Патент на изобретение №2391982
|
||||||||||||||||||||||||||||||||||||
(54) ПРОТИВООПУХОЛЕВОЕ СРЕДСТВО
(57) Реферат:
Изобретение относится к области медицины и фармацевтики и касается противоопухолевого средства для лечения рака желудочно-кишечного тракта, включающего компонент (А) и компонент (В), где компонент (А) представляет собой R-(-)-3-[3-(1-трет-бутилкарбонилметил-2-оксо-5-циклогексил-1,3,4,5-тетрагидро-2Н-1,5-бензодиазепин-3-ил)уреидо]бензойную кислоту или ее фармацевтически приемлемую соль и компонент (В) представляет собой противоопухолевое средство. 3 н. и 18 з.п. ф-лы, 1 табл.
Область техники Настоящее изобретение относится к противоопухолевому средству, а более точно к противоопухолевому средству, полезному при лечении и профилактике рака желудочно-кишечного тракта, лейкемии, опухоли гипофиза, мелкоклеточного рака легких, рака щитовидной железы и нейроастроцитомы. В Японии увеличился процент смертности от рака, и с 1981 года рак является главной причиной смертности в Японии. В 2002 году число жертв рака было 304286 (т.е. 241,5 на каждые 100000), составляя при этом 31,0% от общего числа смертей. В частности, высока частота случаев таких видов рака желудочно-кишечного тракта, как рак поджелудочной железы, рак толстой кишки и рак желудка. Среди этих видов рака желудочно-кишечного тракта рак поджелудочной железы известен как трудноизлечимый рак. В Японии в качестве химиотерапевтического средства для рака поджелудочной железы одобрен только гемцитабин гидрохлорид. Однако химиотерапевтическое средство, такое как гемцитабин гидрохлорид или фторурацил, часто вызывает серьезные побочные эффекты (например, миелосупрессия и интерстициальная пневмония), и, следовательно, на интервал или период приема такого химиотерапевтического средства налагается ограничение. Помимо этого ограничение налагается на лекарственную форму такого химиотерапевтического средства, т.к. средство в основном производится в форме капельного внутривенного вливания. Следовательно, возникла необходимость разработки противоопухолевого средства, которое заменяет такое химиотерапевтическое средство. В качестве противоопухолевого средства в основном применяется химиотерапевтическое средство, проявляющее цитотоксический или разрушающий клетки эффект, а также часто применяется многокомпонентная комплексная химиотерапия, в виду того что применение нескольких химиотерапевтических средств в комплексе уменьшает неблагоприятные побочные эффекты средств и увеличивает противоопухолевый эффект средств. Многокомпонентная комплексная химиотерапия, которая в основном использует фармацевтические средства, проявляющие различные по механизмам действия и различные побочные эффекты, в комплексе, обходит ту проблему, что, когда наблюдается общая токсичность к фармацевтическим средствам (например, миелосупрессия), количества соответствующих фармацевтических средств могут быть уменьшены (Непатентный документ 1). Также многокомпонентная комплексная химиотерапия обходит ту проблему, что фармацевтическое средство может быть заменено другим фармацевтическим средством благодаря толерантности фармацевтического средства. В последние годы механизмы, например, роста, метастазирования, инвазии и злокачественного прогрессирования рака были объяснены на молекулярном уровне и были разработаны несколько селективных фармацевтических средст, направленных на определенные молекулы. Такое фармацевтическое средство для молекулярной мишени в основном проявляет низкую цитотоксичность, и предусматривается, что оно проявляет ослабленные побочные эффекты по сравнению с общепринятым химиотерапевтическим средством, проявляющим цитотоксический эффект. Такое селективное фармацевтическое средство для молекулярной мишени, которое проявляет свой эффект при отдельном использовании, также стало интересно как фармацевтическое средство, используемое в комбинации с химиотерапевтическим средством (Непатентный документ 2). Ранее лечение рака оценивали исключительно на основании уменьшения опухоли благодаря цитотоксическому эффекту используемого химиотерапевтического средства. Однако в последние годы улучшение качества жизни (QOL), подавление метастазирования или увеличение продолжительности жизни являются подходящими критериями оценки лечения рака, и применение химиотерапевтического средства и селективного фармацевтического средства в комплексе считается многообещающим методом лечения рака (Непатентный документ 3). Гастрин является гормоном желудочно-кишечного тракта, который считается фактором роста опухолевых клеток. Как обнаружено, ген рецептора гастрина экспрессируется в раковых клетках поджелудочной железы, толстой кишки или желудка (т.е. рака желудочно-кишечного тракта), благодаря чему при действии гастрина проявляется свойство клеток сильно расти (Непатентные документы 4 и 5). Как сообщено, подобно случаю такого рака желудочно-кишечного тракта, ген рецептора гастрина экспрессируется и в случае лейкемии, опухоли гипофиза, мелкоклеточного рака легких, рака щитовидной железы или нейроастроцитомы, и гастрин может функционировать в качестве фактора роста раковых клеток (Непатентный документ 6). Ранее считалось, что увеличение роста клеток происходит главным образом путем, в котором гастрин стимулирует гастриновый рецептор, находящийся на поверхности клеток. Однако недавние исследования предполагают, что существует путь увеличения роста клеток с помощью гастрина, в котором гастрин связывается с гастриновым рецептором и далее попадает внутрь клеток по средствам эндоцитоза (Непатентный