Патент на изобретение №2396261
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) ПРОИЗВОДНЫЕ ХИНАЗОЛИНОНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ВАНИЛОИДНЫХ АНТАГОНИСТОВ
(57) Реферат:
Настоящее изобретение относится к соединениям общей формулы (I), где R1 – C1-С6алкил, (С1-С6алкил)С1-С6алкил, ди(С1-С6)С1-С6алкил или С3-С6циклоалкил; каждый R2 независимо представляет собой галоген, C1-С6алкил, галогензамещенный C1-С6алкил или циано; R3 – водород, галоген, C1-С6алкил, С2-С6алкенил, С2-С6алкинил, гидроксил, гидроксизамещенный C1-С6алкил, C1-С6алкокси, С3-С6циклоалкил, циано, -С(=O)Н, фенил, (С3-С6циклоалкил)С1-С6алкокси, (С1-С6алкоксикарбониламино)С1-С6алкокси или (С1-С6алкилкарбониламино)С1-С6алкокси; R4 представляет собой гидроксил, C1-С6алкокси, группа -NH(C=O)R4a, где R4a – галогензамещенный C1-С6алкил, или группу -NH(CH2)2OR4b, где R4b – бензил или фенилэтил; и m=1 или 2, в свободной форме или в форме фармацевтически приемлемой соли. Изобретение относится также к фармацевтической композиции на основе соединений (I), предназначенной для лечения или профилактики заболевания или состояния, в котором играет роль или участвует активация ванилоидного рецептора. Кроме того, изобретение относится к применению названных соединений для изготовления лекарственного средства, предназначенного для применения при лечении или профилактике боли или желудочно-кишечного нарушения. 3 н. и 17 з.п. ф-лы.
Настоящее изобретение относится к применению производных хиназолинона в качестве ванилоидных антагонистов, к некоторым новым производным хиназолинона, к способам их получения, к их применению в качестве фармацевтических средств и к содержащим их фармацевтическим композициям. В первом аспекте настоящее изобретение относится к применению соединения хиназолинона формулы:
где R1 представляет собой C1-С6алкил, (C1-С6алкил)C1-С6алкил, ди-(C1-С6алкил)C1-С6алкил, С3-С6циклоалкил, (C1-С6алкил)амино или ди-(C1-С6алкил)амино; каждый R2, независимо, представляет собой галоген, C1-С6алкил, галогензамещенный C1-С6алкил, гидроксиС1-С6алкил, циано или группу -C(=O)-R2a, где R2a представляет собой C1-С6алкил; R3 представляет собой водород, галоген, C1-С6алкил, С2-С6алкенил, С2-С6алкинил, гидроксил, гидроксизамещенный C1-С6алкил, C1-С6алкокси, С3-С6циклоалкил, циано, -С(=O)Н, фенил, (С3-С6циклоалкил)C1-С6алкокси, (C1-С6алкоксикарбониламино)C1-С6алкокси или (C1-С6алкилкарбониламино)С1-С6алкокси; R4 представляет собой гидроксил, гидроксил, этерифицированный в сложный эфир, гидроксил, этерифицированный в простой эфир, амино, (С1-C6aлкил)амино, группу
R5 представляет собой водород или гидроксил; и m обозначает 1 или 2, в свободной форме или в форме соли, и, где возможно, в форме кислотной аддитивной соли, в качестве ванилоидного антагониста. В конкретном варианте осуществления первого аспекта настоящее изобретение относится к применению соединения хиназолинона формулы I, где R1 представляет собой C1-С6алкил, (C1-С6алкил)C1-С6алкил, ди-(C1-С6алкил)С1-С6алкил или С3-С6циклоалкил; каждый R2, независимо, представляет собой галоген, C1-С6алкил, тригалогензамещенный C1-С6алкил, гидроксиС1-С6алкил или группу
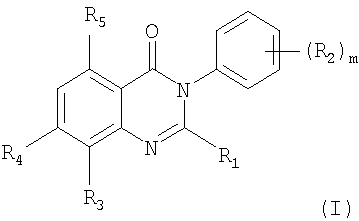
R3 представляет собой водород, галоген, C1-С6алкил, гидроксил, C1-С6алкокси или (С3-С6циклоалкил)C1-С6алкокси; R4 представляет собой гидроксил, гидроксил, этерифицированный в сложный эфир, гидроксил, этерифицированный в простой эфир, амино, (C1-С6алкил)амино, группу R5 представляет собой водород или гидроксил; и m обозначает 1 или 2, в свободной форме или в форме соли и, где возможно, в форме кислотной аддитивной соли, в качестве ванилоидного антагониста. Во втором аспекте настоящее изобретение относится к новым соединениям хиназолинона формулы:
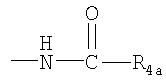
где R1 представляет собой C1-С6алкил, (C1-С6алкил)C1-С6алкил, ди-(C1-С6алкил)C1-С6алкил, С3-С6циклоалкил, (C1-С6алкил)амино или ди-(C1-С6алкил)амино; каждый R2, независимо, представляет собой галоген, C1-С6алкил, галогензамещенный C1-С6алкил, гидроксиС1-С6алкил, циано или группу -C(=O)-R2a, где R2a представляет собой C1-С6алкил; R3 представляет собой водород, галоген, C1-С6алкил, С2-С6алкенил, С2-С6алкинил, гидроксил, гидроксизамещенный C1-С6алкил, C1-С6алкокси, С3-С6циклоалкил, циано, -С(=O)Н, фенил, (С3-С6циклоалкил)C1-С6алкокси, (C1-С6алкоксикарбониламино)C1-С6алкокси или (C1-С6алкилкарбониламино)С1-С6алкокси; R4 представляет собой гидроксил, гидроксил, этерифицированный в сложный эфир, гидроксил, этерифицированный в простой эфир, амино, (С1-C6алкил)амино, группу m обозначает 1 или 2, в свободной форме или в форме соли, и, где возможно, в форме кислотной аддитивной соли. В конкретном варианте осуществления второго аспекта настоящее изобретение относится к новым соединениям хиназолинона формулы Ia, где R1 представляет собой C1-С6алкил, (C1-С6алкил)C1-С6алкил, ди-(C1-С6алкил)C1-С6алкил или С3-С6циклоалкил; каждый R2, независимо, представляет собой галоген, C1-С6алкил, тригалогензамещенный C1-С6алкил, гидроксиС1-С6алкил или группу R3 представляет собой водород, галоген, C1-С6алкил, гидроксил, C1-С6алкокси или (С3-С6Циклоалкил)C1-С6алкокси; R4 представляет собой гидроксил, гидроксил, этерифицированный в сложный эфир, гидроксил, этерифицированный в простой эфир, амино, (С1-С6алкил)амино, группу m обозначает 1 или 2, в свободной форме или в форме соли и, где возможно, в форме кислотной аддитивной соли. Термины, используемые в настоящем описании, имеют следующие значения: “C1-С6алкил” обозначает линейный или разветвленный C1-С6-алкил, например метил этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил или трет-бутил. “C1-С6алкокси” обозначает линейный или разветвленный C1-С6-алкилокси, например метокси, этокси, н-пропокси или изопропокси. “Галоген” обозначает атом галогена, которым может быть I, Br, Cl или F. “Гидроксил, этерифицированный в сложный эфир” обозначает ацилокси, предпочтительно C1-С6алканоилокси, более предпочтительно C1-С4алканоилокси. “Гидроксил, этерифицированный в простой эфир” обозначает C1-С6алкокси, предпочтительно С1-С4алкокси. Соединения хиназолинона по изобретению существуют в свободной форме или в форме соли и, где возможно, в форме кислотной аддитивной соли. Следует понимать, что настоящее изобретение включает соединения формул (I) и (Ia) в свободной форме или в форме соли и, где возможно, в форме кислотной аддитивной соли. В последнем случае подходящие фармацевтически приемлемые кислотные аддитивные соли для фармацевтического применения в соответствии с изобретением включают, в частности, гидрохлоридную соль. В формулах (I) и (Ia) следующие значения являются предпочтительными независимо, вместе или в любой их комбинации или подкомбинации: (а) R1 представляет собой С1-С4алкил, (С1-С4алкил)С1-С4алкил, ди-(С1-С4алкил)С1-С4алкил или циклопропил; (б) каждый R2, независимо, представляет собой хлор, фтор, С1-С4алкил, трифторзамещенный С1-С4алкил, более предпочтительно трифторметил, С1-С4алкилкарбонил, более предпочтительно метилкарбонил или гидроксиС1-С4алкил, более предпочтительно гидроксиметил; (в) R3 представляет собой водород, хлор, бром, С1-С4алкил, гидроксил, С1-С4алкокси или (С3-С6циклоалкил)С1-С4алкокси; и (г) R4 представляет собой гидроксил, амино, (С1-С4алкил)амино или группу В третьем аспекте настоящее изобретение относится к способам получения соединений формулы (Ia), представленным на следующих схемах реакций: А. Для получения соединений формулы (Ia), где R1 и R2 являются такими, как определено выше, R3 является таким, как определено для соединения формулы I, R4 представляет собой амино и m обозначает 1. Схема А Первая стадия:
Общее описание: Первая стадия схемы А включает реакцию конденсации/циклизации амидного соединения формулы 1 с замещенным соединением анилина в присутствии трихлорида фосфора с получением 7-нитрозамещенного соединения хиназолин-4-она формулы 2. Вторая стадия:
Общее описание: Вторая стадия схемы А включает восстановление 7-нитрозамещенного соединения хиназолин-4-она формулы 2 ледяной уксусной кислотой и порошком железа с получением 7-аминозамещенного соединения хиназолин-4-она формулы 3. Соответствующие алкиламины, амиды и карбаматы могут быть получены способами, описанными в уровне техники, используя соединение формулы 3. Более конкретно, алкиламины могут быть получены реакцией соединения формулы 3 восстановительным алкилированием, используя подходящие альдегид или кетон. Альтернативно, соединение формулы 3 может подвергаться реакциия с C1-С6алкилгалогенидом. Амиды могут быть получены ацилированием соединения формулы 3 с подходящим ацилхлоридом. Карбаматы могут быть получены реакцией соединения формулы 3 с подходящим алкилхлорформатом. Б. Для получения соединений формулы (Ia), где R1 и R2 являются такими, как определено выше, R3 является таким, как определено для соединения формулы I, R4 представляет собой гидроксил и m обозначает 1. Схема Б
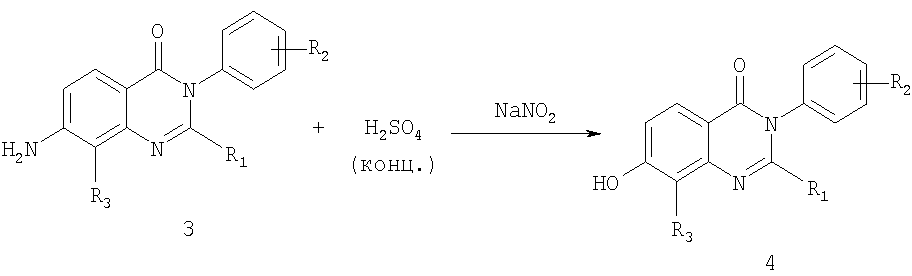
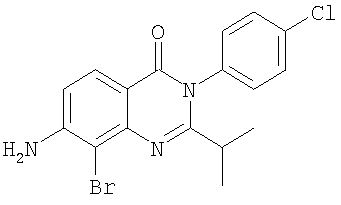
