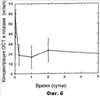
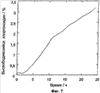
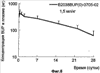
Патент на изобретение №2390331
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
(54) ЖИДКИЕ ДЕПО-ПРЕПАРАТЫ
(57) Реферат:
Изобретение относится к составам, образующим in vivo биологически активную депо-композицию, которые содержат маловязкие нежидкокристаллические смеси диацилглицерина, токоферола, фосфатидилхолина и кислородсодержащего органического растворителя, где биоактивный агент растворен или диспергирован в данной смеси, и где составы образуют жидкокристаллическую фазовую структуру при контакте с жидкостью тела. Указанные составы пригодны для получения парентеральных, непарентеральных и местных депо-композиций для замедленного высвобождения активных агентов. Изобретение относится также к способу получения и применению составов по изобретению и к способу доставки активного агента. Составы по изобретению обеспечивают пролонгированное контролируемое высвобождение активного агента в отсутствие эффектов «взрыва» или «задержки» при начальном высвобождении. 7 н. и 27 з.п. ф-лы, 8 ил., 20 табл.
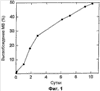
Настоящее изобретение относится к предшественникам препаратов (препаратам-предшественникам) для образования in situ липидных композиций с контролируемым высвобождением. В частности, данное изобретение относится к препаратам-предшественникам в форме маловязких смесей (таких как молекулярные растворы) амфифильных компонентов и по меньшей мере одного биоактивного агента, которые претерпевают по меньшей мере один фазовой переход под действием водных жидкостей, таких как жидкости тела, с образованием, таким образом, матрицы с контролируемым высвобождением, которая возможно является биоадгезивной. Многие биоактивные агенты, включая лекарственные средства, питательные вещества, витамины и так далее, имеют “функциональное окно”. То есть существует интервал концентраций, в котором можно наблюдать, что эти агенты обеспечивают некоторый биологический эффект. Когда концентрация в соответствующей части тела (например, местно или как это демонстрирует концентрация в сыворотке) падает ниже определенного уровня, данному агенту не может быть приписан полезный эффект. Аналогично, обычно существует верхний уровень концентрации, выше которого при увеличении концентрации не получают дополнительной пользы. В некоторых случаях увеличение концентрации выше конкретного уровня приводит к нежелательным или даже опасным эффектам. Некоторые биоактивные агенты имеют длительный период биологического полувыведения и/или широкое функциональное окно и, таким образом, их можно вводить периодически, поддерживая функциональную биологическую концентрацию на протяжении значительного промежутка времени (например, от 6 часов до нескольких суток). В других случаях скорость клиренса является высокой и/или функциональное окно является узким и, таким образом, для того, чтобы поддерживать биологическую концентрацию в рамках этого окна, требуются регулярные (или даже непрерывные) дозы малых количеств. Это может быть особенно затруднительным, когда желательными являются непероральные пути введения (например, парентеральное введение). Кроме того, в некоторых случаях, таких как подборка имплантов (например, заместителей суставов или пероральных имплантов), область желательного действия может оставаться недоступной для повторного введения. В таких случаях одиночное введение должно обеспечивать терапевтический уровень активного агента на протяжении всего промежутка времени, когда требуется эта активность. Использовались и предлагались различные способы замедленного высвобождения биологически активных агентов. Такие способы включают перорально вводимые композиции с медленным высвобождением, такие как покрытые таблетки, препараты, предназначенные для постепенного поглощения, такие как чрескожные пластыри и импланты с медленным высвобождением, такие как “палочки”, имплантированные под кожу. Один способ, посредством которого предлагается осуществить постепенное высвобождение биоактивного агента, представляет собой так называемую “депо”-инъекцию. В этом способе биоактивный агент готовят в виде препарата с носителями, обеспечивающими постепенное высвобождение активного агента в течение периода продолжительностью несколько часов или суток. Они часто основаны на разрушающейся матрице, которая постепенно диспергируется в теле с высвобождением активного агента. Наиболее распространенный из общепризнанных способов депо-инъекции основан на полимерной депо-системе. Обычно она представляет собой биоразлагаемый полимер, такой как поли(молочная кислота) (PLA) и/или поли(сополимер молочной и гликолевой кислоты) (PLGA), и может находиться в форме раствора в органическом растворителе, пред-полимера, смешанного с инициатором, инкапсулированных полимерных частиц или полимерных микросфер. Полимер или полимерные частицы удерживают активный агент и постепенно разрушаются, высвобождая данный агент посредством медленной диффузии и/или по мере поглощения матрицы. Примеры таких систем включают системы, описанные в US 4938763, US 5480656 и US 6113943, и могут приводить к доставке активных агентов на протяжении промежутка времени вплоть до нескольких месяцев. Эти системы, однако, имеют ряд ограничений, включая сложность изготовления и трудность стерилизации (особенно микросфер). Также заметным недостатком является местное раздражение, вызываемое молочной и/или гликолевой кислотой при высвобождении в месте инъекции. Также часто требуется довольно сложная процедура приготовления инъекционной дозы из предшествующего порошка. С точки зрения доставки лекарственного средства, недостатком полимерных депо-композиций также является допустимость только относительно низких загрузок лекарства и наличие профиля высвобождения “взрыв/задержка”. Природа полимерной матрицы, особенно когда ее применяют в виде раствора пред-полимера, вызывает начальный взрыв высвобождения лекарственного средства при первом введении композиции. После этого следует период медленного высвобождения, по мере того как начинается разрушение матрицы, с последующим в конце увеличением скорости высвобождения до желательного замедленного профиля. Такой профиль высвобождения с взрывом/задержкой может приводить к тому, что до достижения поддерживаемой функциональной концентрации концентрация активного агента in vivo непосредственно после введения “взрывается” до значений выше функционального окна, затем на протяжении лаг-периода понижается до значений ниже нижнего значения функционального окна. С функциональной и токсикологической точки зрения очевидно, что такой профиль высвобождения с взрывом/задержкой является нежелательным и может быть опасным. Он также может ограничивать равновесную концентрацию, которую можно обеспечить, из-за угрозы побочных эффектов в точке “пика”. Предпринимались попытки исследовать предыдущие депо-системы с точки зрения проблемы взрывного высвобождения. В частности, предлагалось применение гидролизованной полимолочной кислоты и включение блок-сополимеров полимолочной кислоты-полиэтиленгликоля с получением полимерной системы со “слабым взрывом”, описанной в US 6113943 и US 6630115. Эти системы обеспечивают улучшенные профили, но эффект взрыв/задержка сохраняется, и они не решают других вопросов, таких как раздражение, вызванное применением полимеров, дающих кислые продукты разрушения. Одна альтернатива более традиционным депо-системам на основе полимеров предложена в US 5807573. Там предложена основанная на липидах система диацилглицерина, фосфолипида и, возможно, воды, глицерина, этиленгликоля или пропиленгликоля, образующая систему введения в обращенной мицеллярной фазе “L1” или в кубической жидкокристаллической фазе. Так как эта депо-система образована физиологически хорошо переносимыми диацилглицеринами и фосфолипидами и не дает продуктов разрушения полимерных систем в виде молочной кислоты или гликолевой кислоты, у этой системы имеется меньшая тенденция инициировать воспаление в месте инъекции. Жидкокристаллические фазы, однако, имеют высокую вязкость, и фаза L2 также может быть слишком вязкой для легкости применения. Авторы US 5807573 также не дают какой-либо in vivo оценки профиля высвобождения данного препарата и, таким образом, не ясно, обеспечивается “взрывной” профиль или нет. Применение неламеллярных фазовых структур (таких как жидкокристаллические фазы) в доставке биоактивных агентов в настоящее время является достаточно традиционным. Такие структуры образуются, когда амфифильное соединение подвергают воздействию растворителя, так как амфифил имеет как полярные, так и неполярные группы, которые группируются с образованием полярных и неполярных участков. Эти участки могут эффективно солюбилизировать как полярные, так и неполярные соединения. Кроме того, многие структуры, образованные амфифилами в полярных и/или неполярных растворителях, имеют очень значительную площадь полярной/неполярной границы, на которой могут быть адсорбированы и стабилизированы другие амфифильные соединения. Амфифилы также можно вводить в композицию для защиты активных агентов, по меньшей мере до некоторой степени, от агрессивных биологических сред, включая ферменты, и обеспечивая, таким образом, полезный контроль стабильности и высвобождения активного агента. Образование неламеллярных участков на фазовых диаграммах амфифил/вода, амфифил/масло и амфифил/масло/вода является хорошо известным явлением. Такие фазы включают жидкокристаллические фазы, такие как кубическую Р, кубическую D, кубическую G и гексагональную фазу, которые на молекулярном уровне представляют собой жидкость, но демонстрируют в значительной степени дальный порядок, и фазу L3, которая содержит множественно взаимосвязанную непрерывную в обоих направлениях сеть двухслойных пластов, являющихся неламеллярными, но не имеют дальнего порядка жидкокристаллических фаз. В зависимости от кривизны их амфифильных пластов эти фазы можно описать как нормальные (средняя кривизна в направлении неполярного участка) или обращенные (средняя кривизна в направлении полярного участка). Неламеллярные жидкокристаллические и L3-фазы являются термодинамически устойчивыми системами. То есть они не являются просто метастабильным состоянием, которое будет разделяться и/или преобразовываться в слои, ламеллярные фазы и тому подобное, а представляют собой стабильную термодинамическую форму смеси липид/растворитель. Наряду с тем, что эффективность известных липидных депо-препаратов высока, существуют некоторые аспекты, в которых их эффективность меньше идеальной. В частности, предложенные кубические жидкокристаллические фазы являются относительно вязкими по природе. Это делает введение стандартным шприцем затруднительным и, возможно, болезненным для пациента и делает невозможной стерилизацию посредством фильтрации, так как данная композиция не может пройти через необходимую мелкопористую мембрану. В результате, композиции необходимо готовить в высокостерильных условиях, что увеличивает сложность изготовления. Когда используются фазы L2, они обычно имеют более низкую вязкость, но все же могут вызывать трудности при введении и обеспечивают доступ только к малому участку фазовой диаграммы. В частности, растворители, используемые в известных липидных препаратах, оказывают только ограниченный эффект в снижении вязкости смеси. Например, вода будет индуцировать образование высоковязкой жидкокристаллической фазы, а такие растворители как глицерин и гликоли имеют высокую вязкость и не обеспечивают какого-либо значительного полезного снижения вязкости композиции. Гликоли обычно также являются токсичными и плохо переносимыми in vivo и могут вызывать раздражение при местном применении. Кроме того, известные липидные композиции в фазе L2 с низким содержанием растворителя могут поддерживать только относительно низкий уровень многих биоактивных агентов из-за их ограниченной растворимости в компонентах смеси в отсутствии воды. В присутствии воды, однако, данные препараты принимают вид высоковязкой кубической жидкокристаллической фазы. Было бы очень полезным предложение депо-системы, которую можно было бы инъецировать с низкой вязкостью и которая обеспечила бы высвобождение требуемой концентрации биоактивного агента с использованием меньшего объема депо-композиции. Известные липидные депо-композиции также имеют практический доступ только к определенным фазовым структурам и композициям, потому что другие смеси или являются слишком высоковязкими для введения (например, смеси с высокими концентрациями фосфолипидов) или подвержены риску разделения на две или более отдельные фазы (такие как фаза L2 в равновесии с фазой, обогащенной фосфолипидом). В частности, концентрации фосфолипидов выше 50% недостижимы известными способами, и из фазовой диаграммы, показанной в US 5807573, очевидно, что желательная кубическая фаза стабильна при не более чем 40% фосфолипида. В результате, на практике было невозможно предложить депо-композиции с высокой концентрацией фосфолипидов или имеющие гексагональную жидкокристаллическую фазовую структуру. Авторы настоящего изобретения теперь установили, что путем получения препарата-предшественника, содержащего определенные амфифильные компоненты, по меньшей мере один биоактивный агент и биологически приемлемый растворитель, особенно в маловязкой фазе, такой как молекулярный раствор, можно получить препарат-предшественник, разрешающий многие недостатки предыдущих депо-препаратов. В частности, данный препарат-предшественник является простым в изготовлении, может быть подвергнут стерильной фильтрации, имеет низкую вязкость (делающую возможным легкое и менее болезненное введение), позволяет включить высокий уровень биоактивного агента (позволяя, таким образом, использовать меньшее количество композиции) и/или образует in vivo требуемую неламеллярную депо-композицию, имеющую контролируемый “взрывной” или “невзрывной” профиль высвобождения. Данные композиции также образованы из веществ, которые являются нетоксичными, биоприемлемыми и биоразлагаемыми. Кроме того, данный препарат-предшественник подходит для образования депо-композиций после парентерального введения и также после непарентерального (например, местного) введения в полости тела и/или на поверхности тела, или куда-либо в другое место. В первом аспекте настоящего изобретения, таким образом, предложен препарат-предшественник, содержащий маловязкую смесь: а) по меньшей мере одного нейтрального диациллипида и/или токоферола; б) по меньшей мере одного фосфолипида; в) по меньшей мере одного биосовместимого (предпочтительно кислородсодержащего) органического растворителя; где по меньшей мере один биоактивный агент растворен или диспергирован в маловязкой смеси, и где препарат-предшественник образует или способен образовывать по меньшей мере одну жидкокристаллическую фазовую структуру при контакте с водной жидкостью. В общем случае, водной жидкостью будет жидкость тела, такая как жидкость с поверхности слизистой оболочки, слезы, пот, слюна, желудочно-кишечная жидкость, внесосудистая жидкость, внеклеточная жидкость, интерстициальная жидкость или плазма, и данный препарат-предшественник будет образовывать жидкокристаллическую фазовую структуру при контакте с поверхностью тела, областью или полостью (например, in vivo), при контакте с водной жидкостью тела. Препарат-предшественник по изобретению перед введением обычно не будет содержать какого-либо значительного количества воды. Во втором аспекте изобретения также предложен способ доставки биоактивного агента в организм человека или животного, не являющегося человеком (предпочтительно млекопитающего), включающий введение (предпочтительно парентерально) препарата-предшественника, содержащего маловязкую смесь: а) по меньшей мере одного нейтрального диациллипида и/или токоферола; б) по меньшей мере одного фосфолипида; в) по меньшей мере одного биосовместимого (предпочтительно