документ 7); и существует другой путь, в котором гастрин связывается с гастрин-связывающим белком, присутствующим в клетках, таким образом, регулируя клеточный рост (Непатентные документы 8 и 9). Было также сообщено, что глицин-расширенный гастрин, который является предшественником гастрина, кроме гастринового рецептора, связывается с неопределенным рецептором, таким образом, регулируя клеточный рост (Непатентные документы 10 и 11). Следовательно, считается, что гастрин-зависимый клеточный рост происходит множеством путей. Общепринято, что разработанными антагонистами рецептора гастрина являются соединения, направленные только на гастриновые рецепторы, и таким образом этот общепринятый антагонист рецептора гастрина не проявляет постоянного и надежного противоопухолевого эффекта. Например, сообщено, что L-365,260, который является бензодиазепиновым соединением, подавляет гастрин-индуцированный опухолевый рост в случае рака поджелудочной железы человека PANC-1, ксенотрансплантированного подопытным мышам, но не подавляет опухолевый рост без стимуляции гастрином (Непатентный документ 12). Подобные результаты сообщались в случае CR2093, который является производным глутаминовой кислоты (Непатентный документ 13). Эти данные показывают, что антагонист гастринового рецептора подавляет только рост раковых клеток, вызванный принудительной внешней гастриновой стимуляцией, т.е. рост раковых клеток, вызванный нефизиологической гастриновой стимуляцией. Следовательно, считается, что антагонист гастринового рецептора, который теряет эффект подавления клеточного роста при физиологических условиях, проявляет недостаточный эффект в качестве противоопухолевого средства. CI-988, который является С-терминальным пентапептидным производным CCK, известен как сильнодействующий антагонист гастринового рецептора. Однако, как сообщено, при пероральном приеме мышами с ксенотрансплантированным раком толстой кишки человека дозы 50 мг/кг CI-988 не проявляет эффект подавления клеточного роста, хотя при оральном приеме дозы 25 мг/кг CI-988 проявляет эффект подавления клеточного роста без нефизиологической гастриновой стимуляции (Непатентный документ 14). YF476, который является бензодиазепиновым соединением, известен как избирательный и сильнодействующий антагонист гастринового рецептора. Патентный документ 1 описывает, что YF476 проявляет эффект уменьшения опухоли при раке поджелудочной железы или в модели с ксенотрансплантированным раком толстой кишки. Однако этот патентный документ описывает, что этот эффект наблюдается только в случае, когда YF476 принимается в больших дозах, 200 мг/кг или более, и что не ясно, опосредован ли механизм действия YF476 гастриновым рецептором. Как описано выше, разработаны многочисленные антагонисты гастринового рецептора, но не получен обоснованный вывод относительно противоопухолевого эффекта такого антагониста. В частности, не описано, что антагонистический эффект гастринового рецептора имеет простую взаимосвязь с противоопухолевым эффектом, и роль, которую гастриновый рецептор играет при раке, еще окончательно не выяснена. Между тем, на данный момент не ясно, проявляет ли 1,5-бензодиазепиновое производное, описанное в Патентном документе 2 и имеющее эффект гастринового антагониста, полезный противоопухолевый эффект. Патентный документ 1: WO 02/092096. Патентный документ 2: WO 01/40197. Непатентный документ 1: Nippon Rinsho. 2003, 61, 6, 1015-1020. Непатентный документ 2: Nippon Rinsho. 2004, 62, 7, 1232-1240. Непатентный документ 3: J Clin Oncol. 2003, 21, 7, 1404-1411. Непатентный документ 4: Am J Phisiol. 1985, 249, G761-769. Непатентный документ 5: Am J Phisiol. 1994, 266, R277-283. Непатентный документ 6: Igaku no Ayumi. 1998, 184, 4, 260-261. Непатентный документ 7: Cell Tissue Res. 1997, 287, 325-333. Непатентный документ 8: J Gastroenterol Hepatol. 1995, 10, 215-232. Непатентный документ 9: Eur J Pharmacol. 2000, 388, 9-15. Непатентный документ 10: Science. 1994, 265, 410-412. Непатентный документ 11: Regul Pert. 2000, 93, 37-44. Непатентный документ 12: Am J Physiol. 1995, 268, R135-141. Непатентный документ 13: Br J Cancer. 1992, 65, 879-883. Непатентный документ 14: Clin Exp Pharmacol Physiol. 1996, 23, 438-440. Описание изобретения Проблемы, решаемые изобретением Задача настоящего изобретения – создать противоопухолевое средство, в частности противоопухолевое средство, пригодное для лечения и/или профилактики, например, рака желудочно-кишечного тракта, лейкемии, опухоли гипофиза, мелкоклеточного рака легких, рака щитовидной железы и нейроастроцитомы. Средства для решения проблем Авторы изобретения провели обширные исследования противоопухолевого эффекта 1,5-бензодиазепинового производного, описанного в WO 01/40197, или его фармацевтически приемлемой соли, и в результате было обнаружено, что соединение проявляет хороший противоопухолевый эффект. Таким образом, настоящее изобретение обеспечивает противоопухолевым средством, содержащим в качестве действующего компонента 1,5-бензодиазепиновое производное, представленное следующей формулой (1):
