Патент на изобретение №2396257
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) ПРОИЗВОДНЫЕ 4-АМИНОПИПЕРИДИНА
(57) Реферат:
Изобретение относится к новым соединениям формулы (I)
где R1 выбирают из группы, включающей: фенил, незамещенный или моно-, ди- или тризамещенный независимо низшим алкилом, низшей алкоксигруппой, галогеном или низшим галогеналкилом; нафтил; тетрагидронафтил; С3-7циклоалкил; -(CHR3)m-фенил, где m обозначает 1, 2 или 3; и фенил при этом является незамещенным или моно-, ди- или тризамещенным низшей алкоксигруппой, и где R3 независимо выбирают из водорода и низшего алкила; -(СН2)n-гетероарил, где n обозначает 1, 2 или 3; причем термин «гетероарил» относится к ароматическому 5- или 6-членному кольцу или к бициклическим 9-членным ароматическим группам, которые могут включать 1, 2 или 3 атома, выбранные из азота и/или серы; -(СН2)n-гетероарил, где n обозначает 1, 2 или 3, причем термин «гетероарил» относится к ароматическому 5- или 6-членному кольцу или к бициклическим 9-членным ароматическим группам, которые могут включать 1, 2 или 3 атома, выбранные из азота и/или серы, и гетероарил является моно-, ди- или тризамещенным независимо низшей алкоксигруппой; и R2 выбирают из группы, включающей: н-бутил; фенил, незамещенный или моно-, ди- или тризамещенный независимо низшим алкилом, галогеном или низшей алкоксигруппой; гетероарил, причем термин «гетероарил» относится к ароматическому 5-членному кольцу, которое может включать 1, 2 или 3 атома, выбранные из азота и/или серы, незамещенный или моно-, ди- или тризамещенный независимо низшей алкоксигруппой; -C(O)-NR4R5, где R4 и R5 обозначают низший алкил или вместе с атомом азота, к которому они присоединены, образуют 5-членный гетероцикл, который может кроме того содержать гетероатом, выбранный из N или S, и к их фармацевтически приемлемым солям. Изобретение также относится к фармацевтической композиции. Технический результат – получение новых биологически активных соединений, способных ингибировать DPP-IV. 2 н. и 11 з.п.ф-лы.
Настоящее изобретение относится к новым производным 4-аминопиперидина, их получению и их использованию в качестве лекарственных препаратов. В частности, изобретение относится к соединениям формулы (I):
где R1 выбирают из группы, включающей: фенил, незамещенный или моно-, ди- или тризамещенный независимо низшим алкилом, низшей алкоксигруппой, фенилом, феноксигруппой, галогеном или низшим галогеналкилом; нафтил, незамещенный или моно-, ди- или тризамещенный независимо низшим алкилом, галогеном, низшим галогеналкилом, низшей алкоксигруппой, фенилом или феноксигруппой; тетрагидронафтил; С3–7 циклоалкил; -(CHR3)m-фенил, где m обозначает 1, 2 или 3; и фенил при этом является незамещенным или моно-, ди- или тризамещенным независимо низшим алкилом, галогеном, низшим галогеналкилом, низшей алкоксигруппой, фенилом или феноксигруппой, и где R3 независимо выбирают из водорода, низшего алкила или фенила; -(СН2)n-гетероарил, где n обозначает 1, 2 или 3; -(СН2)n-гетероарил, где n обозначает 1, 2, 3 и гетероарил является моно-, ди- или тризамещенным низшим алкилом, галогеном, низшим галогеналкилом или низшей алкоксигруппой; -С(O)-СН2-фенил, где фенил является незамещенным или моно-, ди- или тризамещенным независимо низшим алкилом, галогеном, низшим галогеналкилом, низшей алкоксигруппой, фенилом или феноксигруппой; -С(O)-СН2-гетероарил; и -С(O)-СН2-гетероарил, где гетероарил является моно-, ди- или тризамещенным независимо низшим алкилом, галогеном, низшим галогеналкилом или низшей алкоксигруппой; и R2 выбирают из группы, включающей: низший алкил, низший галогеналкил; фенил, незамещенный или моно-, ди- или тризамещенный независимо низшим алкилом, галогеном, низшим галогеналкилом или низшей алкоксигруппой; нафтил, незамещенный или моно-, ди- или тризамещенный независимо низшим алкилом, галогеном, низшим галогеналкилом или низшей алкоксигруппой; гетероарил, незамещенный или моно-, ди- или тризамещенный независимо низшим алкилом, галогеном, низшим галогеналкилом или низшей алкоксигруппой; -COOH; -C(O)-NR4R5, где R4 и R5 обозначают низший алкил или вместе с атомом азота, к которому они присоединены, образуют 4-, 5- или 6-членный гетероцикл, который может кроме этого содержать гетероатом, выбранный из N, О или S, и их фармацевтически приемлемым солям. Фермент дипептидилпептидаза IV ЕС.3.4.14.5 (ЕС является аббревиатурой «Enzyme Committee» Международного союза биохимии), обозначаемый далее аббревиатурой DPP-IV, принимает участие в регулировании активности нескольких гормонов. В частности, DPP-IV вызывает эффективную и быструю деградацию глюкагон подобного пептида 1 (GLP-1), являющегося одним из наиболее потенциальных стимуляторов продуцирования и секреции инсулина. Ингибирование DPP-IV может потенцировать эффект эндогенного GLP-1 и приводит к более высоким концентрациям инсулина в плазме. У пациентов, страдающих от пониженной толерантности к глюкозе и сахарного диабета 2 типа, повышенная концентрация плазменного инсулина может смягчать опасную гипергликемию и благодаря этому уменьшать риск повреждения тканей. Следовательно, DPP-IV ингибиторы рассматриваются в качестве возможных лекарственных препаратов для лечения пониженной толерантности к глюкозе и сахарного диабета 2 типа (например, Villhauer, WO 98/19998). Другое родственное описание уровня техники может быть найдено в WO 99/38501, DE 19616486, DE 19834591, WO 01/40180, WO 01/55105, US 6110949, WO 00/34241 и US 6011155. Нами были найдены новые DPP-IV ингибиторы, которые эффективно снижают уровни глюкозы в плазме. Следовательно, соединения по настоящему изобретению применяются для лечения и/или профилактики диабета, в частности инсулиннезависимого сахарного диабета, и/или пониженной толерантности к глюкозе, а также других состояний, где амплификация действия пептида, нормально инактивируемого DPP-IV, приносит терапевтическую пользу. Кроме того, соединения по настоящему изобретению могут быть применены при лечении и/или профилактике ожирения, метаболического синдрома, защите Неожиданно соединения по настоящему изобретению проявили улучшенные терапевтические и фармакологические свойства, сравнимые с другими DPP-IV ингибиторами, известными из области техники, например, в контексте фармакокинетики и биодоступности. Объектами настоящего изобретения являются соединения формулы I и их получение, лекарственные средства на основе соединений формулы I и их производство, а также применение соединений формулы I в соответствии с изобретением при контроле и профилактике заболеваний, упомянутых выше, и соответственно при получении соответствующих лекарственных средств. Если не указано иначе, приведены следующие определения для иллюстрации и определения значения и объема различных терминов, используемых в описании данного изобретения. В этом описании термин “низший” используется для обозначения группы, включающей от одного до шести, предпочтительно от одного до четырех атомов углерода. Термин “галоген” относится к фтору, хлору, брому и йоду, при этом фтор и хлор являются предпочтительными. Наиболее предпочтительным является хлор. Термин “алкил”, один или в комбинации с другими группами, относится к разветвленному или прямолинейному моновалентному алкильному радикалу, содержащему от одного до шести атомов углерода, предпочтительно от одного до четырех атомов углерода. Этот термин далее иллюстрируется такими радикалами, как метил, этил, н-пропил, изопропил, н-бутил, emop-бутил, изобутил, трет-бутил, н-пентил, 3-метилбутил, н-гексил, 2-этилбутил и подобными им. Предпочтительными низшими алкильными радикалами являются метил, этил, н-пропил и н-бутил, при этом метил является особенно предпочтительным. Термин «низший галогеналкил» относится к низшей алкильной группе, где, по крайней мере, один из водородных атомов низшей алкильной группы замещен атомом галогена, предпочтительно фтором или хлором, наиболее предпочтительно фтором. Предпочтительными низшими галогеналкильными группами являются трифторметил, дифторметил, фторметил и хлорметил, при этом особенно предпочтительной является фторметильная группа. Термин “алкоксигруппа” относится к группе R’-O-, где R’ обозначает алкил. Термин “низшая алкоксигруппа” относится к группе R’-O-, где R’ обозначает низший алкил. Примерами низших алкоксигрупп являются метоксигруппа, этоксигруппа, пропоксигруппа, изопропоксигруппа, бутоксигруппа, изобутоксигруппа и гексилоксигруппа, при этом особенно предпочтительной является метоксигруппа. Термин “циклоалкил” или “С3-С7-циклоалкил” относится к моновалентному карбоциклическому радикалу, содержащему от 3 до 7 атомов углерода. Этот термин далее относится к таким радикалам, как циклопропил, циклобутил, циклопентил, циклогексил и циклогептил, при этом предпочтительными являются циклопропил и циклогексил. Термин «гетероарил» относится к ароматическому 5- или 6-членному кольцу, которое может включать 1, 2 или 3 атома, выбранные из азота, кислорода и/или серы, такому, как фурил, пиридил, 1,2-, 1,3- и 1,4-диазинил, тиенил, изоксазолил, оксазолил, имидазолил или пирролил. Термин «гетероарил» относится, кроме того, к бициклическим ароматическим группам, включающим два 5- или 6-членных кольца, в котором одно или оба кольца могут содержать 1, 2 или 3 атома, выбранные из азота, кислорода или серы, таким, как индол или хинолин, или частично гидрированные бициклические ароматические группы такие, как, например, индолинил. Предпочтительными гетероарильными группами являются тиенил, пиридил и индолил, которые необязательно могут быть замещены, как описано выше, предпочтительно низшим алкилом или галогеном. Выражение «R4 и R5 вместе с атомом азота, к которому они присоединены, образуют 4-, 5- или 6-членный гетероциклил, который дополнительно может содержать гетероатом, выбранный из кислорода, азота или серы» подразумевает, что R4 и R5 вместе с атомом азота, к которому они присоединены, образуют кольцо такое, как пирролидинил, дигидропирролил (пирролинил), пиперидил, имидазолидинил, морфолинил, пиперазинил, тиазолидинил, тиоморфолинил, при этом тиазолидинил и дигидропирролил особенно предпочтительны. Термин “фармацевтически приемлемые соли” включает соли соединений формулы (I) с неорганическими или органически кислотами, такими, как хлористоводородная кислота, бромистоводородная кислота, азотная кислота, серная кислота, фосфорная кислота, лимонная кислота, муравьиная кислота, яблочная кислота, уксусная кислота, фумаровая кислота, янтарная кислота, винная кислота, метансульфоновая кислота, салициловая кислота, п-толуолсульфоновая кислота и подобные им, которые являются нетоксичными для живых организмов. Предпочтительными солями являются формиаты, малеаты, цитраты, гидрохлориды, гидробромиды и соли метансульфоновой кислоты, при этом наиболее предпочтительными являются гидрохлориды. Изобретение относится к соединениям формулы (I):
где R1 выбирают из группы, включающей: фенил, незамещенный или моно-, ди- или тризамещенный независимо низшим алкилом, низшей алкоксигруппой, фенилом, феноксигруппой, галогеном или низшим галогеналкилом; нафтил, незамещенный или моно-, ди- или тризамещенный независимо низшим алкилом, галогеном, низшим галогеналкилом, низшей алкоксигруппой, фенилом или феноксигруппой; тетрагидронафтил; С3–7 циклоалкил; -(CHR3)m-фенил, где m обозначает 1, 2 или 3; и фенил при этом является незамещенным или моно-, ди- или тризамещенным независимо низшим алкилом, галогеном, низшим галогеналкилом, низшей алкоксигруппой, фенилом или феноксигруппой, и где R3 независимо выбирают из водорода, низшего алкила или фенила; -(СН2)n-гетероарил, где n обозначает 1, 2 или 3; -(СН2)n-гетероарил, где n обозначает 1, 2, 3 и гетероарил является моно-, ди- или тризамещенным независимо низшим алкилом, галогеном, низшим галогеналкилом или низшей алкоксигруппой; -С(O)-СН2-фенил, где фенил является незамещенным или моно-, ди- или тризамещенным независимо низшим алкилом, галогеном, низшим галогеналкилом, низшей алкоксигруппой, фенилом или феноксигруппой; -С(O)-СН2-гетероарил; и -С(O)-СН2-гетероарил, где гетероарил является моно-, ди- или тризамещенным независимо низшим алкилом, галогеном, низшим галогеналкилом или низшей алкоксигруппой; и R2 выбирают из группы, включающей низший алкил, низший галогеналкил; фенил, незамещенный или моно-, ди- или тризамещенный независимо низшим алкилом, галогеном, низшим галогеналкилом или низшей алкоксигруппой; нафтил, незамещенный или моно-, ди- или тризамещенный независимо низшим алкилом, галогеном, низшим галогеналкилом или низшей алкоксигруппой; гетероарил, незамещенный или моно-, ди- или тризамещенный независимо низшим алкилом, галогеном, низшим галогеналкилом или низшей алкоксигруппой; -СООН; -C(O)-NR4R5, где R4 и R5 обозначают низший алкил или вместе с атомом азота, к которому они присоединены, образуют 4-, 5- или 6-членный гетероцикл, который может кроме этого содержать гетероатом, выбранный из N, О или S, и их фармацевтически приемлемым солям. В предпочтительных соединениях по настоящему изобретению R1 выбирают из группы, включающей: фенил, незамещенный или моно-, ди- или тризамещенный независимо низшим алкилом, галогеном, низшим галогеналкилом, низшей алкоксигруппой, фенилом или феноксигруппой; нафтил, незамещенный или моно-, ди- или тризамещенный независимо низшим алкилом, галогеном, низшим галогеналкилом, низшей алкоксигруппой, фенилом или феноксигруппой; -(CHR3)m-фенил, где m обозначает 1 или 2, и при этом фенил является незамещенным или моно-, ди- или тризамещенным независимо низшим алкилом, галогеном, низшим галогеналкилом, низшей алкоксигруппой, фенилом или феноксигруппой и где R3 обозначает