кислородсодержащего) органического растворителя; и по меньшей мере одного биоактивного агента, растворенного или диспергированного в маловязкой смеси, с образованием, таким образом, по меньшей мере одной жидкокристаллической фазовой структуры при контакте с водной жидкостью in vivo после введения. Предпочтительно, препарат-предшественник, введенный таким способом, представляет собой препарат-предшественник по изобретению, как описано выше. Способ введения, подходящий для указанного выше способа по изобретению, будет являться способом, пригодным для состояния, которое нужно лечить, и для используемого биоактивного агента. Парентеральное депо, таким образом, будет образовано посредством парентерального (например, подкожного или внутримышечного) введения, тогда как биоадгезивная непарентеральная (например, местная) депо-композиция может быть образована путем введения на поверхность кожи, слизистую оболочку и/или ногти, на глазную, назальную, оральную или внутреннюю поверхности или в полости, такие как назальная, ректальная, вагинальная или буккальная полости, периодонтальный карман, или полости, образованные после извлечения природной или имплантированной структуры, или перед введением импланта (например, сустава, стента, косметического импланта, зубного импланта, зубной пломбы или другого импланта). В другом аспекте настоящего изобретения также предложен способ получения жидкокристаллической композиции (особенно депо-композиции), включающий воздействие на препарат-предшественник, содержащий маловязкую смесь: а) по меньшей мере одного нейтрального диациллипида и/или токоферола; б) по меньшей мере одного фосфолипида; в) по меньшей мере одного биосовместимого (предпочтительно кислородсодержащего) органического растворителя; и по меньшей мере одного биоактивного агента, растворенного или диспергированного в маловязкой смеси, водной жидкости (особенно in vivo и/или особенно жидкости тела, как указано в данном описании изобретения). Предпочтительно вводимый препарат-предшественник представляет собой препарат-предшественник по настоящему изобретению, как описано в данном описании изобретения. Воздействие жидкости “in vivo” очевидно может происходить внутри организма или полости тела или может происходить на поверхности тела, такой как поверхность кожи, в зависимости от природы композиции. Жидкокристаллическая композиция, образующаяся таким способом, предпочтительно является биоадгезивной, как описано в данном описании изобретения. В еще одном аспекте настоящего изобретения предложен способ получения препарата-предшественника, подходящего для введения биоактивного агента (предпочтительно млекопитающему) субъекту, причем указанный способ включает получение маловязкой смеси: а) по меньшей мере одного нейтрального диациллипида и/или токоферола; б) по меньшей мере одного фосфолипида; в) по меньшей мере одного биосовместимого (предпочтительно кислородсодержащего) органического растворителя; и растворение или диспергирование по меньшей мере одного биоактивного агента в маловязкой смеси или по меньшей мере в одном из компонентов (а), (б) или (в) перед получением маловязкой смеси. Предпочтительно препарат-предшественник, полученный таким образом, представляет собой препарат по изобретению, как описано в данном описании изобретения. В еще одном аспекте настоящего изобретения предложено применение маловязкой смеси: а) по меньшей мере одного нейтрального диациллипида и/или токоферола; б) по меньшей мере одного фосфолипида; в) по меньшей мере одного биосовместимого (предпочтительно кислородсодержащего) органического растворителя; где по меньшей мере один биоактивный агент растворен или диспергирован в маловязкой смеси, в изготовлении препарата-предшественника для применения в замедленном введении указанного активного агента, где указанный препарат-предшественник способен образовать по меньшей мере одну жидкокристаллическую фазовую структуру при контакте с водной жидкостью. Термин “маловязкая жидкость”, при использовании в данном описании изобретения, указывает смесь, которую можно легко ввести субъекту и, в частности, легко ввести при помощи устройства из стандартного шприца и иглы. На это может указывать, например, возможность дозирования из одноразового шприца на 1 мл через иглу 22 awg (American Wire Gauge, американский сортамент проволоки) (или 23 размера) посредством ручного давления. В особенно предпочтительном воплощении маловязкая смесь должна быть смесью, способной проходить через стандартную мембрану для стерильной фильтрации, такую как фильтрующий шприц 0,22 мкм. В других предпочтительных воплощениях аналогичное функциональное определение подходящей вязкости можно определить как вязкость препарата-предшественника, который можно распылить с использованием компрессорного насоса или устройства для распыления под давлением с использованием традиционного оборудования для распыления. Типичный интервал подходящих вязкостей будет, например, от 0,1 до 5000 мПа·с, предпочтительно от 1 до 1000 мПа·с при 20°С. Обнаружилось, что путем добавления небольших количеств маловязкого растворителя, как указано в данном описании изобретения, можно обеспечить очень значительное изменение вязкости. Как показано на Фиг.2, например добавление только 5% растворителя может снизить вязкость в 100 раз, а добавление 10% может снизить вязкость вплоть до 10000 раз. Для того чтобы добиться такого нелинейного синергетического эффекта в снижении вязкости, важно, чтобы использовался растворитель подходящей низкой вязкости и подходящей полярности. Такие растворители включают растворители, описанные в данном описании изобретения ниже. Особенно предпочтительными примерами маловязких смесей являются молекулярные растворы и/или изотропные фазы, такие как фазы L2 и/или L3. Как описано выше, L3 представляет собой неламеллярную фазу взаимосвязанных пластов, которые имеют некоторую фазовую структуру, но не имеют упорядоченности дальнего порядка, присущей жидкокристаллической фазе. В отличие от жидкокристаллических фаз, которые обычно являются высоковязкими, фазы L3 имеют более низкую вязкость. Очевидно, что смеси фазы L3 и молекулярного раствора и/или частиц фазы L3, суспендированных в основном объеме молекулярного раствора из одного или более компонентов, также являются подходящими. Фаза L2 представляет собой так называемую “обращенную мицеллярную” фазу или микроэмульсию. Наиболее предпочтительные маловязкие смеси представляют собой молекулярные растворы, фазы L3 и их смеси. Фазы L2 являются менее предпочтительными, за исключением набухших фаз L2, как описано ниже. Согласно настоящему изобретению предложен препарат-предшественник, содержащий компоненты (а), (б), (в) и по меньшей мере один биоактивный агент, как указано в данном описании изобретения. Одним из значительных преимуществ препарата-предшественника по изобретению является то, что компоненты (а) и (б) можно ввести в состав в широком интервале соотношений. В частности, можно получить и использовать препараты-предшественники по настоящему изобретению, имеющие значительно более высокое отношение фосфолипида к нейтральному диациллипиду и/или токоферолу, чем это было достижимо ранее, без риска разделения фаз и/или неприемлемо высоких вязкостей в препарате-предшественнике. Массовые соотношения компонентов (а):(б), таким образом, могут быть любыми от 5:95, вплоть до 95:5. Предпочтительные соотношения обычно будут от 90:10 до 20:80 и более предпочтительно от 85:15 до 30:70. В одном предпочтительном воплощении изобретения имеет место большая доля компонента (б), чем компонента (а). То есть массовое соотношение (а):(б) составляет ниже 50:50, например от 48:52 до 2:98, предпочтительно от 40:60 до 10:90, и более предпочтительно от 35:65 до 20:80. Количество компонента (в) в препаратах-предшественниках по изобретению будет по меньшей мере достаточно для обеспечения малой вязкости смеси (например, молекулярного раствора, см. выше) компонентов (а), (б) и (в) и будет легко определимым для любой конкретной комбинации компонентов стандартными способами. Само фазовое поведение можно проанализировать посредством таких методик как визуальное наблюдение в комбинации с поляризационной световой микроскопией, ядерным магнитным резонансом, крио-трансмиссионной электронной микроскопией (крио-ТЕМ), для поиска растворов, фаз L2 или L3 или жидкокристаллических фаз. Вязкость можно измерить непосредственно стандартными способами. Как описано выше, подходящей практической вязкостью является такая, при которой смесь может быть эффективно введена посредством шприца и, особенно, стерильно отфильтрована. Ее легко оценить, как указано в данном описании изобретения. Максимальное количество компонента (в), которое нужно включить, будет зависеть от точного применения препарата-предшественника, но обычно желательные свойства будут обеспечены любым количеством, образующим маловязкую смесь (например, молекулярный раствор, см. выше) и/или раствор с достаточно низкой вязкостью. Так как введение излишне больших количеств растворителя субъекту обычно является нежелательным, количество компонента (в) обычно будет ограничено не более чем десятикратным (например, трехкратным) превышением минимального количества, требующегося для образования маловязкой смеси, предпочтительно не более чем пятикратным, и наиболее предпочтительно не более чем двухкратным превышением этого количества. Композиция по настоящему изобретению, однако, может содержать большее количество растворителя, чем было бы приемлемым в композиции с немедленным дозированием. Это происходит потому, что процесс, посредством которого медленно высвобождаются активные агенты (например, образование оболочек жидкокристаллической фазы, как описано в данном описании изобретения), также служит для задержки выхода растворителя из композиции. В результате, растворитель высвобождается на протяжении некоторого времени (например, минут или часов), а не мгновенно, и, следовательно, может лучше переноситься организмом. Более высокие доли растворителя также можно использовать для непарентеральных (например, местных) применений, особенно на поверхностях тела, где растворитель будет теряться путем испарения, а не поглощаться организмом. Для таких применений можно использовать количества, вплоть до 100 раз превышающие минимальное количество растворителя (например, вплоть до 95% по массе композиции, предпочтительно вплоть до 80% по массе, и более предпочтительно вплоть до 50% по массе), особенно когда желательным является очень тонкий слой образующегося непарентерального депо. Когда композиции по изобретению готовят как (непарентеральный) препарат в виде аэрозольного спрея (например, для местной или системной доставки активного агента), композиция также может содержать пропеллент. Такие композиции также могут включать высокую долю растворителя, компонента (в), как рассмотрено выше, так как большая часть данного растворителя будет испаряться при дозировании композиции. Подходящие пропелленты представляют собой летучие соединения, которые будут смешиваться с композицией по изобретению при давлении спреевого дозатора без образования высоковязких смесей. Очевидно, что они должны иметь приемлемую биосовместимость. Подходящие пропелленты будут легко определены простым тестированием, и примеры включают углеводороды (особенно C1-C4 углеводороды), диоксид углерода и азот. Также могут быть приемлемыми летучие фторуглеводороды, такие как HFC 134, 137а, 227еа и/или 152а. В качестве общего руководства, масса компонента (в) обычно будет составлять примерно от 0,5 до 50% от общей массы раствора (а-б-в). Эта доля предпочтительно (особенно для инъецируемых доз) составляет от 2 до 30%, и более предпочтительно от 5 до 20% по массе. Компонент “а”, как указано в данном описании изобретения, представляет собой нейтральный липидный компонент, содержащий полярную “головную” группу, а также неполярные “хвостовые” группы. Обычно головные и хвостовые участки липида связаны сложноэфирной группировкой, но это присоединение может происходить посредством эфирной, амидной, углерод-углеродной связи или другого присоединения. Предпочтительные полярные головные группы являются неионными и включают полиолы, такие как глицерин, диглицерин, и группировки сахаров (например, на основе инозитольных и глюкозильных группировок); и сложные эфиры полиолов, такие как ацетатные или сукцинатные сложные эфиры. Предпочтительные полярные группы представляют собой глицерин и диглицерин, особенно глицерин. В одном предпочтительном аспекте компонент (а) представляет собой диациллипид, в том смысле, что он имеет две неполярные “хвостовые” группы. Это обычно предпочтительно для применения моноацильных (“лизо”) липидов, так как они обычно хуже переносятся in vivo. Две неполярные группы могут иметь одинаковое или разное число атомов углерода, и каждая из них, независимо, может быть насыщенной или ненасыщенной. Примеры неполярных групп включают С6-С32алкильные и алкенильные группы, которые обычно присутствуют в виде сложных эфиров длинноцепочечных карбоновых кислот. Их часто описывают путем указания числа атомов углерода и числа ненасыщенных связей в углеродной цепи. Таким образом, CX:Z означает углеводородную цепь, имеющую Х атомов углерода и Z ненасыщенных связей. Конкретные примеры включают капроиловые (С6:0), каприлоиловые (С8:0), каприловые (С10:0), лауроиловые (С12:0), миристоиловые (С14:0), пальмитоиловые (С16:0), фитаноиловые (С16:0), пальмитолеоиловые (С16:1), стеароиловые (С18:0), олеоиловые (С18:1), элаидоиловые (С18:1), линолеоиловые (С18:2), линоленоиловые (С18:3), арахидоноиловые (С20:4), бегеноиловые (С22:0) и лигноцероиловые (С24:9) группы. Таким образом, типичные неполярные цепи основаны на жирных кислотах природных сложноэфирных липидов, включая капроновую, каприловую, каприновую, лауриновую, миристиновую, пальмитиновую, фитановую, пальмитолиновую, стеариновую, олеиновую, элаидиновую, линолевую, линоленовую, арахидоновую, бегеновую или лигноцеровую кислоты или соответствующие спирты. Предпочтительными неполярными цепями являются пальмитиновая, стеариновая, олеиновая и линолевая кислоты, особенно олеиновая кислота. Диациллипид, когда он используется как весь компонент “а” или его часть, может быть синтетическим или может происходить из очищенных и/или химически модифицированных природных источников, таких как растительные масла. В качестве компонента (а) можно использовать смеси любого числа диациллипидов. Наиболее предпочтительно этот компонент будет включать по меньшей мере часть диацилглицерина (DAG), особенно глицериндиолеат (GDO). В одном предпочтительном воплощении компонент (а) состоит из DAG. Это может быть один DAG или смесь DAG. Наиболее предпочтительным примером является DAG, содержащий по меньшей мере 50%, предпочтительно по меньшей мере 80%, и даже содержащий по существу 100% GDO. Альтернативным или дополнительным наиболее предпочтительным классом соединений для применения в качестве всего компонента (а) или его части являются токоферолы. Термин “токоферол”, при использовании в данном описании изобретения, означает неионный липидный токоферол, часто известный как витамин Е, и/или его любые подходящие соли и/или аналоги. Подходящими будут аналоги, обеспечивающие фазовое поведение, отсутствие токсичности и фазовый переход при воздействии водных жидкостей, характерное для композиций по настоящему изобретению. Такие аналоги обычно не будут образовывать жидкокристаллических фазовых структур в виде чистого соединение в воде. Наиболее предпочтительным токоферолом является сам токоферол, имеющий указанную ниже структуру. Очевидно что, особенно в случае, когда он очищен из природного источника, могут присутствовать небольшие доли нетокоферольного “загрязнения”, но этого будет недостаточно, чтобы изменить полезное фазовое поведение или отсутствие токсичности. Обычно токоферол будет содержать не более 10% соединений – нетокоферольных аналогов, предпочтительно не более 5%, и наиболее предпочтительно не более 2% по массе.