Общее описание: Схема Б включает реакцию Сандмейера 7-аминозамещенного соединения хиназолин-4-она формулы 3, которое получали методикой, описанной на схеме А, с концентрированной серной кислотой и нитритом натрия с получением 7-гидроксизамещенного соединения хиназолин-4-она формулы 4. Исходные соединения для схемы А являются известными соединениями, которые являются коммерчески доступными. Обработка реакционной смеси указанных выше способов и очистка полученных соединений может осуществляться в соответствии с известными методиками. Кислотные аддитивные соли могут быть получены из свободных оснований известным способом, и наоборот. Соединения формул (I) и (Ia) в оптически чистой форме могут быть получены из соответствующих рацематов в соответствии с хорошо известными методиками, например ВЭЖХ с хиральным носителем. Альтернативно, могут использоваться оптически чистые исходные реагенты. Стереоизомерные смеси, например смеси диастереоизомеров, могут быть разделены на их соответствующие изомеры известным способом с помощью подходящих способов разделения. Диастереомерные смеси, например, могут быть разделены на их индивидуальные диастереоизомеры с помощью фракционной кристаллизации, хроматографии, разделения растворителя и аналогичными способами. Это разделение может иметь место либо для исходного соединения, либо непосредственно для соединения формулы (I) или (Ia). Энантиомеры могут быть разделены с помощью образования диастереомерных солей, например, образованием соли с энантиомерно-чистой хиральной кислотой, или с помощью хроматографии, например ВЭЖХ, используя хроматографические субстраты с хиральными лигандами. В любых дополнительных стадиях способа, проводимых при необходимости, функциональные группы исходных соединений, которые не должны участвовать в реакции, могут присутствовать в незащищенной форме или могут быть защищены, например, одной или несколькими защитными группами, приведенными ниже. Защитные группы затем полностью или частично удаляют в соответствии с одним из описанных здесь способов. Защитные группы уже могут присутствовать в предшественниках и предназначены для защиты функциональных групп от нежелательных побочных реакций. Характеристикой защитных групп является то, что они легко отщепляются сами, то есть без нежелательных побочных реакций, обычно сольволизом, восстановлением, фотолизом или также ферментативной активностью, например, в условиях, аналогичных физиологическим условиям, и что они не присутствуют в конечных продуктах. Специалисту в данной области техники известны или он может легко установить, какие защитные группы подходят для описанных выше и далее реакций. Защита таких функциональных групп защитными группами, сами защитные группы и реакции для их удаления описаны, например, в стандартных источниках, таких как книги J.F.W. McOmie, Protective Groups in Organic Chemistry, Plenum Press, London и NY (1973); T.W. Greene, Protective Groups in Organic Synthesis, Wiley, NY (1981); The Peptides; T. 3, E. Gross и J. Meienhofer, Eds., Academic Press, London и NY (1981); Methoden der organischen Chemie (Methods of organic chemistry), Houben Weyl, 4-ое издание, Т. 15/1, Georg Thieme Verlag, Stuttgart (1974); H.D. Jakubke и H. Jescheit, Aminosauren, Peptide, Proteine (Amino acids, peptides, proteins), Verlag Chemie, Weinheim, Deerfield Beach и Basel (1982); и Jochen Lehmann, Chemie der Kohlenhydrate: Monosaccharide und Derivate (Chemistry ofcarbohydrats: monosaccharides and derivatives), Georg Thieme Verlag., Stuttgart (1974). Все описанные здесь стадии способов могут осуществляться в известных реакционных условиях, предпочтительно в конкретно описанных условиях, в отсутствие или обычно в присутствии растворителей или разбавителей, предпочтительно таких, как растворители, инертные по отношению к используемым реагентам, и способные их растворять, в отсутствие или в присутствии катализаторов, конденсирующих агентов или нейтрализующих агентов, например ионообменников, обычно катионообменников, например, в Н+ форме, в зависимости от типа реакции и/или восстанавливаемых реагентов, при нормальной или повышенной температуре, например, в диапазоне от -100 до около 190°С, предпочтительно от около -80 до около 150°С, например при от -80 до 60°С, при комнатной температуре, при от -20 до 40°С или при температуре кипения используемого растворителя, при атмосферном давлении или в закрытом сосуде, если подходит под давлением, и/или в инертной атмосфере, например в атмосфере аргона или азота. Предпочтительными соединениями формулы (I) являются соединения, где R1 представляет собой С1-С4алкил, (С1-С4алкил)С1-С4алкил или С3-С6циклоалкил; R2 представляет собой хлор, фтор, С1-С4алкил, трифторзамещенный С1-С4алкил, С1-С4алкилкарбонил или гидроксиС1-С4алкил; R3 представляет собой водород, хлор, бром, С1-С4алкил, гидроксил, С1-С4алкокси или (С3-С6циклоалкил)С1-С4алкокси; R4 представляет собой гидроксил, амино или (С1-С4алкил)амино; R5 представляет собой водород или гидроксил; и m обозначает 1 или 2. Более предпочтительными соединениями формулы (I) являются соединения, где R1 представляет собой С1-С4алкил, (С1-С4алкил)С1-С4алкил или С3-С6циклоалкил; R2 представляет собой хлор, фтор, С1-С4алкил, трифторметил, метилкарбонил или гидроксиметил; R3 представляет собой водород, хлор, бром, С1-С4алкил, гидроксил или С1-С4алкокси; R4 представляет собой гидроксил, амино или (С1-С4алкил)амино; R5 представляет собой водород или гидроксил; и m обозначает 1. Предпочтительными соединениями формулы (Ia) являются соединения, где R1 представляет собой С1-С4алкил, (С1-С4алкил)С1-С4алкил или С3-С6циклоалкил; R2 представляет собой хлор, фтор, С1-С4алкил, трифторзамещенный С1-С4алкил, С1-С4алкилкарбонил или гидроксиС1-С4алкил; R3 представляет собой водород, хлор, бром, С1-С4алкил, гидроксил, С1-С4алкокси или (С3-С6циклоалкил)С1-С4алкокси; R4 представляет собой гидроксил, амино или (С1-С4алкил)амино; и m обозначает 1 или 2. Более предпочтительными соединениями формулы (Ia) являются соединения, где R1 представляет собой С1-С4алкил, (С1-С4алкил)С1-С4алкил или С3-С6циклоалкил; R2 представляет собой хлор, фтор, С1-С4алкил, трифторметил, метилкарбонил или гидроксиметил; R3 представляет собой водород, хлор, бром, С1-С4алкил, гидроксил, С1-С4алкокси или (С3-С6циклоалкил)С1-С4алкокси; R4 представляет собой гидроксил, амино или (С1-С4алкил)амино; и m обозначает 1. Еще более предпочтительными соединениями формулы I или Ia являются соединения примеров 1-29, особенно примеров 1-28. Другой аспект настоящего изобретения относится к обнаружению того, что соединения формул (I) и (Ia) и их фармацевтически приемлемые соли и, где возможно, фармацевтически приемлемые кислотные аддитивные соли обладают полезной фармакологической активностью и, следовательно, являются полезными в качестве фармацевтических средств. В частности, соединения формул (I) и (Ia) проявляют антагонистическую активность в отношении ванилоидного рецептора человека. Более конкретно, соединения формул (I) и (Ia) являются активными в отношении рецептора TRPVI, что показано их способностью ингибировать капсаицин и низкой рН активацией ионного канала TRPVI следующим образом: Клетки яичников китайских хомячков K1 (CHO-K1), трансфецированные для экспрессии либо рецептора TRPVI человека, либо крысы, либо морской свинки, выращивали в минимальной питательной среде-альфа (MEM) без нуклеозидов с эмбриональной телячьей сывороткой (10%), 2 мМ L-глутамином, 100 IU/мл пенициллина, 100 мкг/мл стрептомицина и 350-700 мкг/мл генетицина. Все реагенты получали от Invitrogen. Клетки выращивали в колбах Т-175 или в 96-луночных планшетах с возможностью обзора с черными стенками и прозрачным дном Costar и выдерживали при 37°С в инкубаторе с 90% влажностью в атмосфере 5% СО2 и 95% воздуха. Клетки встряхивали дважды в неделю в соотношении от 1:10 до 1:20 для поддержания равномерного роста. Для эксперимента клетки высаживали с плотностью приблизительно 80% и помещали в планшеты с возможностью обзора с концентрацией 40,000 клеток на лунку в 100 мкл среды и выращивали в течение ночи. Анализ иммобилизации кальция В день анализа капсаицина среду аспирировали и клетки промывали 100 мкл 10 мМ раствора N-2-(гидроксиэтилпиперазин-N’-[2-этансульфоновая кислота] (HEPES), обработанного буфером, сбалансированным по соли раствором Хенка (HBSS), рН 7,4. Клетки затем инкубировали в течение 40 минут с 2,3 мкМ раствором красителя, связывающего рациометрический кальций, фура-2/АМ (от Molecular Probes), полученного в HEPES, обработанном буфером HBSS, содержащем 0,01% полиионного F-127. Для анализа рН не добавляли HEPES и уровень рН HBSS доводили до 7,4. После промывания дважды 100 мкл аналитического буфера клетки инкубировали в течение 10 минут со 100 мкл тестируемых соединений (раствор, полученный в HBSS, рН 7,4), в дубликатах, при концентрациях от 0,001 до 30 мкМ. Планшеты затем помещали в Molecular Devices Flexstation. Рецептор TRPV1 стимулировали применением либо капсаицина, либо низкого значения рН. Для испытания действия соединений в отношении возможного антагонизма использовали капсаицин при концентрации EC80, которая составляла 0,05 мкМ для рецептора TRPV1 крысы и 0,1 мкМ для рецептора человека и морской свинки. Для экспериментов установления рН забуференный раствор с низким значением рН [60 мМ 2-[N-морфолино]этансульфоновая кислота (MES) в HBSS] добавляли в анализируемые лунки до конечного значения рН 5,5. Для определения значений IC50 антагониста (концентрации антагониста, которые ингибируют отклики либо на рН 5,5, либо на капсаицин 50%), по крайней мере 10 концентраций антагониста измеряли в дубликате. Отклик в присутствии антагониста рассчитывали как процентное соотношение контрольного отклика на капсаицин или низкое значение рН и строили график зависимости от концентрации антагониста. Значения IC50 определяли анализом нелинейной регрессии на сигмоидально-логистических кривых с помощью программного обеспечения Activity-Base (v5,0,10) или Microcal Origin (v7,03). Эти значения усредняли (значения и стандартная погрешность значения) по крайней мере для трех независимых экспериментов. Соединения формул (I) и (Ia), например, соединения примеров 1-28, проявляющие антагонистическую активность в отношении рецептора TRPV1, показывают значения IC50 в диапазоне 0,004-30 мкМ. Ввиду вышеизложенного соединения формул (I) и (Ia) являются полезными в качестве блокаторов ванилоидного рецептора, например для лечения заболеваний и состояний, в которых играет роль или участвует активация ванилоидного рецептора. Такие состояния включают, в частности, боль, например костную и суставную боль (остеоартрит), раковую боль, миофасцитную боль (мышечная травма, фибромиалгия) и периоперационную боль (общая хирургия, гинекологическая хирургия). Соединения формул (I) и (Ia) являются особенно полезными для лечения или профилактики хронической боли, особенно воспалительной, например хронической воспалительной боли; воспалительных заболеваний, например воспалительного заболевания дыхательных путей, например хронического обструктивного легочного заболевания (COPD), или астмы; кашля; непроизвольного мочеиспускания; мигрени; висцеральных заболеваний, например воспалительного кишечного заболевания; ринита; цистита, например интерстициального цистита; панкреатита; увеита; воспалительных кожных заболеваний и ревматоидного артрита. Соединения формул (I) и (Ia), следовательно, используются в качестве антагонистов ванилоидного рецептора, например, для лечения боли различного генеза или этиологии, и в качестве противовоспалительных и/или противоотечных агентов для лечения воспалительных реакций, заболеваний или состояний, а также для лечения аллергических реакций. Благодаря их болеутоляющему/противовоспалительному профилю они являются полезными для лечения воспалительной боли, для лечения гипералгезии и, в частности, для лечения тяжелой хронической боли. Они, например, являются полезными для лечения боли, воспаления и/или отека, вызванного травмой, например, связанной с ожогами, растяжениями, переломами или им подобными, вследствие хирургического вмешательства, например, в качестве послеоперационных обезболивающих средств, а также для лечения воспалительной боли другого генеза, например, для лечения остеоартрита и ревматоидного артрита и ревматического заболевания, теносиновита и подагры. Они также являются полезными в качестве обезболивающих средств для лечения боли, связанной, например, с ангиной, менструацией или раком. В качестве противовоспалительных/противоотечных агентов они также являются полезными, например, для лечения воспалительных кожных заболеваний, например псориаза и экземы. В качестве блокаторов ванилоидного рецептора соединения формулы (I) и (Ia) также являются полезными в качестве релаксантов гладких миоцитов, например для лечения спазм желудочно-кишечного тракта или матки, например при лечении болезни Крона, язвенного колита или панкреатита. Соединения формулы (I) и (Ia) являются, в частности, полезными в качестве агентов для лечения гиперреактивности дыхательных путей и для лечения воспалительных реакций, связанных с заболеванием дыхательных путей, в частности астмой. Кроме того, агенты по изобретению, например, могут использоваться для контроля, уменьшения или устранения гиперреактивности дыхательных путей при астме. Воспалительные или обструктивные заболевания дыхательных путей, к которым применимо настоящее изобретение, включают астму любого типа или генеза, включая как инфекционно-аллергическую астму, так и, особенно, внешнюю астму. Таким образом, соединения формулы (I) и (Ia) являются полезными для лечения аллергической астмы, а также, например, астмы, вызванной физическими упражнениями, профессиональной астмы, астмы, вызванной бактериальной инфекцией, других неаллергических видов астмы и “синдрома свистящих младенцев”. Эффективность при лечении астмы может быть подтверждена снижением частоты или серьезности симптоматических приступов, например, острого астматического или бронхоконстрикторного приступа, и снижением потребности в другой симптоматической терапии, например, противовоспалительных средств, например кортикостероидов; или бронхорасширяющих средств, например Воспалительные или обструктивные заболевания дыхательных путей, к которым применимо настоящее изобретение, также включают пневмокониоз (воспалительное, обычно профессиональное заболевание легких, часто вызываемое повторяющимся вдыханием пыли) любого типа или генеза, включая, например, алюминоз, антракоз, асбестоз, халькоз, птилоз, сидероз, силикоз, табакоз и, в частности, биссиноз. Другие воспалительные или обструктивные заболевания и состояния дыхательных путей, при которых могут использоваться соединения формул (I) и (Ia), включают респираторный дистрессовый синдром у взрослых (ARDS), хроническое обструктивное заболевание легких или дыхательных путей (COPD или COAD) и бронхиты. Соединения формул (I) и (Ia) также могут использоваться при аллергических и вазомоторных ринитах. В дополнение к вышеизложенному соединения формул (I) и (Ia) также могут использоваться при терапии септического шока, например, в качестве антигиповолемических и/или антигипотензивных агентов; для лечения воспалительного заболевания кишечника; церебрального отека; головной боли; мигрени; воспалительного кожного заболевания, такого как экзема и псориаз; воспалительных заболеваний кишечника, например синдрома воспаленного кишечника; болезни Крона; язвенного колита и цистита, например интерстициального цистита, нефрита и увеита. Агенты по изобретению являются полезными для профилактики и лечения заболеваний и состояний, в которых играет роль или участвует активация рецептора VR1 человека, и следовательно, полезны для лечения путем модулирования (предпочтительно антагонизма) рецепторов VR1. Такие состояния включают хроническую боль с воспалительным компонентом, такую как ревматоидный артрит; костную и суставную боль (остеоартрит); послеоперационную боль; мышечно-скелетную боль, такую как фибромиалгия; миофасцитные болевые синдромы; головную боль, включая мигрень, острую или хроническую головную боль, кластерную головную боль, височно-челюстную боль и верхнечелюстную боль; ушную боль; боль при эпизиотомии; ожоги, и особенно связанную с ними начальную гипералгезию; глубокую и висцеральную боль, такую как сердечную боль, мышечную боль, глазную боль, ротолицевую боль, брюшную боль, гинекологическую боль, такую как менструальная боль и родовые схватки; боль, связанную с мочеполовым трактом, такую как цистит и вульвадиния; воспалительные кожные заболевания, например псориаз и экзема, или зуд неспецифического происхождения; хроническую боль, связанную с травмой нерва и/или заболеваниями нервной системы, такую как нейропатическая боль, связанная с послегерпетической невралгией, диабетической невропатией, вызванной химиотерапией невропатией, ампутациями (“фантомная боль”), связанной с вовлечением нерва в послеоперационный рубец невралгией и невралгиями плечевого сплетения, боли внизу спины, невралгию седалищного нерва и анкилозирующий спондилит, симпатическую рефлекторную дистрофию и другие хронические повреждения нерва; сложные синдромы региональной боли; боли центральной нервной системы, такие как боль вследствие повреждения спинного мозга или ствола мозга, или инсульта; подагру; боль при ране; боль, связанную с карциномой, часто обозначаемую как раковая боль; респираторные заболевания, включая астму, алюминоз, антракоз, воспалительное заболевание дыхательных путей, например хроническое обструктивное легочное заболевание; хронической бронхит, асбестоз, халькоз, птилоз, сидероз, силикоз, табакоз, биссиноз; ринит, аллергические риниты, такие как сезонный и круглогодичный ринит, и неаллергический ринит; кашель, идиопатические или респираторные заболевания, такие как COPD, астма, циститный фиброз, рак или желудочно-кишечные нарушения, такие как гастро-пищеводный рефлюкс; аутоиммунные заболевания; желудочно-кишечные заболевания, включая, но не ограничиваясь ими, синдром воспаленного кишечника, болезнь Крона, язвенный колит, панкреатит, воспалительное кишечное заболевание; заболевания мочеполового тракта, особенно цистит; непроизвольное мочеиспускание, включая гиперрефлексию мочевого пузыря и гиперчувствительность мочевого пузыря. В указанных выше показаниях подходящие дозировки будут, конечно, сильно зависеть, например, от применяемого соединения, пациента, способа введения и природы и степени тяжести излечиваемого заболевания. Однако обычно удовлетворительные результаты на животных наблюдаются при суточных дозировках от около 0,05 до около 150, предпочтительно от около 0,1 до около 100 мг/кг веса тела животного. Для больших