединение или его фармацевтически приемлемая соль может вводиться в виде компонента эликсира, суспензии, сиропа, капсулы, жевательной резинки и тому подобного. Сироп может содержать в дополнение к активным соединениям сахарозу в качестве подсластителя и определенные консерванты, красители и корригенты.
Активное соединение или его фармацевтически приемлемые соли также могут быть смешаны с другими активными материалами, которые не снижают желательного действия, или с материалами, которые поддерживают желательное действие, такими как другие противораковые средства, антибиотики, противогрибковые, противовоспалительные или противовирусные соединения.
Растворы или суспензии, используемые для парентерального, внутрикожного, подкожного или местного применения, могут включать в себя следующие компоненты: стерильный разбавитель, такой как вода для инъекций, физиологический раствор, нелетучие масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические разбавители; антибактериальные средства, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты или фосфаты и агенты для регулирования тоничности, такие как хлорид натрия или декстроза. Препарат для парентерального введения может быть заключен в ампулы, одноразовые шприцы или пузырьки, содержащие множество доз, изготовленные из стекла или пластика.
В случае внутривенного введения предпочтительными носителями являются физиологический солевой раствор или фосфатный солевой буфер (ФСБ).
При одном осуществлении активные соединения готовят с носителями, которые будут предохранять соединения от быстрого вывода их из организма, например, в форме с контролируемым высвобождением, включая имплантаты и микроинкапсулированные системы доставки. Могут применяться поддающиеся биологическому разрушению, биосовместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, сложные полиортоэфиры, полимолочная кислота. Способы получения таких составов будут очевидны специалистам в данной области.
Липосомальные суспензии также могут служить фармацевтически приемлемыми носителями. Они могут быть получены по методам, известным специалистам в данной области, например, как описано в Патенте США N 4522811 (который включен здесь в качестве ссылки полностью). Например, липосомные составы могут быть получены путем растворения подходящего (их) липида(ов) (таких как стеароилфосфатидилэтаноламин, стеароилфосфатидилхолин и холестерин) в неорганическом растворителе, который затем выпаривают, причем на поверхности сосуда остается тонкая пленка высушенного липида. Водный раствор активного соединения вносят затем в сосуд. Сосуд затем вращают рукой для высвобождения липидного материала со стенок сосуда и для измельчения липидных агрегатов, образуя, таким образом, липосомальную суспензию.
III. Биологическая активность.
Среди специалистов в данной области большое количество биологических анализов применялось и является общепринятым для оценки противораковой активности соединений. Каждый из данных методов может быть применен для оценки активности описанных здесь соединений. Один общепринятый способ оценки активности заключается в применении панелей для тестирования линии раковых клеток из National Cancer Institute (“NCI”). Данные тесты оценивают противораковую активность отдельных соединений in vitro и представляют прогностические данные по отношению к применению исследуемых соединений in vivo. Другие анализы включают оценку in vivo эффекта соединения на человеческие или мышиные опухолевые клетки, имплантированные мышам или привитые мышам nude. (-)-OddC исследовали на предмет противораковой активности in vivo на линии лейкозных клеток P388 и линии раковых клеток из толстого кишечника C38. В примерах 9 и 10 представлены детали проведения экспериментов и результаты данных исследований.
Пример 9. Обработка лейкозных клеток P388 (-)-OddC in vivo.
Мышам BDF1, полученным от Southern Research Institute, Alabama, имплантировали внутрибрюшинно 106 лейкозных клеток P388. (-)-OddC вводили внутрибрюшинно дважды в день в течение пяти дней, начиная через один день после имплантации опухолевых клеток. Используя данный протокол, было показано, что 75 мг/кг/доза является токсичным для мышей.
На фиг. 3 и в табл. 1 показаны результаты данных исследований. На фиг. 3 () представлены данные для контроля (животные, не подвергавшиеся лечению), (––) представляет уровень выживаемости животных, которым вводили (-)- OddC по 25 мг/кг массы дважды в день, и (О) представляет уровень выживаемости мышей, которым вводили (-)-OddC один раз в день по 50 мг/кг массы. Из шести мышей, подвергавшихся лечению, 25 мг/кг/доза (-) -OddC, одна мышь прожила долгое время, а срок жизни остальных пяти мышей возрос на 103%.
Пример 10. Обработка опухолевых клеток 38 из толстого кишечника (-)-OddC in vivo.
Опухолевые клетки 38 из толстого кишечника имплантировали подкожно мышам BDF1. (-)-OddC вводили мышам дважды в день в течение пяти дней в дозировке 25 мг/кг/доза. Рост опухолевых клеток из толстого кишечника был остановлен, как показано на фиг. 2. На фиг. 2 () представлены данные по контрольным животным, а () представляет данные по мышам, подвергавшимся лечению (-)- OddC.