Токоферол В другом полезном воплощении изобретения компонент (а) состоит по существу из токоферолов, в частности из токоферола, показанного выше. Предпочтительная комбинация составных частей компонента (а) представляет собой смесь по меньшей мере одного DAG (например, GDO) с по меньшей мере одним токоферолом. Такие смеси включают от 2:98 до 98:2 по массе токоферола:GDO, например от 10:90 до 90:10 токоферола: GDО и, особенно, от 20:80 до 80:20 этих соединений. Также подходящими являются аналогичные смеси токоферола с другими DAG. Компонент “б” в настоящем изобретении представляет собой по меньшей мере один фосфолипид. Как и в случае с компонентом (а), этот компонент содержит полярную головную группу и по меньшей мере одну неполярную хвостовую группу. Различие между компонентами (а) и (б) принципиально лежит в полярной группе. Неполярные части, таким образом, можно подходящим образом получать из жирных кислот или соответствующих спиртов, рассмотренных выше для компонента (а). Обычно, фосфолипид будет содержать две неполярные группы, хотя одна или более чем одна составная часть этого компонента может иметь одну неполярную группировку. Когда присутствует более чем одна неполярная группа, они могут быть одинаковыми или разными. Предпочтительные полярные “головные” группы фосфолипидов включают фосфатидилхолин, фосфатидилэтаноламин, фосфатидилсерин и фосфатидилинозит. Наиболее предпочтительным является фосфатидилхолин (PC). В предпочтительном воплощении компонент (б), таким образом, состоит по меньшей мере из 50% PC, предпочтительно по меньшей мере из 70% PC, и наиболее предпочтительно по меньшей мере из 80% PC. Компонент (б) может по существу состоять из PC. Фосфолипидная часть даже более подходящим образом, чем диациллипидная часть, может происходить из природного источника. Подходящие источники фосфолипидов включают яйцо, сердце (например, коровье), мозг, печень (например, коровью) и растительные источники, включая сою. Такие источники могут обеспечить одну или более чем одну составную часть компонента (б), который может содержать любую смесь фосфолипидов. Так как препараты-предшественники по изобретению следует вводить субъекту для контролируемого высвобождения активного агента, предпочтительно, чтобы компоненты (а) и (б) являлись биосовместимыми. В этом отношении предпочтительно использовать, например, диациллипиды и фосфолипиды, а не моноацильные (лизо) соединения. Заметным исключением из этого является токоферол, как описано выше. Хотя он и имеет только одну алкильную цепь, он не является “лизо” липидом в традиционном смысле. Природа токоферола как хорошо переносимого незаменимого витамина очевидно делает его весьма подходящим по биосовместимости. Кроме того, наиболее предпочтительно, чтобы липиды и фосфолипиды компонентов (а) и (б) являлись встречающимися в природе (происходящими из природного источника или имеющими синтетическое происхождение). Встречающиеся в природе липиды имеют тенденцию вызывать меньшее воспаление и реакцию организма субъекта. Это не только более удобно для данного субъекта, но и может увеличивать время пребывания образующихся депо-композиций, особенно парентеральных депо, так как к месту введения рекрутируется меньшая активность иммунной системы. Однако в некоторых случаях может быть желательным включение в компоненты (а) и/или (б) доли липида, не встречающегося в природе. Это может быть, например, “эфирный липид”, в котором головные и хвостовые группы связаны простой эфирной связью, а не сложноэфирной связью. Такие липиды, не встречающиеся в природе, например, можно использовать для изменения скорости разрушения образующейся депо-композиции путем приобретения большей или меньшей растворимости или уязвимости в отношении механизмов разрушения, присутствующих в сайте высвобождения активного агента. Хотя в объем настоящего изобретения попадают все соотношения, в общем случае по меньшей мере 50% каждого из компонентов (а) и (б) будут липидами, встречающимися в природе. Эта доля предпочтительно будет составлять по меньшей мере 75% и может составлять вплоть до по существу 100%. Две особенно предпочтительные комбинации компонентов (а) и (б) представляют собой GDO с PC и токоферол с PC, особенно в интервале 30-90 мас.% GDO/токоферол, 10-60 мас.% PC и 1-30% растворителя (особенно этанола, NMP (N-метилпирролидона) и/или изопропанола). Помимо амфифильных компонентов (а) и (б) препараты-предшественники по данному изобретению также могут содержать дополнительные амфифильные компоненты в относительно низких содержаниях. В одном воплощении изобретения препарат-предшественник содержит вплоть до 10% (по массе компонентов (а) и (б) заряженного амфифила, особенно анионного амфифила, такого как жирная кислота. Предпочтительные жирные кислоты для этой цели включают капроновую, каприловую, каприновую, лауриновую, миристиновую, пальмитиновую, фитановую, пальмитолиновую, стеариновую, олеиновую, элаидиновую, линолевую, линоленовую, арахидоновую, бегеновую или лигноцеровую или соответствующие спирты. Предпочтительными жирными кислотами являются пальмитиновая, стеариновая, олеиновая и линолевая кислоты, особенно олеиновая кислота. Особенно полезным является то, чтобы данный компонент использовался в комбинации с катионным пептидным активным агентом (смотрите ниже). Полагают, что комбинация анионного липида и катионного пептида обеспечивает композицию с замедленным высвобождением особенной ценности. Отчасти это может быть обусловлено улучшенной защитой пептида от разлагающих его ферментов, присутствующих in vivo. Компонент “в” препаратов-предшественников по изобретению представляет собой кислородсодержащий органический растворитель. Так как данный препарат-предшественник должен образовывать депо-композицию после введения (например, in vivo) при контакте с водной жидкостью, желательно, чтобы этот растворитель переносился субъектом и был способен смешиваться с водной жидкостью и/или диффундировать или растворяться из препарата-предшественника в водную жидкость. Таким образом, предпочтительными являются растворители, имеющие по меньшей мере умеренную растворимость в воде. В предпочтительном варианте растворитель является таким, что относительно малое его добавление к композиции, содержащей (а) и (б), т.е. менее 20%, или более предпочтительно менее 10%, обеспечивает большое снижение величины вязкости на один порядок или более. Как раскрыто в данном описании изобретения, добавление 10% растворителя может обеспечить снижение величины вязкости на два, три или даже четыре порядка по сравнению с композицией, не содержащей растворителя, даже если эта композиция представляет собой раствор или фазу L2, не содержащую растворителя, или содержащую неподходящий растворитель, такой как воду (относится к особому случаю, рассмотренному ниже) или глицерин. Типичные растворители, подходящие для применения в качестве компонента (в), включают по меньшей мере один растворитель, выбранный из спиртов, кетонов, сложных эфиров (включая лактоны), простых эфиров, амидов и сульфоксидов. Примеры подходящих спиртов включают этанол, изопропанол и глицеринформаль. Моноолы (моноспирты) являются предпочтительными по сравнению с диолами и полиолами. Когда используются диолы и полиолы, их предпочтительно используют в комбинации с по меньшей мере равным количеством моноола или другого предпочтительного растворителя. Примеры кетонов включают ацетон и пропиленкарбонат. Подходящие простые эфиры включают диэтиловый эфир, гликофурол, диэтиленгликоля моноэтиловый эфир, диметилизобарбид и полиэтиленгликоли. Подходящие сложные эфиры включают этилацетат и изопропилацетат, а диметилсульфид представляет собой подходящий сульфидный растворитель. Подходящие амиды и сульфоксиды включают диметилацетамид (DMA), N-метилпирролидон (NMP), 2-пирролидон и диметилсульфоксид (DMSO). Менее предпочтительные растворители включают диметилизосорбид, тетрагидрофурфуриловый спирт, диглим и этиллактат. Так как препараты-предшественники предназначены для введения живому субъекту, необходимо, чтобы растворитель, компонент (в) был достаточно биосовместимым. Степень этой биосовместимости будет зависеть от способа применения и, так как компонент (в) может быть любой смесью растворителей, очевидно, что может присутствовать определенное количество растворителя, который в больших количествах не был бы приемлемым. В целом, однако, растворитель или смесь, образующая компонент (в), не должна вызывать неприемлемых реакций у субъекта при введении. Обычно такие растворители будут представлять собой углеводороды или предпочтительно кислородсодержащие углеводороды, причем обе группы возможно будут иметь другие заместители, такие как азотсодержащие группы. Предпочтительно, чтобы компонент (в) не содержал или небольшое количество компонента (в) содержало галогено-замещенные углеводороды, так как они склонны иметь более низкую биосовместимость. Когда необходима часть галогенированного растворителя, такого как дихлорметан или хлороформ, эта доля обычно будет минимизирована. Очевидно, что можно использовать больший диапазон растворителей, когда депо-композиции будут образовываться непарентерально, чем когда депо будет парентеральным. Компонент (в), при использовании в данном описании изобретения, может представлять собой один растворитель или смесь подходящих растворителей, но обычно будет иметь низкую вязкость. Это важно, так как одним из ключевых аспектов настоящего изобретения является то, что оно обеспечивает препараты-предшественники, которые имеют низкую вязкость, и главная роль подходящего растворителя состоит в снижении этой вязкости. Это снижение будет комбинацией эффекта более низкой вязкости растворителя и эффекта молекулярных взаимодействий между растворителем и липидной композицией. Одним наблюдением авторов настоящего изобретения является то, что кислородсодержащие растворители с низкой вязкостью, описанные в данном описании изобретения, осуществляют очень полезные и неожиданные молекулярные взаимодействия с липидными частями композиции, обеспечивая, таким образом, нелинейное снижение вязкости при добавлении небольшого объема растворителя. Вязкость “маловязкого” растворителя, компонента (в) (одиночный растворитель или смесь) обычно должна составлять не более 18 мПа·с при 20°С. Предпочтительно она составляет не более 15 мПа·с, более предпочтительно, не более 10 мПа·с и, наиболее предпочтительно, не более 7 мПа·с при 20°С. Растворитель, компонент (в), обычно, по меньшей мере частично, будет теряться при образовании in vivo депо-композиции или разбавляться путем поглощения воды из окружающего воздуха и/или ткани. Следовательно, предпочтительно, чтобы компонент (в) по меньшей мере до некоторой степени смешивался с водой и/или диспергировался в воде, и по меньшей мере он не должен отталкивать воду до такой степени, чтобы предотвращать поглощение воды. В этом отношении также предпочтительными являются кислородсодержащие растворители с относительно малым числом атомов углерода (например, вплоть до 10 атомов углерода, предпочтительно вплоть до 8 атомов углерода). Очевидно что, когда присутствует больше атомов кислорода, растворитель будет иметь тенденцию оставаться растворимым в воде при наличии большего числа атомов углерода. Отношение углерода к гетероатому (например, N, О, предпочтительно кислороду), таким образом, часто будет составлять приблизительно от 1:1 до 6:1, предпочтительно от 2:1 до 4:1. Когда используется растворитель с отношением, находящимся вне одного из этих предпочтительных интервалов, тогда предпочтительно он будет присутствовать в количестве не более 75%, предпочтительно не более 50%, в комбинации с предпочтительным растворителем (таким как этанол). Это можно использовать, например, для снижения скорости испарения растворителя из препарата-предшественника для того, чтобы контролировать скорость образования жидкокристаллического депо. Дополнительным преимуществом настоящих препаратов-предшественников является то, что в систему можно включить большее количество биоактивного агента. В частности, путем подходящего выбора компонентов (а)-(в) (особенно (в)) в препаратах-предшественниках можно растворить или суспендировать большие количества активного агента. Обычно липидные компоненты в отсутствие воды относительно плохо солюбилизируются, а в присутствии воды образует фазы, слишком вязкие для легкого введения. Более высокие соотношения биоактивного агента можно включить путем применения подходящих растворителей в качестве компонента (в), и этот уровень будет либо растворяться в депо-композиции, когда она образуется in situ, или может образовать микрокапли или микрокристаллы, которые будут постепенно растворяться и высвобождать активный агент. Подходящий выбор растворителя возможно будет осуществить путем традиционного экспериментирования в рамках представленных в данном описании изобретения указаний. Препараты-предшественники по настоящему изобретению обычно не содержат значительных количеств воды. Так как по существу из липидной композиции невозможно удалить все следовые количества воды, данную характеристику следует воспринимать как указание на то, что существует только такое минимальное следовое количество воды, которое не может быть легко удалено. Такое количество обычно будет составлять менее 1% по массе, предпочтительно менее 0,5% по массе препарата-предшественника. В одном предпочтительном аспекте препараты-предшественники по данному изобретению не содержат глицерин, этиленгликоль или пропиленгликоль и содержат не более чем следовое количество воды, как описано только что. Однако существует конкретное воплощение настоящего изобретения, в котором могут быть допустимы более высокие доли воды. Это имеет место, когда вода присутствует как часть компонента-растворителя в комбинации с дополнительным смешивающимся с водой компонентом (в) (одиночным растворителем или смесью). В этом воплощении может присутствовать вплоть до 10 мас.% воды, при условии что также присутствует по меньшей мере 3 мас.%, предпочтительно по меньшей мере 5%, и более предпочтительно по меньшей мере 7 мас.