млекопитающих, например людей, показанная суточная дозировка находится в диапазоне от около 0,5 до около 5,000, предпочтительно от около 1 до около 500 мг соединения формул (I) и (Ia), обычно вводимая, например, несколькими дозами до 4-х раз в сутки или в форме длительного высвобождения. Соединения формул (I) и (Ia) могут быть введены in vivo отдельно или в комбинации с другими фармацевтическими агентами, эффективными для лечения заболеваний и состояний, в которых играет роль или участвует активация ванилоидного рецептора, включая ингибиторы циклооксигеназы-2 (СОХ-2), такие как специфические ингибиторы СОХ-2, например, целекоксиб и рофекоксиб; и нестероидные противовоспалительные лекарственные средства (NSAID), например, производные ацетилсалициловой кислоты и пропионовой кислоты; трициклические антидепрессанты, например, Anafranil®, Asendin®, Aventyl®, Elavil®, Endep®, Norfranil®, Norpramin®, Pamelor®, Sinequan®, Surmontil®, Tipramine®, Tofranil®, Vivactil®, Tofranil-PM®; антиконвульсанты, например, карбамазепин, окскарбазепин и габапентин; антагонисты брадикинина В1 или В2; и агонисты GABAB, например, L-баклофен. Агенты по изобретению могут вводиться in vivo отдельно или в комбинации с другими фармацевтическими агентами, например агентами, эффективными для лечения заболеваний и состояний, в которых играет роль или участвует активация VR1 человека, такими как ингибиторы циклооксигеназы, включая специфические ингибиторы СОХ-2 (например, целекоксиб, лумиракоксиб и вальдекоксиб) или обычно нестероидные противовоспалительные лекарственные средства (NSAID) (например, производные ацетилсалициловой кислоты, пропионовой кислоты), агенты против мигрени, такие как агонисты 5-HTi и антагонисты CGRP, трициклические антидепрессанты (например, кломипрамин, амоксапин, нортрипилин, амитриптилин, имипрамин, десипрамин, доксепин, тримипрамин, протрипилин), селективные ингибиторы захвата серотонина (например, флуоксетин), селективные ингибиторы захвата норадреналина (например, дулоксетин), противосудорожные средства (например, габапентин, прегабалин, окскарбазепин, карбамазепин), агонисты OABAB (например, L-баклофен), опиоиды (например, морфин), агонисты рецептора CB1, антагонисты брадикининового рецептора, антагонисты вещества Р. Фармацевтические композиции для раздельного введения ингредиентов комбинации и для введения в фиксированной комбинации, то есть в одной галеновой композиции, включающей по крайней мере два ингредиента комбинации, в соответствии с изобретением могут быть получены известным способом и пригодны для энтерального, такого как пероральное или ректальное, и парентерального введения млекопитающим, включая человека, и включают терапевтически эффективное количество по крайней мере одного фармакологически активного ингредиента комбинации отдельно или в комбинации с одним или несколькими фармацевтически приемлемыми носителями, особенно подходящими для энтерального или парентерального применения. Фармацевтические композиции содержат, например, от около 0,1 до около 99,9%, предпочтительно от около 20 до около 60% активных ингредиентов. Фармацевтические препараты для комбинированной терапии для энтерального или парентерального введения являются, например, препараты в единичных дозированных формах, такие как покрытые сахаром таблетки, таблетки, капсулы или суппозитории, или ампулы. Их получают известным способом, например обычными способами смешения, гранулирования, покрытия сахаром, растворения или лиофилизации. Ясно, что единичное содержание ингредиента комбинации, включенное в индивидуальную дозировку каждой дозированной формы, не должно составлять эффективное количество, поскольку необходимое эффективное количество может также достигаться введением нескольких дозированных форм. Следующий аспект настоящего изобретения включает “новые” композиции, включающие фармацевтически приемлемый носитель или разбавитель и терапевтически эффективное количество соединения формулы (Ia), в свободной форме или в форме соли и, где возможно, в форме кислотной аддитивной соли. В соответствии с вышеизложенным настоящее изобретение также относится к: (1) соединению формулы (I) или (Ia) в свободной форме или в форме соли и, где возможно, в форме фармацевтически приемлемой кислотной аддитивной соли для применения в качестве блокатора ванилоидного рецептора, например для применения при любом из конкретных показаний, приведенных выше; (2) соединению формулы (I) или (Ia) в свободной форме или в форме соли и, где возможно, в форме фармацевтически приемлемой кислотной аддитивной соли для лечения заболевания или состояния, в котором играет роль или участвует ванилоидный рецептор; (3) способу лечения любого из конкретных показаний, приведенных выше, у пациента, нуждающегося в этом, который включает введение терапевтически эффективного количества соединения формулы (I) или (Ia) в свободной форме или в форме соли и, где возможно, в форме фармацевтически приемлемой кислотной аддитивной соли; (4) способу лечения или профилактики заболевания или состояния, в котором играет роль или участвует ванилоидной рецептор, включающему введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или (Ia) в свободной форме или в форме соли и, где возможно, в форме фармацевтически приемлемой кислотной аддитивной соли; (5) применению соединения формулы (I) или (Ia) в свободной форме или в форме соли и, где возможно, в форме фармацевтически приемлемой кислотной аддитивной соли для изготовления лекарственного средства для лечения или профилактики заболевания или состояний, в котором играет роль или участвует активность ванилоидного рецептора; (6) описанному выше способу, включающему совместное введение, например, одновременно или последовательно, терапевтически эффективного количества антагониста ванилоидного рецептора, например, соединения формулы (I) или (Ia) в свободной форме или в форме соли и, где возможно, в форме фармацевтически приемлемой кислотной аддитивной соли, и второго лекарственного вещества, где указанное второе лекарственное вещество, например, используется для применения в любом из конкретных показаний, приведенных выше; и (7) комбинации, включающей терапевтически эффективное количество соединения формулы (I) или (Ia) в свободной форме или в форме соли и, где возможно, в форме фармацевтически приемлемой кислотной аддитивной соли, и второе лекарственное вещество, где указанное второе лекарственное вещество, например, используется для применения в любом из конкретных показаний, приведенных выше. В следующих примерах, которые никаким образом не предназначены для ограничения объема настоящего изобретения, используются следующие сокращения: EtOAc этилацетат ДХМ дихлорметан Пример 1 Получение 7-амино-3-(4-хлорфенил)-2-изопропил-3H-хиназолин-4-она а) Получение 3-(4-хлорфенил)-2-изопропил-7-нитро-3H-хиназолин-4-она Суспензию изобутирамида 4-нитроантраниловой кислоты (4 г, 15,8 ммоль), 4-хлоранилина (2,2 г, 17,2 ммоль) и трихлорида фосфора (5,6 мл) в толуоле (150 мл) кипятили с обратным холодильником (температура бани 150°С) в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и затем упаривали досуха. Остаток разделяли между водой и EtOAc и водную фазу экстрагировали (2х) EtOAc. Объединенные органические фазы промывали водой, сушили (Na2SO4) и упаривали в вакууме. Растирание с изопропиловым эфиром приводило к получению желаемого соединения в виде коричневого твердого вещества. б) Получение указанного в заголовке соединения Смесь соединения, полученного в примере 1a (2,4 г, 6,98 ммоль), порошка железа (1,16 г, 20,8 ммоль) и ледяной уксусной кислоты (70 мл) перемешивали при 50°С в течение 2,5 часов. Реакционную смесь охлаждали до комнатной температуры и затем досуха упаривали в вакууме. Остаток разделяли между водой и EtOAc и водную фазу экстрагировали (2х) EtOAc. Объединенные органические фазы промывали водой, сушили (Na2SO4) и упаривали в вакууме с получением коричневого твердого вещества. Очистка автоматизированной ускоренной хроматографией (градиент элюирования: EtOAc/ДХМ 0-50%) приводила к получению указанного в заголовке соединения в виде бледно-желтого твердого вещества. (М+Н)+=314,2; время удерживания ВЭЖХ=3,9 минут. Пример 2 Получение 3-(4-хлорфенил)-7-гидрокси-2-изопропил-3Н-хиназолин-4-она К суспензии соединения примера 1 (778 мг, 2,479 ммоль) в смеси концентрированная серная кислота/вода 972 мкл/1,4 мл, охлажденной до температуры ледяной бани, добавляли раствор нитрита натрия (188 мг) в воде (680 мкл). Полученную смесь перемешивали в течение 45 минут при 0-5°С (внутренняя температура) и затем добавляли к смеси серная кислота/вода 3/2 (5 мл), предварительно нагретой до 150°С. После перемешивания в течение 15 минут смесь охлаждали до комнатной температуры, отфильтровывали и экстрагировали EtOAc (3х). Объединенные экстракты EtOAc промывали водой, сушили (Na2SO4) и упаривали с получением желто-оранжевого твердого вещества. Очистка автоматизированной ускоренной хроматографией (градиент элюирования: EtOAc/гексан 0-25%) приводила к получению указанного в заголовке соединения в виде желтого твердого вещества. 1Н ЯМР (400 МГц, MeOH-d4): Примеры 3-28 Соединения примеров 3-28 могут быть получены способом, аналогичным способу, описанному в предыдущих примерах.