(где R1 представляет C1-6-алкильную группу, R2 представляет фенильную группу или циклогексильную группу, и Y представляет простую связь или C1-4-алкиленовую группу), или его фармацевтически приемлемую соль. Настоящее изобретение также обеспечивает использование 1,5-бензодиазепинового производного, представленного формулой (1), или его фармацевтически приемлемой соли для получения противоопухолевого средства. Настоящее изобретение также обеспечивает способ лечения опухоли, который включает введение эффективного количества 1,5-бензодиазепинового производного, представленного формулой (1), или его фармацевтически приемлемой соли. Эффекты, достигаемые при осуществлении изобретения Соединение, соответствующее настоящему изобретению, не проявляет такой разрушающий клетки эффект, который проявляет обычное химиотерапевтическое средство, и не проявляет серьезных побочных эффектов в испытаниях на безопасность на животных, т.е. соединение имеет малый риск серьезных побочных эффектов (например, миелосупрессии и интерстициальной пневмонии), которые в других случаях вызывались бы обычным химиотерапевтическим средством. Следовательно, соединение пригодно в качестве противоопухолевого фармацевтического средства для, например, рака желудочно-кишечного тракта, лейкемии, опухоли гипофиза, мелкоклеточного рака легких, рака щитовидной железы и нейроастроцитомы. Т.к. фармацевтическое средство, соответствующее настоящему изобретению, проявляет низкую токсичность, то это фармацевтическое средство можно принимать оральным способом и в течение продолжительного времени. Следовательно, фармацевтическое средство может изготавливаться в простой, по сравнению со случаем обычного фармацевтического средства, лекарственной форме. Если фармацевтическое средство, соответствующее настоящему изобретению, применяется в многокомпонентной комплексной химиотерапии, доза противоопухолевого средства, проявляющего сильные побочные эффекты, может быть уменьшена, причем, вероятно, осуществляемая многокомпонентная комплексная химиотерапия проявляет хороший противоопухолевый эффект и уменьшает побочные эффекты. При приеме фармацевтического средства в течение продолжительного времени даже после приема общепринятого химиотерапевтического средства предусматривается, что фармацевтическое средство проявляет эффект подавления роста опухоли, т.е. фармацевтическое средство может также применяться в качестве профилактического противоопухолевого средства. Наиболее предпочтительные варианты осуществления изобретения Примеры C1-6-алкильной группы, представленной R1 в формуле (1), включают: метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил и трет-бутил. Из них C1-4-алкильная группа более предпочтительна и С4-алкильная группа наиболее предпочтительна, причем трет-бутильная группа особенно предпочтительна. Особенно предпочтительной R2 группой является циклогексильная группа. Примеры C1-4-алкиленовой группы, представленной Y, включают: метилен, этилен, пропилен, бутилен, метилметилен, диметилметилен, 1-метилэтилен, 1,1-диметилэтилен, 1-метилпропилен и 2-метилпропилен. Из них диметилметиленовая группа особенно предпочтительна. Для Y особенно предпочтительна простая связь. Среди соединений, представленных формулой (1) (здесь далее соединения могут быть собирательно упомянуты как «соединение (1)»), особенно предпочтительными являются R-(-)-3-[3-(1-трет-бутилкарбонилметил-2-оксо-5-циклогексил-1,3,4,5-тетрагидро-2Н-1,5-бензодиазепин-3-ил)уреидо]бензойная кислота или ее фармацевтически приемлемая соль (соединение A) и R-(-)-2-[3-[3-(1-трет-бутилкарбонилметил-2-оксо-5-циклогексил-1,3,4,5-тетрагидро-2Н-1,5-бензодиазепин-3-ил)уреидо]фенил-2-метилпропионовая кислота или ее фармацевтически приемлемая соль (соединение B). Из них соединение A более предпочтительно. Примеры солей соединения (1) включают неорганические соли, такие как натриевая соль, калиевая соль, кальциевая соль и магниевая соль; органические соли, такие как аммониевая соль, пиридиниевая соль, триэтиламмониевая соль, этаноламмониевая соль, (R)- или (S)-формы Здесь «соединение (1)» включает свои оптически активные изомеры, диастереомеры, сольваты (например, гидраты) и кристаллические полиморфные модификации. Соединение (1) может быть получено способом, раскрытым в WO 01/40197. Как описано ниже в примерах, соединение (1) подавляет рост различных опухолей и статистически заметно продлевает время жизни организма – носителя рака. Следовательно, соединение (1) пригодно в качестве фармацевтического средства для профилактики или лечения различных опухолей. Смертельные случаи не наблюдались при приеме соединения (1) (особенно соединения А) крысами и собаками в дозах 1000 мг/кг последовательно в течение 28 дней. Кроме того, отклонений в весе тела, приеме пищи, офтальмологических исследованиях, анализе мочи, весе органа, данных вскрытия и гистопатологических исследованиях обнаружено не было, т.е. соединение (1) проявляет очень высокую безопасность. На рак, в лечении которого применяется противоопухолевое средство, соответствующее настоящему изобретению, не налагается особое ограничение, и примеры рака включают рак желудочно-кишечного тракта, лейкемию, опухоль гипофиза, мелкоклеточный рак легких, рак щитовидной железы и нейроастроцитому. Противоопухолевое средство, соответствующее настоящему изобретению, пригодно для профилактики и/или лечения, среди вышеупомянутых видов рака, рака желудочно-кишечного тракта (в частности, рака поджелудочной железы, рака толстой кишки и рака желудка). Противоопухолевое средство, соответствующее настоящему изобретению, может содержать фармацевтически приемлемый носитель или вспомогательное вещество, и может приниматься перорально и парентерально. Противоопухолевое средство может приниматься перорально в виде твердой формы, такой как таблетки, гранулы, порошки или капсулы. Для приготовления таких твердых форм противоопухолевое средство может быть объединено с подходящим вспомогательным веществом, таким как