% компонента (в), и компонент (в) является смешивающимся с водой, и что образующиеся препараты-предшественники остаются невязкими и, таким образом, не образуют жидкокристаллической фазы. Обычно будет присутствовать большее количество компонента (в) по массе, чем масса воды, включенной в препарат-предшественник. Наиболее подходящие растворители для применения с водой в этом аспекте изобретения включают этанол, изопропиловый спирт, NMP, ацетон и этилацетат. Препараты-предшественники по настоящему изобретению содержат один или более чем один биоактивный агент (указываемый также в данном описании изобретения как “активный агент”). Активные агенты могут представлять собой любое соединение, обладающее нужным биологическим или физиологическим эффектом, такое как белок, лекарственное средство, антиген, питательное вещество, косметическое средство, ароматическое вещество, корригент, диагностический агент, фармацевтический агент, витамин или диетический агент, и будут приготовлены в виде препарата в количестве, достаточном для обеспечения in vivo концентрации на функциональном уровне (включая локальные концентрации для местных композиций). При некоторых обстоятельствах один или более из компонентов (а), (б) и/или (в) также может быть активным агентом, хотя предпочтительно активный агент не должен быть одним из этих компонентов. Наиболее предпочтительными активными агентами являются фармацевтические агенты, включая лекарственные средства, вакцины и диагностические агенты. Лекарственные агенты, которые можно доставлять посредством настоящего изобретения, включают лекарственные средства, которые действуют на клетки и рецепторы, периферические нервы, адренергические рецепторы, холинергические рецепторы, скелетные мышцы, сердечно-сосудистую систему, гладкие мышцы, систему циркуляции крови, эндокринную и гормональную систему, систему кровообращения, синоптические сайты, синаптические сайты нейроэффекторов, иммунную систему, репродуктивную систему, скелетную систему, аутакоидную систему, пищеварительную и экскреторную системы, гистаминную систему и центральную нервную систему. Примеры лекарственных средств, которые можно доставлять посредством композиции по настоящему изобретению, включают, без ограничения ими, антибактериальные агенты, такие как Особенно подходящие активные агенты включают агенты, которые в нормальных условиях имели бы короткое время пребывания в организме из-за быстрого распада или экскреции, и агенты с плохой пероральной биодоступностью. Они включают активные агенты на основе пептидов, белков и нуклеиновых кислот, гормоны и другие встречающиеся в природе агенты в их природной или модифицированной формах. Путем введения таких агентов в виде депо-композиций, образующихся из препарата-предшественника по настоящему изобретению, данные агенты обеспечиваются на постоянном уровне на протяжении промежутка времени, длительность которого может составлять сутки, недели или даже несколько месяцев, несмотря на наличие высоких скоростей клиренса. Это обеспечивает явные преимущества в показателях стабильности и соблюдения пациентами схемы и режима лечения по сравнению с многоразовой ежесуточной дозировкой в течение того же самого промежутка времени. В одном предпочтительном воплощении активный агент, таким образом, имеет время биологического полувыведения (при проникновении в кровяную систему) менее 1 суток, предпочтительно менее 12 часов, и наиболее предпочтительно менее 6 часов. В некоторых случаях это время может быть коротким, например 1-3 часа или менее. Подходящими агентами также являются агенты с плохой пероральной биодоступностью по сравнению с той, которая достигается при инъекции, когда активный агент также или альтернативно имеет биодоступность ниже 0,1%, особенно ниже 0,05% в пероральных препаратах. Активные агенты на основе пептидов и белков включают лекарственные средства для человека и ветеринарии, выбранные из группы, состоящей из адренокортикотропного гормона (АСТН) и его фрагментов, ангиотензина и родственных ему пептидов, антител и их фрагментов, антигенов и их фрагментов, атриальных натрийуретических пептидов, биоадгезивных пептидов, брадикининов и родственных им пептидов, кальцитонинов и родственных им пептидов, фрагментов рецепторных белков клеточной поверхности, хемотаксических пептидов, циклоспоринов, цитокинов, динорфинов и родственных им пептидов, эндорфинов и фрагментов Р-лидотропина, энкефалина и родственных ему белков, ингибиторов ферментов, иммуностимулирующих пептидов и полиаминокислот, фрагментов фибронектина и родственных ему пептидов, желудочно-кишечных пептидов, агонистов и антагонистов гонадотропин-высвобождающего гормона (GnRH), глюкагоноподобных пептидов, пептидов, высвобождающих гормон роста, иммуностимулирующих пептидов, инсулинов и инсулиноподобных факторов роста, интерлейкинов, гормонов, высвобождающих лютенизирующий гормон (LHRH), и родственных им пептидов, меланоцит-стимулирующих гормонов и родственных им пептидов, пептидов, относящихся к сигналу ядерной локализации, нейротензинов и родственных им пептидов, нейротрансмиттерных пептидов, опиоидных пептидов, окситоцинов, вазопрессинов и родственных им пептидов, паращитовидного гормона и его фрагментов, протеинкиназ и родственных им пептидов, соматостатинов и родственных им пептидов, субстанции Р и родственных ей пептидов, трансформирующих факторов роста (TGF) и родственных им пептидов, фрагментов фактора некроза опухолей, токсинов и токсоидов и функциональных пептидов, таких как противораковые пептиды, включая ангиостатины, антигипертензивные пептиды, пептиды, обладающие противосвертывающим действием, и противомикробные пептиды, выбранные из группы, состоящей из белков, таких как иммуноглобулины, ангиогенины, морфогенные белки кости, хемокины, колониестимулирующие факторы (CSF), цитокины, факторы роста, интерфероны (тип I и II), интерлейкины, лептины, факторы, ингибирующие лейкемию, факторы стволовых клеток, трансформирующие факторы роста и факторы некроза опухолей. Дополнительным значительным преимуществом депо-композиций по настоящему изобретению является то, что активные агенты высвобождаются постепенно на протяжении длительных периодов времени без необходимости повторной дозировки. Композиции, таким образом, очень подходят для ситуаций, когда соблюдение пациентом схемы и режима лечения является затруднительным, ненадежным, и когда очень важным является уровень дозировки, как, например, в случае активных агентов, изменяющих настроение, активных агентов с узким терапевтическим окном и активных агентов, вводимых детям или людям, чей стиль жизни не является совместимым с надежным режимом дозировки, а также для активных агентов, влияющих на “стиль жизни”, когда неудобство повторной дозировки может перевесить пользу от активного агента. Конкретные классы активных агентов, для которых этот аспект обеспечивает особое преимущество, включают контрацептивные средства, гормоны, включая контрацептивные гормоны, и особенно гормоны, используемые для детей, такие как гормон роста, антиаддиктивные агенты, добавки, такие как витамины или минеральные добавки, антидепрессанты и противоконвульсивные средства. Катионные пептиды являются особенно подходящими для применения, когда часть препарата-предшественника включает анионный амфифил, такой как жирную кислоту. В этом воплощении предпочтительные пептиды включают октреотид, ланреотид, кальцитонин, окситоцин, интерферон-бета и -гамма, интерлейкины 4, 5, 7 и 8 и другие пептиды, имеющие изоэлектрическую точку выше рН 7, особенно выше рН 8. В одном предпочтительном аспекте настоящего изобретения композиция по изобретению является такой, что фаза I2 или смешанная фаза, включающая фазу I2, образуется при воздействии водных жидкостей и в композицию включен полярный активный агент. Особенно подходящие полярные активные агенты включают пептидные и белковые активные агенты, олигонуклеотиды и небольшие водорастворимые активные агенты, включая перечисленные выше. Особенно интересными в этом аспекте являются пептид октреотид и другие пептиды, родственные соматостатину, интерфероны альфа и бета, глюкагоноподобные пептиды 1 и 2, лупрорелин и другой агонист GnRH, абареликс и другие антагонисты GnRH, интерферон-альфа и -бета, золендронат и ибандронат и другие бифосфонаты и полярный активный хлоргексидин (например, хлоргексидина диглюконат и хлоргексидина дигидрохлорид). Особым преимуществом настоящего изобретения при использовании в комбинации с белковыми/пептидными активными агентами является то, что подавляется агрегация активного агента. В одном предпочтительном воплощении настоящего изобретения, таким образом, предложен депо-предшественник и, в частности, депо-композиция, как описано в данном описании изобретения, которая содержит по меньшей мере один пептидный (например, антитело) или белковый активный агент, где не более 5% активного агента находится в агрегированной форме. Предпочтительно агрегировано не более 3%, и наиболее предпочтительно в агрегированной форме находится не более 2% (особенно менее 2%). Эта стабилизация неагрегированного белка является весьма полезной с точки зрения высокой эффективности, небольших побочных эффектов и предсказуемого профиля поглощения. Кроме того, ожидается, что белковые/пептидные терапевтические агенты будут иметь низкие уровни агрегации белка для того, чтобы обеспечить одобрение регулирующими органами. Количество биоактивного агента, которое нужно приготовить с препаратами-предшественниками по настоящему изобретению, будет зависеть от функциональной дозы и промежутка времени, на протяжении которого депо-композиция, образующаяся при введении, будет обеспечивать замедленное высвобождение. Обычно доза, приготовленная в виде препарата, для конкретного агента будет приблизительно эквивалентна нормальной суточной дозе, умноженной на число суток, на протяжении которых данный препарат должен обеспечить высвобождение. Очевидно, это количество необходимо будет подобрано с учетом любых побочных эффектов большой дозы в начале лечения, и, таким образом, это обычно будет максимальной используемой дозой. Точное количество, подходящее в любом случае, легко будет определено соответствующим экспериментированием. В одном воплощении препараты-предшественники по настоящему изобретению обычно будут вводить парентерально. Это введение обычно не будет осуществляться внутрисосудистым способом, но предпочтительно будет подкожным, внутриполостным или внутримышечным. Обычно такое введение будет осуществляться путем инъекции, причем данный термин используется в данном описании изобретения для указания любого способа, при котором препарат проходит через кожу, например посредством иглы, катетера или безигольного инжектора. В парентеральных (особенно подкожных) депо-предшественниках предпочтительными активными агентами являются активные агенты, подходящие для системного введения, включая антибактериальные агенты (включая амикацин, моноциклин и доксициклин), местные и системные анальгетики (включающие бупивакаин, трамадол, фентанил, морфин, гидроморфон, метадон, оксикодон, кодеин, аспирин, ацетаминофен), NSAIDS (нестероидные противовоспалительные лекарственные средства) (такие как ибупрофен, напроксен, кетепрофен, индометансин, сулиндак, толметин, салициловые кислоты, такие как салициламид, дифлунизал), ингибиторы Сох1 или Сох2 (такие как целекоксиб, рофекоксиб, валдекоксиб), противораковые агенты (включая октреотид, ланреотид, бусерелин, лупрорелин, госерелин, трипторелин, аворелин, деслореин, абареликс, дегареликс, фулвестрант, интерферон-альфа, интерферон-бета, дарбепоэтин-альфа, эпоэтин-альфа, -бета, -дельта и паклитаксел), антипсихотические средства (подобные бромперидолу, рисперидону, оланзапину, илоперидону, палиперадону, пипотиазину и зуклопентиксолу), противовирусные агенты, антиконвульсанты (например, тиагабин, топирамат или габапентин) или никотин, гормоны (такие как тестостерон и тестостерона ундеканоат, медроксипрогестерон, эстрадиол), гормоны роста (подобные гормону роста человека) и факторы роста (подобные гранулоцитарно-макрофагальному колониестимулирующему фактору). В альтернативном воплощении препараты по настоящему изобретению могут образовать непарентеральные депо, где активный агент медленно высвобождается на поверхности тела. В этом воплощении особенно важно, чтобы препараты-предшественники по изобретению и/или жидкокристаллические депо-композиции, образованные из них, предпочтительно были биоадгезивными. То есть композиции должны покрывать поверхность, на которую их нанесли и/или на которой они образуются, как это целесообразно, и должны оставаться на ней, даже когда эта поверхность подвергается воздействию потока воздуха или жидкости, и/или трения. Особенно предпочтительно, чтобы образующиеся жидкокристаллические депо-композиции были устойчивы к промыванию водой. Например, небольшой объем депо-предшественника можно нанести на поверхность тела и в течение 5 минут подвергать воздействию потока воды, величиной в пятьсот раз больше собственного объема депо-предшественника в минуту. После такой обработки композицию можно считать биоадгезивной, если было потеряно менее 50% биоактивного агента. Предпочтительно этот уровень потерь будет сравнимым при пропускании потока воды в 1000 раз и более предпочтительно в 10000 раз превышающего объем композиции в минуту в течение пяти или предпочтительно 10 минут. Хотя непарентеральные депо-композиции по настоящему изобретению могут поглощать некоторое количество или всю воду, необходимую для образования жидкокристаллической фазовой структуры, из биологических поверхностей, с которыми они контактируют, некоторое дополнительное количество воды также может поглощаться из окружающего воздуха. В частности, когда образуется тонкий слой с большой площадью поверхности, тогда сродство данной композиции к воде может быть достаточным для того, чтобы она образовала жидкокристаллическую фазовую структуру при контакте с водой, находящейся в воздухе. В данном воплощении “водная жидкость”, упоминаемая в данном