Пример 11. Исследование (-)-OddC in vitro.
(-)-OddC оценивали по программе NCI скрининга рака. Тест позволяет измерить подавление различных линий раковых клеток при различных концентрациях (-) -OddC. Клеточные линии, которые были исследованы, приведены в табл. 2.
Табл. 2 также дает концентрацию, при которой в тестируемых клеточных линиях наблюдались ИР50 и ПИР. ИР50, ПИР и LC50 являются значениями, представляющими концентрации, при которых ПР (процент ингибирования роста), определенный ниже, равен +50, 0 и -50, соответственно. Данные значения определяли путем интерполяции кривых доза-ответ, полученных для каждой клеточной линии, изображенной в виде функции ПР от log10 концентрации (-)-OddC.
ПР являлся измеряемым эффектом (-)-OddC на клеточную линию и рассчитывался по одному из следующих двух выражений: 11967) (pH 2), 270,0 нм ( 774) (pH 7), 269,0,0 нм ( 8379) (pH 11; Анал. (С8H11N3О4) С, H, N).
II. Фармацевгические композиции.
Люди, лошади, собаки, коровы и другие животные, в частности, млекопитающие, страдающие опухолями и, в частности, раком, могут подвергаться лечению путем введения пациенту эффективного количества (-)-OddC или его производного или его фармацевтически приемлемой соли, необязательно, в фармацевтически приемлемом носителе или разбавителе либо самого по себе или в сочетании с другими известными противораковыми фармацевтическими средствами. Данное лечение также может проводиться в сочетании с другими традиционными способами лечения рака, такими как лучевая терапия или хирургическое лечение.
Данные соединения могут вводиться любым подходящим путем, например, перорально, парентерально, внутривенно, внутрикожно, подкожно или местно в виде жидкости, мази, геля или твердого вещества или в форме аэрозоля. Активное соединение входит в состав с фармацевтически приемлемым носителем или разбавителем в количестве, достаточном для обеспечения пациента терапевтически эффективным количеством по желательному показанию, не вызывая у пациента, подвергающегося лечению, серьезных токсических эффектов. Предпочтительная доза соединения для лечения всех упомянутых здесь состояний колеблется в пределах от около 10 нг/кг до 300 мг/кг, предпочтительно от 0,1 до 100 мг/кг в день, более обычно от 0,5 до около 25 мг на килограмм массы тела реципиента в день. Обычная доза при местном применении будет колебаться от 0,01 до 3% по массе в подходящем носителе.
Соединение удобно вводить в любой подходящей стандартной дозированной форме, включая, но не ограничиваясь, стандартную дозированную форму, содержащую от 1 до 3000 мг, предпочтительно от 5 до 500 мг активного ингредиента в стандартной дозированной форме. При пероральном применении обычно удобной является доза в 25-250 мг.
Активный ингредиент предпочтительно вводится с тем, чтобы получить пиковые концентрации активного соединения в плазме около 0,00001-30 мМ, предпочтительно, около 0,1-30 мкМ. Этого можно достигнуть, например, путем внутривенной инъекции раствора или состава активного ингредиента необязательно в физиологическом растворе или в водной среде или введения в виде болюса с активным ингредиентом.
Концентрация активного соединения в лекарственной композиции будет зависеть от скоростей всасывания, распределения, инактивации и экскреции лекарства, а также от других факторов, известных специалистам в данной области. Также следует отметить, что уровни дозировок будут также варьировать в зависимости от тяжести состояния, подлежащего облегчению. Далее следует учесть, что для каждого отдельного субъекта особая схема введения лекарственного вещества должна устанавливаться по прошествии некоторого времени в соответствии с индивидуальной потребностью и профессиональным суждением лица, вводящего или наблюдающего за введением композиций, и что интервалы концентраций, изложенные здесь, приведены лишь в качестве примера и не направлены на ограничение сферы или применения на практике заявляемой композиции. Активный ингредиент может вводиться сразу или может быть разделен на ряд меньших доз, подлежащих введению через варьирующие интервалы времени.
Композиции для перорального применения будут, как правило, включать в себя инертный разбавитель или съедобный носитель. Они могут быть заключены в желатиновые капсулы или спрессованы в таблетки. В целях терапевтического перорального введения активное соединение или его пролекарственное производное может быть смешано с наполнителями и применено в форме таблеток, пастилок или капсул. Фармацевтически совместимые связывающие агенты и/или адъювантные твердые материалы могут входить в состав в качестве части композиции.