водород; -(СН2)n-гетероарил, где n обозначает 1 или 2; -(СН2)n-гетероарил, где n обозначает 1 или 2, и гетероарил является моно-, ди- или тризамещенным независимо низшим алкилом, галогеном, низшим галогеналкилом или низшей алкоксигруппой; -С(O)-СН2-фенил, при этом фенил является незамещенным или моно-, ди- или тризамещенным независимо низшим алкилом, галогеном, низшим галогеналкилом, низшей алкоксигруппой, фенилом или феноксигруппой; -С(O)-СН2-гетероарил; и -С(O)-СН2-гетероарил, где гетероарил является моно-, ди- или тризамещенным независимо низшим алкилом, галогеном, низшим галогеналкилом или низшей алкоксигруппой. Одной группой предпочтительных соединений формулы I по настоящему изобретению являются такие соединения, где R1 выбирают из группы, включающей: фенил, незамещенный или моно-, ди- или тризамещенный независимо низшим алкилом, галогеном, низшим галогеналкилом, низшей алкоксигруппой, фенилом или феноксигруппой; нафтил, незамещенный или моно-, ди- или тризамещенный независимо низшим алкилом, галогеном, низшим галогеналкилом, низшей алкоксигруппой, фенилом или феноксигруппой; и -(CHR3)m-фенил, где m обозначает 1 или 2, и при этом фенил является незамещенным или моно-, ди- или тризамещенным независимо низшей алкоксигруппой, и где R3 обозначает водород. Особенно предпочтительны соединения формулы I, где R1 обозначает: фенил, моно-, ди- или тризамещенный независимо низшим алкилом, галогеном, низшим галогеналкилом, низшей алкоксигруппой, фенилом или феноксигруппой. Более предпочтительно R1 обозначает фенил, ди- или тризамещенный низшей алкоксигруппой, при этом наиболее предпочтительно, когда R1 является 3,4-диметоксифенилом или 2,3,4-триметоксифенилом. Другой группой предпочтительных соединений формулы I являются такие соединения, где R1 обозначает -(СН2)n-гетероарил, где n обозначает 1 или 2; или -(СН2)n-гетероарил, где n обозначает 1 или 2, и гетероарил является моно-, ди- или тризамещенным независимо низшим алкилом, галогеном, низшим галогеналкилом или низшей алкоксигруппой. Предпочтительным гетероарилом являются индолил или пиридил. Предпочтительно n обозначает 2. Также предпочтительными являются соединения формулы I, где R1 обозначает -С(O)-СН2-фенил, при этом фенил является незамещенным или моно-, ди- или тризамещенным независимо низшим алкилом, галогеном, низшим галогеналкилом, низшей алкоксигруппой, фенилом или феноксигруппой; или -С(O)-СН2-гетероарил. Особенно предпочтительными являются -С(O)-СН2-фенил и -С(O)-СН2-тиенил. В предпочтительных соединениях по настоящему изобретению R2 выбирают из группы, включающей: низший алкил; фенил, незамещенный или моно-, ди- или тризамещенный независимо низшим алкилом, галогеном, низшим галогеналкилом или низшей алкоксигруппой; гетероарил, незамещенный или моно-, ди- или тризамещенный независимо низшим алкилом, галогеном, низшим галогеналкилом или низшей алкоксигруппой;и -C(O)-NR4R5, где R4 и R5 вместе с атомом азота, к которому они присоединены, образуют 5-членный гетероцикл, который может кроме этого содержать гетероатом, выбранный из N, О или S. Особенно предпочтительными являются соединения формулы I, где R2 обозначает низший алкил, при этом наиболее предпочтительным является н-бутил. Другой группой предпочтительных соединений формулы I являются соединения, где R2 обозначает фенил, незамещенный или моно-, ди- или тризамещенный независимо низшим алкилом, галогеном, низшим галогеналкилом или низшей алкоксигруппой. Особенно предпочтительными являются соединения формулы I, где R2 обозначает фенил или фенил, моно-, ди- или тризамещенный независимо низшим алкилом или галогеном. Также предпочтительными являются соединения формулы I, где R2 обозначает: гетероарил, незамещенный или моно-, ди- или тризамещенный независимо низшим алкилом, галогеном, низшим галогеналкилом или низшей алкоксигруппой, при этом гетероарил, являющийся пиридилом или тиенилом, является особенно предпочтительным. Кроме этого, предпочтительными соединениями формулы I являются соединения, где R2 обозначает: -C(O)-NR4R5, где R4 и R5 вместе с атомом азота, к которому они присоединены, образуют 5-членный гетероцикл, который может кроме этого содержать гетероатом, выбранный из N, О или S. Особенно предпочтительными являются соединения формулы I, где R2 обозначает -С(O)-тиазолидинил или -(СО)-дигидропирролил. Примерами соединений формулы I по настоящему изобретению являются следующие: цис-3-бутил-1-фенетилпиперидин-4-иламин, транс-3-бутил-1-фенетилпиперидин-4-иламин, цис-3-бутил-1-бензилпиперидин-4-иламин, транс-3-бутил-1-бензилпиперидин-4-иламин, цис-3-бутил-1-[2-(1Н-индол-3-ил)этил]пиперидин-4-иламин, транс-3-бутил-1-[2-(1Н-индол-3-ил)этил]пиперидин-4-иламин, цис-3-бутил-1-[2-(3,4-диметоксифенил-1-ил)этил]пиперидин-4-иламин, транс-3-бутил-1-[2-(3,4-диметоксифенил-1-ил)этил]пиперидин-4-иламин, гидрохлорид цис-3-бутил-1-(3,4,5-триметоксифенил)пиперидин-4-иламина, гидрохлорид транс-3-бутил-1-(3,4,5-триметоксифенил)пиперидин-4-иламина, цис-3-бутил-1-(4-феноксифенил)пиперидин-4-иламин, транс-3-бутил-1-(4-феноксифенил)пиперидин-4-иламин, гидрохлорид (цис/транс)-3-бутил-1-(5,6,7,8-тетрагидронафталин-1-ил)пиперидин-4-иламина, гидрохлорид цис-3-бутил-1-(3,4-диметоксифенил)пиперидин-4-иламина, гидрохлорид транс-3-бутил-1-(3,4-диметоксифенил)пиперидин-4-иламина, гидрохлорид цис-3-бутил-1-нафталин-2-илпиперидин-4-иламина, гидрохлорид транс-3-бутил-1-нафталин-2-илпиперидин-4-иламина, гидрохлорид (цис/транс)-3-бутил-1-нафталин-1-илпиперидин-4-иламина, гидрохлорид цис-3-бутил-1-(3,4-дихлорфенил)пиперидин-4-иламина, гидрохлорид цис-3-бутил-1-(4-хлор-3-трифторметилфенил)пиперидин-4-иламина, гидрохлорид цис-3-бутил-1-п-толилпиперидин-4-иламина, гидрохлорид транс-3-бутил-1-п-толилпиперидин-4-иламина, гидрохлорид цис-3-бутил-1-(3,5-дихлорфенил)пиперидин-4-иламина, гидрохлорид (цис/транс)-3-бутил-1-(3,5-дихлорфенил)пиперидин-4-иламина, гидрохлорид цис-3-бутил-4-метил-1-фенилпиперидин-4-иламина, гидрохлорид транс-3-бутил-4-метил-1-фенилпиперидин-4-иламина, гидрохлорид цис-3-бутил-1-(3-метокси-5-трифторметилфенил)пиперидин-4-иламина, гидрохлорид (цис/транс)-3-бутил-1-(3-метокси-5-трифторметилфенил)пиперидин-4-иламина, гидрохлорид (цис/транс)-3-бутил-1-циклогексилпиперидин-4-иламина, гидрохлорид (цис/транс)-3-бутил-1-(3,5-бис-трифторметилфенил)пиперидин-4-иламина, гидрохлорид цис-3-бутил-1-(6-метоксибифенил-3-ил)пиперидин-4-иламина, гидрохлорид транс-3-бутил-1-(6-метоксибифенил-3-ил)пиперидин-4-иламина, гидрохлорид (цис/транс)-3-бутил-1-бензгидрил-4-илпиперидин-4-иламина, цис-3-фенил-1-фенетилпиперидин-4-иламин, транс-3-фенил-1-фенетилпиперидин-4-иламин, цис-3-фенил-1-бензилпиперидин-4-иламин, транс-3-фенил-1-бензилпиперидин-4-иламин, (цис/транс)-метил-1′-фенетил-1′,2′,3′,4′,5′,6′-гексагидро[2,3′]бипиридинил-4′-иламин, цис-3-(3-хлорфенил)-1-фенетилпиперидин-4-иламин, (цис/транс)-3-(3-хлорфенил)-1-бензилпиперидин-4-иламин, (цис/транс)-3-(3-метилфенил)-1-бензилпиперидин-4-иламин, цис-3-(3-хлорфенил)-1-[2-(3,4-диметоксифенил)этил]пиперидин-4-иламин, транс-1-бензил-3-тиофен-2-илпиперидин-4-иламин, гидрохлорид цис-3-о-толил-1-(3,4,5-триметоксифенил)пиперидин-4-иламина, гидрохлорид транс-3-о-толил-1-(3,4,5-триметоксифенил)пиперидин-4-иламина, гидрохлорид (цис/транс)-3-о-толил-1-(3,4,5-триметоксифенил)пиперидин-4-иламина, гидрохлорид (цис/транс)-1-(3,4-диметоксифенил)-3-м-толилпиперидин-4-иламина, гидрохлорид (цис/транс)-1-(3,4-диметоксифенил)-3-п-толилпиперидин-4-иламина, гидрохлорид (цис/транс)-1-(3,4-диметоксифенил)-3-(3,4-диметилфенил)пиперидин-4-иламина, гидрохлорид (цис/транс)-1-(3,4-диметоксифенил)-3-(3-метоксифенил)пиперидин-4-иламина, (цис/транс)-1′-(3,4-диметоксифенил)-1′,2′,3′,4′,5′,6′-гексагидро[2,3′]бипиридинил-4′-иламин, ((3R,4S)-4-амино-1-фенетилпиперидин-3-ил)тиазолидин-3-илметанон, ((3S,4R)-4-амино-1-фенетилпиперидин-3-ил)тиазолидин-3-илметанон, [(3S,4R)-4-амино-1-(2-пиридин-2-илэтил)пиперидин-3-ил]тиазолидин-3-илметанон, 1-[(3S,4R)-4-амино-3-(тиазолидин-3-карбонил)пиперидин-1-ил]-2-фенилэтанон, 1-[(3S,4R)-4-амино-3-(тиазолидин-3-карбонил)пиперидин-1-ил]-2-тиофен-2-илэтанон, 3-[(3S,4R) и (3R,4S)-4-амино-1-[2-(3,4-диметоксифенил)этил]пиперидин-3-карбоновая кислота, 3-[(3S,4S) и (3R,4R)-4-амино-1-(3,4-диметоксифенил)пиперидин-3-ил]тиазолидин-3-илметанон 3-[(3S,4S) и (3R,4R)-4-амино-1-(3,4-диметоксифенил)пиперидин-3-ил]-(2,5-дигидропиррол-1-ил)-3-илметанон, 3-[(3S,4R) и (3R,4S)-4-амино-1-(3,4-диметоксифенил)пиперидин-3-ил]-(2,5-дигидропиррол-1-ил)-3-илметанон, и их фармацевтически приемлемые соли. Предпочтительные соединения формулы I выбирают из группы, включающей: цис-3-бутил-1-фенетилпиперидин-4-иламин, цис-3-бутил-1-[2-(1Н-индол-3-ил)этил]пиперидин-4-иламин, гидрохлорид цис-3-бутил-1-(3,4,5-триметоксифенил)пиперидин-4-иламина, цис-3-бутил-1-(4-феноксифенил)пиперидин-4-иламин, гидрохлорид цис-3-бутил-1-(3,4-диметоксифенил)пиперидин-4-иламина, гидрохлорид цис-3-бутил-1-нафталин-2-илпиперидин-4-иламин, гидрохлорид транс-3-бутил-1-нафталин-2-илпиперидин-4-иламина, гидрохлорид (цис/транс)-3-бутил-1-нафталин-1-илпиперидин-4-иламина, гидрохлорид цис-3-бутил-1-(3,4-дихлорфенил)пиперидин-4-иламина, гидрохлорид цис-3-бутил-1-(4-хлор-3-трифторметилфенил)пиперидин-4-иламина, гидрохлорид цис-3-бутил-1-п-толилпиперидин-4-иламина, гидрохлорид цис-3-бутил-1-(3,5-дихлорфенил)пиперидин-4-иламина, гидрохлорид (цис/транс)-3-бутил-1-(3,5-дихлорфенил)пиперидин-4-иламина, гидрохлорид цис-3-бутил-4-метил-1-фенилпиперидин-4-иламина, гидрохлорид цис-3-бутил-1-(3-метокси-5-трифторметилфенил)пиперидин-4-иламина, гидрохлорид (цис/транс)-3-бутил-1-(3-метокси-5-трифторметилфенил)пиперидин-4-иламина, гидрохлорид цис-3-бутил-1-(6-метоксибифенил-3-ил)пиперидин-4-иламина, цис-3-фенил-1-фенетилпиперидин-4-иламин, (цис/транс)-метил-1′-фенетил-1′,2′,3′,4′,5′,6′-гексагидро[2,3′]бипиридинил-4′-иламин, цис-3-(3-хлорфенил)-1-фенетилпиперидин-4-иламин, (цис/транс)-3-(3-хлорфенил)-1-бензилпиперидин-4-иламин, (цис/транс)-3-(3-метилфенил)-1-бензилпиперидин-4-иламин, цис-3-(3-хлорфенил)-1-[2-(3,4-диметоксифенил)этил]пиперидин-4-иламин, гидрохлорид транс-3-о-толил-1-(3,4,5-триметоксифенил)пиперидин-4-иламина, гидрохлорид (цис/транс)-3-о-толил-1-(3,4,5-триметоксифенил)пиперидин-4-иламина, гидрохлорид (цис/транс)-1-(3,4-диметоксифенил)-3-м-толилпиперидин-4-иламина, (цис/транс)-1′-(3,4-диметоксифенил)-1′,2′,3′,4′,5′,6′-гексагидро[2,3′]бипиридинил-4′-иламин, ((3R,4S)-4-амино-1-фенетилпиперидин-3-ил)тиазолидин-3-илметанон, ((3S,4R)-4-амино-1-фенетилпиперидин-3-ил)тиазолидин-3-илметанон, [(3S,4R)-4-амино-1-(2-пиридин-2-илэтил)пиперидин-3-ил]тиазолидин-3-илметанон, 1-[(3S,4R)-4-амино-3-(тиазолидин-3-карбонил)пиперидин-1-ил]-2-фенилэтанон, 1-[(3S,4R)-4-амино-3-(тиазолидин-3-карбонил)пиперидин-1-ил]-2-тиофен-2-ил-этанон, 3-[(3S,4S) и (3R,4R)-4-амино-1-(3,4-диметоксифенил)пиперидин-3-ил] тиазолидин-3-илметанон 3-[(3S,4S) и (3R,4R)-4-амино-1-(3,4-диметоксифенил)пиперидин-3-ил]-(2,5-дигидропиррол-1-ил)-3-илметанон, 3-[(3S,4R) и (3R,4S)-4-амино-1-(3,4-диметоксифенил)пиперидин-3-ил]-(2,5-дигидропиррол-1-ил)-3-илметанон, и их фармацевтически приемлемые соли. Наиболее предпочтительные соединения выбирают из группы, включающей: цис-3-(3-хлорфенил)-1-фенетилпиперидин-4-иламин, (цис/транс)-3-(3-хлорфенил)-1-бензилпиперидин-4-иламин, (цис/транс)-3-(3-хлорфенил)-1-[2-(3,4-диметоксифенил)этил]пиперидин-4-иламин, гидрохлорид цис-1-(3,4-диметоксифенил)-3-м-толилпиперидин-4-иламина, ((3S,4R)-4-амино-1-фенетилпиперидин-3-ил)тиазолидин-3-илметанон, [(3S,4R)-4-амино-1-(2-пиридин-2-илэтил)пиперидин-3-ил]тиазолидин-3-илметанон, 3-[(3S,4R) и (3R,4S)-4-амино-1-(3,4-диметоксифенил)пиперидин-3-ил]-тиазолидин-3-илметанон, 3-[(3S,4S) и (3R,4R)-4-амино-1-(3,4-диметоксифенил)пиперидин-3-ил]-тиазолидин-3-илметанон, и их фармацевтически приемлемые соли. Соединения формулы I имеют два асимметрических атома и могут существовать в форме оптически чистых энантиомеров, смесей диастереоизомеров, рацематов или смесей диастереоизомерных рацематов. Изобретение включает все эти формы. В предпочтительном варианте R2 и аминогруппа пиперидиновой структуры находятся в транс-конфигурации, например,
В следующем предпочтительном варианте R2 и аминогруппа пиперидиновой структуры находятся в цис-конфигурации, например,
В следующем варианте изобретение предлагает способ получения соединений формулы I, включающий или а) превращение соединения формулы
где R1 и R2 определены выше, путем взаимодействия с гидроксиламином или его солью в оксим формулы
где R1 и R2 определены выше, и дальнейшее восстановление оксима формулы III посредством каталитического гидрирования или альтернативно восстановление с помощью гидрида металла, с образованием соединения формулы I, или б) снятие защиты у производного 4-аминопиперидина формулы
где R1 и R2 определены выше и Rp обозначает амино-защитную группу. Rp обозначает соответствующую группу, защищающую аминогруппу, такую как бензилоксикарбонил (Z или Cbz), аллилоксикарбонил (Алок), 9-флюоренилметоксикарбонил (Фмок), и предпочтительно трет-бутоксикарбонил (Бок). Конкретнее соединения формулы (I) могут быть получены методами, приведенными ниже, методами, приведенными в примерах, или аналогичными методами. Соответствующие реакционные условия для отдельных реакционных стадий известны специалистам в области техники. Исходные вещества являются либо коммерчески доступными либо могут быть получены методами, аналогичными методам, приведенным ниже, либо методами, приведенными в примерах, или методами, известными из уровня техники. Соединения формулы I по настоящему изобретению могут быть получены, как указано на схемах 1-6, приведенных ниже. 4-Аминопиперидины формулы I с R1, имеющим значения -(CHR3)m – фенил и -(СН2)n – гетероарил, где R3, n и m определены выше, и R2 обозначает низший алкил, могут быть получены в соответствии со схемой 1: Схема 1