описании изобретения, таким образом, по крайней мере частично представляет собой воздух, содержащий некоторое количество влаги. Непарентеральные депо-композиции обычно будут образовываться путем местного нанесения препарата-предшественника на поверхность тела или в естественную или искусственно полученную полость тела и/или на поверхность импланта. Это нанесение может представлять собой непосредственное нанесение жидкости, например путем распыления, окунания, промывания, нанесения с помощью прижимного или шарикового ролика, внутриполостной инъекции (например, в открытую полость с применением или без применения иглы), смазывания, закапывания (особенно в глаза), намазывания и аналогичными способами. Высокоэффективный способ представляет собой аэрозольное распыление или распыление нагнетанием, и очевидно требуется, чтобы вязкость препарата-предшественника была настолько низкой, насколько это возможно, и, таким образом, очень подходящей для композиций по изобретению. Непарентеральные депо, однако, можно использовать для введения системных агентов, например, трансмукозально или трансдермально. Непарентеральные депо также можно использовать для нанесения на поверхности, в частности имплантов и веществ, которые будут находиться в контакте с телом или с частью или с жидкостью тела. Приспособления, такие как импланты, катетеры и прочее, таким образом, можно обрабатывать путем окунания в препараты-предшественники по изобретению или опрыскивания препаратами-предшественниками по изобретению, которые будут формировать устойчивый слой, уменьшающий попадание инфекционного агента. Противоинфекционные активные агенты особенно подходят для этого аспекта. Состояния, особенно подходящие для каузального или симптоматического лечения при помощи местных биоадгезивных депо-композиций по настоящему изобретению, включают состояния кожи (такие как болезненность, возникающую по любой причине, включая образование трещин, царапин) и состояния кожи, включая экзему и герпес), состояния глаз, болезненность в гениталиях (включая болезненность, обусловленную генитальной инфекцией, такой как генитальный герпес), инфекции и состояния ногтей пальцев рук и/или ног (такие как бактериальные или грибковые инфекции ногтей, например, онихомикоз или паронихия). Биоадгезивные препараты местного типа также можно использовать для введения системных активных агентов (например, препарата), в частности путем поглощения кожей, пероральным, чрескожным или ректальным путями. Предпочтительным примером является лекарство против укачивания, а также никотин (например, для помощи против курения). Там, где это позволяет контекст “местное введение”, как оно упоминается в данном описании изобретения, включает системные агенты, нанесенные непарентерально на конкретный участок тела. Периодонтальные инфекции особенно подходят для лечения композициями по настоящему изобретению. В частности, известные композиции для лечения периодонтальной инфекции трудны в нанесении или, как правило, неэффективны. Наиболее широко используемая периодонтальная депо-композиция включает вставку в периодонтальное пространство коллагенового “чипа”, из которого высвобождается противоинфекционный агент. Этот чип трудно вставить, и он не соответствует по форме и объему периодонтальному пространству, так что могут остаться необработанными карманы с инфекцией. В отличие от этого, композиции по настоящему изобретению, наносимые в виде маловязких препаратов-предшественников, можно легко и быстро инъецировать в периодонтальное пространство, и они будут растекаться, точно адаптируясь к этому пространству и заполняя доступный объем. Данные композиции затем быстро поглощают воду с образованием прочного геля, устойчивого к водным условиям во рту. Единственная известная предыдущая попытка такого инъекционного периодонтального лечения основывалась на дисперсиях относительно высокой вязкости, которые было трудно наносить и которые были подвержены нежелательному разделению фаз. Теперь все эти недостатки исправлены в композициях по настоящему изобретению, как описано в данном описании изобретения. Очень подходящими активными агентами для периодонтального введения являются противоинфекционные агенты, особенно бензидамин, трамадол и хлоргексидин. Непарентеральные депо-композиции также очень полезны в комбинации с нефармацевтическими активными агентами, такими как косметические активные агенты, ароматизаторы, эфирные масла и пр. Такие нефармацевтические депо будут сохранять важные аспекты биоадгезии и замедленное высвобождение, обеспечивая продолжительные косметические эффекты, но и могут быть легко нанесены распылением или натиранием. Это также применимо к агентам, которые имеют как косметическую, так и медицинскую (особенно профилактическую) пользу, например солнцезащитным агентам. Так как депо-композиции местного типа обеспечивают прочные водостойкие барьеры, которые могут солюбилизировать высокие уровни активных агентов, они особенно подходят для солнцезащитных кремов в комбинации с агентами, поглощающими и/или рассеивающими ультрафиолетовый свет (UV, например UVa, UVb и/или UVc), особенно когда желательными являются высокие уровни защиты. Данные композиции, кроме того, являются высоко биосовместимыми и могут действовать, увлажняя и смягчая кожу в процессе солнечного воздействия. Композиции по изобретению, содержащие смягчающие агенты, такие как Aloe vera, также являются очень подходящими для применения с целью смягчения и увлажнения после воздействия солнечного света или на коже, которая является сухой, воспаленной или поврежденной, например из-за раздражения, ожога или ссадин. Активные агенты, особенно подходящие для непарентерального (например, местного) введения в виде депо, включающего пероральный, буккальный, назальный, глазной, кожный и вагинальный пути доставки, включают антибактериальные агенты, такие как хлоргексидин, хлорамфеникол, триклозан, тетрациклин, тербинафин, тобрамицин, фузидат натрия, бутенафин, метронидазол (последний особенно для (например, симптоматического) лечения розовых угрей – акне взрослых и некоторых вагинальных инфекций), антивирусные агенты, включая ацикловир, противоинфекционные агенты, такие как биброкатол, ципрофлоксацин, левофлоксацин, местные анальгетики, такие как бензидамин, лидокаин, прилокаин, ксилокаин, бупивакаин, анальгетики, такие как трамадол, фентанил, морфин, гидроморфон, метадон, оксикодон, кодеин, аспирин, ацетаминофен, NSAIDS, такие как ибупрофен, флурбипрофен, напроксен, кетопрофен, фенопрофен, диклофенак, этодалак, дифлунизал, оксапроксин, пироксикам, индометансин, сулиндак, толметин, салициловые кислоты, такие как салициламид и дифлунизал, ингибиторы Сох1 или Сох2, такие как целекоксиб, рофекоксиб или валдекоксиб, кортикостероиды, противораковые и иммуностимулирующие агенты (например, метиламинолевулината гидрохлорид, интерферон-альфа и -бета), антиконвульсанты (например, тиагабина топирамат или габапентин), гормоны (такие как тестостерон и тестостерона ундеканоат, медроксипрогестерон, эстрадиол), гормоны роста (подобные гормону роста человека) и факторы роста (подобные гранулоцитарно-макрофагальному колониестимулирующему фактору), иммунодепрессанты (циклоспорин, сиролимус, такролимус), никотин и противовирусные агенты (например, ацикловир). Некоторые конкретные активные агенты, которые, как обнаружили авторы изобретения, образуют высокоэффективные депо по изобретению, включают следующие: Для инъецируемых депо-продуктов длительного действия с гидрофильными активными агентами; 1) октреотид (или другие аналоги соматостатина, такие как ланреотид, для лечения саркоидных и VIР(вазоактивный интестинальный пептид)-продуцирующих опухолей и акромегалии). Формируемые подкожные депо, особенно с GDO и PC (фосфатидилхолином), имеющие продолжительность замедленного высвобождения более одного месяца, у которых выявляется менее 20% октреотида, распавшегося за один месяц в депо, разбухающем в воде при 37°С. Наблюдалась неожиданно хорошая стабильность, и обнаружилось, что она лучше, чем у октреотида, приготовленного в виде препарата в микросферах. Депо демонстрировало менее чем 5%-ное разрушение продукта препарата-предшественника за восемь недель при 4°С. 2) гормон роста человека. Для лечения нарушений роста и дефицитов гормона роста. Формируемое подкожное депо, особенно с GDO и PC, имеющее продолжительность замедленного высвобождения более двух недель. 3) интерферон-альфа для лечения рака и вирусных инфекций. Формируемое подкожное депо, особенно с GDO и PC, имеющее продолжительность замедленного высвобождения более одного месяца. 4) лейпролид. Формируемое депо имеют непрерывную доставку (предпочтительно непрерывную доставку в пределах терапевтического окна) в течение минимум одного месяца. Для инъекционных депо длительного действия липофильных/амфифильных активных агентов; 1) рисперидон, 2) оланзапин, 3) тестостерона ундеканоат. Формируемые депо (1)-(3) имеют непрерывную доставку (предпочтительно непрерывную доставку в пределах терапевтического окна) в течение минимум двух недель. Для местных биоадгезивных продуктов с контролируемым высвобождением для перорального (включая буккальное и периодонтальное) введения; 1) бензидамин (местный анальгетик, противовоспалительное средство) или другой местный анальгетик, анальгетик, противовоспалительное, антибактериальное, антигрибковое средство или их комбинация. Композиция обеспечивает устойчивый эффект на слизистой оболочке рта, в частности на поврежденной, имеющей повышенную чувствительность инфицированной слизистой оболочке, например у пациентов, страдающих мукозитом полости рта (индуцированным, например, химио- и радиотерапией). В частности для лечения мукозита полости рта. 2) трамадол (анальгетик). Обеспечивает композицию с устойчивым системным обезболивающим эффектом. 3) хлоргексидина глюконат (антибактериальное средство) для лечения периодонтальных и местных инфекций. Особенно для долговременного эффекта в периодонтальном кармане. Композиции приводят к образованию депо, высвобождающих хлоргексидин в течение более 1 ч, предпочтительно более 6 ч, наиболее предпочтительно более 24 ч, при нанесении в виде жидкости, образующей биоадгезивный гель in situ. Наблюдаемое время образования поверхностного геля составляло от 1 секунды до 5 мин. Формируемые депо (1)-(3) имеют высокую степень включения активного агента и высокую степень устойчивости к смыванию. Препараты-предшественники находятся в форме жидкости, вводимые в виде спрея или струи/промывки жидкостью в случае (1) и (2), и гелеобразующей жидкости в случае (3), когда жидкость наносят в периодонтальный карман, например, путем инъекции. Для непарентеральных (например, местных или системных) биоадгезивных продуктов с контролируемым высвобождением для назального введения; 1) фентанил (анальгетик) обеспечивает быстрое начало и постоянную продолжительную анальгезию при введении в виде спрея, 2) диазепам (средство против тревоги) обеспечивает непарентеральное назальное депо с системным эффектом, дающее быстрое начало и постоянную продолжительность. Вводится в виде спрея. Для местных биоадгезивных средств с контролируемым высвобождением для введения в глаза: 1) диклофенак (NSAID) с постоянной продолжительностью действия. Вводится в виде жидкости, образующей фазы in situ.,2) пилокарпин (парасимптомиметик, холинергический агонист) для лечения глаукомы, 3) левокабастина гидрохлорид, кетотифена фумарат, обеспечивающие получение жидкости для закапывания в глаза с целью обеспечения долговременного облегчения аллергического конъюнктивита с длительным промежутком времени между повторным нанесением, 4) пилокарпина гидрохлорид для лечения синдрома Шегрена, 5) дексаметазон (кортикостероид), 6) хлорамфеникол (главным образом бактериостатическое противоинфекционное средство), 7) индометацин (NSAID). Депо (1)-(7) приготовлены в виде жидкого спрея или, более предпочтительно, капель для непосредственного нанесения на поверхность глаза и обеспечивают in situ образование депо с высокой устойчивостью к смыванию слезами или истиранию из-за моргания/потирания глаз. Другие активные агенты, подходящие для офтальмологических композиций, включают антигистаминные препараты, стабилизаторы тучных клеток, нестероидные противовоспалительные лекарства (NSAIDs), кортикостероиды (например, для лечения аллергического конъюнктивита), противоглаукомные активные агенты, включая агенты, подавляющие/ингибирующие поступление (бета-блокирующие агенты: тимолол, бетаксолол, картеолол, левобунолол и др., местные ингибиторы карбоангидразы: дорзоламид, бринзоламид, симпатомиметики: эпинефрин, дипивефрин, клонидин, апраклонидин, бримонидин), агенты, облегчающие выделение (парасимпатомиметики (холинергические агонисты): пилокарпиновые аналоги простагландина и родственные соединения; атанопрост, травопрост, биматопрост, унопростон). Для непарентеральных (например, местных или системных) биоадгезивных продуктов с контролируемым высвобождением для дерматологического введения: 1) ацикловир (противовирусный препарат). Композиция образует биоадгезивный продукт, образующий пленку, с устойчивой продолжительностью действия. Наносится в виде спрея или жидкости. 