Патент на изобретение №2397986
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) АНТИБИОТИКИ, СОДЕРЖАЩИЕ КОМПЛЕКСЫ БОРИНОВОЙ КИСЛОТЫ, И СПОСОБЫ ИХ ПОЛУЧЕНИЯ
(57) Реферат:
Описаны структура и получение антибиотиков, включающих комплексы бориновой кислоты, в частности производные гидроксихинолина, имидазола и пиколиновой кислоты, вместе с композициями данных антибиотиков и способами применения антибиотиков и композиций в качестве бактерицидных и фунгицидных средств так же, как и терапевтических средств для лечения заболеваний, вызываемых бактериями и грибками. 8 н. и 43 з.п. ф-лы, 4 табл.
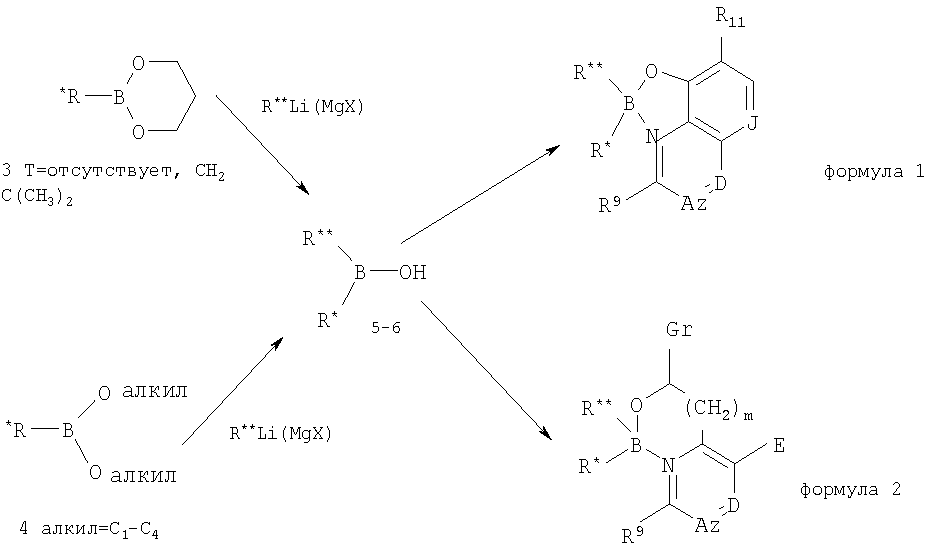
Данная заявка претендует на приоритет предварительной заявки США Serial Область техники Данное изобретение относится к области антибиотиков, в частности к области антибактериальных и противогрибковых соединений и их применению. Также представлены способы получения и применения данных антибиотиков и фармацевтические композиции на их основе. Уровень техники Одним из отличительных признаков современного этапа развития медицины стало снижение заболеваемости и смертности, связанных с бактериальными и грибковыми инфекциями. Однако неправильное употребление антибиотиков и природная селекция инфекционных бактериальных популяций привели к развитию различных уровней устойчивости к лекарственным средствам у большинства инфекционных бактериальных агентов к большинству антибиотиков. Для некоторых случаев, таких как MRSA (устойчивость стафилококка А к различным лекарственным средствам/Multidrug-Resistant StaphA), в настоящее время имеется один или лишь несколько эффективных антибиотиков. Кроме того, существование синдромов иммунодефицита приводит к дополнительному возникновению условно-патогенных инфекций, для лечения которых требуется интенсивное применение антибиотиков. Таким образом, в области медицины существует острая потребность в новых, более эффективных антибиотиках, особенно для лечения бактериальных инфекций, которые являются устойчивыми при применении современных доступных терапевтических средств. Краткая сущность изобретения В одном из аспектов настоящее изобретение относится к соединениям, обладающим активностью антибиотиков. Соединения данного изобретения, обладающие активностью антибиотиков, представляют собой производные боринатов, в частности комплексы бориновой кислоты, и включают такие соединения, как производные гидроксихинолинов, пиколиновых кислот и имидазолов. Соединения, обладающие активностью антибиотиков, также представлены в виде фармацевтических композиций, которые могут быть введены животному, особенно предпочтительно человеку, для лечения заболевания с бактериальной или грибковой этиологией или условно-патогенных инфекций, связанных с бактериями или грибками, у животного, особенно предпочтительно у человека, при нарушении иммунитета или ослабленном состоянии здоровья. В предпочтительных вариантах осуществления соединения данного изобретения представляют собой соединения, строение которых соответствует формулам 1 или 2 с предпочтительными заместителями, раскрытыми здесь. Изобретение также относится к способам получения соединений, обладающих активностью антибиотиков, к приготовлению фармацевтических композиций на их основе и способам применения указанных антибиотиков в терапевтических целях. Рассмотрены также варианты осуществления, относящиеся к наборам и упаковке антибиотиков и фармацевтических композиций данного изобретения. Изобретение также относится к способам лечения инфекций, предпочтительно бактериальных и/или грибковых инфекций, с применением соединений, обладающих активностью антибиотиков, которые раскрыты здесь. Подробное описание изобретения Данное изобретение относится к антибиотикам, в частности к антибактериальным и противогрибковым соединениям, применимым для лечения и/или профилактики бактериальных инфекций. Изобретение относится к соединению, строение которого соответствует формуле
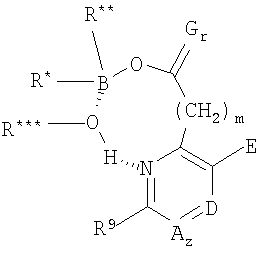
где В представляет собой бор, О представляет собой кислород, m равно 0, 1 или 2, где R и где z равно 0 или 1, и когда z равно 1, А представляет собой СН, CR10 или N, и где D представляет собой N, СН или CR12, и где Е представляет собой Н, ОН, алкокси или N-(морфолинил)этокси, и где r равно 1 или 2, и если r равно 1, то G представляет собой =О (кислород с двойной связью), и если r равно 2, то каждый G независимо представляет собой Н, метил, этил или пропил, где R12 выбран из (CH2)kOH (где k=1, 2 или 3), СН2NН2, CH2NH-алкила, СН2N(алкила)2, СО2Н, СО2алкила, CONH2, ОН, алкокси, арилокси, SH, S-алкила, S-арила, SО2алкила, SО3Н, SСF3, CN, галогена, СF3, NО2, NH2, 2 и где J представляет собой CR10 или N, и где R9, R10 и R11 каждый независимо выбран из группы, содержащей водород, алкил, (CH2)nOH (n=1-3), CH2NH2, СН2NH-алкил, СН2N(алкил)2, галоген, СНО, CH=NOH, CO2H, СО2-алкил, S-алкил, SO2-алкил, S-арил, NН2, алкокси, СF3, SСF3, NO2, SО3Н и ОН, включая его соли, особенно все фармацевтически приемлемые соли. В предпочтительном варианте осуществления в формуле 1 или 2 один из R В предпочтительном варианте осуществления в формуле 1 или 2 один из R В предпочтительном варианте осуществления в формуле 1 или 2 один из R
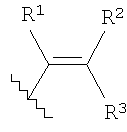
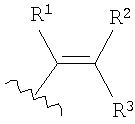
в которой R1, R2 и R3 каждый независимо выбран из группы, содержащей водород, алкил, арил, замещенный арил, бензил, замещенный бензил, (CH2)kOH (где k=1, 2 или 3), CH2NH2, СН2NН-алкил, СН2N(алкил)2, СО2Н, СО2алкил, CONH2, S-алкил, S-арил, SO2алкил, SО3Н, SСF3, CN, галоген, СF3 и NO2. В предпочтительном варианте осуществления в формуле 1 или 2 один из R
в которой R1 выбран из группы, содержащей водород, алкил, арил, замещенный арил, бензил, замещенный бензил, (CH2)kOH (где k=1, 2 или 3), CH2NH2, CH2NH-алкил, CH2N (алкил)2, СО2Н, СО2алкил, CONH2, S-алкил, S-арил, SО2алкил, SО3Н, SСF3, CN, галоген, СF3 и NО2. В предпочтительном варианте осуществления в формуле 1 или 2 один из R
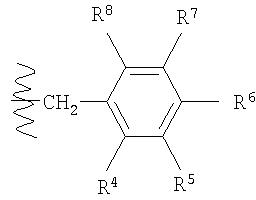
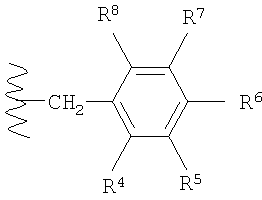
в которой R4, R5, R6, R7 и R8 каждый независимо выбран из группы, содержащей водород, алкил, арил, замещенный арил, бензил, замещенный бензил, (CH2)kOH (где k=1, 2 или 3), СH2NH2, СН2NН-алкил, СН2N(алкил)2, СO2Н, СO2алкил, CONH2, СОNH-алкил, CON(алкил)2, ОН, алкокси, арилокси, SH, S-алкил, S-арил, SО2алкил, SО3Н, SCF3, CN, галоген, СF3, NO2, NH2, 2°-амино, 3°-амино, NH2SO2, OCH2CH2NH2, ОСН2СН2NH-алкил, ОСН2СН2N(алкил)2, оксазолидин-2-ил или алкилзамещенный оксазолидин-2-ил. В предпочтительном варианте осуществления в формуле 1 или 2 один из R
в которой R4, R5, R6, R7 и R8 каждый независимо выбран из группы, содержащей алкил, арил, замещенный арил, бензил, замещенный бензил, (CH2)kOH (где k=1, 2 или 3), CH2NH2, СH2NН-алкил, СН2N(алкил)2, СO2Н, СO2алкил, СОNН2, СОNH-алкил, CON (алкил)2, ОН, алкокси, арилокси, SH, S-алкил, S-арил, SO2алкил, SО3Н, SСF3, CN, галоген, СF3, NO2, NH2, 2°-амино, 3°-амино, NH2SO2, ОСН2СН2NН2, ОСН2СН2NH-алкил, ОСН2СН2N(алкил)2, оксазолмдин-2-ил или алкилзамещенный оксазолидин-2-ил. В предпочтительном варианте осуществления в формуле 1 или 2 один из R
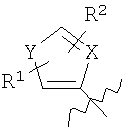
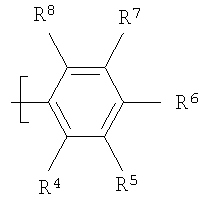
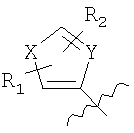
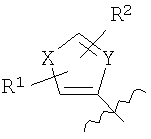
где Х=СН=СН, N=CH, NR13 (где R13=H, алкил, арил или бензил), О или S, и где Y=СН или N, и где R1 и R2 каждый независимо выбран из группы, содержащей водород, алкил, арил, замещенный арил, бензил, замещенный бензил, (CH2)kOH (где k=1, 2 или 3), CH2NH2, СH2NН-алкил, CH2N (алкил)2, СO2Н, СO2алкил, CONH2, 3-алкил, S-арил, SO2алкил, SО3Н, SСF3, CN, галоген, СF3 и NO2. Структура соединений данного изобретения также допускает взаимодействие с растворителем, что приводит к образованию структур (формулы 1В и 2В) с включением атомов из растворителя, который захватывается соединением по ходу синтеза или терапевтического применения. Так, подобные структуры растворителей могут, в частности, встраиваться в соединения изобретения между атомами бора и азота, увеличивая таким образом размер цикла на один или два атома в сравнении с тем, что раскрывается в структурах данного изобретения. В случае когда борсодержащая циклическая структура данного изобретения содержит 5 атомов, включая, например, бор, азот, кислород и 2 атома углерода, встраивание атома растворителя между бором и азотом должно привести к образованию 7-членного цикла. Например, применение гидроксил- и аминосодержащих растворителей может привести к образованию структур, содержащих кислород или азот между атомами бора и азота в цикле с увеличением его размера. Подобные структуры специально рассмотрены в настоящем изобретении, где R
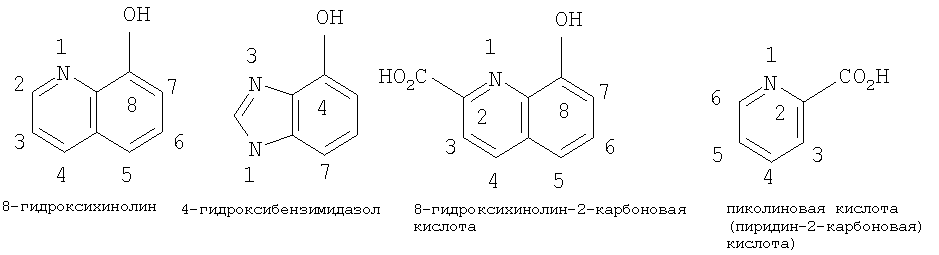
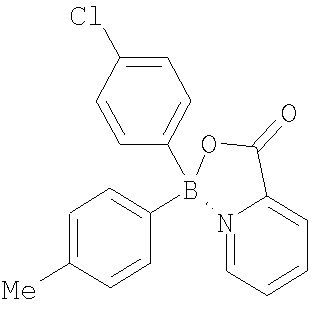
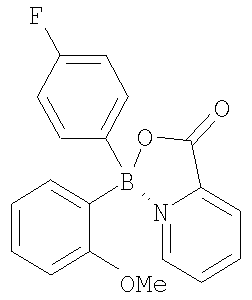