Таблетки, пилюли, капсулы, пастилки и тому подобное могут содержать любые из следующих ингредиентов или соединения сходной природы: связывающее вещество, такое как микрокристаллическая целлюлоза, смола трагаканта или желатин; наполнитель, такой как крахмал или лактоза, диспергирующее средство, такое как альгиновая кислота, Primogel или кукурузный крахмал; смазывающее вещество, такое как стеарат магния или Sterotes; скользящее вещество, такое как коллоидный диоксид кремния; подсластитель, такой как сахароза или сахарин; или корригент, такой как перечная мята, метилсалицилат или корригент со вкусом апельсина. Когда стандартной дозированной формой является капсула, она может содержать в дополнение к материалам выше перечисленного типа жидкий носитель, такой как нелетучее масло. Кроме этого, стандартные дозированные формы могут содержать другие разнообразные материалы, которые меняют физическую форму дозированной единицы, например, оболочки из сахара, шеллака или энтеросолюбильных веществ.
Активное соединение или его фармацевтически приемлемая соль может вводиться в виде компонента эликсира, суспензии, сиропа, капсулы, жевательной резинки и тому подобного. Сироп может содержать в дополнение к активным соединениям сахарозу в качестве подсластителя и определенные консерванты, красители и корригенты.
Активное соединение или его фармацевтически приемлемые соли также могут быть смешаны с другими активными материалами, которые не снижают желательного действия, или с материалами, которые поддерживают желательное действие, такими как другие противораковые средства, антибиотики, противогрибковые, противовоспалительные или противовирусные соединения.
Растворы или суспензии, используемые для парентерального, внутрикожного, подкожного или местного применения, могут включать в себя следующие компоненты: стерильный разбавитель, такой как вода для инъекций, физиологический раствор, нелетучие масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические разбавители; антибактериальные средства, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты или фосфаты и агенты для регулирования тоничности, такие как хлорид натрия или декстроза. Препарат для парентерального введения может быть заключен в ампулы, одноразовые шприцы или пузырьки, содержащие множество доз, изготовленные из стекла или пластика.
В случае внутривенного введения предпочтительными носителями являются физиологический солевой раствор или фосфатный солевой буфер (ФСБ).
При одном осуществлении активные соединения готовят с носителями, которые будут предохранять соединения от быстрого вывода их из организма, например, в форме с контролируемым высвобождением, включая имплантаты и микроинкапсулированные системы доставки. Могут применяться поддающиеся биологическому разрушению, биосовместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, сложные полиортоэфиры, полимолочная кислота. Способы получения таких составов будут очевидны специалистам в данной области.
Липосомальные суспензии также могут служить фармацевтически приемлемыми носителями. Они могут быть получены по методам, известным специалистам в данной области, например, как описано в Патенте США N 4522811 (который включен здесь в качестве ссылки полностью). Например, липосомные составы могут быть получены путем растворения подходящего (их) липида(ов) (таких как стеароилфосфатидилэтаноламин, стеароилфосфатидилхолин и холестерин) в неорганическом растворителе, который затем выпаривают, причем на поверхности сосуда остается тонкая пленка высушенного липида. Водный раствор активного соединения вносят затем в сосуд. Сосуд затем вращают рукой для высвобождения липидного материала со стенок сосуда и для измельчения липидных агрегатов, образуя, таким образом, липосомальную суспензию.
III. Биологическая активность.
Среди специалистов в данной области большое количество биологических анализов применялось и является общепринятым для оценки противораковой активности соединений. Каждый из данных методов может быть применен для оценки активности описанных здесь соединений. Один общепринятый способ оценки активности заключается в применении панелей для тестирования линии раковых клеток из National Cancer Institute (“NCI”). Данные тесты оценивают противораковую активность отдельных соединений in vitro и представляют прогностические данные по отношению к применению исследуемых соединений in vivo. Другие анализы включают оценку in vivo эффекта соединения на человеческие или мышиные опухолевые клетки, имплантированные мышам или привитые мышам nude. (-)-OddC исследовали на предмет противораковой активности in vivo на линии лейкозных клеток P388 и линии раковых клеток из толстого кишечника C38. В примерах 9 и 10 представлены детали проведения экспериментов и результаты данных исследований.
Пример 9. Обработка лейкозных клеток P388 (-)-OddC in vivo.
Мышам BDF1, полученным от Southern Research Institute, Alabama, имплантировали внутрибрюшинно 106 лейкозных клеток P388. (-)-OddC вводили внутрибрюшинно дважды в день в течение пяти дней, начиная через один день после имплантации опухолевых клеток. Используя данный протокол, было показано, что 75 мг/кг/доза является токсичным для мышей.
На фиг. 3 и в табл. 1 показаны результаты данных исследований. На фиг. 3 () представлены данные для контроля (животные, не подвергавшиеся лечению), (––) представляет уровень выживаемости животных, которым вводили (-)- OddC по 25 мг/кг массы дважды в день, и (О) представляет уровень выживаемости мышей, которым вводили (-)-OddC один раз в день по 50 мг/кг массы. Из шести мышей, подвергавшихся лечению, 25 мг/кг/доза (-) -OddC, одна мышь прожила долгое время, а срок жизни остальных пяти мышей возрос на 103%.