наполнитель (например, лактоза, маннитол, кукурузный крахмал или кристаллическая целлюлоза), связующее (например, производное целлюлозы, аравийская камедь или желатин) и дезинтегрирующее средство (например, кальций карбоксиметилцеллюлоза) или смазка (например, тальк или стеарат магния). Такие твердые формы могут быть приготовлены в виде форм с контролируемым высвобождением с помощью использования такого основного материала покрытия, как фталат гидроксиметилцеллюлозы, сукцинат ацетата гидроксипропилметилцеллюлозы, фталат ацетата целлюлозы или метакрилатный сополимер. Противоопухолевое средство может также быть приготовлено в виде жидкой формы, такой как раствор, суспензия или эмульсия. Противоопухолевое средство, соответствующее настоящему изобретению, может приниматься парентерально в форме инъекции. Для приготовления инъекции противоопухолевое средство может быть объединено с, например, водой, этанолом, глицерином или общеупотребимым поверхносто-активным веществом. Противоопухолевое средство может быть приготовлено в виде суппозитория с помощью использования подходящего основного материала. Доза соединения (1), содержащаяся в противоопухолевом средстве, соответствующем настоящему изобретению, соответствующим образом определяется с учетом метода приема и формы продукта, а также признака болезни, возраста, пола и т.д. конкретных пациентов, которые в нем нуждаются. Ежедневная пероральная доза соединения (1) для взрослых обычно от 10 до 1000 мг, предпочтительно от 50 до 600 мг, более предпочтительно от 180 до 500 мг. Предпочтительно ежедневную пероральную дозу принимать один раз в день или дробным способом (два или три раза в день). Противоопухолевое средство, соответствующее настоящему изобретению, может приниматься в комбинации с противоопухолевым средством, применяемым в многокомпонентной комплексной терапии (например, по крайней мере одно противоопухолевое средство помимо противоопухолевого средства, соответствующего настоящему изобретению), или с радиотерапией, в которой эти противоопухолевые средства могут приниматься одновременно или раздельно с одинаковой периодичностью дозировки или с разными периодичностями, с помощью одинакового метода приема или различных методов приема. Таким образом, противоопухолевое средство, соответствующее настоящему изобретению, может применяться в комбинации с многокомпонентной комплексной химиотерапией или радиотерапией для того, чтобы лечить раковых пациентов. Если противоопухолевое средство, соответствующее настоящему изобретению, применяется в многокомпонентной комплексной терапии, то противоопухолевое средство может добавляться к различным используемым в комплексной терапии фармацевтическим средствам или может замещаться одним или двумя противораковыми средствами среди фармацевтических средств. Примеры противоопухолевых средств, которые предпочтительно применяются в комбинации с противоопухолевым средством, соответствующим настоящему изобретению, включают, но не ограничиваются, антиметаболиты, такие как фторурацил, гемцитабин гидрохлорид, метотрексат, цитарабин, флударабин; противоопухолевые антибиотики, такие как блеомицин гидрохлорид, митомицин С, доксорубицин гидрохлорид, даунорубицин гидрохлорид и идарубицин гидрохлорид; алкилирующие средства, такие как бусульфан, координационные металлокомплексы (карбоплатин и цисплатин), циклофосфамид, дакарбазин и мелфалан; нестероидные ингибиторы ароматазы, такие как анастрозол и эксеместан; иммунотерапевтические средства, такие как трастузумаб и ритуксимаб; ингибиторы митоза, такие как паклитаксел, доцетоксел гидрат, винкристин сульфат и винбластин сульфат; ингибиторы топоизомеразы, такие как иринотекан гидрохлорид; средства для гормональной терапии, такие как тамоксифен цитрат и лейпрорелин ацетат; и другие противоопухолевые средства, такие как левофолинат кальция, ингибиторы тирозинкиназы (например, гефитиниб), моноклональные антитела (например, цетуксимаб и бевацизумаб), ингибиторы матричных металлопротеаз и ингибиторы фарнезил трансфераз. Особенно предпочтительно прибавлять противоопухолевое средство, соответствующее настоящему изобретению, в течение использования гемцитабина гидрохлорида, который, как известно, проявляет эффект лечения рака поджелудочной железы, или прибавлять противоопухолевое средство к комбинированной терапии, использующей гемцитабин гидрохлорид и другие химиотерапевтические средства (например, фторурацил, левофолинат кальция, иринотекан гидрохлорид или координационные металлокомплексы). Если соединение (1) применяется в комбинации с другими противоопухолевыми средствами, доза соединения (1) или противоопухолевых средств соответственно определяется с учетом, например, идентичности каждого из противоопухолевых средств, симптома нуждающегося в них пациента и их метода приема. В многокомпонентной комплексной терапии доза соединения (1) равна описанной выше дозе. Оптимальные период приема, частота приема и лекарственная форма соединения (1) выбираются с учетом идентичности каждого из противоопухолевых средств, используемых в комбинации с соединением (1). Особенно, соединение (1) и по меньшей мере одно противоопухолевое средство (предпочтительно от одного до четырех противоопухолевых средств) принимаются одновременно или раздельно с одинаковой периодичностью или с различными периодичностями, в виде одинаковых или различных лекарственных форм. В многокомпонентной комплексной химиотерапии соединение (1) предпочтительно принимается перорально или внутривенно один или более раз в день. Противоопухолевое