Термины, применяемые в данном изобретении, имеют следующее значение. Термином “алкил”, “низший алкил” и “C1-С6алкил” в настоящем изобретении обозначены алкильные группы с прямой или разветвленной цепью, содержащие 1-6 атомов углерода, такие как метил, этил, пропил, изопропил, н-бутил, втор-бутил, трет-бутил, пентил, 2-пентил, изопентил, неопентил, гексил, 2-гексил, 3-гексил и 3-метилпентил. Термином “алкокси”, “низший алкокси” и “С1-С6алкокси” в настоящем изобретении обозначены алкокси-группы с прямой или разветвленной цепью, содержащие 1-6 атомов углерода, такие как, например, метокси, этокси, пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, пентокси, 2-пентокси, изопентокси, неопентокси, гексокси, 2-гексокси, 3-гексокси и 3-метилпентокси. Термином “галоген” в настоящем изобретении обозначены фтор, бром, хлор и йод. Термином “циклоалкил”, например С3-С7 циклоалкил, в настоящем изобретении обозначены циклоалкильные группы, содержащие 3-7 атомов, такие как, например, циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. В С3-С7 циклоалкильных группах, предпочтительно в C5-C7 циклоалкильных группах, один или два атома углерода, образующие цикл, необязательно могут быть замещены гетероатомом, таким как сера, кислород или азот. Примерами подобных групп являются пиперидинил, пиперазинил, морфолинил, пирролидинил, имидазолидинил, оксазолидинил, пергидроазепинил, пергидрооксазапинил, оксепанил и пергидрооксепанил. С3 и С4 циклоалкильные группы с атомом углерода, замещенным на азот или кислород, включают азиридинил, азетидинил, оксетанил и оксиранил. Термином “арил” обозначена ароматическая карбоциклическая группа, содержащая одно кольцо (например, фенил), несколько колец (например, бифенил) или поликонденсированные кольца, в которых, по меньшей мере, одно кольцо является ароматическим (например, 1,2,3,4-тетрагидронафтил, нафтил, антрил или фенантрил), и которая необязательно является моно-, ди- или тризамещенной, например, галогеном, низшим алкилом, низшим алкокси, низшим алкилтио, трифторметилом, низшим ацилокси, арилом, гетероарилом и гидрокси. Предпочтительные арильные группы включают фенил и нафтил, каждый из которых необязательно замещен, как указано выше. Термином “гетероарил” обозначена одна или несколько ароматических циклических систем, состоящих из 5-, 6-или 7-членных колец, содержащих, по меньшей мере, от одного до четырех гетероатомов, выбранных из азота, кислорода или серы. Такие гетероарильные группы включают, например, тиенил, фуранил, тиазолил, имидазолил, (из)оксазолил, пиридил, пиримидинил, (изо)хинолинил, нафтиридинил, бензимидазолил и бензоксазолил. Предпочтительные гетероарилы представляют собой тиазолил, пиримидинил, предпочтительно пиримидин-2-ил, и пиридил. Другие предпочтительные гетероарильные группы включают 1-имидазолил, 2-тиенил, 1- или 2-хинолинил, 1- или 2-изохинолинил, 1-или 2-тетрагидроизохинолинил, 2- или 3-фуранил и 2-тетрагидрофуранил. Термином “лиганд” обозначена азотсодержащая ароматическая система, которая способна образовывать обратную донорно-акцепторную связь с льюисовским кислотным центром бора и присоединяется как фрагмент боринатного эфира. Такие лиганды известны специалистам в данной области. Примеры их структур приведены ниже.
Соединения настоящего изобретения участвуют в ингибировании ключевых микробных ферментов, таких как метилтрансфераза бактериальной ДНК(DNА). Многие из соединений, раскрываемых в данном описании, являются селективными ингибиторами метилтрансфераз микробов, но не ингибируют метилтрансферазы млекопитающих. Однако антибактериальная и противогрибковая активность соединений изобретения не ограничена активностями, относящимися к ингибированию указанного фермента, не являющимся конечным эффектом, существенно необходимым для указанной терапевтической активности. Данное изобретение также относится к вариантам осуществления соединений, раскрываемых здесь, в виде фармацевтических композиций. Фармацевтические композиции настоящего изобретения могут быть приготовлены хорошо известными способами, например посредством обычного смешивания, растворения, гранулирования, дражирования, растирания порошка, эмульгирования, инкапсулирования, включения или лиофилизации. Фармацевтические композиции для применения в соответствии с настоящим изобретением могут быть приготовлены обычным способом с использованием одного или нескольких физиологически приемлемых носителей, включающих наполнители и вспомогательные добавки, которые облегчают превращение активных соединений в препараты, предназначенные для фармацевтического применения. Подходящий препарат зависит от выбранного пути введения. Нетоксичные фармацевтические соли включают соли кислот, таких как хлористоводородная, фосфорная, бромистоводородная, серная, сульфиновая, муравьиная, толуолсульфоновая, метансульфоновая, гидроксиэтансульфоновая, азотная, бензойная, лимонная, винная, малеиновая, йодистоводородная, алкановая, такая как уксусная, НООС-(СН2)n-СН3, где n равно 0-4, и т.п. Нетоксичные фармацевтические основно-аддитивные соли включают соли оснований, таких как основания натрия, калия, кальция, аммония и функциональных эквивалентов. Специалистам в данной области будет очевидно многообразие нетоксичных фармацевтически приемлемых аддитивных солей. Для инъекции соединения данного изобретения могут быть приготовлены в виде соответствующих водных растворов, таких как физиологически совместимые буферы, например раствор Хенкса (Hanks’ solution), раствор Рингера (Ringer’s solution) или физиологический солевой буфер. Для трансмукозального и чрескожного введения в препарате используют соответствующие смачивающие вещества, предназначенные для преодоления необходимого барьера. Такие вещества хорошо известны в данной области. Для перорального введения композиции могут быть легко приготовлены сочетанием активных соединений с фармацевтически приемлемыми наполнителями, хорошо известными в данной области. С такими носителями композиции соединений изобретения могут быть приготовлены в виде таблеток, пилюль, капсул, жидкостей, гелей, сиропов, взвесей, суспензий и т.п. для перорального введения пациенту, нуждающемуся в лечении. Фармацевтические препараты для перорального применения могут быть получены с твердым наполнителем, необязательно с размолом образующейся смеси, переработкой смеси в гранулы после добавления дополнительных подходящих вспомогательных компонентов, если желательно, для получения таблеток. Подходящие наполнители представляют собой, в частности, такие наполнители, как сахара, включающие лактозу, сахарозу, маннит или сорбит; препараты клетчатки, такие как, например, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакант, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрий карбоксиметилцеллюлоза, и/или поливинилпирролидон (PVP). Если желательно, то могут быть добавлены диспергирующие средства, такие как поперечно-сшитый поливинилпирролидон, агар или альгиновая кислота или ее соль, такая как альгинат натрия. Фармацевтические препараты, которые могут быть применены перорально, включают “push-fit” капсулы, сделанные из желатина, а также мягкие заплавленные капсулы, сделанные из желатина и пластификатора, такого как глицерин или сорбит. “Push-fit” капсулы могут содержать активные ингредиенты в смеси с наполнителем, таким как лактоза, связующими веществами, такими как крахмалы, и/или смазками, такими как тальк или стеарат магния, и, необязательно, стабилизаторами. В мягких капсулах активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как нелетучие жидкие масла, жидкий парафин или жидкие полиэтиленгликоли. Кроме того, могут быть добавлены стабилизаторы. Все препараты для перорального введения должны иметь дозировку, подходящую для такого введения. Для трансбуккального введения препараты могут быть в форме таблеток или пастилок, приготовленных обычным способом. Для введения ингаляцией композиции соединений для применения в соответствии с настоящим изобретением обычно поставляются путем подачи аэрозоля из упаковок под давлением или из распылителя с использованием подходящего распыляющего аэрозольного вещества, например дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа. В случае аэрозоля под давлением стандартная доза может быть определена при наличии вентиля для поступления отмеренного количества. Для применения в ингаляторе могут быть приготовлены капсулы и картриджи из, например, желатина, содержащие порошкообразную смесь соединения, и подходящей порошкообразной основы, такой как лактоза или крахмал. Композиции соединений могут быть приготовлены для парентерального введения путем инъекции, например болюс-инъекции или непрерывного вливания. Препараты для инъекций могут быть сделаны в стандартной лекарственной форме, например, в ампулах или в контейнерах с множеством доз с добавкой консерванта. Подобные композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных наполнителях, и могут содержать адъюванты, такие как суспендирующие, стабилизирующие и/или диспергирующие средства. Фармацевтические препараты для парентерального введения включают водные растворы активных соединений в водорастворимой форме. Кроме того, суспензии активных соединений могут быть приготовлены как соответствующие масляные суспензии для инъекций. Подходящие липофильные растворители или наполнители включают нелетучие жидкие масла, такое как кунжутное масло, или синтетические эфиры жирных кислот, такие как этилолеат или триглицериды, или липосомы. Водные суспензии для инъекций могут содержать вещества, которые повышают вязкость суспензии, такие как натрий карбоксиметилцеллюлоза, сорбит или декстран. Необязательно, но суспензия может также содержать подходящие стабилизаторы или средства, которые повышают растворимость соединений для создания в препарате высококонцентрированных растворов. Альтернативно активный ингредиент может быть в порошкообразной форме для соединения с подходящим наполнителем, например со стерильной антипирогенной водой перед применением. Соединения могут быть также