Пример 10. Обработка опухолевых клеток 38 из толстого кишечника (-)-OddC in vivo.
Опухолевые клетки 38 из толстого кишечника имплантировали подкожно мышам BDF1. (-)-OddC вводили мышам дважды в день в течение пяти дней в дозировке 25 мг/кг/доза. Рост опухолевых клеток из толстого кишечника был остановлен, как показано на фиг. 2. На фиг. 2 () представлены данные по контрольным животным, а () представляет данные по мышам, подвергавшимся лечению (-)- OddC.
Пример 11. Исследование (-)-OddC in vitro.
(-)-OddC оценивали по программе NCI скрининга рака. Тест позволяет измерить подавление различных линий раковых клеток при различных концентрациях (-) -OddC. Клеточные линии, которые были исследованы, приведены в табл. 2.
Табл. 2 также дает концентрацию, при которой в тестируемых клеточных линиях наблюдались ИР50 и ПИР. ИР50, ПИР и LC50 являются значениями, представляющими концентрации, при которых ПР (процент ингибирования роста), определенный ниже, равен +50, 0 и -50, соответственно. Данные значения определяли путем интерполяции кривых доза-ответ, полученных для каждой клеточной линии, изображенной в виде функции ПР от log10 концентрации (-)-OddC.
ПР являлся измеряемым эффектом (-)-OddC на клеточную линию и рассчитывался по одному из следующих двух выражений:Если (Средняя ОПтест – Средняя ОПтнол) 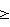 0, 0,тогда ПР = 100 (Средняя ОПтест – Средняя ОПтнол)/(Средняя ОПконтр – Средняя ОПтнол).
Если (Средняя ОПтест – Средняя ОПтнол) < 0,тогда ПР = 100 (Средняя ОПтест – Средняя ОПтнол)/(Средняя ОПтнол).
Где:Средняя ОПтнол = Измеренные значения средней оптической плотности цвета, который дает SRB, непосредственно перед воздействием на клетки тестируемого соединения. Средняя ОПтест = Измеренные значения средней оптической плотности цвета, который дает SRB, после 48 часов воздействия на клетки тестируемого соединения. Средняя ОПконтр = Измеренные значения средней оптической плотности цвета, который дает SRB, через 48 часов при отсутствии воздействия на клетки тестируемого соединения. В табл. 2 первые два столбца описывают субпанель (например, лейкоз) и клеточную линию (например, CCRF-CEM), которые обрабатывали (-)-OddC. Столбец 3 показывает log10, при котором наблюдается ИР50, а столбец 4 показывает log10, при котором наблюдается ПИР. Если данные параметры ответа не могут быть получены путем интерполяции, значение, даваемое для каждого параметра ответа, представляет собой наивысшую тестированную концентрацию, и ему предшествует значок “>”. Например, если все значения ИР при всех концентрациях (-)-OddC, данных для отдельной клеточной линии, превышают +50, данный параметр не может быть получен путем интерполяции. Фиг. 4 представляет собой график, который показывает относительную избирательность (-)-OddC по отношению к конкретной клеточной линии. Столбцы, вытянутые направо, представляют чувствительность клеточной линии к (-)-OddC, большую, чем средняя чувствительность всех протестированных клеточных линий. Поскольку масштаб столбцов является логарифмическим, столбец в 2 единицы вправо означает, что соединение достигает ИР50 на клеточной линии в концентрации, равной одной сотой от средней концентрации, требуемой на других клеточных линиях, и, таким образом, клеточная линия необычно чувствительна к (-)-OddC. Столбцы, вытянутые налево, соответственно означают чувствительность меньшую, чем среднюю. Данные клеточные линии могут быть легко определены из табл. 2, поскольку значению log10 концентрации будет предшествовать “>”. На фиг. 4 можно заметить, что, по крайней мере, одна клеточная линия из каждого протестированного типа раковых клеток проявляла чувствительность к (-) -OddC. Определенные клеточные линии рака предстательной железы, лейкозных клеточных линий и клеточных линий из толстого кишечника проявляют чрезвычайную чувствительность к (-)-OddC. Пример 12. Сравнение (-)-OddC и araC. Как обсуждалось в Предпосылках изобретения, цитозинарабинозид (на который также ссылаются как на Цитарабин, araC и Цитозар) является нуклеозидным аналогом дезоксицитидина, применяемым для лечения острого миелолейкоза. Он также проявляет активность против острого лимфолейкоза и, в меньшей степени, применим в случае хронического миелолейкоза и не-Ходжкинских лимфом. Основным механизмом действия araC является подавление синтеза ДНК в ядре. Интересно было сравнить токсичность (-)-OddC и AraC по отношению к опухолевым клеткам. Клетки в логарифмической фазе роста высевали с плотностью 5000 клеток/мл/лунка на 24-луночные планшеты. К клеткам в различных дозах добавляли препараты и культуры поддерживали в течение трех поколений. По окончании данного времени проводили анализы с помощью метиленового синего и/или количества клеток подсчитывали непосредственно. Метиленовый синий представляет собой краситель, который стехиометрически связывается с белками жизнеспособных клеток и может применяться для непрямого подсчета количества клеток (Finlay, 1984). Значения IC50 определяли путем интерполяции данных на графике. Каждое показанное значение представляет собой среднее стандартное отклонение по пяти опытам, причем каждая точка данных найдена в двух экземплярах.