средство в основном принимается с помощью внутривенного вливания, но более предпочтительно принимать с помощью перорального пути ввиду того, что может быть выбрана простая лекарственная форма. Как описано ниже в примерах, при использовании противоопухолевого средства, соответствующего настоящему изобретению, в комбинации с другими противоопухолевыми средствами наилучший противоопухолевый эффект достигается без усиления побочных эффектов. Следовательно, при использовании противоопухолевого средства, соответствующего настоящему изобретению, в многокомпонентной комплексной химиотерапии доза другого противоопухолевого средства, проявляющего сильные побочные эффекты, может быть уменьшена. Противоопухолевое средство, соответствующее настоящему изобретению может продолжительно приниматься даже после химиотерапии, и таким образом предусматривается получение дальнейшего наилучшего противоопухолевого эффекта. Сообщается, что соединение (1) имеет высокое сродство связывания с крысиным гастриновым рецептором (значение Ki=0,24 нМ), и интрадуоденальный прием соединения (1) в дозах 0,17 мг/кг подавляет возбуждаемую гастрином секрецию гастриновой кислоты в крысах до 50% (гастроэнтерология 2001; А-311:1605). Напротив, противоопухолевое средство из настоящего изобретения требовало более высоких доз для проявления противоопухолевого эффекта. Примеры Настоящее изобретение будет далее описываться подробно, с ссылками на примеры и сравнительные примеры, но изобретение не ограничивается этими примерами. Противоопухолевый эффект и токсичность соединения (1) будут описаны в примерах 1-6. Получение противоопухолевого средства для настоящего изобретения будет описываться в примерах приготовления лекарственного средства 1-3. Пример 1 3 × 106 раковых клеток поджелудочной железы человека (MIAPaCa 2) были имплантированы подкожно в правую брюшную полость женских особей Balb/c голых мышей. После того как объем опухоли стал 100 мм3 и более, мышам из группы приема давали принимать перорально (R)-(-)-3-[3-(1-трет-бутилкарбонилметил-2-оксо-5-циклогексил-1,3,4,5-тетрагидро-2H-1,5-бензодиазепин-3-ил)уреидо]бензоат кальция (ранее названного здесь Пример 2 1 × 106 раковых клеток поджелудочной железы человека (PAN1VC) были имплантированы в поджелудочную железу мужских особей голых мышей. Со дня, следующего за днем имплантации опухоли, соединение A1 давали принимать перорально в дозах 30 мг/кг и 100 мг/кг ежедневно в течение 36 дней. В первый, третий и шестой день после имплантации опухоли вводили внутривенно гемцитабин гидрохлорид (Gemzar Injection (R)) в количестве 5 мг/кг. Процент подавления роста опухоли рассчитывался, основываясь на весах опухоли в каждой группе приема в сопоставлении с весами опухоли в контрольной группе. В результате, процент подавления одной дозы гемцитабина гидрохлорида (Gemzar Injection (R)), 30 мг/кг соединения A1 и 100 мг/кг соединения A1 были 32%, 19% и 23% соответственно. Напротив, при приеме гемцитабина гидрохлорида (Gemzar Injection (R)) и соединения A1 в комбинации процент подавления при 30 мг/кг и 100 мг/кг соединения A1 были 73% и 84% соответственно. Эти данные показывают, что комбинация соединения A1 и гемцитабина гидрохлорида (Gemzar Injection (R)) проявляет наилучший противоопухолевый эффект. Пример 3 1,5 × 106 раковых клеток прямой кишки человека (C170HM2) были введены внутрибрюшинно мужским особям голых мышей. После имплантации в группе приема мышам давали однократно ежедневно принимать перорально соединение A1 в количествах 3 мг/кг и 30 мг/кг. Тем временем, в положительной контрольной группе внутривенно вводили комбинацию 5-фторурацила (ранее названного здесь Напротив, процент подавления метастазирования в положительной контрольной группе был 63%. Эти данные показывают, что соединение A1 проявляет противометастазный эффект, сравнимый или больший, чем эффект химиотерапевтического средства. Пример 4 5 × 105 раковых клеток желудка человека (MGLVA1) были введены внутрибрюшинно женским особям SCID голых мышей. После имплантации мышам давали однократно ежедневно принимать перорально соединение A1 в дозах 3 мг/кг и 30 мг/кг. Продление времени жизни с помощью соединения A1 оценивалось потому, что эта модель, использующая MGLVA1, была моделью со смертельным исходом. На шестой день после начала приема коэффициент выживаемости был 6,7% в контрольной группе, тогда как коэффициент выживаемости в группе приема соединения А1 в дозе 30 мг/кг был 46,7%. Эти данные свидетельствуют, что соединение А1 оказывает эффект продления времени жизни после имплантации опухоли. Пример 5 1 × 106 раковых клеток прямой кишки человека (HT-29) были имплантированы подкожно в правую брюшную полость женских особей Balb/c голых мышей. Через четыре дня после имплантации опухоли соединение А1 давали принимать перорально мышам из группы приема в дозах 10, 30 и 100 мг/кг однократно ежедневно в течение 17 дней. В день, следующий за последним приемом, опухоль удалялась и взвешивалась. В контрольной группе мышам давали принимать плацебо, и вес опухоли измерялся подобным описанному выше способом. Процент подавления роста опухоли рассчитывался, основываясь на весе опухоли в каждой группе приема в сопоставлении с весом опухоли в контрольной группе. В результате, проценты подавления при 30 мг/кг и 100 мг/кг соединения A1 были 44% и 50% соответственно. Прием соединения A1 значительно подавлял рост опухоли дозозависимым способом. Пример 6 1 × 106 раковых клеток прямой кишки человека (HT-29) были имплантированы подкожно в правую брюшную