приготовлены в композициях для ректального применения, в таких как суппозитории или удерживающие клизмы, например, содержащие обычные основы для суппозиториев, такую как масло какао или другие глицериды. В дополнение к препаратам, описанным ранее, соединения могут быть также приготовлены в виде депо-препарата. Такие долго действующие препараты могут быть введены имплантацией (например, подкожно или внутримышечно) или внутримышечной инъекцией. Так, например, соединения могут быть приготовлены с подходящими полимерными или гидрофобными продуктами (например, в виде эмульсии в приемлемом масле) или ионообменными смолами или как умеренно растворимые производные, например в виде умеренно растворимой соли. Фармацевтический носитель для гидрофобных соединений данного изобретения представляет собой систему сорастворителей, содержащую бензиловый спирт, неполярное поверхностно-активное вещество, органический полимер, не смешивающийся с водой, и водную фазу. Система сорастворителей может быть системой сорастворителей VPD. VPD представляет собой раствор 3% маc./об. бензилового спирта, 8% мас./об. неполярного поверхностно-активного вещества, полисорбата 80 и 65% мас./об. полиэтиленгликоля 300, в абсолютном этаноле. Система сорастворителей VPD (VPD:5W) содержит VPD, разбавленный в соотношении 1:1 5% декстрозой в водном растворе. Такая система сорастворителей хорошо растворяет гидрофобные соединения и сама по себе обладает низкой токсичностью при систематическом введении. Естественно, пропорции в системе сорастворителей могут быть значительно изменены без нарушения характеристик ее растворимости и токсичности. Кроме того, может быть изменена идентичность компонентов-сорастворителей: например, вместо полисорбата 80 могут быть применены другие малотоксичные неполярные поверхностно-активные вещества; может быть изменен фракционный размер полиэтиленгликоля; другие биосовместимые полимеры могут заменять полиэтиленгликоль, например поливинилпирролидон; другие сахара или полисахариды могут заменять декстрозу. Альтернативно могут быть применены другие системы доставки гидрофобных фармацевтических соединений. Липосомы и эмульсии представляют собой хорошо известные примеры наполнителей или носителей для доставки гидрофобных лекарственных средств. Некоторые органические растворители, такие как диметилсульфоксид, также могут быть применены, хотя с некоторым риском из-за повышенной токсичности. Дополнительно соединения могут быть доставлены с применением системы регулируемого высвобождения действующего вещества, такой как полупроницаемые матрицы твердых гидрофобных полимеров, содержащих терапевтическое средство. Найдены различные материалы для регулируемого высвобождения, которые хорошо известны специалистам в данной области. Капсулы для регулируемого высвобождения в зависимости от их химической природы высвобождают соединения в течение периода от нескольких недель до 100 дней. В зависимости от химической природы и биологической стабильности терапевтического реагента могут быть применены дополнительные стратегии по стабилизации протеина и нуклеиновой кислоты. Фармацевтические композиции могут также содержать твердую или гелевую фазу наполнителей или носителей. Примеры таких наполнителей или носителей включают, но без ограничения только ими, карбонат кальция, фосфат кальция, различные сахара, крахмалы, производные целлюлозы, желатин и полимеры, такие как полиэтиленгликоли. Соединения данного изобретения могут быть представлены в виде солей с фармацевтически совместимыми противоионами. Фармацевтически совместимые соли могут быть образованы со многими кислотами, включающими, но без ограничения только ими, хлористоводородную, серную, уксусную, молочную, винную, яблочную, янтарную, фосфорную, бромистоводородную, сульфиновую, муравьиную, толуолсульфоновую, метансульфоновую, азотную, бензойную, лимонную, малеиновую, йодистоводородную, алкановую, такую как уксусная, НООС-(СН2)n-СН3, где n равно 0-4, и т.п. Соли, как правило, более растворимы в водных или других протонных растворителях, чем соответствующие формы свободного основания. Нетоксичные фармацевтические основно-аддитивные соли включают соли оснований, таких как основания натрия, калия, кальция, аммония и т.п. Специалистам в данной области будет очевидно многообразие нетоксичных фармацевтически приемлемых аддитивных солей. Фармацевтические композиции соединений настоящего изобретения могут быть приготовлены и введены различными способами, включающими системное, локальное или местное введение. Методики приготовления препаратов и их введения могут быть найдены в издании “Remington’s Pharmaceutical Sciences”, Mack Publishing Co., Easton, PA. Способ введения может быть выбран с целью максимальной доставки к желаемому целевому сайту в организме. Подходящие пути введения могут включать, например, пероральное, ректальное, трансмукозальное, чрескожное или кишечное введение; парентеральную доставку, включающую внутримышечные, подкожные, интрамедуллярные инъекции, также как и внутриоболочечные, прямые интравентрикулярные, внутривенные, интраперитонеальные, внутриносовые или внутриглазные инъекции. Альтернативно можно вводить соединение локальным, а не системным образом, например путем инъекции соединения непосредственно в специфическую ткань, часто с помощью депо-препарата или препарата с регулируемым высвобождением действующего вещества. Фармацевтические композиции, подходящие для применения согласно настоящему изобретению, включают композиции, в которых активные ингредиенты содержатся в эффективном количестве для достижения намеченной цели. Более конкретно, терапевтически эффективное количество означает количество, эффективное для предотвращения или ослабления существующих симптомов у пациента, подлежащего лечению. Определение эффективных количеств находится в компетенции квалифицированных специалистов в данной области, в частности в рамках подробного раскрытия, представленного здесь. Для любого соединения, примененного по способу данного изобретения, терапевтически эффективная доза может быть первоначально оценена по данным анализа культур клеток. Например, на моделях животных доза может быть составлена для достижения повторяющегося диапазона концентраций, который включает EC50 (эффективная доза для 50% повышения), определенную на культуре клеток, т.е. концентрацию испытанного соединения, при которой достигается половина максимального ингибирования роста бактериальных клеток. Такая информация может быть использована для более точного определения доз, применимых для человека. Следует представлять, однако, что специфический уровень дозы для любого конкретного пациента будет зависеть от множества факторов, включающих природу соединения, возраст, массу тела, общее состояние здоровья, пол, диету, время введения, путь введения и скорость выведения, комбинацию лекарственных средств, тяжесть конкретного заболевания в процессе терапии и мнение лечащего врача. Для введения животным, не принадлежащим к человеческому роду, лекарственное средство или фармацевтическая композиция, содержащая лекарственное средство, могут быть добавлены в пищу животного или питьевую воду. Удобнее всего подготавливать пищевые продукты и питьевую воду для животного с предварительно определенной дозой лекарственного средства, таким образом животное потребляет соответствующее количество лекарственного средства с пищей. Также удобно добавлять предварительно приготовленную смесь, содержащую лекарственное средство, в пищу или питьевую воду непосредственно перед потреблением продуктов животным. Предпочтительные соединения данного изобретения будут характеризоваться некоторыми фармакологическими свойствами. Такие свойства включают, но без ограничения только ими, пероральную биодоступность, низкую токсичность, низкое сывороточное связывание белков и приемлемые периоды полураспада in vitro и in vivo. Биологические испытания могут быть проведены для прогноза указанных желательных свойств. Биологические испытания, проводимые для прогноза биодоступности, включают транспорт через монослои кишечных клеток, включающие Сасо-2 клеточные монослои. Сывороточное связывание белков может быть предсказано по результатам биопробы на связывание альбумина. Такие биологические испытания описаны в обзоре Oravcova et al. (1996, J. Chromat. В 677: 1-27). Период полураспада соединения обратно пропорционален частоте дозировки соединения. In vitro периоды полураспада соединений могут быть предсказаны по результатам микросомального периода полураспада, как описано в литературе: Kuhnz, Gieschen (Drug Metabolism and Disposition, (1998) volume 26, pages 1120-1127). Токсичность и терапевтическая эффективность соединений может быть установлена по стандартным фармацевтическим методикам на культурах клеток или на экспериментальных животных, например, по определению LD50 (доза, летальная для 50% популяции) и ED50 (доза, терапевтически эффективная для 50% популяции). Отношение доз, характеризующих токсические и терапевтические эффекты, представляет собой терапевтический индекс, и он может быть выражен как отношение между LD50 и ED50. Соединения, которые обладают высокими терапевтическими индексами, являются предпочтительными. Данные, полученные из испытаний на культуре клеток и на животных, могут быть использованы для определения диапазона доз применительно к людям. Дозировка таких соединений находится предпочтительно в диапазоне циркулирующих концентраций, которые