Во всех исследованных клеточных линиях (-)-OddC был более токсичным, чем AraC. (-)-OddC был значительно более эффективен, чем AraC, в случае клеточной линии KB карциномы носоглотки и в двух линиях DU-145 и PC-3 карциномы предстательной железы. Клетки HepG2 происходят из печеночно-клеточного рака, а линия 2.2.15 происходит от клеток HepG2, трансфицированных копией генома вируса гепатита В. Клетки СЕМ происходят из клеток острого лимфобластного лейкоза. (-)-OddU, соединение, которое будет образовываться путем дезаминирования (-)-OddC, не проявлял токсичности по отношению к какой-либо протестированной клеточной линии. Ферментативные исследования показывают, что в отличие от AraC, клиническая эффективность которого чрезвычайно снижается из- за чувствительности к дезаминированию, (-)-OddC не является субстратом дезаминазы.
Было определено, что (-)-OddC может быть фосфорилирован до моно-, ди- и трифосфатнуклеотида in vivo. Оказывается, что (-) -OddC проявляет клеточную токсичность в фосфорилированной форме, поскольку клетки, не способные к фосфорилированию соединения, гораздо менее чувствительны к соединению. Первым ферментом, ответственным за фофсорилирование, является человеческая дезоксицитидинкиназа. Ферментативные исследования in vitro показывают, что (-) -OddC может быть фосфорилирован данным ферментом.
В отличие от araC, (-) -OddC не подвергается дезаминированию цитидиндезаминазой. Присутствие цитидиндезаминазы в тканях солидных опухолей может быть ключевым способствующим фактором, ответственным за отсутствие активности araC в солидных опухолях. Это может отчасти объяснить, почему (-)-OddC проявляет активность против клеток HepG2 у мышей nude, тогда как araC такой активности не проявляет. Это также объясняет, почему (-)-OddC имеет спектр противоопухолевой активности, отличный от такового araC. Кроме того, наличие цитидиндезаминазы в желудочно-кишечном тракте может играть важную роль в том факте, что araC нельзя принимать перорально.
Биохимические исследования (-)-OddC.
Цитотоксичность AraC, (-)-OddC и (-)-OddU in vitro (см. табл.3).
Пример 13. Исследования in vivo.
Трех-шестинедельных мышей NCr nude (мыши и крысы с иммунодефицитом Taconic) прививали подкожно на каждый бок 2106 клеток HepG2 или DU-145 и опухоли позволяли расти. Лечение начинали, когда опухоли достигали 100-250 мг, что определялось путем измерения с помощью штанген-циркуля и рассчитывалось по формуле:Масса опухоли (мг) = длина (мм) х ширина (мм) – 2 Лекарства давали в указанных дозах в дни от 0 до 4 включительно и размеры опухолей измеряли каждые несколько дней. Кривые роста опухолей строили, как описано в Bell, et al. Cancer (phila.)/ 36:2437-2440 (1975), и показано на фиг. 5 (a) и 5(b). Токсичность оценивали по изменениям массы тела. Хотя токсичность AraC in vitro была сходна с таковой (L)- OddC, AraC оказывался неэффективным в данной животной модели. Ферментный анализ экстракта опухоли показал, что это происходило не благодаря повышенной активности дЦД или пониженной активности дЦК, но могло быть результатом активного метаболизма AraC в печени, которая имеет высокие уровни дЦД. В отличие от AraC (L)- OddC был эффективен, как в случае ксенотрансплантата HepG2, так и в случае ксенотрансплантата DU-145. Величина чистой клеточной гибели (log 10), рассчитанная для опухолей HepG2, составляла 0,67 и 0,87 для внутрибрюшинного и перорального лечения соответственно. Опухоли DU-145 уменьшались в размерах, причем половина из них полностью регрессировала на 15 день. Опухоли начали рецидивировать приблизительно через 25 дней после последнего введения лекарства, но рост вновь прекращался после 47 дня. На 60 день животных умерщвляли и опухоли удаляли. Опухоли имели некротическую морфологию с очень небольшим числом клеток, способных не включать трипановый синий. В дополнение к этому в данной ткани нельзя было определить никакой ферментативной активности. Введенные дозы AraC и (L)-OddC были токсичны в равной степени, на что указывала потеря животными веса, и предварительные эксперименты по токсичности предполагают, что 25 мг/кг дважды в день может быть максимальной переносимой дозой для пяти дней непрерывного лечения. Может быть предпочтительной такая схема проведения эксперимента, когда лекарство