2) тестостерона ундеканоат (гормональная недостаточность). Биоадгезивная композиция, образующая пленку, с устойчивой продолжительностью действия. Может наноситься в виде аэрозольного или нагнетаемого спрея из пульверизатора, или в виде жидкости. Особенно подходящими применениями дерматологических препаратов являются противоинфекционные дерматологические биоадгезивные депо для защиты в условиях окружающей среды, где вероятен контакт с инфекционными агентами (например, хирургия человека и ветеринарная хирургия, работа на скотобойне, определенные типы чистки и пр.). Биоадгезивные депо, образующиеся из композиции по изобретению, обеспечивают надежную и устойчивую защиту того, на что их наносят. Композиции с противоинфекционными агентами также можно использовать в ситуациях, когда стерильность кожи того, кто их наносит, важна для здоровья других, как, например, в случае медицинских сестер или докторов, посещающих в больнице многих пациентов, где необходимо избегать перекрестной инфекции. Предварительное покрытие композицией по настоящему изобретению может обеспечивать противодействие подхватыванию инфекционных агентов в одном месте и, таким образом, может предотвращать их перенос в другое место. Препараты-предшественники по настоящему изобретению образуют неламеллярные жидкокристаллические депо-композиции при воздействии водных жидкостей, особенно in vivo и при контакте с поверхностями тела. Термин “неламеллярный”, как он использован в данном описании изобретения, означает нормальную или обращенную жидкокристаллическую фазу (такую как кубическая или гексагональная фаза), или фазу L3, или их любую комбинацию. Термин “жидкокристаллический” означает все гексагональные, все кубические жидкокристаллические фазы и/или все их смеси. Термин “гексагональный”, как он использован в данном описании изобретения, означает “нормальную” или “обращенную” гексагональную (предпочтительно обращенную), а “кубический” означает любую кубическую жидкокристаллическую фазу, если не определено иное. Путем использования препаратов-предшественников по настоящему изобретению можно получить любую фазовую структуру, присутствующую в фазовой диаграмме компонентов (а) и (б) с водой. Это обусловлено тем, что могут быть получены препараты-предшественники с более широким интервалом относительных концентраций компонента, чем в предыдущих липидных депо-системах без риска разделения фаз, или приводящие к высоковязким растворам для инъекции. В частности, в настоящем изобретении предложено применение концентраций фосфолипидов выше 50% от общего содержания амфифила. Это обеспечивает доступ к фазам, наблюдаемым только при высоких концентрациях фосфолипидов, особенно к гексагональным жидкокристаллическим фазам. Для многих комбинаций липидов существуют только определенные неламеллярные фазы или существуют в любом стабильном состоянии. Неожиданным свойством настоящего изобретения является то, что композиции, как описано в данном описании изобретения, часто имеют неламеллярные фазы, которые отсутствуют со многими другими комбинациями компонентов. Следовательно, в одном особенно полезном воплощении настоящее изобретение относится к композициям, имеющим такую комбинацию компонентов, для которой существует область фазы I2 и/или L2, при разведении водным растворителем. Присутствие или отсутствие таких областей может быть легко протестировано для любой конкретной комбинации путем простого разбавления данной композиции водным растворителем и исследования образующихся фазовых структур способами, описанными в данном описании изобретения. В очень полезном воплощении композиции по данному изобретению могут образовать фазу I2 или смешанную фазу, включающую фазу I2, при контакте с водой. Фаза I2 представляет собой обращенную кубическую жидкокристаллическую фазу, имеющую дискретные водные области. Эта фаза обладает особым преимуществом при контролируемом высвобождении активных агентов и особенно в комбинации с полярными активными агентами, такими как водорастворимые активные агенты, так как дискретные полярные домены предотвращают быструю диффузию активных агентов. Депо-предшественники в L2 являются высокоэффективными в комбинации с образованием депо с фазой I2. Это обусловлено тем, что фаза L2 является так называемой “обращенной мицеллярной” фазой, имеющей непрерывную гидрофобную область, окружающую дискретные полярные ядра. L2, таким образом, обладает аналогичными преимуществами с гидрофильными активными агентами. На переходных стадиях после контакта с жидкостью тела данная композиция может содержать множественные фазы, так как образование начальной поверхностной фазы будет задерживать прохождение растворителя в ядро депо, особенно при введениях внутренних депо существенных размеров. Без привязки к теории предполагается, что это временное образование поверхностной фазы, особенно жидкокристаллической поверхностной фазы, обеспечивает значительное сглаживание профиля “взрыв/задержка” у настоящих композиций путем немедленного ограничения скорости обмена между композицией и ее окружением. Временные фазы могут включать (обычно в направлении снаружи к центру депо): Н11 или L Важно понимать, что препараты-предшественники по настоящему изобретению имеют низкую вязкость. В результате эти препараты-предшественники не должны находиться в насколько-либо объемной жидкокристаллической фазе, так как все жидкокристаллические фазы обладают значительно более высокой вязкостью, допустимой при введении посредством шприца или спреевого дозатора. Препараты-предшественники по настоящему изобретению, таким образом, будут находиться в не- жидкокристаллическом состоянии, таком как раствор, фаза L2 или L2, особенно раствор или L2. Фаза L2, как она используется в данном описании изобретения, предпочтительно представляет собой “разбухшую” фазу Ls, содержащую более 10 мас.% растворителя (компонент (в)), обладающего эффектом снижения вязкости. Она отличается от “концентрированной” или “неразбухшей” фазы L2, не содержащей растворителя или содержащей меньшее количество растворителя или растворитель (или смесь), которые не обеспечивают уменьшения вязкости, связанного с кислородсодержащими маловязкими растворителями, определенными в данном описании изобретения. При введении препараты-предшественники по настоящему изобретению претерпевают переход фазовой структуры от маловязкой смеси к высоковязкой (обычно прилипающей к ткани) депо-композиции. Обычно это будет переход от молекулярной смеси, разбухшей фазы L2 и/или L3 к одной или более (высоковязкими) жидкокристаллическими фазами, такими как нормальная или обращенная гексагональная или кубическая жидкокристаллическая фаза, или к их смесям. Как указано выше, после введения также могут иметь место дополнительные фазовые переходы. Очевидно, что полный фазовый переход не является необходимым для функционирования изобретения, но по меньшей мере поверхностный слой введенной смеси будет образовывать жидкокристаллическую структуру. Обычно этот переход будет быстрым по меньшей мере для поверхностного участка введенного препарата (эта часть в непосредственном контакте с воздухом, поверхностями тела и/или жидкостями тела). Наиболее предпочтительно, это будет происходить в течение нескольких секунд или минут (например, вплоть до 30 минут, предпочтительно вплоть до 10 минут, более предпочтительно 5 минут или менее). У остальной части композиции фаза может измениться до жидкокристаллической медленнее путем диффузии и/или по мере диспергирования поверхностного участка. В одном предпочтительном воплощении согласно настоящему изобретению, таким образом, предложен препарат-предшественник, как описано в данном описании изобретения, по меньшей мере часть которого образует гексагональную жидкокристаллическую фазу при контакте с водной жидкостью. Образованная таким образом гексагональная фаза может постепенно диспергироваться, высвобождая активный агент, или может впоследствии превращаться в кубическую жидкокристаллическую фазу, которая в свою очередь затем постепенно диспергируется. Полагают, что гексагональная фаза будет обеспечивать более быстрое высвобождение активного агента, в частности гидрофобного активного агента, чем кубическая фазовая структура, особенно фаза I2 и L2. Таким образом, когда гексагональная фаза образуется до кубической фазы, это будет приводить к первоначальному высвобождению активного агента для быстрого доведения концентрации вплоть до эффективного уровня с последующим постепенным высвобождением “поддерживающей дозы” по мере разрушения кубической фазы. Таким способом можно контролировать профиль высвобождения. Не будучи связанными теорией, полагают, что при воздействии (например, жидкостей тела) препараты-предшественники по настоящему изобретению теряют некоторое количество или весь органический растворитель, включенный в них (например, путем диффузии и/или испарения), и поглощают водную жидкость из окружающей среды тела (например, из влажного воздуха вблизи тела или из in vivo окружения) так, что по меньшей мере часть данного препарата образует неламеллярную, в высокой степени жидкокристаллическую фазовую структуру. В большинстве случаев эти неламеллярные структуры являются высоковязкими и являются легкорастворимыми или диспергируемыми в in vivo окружении, а также являются биоадгезивными и, таким образом, не легко отмываются или смываются. Кроме того, так как неламеллярная структура имеет большие полярные, неполярные и пограничные области, она является высокоэффективной в солюбилизации и стабилизации многих типов активных агентов и в их защите от механизмов разрушения. По мере того как депо-композиция, образующаяся из препарата-предшественника, постепенно разрушается на протяжении суток, недель или месяцев, активный агент постепенно высвобождается и/или диффундирует из композиции. Так как окружающая среда в рамках депо-композиции является относительно защищенной, препараты-предшественники по изобретению являются очень подходящими для активных агентов с относительно низким биологическим периодом полувыведения (см. выше). Неожиданным открытием авторов настоящего изобретения является то, что препараты-предшественники образуют депо-композицию, которая обладает очень незначительным эффектом “взрыва” в профиле высвобождения активного агента. Это является неожиданным, так как можно было бы ожидать, что маловязкая смесь (особенно если она представляет собой раствор) препарата-предшественника будет быстро терять активный агент при воздействии воды. На самом деле, препараты-предшественники по изобретению демонстрировали значительно меньший первоначальный “взрыв”, чем известные ранее депо-композиции на полимерной основе. Это проиллюстрировано в примерах, приведенных ниже, и на фигурах, прилагаемых к этому описанию изобретения. В одном воплощении согласно данному изобретению, таким образом, предложены инъецируемые препараты-предшественники и образующиеся в результате депо-композиции, где максимальная концентрация активного агента в плазме после введения не более чем в 5 раз превышает среднюю концентрацию за промежуток от 24 часов до 5 суток после введения. Это отношение предпочтительно превышает среднюю концентрацию не более чем в 4 раза и наиболее предпочтительно не более чем в 3 раза. В дополнительном аспекте данного изобретения местные композиции можно использовать для обеспечения физического барьера на поверхностях тела в отсутствие какого-либо активного агента. В частности, из-за очень сильного биоприлипания данных композиций из настоящих композиций путем распыления или нанесения жидкости могут образовываться “барьерные” покрытия так, чтобы снизить контакт с потенциальными инфекционными или раздражающими агентами или чтобы снизить загрязнение поверхностей тела. Стойкая природа данных композиций и устойчивость к смыванию обеспечивают таким барьерам, которые можно удобно наносить в виде жидкости или путем распыления, полезные характеристики. Данное изобретение далее будет дополнительно проиллюстрировано ссылкой на следующие неограничивающие примеры и прилагающиеся фигуры, где на Фиг.1 показано кумулятивное высвобождение метиленового синего (MB) из депо-препарата, содержащего PC/GDO/EtOH (45/45/10 мас.%) и образующего обращенную гексагональную фазу НII, при инъекции в избыток воды. На Фиг.2 показано снижение вязкости депо-предшественника при добавлении растворителей. PC/GDO (6/4) представляет собой предшественник обращенной гексагональной фазы НII и PC/GDO (3/7) представляет собой предшественник обращенной кубической фазы I2. Показано нелинейное снижение вязкости препарата-предшественника при добавлении N-метилпирролидона (NMP) и ЕtOН. На Фиг.3 показана концентрация в плазме кальцитонина лосося (sCT), в крысиной модели после подкожной инъекции различных PC/GDO/EtOH депо-предшественников (препаратов Е-I), содержащих 500 мкг sCT/г препарата. В качестве эталона использовали депо на основе кунжутного масла. На Фиг.4 показано начальное высвобождение in vivo (вплоть до 48 часов) sCT (то есть концентрация в плазме в крысиной модели) из двух разных депо-препаратов F и G после подкожной инъекции. На Фиг.5 показана концентрация в плазме (у крыс) октреотида (ОСТ) после подкожной инъекции депо-предшественника, содержащего PC/GDO/ЕtOН (36/54/10% масс.), содержащего 5 мг ОСТ/г препарата, соответствующих 0,5%-ной по массе загрузке октреотида. На Фиг.6 показана концентрация в плазме (у крыс) октреотида (ОСТ) после подкожной инъекции депо-препарата Р (см. пример 45), содержащего PC/GDO/EtOH (47,5/47,5/5,0 мас.%), содержащего 30 мг ОСТ/г препарата, соответствующих 3%-ной загрузке лекарственного средства. На Фиг.7 показано in vitro высвобождение хлоргексидина в избыток водной фазы из депо-препарата А (см. пример 47), содержащего PC/GDO/EtOH (36/54/10 мас.%), содержащего 50 мг хлоргексидина/г препарата, соответствующих 5%-ной загрузке лекарственного средства. На Фиг.8 показана концентрация бупренорфина в плазме (у крыс) после подкожного введения депо-препарата, содержащего этанол (ЕtOН) и диметилсульфоксид (DMSO). Данные представляют собой средние значения со стандартными отклонениями (пример 48). Примеры: Пример 1. Доступность различных жидкокристаллических фаз в депо путем выбора композиции Готовили инъецируемые препараты, содержащие различные доли фосфатидилхолина (“PC” – Epikuron 200), глицериндиолеата (GDO) и с ЕtOН в качестве растворителя для иллюстрации того, что после уравновешивания предшественника депо-препарата с избытком воды можно получить различные жидкокристаллические фазы. Подходящие количества PC и ЕtOН взвешивали в стеклянных сосудах, и смесь помещали на шейкер, пока PC не растворялся полностью с образованием прозрачного жидкого раствора. Затем добавляли GDO с образованием инъецируемого гомогенного раствора. Каждый препарат инъецировали в сосуд и уравновешивали с избытком воды. Фазовое поведение оценивали визуально и между скрещенными поляризаторами при 25°С. Результаты приведены в таблице 1.