включают ED50 с низкой токсичностью или с ее отсутствием. Доза может меняться внутри данного диапазона в зависимости от примененной лекарственной формы и способа введения. Точная технология приготовления лекарственного средства, способ введения и доза могут быть выбраны индивидуально врачом с учетом состояния пациента. (См., например, Fingi et al., 1975, in “The Pharmacological Basis of Therapeutics”, Ch.l, p.1.) Количество доз и интервалы между ними могут быть установлены индивидуально для обеспечения такого содержания в плазме активного вещества, которое достаточно для проявления ростингибирующего воздействия на бактериальные клетки. Дозировки для обычного пациента при системном введении колеблются от 100 до 2000 мг/сутки. С учетом площади поверхности тела пациента обычные дозировки колеблются от 50 до 910 мг/м2/сутки. Обычные средние уровни в плазме должны находиться в пределах 0,1-1000 мкМ. В случаях локального введения или селективного поглощения эффективная локальная концентрация соединения не может быть связана с концентрацией в плазме. Соединения данного изобретения применимы как антибиотики для лечения болезней как у животных, так и у людей, включающих, но без ограничения только ими, актиномикоз, сибирскую язву, бактериальную дизентерию, ботулизм, бруцеллез, целлюлит, холеру, конъюнктивит, цистит, дифтерию, бактериальный эндокардит, эпиглоттит, гастроэнтерит, сап, гонорею, “болезнь легионеров”, лептоспироз, септический менингит, чуму, бактериальную пневмонию, послеродовой сепсис, ревматическую лихорадку, пятнистую лихорадку Скалистых гор, скарлатину, стрептококковый фарингит, сифилис, столбняк, туберкулез, туляремию, брюшной тиф, сыпной тиф и коклюш. Содержание в данной заявке всех статей и отсылочных материалов, включая патенты, представлено здесь в качестве ссылок. Соединения данного изобретения включают новый класс антибиотиков широкого спектра действия. Виды бактерий, важных с медицинской точки зрения, которые являются соответствующими мишенями для проявления антибактериальной активности ингибиторов данного изобретения, включают грамположительные бактерии, включающие кокки, такие как виды Staphylococcus и виды Streptococcus; кислотоустойчивые бактерии, включающие виды Mycobacterium; бациллы, включающие виды Bacillus, виды Corynebacterium и виды dostr-idlum; нитчатые бактерии, включающие виды Actinomyces и виды Streptomyces; грамотрицательные бактерии, включающие кокки, такие как виды Neisseria и виды Acinetobacter; бациллы, такие как виды Pseudomonas, виды Brucella, виды Agrobacterium, виды Bordetella, виды Escherichia, виды Shigella, виды Yersinia, виды Salmonella, виды Klebsiella, виды Enterobacter, виды Haemophilus, виды Pasteurella и виды Streptobacillus; виды спирохет, виды Campylobacter, виды Vibrio; и внутриклеточные бактерии, включающие виды Rickettsiae и виды Chlamydia. Специфические виды бактерий, которые являются мишенями антибиотиков данного изобретения, включают Staphylococcus aureus, Staphylococcus epidermidis, Staphylococcus saprophyticus, Streptococcus pyogenes. Streptococcus agalactiae, Streptococcus pneumoniae, Enterococcus faecalis, Enterococcus Saecium, Bacillus anthracis, Mycobacterium avium, Mycobacterium tuberculosis, Acinetobacter baumannii, Corynebacterium diphtheria, Clostridium perfringens, Clostridium botulinum, Clostridium tetani, Neisseria gonorrhoeae, Neisseria meningitidis, Pseudomonas aeruginosa, Legionella pneumophila, Escherichia coli, Yersinia pestis, Haemophilus influenzae, Helicobacter pylori, Campylobacter fetus, Campylobacter jejuni, Vibrio cholerae. Vibrio parahemolyticus, Trepomena pallidum, Actinomyces Israelii, Rickettsia prowazekii, Rickettsia rickettsii, Chlamydia trachomatis, Chlamydia psittaci, Brucella abortus, Agrobacterium tumefaciens и Francisella tularensis. При выполнении процедур настоящего изобретения, конечно, следует понимать, что отсылка к конкретным буферам, средам, реагентам, клеткам, условиям культивирования и т.п. не предназначена для ограничения только ими, но должна быть прочитана таким образом, что включаются все родственные материалы, которые мог бы рассматривать обычный специалист в данной области, как представляющие интерес или значимость в конкретном контексте, в котором такое обсуждение приведено. Например, часто возможно заменить одну буферную систему или среду для культивирования на другую и тем не менее получить подобные, если не идентичные, результаты. Специалистам в данной области будет достаточно знаний таких систем и методологий, чтобы быть способными без чрезмерного экспериментирования осуществить подобные замены, что будет оптимально служить их целям по применению способов и методик, раскрытых в данной работе. Изобретение описано более детально в следующих далее примерах, но без ограничения только ими. Следует представлять, что данные способы и примеры никоим образом не ограничивают изобретение вариантами осуществления, описанными здесь, и что другие варианты осуществления и применения, несомненно, будут очевидны для специалистов в данной области. Соединения данного изобретения оценивали на антибактериальную активность по рекомендациям и методикам, предписанным Национальным комитетом клинических лабораторных стандартов/National Committee for Clinical Laboratory Standards (NCCLS) (cf., NCCLS Document M7-A3, 1993 – Antimicrobial Susceptibility Testing). Протокол определения MIC Использованный протокол определения MIC представляет собой следующее: 1. Приблизительно по 2,5 мг соединений, предназначенных для испытания, отвешивали в криососуды. 2. Основные растворы с концентрацией 5 мг/мл готовили добавлением ДМСО к образцам. 3. Рабочие растворы с концентрацией 256 мкг/мл готовили из 5 мг/мл основных растворов добавлением стерильной дистиллированной воды соответственно. 4. Установку, Beckman 2000 Automated Workstation, программировали на загрузку 96-луночных планшетов питательной средой и соединениями следующим образом: – 100 мкл соответствующей среды добавляли в колонки 1-11, – 200 мкл соответствующей среды добавляли в колонку 12, – 100 мкл соединения из 256 мкг/мл рабочего раствора добавляли в колонку 1 (одно соединение на ряд), – двукратное серийное разбавление проводили от колонки 1 до 10, – колонка 11 служила в качестве контроля роста. 5. Высевали набор 10 организмов из основных сосудов, хранившихся при -80°С, и инкубировали 24 часа при 34°С. Организмы затем пересевали и инкубировали 24 часа при 34°С. – Инокуляты сначала готовили в стерильной дистиллированной воде для достижения 0,09-0,11 поглощения при длине волны 620 нм, – проводили 1/100 разбавление в соответствующей среде, – 100 мкл среды с организмом добавляли в колонки 1-11, – колонка 12 служила в качестве чистого контроля. 6. Укомплектованные 96-луночные планшеты инкубировали 24 часа при 34°С. С 96-луночных планшетов снимали данные с помощью считывающего устройства, Beckman Automated Plate Reader, при длине волны 650 нм. MIC определяли вычислениями, включающими данные по контролю роста (колонка 11) и чистому контролю (колонка 12). Боринатные комплексы Данная процедура была применена для получения результатов, представленных далее в таблицах. Приведенные микробиологические данные для соединений 10-123 показаны в таблицах 1-4 в виде MIC (минимальная ингибирующая концентрация) с размерностью микрограммы на мл. Таким образом, изобретение относится к антибиотикам, которые в общем виде названы комплексами бориновой кислоты, наиболее предпочтительно являются производными замещенных бориновых кислот. Синтез соединений данного изобретения проведен несколькими путями. Схема реакции Как показано на схеме 1, комплексы бориновой кислоты получают из предшественников – бориновых кислот взаимодействием с 1 эквивалентом желаемого гетероциклического лиганда в подходящих растворителях (таких, как этанол, изопропанол, диоксан, эфир, толуол, диметилформамид, N-метилпирролидон или тетрагидрофуран). Схема 1
В некоторых случаях соединения изобретения могут содержать один или несколько асимметрических атомов углерода, т.е. соединения могут существовать в различных стереоизомерных формах. Подобные соединения могут быть, например, рацематами или оптически активными формами. В таких ситуациях индивидуальные энантиомеры, т.е. оптически активные формы, могут быть получены асимметрическим синтезом или разделением рацематов. Разделение рацематов может быть проведено, например, традиционными способами, такими как кристаллизация в присутствии разделяющего средства или хроматография с применением, например, хиральной ВЭЖХ колонки. Представленные соединения настоящего изобретения включают, но без ограничения только ими, соединения, раскрытые в данной работе, и их фармацевтически приемлемые кислотно- и основно-аддитивные соли. Кроме того, если соединение изобретения получено в виде кислотно-аддитивной соли, то свободное основание может быть получено подщелачиванием раствора кислотной соли. Наоборот, если продукт представляет собой свободное основание, то аддитивная соль, особенно фармацевтически приемлемая аддитивная соль, может быть получена растворением свободного основания в подходящем органическом растворителе и обработкой полученного раствора кислотой в соответствии с обычными процедурами для приготовления кислотно-аддитивных солей из соединений-оснований. В предпочтительном воплощении соединения данного изобретения включают любое из соединений 10-123 (таблицы 1, 2, 3 и 4) и их варианты.