Пример 29 Соединения 29,1-29,54 могут быть получены способом, аналогичным способу, описанному в предыдущих примерах.
Пример 30 Получение мягких желатиновых капсул 5’000 мягких желатиновых капсул, каждая включающая в качестве активного ингредиента 0,05 г одного из соединений формулы (Ia), указанного в приведенных выше примерах, получали следующим образом: Композиция Активный ингредиент 250 г Лаурогликоль® 2 л Измельченный активный ингредиент суспендировали в лаурогликоле® (пропиленгликольлаурат, Gattefosse S.A., Saint Priest, France) и распыляли влажным пульверизатором с получением частиц размером около 1-3 мкм. Порцию 0,419 г смеси затем наполняли в мягкую желатиновую капсулу с помощью устройства для наполнения капсул.
Формула изобретения
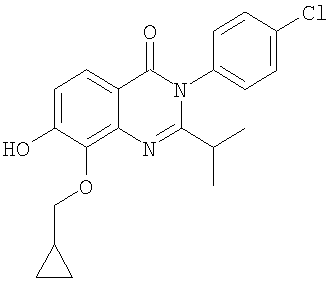
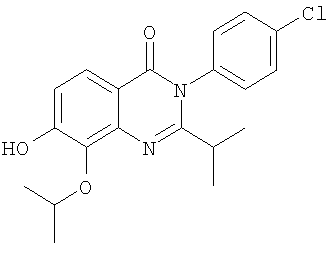
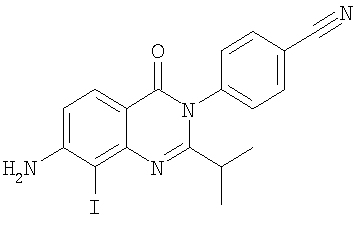
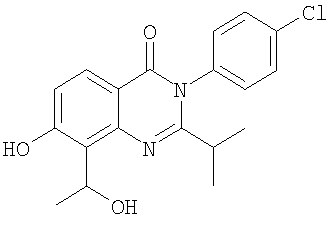
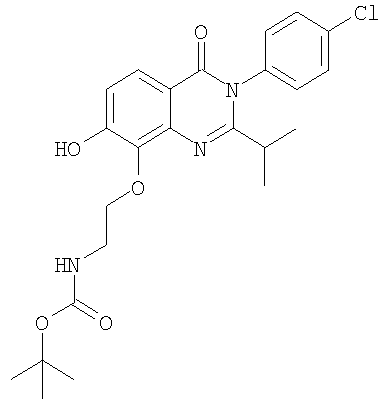
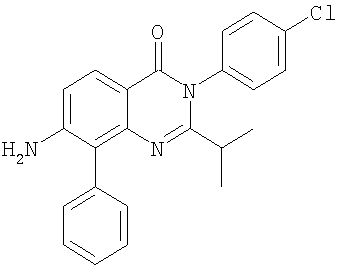
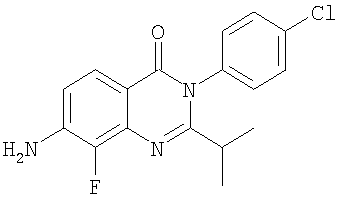
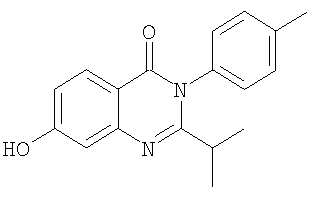
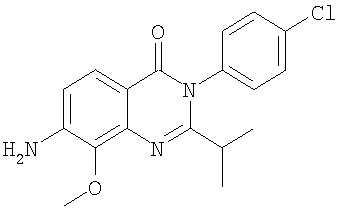
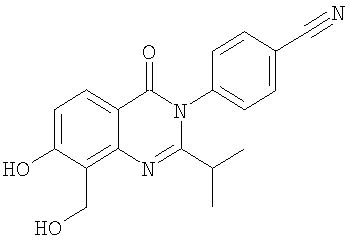
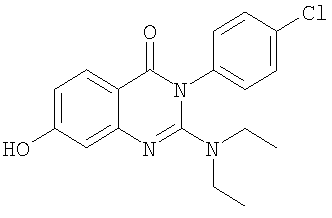
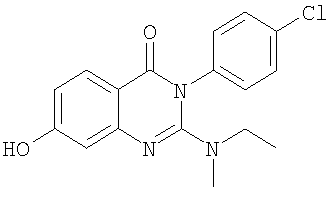
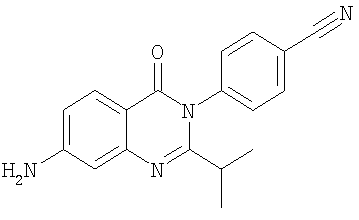
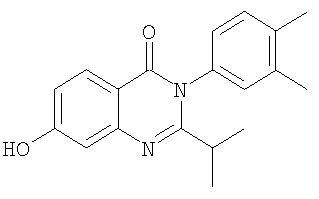
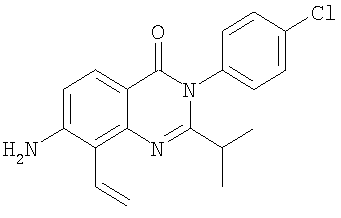
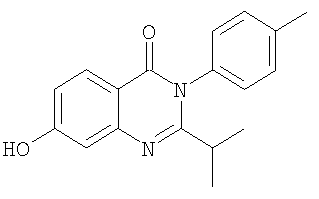
1. Соединение формулы: 2. Соединение по п.1, выбранное из группы, включающей следующие соединения: 3. Соединение по п.2, выбранное из группы, включающей следующие соединения: 4. Соединение по п.2, представляющее собой 5. Соединение по п.2, представляющее собой 6. Соединение по п.2, представляющее собой 7. Соединение по п.2, представляющее собой 8. Соединение по п.2, представляющее собой 9. Соединение по п.2, представляющее собой 10. Соединение по п.2, представляющее собой 11. Соединение по п.2, представляющее собой 12. Соединение по любому из пп.1-11 в свободной форме или в форме фармацевтически приемлемой соли, предназначенное для применения при лечении или профилактике боли. 13. Соединение по любому из пп.1-11 в свободной форме или в форме фармацевтически приемлемой соли, предназначенное для применения при лечении или профилактике желудочно-кишечного нарушения. 