На первой стадии R2, обозначающий низший алкил, вводится в 3 положение посредством взаимодействия N-защищенного алкилового эфира 4-оксопиперидинкарбоновой кислоты (Alk обозначает низший алкил) с соответствующим алкилйодидом. N-защита может быть легко осуществлена с помощью бензильной группы (Bz). На второй стадии N-защиту удаляют, например, посредством каталитического гидрирования. На третьей стадии R1 – галогенид с R1, имеющим значения -(CHR3)m – фенил и -(СН2)n – гетероарил, вводят в реакцию с лишенным защиты пиперидином, получая соответствующий 3-алкил-1-арил-4-оксопиперидин. В случае, когда R1 обозначает бензил, эти две последние стадии могут быть пропущены. Превращение 3-алкил-1-арил-4-оксопиперидина в требуемый 4-аминопиперидин может быть проведено через образование оксима взаимодействием с гидроксиламином с последующим восстановлением, например, литийалюминийгидридом (ЛАГ) в соответствующем растворителе. 4-Аминопиперидины формулы I с R1, обозначающим незамещенный или моно-, ди- или тризамещенный фенил, незамещенный или моно-, ди- или тризамещенный нафтил, тетрагидронафтил или С3-7 – циклоалкил, и R2, обозначающим при этом низший алкил, могут быть получены в соответствии со схемой 2: Схема 2
На первой стадии 4-оксопиперидиний йодид образуется из соответствующего N-бензилзащищенного 3-алкил-4-оксопиперидина взаимодействием его с метилйодидом в соответствующем растворителе. R1 вводят реакцией 4-оксопиперидиний йодида с соответствующим анилином R1NH2, в результате чего получается соответствующий 3-алкил-1-арил-4-оксопиперидин. Превращение этого промежуточного соединения в требуемый 4-аминопиперидин может происходить через образование оксима в результате взаимодействия с гидроксиламином с последующим каталитическим гидрированием в присутствии обычного катализатора гидрирования такого, как никель Ренея или палладий на активированном угле, в соответствующем растворителе. 4-Аминопиперидины формулы I с R1, имеющим значения -(CHR3)m – фенил и -(СН2)n – гетероарил, где R3, n и m определены выше, и R2 обозначает незамещенный или моно-, ди- или тризамещенный фенил, незамещенный, или моно-, ди- или тризамещенный нафтил, или незамещенный или моно-, ди- или тризамещенный гетероарил, могут быть получены в соответствии со схемой 3: Схема 3
На первой стадии синтеза эфир акриловой кислоты вводят в реакцию с соответствующим арилалкиламином R1NH2 с получением соответствующего эфира арилалкиламинопропионовой кислоты, который затем превращают в результате взаимодействия с арилуксусной кислотой или галогенангидридом арилуксусной кислоты (R2COOH или R2COHal) в эфир R2 ацетиламинопропионовой кислоты. Последующий цикл взаимодействия, например, с щелочным бутилатом или гидридом натрия, приводит к пиперидин-2, 4-диону, который затем превращается с помощью гидроксиламина в пиперидин-2, 4-дион-4-оксим. Этот интермедиат может быть в конце восстановлен, например, с использованием литийалюминийгидрида в соответствующем растворителе до требуемого 4-аминопиперидина. 4-Аминопиперидины формулы I с R1, обозначающим незамещенный или моно-, ди – или тризамещенный независимо фенил, незамещенный или моно-, ди – или тризамещенный независимо нафтил, тетрагидронафтил или С3-7-циклоалкил, и R2, обозначающим незамещенный или моно-, ди- или тризамещенный независимо фенил, незамещенный или моно-, ди – или тризамещенный независимо нафтил, или незамещенный или моно-, ди – или тризамещенный независимо гетероарил, могут быть получены в соответствии со схемой 4: Схема 4
Синтез 1-арил-3-арил-4-аминопиперидинов может быть проведен либо согласно схеме 2 из N-бензилзащищенного 3-алкил-4-оксопиперидиниййодида, который вводят в реакцию с анилином R1NH2, в результате чего получается соответствующий 3-алкил-1-арил-4-оксопиперидин. Обработка этого соединения гидроксиламином дает соответствующий оксим, который может затем быть восстановлен до требуемого 4-аминопиперидина. Альтернативно N-бензилзащищенный йодид 3-алкил-4-оксопиперидиния на первой стадии вводят в реакцию с ариланилином R1NH2, в результате чего получается соответствующий 1-арил-4-оксопиперидин. Арильная группа может затем быть введена в положение 3 с помощью арилгалогенида R1Ha2 в присутствии ацетата палладия, трет-бутоксида натрия и Р(tBu)3. Образовавшийся 3-арил-1-арил-4-оксопиперидин затем превращают в требуемый 4-аминопиперидин, как описано выше. 4-Аминопиперидины формулы I с R1, имеющим значения -(CHR3)m – фенил и -(СН2)n – гетероарил, где R3, n и m определены выше, и R2 обозначает -С(O)-NR4R5, где R4 и R5 обозначены выше, могут быть получены в соответствии со схемой 5: Схема 5
На первой стадии аминогруппу диэфира 4-аминопиперидин-1,3-дикарбоновой кислоты защищают бензилоксикарбонильной группой (БОК). Эфирную группу в положении 3 затем гидролизуют, после чего следует образование амида с использованием стандартных условий пептидной конденсации. В последующих стадиях БОК-защитную группу в положении 1 удаляют, например, с помощью трифторуксусной кислоты, а затем арилалкильная группа может быть введена при N1 путем обработки соответствующим арилалкилгалогенидом R1Hal. В конце требуемый 4-аминопиперидин получают удалением бензилоксикарбонильной защитной группы в присутствии HBr. 4-Аминопиперидины формулы I с R1, обозначающим незамещенный или моно-, ди- или тризамещенный фенил, незамещенный или моно-, ди- или тризамещенный нафтил, тетрагидронафтил или С3-7-циклоалкил, и R2, обозначающим незамещенный или моно-, ди- или тризамещенный фенил, незамещенный или моно-, ди- или тризамещенный нафтил, или незамещенный или моно-, ди- или тризамещенный гетероарил, могут быть получены в соответствии со схемой 6: Схема 6
Эфир 1-бензил-4-оксопиперидин-3-карбоновой кислоты вначале вводят в реакцию с аммиаком, получая соответствующий тетрагидропиридин, который затем восстанавливают, например, боргидридом натрия с получением эфира 4-аминопиперидинкарбоновой кислоты. БОК-защиту аминогруппы в 4 положении и последущее удаление бензильной группы, например, посредством каталитического гидрирования в присутствии катализатора гидрирования, приводит к 4-N-БОК-защищенному эфиру пиперидин-3-карбоновой кислоты. Арилалкильная группа R1 может быть введена с использованием соответствующего альдегида R1CH=O с последующим восстановлением. Удаление БОК-защитной группы и гидролиз эфира соответствующим гидроксидом щелочного металла приводит к требуемой 1-арилалкил-4-аминопиперидинкарбоновой кислоте. 4-Аминопиперидины формулы I с R1, имеющим значения -(CHR3)m – фенил и -(СН2)n – гетероарил, где R3, n и m определены выше, и R2 обозначает -С(O)-NR4R5, где R4 и R5 обозначены выше, могут быть получены в соответствии со схемой 7: Схема 7
На первой стадии синтеза ариламин R1 вводят в реакцию с эфиром акриловой кислоты в присутствии CuCl и уксусной кислоты, получая соответствующий эфир 1-арил-4-оксопиперидин-3-карбоновой кислоты. Обработка этого промежуточного соединения ацетатом аммония и цианборгидридом натрия дает эфир 1-арил-4-аминопиперидин-3-карбоновой кислоты. БОК-защита свободной аминогруппы в 4 положении и последующий гидролиз приводят к свободной карбоновой кислоте, которая используется затем для конденсации с амином R4R5NH. Требуемый 4-аминопиперидин затем получают удалением БОК-защитной группы, например, с помощью трифторуксусной кислоты. Изобретение относится далее к соединениям формулы I по определению выше, получаемых способом по определению выше. Изобретение относится также к соединениям формулы I по определению выше для применения в качестве терапевтически активных субстанций для лечения и/или профилактики болезней, ассоциируемых с DPP-IV. К таким болезням, которые ассоциируются с DPP-IV, относятся диабет, в частности инсулиннезависимый сахарный диабет, и/или пониженная толерантность к глюкозе, а также другие состояния, где амплификация действия пептида, нормально инактивируемого DPP-IV, приводит к терапевтическому эффекту. Кроме того, соединения по настоящему изобретению могут быть применены также при лечении и/или профилактике ожирения, метаболического синдрома, защите Как описано выше, соединения формулы I по настоящему изобретению могут быть применены в качестве лекарственных средств для лечения и/или профилактики болезней, ассоциируемых с DPP-IV, описанных выше. Изобретение вследствие этого относится также к фармацевтическим композициям, включающим соединение формулы I по настоящему изобретению и фармацевтически приемлемый носитель и/или адъювант. Далее изобретение относится также к соединениям формулы I по настоящему изобретению для применения в качестве терапевтически активных субстанций, в частности в качестве терапевтически активных субстанций для лечения и/или профилактики болезней, ассоциируемых с DPP-IV, описанных выше. В другом варианте изобретение относится к способу лечения и/или профилактики болезней, ассоциируемых с DPP-IV, по определению выше, включающему введение соединения формулы I человеку или животному. Изобретение относится также к применению соединений формулы I по настоящему изобретению для лечения и/или профилактики болезней, ассоциируемых с DPP-IV, по определению выше. В контексте со способами и применением по определению выше предпочтительными являются следующие болезни: диабет, в частности инсулиннезависимый сахарный диабет, пониженная толерантность к глюкозе, ожирение и/или метаболический синдром или защита Для определения активности соединений формулы I были проведены следующие тесты. Активность DPP-IV ингибиторов протестирована на натуральном человеческом DPP-IV, выделенном из пула человеческой плазмы, или с рекомбинантным человеческим DPP-IV. Человеческая цитратная плазма от различных доноров собирается, фильтруется через мембрану толщиной 0,2 мкм в стерильных условиях и аликвоты в 1 мл замораживают и хранят до использования при температуре -120°С. В качестве источника фермента используют в калориметрическом DPP-IV анализе от 5 до 10 мкл человеческой плазмы, а в флюорометрическом анализе используют 1,0 мкл человеческой плазмы с общим анализируемым объемом 100 мкл. Последовательность аминокислот цДНК человеческого DPP-IV от 31 до 766, подвергнутая рестрикции на N-терминальный и трансмембранный домен, клонируют в Pichia pastoris. Человеческую DPP-IV экспрессируют и выделяют из культуральной среды с использованием стандартной колоночной хроматографии, включающей вытеснительную, анионную и катионную хроматографию. Чистота полученного на Coomassie blue SDS-PAGE конечного фермента составляет >95%. В колориметрическом DPP-IV анализе используют в качестве источника фермента 20 нг rec-h DPP-IV, а в флюорометрическом анализе 2 нг rec-h DPP-IV с общим анализируемым объемом 100 мкл. В флюорометрическом анализе Ala-Pro-7-амидо-4-трифторметилкумарин (Calbiochem No 125510) используется в качестве субстрата. 20 мМ исходного раствора в 10% ДМФ/Н2О хранят при температуре -20°С до использования. При определениях IC50 используют конечную концентрацию субстрата 50 мкМ. В анализах по определению кинетических параметров таких, как Km, Vmax, Ki, концентрацию субстрата варьируют от 10 мкМ до 500 мкМ. В колориметрическом анализе в качестве субстрата используют H-Ala-Pro-pNA.HCl (Bachem L-1115). 10 мМ исходного раствора в 10% МеОН/НзО хранят при температуре -20°С до использования. При IC50 определениях используют конечную концентрацию субстрата 200 мкМ. В анализах по определению кинетических параметров таких, как Km, Vmax, Ki, концентрацию субстрата варьируют от 100 мкМ до 2000 мкМ Флюоресценцию детектируют на Perkin Elmer люминесцентном спектрометре LS 50 В при длине волны возбуждения 400 нм и эмиссионной длине волны 505 нм непрерывно каждые 15 с в течение от 10 до 30 мин. Константа скорости возбуждения вычисляется с помощью метода линейной регрессии. Абсорбция pNA, высвобождаемого из колориметрического субстрата, детектируется на Packard SpectraCount при 405 нм непрерывно каждые 2 мин в течение от 30 до 120 мин. Константа скорости возбуждения вычисляется с помощью метода линейной регрессии. Анализ DPP-IV активности осуществляют в 96-ячеистых планшетах при температуре 37°С с общим анализируемым объемом 100 мкл. Буфер для анализа состоит из 50 мМ Tris/HCl, pH 7,8, содержащей 0,1 мг/мл БСА и 100 мМ NaCl. Тестируемые соединения растворяют в 100% ДМСО, разбавляя до необходимой концентрации в 10% ДМСО/Н2О. Конечная концентрация ДМСО при анализе составляет 1% (об./об.). При этой концентрации инактивация фермента ДМСО составляет <5%. Соединения берутся с предварительным инкубированием с ферментом (10 мин при 37°С) и без него. Ферментные реакции начинаются с применением субстрата с последующим немедленным смешением. IC50 значения тестируемых соединений вычисляют с помощью метода нелинейной регрессии DPP-IV ингибирования по крайней мере при 5 различных концентрациях соединения. Кинетические параметры ферментной реакции вычисляют по крайней мере при 5 различных концентрациях субстратов и по крайней мере при 5 различных концентрациях тестируемого соединения. Соединения по настоящему изобретению обладают IC50 величинами от 0,1 мкМ до 50 мкМ, более предпочтительно от 0,1 мкМ до 1 мкМ, как представлено в следующей таблице:
Соединения формулы I и/или их фармацевтически приемлемые соли могут быть использованы в качестве лекарственных средств, например в форме фармацевтических препаратов для энтерального, парентерального или топического введения. Они могут быть введены перорально, например, в форме таблеток, таблеток в оболочке, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий, ректально, например, в форме суппозиториев, парентерально, например, в форме инъекционных растворов или инфузионных растворов, или топически, например, в форме мазей, кремов или масел. Предпочтительным является оральное введение. Приготовление фармацевтических препаратов осуществляют методом, известным любому специалисту в данной области техники, включающим внесение описанных соединений формулы I и/или их фармацевтически приемлемых солей, необязательно в комбинации с другими фармацевтичеки ценными субстанциями, в фармацевтически принятые вносимые формы вместе с подходящими, нетоксичными, инертными терапевтически совместимыми твердыми или жидкими носителями и, если желательно, с обычными фармацевтическими адъювантами. Подходящими носителями могут служить не только неорганические материалы, но также и органические носители. Так, например, лактоза, кукурузный крахмал или их производные, тальк, стеариновая кислота или ее соли могут быть использованы в качестве носителей при изготовлении таблеток, таблеток в оболочке, драже и твердых желатиновых капсул. Подходящими носителями для мягких желатиновых капсул служат, например, растительные масла, воски, жиры и полутвердые и жидкие полиолы (однако вне зависимости от природы активного ингредиента, в случае твердых желатиновых капсул никакие носители могут не потребоваться). Подходящими носителями для изготовления растворов и сиропов являются, например, вода, полиолы, сахароза, инвертный сахар и им подобные. Подходящими носителями для инъекционных растворов, являются, например, вода, спирты, полиолы, глицерин и растительные масла. Подходящими носителями для суппозиториев являются, например, натуральные или гидрированные масла, воски, жиры и полутвердые или жидкие полиолы. Подходящими носителями для топических препаратов являются, например, глицериды, полусинтетические и синтетические глицериды, гидрированные масла, жидкие воска, жидкие парафины, жидкие жирные спирты, стеролы, полиэтиленгликоли и производные целлюлозы. Обычные стабилизаторы, консерванты, смачивающие и эмульсионные агенты, агенты, улучшающие консистенцию, вкусовые агенты, соли для варьирования осмотического давления, буферные субстанции, растворители, красители и маскирующие агенты и антиоксиданты рассматриваются в качестве фармацевтических адъювантов. Дозирование соединения формулы (I) может варьироваться в широких пределах в зависимости от контролируемой болезни, возраста и индивидуального состояния пациента и способа введения, и должно соответствовать, конечно, индивидуальным требованиям в каждом отдельном случае. Для взрослого пациента суточная доза составляет приблизительно от 1 мг до 1000 мг, в частности приблизительно от 1 мг до 100 мг. В зависимости от тяжести заболевания и определенного фармакокинетического профиля соединение может быть введено в виде одной или нескольких дневных единичных доз, например от 1 до 3 единичных доз. Фармацевтические препараты обычно содержат приблизительно 0,1-500 мг, предпочтительно 1-100 мг соединения формулы (I). Следующие примеры более детально иллюстрируют настоящее изобретение. Они, однако, ни в коей мере не лимитируют его объема. Примеры Используемые аббревиатуры: ДХМ: дихлорметан; НОАс: уксусная кислота; AcOEt: этилацетат; ДМФ: диметилформамид; ДИПЭА: диизопропилэтиламин (основание Хюнига); ТГФ: тетрагидрофуран; LAH: литийалюминийгидрид; КДИ: карбонилдиимидазол; ТФК: трифторуксусная кислота; ЭДС1: N-(3-диметиламинопропил)-N’-этилкарбодиимид HCl; ХОБТ: 1-гидроксибензотриазол. КТ: комнатная температура; ВВ: высокий вакуум; ТСХ: тонкослойная хроматография. Пример 1: 3-Бутил-1-фенетилпиперидин-4-иламин Стадия А: 1-Бензил-3-бутил-пиперидин-4-он К суспензии безводного карбоната калия (22,8 г) и этилового эфира 1-бензил-4-оксопиперидин-3-карбоновой кислоты (10 г) в ацетоне (125 мл) добавляют раствор бутилйодида (13,4 г, 8,3 мл) в ацетоне (50 мл) в атмосфере аргона в течение 30 мин. Образовавшуюся суспензию перемешивают при комнатной температуре в течение 30 мин, а затем нагревают с обратным холодильником в течение 12 ч. Суспензию охлаждают, фильтруют и концентрируют в вакууме. Остаток растворяют в ДХМ, раствор промывают водой и рассолом, высушивают над Na2SO4 и выпаривают. К остатку добавляют водный раствор НС1 (20%-ный, 100 мл) и раствор нагревают с обратным холодильником в течение 24 ч. Растворитель выпаривают, а остаток растворяют в ДХМ, промывают 10%-ным водным раствором Na2CO3 и рассолом, высушивают и выпаривают. Сырой продукт очищают с помощью ускоренной хроматографии (этилацетат/гексан в соотношении 1:2), получая продукт в виде слегка желтого масла (6,4 г). МС (ЭСИ): 246,4 (МН). Стадия Б: 3-Бутил-пиперидин-4-он К раствору 1-бензил-3-бутил-пиперидин-4-она (1,3 г) в смеси НОАс/вода в соотношении 3:1 (25 мл) добавляют 10%-ный Pd на угле (130 мг). Водородную атмосферу создают попеременным введением/удалением водорода. Суспензию энергично перемешивают в течение 5 ч. Катализатор удаляют путем фильтрования через дикалит, а фильтрат концентрируют в вакууме. Маслянистый остаток обрабатывают 10%-ным водным раствором Na2CO3 и затем экстрагируют ДХМ. Органический слой отделяют, высушивают над Na2SO4, фильтруют и выпаривают, получая сырой продукт в виде слегка желтого масла (680 мг), который используют без дальнейшей очистки. МС (ЭСИ): 156,2 (МН+). Стадия В: 3-Бутил-1-фенетилпиперидин-4-он К раствору 3-бутилпиперидин-4-она (250 мг) в ДМФ (5 мл) добавляют ДИПЭА (468 мг), а затем раствор (2-бромэтил)бензола (373 мг) в ДМФ (5 мл) в течение 45 мин. Образовавшуюся смесь перемешивают при комнатной температуре в течение 1 ч, а затем нагревают до 60°С в течение 6 ч. Реакционную смесь охлаждают, разбавляют эфиром и органический раствор дважды промывают 10%-ным водным раствором Na2CO3 и рассолом. Органический слой высушивают над MgSO4, фильтруют и выпаривают, получая остаток, который очищают с помощью ускоренной хроматографии (этилацетат/гексан в соотношении 1:3), получая продукт в виде бесцветной жидкости (355 мг). МС (ЭСИ): 260,4 (МН+). Стадия Г: Оксим 3-бутил-1-фенетилпиперидин-4-она 3-Бутил-1-фенетилпиперидин-4-он (300 мг), NaOAc (878 мг) и гидрохлорид гидроксиламина (708 мг) суспендируют в смеси этанол/вода в соотношении 1:1 (10 мл), нагревают смесь с обратным холодильником в течение 5 ч. Полученный после этой обработки прозрачный раствор охлаждают, разбавляют водой и подщелачивают 10%-ным водным раствором Na2CO3 до рН 10. Суспензию экстрагируют ДХМ и органический слой промывают рассолом, высушивают над MgSO4 и выпаривают. Продукт тщательно очищают для использования на следующей стадии, получая желтоватую стекловидную смесь цис- и транс-диастереомеров (332 мг). МС (ЭСИ): 261,4 (МН+). Стадия Д: 3-Бутил-1-фенетилпиперидин-4-иламин Оксим 3-бутил-1-фенетилпиперидин-4-она (218 мг) растворяют в ТГФ (10 мл) и добавляют одной порцией LAH (261 мг). Суспензию перемешивают затем при комнатной температуре в течение 24 ч. Реакционную смесь осторожно с помощью пипетки переносят в 5%-ный водный раствор NaHCO3 и водный слой экстрагируют ДХМ. Органический слой промывают рассолом, высушивают над MgSO4 и выпаривают, получая бесцветное масло, которое очищают с помощью ускоренной хроматографии (градиент МеОН в ДХМ, содержащий 1% NH4OH), получая продукт в виде цис- и транс-диастереомеров (цис: 127 мг; транс: 36 мг). MC (ЭСИ): 261,4(MH+). Пример 2: 3-Бутил-1-бензилпиперидин-4-иламин Данное соединение было синтезировано аналогично примеру 1, за исключением того, что стадии Б и В были целиком исключены, а бензильная группа участвовала во всем синтезе, в результате чего были получены цис- и транс-диастереомеры в виде бесцветных масел. МС (ЭСИ): 247,4 (МН+). Пример 3: 3-Бутил-1-[2-(1Н-индол-3-ил)этил]пиперидин-4-иламин Данное соединение было синтезировано, как описано в примере 1, из этилового эфира 1-бензил-4-оксопиперидин-3-карбоновой кислоты и 3-(2-бромэтил)-1Н-индола в качестве алкилирующего агента на стадии В, в результате чего были получены цис- и транс-диастереомеры в виде бесцветных масел. МС (ЭСИ): 300,5 (МН+). Пример 4: 3-Бутил-1-[2-(3,4-диметоксифенил-1-ил)этил]пиперидин-4-иламин Данное соединение было синтезировано, как описано в примере 1, из этилового эфира 1-бензил-4-оксопиперидин-3-карбоновой кислоты и (2-бромэтил)-3,4-диметоксибензола в качестве алкилирующего агента на стадии В, в результате чего были получены цис- и транс-диастереомеры в виде бесцветных масел. МС (ЭСИ): 321,4 (МН+). Пример 5: Гидрохлорид 3-бутил-1-(3,4,5-триметоксифенил)пиперидин-4-иламина Стадия А: 1-Бензил-3-бутил-1-метил-4-оксопиперидиний йодид К раствору 3-бутил-1-бензилпиперидин-4-она (9080 мг) в ацетоне (40 мл) медленно добавляют метилйодид (6303 мг) при комнатной температуре, а затем раствор перемешивают при комнатной температуре в течение 3 ч. Выпавшее в осадок белое твердое вещество отфильтровывают, а затем четыре раза промывают 50 мл ацетона и высушивают при пониженном давлении. Фильтрат выпаривают, а остаток перемешивают с этилацетатом. Белое твердое вещество фильтруют, промывают этилацетатом и высушивают в вакууме. Два твердых вещества объединяют, получая 1-бензил-3-бутил-1-метил-4-оксопиперидиний йодид в виде белого порошка (11200 мг). МС (ЭСИ): 204,2 (М-Г). Стадия Б: 3-Бутил-1-(3,4,5-триметоксифенил)пиперидин-4-он Суспензию 1-бензил-3-бутил-1-метил-4-оксопиперидиний йодида (306 мг) в воде (2 мл) добавляют одной порцией к нагреваемому с обратным холодильником раствору 3, 4, 5-триметоксианилина (929 мг) и безводного карбоната калия (33 мг) в этаноле (4 мл). Темный раствор нагревают с обратным холодильником в течение 3 ч, добавляют воду и дважды экстрагируют реакционную смесь ДХМ. Объединенные органические слои высушивают над сульфатом натрия, фильтруют и выпаривают растворитель, получая темное масло, которое очищают с помощью ускоренной хроматографии (этилацетат/гексан в соотношении 1:1), получая продукт в виде бесцветного масла (520 мг). МС (ЭСИ): 322,4 (МН+). Стадия В: Гидрохлорид 3-бутил-1-(3,4,5-триметоксифенил)пиперидин-4-иламина Кетон, полученный на предыдущей стадии (460 мг), растворяют в этаноле (60 мл), добавляют гидрохлорид гидроксиламина (109 мг) и ацетат натрия (129 мг), после чего раствор перемешивают при комнатной температуре в течении е 1 ч. После контроля с помощью ТСХ добавляют никель Ренея (Nr 313 Degussa В 1132) и перемешивают реакционную смесь при комнатной температуре в атмосфере водорода в течение ночи. Катализатор отфильтровывают, а фильтрат концентрируют. Остаток очищают с помощью хроматографии на силикагеле (ДХМ/МеОН/25%-ный водный раствор NH4OH в соотношении 95:5:1). Выделенные продукты растворяют в этаноле и добавляют 1 мл насыщенного этанольного раствора хлористого водорода. Растворы выпаривают, получая цис- (150 мг) и транс-диастереомеры (160 мг) в виде слегка желтоватых твердых веществ. МС (ЭСИ): 323,4 (МН+). Пример 6: Гидрохлорид 3-бутил-1-(4-феноксифенил)пиперидин-4-иламина Данное соединение было синтезировано аналогично примеру 5 с использованием стадиий Б и В из 1-бензил-3-бутил-1-метил-4-оксопиперидиний йодида и 4-феноксианилина, за исключением того, что стадия В была заменена другим методом. Стадия В (модифицированная версия)]: гидрохлорид 3-бутил-1-(4-феноксифенил)пиперидин-4-иламина 3-Бутил-1-(4-феноксифенил)пиперидин-4-он (320 мг) растворяют в этаноле (8 мл) и добавляют гидрохлорид гидроксиламина (76 мг) и ацетат натрия (89 мг). Реакционная смесь приобретает желтую окраску и ее перемешивают при комнатной температуре в течение 2 ч. Контроль с помощью ТСХ указывает на образование E/Z-оксимов. После добавления воды (8 мл) образуется суспензия, к которой добавляют Al-Ni-сплав (300 мг), а затем медленно добавляют 32%-ный водный раствор гидроксида натрия (1,4 мл), при этом наблюдается разогревание реакционной смеси. После окончательного добавления реакционную смесь перемешивают в течение 2 ч при комнатной температуре и отфильтровывают твердое вещество. Осадок промывают ДХМ. Водный раствор экстрагируют ДХМ. Объединенные органические слои промывают рассолом, высушивают над сульфатом натрия, фильтруют, а растворитель выпаривают. Остаток очищают с помощью ускоренной хроматографии (ДХМ/МеОН/насыщ. водн. раствор NH4OH в соотношении 100:5:1). Выделенные продукты растворяют в этаноле и добавляют 1 мл насыщенного этанольного раствора хлористого водорода. Растворы выпаривают, получая цис- (117 мг) и транс-диастереомеры (52 мг), каждый в виде слегка желтоватого твердого вещества. МС (ЭСИ): 325,5 (МН+) транс- и цис-диастереомеры. Пример 7: Гидрохлорид 3-бутил-1-(5,6,7,8-тетрагидронафталин-1-ил)пиперидин-4-иламина Данное соединение было синтезировано аналогично примеру 6 из 1-бензил-3-бутил-1-метил-4-оксопиперидиний йодида и 5-аминотетралина с образованием смеси цис- и транс-диастереомеров в виде белого твердого вещества. Попытка разделить диастереомеры (в виде свободных аминов) с использованием ускоренной хроматографии не удалась. МС (ЭСИ): 287,3 (МН+). Пример 8: Гидрохлорид 3-бутил-1-(3,4-диметоксифенил)пиперидин-4-иламина Данное соединение было синтезировано аналогично примеру 6 из 1-бензил-3-бутил-1-метил-4-оксопиперидиний йодида и 3,4-диметоксианилина с образованием цис- и транс-диастереомеров в виде белых твердых веществ. МС (ЭСИ): 293.4 (МН) цис- и транс-диастереомеры. Пример 9: Гидрохлорид 3-бутил-1-нафталин-2-илпиперидин-4-иламина Данное соединение было синтезировано аналогично примеру 6 из 1-бензил-3-бутил-1-метил-4-оксопиперидиний йодида и 2-нафтиламина с образованием смеси цис- и транс-диастереомеров в соотношении 5:2 и чистого транс-диастереомера в виде белого твердого вещества. МС (ЭСИ): 283,2 (МН+) цис- и транс-диастереомеры. Пример 