Настоящее изобретение также включает ацилированные пролекарства соединений изобретения. Специалистам в данной области будут предоставлены различные синтетические методологии, которые могут быть применены для приготовления нетоксичных фармацевтически приемлемых аддитивных солей и ацилированных пролекарств изобретенных соединений. Таблицы 1, 2 и 3 содержат также ингибирующую активность известных антибиотиков, приведенных в конце каждой из таблиц. Примеры Протонный ЯМР зарегистрирован на спектрометре Varian AS 400, и химические сдвиги выражены в виде Образование этиленгликолевого эфира бороновой кислоты (соединение 3 Т=отсутствует) Общая процедура Бороновую кислоту растворяли в безводном ТГФ или безводном диэтиловом эфире (~10 мл/г) в атмосфере азота. Этиленгликоль (1 молярный эквивалент) добавляли к реакционной смеси и смесь кипятили с обратным холодильником от 1 до 4 часов. Реакционную смесь охлаждали до комнатной температуры и растворитель удаляли при пониженном давлении, получая в остатке этиленгликолевый эфир в виде масла или твердого вещества. В случаях когда получали масло или твердое вещество, которые растворялись в гексане, добавляли безводный гексан и удаляли его при пониженном давлении. Продукт затем выдерживали несколько часов в высоком вакууме. В случаях когда получали твердое вещество, нерастворимое в гексане, твердое вещество отделяли фильтрованием и промывали холодным гексаном. Этиленгликолевый эфир 3-цианофенилбороновой кислоты (3а) 3-Цианофенилбороновую кислоту (1 г, 6,8 ммоль) растворяли в безводном ТГФ (10 мл) в атмосфере азота. Добавляли этиленгликоль (379 мкл, 422 мг, 6,8 ммоль) и реакционную смесь кипятили с обратным холодильником 4 часа, затем охлаждали до комнатной температуры. ТГФ удаляли с помощью роторного испарителя, получая белое твердое вещество, к которому добавляли холодный гексан. Продукт отделяли фильтрованием, получая белое твердое вещество (1,18 г, колич. выход). 1H-ЯМР (300,058 МГц, ДМСО-d6) Этиленгликолевый эфир тиофен-3-бороновой кислоты (3b) Тиофен-3-бороновую кислоту (1 г, 7,8 ммоль) растворяли в безводном ТГФ (10 мл) в атмосфере азота. Добавляли этиленгликоль (435 мкл, 484 мг, 7,8 ммоль) и реакционную смесь кипятили с обратным холодильником 1 час, затем охлаждали до комнатной температуры. ТГФ удаляли с помощью роторного испарителя, получая белое твердое вещество. Добавляли гексан, растворяя твердое вещество, и удаляли растворитель с помощью роторного испарителя. Продукт помещали в высокий вакуум, получая желтовато-коричневое твердое вещество (1,17 г, 97%). 1H-ЯМР (300,058 МГц, CDCl3) Образование несимметричной бориновой кислоты (6) из этиленгликолевого эфира бороновой кислоты Общая процедура А: методика реакции Гриньяра Этиленгликолевый эфир бороновой кислоты растворяли в безводном ТГФ (10-20 мл/г) в атмосфере азота. Раствор охлаждали до -78°С на бане ацетон/сухой лед или до 0°С на бане лед/вода. К охлажденному раствору добавляли по каплям реактив Гриньяра (от 0,95 до 1,2 молярного эквивалента). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 3-18 часов. К смеси добавляли 6N НСl (2 мл/г) и растворитель удаляли в вакууме. Продукт экстрагировали диэтиловым эфиром (40 мл/г) и промывали водой (3 × равный объем). Органический слой сушили (MgSO4), фильтровали и растворитель удаляли с помощью роторного испарителя, получая неочищенный продукт, который очищали либо колоночной хроматографией, либо использовали на следующей стадии без очистки. Альтернативная процедура: если продукт бориновой кислоты содержал основную группу, такую как аминная или пиридиновая, то после перемешивания при комнатной температуре в течение 3-18 часов добавляли воду (2 мл/г) и рН устанавливали до 5-7. Продукт экстрагировали диэтиловым эфиром (40 мл/г) и промывали водой (3 × равный объем). Органический слой сушили (MgSO4), фильтровали и растворитель удаляли с помощью роторного испарителя, получая неочищенный продукт, который очищали либо колоночной хроматографией, либо использовали на следующей стадии без очистки. (4-Цианофенил)(3-фторфенил)бориновая кислота (6а) Этиленгликолевый эфир 4-цианофенилбороновой кислоты (500 мг, 2,89 ммоль) растворяли в безводном ТГФ в атмосфере азота. Раствор охлаждали до -78°С на бане ацетон/сухой лед и к холодному раствору добавляли по каплям 3-фторфенилмагнийбромид (1М в ТГФ) (2,74 мл, 2,74 ммоль, 0/95 молярного эквивалента). Реакционной смеси позволяли медленно нагреться до комнатной температуры и перемешивали в течение 18 часов. К реакционной смеси добавляли 6N НСl (1 мл), что вызывало помутнение, и растворитель удаляли с помощью роторного испарителя. Продукт экстрагировали диэтиловым эфиром (20 мл) и промывали водой (3×20 мл). Органический слой сушили (МgSO4), фильтровали и растворитель удаляли с помощью роторного испарителя, получая неочищенный продукт в виде маслянистого твердого вещества. Продукт использовали на следующей стадии без очистки. Общая процедура В: методика реакции с (гетеро)ариллитием (Гетеро)арилбромид или йодид растворяли в безводном ТГФ (20-30 мл/г) в атмосфере азота и дегазировали. Раствор охлаждали до -78°С на бане ацетон/сухой лед и к охлажденному раствору добавляли по каплям н-, втор- или трет-бутиллитий в ТГФ или другом растворителе (1,5-2,4 молярных эквивалентов), что обычно вызывало интенсивное пожелтение раствора. Этиленгликолевый эфир бороновой кислоты (1 молярный эквивалент) растворяли в безводном ТГФ или диэтиловом эфире (2-5 мл/г) в атмосфере азота. Этиленгликолевый эфир бороновой кислоты в ТГФ добавляли по каплям к охлажденному раствору ариллития, что обычно приводило к бледно-желтой окраске раствора. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 3-18 часов. К смеси добавляли 6N НСl (2-4 мл/г) и растворитель удаляли в вакууме. Продукт экстрагировали диэтиловым эфиром (40 мл/г) и промывали водой (3 × равный объем). Органический слой сушили (MgSO4), фильтровали и растворитель удаляли с помощью роторного испарителя, получая неочищенный продукт, который очищали либо колоночной хроматографией, либо использовали на следующей стадии без очистки. Альтернативная процедура: если продукт бориновой кислоты содержал основную группу, такую как аминная или пиридиновая, то после перемешивания при комнатной температуре в течение 3-18 часов добавляли воду (2 мл/г) и рН устанавливали до 5-7. Продукт экстрагировали диэтиловым эфиром (40 мл/г) и промывали водой (3 × равный объем). Органический слой сушили (MgSO4), фильтровали и растворитель удаляли с помощью роторного испарителя, получая неочищенный продукт, который очищали либо колоночной хроматографией, либо использовали на следующей стадии без очистки. (3-Тиофен) (3-хлорфенил)бориновая кислота (6b) 3-Хлор-бромбензол (447 мкл, 728 мг, 3,8 ммоль) растворяли в безводном ТГФ (15 мл) в атмосфере азота. Раствор дегазировали и охлаждали до -78°С на бане ацетон/сухой лед. К охлажденному раствору добавляли по каплям трет-бутиллитий (1,7 М в ТГФ) (4,47 мл, 7,6 ммоль, 2 молярных эквивалента), что вызывало интенсивное пожелтение раствора. Раствор перемешивали при -78°С и одновременно растворяли этиленгликолевый эфир 3-тиофенбороновой кислоты (586 мг) в безводном диэтиловом эфире (1 мл). Раствор эфира бороновой кислоты затем добавляли по каплям к охлажденному раствору, что приводило к изменению окраски раствора до бледно-желтой. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 18 часов. К смеси добавляли 6N НСl (2 мл) и реакционную смесь перемешивали 1 час. Растворитель удаляли с помощью роторного испарителя. Продукт экстрагировали диэтиловым эфиром (10 мл) и промывали водой (2×10 мл). Органический слой сушили (МgSO4), фильтровали и растворитель удаляли с помощью роторного испарителя, получая неочищенный продукт в виде оранжевого масла. Продукт очищали колоночной хроматографией с применением силикагеля и элюента гексан: этилацетат 5:1 и получали чистый продукт в виде прозрачного масла (614 мг, 73%). (3-Хлорфенил)винилбориновая кислота (6с) Соединение получали способом, аналогичным описанному для 6b, взаимодействием этиленгликолевого эфира 3-хлорфенилбороновой кислоты с виниллитием. (3-Фтор-5-хлорфенил)этинилбориновая кислота (6d) Соединение получали способом, аналогичным описанному для 6b, взаимодействием этиленгликолевого эфира 3-фтор-5-хлорфенилбороновой кислоты с этиниллитием. (4-Метил-3-хлорфенил)(2-тиенил)бориновая кислота (6е) Соединение получали способом, аналогичным описанному для 6b, взаимодействием этиленгликолевого эфира 2-тиенилбороновой кислоты с 4-метил-3-хлорфениллитием. (4-Цианофенил)этинилбориновая кислота (6f) Соединение получали способом, аналогичным описанному для 6b, взаимодействием этиленгликолевого эфира 4-цианофенилбороновой кислоты с этиниллитием. (3-Фторфенил)циклопропилбориновая кислота (6g) Соединение получали способом, аналогичным описанному для 6b, взаимодействием этиленгликолевого эфира 3-фторфенилбороновой кислоты с циклопропиллитием. (3-Тиенил)метилбориновая кислота (6h) Соединение получали способом, аналогичным описанному для 6b, взаимодействием этиленгликолевого эфира 3-тиенилбороновой кислоты с метиллитием. (4-Пиридил)фенилбориновая кислота (6i) Соединение получали способом, аналогичным описанному для 6b, взаимодействием этиленгликолевого эфира фенилбороновой кислоты с 4-пиридиллитием. (3-Цианофенил)(2-фторфенил)бориновая кислота (6j) Соединение получали способом, аналогичным описанному для 6b, взаимодействием этиленгликолевого эфира 3-цианофенилбороновой кислоты с 2-фторфениллитием. Образование симметричной бориновой кислоты (5) взаимодействием металлоорганических соединений с триалкилборатами Бис(4-хлорфенил)бориновая кислота (5а) (Процедура С) К холодному раствору (-78°С) Гриметилбората (0,37 мл) в безводном тетрагидрофуране (ТГФ, 25 мл) добавляли по каплям 4-хлорфенилмагнийбромид (6,75 мл, 1М раствор в эфире). Реакционную смесь перемешивали при -78°С в течение 1 час и затем 18 час при комнатной температуре. Растворитель удаляли при пониженном давлении. Полученный остаток перемешивали с 100 мл эфира и 15 мл 6N хлористоводородной кислоты. Органический слой отделяли и водный слой экстрагировали эфиром (2×100 мл). Объединенные органические экстракты промывали насыщенным раствором соли и сушили над безводным сульфатом магния. Растворитель удаляли и получали светло-желтоватое твердое вещество. Продукт хроматографировали на силикагеле (гексан: эфир=1:1) и получали 420 мг бориновой кислоты. 1Н-ЯМР (400 МГц, DОСl3) Указанное в заголовке соединение получали аналогично способу, описанному для 5а, взаимодействием 3-хлор-4-метилфенилмагнийбромида с триметилборатом. Продукт выделяли хроматографией на силикагеле. Бис(3-фтор-4-метилфенил)бориновая кислота (5с) Указанное в заголовке соединение получали аналогично способу, описанному для 5а, взаимодействием 3-фтор-4-метилфениллития с триметилборатом. Продукт выделяли хроматографией на силикагеле. Бис(3-хлор-4-метоксифенил)бориновая кислота (5d) Указанное в заголовке соединение получали аналогично способу, описанному для 5а, взаимодействием 3-хлор-4-метоксифениллития с триметилборатом. Продукт выделяли хроматографией на силикагеле. Бис(3-фтор-4-метоксифенил)бориновая кислота (5е) Указанное в заголовке соединение получали аналогично способу, описанному для 5а, взаимодействием 3-фтор-4-метоксифениллития с триметилборатом. Продукт выделяли хроматографией на силикагеле. Образование несимметричной бориновой кислоты (6) взаимодействием металлоорганических соединений с алкил(арил)диалкоксиборанами (4-Хлорфенил)метилбориновая кислота (6k) (Процедура D) К 4-хлорфенилмагнийбромиду (5,5 мл, 1М раствор в эфире) при -78°С добавляли по каплям с помощью шприца ди(изопропокси)метилборан (1 мл, 0,78 г). Реакционную смесь перемешивали при -78°С в течение 1 час и затем перемешивали в течение ночи при температуре окружающей среды. К реакционной смеси добавляли по каплям 100 мл эфира и 15 мл 6N хлористоводородной кислоты и затем смесь перемешивали 1 час. Органический слой отделяли и водный слой экстрагировали эфиром (2×100 мл). Объединенные органические экстракты промывали насыщенным раствором соли и сушили над безводным сульфатом магния. Растворитель удаляли при пониженном давлении и получали 1,1 г масла. 