14. Фармацевтическая композиция, предназначенная для лечения или профилактики заболевания или состояния, в котором играет роль или участвует активация ванилоидного рецептора, включающая в эффективном количестве соединение по любому из пп.1-11 в свободной форме или в форме фармацевтически приемлемой соли в качестве активного ингредиента, и фармацевтически приемлемый носитель или разбавитель. 15. Фармацевтическая композиция по п.14, предназначенная для применения при лечении или профилактике боли. 16. Фармацевтическая композиция по п.14, предназначенная для применения при лечении или профилактике желудочно-кишечного нарушения. 17. Фармацевтическая композиция по п.14, дополнительно включающая другой активный ингредиент, выбранный из ингибиторов циклооксигеназы-2 (СОХ-2), нестероидных противовоспалительных лекарственных средств, агентов против мигрени, трициклических антидепрессантов, селективных ингибиторов захвата серотонина, селективных ингибиторов захвата норадреналина, противосудорожных средств, агонистов OABAB, опиоидов, агонистов рецептора CB1, антагонистов брадикининового рецептора и антагонистов вещества Р. 18. Фармацевтическая композиция по п.17, предназначенная для применения при лечении или профилактике боли. 19. Фармацевтическая композиция по п.17, предназначенная для применения при лечении или профилактике желудочно-кишечного нарушения. 20. Применение соединение по любому из пп.1-11 в свободной форме или в форме фармацевтически приемлемой соли для изготовления лекарственного средства, предназначенного для применения при лечении или профилактике боли или желудочно-кишечного нарушения.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
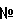 11
11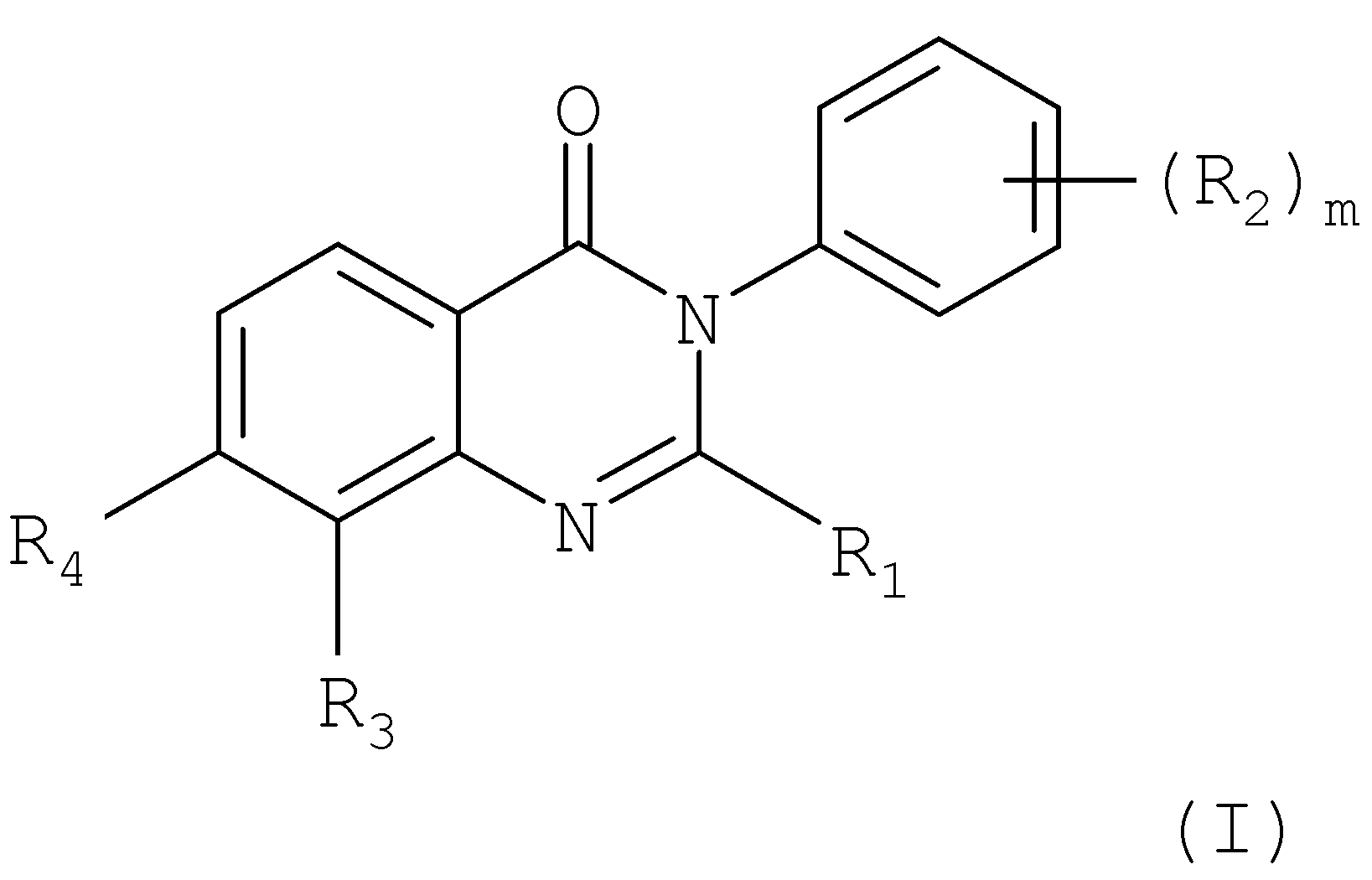
 или группу
или группу 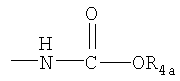 , где R4a представляет собой C1-С6алкил или галогензамещенный C1-С6алкил, или группу
, где R4a представляет собой C1-С6алкил или галогензамещенный C1-С6алкил, или группу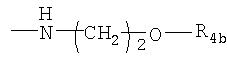 , где R4b представляет собой бензил или фенилэтил;
, где R4b представляет собой бензил или фенилэтил;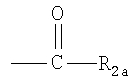 , где R2a представляет собой C1-С6алкил;