10: Гидрохлорид 3-бутил-1-нафталин-1-илпиперидин-4-иламина Данное соединение было синтезировано аналогично примеру 6 из 1-бензил-3-бутил-1-метил-4-оксопиперидиний йодида и 1-нафтиламина с образованием смеси цис- и транс-диастереомеров в соотношении 1:2 в виде белого твердого вещества. Попытка разделить диастереомеры (в виде свободных аминов) с использованием ускоренной хроматографии не удалась. МС (ЭСИ): 283,2 (МН+). Пример 11: Гидрохлорид 3-бутил-1-(3,4-дихлорфенил)пиперидин-4-иламина Данное соединение было синтезировано аналогично примеру 6 из 1-бензил-3-бутил-1-метил-4-оксопиперидиний йодида и 3,4-дихлоранилина с образованием цис-диастереомера в виде белого твердого вещества. МС (ЭСИ): 301,3 (МН+). Пример 12: Гидрохлорид 3-бутил-1-(4-хлор-3-трифторметилфенил)пиперидин-4-иламина Данное соединение было синтезировано аналогично примеру 6 из 1-бензил-3-бутил-1-метил-4-оксопиперидиний йодида и 4-хлор-3-трифторметиланилина с образованием цис-диастереомера и смеси цис- и транс-диастереомеров в соотношении 1:1 в виде белых твердых веществ. МС (ЭСИ): 335,3 (МН+) цис- и транс-диастереомеры. Пример 13: Гидрохлорид 3-бутил-1-п-толил-пиперидин-4-иламина Данное соединение было синтезировано аналогично примеру 6 из 1-бензил-3-бутил-1-метил-4-оксопиперидиний йодида и 4-метиланилина с образованием цис- и транс-диастереомеров в виде белых твердых веществ. МС (ЭСИ): 247,4 (МН+). Пример 14: Гидрохлорид 3-бутил-1-(3,5-дихлорфенил)пиперидин-4-иламина и гидрохлорид 3-бутил-1-фенилпиперидин-4-иламина Данное соединение было синтезировано аналогично примеру 6 из 1-бензил-3-бутил-1-метил-4-оксопиперидиний йодида и 3,5-дихлор-4-метиланилина с образованием цис-диастереомера и смеси цис- и транс-диастереомеров в соотношении 1:1 в виде твердых белых веществ. МС (ЭСИ): 301,3 (МН+). Добавочное восстановление приводит к образованию дегалогенированных цис- и транс-фенилпроизводных, которые были разделены в виде аминов с помощью ускоренной хроматографии. Гидрохлориды цис-и транс-диастереомеров были выделены в виде белых твердых веществ. МС (ЭСИ): 233,3 (МН+). Пример 15: Гидрохлорид 3-бутил-1-(3-метокси-5-трифторметилфенил)пиперидин-4-иламина Данное соединение было синтезировано аналогично примеру 6 из 1-бензил-3-бутил-1-метил-4-оксопиперидиний йодида и 3-метокси-5-трифторметиланилина с образованием смеси цис- и транс-диастереомеров и цис-диастереомера в виде белых твердых веществ. МС (ЭСИ): 331,4 (МН+). Пример 16: Гидрохлорид 3-бутил-1-циклогексилпиперидин-4-иламина Данное соединение было синтезировано аналогично примеру 6 из 1-бензил-3-бутил-1-метил-4-оксопиперидиний йодида и циклогексиламина с образованием смеси цис- и транс-диастереомеров в виде белого твердого вещества. 1Н ЯМР (ДМСО): Пример 17: Гидрохлорид 1-(3.5-бис-трифторметилфенил)-3-бутилпиперидин-4-иламина Данное соединение было синтезировано аналогично примеру 6 из 1-бензил-3-бутил-1-метил-4-оксопиперидиний йодида и 3,5-бис-(трифторметил)анилина с образованием смеси цис- и транс-диастереомеров в виде белого твердого вещества. МС (ЭСИ): 369,3 (МН+). Пример 18: Гидрохлорид 3-бутил-1-(6-метоксибифенил-3-ил)пиперидин-4-иламина Данное соединение было синтезировано аналогично примеру 6 из 1-бензил-3-бутил-1-метил-4-оксопиперидиний йодида и 6-метоксибифенил-3-иламина с образованием цис- и транс-диастереомеров в виде белых твердых веществ. 1Н ЯМР (ДМСО, цис-диастереомер): Пример 19: Гидрохлорид 1-бензгидрил-3-бутилпиперидин-4-иламина Данное соединение было синтезировано аналогично примеру 6 из (рац)-1-бензил-3-бутил-1-метил-4-оксопиперидиний йодида и дифенилметиламина как смесь цис/транс-диастереомеров в соотношении 1:1 в виде белого твердого вещества. 1Н ЯМР (ДМСО): Пример 20: 3-Фенил-1-фенетилпиперидин-4-иламин Стадия А: Этиловый эфир 3-фенетиламинопропионовой кислоты Фенилэтиламин (10 г) растворяют в EtOH (50 мл) и затем по каплям прибавляют этиловый эфир акриловой кислоты (8,3 г) в атмосфере аргона при комнатной температуре. Образовавшуюся смесь перемешивают в течение ночи и после выпаривания растворителя высушивают в вакууме. Остаток в виде бесцветной жидкости (18,9 г) используют без дальнейшей очистки. МС (ЭСИ): 222,3 (МН+). Стадия Б: Этиловый эфир 3-(фенетилфенилацетиламино)пропионовой кислоты Этиловый эфир 3-фенетиламинопропионовой кислоты (8,0 г) растворяют в абсолютном пиридине (12 мл) и охлаждают до 0°С на ледяной бане. Хлорангидрид фенилуксусной кислоты (1,5 мл) добавляют затем по каплям в течение 10 мин, получая желтую суспензию, которую затем нагревают до 60°С в течение 2,5 ч, охлаждают до комнатной температуры, после чего перемешивают в течение ночи. Реакционную смесь переносят затем в ледяную воду, содержащую 25% HCl, и водный слой экстрагируют этилацетатом. Органический слой отделяют, промывают рассолом, высушивают над Na2SO4 и выпаривают. Остаток очищают с помощью ускоренной хроматографии (этилацетат/гексан в соотношении 1:4), получая продукт в виде желтого масла (3,9 г). МС (ЭСИ): 340,4 (МН+). Стадия В: 1-Фенетил-3-фенилпиперидин-2.4-дион Гидрид натрия (1,1 г, 50%-ный раствор в минеральном масле) суспендируют в эфире (40 мл) в атмосфере аргона при КТ. Этиловый эфир 3-(фенетилфенилацетил-амино)пропионовой кислоты (2,5 г) добавляют по частям с последующим добавлением абс. EtOH (0,5 мл). Образовавшуюся смесь нагревают с обратным холодильником в течение 2 ч, охлаждают и переносят в смесь вода/лед/1-нормальная HCl. Водный слой экстрагируют этилацетатом, органический слой отделяют, промывают рассолом, высушивают над Na2SO4, фильтруют и выпаривают. Остаток очищают с помощью ускоренной хроматографии (этилацетат/гексан в соотношении 1:1, а затем ДХМ/МеОН в соотношении 95:5), получая заданный продукт в виде слегка желтоватой пены (801 мг). МС (ЭСИ: 294,4 (МН+). Стадия Г: 1-Фенетил-3-фенилпиперидин-2.4-дион-4-оксим 1-Фенетил-3-фенилпиперидин-2,4-дион (781 мг), ацетат натрия (1,15 г) и гидрохлорид гидроксиламина (925 мг) суспендируют в смеси EtOH/вода (в соотношении 1:1, 25 мл) и нагревают суспензию с обратным холодильником в течение 4 ч. Затем реакционную смесь переносят в смесь лед/вода/1-нормальный раствор NaOH и экстрагируют этилацетатом. Органический слой промывают рассолом, высушивают и выпаривают, получая сырой продукт, который очищают с помощью ускоренной хроматографии ДХМ/MeOH/NH4OH в соотношении 98:2:0.25), получая соединение в виде белой пены (489 мг). МС (ЭСИ): 309,4 (МН+). Стадия Д: 1-Фенетил-3-фенилпиперидин-4-иламин 1-Фенетил-3-фенилпиперидин-2,4-дион-4-оксим (480 мг) растворяют в абсолютном эфире (30 мл) в атмосфере аргона. LAH (473 мг) добавляют одной порцией, и образовавшуюся суспензию нагревают с обратным холодильником в течение ночи. Смесь осторожно переносят в 1-молярный водный раствор Na/K-соли винной кислоты, и водный слой экстрагируют этилацетатом. Органический слой промывают рассолом, высушивают (Na2SO4), фильтруют и выпаривают. Остаток очищают с помощью ускоренной хроматографии (градиент МеОН в ДХМ, содержащий 0,25% NH4OH, а затем ДХМ/МеОН/NH4OH в соотношении 85/15/0.25), получая соединение, состоящее из цис-диастереомера (95 мг) и транс-диастереомера (101 мг), в виде бесцветного масла. МС (ЭСИ): 281,4 (МН+). Пример 21: 3-Фенил-1-бензилпиперидин-4-иламин Данное соединение получено согласно примеру 20 (стадии Б-Д) из этилового эфира 3-бензиламинопропионовой кислоты и хлорангидрида фенилуксусной кислоты с образованием цис- и транс-диастереомеров в виде желтого масла. МС (ЭСИ): 326,4 (МН+). Пример 22: 4-Метил-1′-фенетил-1′,2′,3′,4′,5′,6′-гексагидро[2,3′]бипиридинил-4′-иламин Данное соединение получено согласно методике примера 20 (стадии А-Д) из фенетиламина и этилацетата, но с применением модифицированной конденсирующей стадии Б, где использована (4-метилпиридин-2-ил)уксусная кислота вместо ее хлорангидрида. Стадия А: (4-Метилпиридин-2-ил)уксусная кислота Этиловый эфир (4-метилпиридин-2-ил)уксусной кислоты (1,0 г), полученный согласно Chem. Farm. Bull. 32 (12), 1984, 4866-4872, растворяют в этаноле (30 мл) и обрабатывают 1-молярным этанольным раствором NaOH (5,83 мл). Смесь нагревают с обратным холодильником в течение 4 ч, охлаждают и выпаривают в вакууме. Остаток растворяют в воде (50 мл) и доводят значение рН до 3,0 с помощью 1-молярного раствора HCl. Растворитель выпаривают, а остаток суспендируют в этаноле (150 мл) и фильтруют. Чистый фильтрат выпаривают до сухого состояния, а остаток досушивают в вакууме, получая слегка желтое твердое вещество (1,0 г). ЯМР (ДМСО-d6): 8,65 (d, 1H); 7,72 (s, 1H), 7,68 (d, 1H), 4,04 (s, 2H), 2,49 (s, 3Н). Стадия Б: Этиловый эфир 3-{[2-(4-метилпиридин-2-ил)ацетил]-фенетиламино}пропионовой кислоты (4-Метилпиридин-2-ил)уксусная кислота (1,0 г), карбонилдиимидазол (1,07 г) и ДИПЭА (0,856 г), добавленные к ТГФ (15 мл), образуют коричневатую суспензию, которую перемешивают при комнатной температуре в течение 1 ч. К смеси по каплям добавляют этиловый эфир 3-фенетиламинопропионовой кислоты (0,898 г) и образовавшуюся смесь перемешивают при 55°С в течение ночи. Смесь переносят затем в лед с водой, доводят значение рН до 5,0 и экстрагируют этилацетатом. Органический слой отделяют, промывают рассолом, высушивают над Na2SO4, фильтруют и выпаривают. Остаток очищают с помощью ускоренной хроматографии (этилацетат/гексан в соотношении 2:1), получая заданный продукт в виде слегка коричневой жидкости (0,462 г). МС (ЭСИ): 355,0 (МН+). Конечный продукт этого синтеза в виде желтого масла представляет собой смесь цис- и транс-диастереомеров. МС (ЭСИ): 296,4 (МН+). Пример 23: 3-(3-Хлорфенил)-1-фенетилпиперидин-4-иламин Данное соединение получено согласно примеру 22 из фенетиламина, этилакрилата и (3-хлорфенил)уксусной кислоты. Полученное соединение в виде желтого масла представляет собой цис-диастереомер. МС (ЭСИ): 315,1 (МН+). Пример 24: 3-(3-Хлорфенил)-1-бензилпиперидин-4-иламин Данное соединение получено согласно примеру 20 (стадии Б-Д) из этилового эфира 3-бензиламинопропионовой кислоты и хлорангидрида (3-хлорфенил)уксусной кислоты. Полученное соединение в виде желтого масла представляет собой смесь цис- и транс-диастереомеров. МС (ЭСИ): 301,2 (МН+). Пример 25: 3-(3-Метилфенил)-1-бензилпиперидин-4-иламин Данное соединение получено согласно примеру 20 (стадии Б-Д) из этилового эфира 3-бензиламинопропионовой кислоты и хлорангидрида (3-метилфенил)уксусной кислоты. Полученное соединение в виде желтого масла представляет собой смесь цис- и транс-диастереомеров. МС (ЭСИ): 281,3 (МН+). Пример 26: 3-(3-Хлорфенил)-1-[2-(3,4-диметоксифенил)этил]пиперидин-4-иламин Данное соединение получено согласно примеру 20 из этилакрилата, 2-(3, 4-диметоксифенил)этиламина и хлорангидрида (3-хлорфенил) уксусной кислоты. Полученное соединение в виде желтого масла представляет собой смесь цис- и транс-диастереомеров. МС (ЭСИ): 375.2 (МН+). Пример 27: 1-Бензил-3-тиофен-2-илпиперидин-4-иламин Данное соединение получено аналогично примеру 20 (стадии Б-Д) из этилового эфира 3-бензиламинопропионовой кислоты и тиофен-2-ацетилхлорида. Полученное соединение в виде желтого масла представляет собой транс-диастереомер МС (ЭСИ): 273,2 (МН+). Пример 28: Гидрохлорид 3-о-толил-1-(3,4,5-триметоксифенил)пиперидин-4-иламин Стадия А: 1-Бензил-1-метил-4-оксо-3-о-толилпиперидиний йодид К раствору 1-бензил-3-о-толилпиперидин-4-она (5165 мг) в ацетоне (25 мл) медленно добавляют метилйодид (3048 мг) при комнатной температуре. Раствор перемешивают при комнатной температуре в течение ночи. Выпавшее в осадок белое твердое вещество отфильтровывают, четыре раза промывают 50 мл ацетона и высушивают при пониженном давлении. Фильтрат выпаривают, а остаток перемешивают с этилацетатом. Белое твердое вещество отфильтровывают, промывают этилацетатом и высушивают в вакууме. Оба полученных твердых вещества объединяют, получая 1-бензил-3-бутил-1-метил-4-оксопиперидиний йодид в виде желтоватого твердого вещества (4400 мг). МС (ЭСИ): 294,3 (М-I–). Стадия Б: 3-о-толил-1-(3,4,5-триметоксифенил)пиперидин-4-он Суспензию 1-бензил-1-метил-4-оксо-3-о-толилпиперидиний йодида (1000 мг) в воде (5 мл) добавляют целиком к нагреваемому с обратным холодильником раствору 3,4,5-триметоксианилина (395 мг) и безводного карбоната калия (37 мг) в этаноле (10 мл). Реакционную смесь нагревают с обратным холодильником в течение ночи, затем добавляют воду и четыре раза экстрагируют ДХМ. Объединенные органические фазы высушивают над сульфатом натрия, фильтруют, после чего выпаривают растворитель, получая красно-коричневое масло, которое очищают с помощью ускоренной хроматографии (диэтиловый эфир), получая продукт в виде желтого масла (600 мг). МС (ЭСИ): 356,2 (МН+). Стадия В: Гидрохлорид 3-о-толил-1-(3,4,5-триметоксифенил)пиперидин-4-иламина 3-о-Толил-1-(3,4,5-триметоксифенил)пиперидин-4-он (300 мг) растворяют в этаноле (8 мл). После добавления гидрохлорида гидроксиламина (64 мг) и ацетата натрия (76 мг) желтый раствор превращается в коричневую суспензию. Затем в течение трех часов перемешивают смесь при комнатной температуре и добавляют воду (8 мл), в результате чего образуется суспензия, в которую вносят Al-Ni-сплав (300 мг), а затем медленно добавляют 32%-ный водный раствор гидроксида натрия (1,4 мл), при этом наблюдается нагревание реакционной смеси. После окончательного добавления реакционную смесь перемешивают в течение 2 дней при комнатной температуре, а затем отфильтровывают твердое вещество. Осадок промывают ДХМ. Водный раствор экстрагируют ДХМ. Объединенные органические слои промывают рассолом, высушивают над сульфатом натрия, фильтруют, а растворитель выпаривают. Остаток очищают с помощью ускоренной хроматографии (ДХМ/МеОН/насыщ. водн. раствор NH3 в соотношении 100:5:1). Выделенные продукты