1H-ЯМР продукта соответствовал (4-хлорфенил)метилбориновой кислоте. (4-Фторфенил)метилбориновая кислота (6m) Указанное в заголовке соединение получали аналогично способу, описанному для 6k, взаимодействием 4-фторфенилмагнийбромида с ди(изопропокси)метилбораном. Продукт выделяли хроматографией на силикагеле. (4-Бифенил)метилбориновая кислота (6n) Указанное в заголовке соединение получали аналогично способу, описанному для 6k, взаимодействием 4-бифениллития с ди(изопропокси)метилбораном. Продукт выделяли хроматографией на силикагеле. (3-Хлор-4-метилфенил)метилбориновая кислота (6о) Указанное в заголовке соединение получали аналогично способу, описанному для 6k, взаимодействием 3-хлор-4-метилфениллития с ди(изопропокси)метилбораном. Продукт выделяли хроматографией на силикагеле. (3-Хлор-4-метоксифенил)метилбориновая кислота (6р) Указанное в заголовке соединение получали аналогично способу, описанному для 6k, взаимодействием 3-хлор-4-метоксифениллития с ди(изопропокси)метилбораном. Продукт выделяли хроматографией на силикагеле. (4-Диметиламинофенил)метилбориновая кислота (6q) Указанное в заголовке соединение получали аналогично способу, описанному для 6k, взаимодействием 4-диметиламинофениллития с ди(изопропокси)метилбораном. Продукт выделяли хроматографией на силикагеле. (3-Хлор-4-диметиламинофенил)винилбориновая кислота (6r) Указанное в заголовке соединение получали аналогично способу, описанному для 6k, взаимодействием 3-хлор-4-диметиламинофениллития с ди(бутоксивинил)бораном. Продукт выделяли хроматографией на силикагеле. Эфир бис(3-хлорфенил)бориновой кислоты и 4-(гидроксиэтил)имидазола (121) К раствору бис(3-хлорфенил)бориновой кислоты (0,4 г, 1,428 ммоль) в этаноле (10 мл) добавляли гидрохлорид 4-(гидроксиэтил)имидазола (0,191 г, 1,428 ммоль) и бикарбонат натрия (0,180 г, 2,143 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 18 час. Соль удаляли фильтрованием. Фильтрат концентрировали, обрабатывали гексаном и получали продукт в виде твердого вещества, который отделяли фильтрованием (450 мг, 84,9%). Эфир бис(4-хлорфенил)бориновой кислоты и 4-(гидроксиметил)имидазола (126) Указанное в заголовке соединение получали аналогично способу примера 121 взаимодействием бис(4-хлорфенил)бориновой кислоты с гидрохлоридом 4-(гидроксиметил)имидазола. Продукт получали в виде белых кристаллов. Эфир бис(3-хлор-4-метилфенил)бориновой кислоты и 1-бензил-4- (гидроксиметил)имидазола (127) К раствору 1-бензил-4-(гидроксиметил)имидазола (96 мг, 0,521 ммоль) в метаноле (5 мл) добавляли бис(3-хлор-4-метилфенил)бориновую кислоту (121 мг, 0,521 ммоль) и реакционную смесь перемешивали при комнатной температуре 2 час. Растворитель удаляли при пониженном давлении и остаток обрабатывали гексаном, что приводило к образованию твердого вещества. Осадок отделяли фильтрованием, промывали гексаном и получали продукт (193 г, 83%). 1H-ЯМР (CDCl3) Эфир бис(3-хлор-4-метилфенил)бориновой кислоты и 1-метил-2- (гидроксиметил)имидазола (128) Указанное в заголовке соединение получали аналогично способу примера 127 взаимодействием бис(3-хлор-4-метилфенил)бориновой кислоты с гидрохлоридом 1-метил-2-(гидроксиметил)имидазола. Продукт получали в виде белых кристаллов. Эфир бис(3-хлор-4-метилфенил)бориновой кислоты и 1-этил-2- (гидроксиметил)имидазола (129) Указанное в заголовке соединение получали аналогично способу примера 127 взаимодействием бис(3-хлор-4-метилфенил)бориновой кислоты с гидрохлоридом 1-этил-2-(гидроксиметил)имидазола. Продукт получали в виде белых кристаллов. Эфир бис(3-хлор-4-метилфенил)бориновой кислоты и 1-метил-4- (гидроксиметил)имидазола (130) Указанное в заголовке соединение получали аналогично способу примера 127 взаимодействием бис(3-хлор-4-метилфенил)бориновой кислоты с гидрохлоридом 1-метил-4-(гидроксиметил)имидазола. Продукт получали в виде белых кристаллов. 2-Пиридилэтиловый эфир бис(3-хлор-4-метилфенил)бориновой кислоты (131) Указанное в заголовке соединение получали аналогично способу примера 121 взаимодействием бис(3-хлор-4-метилфенил)бориновой кислоты с 2-пиридилэтанолом. Продукт получали в виде белых кристаллов. 2-Пиридилметиловый эфир бис(4-хлорфенил)бориновой кислоты (132) Указанное в заголовке соединение получали аналогично способу примера 121 взаимодействием бис(4-хлорфенил)бориновой кислоты с 2-пиридилметанолом. Продукт получали в виде белых кристаллов. 2-Пиридилметиловый эфир бис(4-фторфенил)бориновой кислоты (133) Указанное в заголовке соединение получали аналогично способу примера 121 взаимодействием бис(4-фторфенил)бориновой кислоты с 2-пиридилметанолом. Продукт получали в виде белых кристаллов. Производные гидроксихинолина Эфир бис(3-хлорфенил)бориновой кислоты и 8-гидроксихинолина (10) Раствор бис(3-хлорфенил)бориновой кислоты (0,18 г) в этаноле (1 мл) и 8-гидроксихинолин (0,105 г) перемешивали при 5°С. Реакционную смесь затем перемешивали при температуре окружающей среды, при этом образовывался желтый осадок. Реакционную смесь перемешивали дополнительно 4 часа. Продукт выделяли фильтрованием, промывали гексаном, сушили на воздухе и получали 160 мг комплекса. Эфир бис(3-хлорфенил)бориновой кислоты и 5-фтор-8-гидроксихинолина (12) Указанное в заголовке соединение получали аналогично способу примера 10 взаимодействием бис(3-хлорфенил)бориновой кислоты с 5-фтор-8-гидроксихинолином. Продукт получали в виде желтых кристаллов. Эфир бис(3-хлорфенил)бориновой кислоты и 5-хлор-8-гидроксихинолина (13) Указанное в заголовке соединение получали аналогично способу примера 10 взаимодействием бис(3-хлорфенил)бориновой кислоты с 5-хлор-8-гидроксихинолином. Продукт получали в виде желтых кристаллов. Эфир бис(3-хлорфенил)бориновой кислоты и 5-циано-8-гидроксихинолина (19) Указанное в заголовке соединение получали аналогично способу примера 10 взаимодействием бис(3-хлорфенил)бориновой кислоты с 5-циано-8-гидроксихинолином. Продукт получали в виде желтых кристаллов. Эфир (2-тиенил)метилбориновой кислоты и 8-гидроксихинолина (26) Указанное в заголовке соединение получали аналогично способу примера 10 взаимодействием (2-тиенил)метилбориновой кислоты с 8-гидроксихинолином. Продукт получали в виде желтых кристаллов. Эфир (3-хлорфенил)(2-тиенил)бориновой кислоты и 8-гидроксихинолина (36) Указанное в заголовке соединение получали аналогично способу примера 10 взаимодействием (3-хлорфенил)(2-тиенил)бориновой кислоты с 8-гидроксихинолином. Продукт получали в виде желтых кристаллов. Эфир (3-цианофенил)винилбориновой кислоты и 8-гидроксихинолина (40) Указанное в заголовке соединение получали аналогично способу примера 10 взаимодействием (3-цианофенил)винилбориновой кислоты с 8-гидроксихинолином. Продукт получали в виде желтых кристаллов. Эфир (2-хлорфенил)этинилбориновой кислоты и 8-гидроксихинолина (43) Указанное в заголовке соединение получали аналогично способу примера 10 взаимодействием (2-хлорфенил)этинилбориновой кислоты с 8-гидроксихинолином. Продукт получали в виде желтых кристаллов. Эфир бис(этинил)бориновой кислоты и 8-гидроксихинолина (44) (XXI) Указанное в заголовке соединение получали аналогично способу примера 10 взаимодействием бис(этинил)бориновой кислоты с 8-гидроксихинолином. Продукт получали в виде светло-желтых кристаллов. Эфир (3-фторфенил)циклопропилбориновой кислоты и 8-гидроксихинолина (70) Указанное в заголовке соединение получали аналогично способу примера 10 взаимодействием (3-фторфенил)циклопропилбориновой кислоты с 8-гидроксихинолином. Продукт получали в виде светло-желтых кристаллов. В предпочтительном варианте осуществления настоящее изобретение включает все соединения, специально перечисленные здесь, их фармацевтически приемлемые соли и композиции любого из данных соединений в тех случаях, когда они содержат фармацевтически приемлемый носитель. Самыми предпочтительными являются соединения, имеющие структуру любого соединения из перечисленных в таблицах 1, 2, 3 или 4, особенно соединения 10-108, соединения 111-112 или соединения 116-120. В таких соединениях лиганд, как описано здесь в другом месте, присоединен к бору через указанные реакционноспособные группы. Настоящее изобретение также относится к способу лечения заболеваний, вызываемых микробами, при поражении ими пациента и/или профилактики такой инфекции у пациента при опасности заражения, к способу, который включает введение указанному пациенту терапевтически эффективного количества любого из соединений данного изобретения, предпочтительно одного или нескольких из перечисленных в таблицах 1-4. В одном из аспектов соединения изобретения проявляют антибактериальную (т.е. бактерицидную) и противогрибковую (т.е. фунгицидную) активность. В предпочтительном варианте осуществления микроб представляет собой бактерию, предпочтительно грамположительную, при этом указанная грамположительная бактерия относится к группе, включающей виды Staphylococcus, виды Streptococcus, виды Bacillus, виды Mycobacterium, виды Corynebacterium, виды Clostridium, виды Actinomyces, виды Enterococcus и виды Streptomyces. В другом предпочтительном варианте осуществления указанного способа бактерия представляет собой грамотрицательную бактерию, предпочтительно выбранную из группы, включающей виды Acinetobacter, виды Neisseria, виды Pseudomonas, виды Brucella, виды Agrobacterium, виды Bordetella, виды Escherichia, виды Shigella, виды Yersinia, виды Salmonella, виды Klebsiella, виды Enterobacter, виды Haemophilus, виды Pasteurella, виды Streptobacillus, виды спирохет, виды Campylobacter, виды Vibrio и виды Helicobacter. В самом предпочтительном варианте осуществления настоящего изобретения бактерия выбрана из группы, включающей Staphylococcus aureus, Staphylococcus epidermidis, Staphylococcus saprophyticus, Streptococcus pyogenes, Streptococcus agalactiae. Streptococcus pneumoniae, Enterococcus faecalis, Enterococcus faecium. Bacillus anthracis, Mycobacterium avium, Mycobacterium tuberculosis, Acinetobacter baumannii, Corynebacterium diphtheria, Clostridium perfringens, Clostridium botulinum, Clostridium tetani, Neisseria gonorrhoeae, Neisseria meningitidis, Pseudomonas aeruginosa, Legionella pneumophila, Escherichia coli, Yersinia pestis, Haemophilus influenzae, Helicobacter pylori, Campylobacter fetus, Campylobacter jejuni. Vibrio cholerae. Vibrio parahemolyticus, Trepomena pallidum, Actinomyces israelii, Rickettsia prowazekii, Rickettsia rickettsii, Chlamydia trachomatis, Chlamydia psittaci, Brucella abortus, Agrobacterium tumefaciens и Francisella tularensis.