, где R2a представляет собой C1-С6алкил;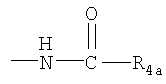 или группу
или группу 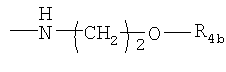 , где R4b представляет собой бензил или фенилэтил;
, где R4b представляет собой бензил или фенилэтил;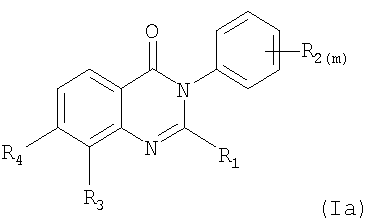
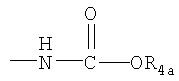 , где R4a представляет собой C1-С6алкил или галогензамещенный C1-С6алкил, или группу
, где R4a представляет собой C1-С6алкил или галогензамещенный C1-С6алкил, или группу 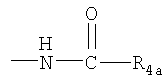 , где R4a представляет собой С1-С4алкил.
, где R4a представляет собой С1-С4алкил.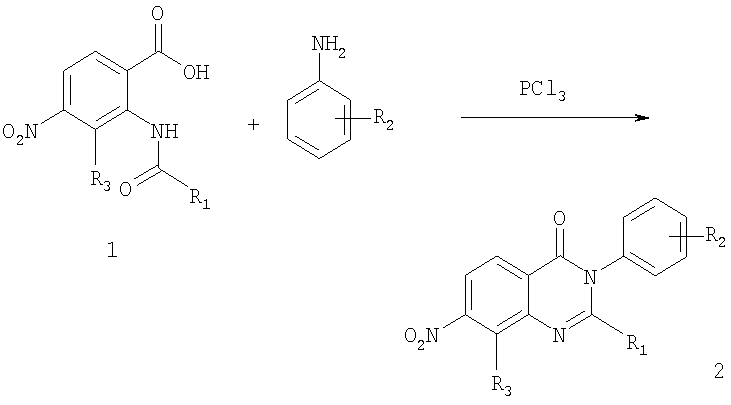
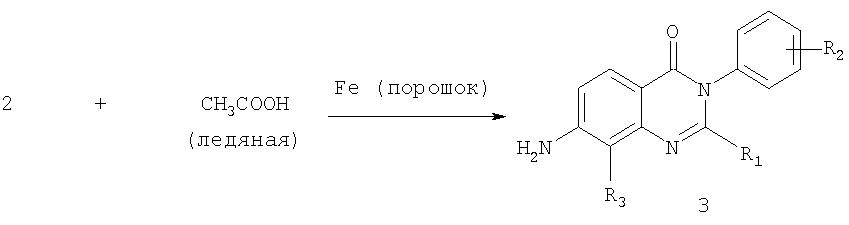
 2 адренергетиков.
2 адренергетиков. 7,91 (1Н, d,J=8,7 Гц), 7,49 (2Н, d,J=9,5 Гц), 7,25 (2Н,J=9,5 Гц), 6,93 (1Н, d,J=2,3 Гц), 6,86 (1Н, dd,J=2,3, 8,7 Гц), 2,56 (1Н, quint,J=6,7 Гц), 1,11 (6Н, d,J=6,7 Гц); (М+Н)+=315,8; время удерживания ВЭЖХ=4,2 минут.
7,91 (1Н, d,J=8,7 Гц), 7,49 (2Н, d,J=9,5 Гц), 7,25 (2Н,J=9,5 Гц), 6,93 (1Н, d,J=2,3 Гц), 6,86 (1Н, dd,J=2,3, 8,7 Гц), 2,56 (1Н, quint,J=6,7 Гц), 1,11 (6Н, d,J=6,7 Гц); (М+Н)+=315,8; время удерживания ВЭЖХ=4,2 минут.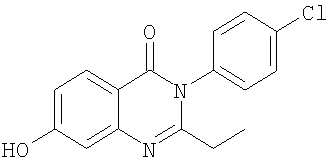
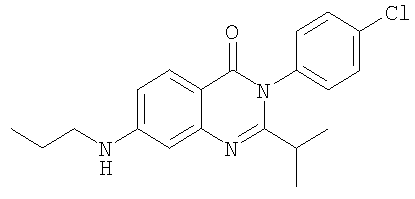
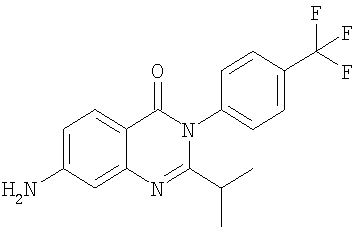
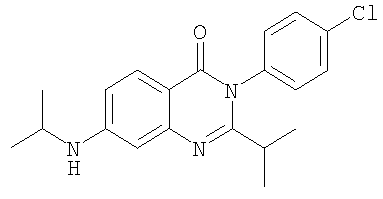
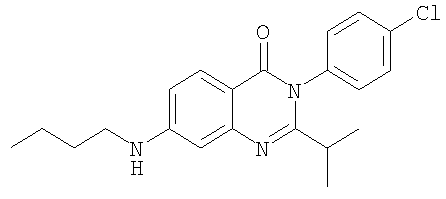
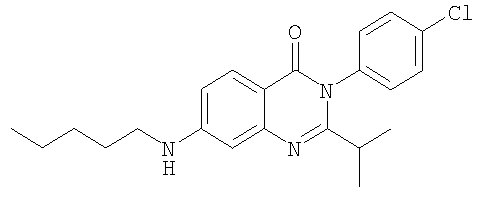
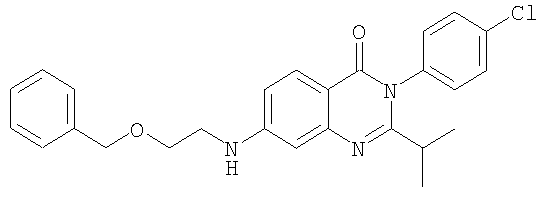
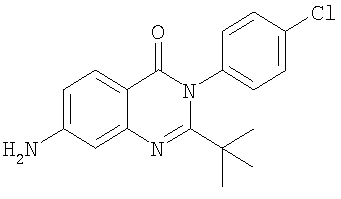
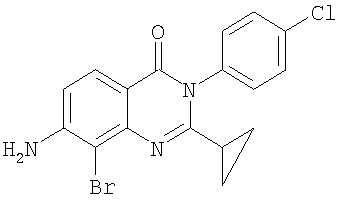
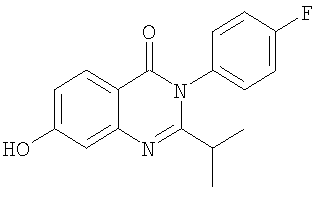
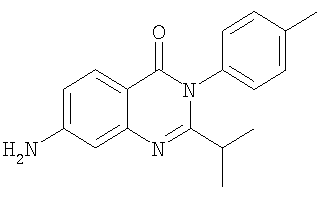
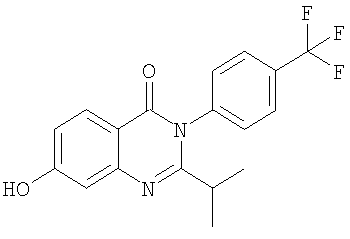
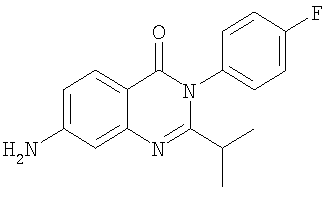
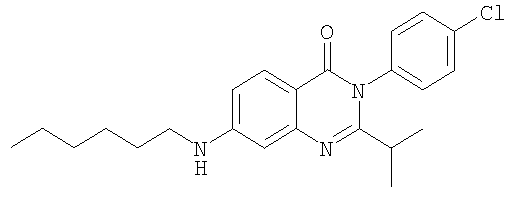
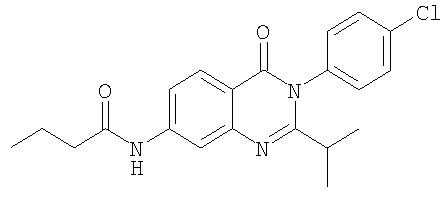
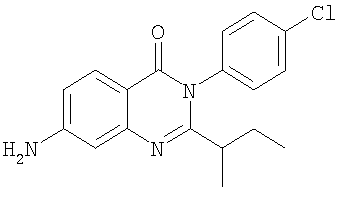
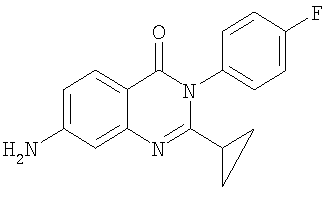
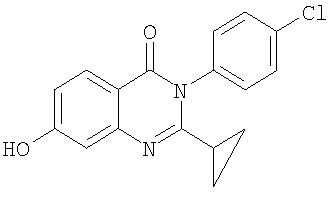
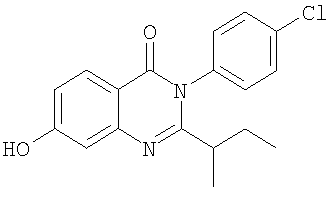
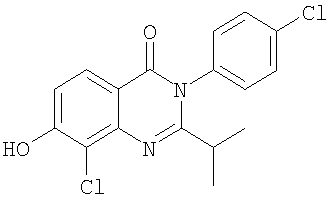
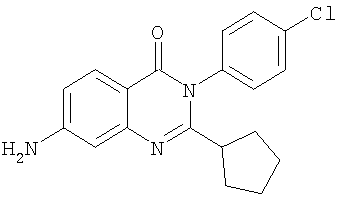
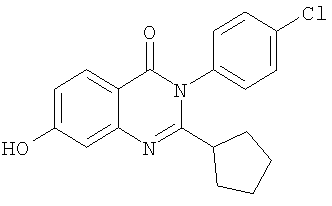
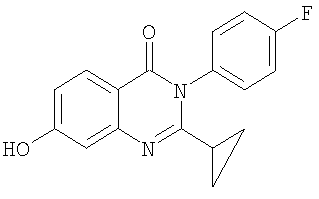
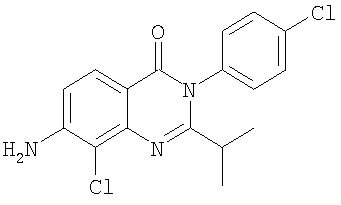
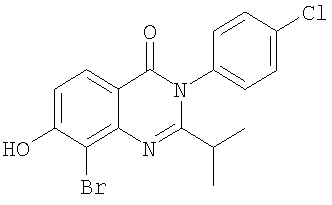
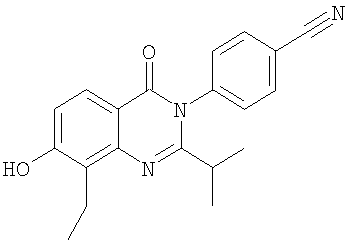
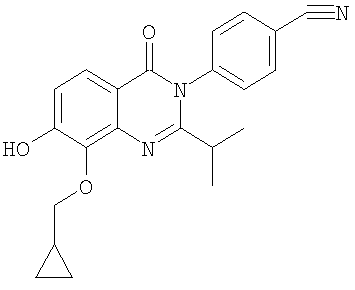