растворяют в этаноле и добавляют 1 мл насыщенного этанольного раствора хлористого водорода. Растворы выпаривают, получая цис-диастереомер (76 мг), смесь цис- и транс-диастереомеров (170 мг) и транс-диастереомер (40 мг), каждый в виде белого твердого вещества. МС (ЭСИ): 357,3 (МН+). Пример 29: Гидрохлорид 1-(3,4-диметоксифенил)-3-м-толилпиперидин-4-иламин Стадия А: 1-(3, 4-Диметоксифенил)пиперидин-4-он Суспензию 1-бензил-1-метил-4-оксопиперидиний йодида (9270 мг), 3, 4-диметоксианилина (3900 мг) и безводного карбоната калия (437 мг) в смеси этанол (90 мл)/вода (45 мл) нагревают с обратным холодильником в течение 6 ч. Затем дополнительно добавляют карбонат калия (200 мг) и реакционную смесь нагревают при 100°С в течение ночи. К реакционной смеси добавляют воду (50 мл) и трижды экстрагируют ДХМ. Объединенные органические фазы промывают водой и рассолом, высушивают над сульфатом натрия, фильтруют, а затем удаляют растворитель в вакууме, получая сырой продукт в виде темного масла. Очистка с помощью колоночной хроматографии (этилацетат/гексан в соотношении 1:1) дает продукт в виде желтого твердого вещества (3700 мг). МС (ЭСИ): 236,1 (МН+). Стадия Б: 1-(3.4-Диметоксифенил)-3-м-толилпиперидин-4-он Ацетат палладия (23,8 мг), трет-бутоксид, натрия (306 мг) и 1-(3,4-диметоксифенил)пиперидин-4-он растворяют в свободном от кислорода тетрагидрофуране (3 мл) в атмосфере аргона. Смесь затем немедленно дегазируют. После добавления 3-бромтолуола (363 мг) и три(трет-бутил)фосфина (25,8 мг) смесь перемешивают при 50°С в течение ночи. Так как при этом остается еще некоторое количество исходных веществ, реакционную смесь дополнительно нагревают до 70°С в течение 2 ч. После охлаждения смесь разбавляют этилацетатом, промывают 1 – нормальным водным раствором хлористого водорода, водой и рассолом. Органический слой высушивают над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают с помощью колоночной хроматографии (гексан/этилацетат в соотношении 2:1), получая продукт в виде желтого масла (186 мг). МС (ЭСИ): 326,3 (МН+). Стадия В: Гидрохлорид 1-(3, 4-диметоксифенил)-3-м-толилпиперидин-4-иламина 1-(3,4-Диметоксифенил)-3-м-толилпиперидин-4-он (169 мг) растворяют в этаноле (4 мл). После добавления гидрохлорида гидроксиламина (40 мг) и ацетата натрия (47 мг) прозрачный желтый раствор превращается в суспензию. Затем в течение трех часов перемешивают смесь при комнатной температуре и добавляют воду (4 мл), в результате чего образуется суспензия, в которую вносят Al-Ni-сплав (150 мг), а затем медленно добавляют 32%-ный водный раствор гидроксида натрия (0,7 мл), при этом наблюдается нагревание реакционной смеси. После окончательного добавления реакционную смесь перемешивают в течение 2 дней при комнатной температуре, а затем отфильтровывают твердое вещество. Осадок промывают ДХМ. Водный раствор экстрагируют ДХМ. Объединенные органические слои промывают рассолом, высушивают над сульфатом натрия, фильтруют, а растворитель выпаривают. Остаток очищают с помощью ускоренной хроматографии (ДХМ/МеОН/насыщ. водн. раствор NH3 в соотношении 100:5:1). Выделенные продукты растворяют в этаноле и добавляют 1 мл насыщенного этанольного раствора хлористого водорода. Растворы выпаривают, получая цис-диастереомер (15 мг), смесь транс- и цис-диастереомеров (144 мг) и транс-диастереомер (6 мг), каждый в виде белого твердого вещества. МС (ЭСИ): 327,3 (МН). Пример 30: Гидрохлорид 1-(3,4-диметоксифенил)-3-п-толилпиперидин-4-иламина Данное соединение было синтезировано аналогично примеру 29 (стадии Б и В) из 1-(3,4-диметоксифенил)пиперидин-4-она и 4-бромтолуола в виде белого твердого вещества, состоящего из смеси цис- и транс-диастереомеров. На стадии Б арилбромид и три(трет-бутил)фосфин добавляют в виде раствора в тетрагидрофуране и нагревают при 70°С в течение 5 ч. Разделение диастереомеров (в виде свободных оснований) не удается провести с помощью ускоренной хроматографии. МС (ЭСИ): 327,3 (МН+). Пример 31: Гидрохлорид 1-(3,4-диметоксифенил)-3-(3,4-диметилфенил) пиперидин-4-иламина Данное соединение было синтезировано аналогично примеру 30 из 1-(3,4-диметоксифенил)пиперидин-4-она и 4-бром-о-ксилола в виде белого твердого вещества, состоящего из смеси цис- и транс-диастереомеров. Разделение диастереомеров (в виде свободных оснований) не удается провести с помощью ускоренной хроматографии. МС (ЭСИ): 341,3 (МН+). Пример 32: Гидрохлорид 1-(3,4-диметоксифенил)-3-(3-метоксифенил)пиперидин-4-иламина Данное соединение было синтезировано аналогично примеру 30 из 1-(3,4-диметоксифенил)пиперидин-4-она и 4-броманизола в виде белого твердого вещества, состоящего из смеси цис- и транс-диастереомеров. Разделение диастереомеров (в виде свободных оснований) не удается провести с помощью ускоренной хроматографии. МС (ЭСИ): 343,3 (МН+). Пример 33: 1′-(3,4-диметоксифенил)-1′,2′,3′,4′,5′,6′-гексагидро[2,3′]бипиридинил-4′-иламин Данное соединение было синтезировано аналогично примеру 30 из 1-(3,4-диметоксифенил)пиперидин-4-она и 2-бромпиридина в виде белого твердого вещества, состоящего из смеси цис- и транс-диастереомеров. Разделение диастереомеров не удается провести с помощью ускоренной хроматографии. МС (ЭСИ): 314,3 (МН+). Пример 34: ((3R,4S)-4-Амино-1-фенетилпиперидин-3-ил)тиазолидин-3-илметанон Стадия А: 1-трет-Бутиловый эфир/3-метиловый эфир (3R,4S)-4-бензилоксикарбониламинопиперидин-1,3-дикарбоновой кислоты 1-трет-Бутиловый эфир/3-метиловый эфир (3R,4S)-4-аминопиперидин-1, 3-дикарбоновой кислоты (540 мг, синтезированный согласно Duan, Jingwu et al, PCT Int. Appl. (2001), WO 2001070673 A2 и WO 2002002525) растворяют в абс. ДХМ и добавляют NEt3 (0,39 мл), а затем бензилхлорформиат (0,33 мл) и смесь оставляют при перемешивании при комнатной температуре в течение ночи. Затем реакционную смесь переносят в смесь лед/рассол, и водный слой экстрагируют этилацетатом. Органический слой промывают рассолом, высушивают и выпаривают, а остаток очищают с помощью ускоренной хроматографии (градиент этилацетата в гептане), получая бесцветную смолу (598 мг). МС (ЭСИ): 393,2 (МН+). Стадия Б: 1 трет-Бутиловый эфир (3R,4S)-4-метоксикарбониламинопиперидин-1,3-дикарбоновой кислоты 1-трет-Бутиловый эфир/3-метиловый эфир (3R,4S)-4-бензилоксикарбониламинопиперидин-1,3-дикарбоновой кислоты (580 мг) растворяют в ТГФ (20 мл) и добавляют 1-молярный водный раствор LiOH (3,0 мл). Смесь оставляют при перемешивании при комнатной температуре в течение 20 ч. Затем реакционную смесь переносят в смесь лед/рассол, содержащую 2-молярный раствор HCl (2,5 мл). Водный слой экстрагируют этилацетатом, органический слой отделяют, промывают рассолом, высушивают (Na2SO4) и выпаривают. Остаток высушивают в высоком вакууме, получая бесцветную пену (558 мг). МС (ЭСИ): 377,3 (МН+). Стадия В: трет-Бутиловый эфир (3R,4S)-4-бензилоксикарбониламино-3-(тиазолидин-3-карбонил)пиперидин-1-карбоновой кислоты 1-трет-Бутиловый эфир (3R,4S)-4-метоксикарбониламинопиперидин-1,3-дикарбоновой кислоты (557 мг) растворяют в абс. ДХМ (20 мл). К данному раствору последовательно добавляют (бензотриазол-1-илокси)-трипирролидинфосфоний-гексафторфосфат (919 мг), триэтиламин (0,47 мл) и через 5 мин тиазолидин (151 мг). Смесь оставляют при перемешивании при комнатной температуре в течение 5 ч. Затем реакционную смесь переносят в смесь лед/рассол, и водный слой экстрагируют этилацетатом. Органический слой промывают рассолом, высушивают (Na2SO4) и выпаривают. Остаток очищают с помощью ускоренной хроматографии (градиент этилацетата в гептане), получая продукт в виде белой пены (403 мг). МС (ЭСИ): 450,3 (МН+). Стадия Г: Бензиловый эфир (3R,4S)-[3-(тиазолидин-3-карбонил)пиперидин-4-ил]карбаминовой кислоты 1-трет-Бутиловый эфир (3R,4S)-4-бензилоксикарбониламино-3-(тиазолидин-3-карбонил)пиперидин-1-карбоновой кислоты (325 мг) растворяют в абс. ДХМ (12 мл) и по каплям добавляют ТФК (0,44 мл). Смесь оставляют при перемешивании при комнатной температуре в течение ночи, а затем вакуумируют. Остаток переносят в смесь лед/рассол, подщелачивают до рН 10 с помощью 2-нормального раствора NaOH и экстрагируют этилацетатом. Органический слой промывают рассолом, высушивают (Na2SO4) и выпаривают. Остаток очищают с помощью ускоренной хроматографии, получая заданный продукт в виде белой пены (260 мг). МС (ЭСИ): 350,1 (МН+). Стадия Д: Бензиловый эфир (3R-4S)[1-фенетил-3-(тиазолидин-3-карбонил)пиперидин-4-ил]карбаминовой кислоты Бензиловый эфир (3R,4S)-3-(тиазолидин-3-карбонил)пиперидин-4-ил]карбаминовой кислоты (255 мг) растворяют в ДМФ (5 мл) и добавляют ДИПЭА (0,28 мл). Через 20 мин добавляют в течение 15 мин раствор 2-(бромэтил)бензола (169 мг) в ДМФ (4 мл). Смесь перемешивают при КТ в течение ночи. Реакционную смесь переносят в смесь лед/вода/насыщенный раствор Na2CO3, и водный слой экстрагируют этилацетатом. Органический слой промывают рассолом, высушивают над Na2SO4 и выпаривают. Остаток очищают с помощью ускоренной хроматографии, получая заданный продукт в виде слегка желтого масла (308 мг). МС (ЭСИ): 454,2 (МН+). Стадия Е: (3R,4S)-(4-Амино-1-фенетилпиперидин-3-ил)тиазолидин-3-илметанон Бензиловый эфир (3R,4S)-[1-фенетил-3-(тиазолидин-3-карбонил)пиперидин-4-ил]карбаминовой кислоты (216 мг) обрабатывают раствором 33%-ной HBr в уксусной кислоте (2,5 мл) в течение 2 ч при КТ в атмосфере аргона. Затем добавляют эфир (15 мл), образовавшуюся суспензию охлаждают до -10°С и перемешивают в течение 1 ч. Растворитель затем удаляют декантированием и твердое вещество промывают небольшим количеством EtOH. Остаток растворяют в воде (рН доводят до 10 с помощью конц. NH4OH), водный слой насыщают NaCl и экстрагируют этилацетатом. Органический слой отделяют, промывают рассолом, высушивают (Na2SO4) и выпаривают. Остаток очищают с помощью ускоренной хроматографии (градиент МеОН в ДХМ, содержащий 0,5% NH4OH), получая заданный продукт в виде слегка желтого масла (92 мг). МС (ЭСИ): 320,1 МН+. Пример 35: ((3S,4R-4-Амино-1-фенетилпиперидин-3-ил)тиазолидин-3-илметанон Данное соединение получено, как описано в примере 34, из противоположного энантиомера 1-трет-бутилового эфира/3-метилового эфира (3S,4R)-4-аминопиперидин-1,3-дикарбоновой кислоты, тиазолидина и 2-(бромэтил)бензола в виде желтого масла. МС (ЭСИ): 320,4 (МН+). Пример 36: [((3S,4R-4-Амино-1-(2-пиридин-2-илэтил)пиперидин-3-ил1 тиазолидин-3-илметанон Данное соединение получено, как описано в примере 34, из противоположного энантиомера 1-трет-бутилового эфира/3-метилового эфира (3S,4R)-4-аминопиперидин-1,3-дикарбоновой кислоты, тиазолидина и 2-(бромэтил)пиридина (полученного по методике: Synthesis, 5, 1987, 452-455) в виде желтого масла. МС (ЭСИ): 321,4 (МН+). Пример 37: 1-[(3S,4R)-4-Амино-3-(тиазолидин-3-карбонил)пиперидин-1-ил]-2-фенилэтанон] Данное соединение получено, как описано в примере 34, но с модификацией стадий Д и Е: Стадия Д: Метиловый эфир [1-фенилацетил-3-(тиазолидин-3-карбонил)пиперидин-4-ил]карбаминовой кислоты Бензиловый эфир 3-(тиазолидин-3-карбонил)пиперидин-4-ил]карбаминовой кислоты (250 мг) растворяют в абс. ДХМ (8 мл) и добавляют ДИПЭА (0,184 мл). Смесь оставляют при перемешивании при -15°С (баня лед/соль) в течение 20 мин, а затем добавляют по каплям фенилацетилхлорид (0,104 мл). Смесь нагревают до 0°С и перемешивают в течение 30 мин. Реакционную смесь переносят в смесь лед/вода/насыщенный раствор NaHCO3 и водный слой экстрагируют этилацетатом. Органический слой отделяют, промывают рассолом, высушивают над Na2SO4 и выпаривают в вакууме. Остаток очищают с помощью ускоренной хроматографии, получая заданный продукт – метиловый эфир [1-фенилацетил-3-(тиазолидин-3-карбонил)пиперидин-4-ил]карбаминовой кислоты (304 мг) в виде белой пены. МС (ЭСИ): 468,1 (МН+). Стадия Е: 1-[(3S,4R)-4-Амино-3-(тиазолидин-3-карбонил)пиперидин-1-ил]-2-фенилэтанон Реакцию проводят аналогично примеру 34 (стадия Е) из метилового эфира [1-фенилацетил-3-(тиазолидин-3-карбонил)пиперидин-4-ил]карбаминовой кислоты (295 мг) и HBr в НОАс (3 мл), но с модифицированной стадией очистки: продукт очищают с помощью препаративной ВЭЖХ (RPC18, градиент CH3CN в воде, содержащий 0,05% муравьиной кислоты). Выпаривание соответствующих фракций дает заданный продукт в виде соли муравьиной кислоты (белое твердое вещество, 12 мг). МС (ЭСИ): 334,4 (МН, свободное основание). Пример 38: 1-[(3S-4R-4-Амино-3-(тиазолидин-3-карбонил)пиперидин-1-ил]-2-тиофен-2-илэтанон Данное соединение получено согласно примеру 37 с соответствующими реагентами в виде бесцветной смолы: МС (ЭСИ): 340,4 (МН+) свободное основание). Пример 39: 3[(3S,4R) и (3R,4S)-4-Амино-1-(3,4-диметоксифенил)пиперидин-3-ил]тиазолидин-3-илметанон Стадия А: Метиловый эфир 4-амино-1-бензил-1,2,5,6-тетрагидропиридин-3-карбоновой кислоты Метиловый эфир рац-1-бензил-4-оксопиперидин-3-карбоновой кислоты (25 г) суспендируют в 100 мл 25%-ного гидроксида аммония и нагревают при 50°С в течение 18 ч. Смесь охлаждают льдом, а затем по частям добавляют NaBH4 (1 г). Перемешивают в течение 18 ч при комнатной температуре, разбавляют водой со льдом при охлаждении и экстрагируют AcOEt. Органический экстракт промывают рассолом и высушивают над сульфатом натрия. Удаление растворителя при пониженном давлении дает коричневое масло, которое очищают с помощью колоночной хроматографии (силикагель, гептан/AcOEt в соотношении 1:1) и осаждают (AcOEt/гептан), получая 12,1 г (56%) названного в заголовке соединения в виде белого твердого вещества, и 6 г (25%) названного в заголовке соединения в виде его бороводородной соли. Метиловый эфир 4-амино-1-бензил-1,2,5,6-тетрагидропиридин-3-карбоновой кислоты: МС: 247,2 (М+Н)+ ЯМР: (ДМСО, 1Н, 400 МГц, Метиловый эфир 4-амино-1-бензил-1,2,5,6-тетрагидропиридин-3-карбоновой кислоты; ВН3 