вводят прерывисто. Данные, полученные in vitro и in vivo, показанные здесь, демонстрируют, что (L)-OddC имеет значительную противораковую активность и может по многим параметрам превосходить доступные в настоящее время аналоги дезоксицитидина. Он является не только первым L-нуклеозидным аналогом, для которого показано наличие противораковой активности, но и является также первым истинным терминатором, способным подавлять рост опухоли, хотя его неприродная стереохимия не предотвращает активацию (L)-OddC метаболическими ферментами или его включения в ДНК, это может быть фактором, который предохраняет данное соединение от разрушения дЦД. (L)-OddC также уникален в том отношении, что он проявляет активность в солидных опухолях, которые обычно не восприимчивы к терапии нуклеозидными аналогами. Лекарство 2′, 2′-дифтордезоксицитидин (гемцитидин), которое в настоящее время проходит клинические испытания для лечения солидных опухолей, все еще чувствительно к инактивации дЦД (16). Поскольку повышение уровней дЦД является механизмом, по которому клетки приобретают устойчивость к аналогам дЦид, таким как AraC (17), (L)-OddC может применяться в лечении пациентов, которые стали невосприимчивы к данным лекарствам. IV. Применение (-)-OddC в олигонуклеотидной и в антисмысловой технологии. Понятие антисмысловой технологии, вообще, относится к модуляции экспрессии генов посредством процесса, в котором синтетические олигонуклеотиды гибридизуют с комплементарной последовательностью нуклеиновой кислоты в целях ингибирования транскрипции или репликации (если последовательностью-мишенью является ДНК), ингибирования трансляции (если последовательностью-мишенью является PHK) или ингибирования процессинга (если последовательностью-мишенью является пре-PHK). Применяя данную технологию, можно модулировать широкий спектр клеточной активности. Простым примером является ингибирование биосинтеза белка с помощью антисмыслового олигонуклеотида, присоединенного к мPHK. В другом осуществлении синтетический олигонуклеотид гибридизуют со специфической генной последовательностью в двухцепочечной ДНК с образованием трехцепочечного комплекса (триплекса), который ингибирует экспрессию данной генной последовательности. Антисмысловые олигонуклеотиды могут также применяться для косвенной активации экспрессии генов путем подавления биосинтеза естественного репрессора или для непосредственной активации путем сокращения терминации транскрипции. Антисмысловая олигонуклеотидная терапия (AOT) может применяться для ингибирования экспрессии патологических генов, включая гены, которые вовлечены в неконтролируемый рост клеток доброкачественных или злокачественных опухолей или которые вовлечены в репликацию вирусов, включая ВИЧ и вирус гепатита В. Устойчивость олигонуклеотидов против нуклеаз является важным фактором для применений in vivo. Известно, что 3′-экзонуклеазная активность ответственна за большую часть разрушения немодифицированных антисмысловых олигонуклеотидов в сыворотке. Vlassov, V.V., Yakubov L.A., in Prospects for Antisense Nucleic Acid Therapy of Cancers and AIDS, 1991, 243-266, Viley-Liss, Inc., New York; Nucleic Acids Res., 1993, 21, 145. Замещение нуклеотида на 3′-конце олигонуклеотида (-)-OddC или его производным может повысить устойчивость олигонуклеотида против разрушения 3′-экзонуклеазами. Альтернативно или в дополнение к этому, внутренний нуклеотид может быть замещен (-)-OddC или его производным для препятствования разрушению олигонуклеотида эндонуклеазами. Используя данное здесь описание, специалист в данной области сможет применить (-)-OddC или его производные для стабилизации широкого спектра олигонуклеотидов против разрушения как экзонуклеазами, так и эндонуклеазами, включая нуклеозиды, применяемые в антисмысловой олигонуклеотидной терапии. Считается, что все данные осуществления попадают в сферу данного изобретения. Пример 13 описывает один, неограничивающий, пример применения (-)-OddC для препятствования активности 3′-экзонуклеазы. Пример 14. Препятствование 3′-экзонуклеазной активности с помощью (-)-OddC. Цитозольную экзонуклеазную активность человека в человеческих H9 (T-лимфоцитарных лейкозных клетках) определяли путем секвенирующего анализа в геле. Вкратце, 3′ -концевой