Формула изобретения
1. Соединение структурной формулы 2 2. Соединение по п.1, где по меньшей мере один из R* и R** соответствует формуле 3. Соединение по п.1, где по меньшей мере один из R* и R** соответствует формуле 4. Соединение по п.1, где по меньшей мере один из R* и R** соответствует формуле 5. Соединение по п.1, где по меньшей мере один из R* и R** соответствует формуле 6. Соединение по п.1, где по меньшей мере один из R* и R** соответствует формуле 7. Соединение по п.1, где R9 представляет собой Н; m представляет собой 0; А представляет собой СН; D представляет собой СН; Е представляет собой ОН; R* выбран из 4-Ме-3-СlС6Н3, 3-ClC6H4, 4-EtO-3-ClC6H3, 2-Cl-5-Br-6-F-С6Н2, 2-Ме-4-ClC6H3; и R** выбран из 4-Ме-3-ClC6H3, 4-Ме-С6Н4; фенилэтила; 3-ClC6H4, 4-EtO-3-ClC6H3, 2-F-4-ClC6H3, 2-Ме-4-ClC6H3. 8. Соединение по п.1, где R9 представляет собой Н; m представляет собой 0; А представляет собой СН; D представляет собой СН; Е представляет собой ОН; 9. Соединение по п.1, где R9 представляет собой Н; m представляет собой 0; А представляет собой СН; D представляет собой СН; Е представляет собой ОН; R* представляет собой 4-Ме-3-ClC6H3; R** представляет собой 4-Ме-3-ClC6H3. 10. Соединение по п.1, где R9 представляет собой Н; m представляет собой 0; А представляет собой СН; D представляет собой СН; Е представляет собой ОН; R* представляет собой 2-Ме-4-ClC6H3; R** представляет собой 2-Ме-4-ClC6H3. 11. Соединение по п.1, где R9 представляет собой Н; m представляет собой 0; А представляет собой СН; D представляет собой СН; Е представляет собой 2-гидроксиэтокси, N-морфолинилэтокси, ОСН2СН2СН2СООН, СООН или ОАс; R* представляет собой 4-Ме-3-ClC6H3; R** представляет собой 4-Ме-3-ClC6H3. 12. Соединение по п.1, где R9 представляет собой Н; m представляет собой 1; А представляет собой СН; D представляет собой СН; Е представляет собой Н; R* представляет собой 4-Ме-3-ClC6H3 и R** представляет собой 4-Ме-3-ClC6H3. 13. Соединение по п.1, где растворитель в составе аддукта с растворителем выбран из гидроксильного растворителя и аминорастворителя. 14. Соединение, выбранное из комплекса бис(3-хлор-4-метилфенил)бориновой кислоты с 3-(2-гидроксиэтокси)пиколиновой кислотой (107), комплекса бис(3-хлор-4-метилфенил)бориновой кислоты с 3-(N-морфолинилэтокси)пиколиновой кислотой (108), комплекса бис(3-хлор-4-метилфенил)бориновой кислоты с 3-(OCH2CH2CH2CO2H) пиколиновой кислотой (109), комплекса бис(3-хлор-4-метилфенил) бориновой кислоты с 3-карбоксипиколиновой кислотой (110), комплекса бис(3-хлор-4-метилфенил)бориновой кислоты с 3-гидроксипиколиновой кислотой (111), комплекса 3-хлор-4-метилфенил, 4-метилфенил-бориновой кислоты с 3-гидроксипиколиновой кислотой (112), комплекса 3-хлор-4-метилфенил, фенилэтил-бориновой кислоты с 3-гидроксипиколиновой кислотой (113), комплекса бис(3-хлорфенил)бориновой кислоты с 3-гидроксипиколиновой кислотой (114), комплекса бис(3-хлор-4-этоксифенил)бориновой кислоты с 3-гидроксипиколиновой кислотой (115), комплекса 5-бром-2-хлор-6-фторфенил-4-хлор-2-фторфенилбориновой кислоты с 3-гидроксипиколиновой кислотой (116), комплекса 4-хлор-2-метилфенил-3-хлорфенилбориновой кислоты с 3-гидроксипиколиновой кислотой (117), комплекса бис(4-хлор-2-метилфенил)бориновой кислоты с 3-гидроксипиколиновой кислотой (118), комплекса бис(3-хлор-4-метилфенил)бориновой кислоты с 3-ОАс-пиколиновой кислотой (119) и комплекса бис(3-хлорфенил)бориновой кислоты с 6-амино-3-гидроксипиколиновой кислотой (122) и их солей и аддуктов с растворителем. 15. Соединение, выбранное из комплекса бис(3-хлорфенил)бориновой кислоты с 1-(2-морфолино-4-илэтил)-имидазолацетатом (101), комплекса бис(3-хлорфенил)бориновой кислоты с 2-(гидроксиизопропил)-3-гидроксипиридином (102), комплекса бис(4-хлорфенил)бориновой кислоты с 2-(гидроксиизопропил)-3-гидроксипиридином (103), комплекса бис(4-метил-3-хлорфенил) бориновой кислоты с 2-гидроксиметил-1N-бензилимидазолом (104), комплекса бис(3-хлорфенил)бориновой кислоты с 2-(гидроксиметил)пиридином (105), эфира бис(3-хлор-4-метилфенил)бориновой кислоты и 4-(гидрокси)бензимидазола (120), эфира бис(3-хлорфенил)бориновой кислоты и 4-(гидроксиэтил)имидазола (121), комплекса бис(3-хлорфенил)бориновой кислоты с 6-амино-3-гидроксипиколинатом (122), комплекса бис(3-хлорфенил)бориновой кислоты с имидазолацетатом (123), эфира бис(4-хлорфенил)бориновой кислоты и 4-(гидроксиметил)имидазола (126), эфира бис(3-хлор-4-метилфенил)бориновой кислоты и 1-бензил-4-(гидроксиметил)-имидазола (127), эфира бис(3-хлор-4-метилфенил)бориновой кислоты и 1-метил-2-(гидроксиметил)имидазола (128), эфира бис(3-хлор-4-метилфенил) бориновой кислоты и 1-этил-2-(гидроксиметил)-имидазола (129), эфира бис(3-хлор-4-метилфенил)бориновой кислоты и 1-метил-4-(гидроксиметил)-имидазола (130), комплекса бис(3-хлор-4-метилфенил)бориновой кислоты с 2-пиридилэтанолом (131), комплекса бис(4-хлорфенил)бориновой кислоты с 2-пиридилметанолом (132), комплекса бис(4-фторфенил)бориновой кислоты с 2-пиридилметанолом (133) и их солей и аддуктов с растворителем. 16. Соединение по любому из пп.1, 7-12, 15, имеющее следующую структуру: 17. Фармацевтическая композиция, обладающая антибактериальной активностью, содержащая соединение по любому из пп.1, 9, 14 или 15 или его соль в фармацевтически приемлемом носителе. 18. Композиция по п.17 в форме, выбранной из таблетки, пилюли, капсулы, жидкости, геля, сиропа, взвеси и суспензии. 19. Композиция по п.17, которая дополнительно содержит наполнитель, выбранный из сахара, препарата клетчатки и диспергирующего средства. 20. Композиция по п.19, в которой указный сахар выбран из лактозы, сахарозы, маннита и сорбита. 21. Композиция по п.19, в которой указанный препарат клетчатки выбран из кукурузного крахмала, пшеничного крахмала, рисового крахмала, картофельного крахмала, желатина, трагаканта, метилцеллюлозы, гидроксипропилметилцеллюлозы, натрий карбоксиметилцеллюлозы и поливинилпирролидона. 22. Композиция по п.19, в которой диспергирующее средство выбрано из агара, альгиновой кислоты и ее солей. 23. Композиция по п.17, в которой носитель содержится в твердой фазе или гелевой фазе. 24. Композиция по п.23, в которой носитель выбран из карбоната кальция, фосфата кальция, сахара, крахмала, производных целлюлозы, желатина и полиэтиленгликоля. 25. Композиция по п.17, в которой соль указанного соединения представляет собой соль, образованную с фармацевтически совместимой кислотой или основанием. 26. Композиция по п.25, в которой фармацевтически совместимая соль образована с кислотой, выбранной из хлористо-водородной, серной, уксусной, молочной, винной, яблочной, янтарной, фосфорной, бромисто-водородной, сульфиновой, муравьиной, толуолсульфоновой, метансульфоновой, азотной, бензойной, лимонной, малеиновой, йодисто-водородной и алкановой кислот. 27. Композиция по п.26, в которой алкановая кислота, образующая соль, имеет формулу НООС-(СН2)n-СН3, где n равно целому числу от 0 до 4. 28. Композиция по п.25, в которой фармацевтически совместимая соль, образованная с основанием, выбрана из солей натрия, калия, кальция и аммония. 29. Способ лечения заболевания, вызываемого микробами, у пациента, пораженного им, включающий введение указанному пациенту терапевтически эффективного количества соединения по по любому из пп.1, 9, 14 или 15. 