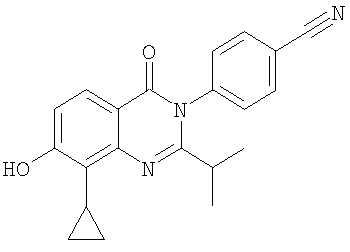
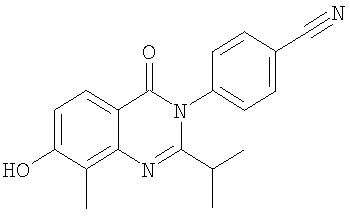
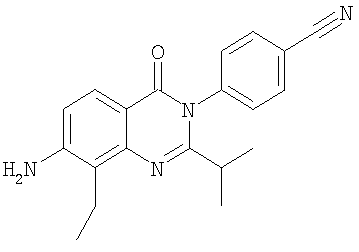
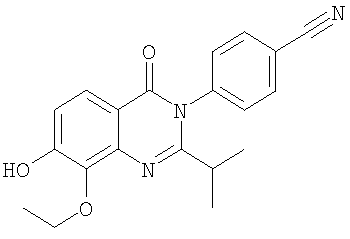
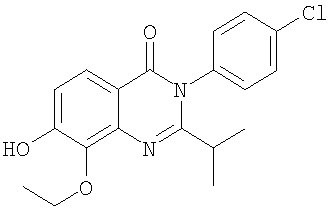
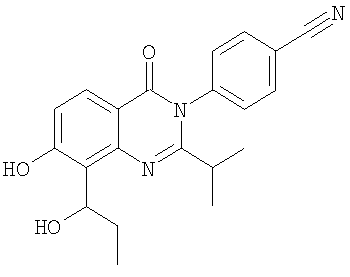
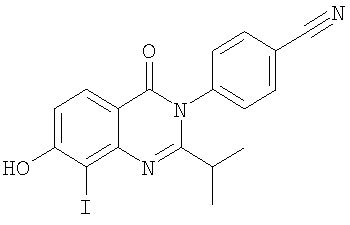
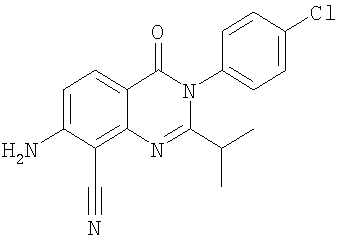

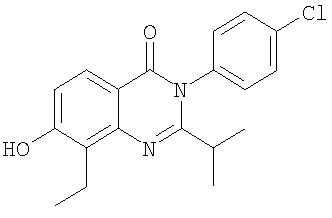
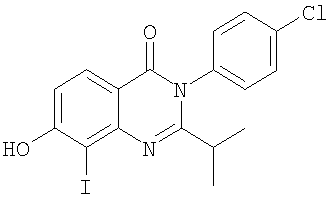
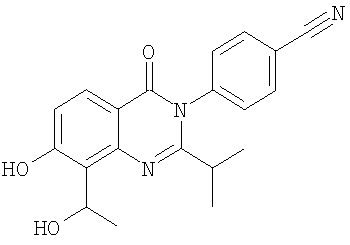
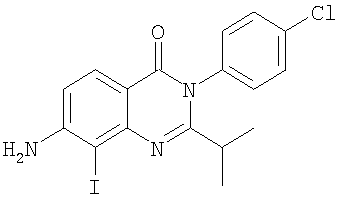
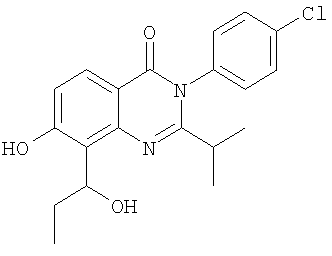
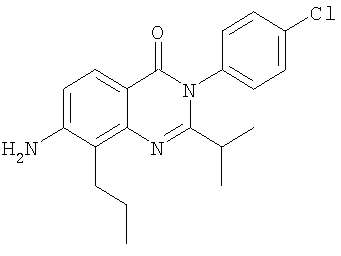
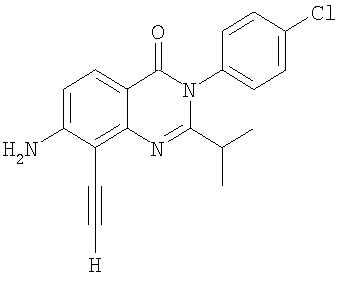
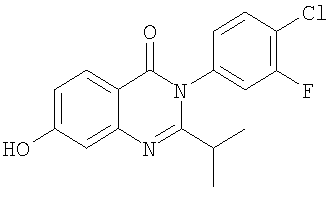
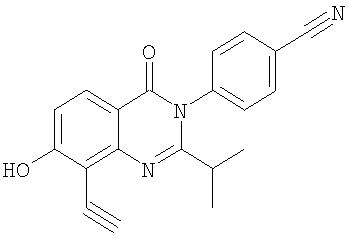
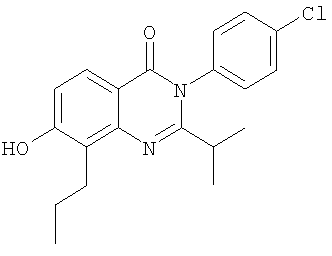
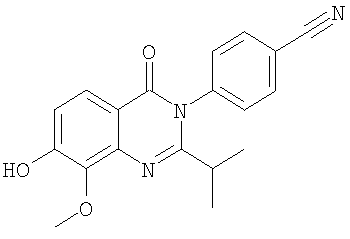
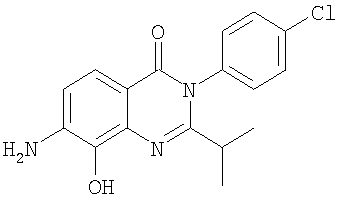
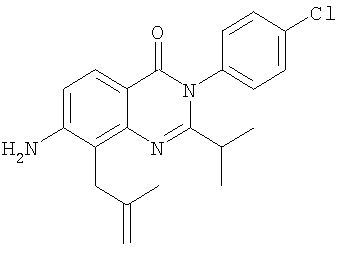
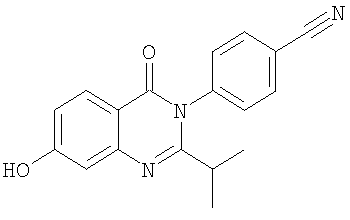
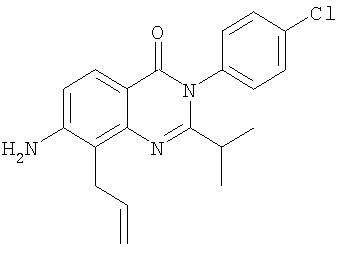
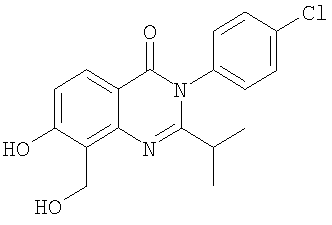
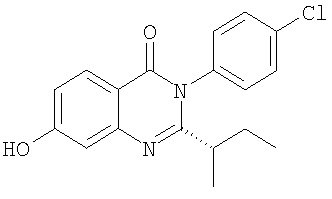
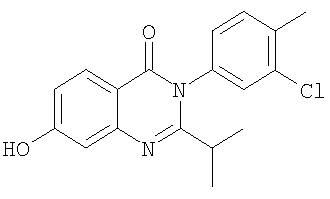
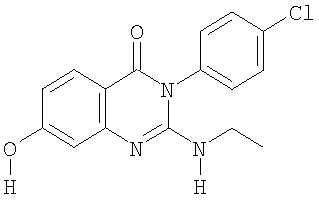
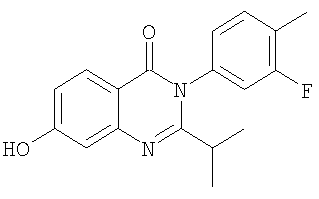
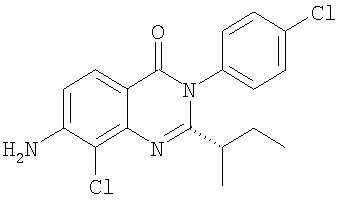
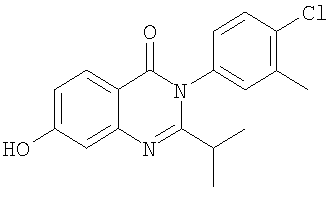
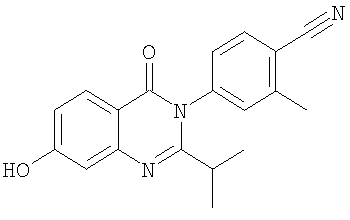
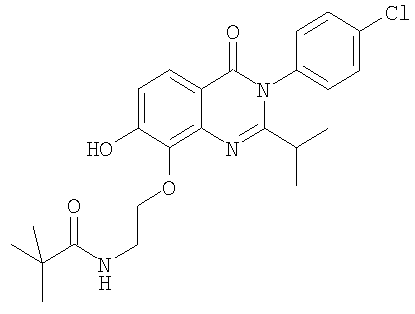
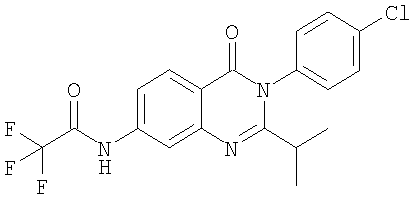
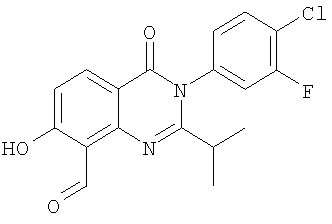
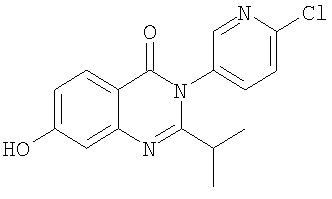
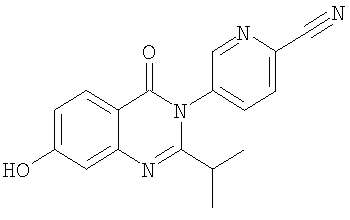
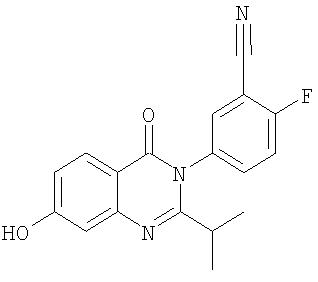
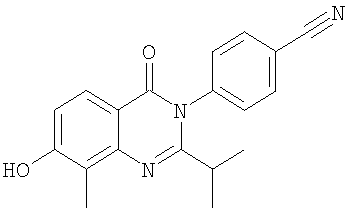 ,
,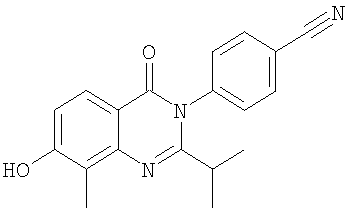 ,
,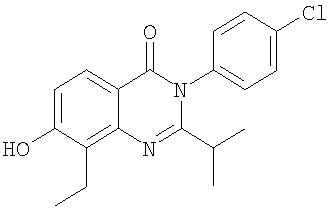 ,
,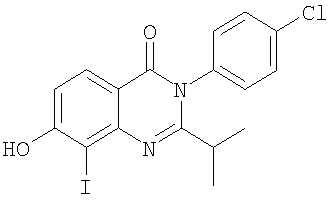 ,
,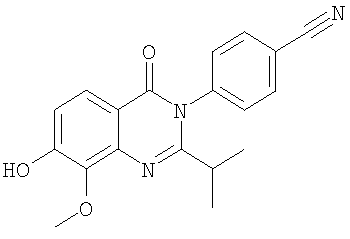 ,
,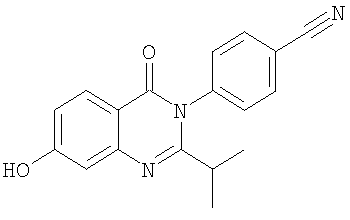 ,
,
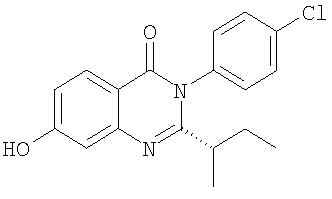
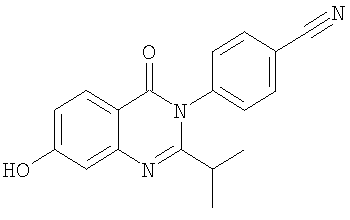 ,
,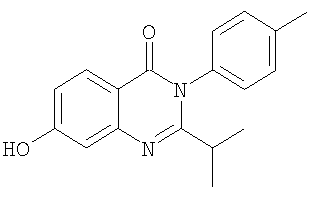
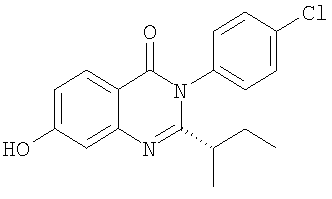 .
.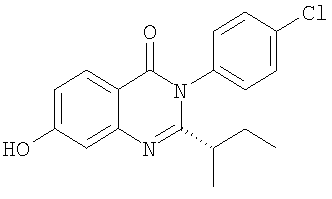
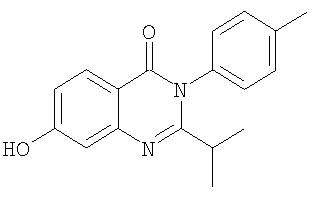
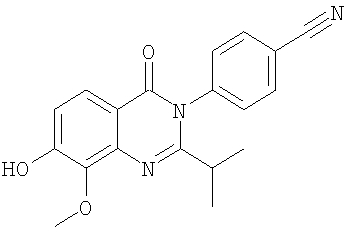 .
.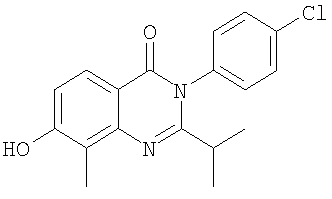 .
.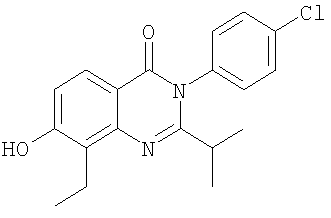 .
.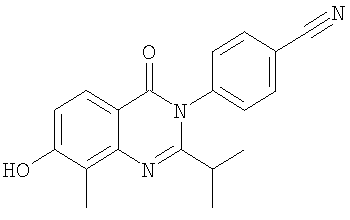 .
.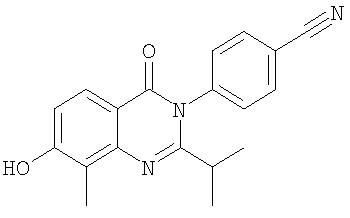 .
.