Пример 2 In vitro высвобождение водорастворимого вещества Водорастворимый краситель, метиленовый синий (MB), диспергировали в препарате С (см. пример 1) в концентрации 11 мг/г препарата. Когда инъецировали 0,5 г препарата в 100 мл воды, образовывалась густая обращенная гексагональная фаза НII. Оптическую плотность MB, высвобождаемого в водную фазу, отслеживали при 664 нм в течение промежутка времени 10 суток. Изучение высвобождения проводили в колбе Эрленмейера при 37°С и слабом перемешивании магнитной мешалкой. Профиль высвобождения MB (см. Фиг.1) из гексагональной фазы показывает, что этот (и аналогичные) препараты представляют собой многообещающие депо-системы. Кроме того, данный препарат, по-видимому, дает слабый начальный взрыв, и профиль высвобождения показывает, что данное вещество может высвобождаться в течение нескольких недель; только примерно 50% MB высвобождается через 10 суток. Пример 3 Вязкость в PC/GDO (6:4) или PC/GDO (3:7) при добавлении растворителя (EtOH, PG и NMP) Смесь PC/GDO/ЕtOН получали согласно способу в примере 1. Весь или почти весь EtOH удаляли из смеси с помощью ротационного испарителя (вакуум, 40°С, 1 ч), и полученную твердую смесь взвешивали в стеклянном сосуде, после чего добавляли 2, 5, 10 или 20% растворителя (EtOH, пропиленгликоль (PG) или N-метилпирролидон (NMP)). Пробы оставляли уравновешиваться в течение нескольких суток, затем измеряли вязкость при скорости сдвига 0,1 с-1 реометром Physica UDS 200 при 25°С. Этот пример ясно показывает необходимость растворителя в случае некоторых депо-предшественников для того, чтобы получить инъецируемый препарат (см. Фиг.2). Вязкость смесей PC/GDO, не содержащих растворитель, возрастает с увеличением доли PC. Системы с низким отношением PC/GDO (больше GDO) являются инъецируемыми при более низкой концентрации растворителя. Пример 4 Композиция и in vitro фазовое исследование Препараты получали согласно способу, описанному в примере 1, с композициями согласно таблице 2. К каждому препарату добавляли активное вещество (пептид), кальцитонин лосося (sCT) в концентрации 500 мкг sCT/г препарата. Препараты разрабатывались в виде гомогенных суспензий для парентерального введения (незадолго до применения требовалось перемешивание, так как лекарственное средство не полностью растворено в системе PC/GDO/ЕtOН). Фазовое исследование в данном примере проводят в избытке крысиной сыворотки при 37°С для того, чтобы имитировать ситуацию in vivo. Таблица 2 показывает, что образуются те же самые фазы, что и в воде (сравните с таблицей 1).
Пример 5 Стерильная фильтрация препаратов с пониженной вязкостью Иногда для того, чтобы получить инъецируемый препарат и иметь возможность вводить данную систему обычным шприцем (см. пример 3), необходимо понизить вязкость различными растворителями. Другим важным эффектом понижающего вязкость растворителя является то, что препараты можно стерилизовать фильтрацией. Препараты Е-I в примере 4 исследовали в тесте стерильной фильтрации, используя фильтр 0,22 мкм (перед добавлением активного вещества). Препараты Е-Н успешно фильтровались, а препарат 1 нет, так как вязкость была слишком высокой. Следовательно, для этого препарата требовалась асептическая методика изготовления. Пример 6 Исследование in vivo высвобождения из депо-препаратов, введенных подкожно В исследовании in vivo высвобождения лекарственного средства у крыс использовали препараты Е-I из примера 4. Данные препараты вводили подкожно между лопатками, используя шприц (21 G, 0,6 мм × 30 мм), и доза sCT составляла 500 мкг/кг массы тела. Профиль высвобождения отслеживали в течение промежутка времени 13 суток. Концентрацию sCT в образцах плазмы крыс анализировали посредством иммуноферментного сэндвич-анализа, используя имеющийся в продаже набор от DSLabs. На Фиг.3 показаны результаты (n=4). В качестве эталонной липидной системы выбирали чистый триглицеридный носитель на основе кунжутного масла. Пример 7 Исследование in vivo высвобождения в начальной фазе Препараты F и G, как в примере 6, использовали в in vivo исследовании на крысах, предназначенном для изучения первоначального “взрывного эффекта”. Из Фиг.4 (n=8) видно, что ни один из исследованных препаратов не обладает сильным взрывным эффектом. Пример 8: Получение композиций депо-предшественника с различными растворителями В зависимости от композиции препарата, а также природы и концентрации активного вещества, предпочтительными могут быть определенные растворители. Препараты депо-предшественника (PC/GDO/растворитель (36/54/10)) готовили с различными растворителями; NMP, PG, PEG400, глицерином/ЕtOН (90/10) способом примера 1. Все композиции депо-предшественника представляли собой гомогенные однофазные растворы с вязкостью, которая делала возможной инъекцию через шприц (23G – т.е. игла 23-го размера; 0,6 мм × 30 мм). После инъекции предшественников препарата в избыток воды с предшественниками, содержащими NMP и PG, быстро формировалась жидкокристаллическая фаза в виде высоковязкого монолита. Жидкокристаллическая фаза имела обращенную кубическую мицеллярную (I2) структуру. С PEG400, глицерином/ЕtOН (90/10) процесс увеличения вязкости/отверждения был значительно более медленным, и первоначально жидкий предшественник трансформировался в мягкий, до некоторой степени клейкий кусок. Различие во внешнем виде возможно отражает более медленное растворение PEG400 и глицерина в избытке водной фазы по сравнению с ЕtOН, NMP и PG. Пример 9: Получение депо-композиции, содержащей гормон роста человека (hGH) Гормон роста человека (hGH) играет критическую роль в стимулировании роста и развития организма и вовлечен в образование мышечного белка и в разрушение жиров. Недостаток данного гормона неблагоприятно влияет на многочисленные процессы в теле, такие как липидный профиль, инсулиновый статус, физическая работоспособность, плотность минеральных компонентов костей и качество жизни. Целевая доза каждые 2 недели оценивается как 0,10-0,24 мг/кг массы тела. 1 мл предшественника 2-недельного депо-препарата получали путем последовательного смешивания 10 мг hGH и 360 мг PC в 0,1 мл NMP. Добавляли к смеси 540 мг GDO с получением маловязкого предшественника депо-препарата. Инъекция данного предшественника препарата в избыток воды (шприц 23G; 0,6×30 мм) приводила к монолитной жидкокристаллической фазе (структура I2). Пример 10: Получение депо-композиции, содержащей умеренно растворимое активное вещество Рисперидон представляет собой нейролептическое лекарственное средство, принадлежащее к химическому классу производных бензизоксазола. Он является очень сильным дофаминовым блокатором (антагонистом); т.е. он ингибирует функционирование дофаминовых рецепторов, он является практически нерастворимым в воде и имеет lоg(Р)=3,49. 1 г депо-препарата, содержащего 50 мг рисперидона, получали путем растворения активного вещества в 0,7 г смеси 95 мас.% ЕtOН (99,5%) и 5 мас.% уксусной кислоты. В этом растворе затем растворяли 0,34 г PC и 0,51 г GDO с последующим уменьшением количества растворителя до 0,15 г (0,55 г выпаривали под вакуумом). Композиция конечного гомогенного и прозрачного депо-препарата с 50 мг рисперидона представляла собой PC/GDO/растворитель/рисперидон (32/49/14/5). Инъекция данного предшественника препарата в избыток воды (шприц 23G; 0,6 мм × 30 мм) приводила к образованию монолитной жидкокристаллической фазы (структура I2). =To есть данное количество активного вещества (5%) не меняло образование монолита и фазовое поведение после воздействия водной среды. Пример 11: Альтернативное получение депо-композиции, содержащей рисперидон Препарат депо-предшественника с рисперидоном также можно приготовить, используя смесь растворителей, состоящую из 90 мас.% ЕtOН (99,5%) и 10 мас.% уксусной кислоты. 50 мг рисперидона растворяли в 0,7 г смеси растворителей, после чего в этом растворе растворяли 0,36 г PC и 0,54 г GDO. 0,60 г смеси растворителей упаривали под вакуумом до гомогенного и прозрачного предшественника депо-препарата с 50 мг рисперидона (PC/GDO/растворитель/рисперидон (34/51/10/5)). Инъецирование предшественника препарата в избыток воды (шприц 23G; 0,6 мм × 30 мм) приводило к образованию монолитной жидкокристаллической фазы (структура I2). То есть данное количество активного вещества (5%) не меняло образование монолита и фазовое поведение после воздействия водной среды. Пример 12: Температурная стабильность депо-композиции, содержащей труднорастворимое активное вещество. Препараты депо-предшественника с рисперидоном в примерах 10 и 11 тестировали на устойчивость против кристаллизации при хранении. Каждый препарат оставался стабильным при 25°С по меньшей мере в течение двух недель и при +8°С в течение по меньшей мере одной недели. Пример 13: Получение депо-композиции, содержащей бензидамин Бензидамин представляет собой нестероидное противовоспалительное лекарственное средство и широко используется в качестве местного лекарственного средства при воспалительных состояниях. 1 г депо-препарата, содержащего 1,5 мг бензидамина, получали путем растворения активного вещества в смеси PC/GDO/EtOН (36/54/10), полученной как описано в примере 1. Депо-композиция была устойчивой против кристаллизации в процессе хранения при 25°С в течение по меньшей мере двух недель. Уравновешивание предшественника препарата с избытком воды приводило к высоковязкой монолитной жидкокристаллической фазе (структура I2). Пример 14: Устойчивость поведения препарата в отношении изменений качества эксципиента Препараты депо-предшественников получали с использованием нескольких различных сортов GDO (поставленных Danisco, Dk), таблица 3, используя способ по примеру 1. Конечные депо-предшественники содержали 36 мас.% PC, 54 мас.% GDO и 10 мас%. ЕtOН. Внешний вид депо-предшественников был нечувствительным к изменению используемого сорта, и после контакта с избытком воды образовывался монолит с поведением обращенной мицеллярной кубической фазы (структура I2).
Пример 15: Получение депо-композиции, содержащей насыщенный PC (Epikuron 200SH). Препараты депо-предшественника получали с различными количествами PC, содержащего насыщенные углеводородные цепи, путем добавления Epicuron 200SH непосредственно к смеси PC/GDO/EtOH, полученной как в примере 1. Данные препараты представлены в таблице 4. Все препараты предшественников при комнатной температуре представляли собой гомогенные однофазные образцы, тогда как с увеличением количества Epicuron 200SH они становились более вязкими. Инъецирование депо-предшественника в избыток воды давало монолит, содержащий обращенную мицеллярную кубическую структуру (I2). Монолиты, образовавшиеся из образцов, содержащих более значительные количества Epicuron 200SH, становились мутными, что, возможно, указывает на сегрегацию между Epicuron 200SH и другими компонентами при воздействии воды и образовании фазы I2.
Пример 16: Получение депо-предшественника, являющегося дисперсией или раствором пептида, кальцитонина лосося Дисперсия sCT образовывалась путем добавления 500 мкг sCT/г препарата к раствору PC/GDO/EtOH (36/54/10), полученному как в примере 1. При альтернативном способе 500 мкг sCT растворяли в избытке ЕtOН, затем добавляли PC и GDO. Затем концентрацию растворителя снижали (упаривание ЕtOН) с получением гомогенного (активное лекарство в растворе) препарата. Эту последнюю методику можно использовать для получения более высоких загрузок лекарственного средства. Этим способом можно было получить композиции предшественника, соответствующие по меньшей мере 1500 мкг растворенного sCT на грамм конечной композиции депо-предшественника. Пример 17: Изучение in vivo высвобождения из депо-препарата, введенного подкожно Две композиции sCT, описанные в примере 16, вводили в in vivo крысиной модели путем подкожной инъекции (между лопаток). Обнаружено, что первый депо-предшественник, содержащий диспергированный sCT, дает в некоторой степени нестабильные начальные концентрации в плазме, тогда как второй депо-предшественник, содержащий растворенный в нем sCT, давал намного более стабильные начальные уровни в плазме (см. таблицу 5).
Пример 18: Получение депо-композиции, содержащей пептид октреотид Октреотид представляет собой ацетатную соль синтетического октапептида и аналогичен гормону соматостатину. Октреотид снижает образование таких веществ как гормон роста, инсулин и глюкагоны. Его используют в лечении акромегалии и для ослабления приливов и водной диареи, вызванной метастатическими раковыми опухолями (карциноидный синдром) или опухолями, названными опухолями, вырабатывающими вазоактивный интестинальный пептид (випомы). 24 мг или 60 мг октреотида растворяли в 0,1 г ЕtOН. Затем растворяли в этом растворе 0,36 г PC и 0,54 г GDO и получали предшественник депо-препарата. Инъекция данного предшественника препарата в избыток водной фазы (шприц 23G; 0,6 мм × 30 мм) приводила к образованию монолитной жидкокристаллической фазы (структура h). To есть октреотид (2,4% или 6,0%) не изменял образование монолита и фазовое поведение после воздействия водного окружения. Препараты депо-предшественника с октреотидом в этом примере тестировали на устойчивость против кристаллизации во время хранения. Каждый препарат оставался стабильным при 4-8°С в течение по меньшей мере двух недель. Пример 19: Изучение in vivo высвобождения из депо-препарата, содержащего октреодид, введенного подкожно В in vivo крысиной модели высвобождение лекарственного средства октреотида отслеживали на протяжении 28 суток. Данные препараты вводили подкожно между лопаток, используя шприц (23G; 0,6 мм × 30 мм). Концентрацию октреотида в плазме крыс отслеживали в течение промежутка времени 28 суток (см. Фиг.5). Доза составляла 5 мг/кг, и объем составлял 1 мл/кг, что соответствовало загрузке лекарственного средства – 0,5% октреотида – в предшественнике депо-препарата (PC/GDO/EtOH (36/54/10)). Из Фиг.5 (n=3) видно, что исследованный препарат дает профиль высвобождения по существу без взрывного эффекта. Фиг.5 показывает уровни октреотида в плазме у крысиной модели после введения предшественника препарата октреотида (0,5% по октреотиду). Пример 20: Разрушение депо-препарата у крысы Крысе инъецировали разные объемы (1, 2, 6 мл/кг) депо-предшественника (36 мас.% PC, 54 мас.% GDO и 10 мас.% ЕtOН) и снова удаляли через промежуток времени 14 суток. Обнаружено, что по истечении этого времени у крысы подкожно все еще присутствовали значительные количества данных препаратов, см. таблицу 6.