соль: МС: 247,2 (М+Н)+ ЯМР: (ДМСО, 1Н, 400 МГц, Стадия Б: Этиловый эфир рац-4-амино-1-бензилпиперидин-3-карбоновой кислоты ВН3 соль метилового эфира 4-амино-1-бензил-1,2,5,6-тетрагидропиридин-3-карбоновой кислоты нагревают в этаноле с 25% NaOH. После 72 ч нагревания при 50°С смесь охлаждают, промывают водой со льдом и экстрагируют AcOEt. Экстракт промывают рассолом и высушивают над сульфатом натрия. Удаление растворителя при пониженном давлении дает желтое масло. К суспензии метилового эфира 4-амино-1-бензил-1, 2, 5, 6-тетрагидропиридин-3-карбоновой кислоты (21 г) в 200 мл ТГФ добавляют 50 мл ТФК при 10°С в атмосфере аргона. После перемешивания в течение 15 мин при 0°С добавляют NaBH4 (6,43 г) в течение 75 мин при 10°С. Смесь перемешивают дополнительно в течение 90 мин при 0°С. После дополнительного насыщения 100 мл NH4Cl раствор дважды экстрагируют CH2Cl2. Объединенные экстракты промывают водой со льдом и рассолом и высушивают над сульфатом натрия. Удаление растворителя дает названное в заголовке соединение в виде желтого масла, которое используется на следующей стадии без очистки. Стадия В: Этиловый эфир (3S,4R) и (3R,4S)-1-бензил-4-трет-бутоксикарбониламинопиперидин-3-карбоновой кислоты и этиловый эфир (3S,4S) и (3R,4R-1-бензил-4-трет-бутоксикарбониламинопиперидин-3-карбоновой кислоты Этиловый эфир рац-4-амино-1-бензилпиперидин-3-карбоновой кислоты (80,7 ммолей, 21 г) и ВОС2О (20,5 г) в 200 мл CH2Cl2 перемешивают в течение 17 ч при комнатной температуре. Раствор выпаривают и хроматографируют (силикагель, AcOEt/гептан в соотношении 1:1, получая 7,05 г рацемического транс-изомера, 1,88 г рацемического цис-изомера и 13,87 г рацемической смеси диастереомеров. Этиловый эфир (3S,4R) и (3R,4S)-1-бензил-4-трет-бутоксикарбониламинопиперидин-3-карбоновой кислоты: МС: 363,3 (М+Н)+. ЯМР: (ДМСО, 1Н, 400 МГц, Этиловый эфир (3S,4S) и (3R,4R)-1-бензил-4-трет-бутоксикарбониламинопиперидин-3-карбоновой кислоты: МС: 363,3 (М+Н)+. ЯМР: (ДМСО, 1Н, 400 МГц, Стадия Г: Этиловый эфир (3R,4R) и (3S,4S)-4-трет-бутоксикарбониламино-1-[2-(3,4-диметоксифенил)этил]пиперидин-3-карбоновой кислоты Суспензию этилового эфира (3S,4S) и (3R,4R)-1-бензил-4-трет-бутоксикарбониламинопиперидин-3-карбоновой кислоты (700 мг) и 200 мг Pd/C (10%-ный) в 10 мл EtOH и 1 мл основания Хюнига гидрируют при 22°С/1 бар в течение ночи. Суспензию фильтруют, фильтрат выпаривают, а остаток очищают с помощью колоночной хроматографии (силикагель, AcOEt), получая бледно-желтое твердое вещество (538 мг). К раствору названного выше продукта (272 мг) и 3, 4-диметоксибензолацетальдегида (CAS 5703-21-9, 191 мг) в этаноле при 0°С добавляют 8-молярный раствор пиридин-боран-комплекса (0,5 мл). После 2 ч перемешивания при 0°С и 3 ч перемешивания при комнатной температуре смесь разбавляют водой со льдом и дважды экстрагируют этилацетатом. Объединенные органические слои промывают рассолом и высушивают над сульфатом натрия. Удаление растворителя при пониженном давлении дает масло, которое очищают с помощью колоночной хроматографии (силикагель, AcOEt), получая 196 мг (45%) названного в заголовке соединения в виде желтого масла. МС: 437,4 (М+H)+ ЯМР: (ДМСО, 1Н, 400 МГц, Стадия Д: Этиловый эфир (3R,4R) и (3S,4S)-4-амино-1-[2-(3,4-диметоксифенил)этил]пиперидин-3-карбоновой кислоты Раствор (3R,4R) и (3S,4S)-4-трет-бутоксикарбониламино-1-[2-(3,4-диметоксифенил)этил]пиперидин-3-карбоновой кислоты (24 мг) в 1 мл ТФК перемешивают в течение 1 ч при 0°С.Смесь переносят в 1-молярный раствор NaOH/лед и экстрагируют AcOEt. Объединенные органические слои промывают рассолом и высушивают над сульфатом натрия. Удаление растворителя при пониженном давлении дает масло, которое очищают с помощью колоночной хроматографии (Isolute SPE Flash NH2, 10 г, AcOEt/гептан в соотношении 1:2), получая 9 мг (50%) названного в заголовке соединения. МС: 337,4 (M+H)+ Стадия Е: (3R,4R) и (3S,4S)-4-Амино-1-[2-(3,4-диметоксифенил)этил]пиперидин-3-карбоновая кислота Этиловый эфир (3R,4R) и (3S,4S)-4-амино-1-[2-(3,4-диметоксифенил)этил]пиперидин-3-карбоновой кислоты (262 мг) и гидроксид лития (166 мг) в 5 мл ТГФ, 5 мл МеОН и 1 мл Н2О перемешивают в течение 24 ч при комнатной температуре. Смесь разбавляют буфером рН 7 и экстрагируют AcOEt. Объединенные органические слои промывают рассолом и высушивают над сульфатом натрия. Удаление растворителя при пониженном давлении дает масло, которое очищают с помощью колоночной хроматографии (Isolute SPE Flash NH2, AcOEt) и осаждают (AcOEt/гептан), получая 90 мг (37%) названного в заголовке соединения в виде белого твердого вещества. МС: 309,2 (М+Н)+ ЯМР: (ДМСО, 1Н, 400 МГц, Пример 40: 3[(3S,4R) и (3R,4S)-4-Амино-1-(3,4-диметоксифенил)пиперидин-3-ил]тиазолидин-3-илметанон Стадия А: Этиловый эфир рац-1-(3,4-диметоксифенил)-4-оксопиперидин-3-карбоновой кислоты Смесь 3,4-диметоксианилина (12 г), хлорида меди (1) (1,7 г), уксусной кислоты (9 мл) и этилакриловой кислоты (26 мл) нагревают в течение 17 ч при 140°С, охлаждают льдом, разбавляют CH2Cl2 и последовательно промывают водой, 10%-ным раствором гидроксида аммония, водой и рассолом. Органические слои высушивают над MgSO4, фильтруют и выпаривают. Хроматография (силикагель, AcOEt/гептан в соотношении 1:1) дает желтое масло (22,8 г), которое растворяют в 50 мл ксилола и добавляют этоксид натрия (4,42 г). Затем суспензию перемешивают в течение 2 ч при 140°С, охлаждают, разбавляют AcOEt и промывают водой и рассолом. Водные фазы дважды экстрагируют AcOEt, органические слои высушивают над MgSO4, фильтруют, выпаривают и хроматографируют (силикагель, AcOEt/гептан в соотношении 1:3), получая названное в заголовке соединение в виде белого твердого вещества (выход 13,53 г, 68%). МС: 308,2 (M+H)+ Стадия Б: Этиловый эфир рац-4-амино-1-(3,4-диметоксифенил)пиперидин-3-карбоновой кислоты Суспензию этилового эфира рац-1-(3,4-диметоксифенил)-4-оксопиперидин-3-карбоновой кислоты (640 мг) и ацетата аммония (3 г) в 10 мл метанола перемешивают в течение 18 ч при КТ, а затем добавляют цианборгидрид натрия (2 г). После перемешивания в течение 18 ч при КТ смесь разбавляют AcOEt, дважды промывают водой и рассолом. Органические слои высушивают над MgSO4, фильтруют, выпаривают и хроматографируют (силикагель, AcOEt/гептан в соотношении 1:1), получая названное в заголовке соединение (выход 450 мг, 70%). Стадия В: Этиловый эфир рац-4-трет-бутоксикарбониламино-1-(3,4-диметоксифенил)пиперидин-3-карбоновой кислоты Раствор этилового эфира рац-4-амино-1-(3,4-диметоксифенил)пиперидин-3-карбоновой кислоты (6,47 г) и Вок2О (5,02 г) в 50 мл CH2Cl2 перемешивают в течение 24 ч при КТ, затем разбавляют AcOEt, промывают водой и рассолом. Органические слои высушивают над MgSO4, фильтруют, выпаривают и хроматографируют (силикагель, AcOEt/гептан в соотношении 1:1), получая названное в заголовке соединение в виде смеси цис- и транс-диастереомеров (выход 4,50 г, 53%). МС: 409,4 (M+H)+ Стадия Г: рац-4-трет-Бутоксикарбониламино-1-(3,4-диметоксифенил)пиперидин-3-карбоновая кислота К раствору этилового эфира рац-4-трет-бутоксикарбониламино-1-(3,4-диметоксифенил)пиперидин-3-карбоновой кислоты (170 мг) в 2 мл ТГФ добавляют 1 мл 1-молярного раствора NaOH и гидроксид лития (100 мг). Суспензию перемешивают в течение 24 ч при КТ, разбавляют водой и дважды экстрагируют трет-бутилметиловым эфиром. Водный слой подкисляют до рН 4, насыщают NaCl и затем продукт трижды экстрагируют AcOEt. Органические слои высушивают над MgSO4, фильтруют, выпаривают и хроматографируют (силикагель, AcOEt), получая названное в заголовке соединение (выход 38 мг, 55%). MC: 381,3 (M+H)+ Стадия Д: трет-Бутиловый эфир [(3S,4R) и (3R,4S)-1-(3,4-диметоксифенил)-3-(тиазолидин-3-карбонил)пиперидин-4-ил]карбаминовой кислоты и трет-бутиловый эфир [3S,4S) и (3R,4R)-1-(3,4-диметоксифенил)-3-(тиазолидин-3-карбонил)пиперидин-4-ил]-карбаминовой кислоты К раствору рац-4-трет-бутоксикарбониламино-1-(3,4-диметоксифенил)пиперидин-3-карбоновой кислоты (200 мг) в ацетонитриле (5 мл) при охлаждении льдом добавляют основание Хюнига (0,5 мл), ЭДС1 (191 мг), НОВТ (135 мг) и тиазолидин (0,3 мл). Реакционную смесь перемешивают в течение 3 ч при 0°С и выдерживают в течение 24 ч при КТ, разбавляют AcOEt, промывают 5%-ным раствором NaHCO3 и рассолом, высушивают над сульфатом магния и концентрируют. Сырой продукт очищают с помощью хроматографии на силикагеле с использованием AcOEt, получая трет-бутиловый эфир [(3S,4R) и (3R,4S)-1-(3,4-диметоксифенил)-3-(тиазолидин-3-карбонил)пиперидин-4-ил]карбаминовой кислоты (73 мг) и трет-бутиловый эфир [(3S,4S) и (3R,4R)-1-(3,4-диметоксифенил)-3-(тиазолидин-3-карбонил)пиперидин-4-ил]карбаминовой кислоты (81 мг). МС: (М+Н)+ 452,4. Стадия Е: [(3S,4R) и (3R,4S)-4-Амино-1-(3,4-диметоксифенил)пиперидин-3-ил]тиазолидин-3-илметанон трет-Бутиловый эфир [(3S,4R) и (3R,4S)-1-(3,4-диметоксифенил)-3-(тиазолидин-3-карбонил)пиперидин-4-ил]карбаминовой кислоты (50 мг) обрабатывают 2 мл ТФК в течение 1 ч при 0°С. Реакционную смесь разбавляют AcOEt, промывают 1-молярным раствором NaOH и рассолом, органические слои высушивают над MgSO4, фильтруют, выпаривают и хроматографируют (Isolute Flash SPE NH2, AcOEt), получая [(3S,4R) и (3R,4S)-4-амино-1-(3,4-диметоксифенил)пиперидин-3-ил]тиазолидин-3-илметанон (23 мг). МС: 352,1 (М+H)+ ЯМР: (ДМСО, 1Н, 400 МГц, Пример 41: 3[(3S,4S) и (3R,4R)-4-Амино-1-(3,4-диметоксифенил)пиперидин-3-ил]тиазолидин-3-илметанон Данное соединение получено из рацемического транс-изомера, выделенного в примере 40 (стадия Д), с использованием методик, описанных в примере 40 (стадия Е). При этом получено 28 мг названного в заголовке соединения. МС: 352,1 (M+H)+ ЯМР: (ДМСО, 1H, 400 МГц, Пример 42: 3[(3S,4S) и (3R,4R)-4-Амино-1-(3,4-диметоксифенил)пиперидин-3-ил]-(2,5-дигидропиррол-1-ил)-3-илметанон Данное соединение получено аналогично примеру 40 (стадии Д и Е), с использованием 2,5-дигидропиррола. МС: 332,2 (М+1)+ Пример 43: 3[(3S,4R) и (3R,4S)-4-Амино-1-(3,4-диметоксифенил)пиперидин-3-ил]-(2,5-дигидропиррол-1-ил)-3-илметанон Данное соединение получено аналогично примеру 40 (стадии Д и Е) с использованием 2, 5-дигидропиррола. МС: 332,2 (М+Н)+ Фармацевтические примеры Пример А Таблетки, покрытые оболочкой, содержащие следующие ингредиенты, могут быть получены стандартным методом:
Активные ингредиенты просеивают, смешивают с микрокристаллической целлюлозой и гранулируют смесь в растворе поливинилпирролидона в воде. Гранулят смешивают с щелочным гликолятом крахмала и стеаратом магния и прессуют, получая сердцевину таблеток массой 120 или 350 мг соответственно. Сердцевину лакируют с помощью водного раствора / суспензии упомянутой выше пленочной оболочки. Пример Б Капсулы, содержащие следующие ингредиенты, могут быть получены стандартным методом:
Компоненты просеивают, смешивают и вносят в капсулы размера 2. Пример В Инъекционные растворы могут иметь следующий состав:
Активный ингредиент растворяют в смеси полиэтиленгликоля 400 и воды для инъекций (часть). Доводят рН до 5,0 с помощью уксусной кислоты. Объем доводят до 1,0 мл добавлением оставшегося количества воды. Раствор отфильтровывают, заполняют в пузырьки, используя соответствующее устройство, и стерилизуют. Пример Г Мягкие желатиновые капсулы, содержащие следующие ингредиенты, могут быть получены стандартным методом:
Активный ингредиент растворяют в теплом расплаве других ингредиентов и смесью заполняют мягкие желатиновые капсулы соответствующего размера. Заполненные мягкие желатиновые капсулы обрабатывают с использованием обычных методов. Пример Д Облатки, содержащие следующие ингредиенты, могут быть получены стандартным методом:
Активный ингредиент смешивают с лактозой, микрокристаллической целлюлозой и натрийкарбоксиметилцеллюлозой, а затем гранулируют со смесью поливинилпирролидона в воде. Гранулят смешивают со стеаратом магния и вкусовыми добавками и вносят в саше.
Формула изобретения
1. Соединения формулы (I) 2. Соединения формулы I по п.1, где R1 выбирают из группы, 3. Соединения формулы I по п.1, где R1 выбирают из группы, включающей: 4. Соединения формулы I по п.1, где R1 обозначает 5. Соединения формулы I по п.1, где R1 обозначает 6. Соединения формулы I по п.1, где R2 выбирают из группы, включающей: 7. Соединения формулы I по п.1, где R2 обозначает н-бутил. 8. Соединения формулы I по п.1, где R2 обозначает 9. Соединения формулы I по п.1, где R2 обозначает тиенил, который является незамещенным или моно-, ди- или тризамещенным независимо низшей алкоксигруппой. 10. Соединения формулы I по п.1, где R2 обозначает 11. Соединения формулы I по п.1, выбранные из группы: 12. Соединения по п.1, предназначенные для использования в качестве терапевтически активных субстанций, способных ингибировать DPP-IV. 13. Фармацевтическая композиция, способная ингибировать DPP-IV, включающая соединение по одному из пп.1-11 и фармацевтически приемлемый носитель и/или адъювант.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
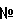 11
11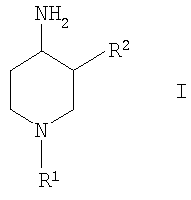
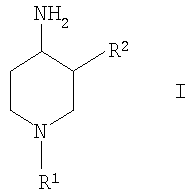
 -клеток, аутоиммунных болезней, таких, как воспалительное заболевание кишечника, энцефалитный периаксиальный склероз и ревматоидный артрит, неспецифический язвенный колит, болезнь Крона, псориаз, красный плоский лишай и/или доброкачественная гипертрофия простаты. Соединения могут быть также использованы для профилактики СПИД (синдром приобретенного иммунодефицита) или для профилактики метастаза, в частности метастаза рака груди и простаты в легкие. Кроме того, соединения по настоящему изобретению могут быть использованы в качестве диуретических агентов и для лечения и/или профилактики гипертензии.