полость женских особей Balb/c голых мышей. Через десять дней после имплантации опухоли соединение А1 давали принимать перорально мышам из группы приема в дозе 30 мг/кг однократно ежедневно в течение 12 дней. Для сравнения, мышам из положительных контрольных групп вводили внутрибрюшинно 5-FU в дозах 3, 10 и 30 мг/кг однократно ежедневно в течение 12 дней. Кроме того, смесь соединения А1 (30 мг/кг) и 5-FU (3, 10 и 30 мг/кг) давали принимать мышам в каждой комбинационной группе. В день, следующий за последним приемом, опухоль удалялась и взвешивалась. В контрольной группе мышам давали принимать плацебо и вес опухоли измерялся подобным описанному выше способом. Процент подавления роста опухоли рассчитывался, основываясь на весе опухоли в каждой группе приема в сопоставлении с весом опухоли в контрольной группе. Процент подавления при единичном приеме 30 мг/кг соединения A1 был 34%. Проценты подавления при единичном приеме 5-FU в дозах 3, 10 и 30 мг/кг были 24%, 30% и 58% соответственно. Напротив, при приеме соединения А1 в дозе 30 мг/кг и 5-FU в дозах 3, 10 и 30 мг/кг в комплексе проценты подавления были 31%, 54% и 76% соответственно. Эти данные свидетельствуют, что смесь соединения А1 и 5-FU проявляет наилучший противоопухолевый эффект. Пример 7 Небольшой участок (70-80 мг) раковых клеток поджелудочной железы человека (PANC-1) был имплантирован в поджелудочную железу женских особей SCID мышей (15 мышей на каждую группу). Через семь дней после имплантации в одной группе мышей давали принимать перорально соединение А1 в дозе 100 мг/кг однократно ежедневно. В другой группе мышей на седьмой, десятый и четырнадцатый день после имплантации вводили внутривенно гемцитабин гидрохлорид (Gemzar Injection (R)), в качестве достоверного контроля, в дозе 100 мг/кг. Продление времени жизни с помощью соединения A1 оценивалось, потому что эта модель, использующая PANC-1, была моделью со смертельным исходом. На сороковой день после начала приема (на 46 день после имплантации) коэффициент выживаемости был 46,7% в контрольной группе (группа приема плацебо), тогда как коэффициент выживаемости в группе приема соединения А1 (100 мг/кг) был 86,7%. Между тем, коэффициент выживаемости в группе приема гемцитабина был 93,3%. Эти данные свидетельствуют, что соединение А1 оказывает положительный эффект продления времени жизни после имплантации опухоли, сравнимый с химиотерапевтическим средством. Пример 8 Небольшой участок (70-80 мг) раковых клеток поджелудочной железы человека (PANC-1) был имплантирован в поджелудочную железу женских особей SCID мышей (15 мышей на каждую группу). Через семь дней после имплантации в одной группе мышей давали принимать перорально соединение А1 в дозе 100 мг/кг однократно ежедневно. В другой группе на седьмой, десятый и четырнадцатый день после имплантации мышей вводили внутривенно гемцитабин гидрохлорид (Gemzar Injection (R)) в дозе 100 мг/кг. Продление времени жизни с помощью соединения A1 оценивалось, потому что эта модель, использующая PANC-1, была моделью со смертельным исходом. Как показано в таблице 1, прием гемцитабина гидрохлорида («GEM» в таблице 1) (100 мг/кг) и соединения А1 (100 мг/кг) в комплексе продлевает время жизни. Эти данные свидетельствуют, что прием в комплексе соединения А1 и химиотерапевтического средства оказывает положительный эффект продления времени жизни после имплантации опухоли.
Тестовый пример 1 Тест на токсичность с помощью 28-дневного повторяющегося перорального приема крысами Соединение А1 давали принимать перорально шестинедельным SD крысам мужского и женского пола в дозах 30, 100, 300 и 1000 мг/кг в течение 28 дней повторяющимся способом. Ни в одной группе не наблюдались случаи смерти и не было обнаружено отклонения в весе тела, приеме пищи, офтальмологических исследованиях, анализе мочи, весе органа, данных вскрытия и гистопатологических исследованиях. Тестовый пример 2 Тест на токсичность с помощью 28-дневного повторяющегося перорального приема собаками Соединение А1 давали принимать перорально восьмимесячным собакам породы бигль мужского и женского пола в дозах 30, 100, 300 и 1000 мг/кг в течение 28 дней повторяющимся способом. Ни в одной группе не наблюдались случаи смерти и не было обнаружено отклонения в весе тела, приеме пищи, офтальмологических исследованиях, анализе мочи, весе органа, данных вскрытия и гистопатологических исследованиях. Пример выпуска 1 Соединение А1 (20 г), лактоза (315 г), кукурузный крахмал (125 г) и кристаллическая целлюлоза (25 г) смешиваются до однородной массы, и к получающейся в результате смеси прибавляют 7,5% водный раствор гидроксипропилцеллюлозы (200 мл). Смесь гранулируют при помощи экструзионного гранулятора, применяя решетку (диаметр отверстия 0,5 мм), и сразу же после этого результирующему продукту придают сферическую форму при помощи марумеризора с последующей сушкой, в результате чего получают гранулы. Пример выпуска 2 Соединение А1 (20 г), лактоза (100 г), кукурузный крахмал (36 г) и кристаллическая целлюлоза (30 г), кальций карбоксиметилцеллюлоза (10 г) и стеарат магния (4 г) смешивают до однородной массы. Полученной в результате смеси придают форму таблеток (200 мг каждая) при помощи одноштамповочной таблетирующей машины, имеющей пестик 7,5 мм в диаметре. Пример выпуска 3 Соединение А1 (100 мг), ацетат натрия (2 мг), уксусную кислоту (для регуляции pH до 5,8) (соответствующее количество) и дистиллированную воду (баланс) (составляя в итоге 10 мл на пробирку) выпускали в виде инъекции с помощью стандартного метода.