субстрат получали из праймера ДНК длиной от 20 до 23 оснований с последовательностью 1 (см. в конце описания). Праймеры метили на 5′-конце с помощью [Y-32P] АТФ, гибридизовали с комплементарными матрицами PHK и терминировали на 3′-конце dTTP (20 mer), dCTP (23 mer) или (-)-OddCTP (23 mer) в реакции, катализируемой обратной транскриптазой ВИЧ-1. В данных условиях 20 mer терминировался с помощью dTMP (А) 23 mer терминировался dCMP (В) или (-)-О-ddCMP (С). Данные одноцепочечные ДНК-субстраты использовали для исследования их чувствительности к цитоплазматической экзонуклеазе. Анализы проводили в реакционных смесях по 10 мкл, содержащих 50 мМ Трис-HCl, pH 8,0, 1 мМ MgCl2, 1 мМ дитиотреитол, 0,1 мг/мл бычий сывороточный альбумин, 0,18 мкКи/мл 3′ -концевой субстрат и 2 мкл экзонуклеазы (0,03 единицы). Реакции инкубировали при 37oC в течение указанных промежутков времени и останавливали путем добавления 4 мкл 98% формамида, 10 мМ ЭДТА и 0,025% бромфенолового синего. Образцы денатурировали при 100oC в течение 5 минут с последующим быстрым охлаждением на льду. Непрореагировавшее вещество, так же как и продукты реакции, разделяли на 15% полиакриламид/мочевина секвенирующих гелях и визуализировали с помощью авторадиографии. Олигонуклеотид с (-)-OddC на 3′-конце оставался устойчив к 3′-экзонуклеазе, по крайней мере, в пять раз дольше, чем другие олигонуклеотиды. Модификации и вариации настоящего изобретения в лечении злокачественных опухолей будут очевидны специалистам в данной области из предшествующего детального описания изобретения. Имеется в виду, что такие модификации и вариации входят в сферу прилагаемой формулы изобретения. Формула изобретения
-L-энантиомера формулы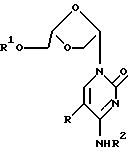 где R1 и R2 выбраны из группы, содержащей водород, ацил и C1 – C18 алкил; R представляет собой H или F, которое по меньшей мере на 95% свободно от соответствующего -D-энантиомера,или его фармацевтически приемлемой соли в фармацевтически приемлемом носителе. 2. Композиция по п.1, где R, R1 и R2 представляют собой водород. 3. Композиция по п.1, где алкильная группа выбрана из группы, содержащей метил, этил, пропил, бутил, пентил, гексил, изопропил, изобутил, втор-бутил, трет-бутил и изопентил. 4. Композиция по п.1, где ацильная группа представляет собой -C(O)R, где R обозначает C1 – C5 алкильную группу, фенил или бензил. 5. Композиция по п.1 или 2, где животным хозяином является человек. 6. Фармацевтическая композиция по пп.1 – 5, где носитель является приемлемым для перорального введения. 7. Фармацевтическая композиция по пп.1 – 5, где носитель представлен в виде капсулы. 8. Фармацевтическая композиция по пп.1 – 5, где носитель представлен в виде таблетки. 9. Фармацевтическая композиция по пп.1 – 5, где носитель является приемлемым для внутривенного введения. 10. Фармацевтическая композиция по пп.1 – 5, где носитель является приемлемым для местного или чрескожного введения. 11. Фармацевтическая композиция по пп.1 – 5, где введение является парентеральным. 12. Фармацевтическая композиция по пп.1 – 5, где опухоль является раковой. 13. Фармацевтическая композиция по п.12, где рак представляет собой рак предстательной железы. 14. Фармацевтическая композиция по п.12, где рак представляет собой лейкемию. 15. Фармацевтическая композиция по п.12, где рак представляет собой рак толстой кишки. 16. Фармацевтическая композиция по п.12, где рак представляет собой рак мочевого пузыря. 17. Фармацевтическая композиция по п.12, где рак представляет собой печеночно-клеточный рак. 18. Фармацевтическая композиция по п.12, где рак представляет собой рак молочной железы. 19. Фармацевтическая композиция по п.12, где рак представляет собой рак легкого. 20. Фармацевтическая композиция по п.12, где рак представляет собой назофарингеальный рак. 21. Фармацевтическая композиция по п.12, где рак представляет собой рак поджелудочной железы. 22. Фармацевтическая композиция по п.12, где рак представляет собой рак яичника. 23. Фармацевтическая композиция по п.12, где рак представляет собой лимфому. 