30. Способ по п.29, в котором указанный микроб представляет собой бактерию. 31. Способ по п.30, в котором указанная бактерия представляет собой грамположительную бактерию. 32. Способ по п.31, в котором указанная грамположительная бактерия выбрана из группы, включающей виды Staphylococcus, виды Streptococcus, виды Bacillus, виды Mycobacterium, виды Corynebacterium, виды Clostridium, виды Actinomyces, виды Enterococcus и виды Streptomyces. 33. Способ по п.30, в котором указанная бактерия представляет собой грамотрицательную бактерию. 34. Способ по п.33, в котором указанная грамотрицательная бактерия, выбрана из группы, включающей виды Acinetobacter, виды Neisseria, виды Pseudomonas, виды Brucella, виды Agrobacterium, виды Bordetella, виды Escherichia, виды Shigella, виды Yersinia, виды Salmonella, виды Klebsiella, виды Enterobacter, виды Haemophillus, виды Pasteurella, виды Streptobacillus, виды спирохет, виды Campylobacter, виды Vibrio и виды Helicobacter. 35. Способ по п.30, в котором указанная бактерия выбрана из группы, включающей Staphylococcus aureus, Staphylococcus epidermidis, Staphylococcus saprophyticus, Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus pneumoniae, Enterococcus faecalis, Enterococcus laecium. Bacillus anthracis, Mycobacterium avium, Mycobacterium tuberculosis, Acinetobacter baumanii, Corynebacterium diphtheria, Clostridium perfringens, Clostridium botulinum, Clostridium tetani, Neisseria gonorrhoeae, Neisseria meningitidis, Pseudomonas aeruginosa, Legionella pneumophila, Escherichia coli, Yersinia pestis, Haemophilus influenzae, Helicobacter pylori, Campylobacter fetus, Campylobacter jejuni. Vibrio cholerae. Vibrio parahemolyticus, Trepomena pallidum, Actinomyces israelii, Rickettsia prowazekii, Rickettsia rickettsii, Chlamydia trachomatis, Chlamydia psittaci, Brucella abortus, Agrobacterium tumefaciens и Francisella tularensis. 36. Способ по п.29, в котором указанное заболевание выбрано из актиномикоза, сибирской язвы, бактериальной дизентерии, ботулизма, бруцеллеза, целлюлита, холеры, конъюнктивита, цистита, дифтерии, бактериального эндокардита, эпиглоттита, гастроэнтерита, сапа, гонореи, “болезни легионеров”, лептоспироза, септического менингита, чумы, бактериальной пневмонии, послеродового сепсиса, ревматической лихорадки, пятнистой лихорадки Скалистых гор, скарлатины, стрептококкового фарингита, сифилиса, столбняка, туберкулеза, туляремии, брюшного тифа, сыпного тифа и коклюша. 37. Способ лечения заболевания, вызываемого грибками или дрожжами, у пациента, пораженного им, включающий введение указанному пациенту терапевтически эффективного количества соединения по любому из пп.1, 9, 14 или 15. 38. Способ уничтожения микроорганизма или ингибирования роста микроорганизма, включающий контактирование указанного микроорганизма с соединением, выбранным из соединений по любому из пп.1, 9, 14 или 15. 39. Способ по п.38, в котором указанное соединение используют в терапевтически эффективном количестве. 40. Способ по п.38, в котором указанный микроорганизм представляет собой бактерию. 41. Способ по п.40, в котором указанная бактерия представляет собой грамположительную бактерию. 42. Способ по п.41, в котором указанная грамположительная бактерия выбрана из группы, включающей виды Staphylococcus, виды Streptococcus, виды Bacillus, виды Mycobacterium, виды Corynebacterium, виды Clostridium, виды Actinomyces, виды Enterococcus и виды Streptomyces. 43. Способ по п.40, в котором указанная бактерия представляет собой грамотрицательную бактерию. 44. Способ по п.43, в котором указанная грамотрицательная бактерия выбрана из группы, включающей виды Acinetobacter, виды Neisseria, виды Pseudomonas, виды Brucella, виды Agrobacterium, виды Bordetella, виды Escherichia, виды Shigella, виды Yersinia, виды Salmonella, виды Klebsiella, виды Enterobacter, виды Haemophilus, виды Pasteurella, виды Streptobacillus, виды спирохет, виды Campylobacter, виды Vibrio и виды Helicobacter. 45. Способ по п.40, в котором указанная бактерия выбрана из группы, включающей Staphylococcus aureus, Staphylococcus epidermidis, Staphylococcus saprophyticus, Streptococcus pyogenes, Streptococcus agalactiae. Streptococcus pneumoniae, Enterococcus faecalis, Enterococcus faecium, Bacillus anthracis, Mycobacterium avium, Mycobacterium tuberculosis, Acinetobacter baumanii, Corynebacterium diphtheria, Clostridium perfringens, Clostridium botulinum, Clostridium tetani, Neisseria gonorrhoeae, Neisseria meningitidis, Pseudomonas aeruginosa, Legionella pneumophila, Escherichia coli, Yersinia pestis, Haemophilus influenzae, Helicobacter pylori, Campylobacter fetus, Campylobacter jejuni. Vibrio cholerae. Vibrio parahemolyticus, Trepomena pallidum, Actinomyces israelii, Rickettsia prowazekii, Rickettsia rikettsii, Chlamydia trachomatis, Chlamydia psittaci, Brucella abortus, Agrobacterium tumefaciens и Francisella tularensis. 46. Способ по п.38, в котором указанный микроорганизм представляет собой грибок или дрожжи. 47. Способ по п.46, в котором указанные дрожжи выбраны из группы, состоящей из Candida albicans, Candida glabrata, Candida krusei, Candida tropicalis и Candida parapsilosis. 48. Способ по п.46, в котором указанный грибок выбран из группы, состоящей из видов Aspergilius, видов Trichophyton, видов Microsporum, Cryptococcus neoformans, Blastomyces dermatitidis, Coccidiodes immitis, Histoplasma capsulatum и Paracoccidioies brasiliensis. 49. Способ по п.41, в котором указанная грамположительная бактерия выбрана из видов Staphylococcus, видов Streptococcus, видов Bacillus, видов Clostridium и видов Enterococcus. 50. Способ по п.49, в котором указанная грамположительная бактерия относится к виду Staphylococcus aureus. 51. Способ по п.50, в котором указанная грамположительная бактерия относится к виду Staphylococcus aureus.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 60/434375, поданной 18 декабря 2002, Serial
60/434375, поданной 18 декабря 2002, Serial 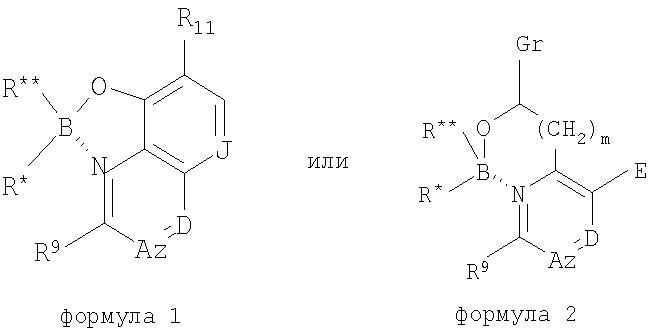
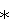 и R
и R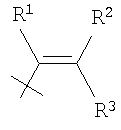





 (м.д.) относительно области тетраметилсилана. Масс-спектры определены на приборе Micromass Quattro II. Номера примеров относятся к соединениям.
(м.д.) относительно области тетраметилсилана. Масс-спектры определены на приборе Micromass Quattro II. Номера примеров относятся к соединениям.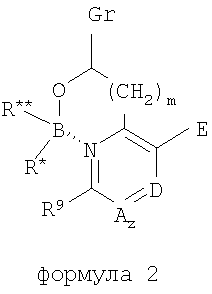
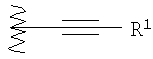 где R1 выбран из группы, содержащей водород, алкил, фенил, замещенный фенил, бензил, замещенный бензил, (СН2)kOH (где k=1, 2 или 3), CH2NH2, CH2NH-алкил, CH2N(алкил)2, CO2H, CO2алкил, CONH2, S-алкил, S-фенил, SO2алкил, SO3H, SCF3, CN, галоген, CF3 и NO2;
где R1 выбран из группы, содержащей водород, алкил, фенил, замещенный фенил, бензил, замещенный бензил, (СН2)kOH (где k=1, 2 или 3), CH2NH2, CH2NH-алкил, CH2N(алкил)2, CO2H, CO2алкил, CONH2, S-алкил, S-фенил, SO2алкил, SO3H, SCF3, CN, галоген, CF3 и NO2;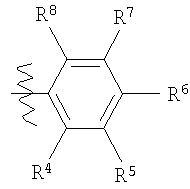
 или
или 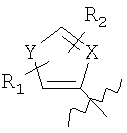
 ,
,  и
и 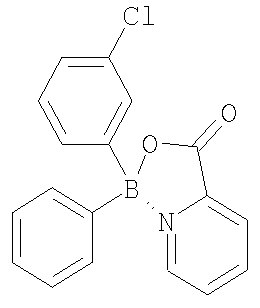 ,
,
 или
или