-клеток, аутоиммунных болезней, таких, как воспалительное заболевание кишечника, энцефалитный периаксиальный склероз и ревматоидный артрит, неспецифический язвенный колит, болезнь Крона, псориаз, красный плоский лишай и/или доброкачественная гипертрофия простаты. Соединения могут быть также использованы для профилактики СПИД (синдром приобретенного иммунодефицита) или для профилактики метастаза, в частности метастаза рака груди и простаты в легкие. Кроме того, соединения по настоящему изобретению могут быть использованы в качестве диуретических агентов и для лечения и/или профилактики гипертензии.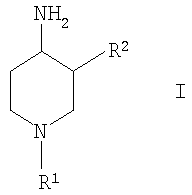
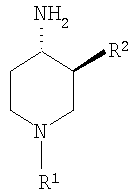 или
или 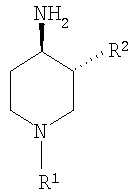
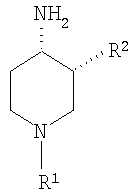 или
или 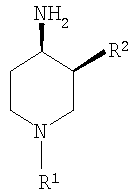
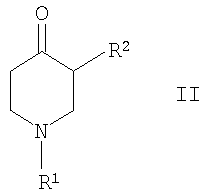
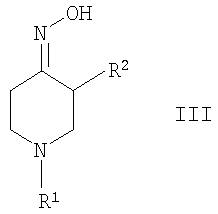
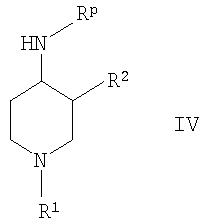
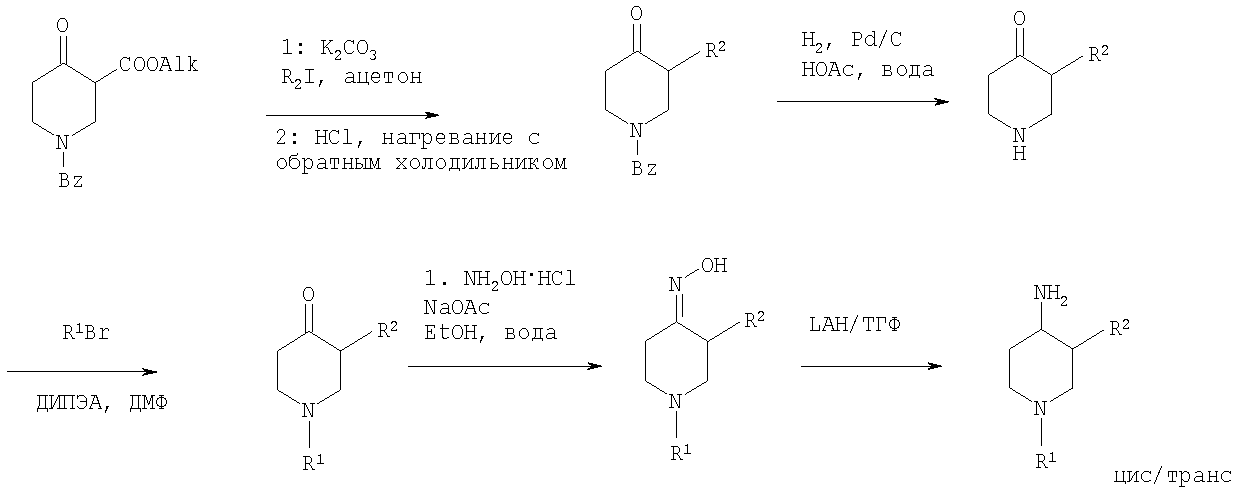
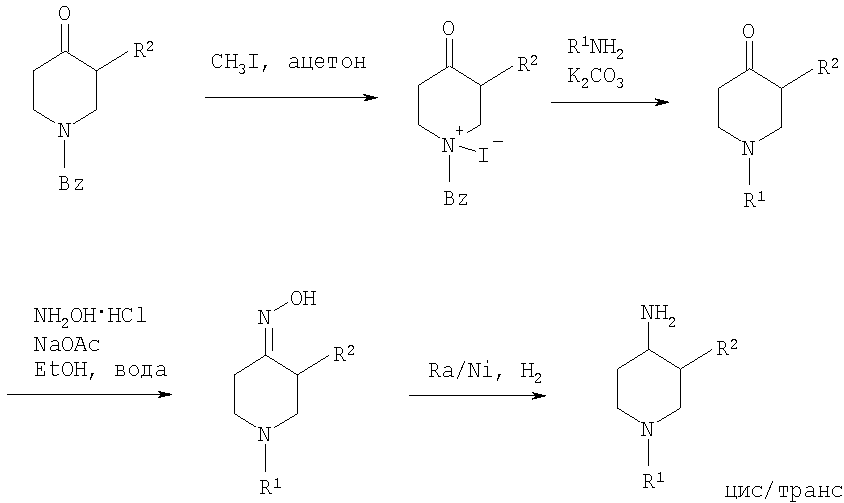
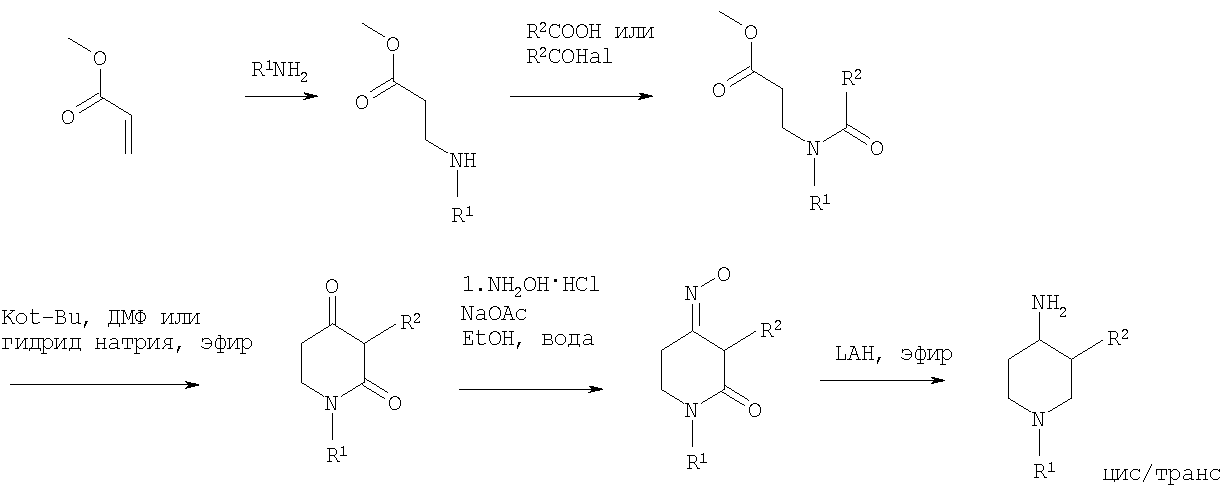
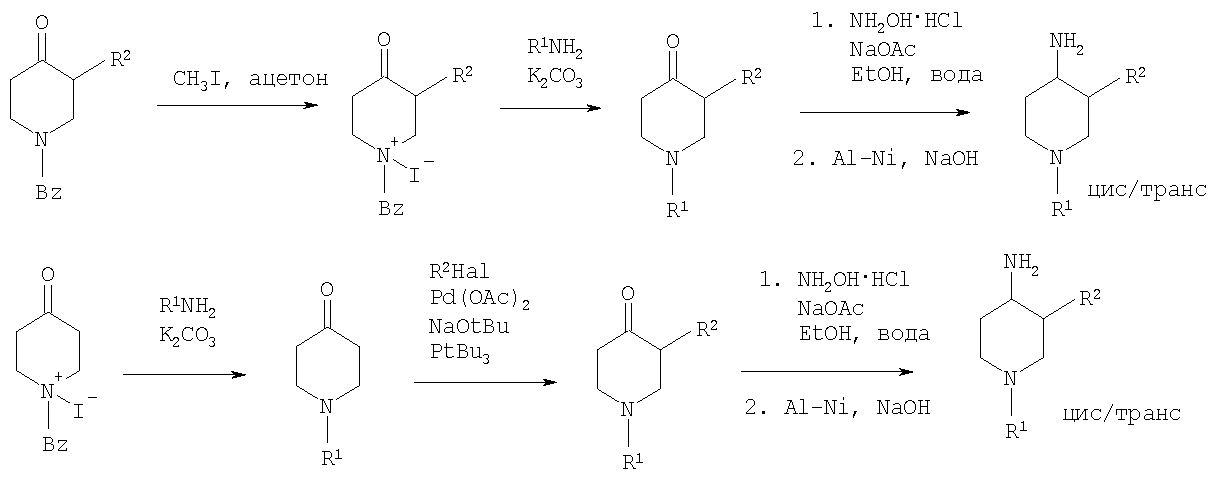
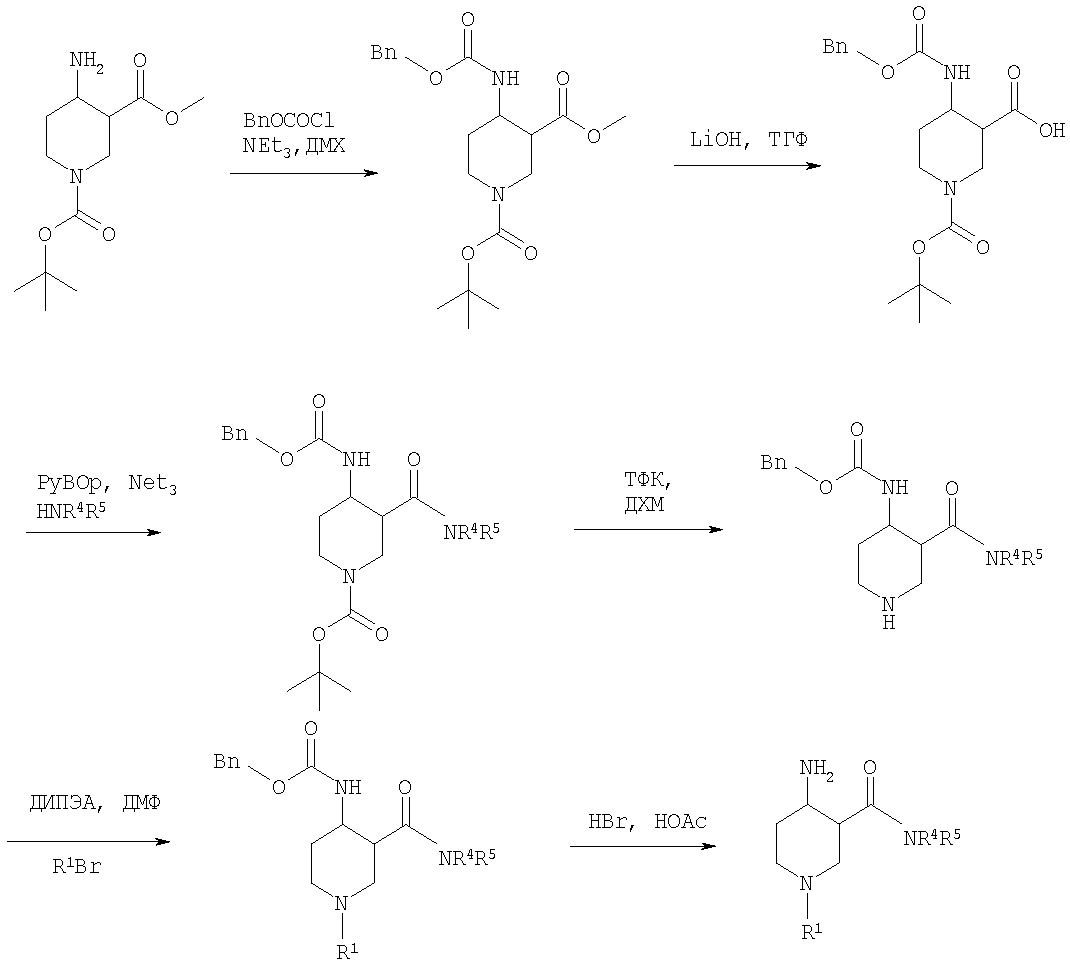
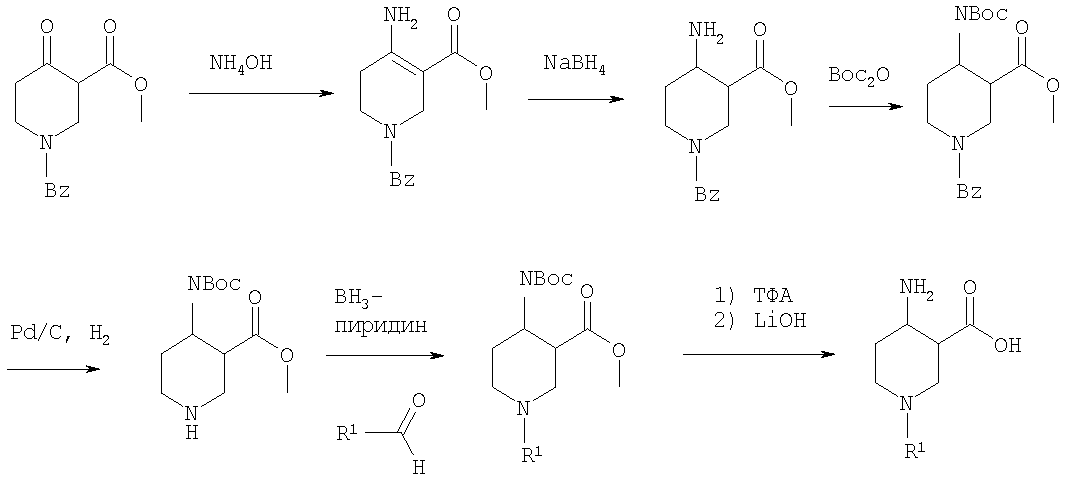
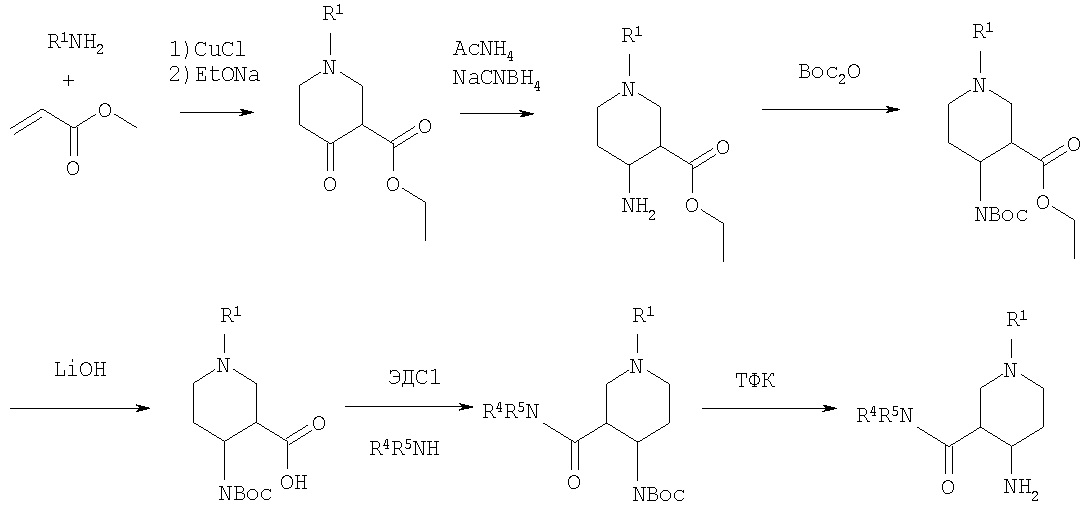
 2,81-2,65 (m, 2H), 2,60-2,00 (m, 5H), 1.74-1,60 (m, 5H), 1,60-1,40 (m, 3H), 1,40-1,00 (10 Н), 0,87 (t, 3H).
2,81-2,65 (m, 2H), 2,60-2,00 (m, 5H), 1.74-1,60 (m, 5H), 1,60-1,40 (m, 3H), 1,40-1,00 (10 Н), 0,87 (t, 3H).