Формула изобретения
1. Противоопухолевое средство для лечения рака желудочно-кишечного тракта, включающее сочетание компонентов (А) и (В), где компонент (А) представляет собой 1,5-бензодиазепиновое производное, а именно, R-(-)-3-[3-(1-трет-бутилкарбонилметил-2-оксо-5-циклогексил-1,3,4,5-тетрагидро-2Н-1,5-бензодиазепин-3-ил)уреидо]бензойную кислоту или ее фармацевтически приемлемую соль, и компонент (В) представляет собой противоопухолевое средство. 2. Противоопухолевое средство по п.1, в котором компонент (А) представляет собой R-(-)-3-[3-(1-трет-бутилкарбонилметил-2-оксо-5-циклогексил-1,3,4,5-тетрагидро-2Н-1,5-бензодиазепин-3-ил)уреидо]бензойную кислоту или ее кальциевую соль. 3. Противоопухолевое средство по п.1 или 2, содержащее в качестве компонента (В) противоопухолевое средство, выбранное из группы, состоящей из антиметаболитов, противоопухолевых антибиотиков, алкилирующих средств, координационных металлокомплексов, нестероидных ингибиторов ароматазы, иммунотерапевтических средств, ингибиторов митоза, ингибиторов топоизомеразы, средств для гормональной терапии, ингибиторов тирозинкиназы, моноклональных антител, ингибиторов матричных металлопротеаз и ингибиторов фарнезил трансфераз. 4. Противоопухолевое средство по п.1 или 2, содержащее в качестве компонента (В) противоопухолевое средство, выбранное из группы, состоящей из антиметаболитов, координационных металлокомплексов, ингибиторов топоизомеразы, ингибиторов тирозинкиназы и моноклональных антител. 5. Противоопухолевое средство по п.1 или 2, содержащее в качестве компонента (В) противоопухолевое средство, представляющее собой антиметаболит. 6. Противоопухолевое средство по п.1 или 2, представляющее собой средство для лечения рака поджелудочной железы, рака толстой кишки и рака желудка. 7. Противоопухолевое средство по п.1 или 2, представляющее собой средство для перорального приема. 8. Применение 1,5-бензодиазепинового производного, представляющего собой R-(-)-3-[3-(1-трет-бутилкарбонилметил-2-оксо-5-циклогексил-1,3,4,5-тетрагидро-2Н-1,5-бензодиазепин-3-ил)уреидо]бензойную кислоту или ее фармацевтически приемлемую соль, в качестве компонента (А) для получения противоопухолевого средства для лечения рака желудочно-кишечного тракта, содержащего в качестве компонента (В) другое противоопухолевое средство. 9. Применение по п.8, где компонент (А) представляет собой R-(-)-3-[3-(1-трет-бутилкарбонилметил-2-оксо-5-циклогексил-1,3,4,5-тетрагидро-2Н-1,5-бензодиазепин-3-ил)уреидо]бензойную кислоту или ее кальциевую соль. 10. Применение по п.8 или 9, где компонент (В) представляет собой противоопухолевое средство, выбранное из группы, состоящей из антиметаболитов, противоопухолевых антибиотиков, алкилирующих средств, координационных металлокомплексов, нестероидных ингибиторов ароматазы, иммунотерапевтических средств, ингибиторов митоза, ингибиторов топоизомеразы, средств для гормональной терапии, ингибиторов тирозинкиназы, моноклональных антител, ингибиторов матричных металлопротеаз и ингибиторов фарнезил трансфераз. 11. Применение по п.8 или 9, где компонент (В) представляет собой противоопухолевое средство, выбранное из группы, состоящей из антиметаболитов, координационных металлокомплексов, ингибиторов топоизомеразы, ингибиторов тирозинкиназы и моноклональных антител. 12. Применение по п.8 или 9, где компонент (В) противоопухолевое средство в качестве компонента (В) представляет собой антиметаболит. 13. Применение по п.8 или 9, где противоопухолевое средство представляет собой средство для лечения рака поджелудочной железы, рака толстой кишки и рака желудка. 14. Применение по п.8 или 9, где противоопухолевое средство представляет собой средство для перорального приема. 15. Способ лечения рака желудочно-кишечного тракта, включающий введение эффективного количества сочетания компонентов (А) и (В), где компонент (А) представляет собой 1,5-бензодиазепиновое производное, а именно, R-(-)-3-[3-(1-трет-бутилкарбонилметил-2-оксо-5-циклогексил-1,3,4,5-тетрагидро-2Н-1,5-бензодиазепин-3-ил)уреидо]бензойную кислоту или ее фармацевтически приемлемую соль, и компонент (В) представляет собой противоопухолевое средство. 16. Способ по п.15, в котором компонент (А) представляет собой R-(-)-3-[3-(1-трет-бутилкарбонилметил-2-оксо-5-циклогексил-1,3,4,5-тетрагидро-2Н-1,5-бензодиазепин-3-ил)уреидо]бензойную кислоту или ее кальциевую соль. 17. Способ по п.15 или 16, в котором в качестве компонента (В) используют противоопухолевое средство, выбранное из группы, состоящей из антиметаболитов, противоопухолевых антибиотиков, алкилирующих средств, координационных металлокомплексов, нестероидных ингибиторов ароматазы, иммунотерапевтических средств, ингибиторов митоза, ингибиторов топоизомеразы, средств для гормональной терапии, ингибиторов тирозинкиназы, моноклональных антител, ингибиторов матричных металлопротеаз и ингибиторов фарнезил трансфераз. 18. Способ по п.15 или 16, в котором в качестве компонента (В) используют противоопухолевое средство, выбранное из группы, состоящей из антиметаболитов, координационных металлокомплексов, ингибиторов топоизомеразы, ингибиторов тирозинкиназы и моноклональных антител. 19. Способ по п.15 или 16, в котором в качестве компонента (В) используют противоопухолевое средство, представляющее собой антиметаболит. 20. Способ по п.15 или 16, рак представляет собой рак поджелудочной железы, рак толстой кишки и рак желудка. 21. Способ по п.15 или 16, где используют пероральное введение.