24. Фармацевтическая композиция для лечения рака у животного-хозяина, содержащая эффективное количество -L-энантиомера формулы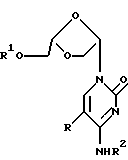 где R1 и R2 выбраны из группы, содержащей водород, ацил и C1 – C18 алкил; R представляет собой H или F, которое по меньшей мере на 95% свободно от соответствующего -D-энантиомера,или его фармацевтически приемлемой соли в фармацевтически приемлемом носителе. 25. Фармацевтическая композиция по п.24, где R обозначает фтор и R1 и R2 обозначают водород. 26. Фармацевтическая композиция по п.24 или 25, где животным-хозяином является человек. 27. Фармацевтическая композиция по п.26, где носитель является приемлемым для перорального введения. 28. Фармацевтическая композиция по п.26, где носитель представлен в виде капсулы. 29. Фармацевтическая композиция по п.26, где носитель представлен в виде таблетки. 30. Фармацевтическая композиция по п.26, где введение является парентеральным. 31. Фармацевтическая композиция по п.26, где R обозначает водород. 32. Фармацевтическая композиция по п.31, где рак представляет собой рак предстательной железы. 33. Фармацевтическая композиция по п.31, где рак представляет собой лейкемию. 34. Фармацевтическая композиция по п.31, где рак представляет собой рак толстой кишки. 35. Фармацевтическая композиция по п.31, где рак представляет собой рак мочевого пузыря. 36. Фармацевтическая композиция по п.31, где рак представляет собой печеночно-клеточный рак. 37. Фармацевтическая композиция по п.31, где рак представляет собой рак молочной железы. 38. Фармацевтическая композиция по п.31, где рак представляет собой рак легкого. 39. Фармацевтическая композиция по п.31, где рак представляет собой назофарингеальный рак. 40. Фармацевтическая композиция по п.31, где рак представляет собой рак поджелудочной железы. 41. Фармацевтическая композиция по п.31, где рак представляет собой рак яичника. 42. Фармацевтическая композиция по п.31, где рак представляет собой лимфому. 43. Фармацевтическая композиция для лечения опухоли у животного-хозяина, содержащая эффективное количество соединения формулы 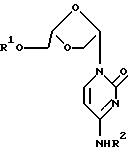 где R1 и R2 выбраны из группы, содержащей водород, алкил, ацил, монофосфат, дифосфат и трифосфат, или его фармацевтически приемлемой соли, необязательно в фармацевтически приемлемом носителе. 44. Фармацевтическая композиция по п.43, где R1 и R2 обозначают водород. 45. Способ получения 2-гидроксиметил-5-(цитозин-1-ил)-1,3-диоксолана, включающий взаимодействие соединения формулы 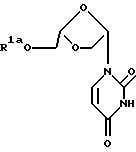 где R1a обозначает гидроксизащитную группу, с реагентом, который преобразует оксогруппу в 4-ом положении урацильного кольца в аминогруппу, и затем удаление любой гидроксизащитной группы. 46. Способ получения (2S, 4S)-1-(2-гидроксиметил-1,3-диоксолан-4-ил)цитозина, отличающийся тем, что проводят взаимодействие защищенного 1,3-диоксолана формулы 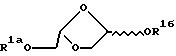 где R1a – бензоил, R1б – ацетил, с защищенным, например, силилированным, цитозиновым основанием с использованием кислоты Льюиса, такой, как триметилсилилфталат, которая не рацемизирует целевой продукт. 47. (-)-(2S, 4S)-1-(2-Гидроксиметил-1,3-диоксолан-4-ил)цитозина или его производные или соли, или (+)-энантиомер, или его рацемическая смесь и фармацевтически приемлемые производные и соли, в качестве активного ингредиента лекарственного средства для лечения или профилактики опухолей, включая раковую опухоль, или для лечения состояний с патологической или нежелательной пролиферацией клеток. 48. (-)-(2S,4S)-1-(2-Гидроксиметил-1,3-диоксолан-4-ил)цитозина, или его производные и соли, или (+)-энантиомер, или его рацемическая смесь и его фармацевтически приемлемые производные ингредиента и солей в качестве активного средства в лекарственной терапии, например, для лечения или профилактики опухолей, включая раковую опухоль, или для лечения состояний с патологической или нежелательной пролиферацией клеток. 49. Фармацевтическая композиция для лечения псориаза, содержащая эффективное количество соединения формулы 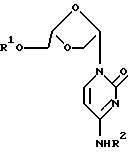 где R1 и R2 выбраны из группы, содержащей водород, алкил, ацил, монофосфат, дифосфат и трифосфат, или его фармацевтически приемлемой соли, необязательно в фармацевтически приемлемом носителе. Приоритет по пунктам: 06.09.94 по пп.1-46; 17.02.95 по пп.1-46; 05.09.95 по пп.47-49. РИСУНКИ
|
||||||||||||||||||||||||||