|
||||||||||||||||||||||||||||||||||||
 364
364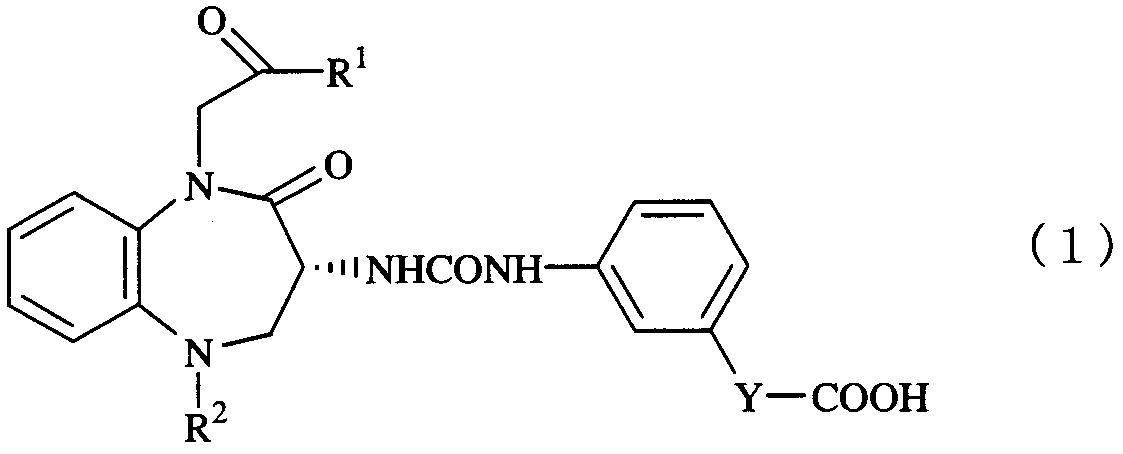
 -фенэтиламмониевой соли, бензиламмониевая соль и 4-метилбензиламмониевая соль, и соли органических и неорганических кислот, получаемые в результате присоединения кислоты. Из них основные соли предпочтительны. Среди основных солей неорганические соли более предпочтительны. Среди неорганических солей предпочтительны соли щелочноземельных металлов, причем кальциевая соль особенно предпочтительна.
-фенэтиламмониевой соли, бензиламмониевая соль и 4-метилбензиламмониевая соль, и соли органических и неорганических кислот, получаемые в результате присоединения кислоты. Из них основные соли предпочтительны. Среди основных солей неорганические соли более предпочтительны. Среди неорганических солей предпочтительны соли щелочноземельных металлов, причем кальциевая соль особенно предпочтительна. соединение А1
соединение А1 ) в дозах 10, 30 и 100 мг/кг ежедневно в течение 21 дня. В день, следующий за последним приемом, опухоль удаляли и взвешивали. Для сравнения, мышам из контрольной группы давали принимать перорально плацебо и вес опухоли измерялся подобным описанному выше способом. Процент подавления роста опухоли рассчитывался, основываясь на весе опухоли в каждой группе приема, в сопоставлении с весом опухоли в контрольной группе. В результате, проценты подавления при 30 мг/кг и 100 мг/кг соединения A1 были 40% и 42% соответственно. Прием соединения A1 значительно подавлял рост опухоли MIAPaCa 2 дозозависимым способом.
) в дозах 10, 30 и 100 мг/кг ежедневно в течение 21 дня. В день, следующий за последним приемом, опухоль удаляли и взвешивали. Для сравнения, мышам из контрольной группы давали принимать перорально плацебо и вес опухоли измерялся подобным описанному выше способом. Процент подавления роста опухоли рассчитывался, основываясь на весе опухоли в каждой группе приема, в сопоставлении с весом опухоли в контрольной группе. В результате, проценты подавления при 30 мг/кг и 100 мг/кг соединения A1 были 40% и 42% соответственно. Прием соединения A1 значительно подавлял рост опухоли MIAPaCa 2 дозозависимым способом.