Пример 21: Изучение in vitro образования депо-монолита после инъекции предшественника депо-препарата между костью и надкостницей. Предшественник (36 мас.% PC, 54 мас.% GDO и 10 мас.% ЕtOН, полученный как описано в примере 1) инъецировали посредством шприца между костью и надкостницей. Наблюдали, что данная композиция распространялась с заполнением пустот и после захвата водных жидкостей образовывала монолит, который был биоадгезивным как по отношению к кости, так и к надкостнице. Пример 22: Биоадгезивный спрей препарата депо-предшественника Обнаружено, что пульверизатор является удобным способом местного нанесения препарата, например, на кожу или на слизистую оболочку полости рта. Препарат депо-предшественника, полученный как в примере 1 (36 мас.% PC, 54 мас.% GDO и 10 мас.% ЕtOН), распыляли при помощи пульверизатора на кожу и слизистую оболочку рта. Вскоре после нанесения образовывалась пленка с механическими свойствами твердого тела. Пример 23: Устойчивость пленки местного типа После нанесения препарата депо-предшественника на кожу, как описано в примере 22 (36 мас.% PC, 54 мас.% GDO и 10 мас.% ЕtOН), нанесенный препарат подвергали воздействию промывной воды (10 л/мин) в течение 10 минут. Препарат демонстрировал превосходные биоадгезивные свойства и устойчивость против смывания, и не было замечено никакой потери препарата. Пример 24: Образование кубической фазы со свойствами твердого тела после воздействия воздуха на препарат депо-предшественника После воздействия воздуха (комнатная температура, относительная влажность 40%) на препарат депо-предшественника, полученный, как описано в примере 1 (36 мас.% PC, 54 мас.% GDO и 10 мас.% ЕtOН), в течение по меньшей мере 3 часов образовывалась твердая кубическая фаза. Это образование кубической фазовой структуры демонстрирует, что пленка местного типа после нанесения будет приобретать в массе свойства неламеллярного депо без необходимости непосредственного воздействия избытка водной жидкости. Пример 25: Препарат для лечения периодонтита и перимплантита Для того чтобы лечить периодонтит или перимплантит, в периодонтальный карман инъецируют антибактериальный препарат, и обычно желательным является длительный эффект данного препарата. 100 мкл препарата, такого как получен в примере 1, с добавлением антибиотика хлоргексидина (PC/GDO/ЕtOН/хлоргексидин (35/53/10/2)) инъецируют через шприц в периодонтальный карман крысы. Наблюдают, что инъецированная композиция трансформируется из маловязкого препарата, который первоначально распространяется с заполнением пустот, с образованием твердой массы путем захвата десневых жидкостей. Таким образом, получается антибактериальная депо-система. Хлоргексидин остается в GCF периодонтальных карманов на клинически эффективных уровнях (MIC 125 мкг/мл) в течение более 1 недели. Данная депо-система полностью разрушается ферментами в пределах 7-10 суток, и ее не требуется удалять. Пример 26: Альтернативный антибактериальный препарат для лечения периодонтита или перимплантита Альтернативный антибактериальный препарат был получен при помощи препарата, приготовленного как описано в примере 1 и содержащего антибактериальный детергент Gardol (глицин, N-метил-N-(1-оксододецил)-, натриевая соль) (PC/GDO/EtOH/Gardol (34/51/10/5)). Этот препарат инъецируют в периодонтальный карман крысы. Наблюдали, что Gardol остается в GCF периодонтальных карманов на клинически эффективных уровнях на протяжении длительного промежутка времени (несколько суток). Данная депо-система полностью разрушается ферментами в пределах 7-10 суток, и ее не требуется удалять. Пример 27: Адгезия препарата к высокоэнергетическим поверхностям Для того чтобы вылечить перимплантит, важна адгезия не только к биологическим поверхностям, но также и к высокоэнергетическим поверхностям, таким как золотой или титановый имплант. Также важно, чтобы препарат приклеивался к керамическим и пластмассовым поверхностям. Препарат (PC/GDO/ЕtOН (36/54/10)), такой как получен в примере 1, наносили на различные поверхности в полости рта. Данная композиция демонстрировала превосходную адгезию к керамическим, пластмассовым, золотым поверхностям, а также к нормальной поверхности зуба, и не могла быть смыта избытком водной жидкости. Депо, образующееся из данной композиции, оставалось в полости рта на том месте, куда его наносили, в течение по меньшей мере 6 ч. Пример 28: Биоадгезивный препарат замедленного высвобождения фторида натрия для применения на зубах Фторидсодержащие соединения часто нужны для противодействия поражению кариесом, и из смеси PC/GDO/ЕtOН/фторид натрия (35/53/10/2) готовили предшественник биоадгезивного препарата с эффектом депо, как показано в примере 1. Данный препарат представлял собой дисперсию фторида натрия, так как его нельзя было растворить в предшественнике. Жидкий препарат наносили на зубы при помощи кисточки. Данный препарат затвердевал при захвате слюны и образовывал депо, обеспечивающее замедленное высвобождение фторида натрия в течение длительного промежутка времени (несколько часов). Пример 29: Депо-композиция для спрея для полости рта Чтобы быть пригодной в качестве депо-системы местного типа в полости рта, механические свойства данной системы были скорректированы путем уменьшения отношения PC/GDO. Смесь, содержащую PC/GDO/EtOH (27/63/10), получали согласно примеру 1. Добавляли каплю патентованного синего, чтобы визуализировать образование после нанесения. Примерно 300 мкл данного препарата распыляли в полость рта с помощью пульверизатора. Вскоре после нанесения происходило увеличение вязкости/отверждение препарата, так как он претерпевал фазовый переход при захвате водной жидкости (слюны) и потере растворителя (ЕtOН). Данный препарат имел превосходную биоадгезию к кератинизированным поверхностям, таким как твердое небо и десна. В этом случае пленка сохранялась в течение нескольких часов, несмотря на секрецию слюны и механическое истирание языком. На мягких поверхностях слизистых длительность была значительно короче (минуты). Пример 30: Жидкая депо-композиция для полости рта Чтобы годиться для нанесения пипеткой в полость рта, отверждение/увеличение вязкости у препарата должно быть задержано по сравнению с препаратом в виде спрея. Это требуется для того, чтобы сделать возможным удобное распределение данного препарата языком с образованием тонкой пленки в полости рта после нанесения. К препарату, полученному как в примере 1, добавляли пропиленгликоль (PG) и ЕtOН до получения конечной композиции PC/GDO/EtOH/PG (24/56/10/10). 300 мкл данного препарата без труда наносили пипеткой в полость рта, и они распределялись в полости рта посредством языка с образованием тонкой пленки. Примерно через 20 секунд начиналось увеличение вязкости данного препарата, так как он подвергался фазовой трансформации при захвате водной жидкости (слюны) и потере растворителя (ЕtOН и PG). Примерно через одну минуту отверждение/увеличение вязкости, по-видимому, заканчивалось. Данный препарат обладал превосходной биоадгезией к керанизированным поверхностям, таким как твердое небо и десна. В данном случае пленка сохранялась в течение нескольких часов несмотря на секрецию слюны и механическое истирание языком. На мягких слизистых поверхностях длительность была значительно короче (минуты). Пример 31: Биоадгезивные депо для ногтей Смесь по примеру 29 распыляли на ногтевое ложе и между пальцами ног. Происходит медленное отверждение/увеличение вязкости данного препарата посредством захвата водных жидкостей (например, пота). Отверждение можно ускорить путем добавления воды после нанесения спрея. Данный препарат имеет превосходные биоадгезивные свойства и имеет продолжительность действия несколько часов. Пример 32: Допускаемая нагрузка биоактивного агента бензидамина в предшественниках препаратов Препараты с композициями, как они определены в таблице 7, получали, используя способ примера 1. К 0,5 г препаратов добавляли избыточное количество бензидамина (50 мг). Сосуды помещали на шейкер на трое суток при 15°С, после чего растворы фильтровали через фильтр (0,45 мкм), чтобы избавиться от кристаллов нерастворившегося бензидамина. Концентрацию бензидамина в каждом препарате определяли при помощи градиентной HPLC (жидкостной хроматографии высокого давления) на обращенной фазе и УФ-детекции при 306 нм, результаты приведены в таблице 7.
Пример 33: Композиции, содержащие PC и токоферол Препараты депо-предшественников получали с несколькими разными композициями РС/ Каждый препарат инъецировали в сосуд и уравновешивали с избытком воды. Фазовое поведение оценивали визуально и между скрещенными поляризаторами при 25°С. Результаты представлены в таблице 8.
Пример 34: Композиция, содержащая октреотид 60 мг октреотида растворяли в 0,1 г ЕtOН. Затем в этом растворе растворяли 0,25 г PC и 0,59 г Препарат депо-предшественника с октреотидом в этом примере тестировали на устойчивость против кристаллизации на протяжении хранения. Данный препарат был стабильным при 4-8°С в течение по меньшей мере двух недель. Пример 35: Высвобождение in vitro водорастворимого динатрия флуоресцеина Водорастворимый краситель, динатрия флуоресцеин (Fluo), растворяли в препарате, содержащем РС/ Высвобождение Fluo из РС/
Пример 36: Препараты обезболивающего/противовоспалительного бензидамина Препараты получали как в примере 1 путем смешивания бензидамина со смесью GDO, PC, этанола и возможно RG/AP в следующих соотношениях,
где BZD представляет собой бензидамин, EtOН представляет собой этанол, PC представляет собой фосфатидилхолин сои LIPOID S100, GDO представляет собой глицериндиолеат, PG представляет собой пропиленгликоль и АР представляет собой аскорбилпальмитат. Все препараты представляют собой маловязкие жидкости, которые при воздействии водных условий образуют композиции с жидкокристаллической фазой. Пример 37: Назальный препарат фентанила Препараты получили как в примере 1 путем смешивания наркотического анальгетика фентанила со смесью GDO, PC, этанола и, возможно, PG в следующих соотношениях,
где ЕtOН представляет собой этанол, PC представляет собой фосфатидилхолин сои LIPOID S100, GDO представляет собой глицериндиолеат и PG представляет собой пропиленгликоль. Все препараты представляют собой маловязкие жидкости, подходящие для введения посредством назального спрея, которые при воздействии водных условий образуют композиции с жидкокристаллической фазой. Пример 38: Назальные препараты диазепама Препараты получали как в предыдущих примерах путем смешивания бензодиазепинового анксиолитического агента диазепама со смесью GDO, PC, этанола и возможно PG в следующих соотношениях,
где ЕtOН представляет собой этанол, PC представляет собой фосфатидилхолин сои LIPOID S100, GDO представляет собой глицериндиолеат, PG представляет собой пропиленгликоль. Все препараты представляют собой маловязкие жидкости, подходящие для введения посредством назального спрея, которые при воздействии водных условий образуют композиции с жидкокристаллической фазой. Пример 39: Интерферон Альфа-2а Интерфероны (INFs) используют для лечения многих типов системного рака, часто в комбинации с химиотерапией или облучением. Недавние данные свидетельствуют о том, что IFN-альфа представляет собой мультифункциональный иммуномодулирующий цитокин с сильным влиянием на цитокиновый каскад, включая некоторые противовоспалительные свойства. Эти недавно установленные иммунорегуляторные и противовоспалительные функции также могут быть важными в лечении таких заболеваний как хронический вирусный гепатит и могут помочь объяснить некоторые IFN-механизмы. Препарат безводного предшественника получали путем растворения PC (360 мг) и GDO (540 мг) в ЕtOН (100 мг). Интерферон Альфа-2а (4 мг) растворяли в воде (76 мг), и затем этот раствор добавляли к неводному препарату предшественника с образованием маловязкого предшественника депо-препарата. Инъецирование депо-предшественника в избыток воды (шприц 23 G; 0,6 мм × 30 мм) приводило к монолитной жидкокристаллической фазе (структура I2). Пример 40: Лейпрорелин (лейпролид) Лейпрорелина ацетат (или лейпролида ацетат) представляет собой синтетический нонапептидный аналог встречающегося в природе гонадотропин-высвобождающего гормона (GnRH или LH-RH), который при непрерывном введении (например, в виде депо-препарата) ингибирует секрецию гипофизарного гонадотропина и подавляет тестикулярный и яичниковый стероидогенез. Лейпрорелин используют для лечения прогрессирующего рака простаты. Предшественник депо-препарата получали путем последовательного растворения 22,5 мг лейпрорелина ацетата и 360 мг PC в 100 мг NMP. К смеси добавляли 540 мг GDO с получением маловязкого молекулярного раствора предшественника депо-препарата. Инъецирование предшественника препарата в избыток воды (шприц 23 G; 0,6 мм × 30 мм) приводило к образованию монолитной жидкокристаллической фазы (структура I2). Пример 41: Алендронат Бисфосфонаты представляют собой структурные аналоги пирофосфатов и имеют специфическую фармакологическую активность в отношении костей благодаря сильному сродству бисфосфонатов к гидроксиапатиту, главному неорганическому компоненту кости. Соединения используют для лечения постменопаузного остеопороза, гиперкальцемии злокачественных новообразований и метастатической болезни костей (MBD). Неводный препарат предшественника получали путем растворения PC (360 мг) и GDO (540 мг) в ЕtOН (100 мг). Алендронат (12 мг) растворяли в воде (80 мг), и затем этот раствор добавляли к неводному препарату предшественника с образованием маловязкого предшественника депо-препарата. Инъецирование депо-предшественника в избыток воды (шприц 23 G; 0,6 мм × 30 мм) приводило к образованию монолитной жидкокристаллической фазы (структура I2). Пример 42: Оланзапин Оланзапин представляет собой низкомолекулярное лекарственное средство, используемое для лечения пациентов с шизофренией. Предшественник депо-препарата получали путем последовательного смешивания 50 мг оланзапина, 360 мг PC и 100 мг ЕtOН. К данной смеси добавляли 540 мг GDO, что приводило к образованию предшественника конечного депо-препарата. Инъецирование данного предшественника препарата в избыток воды (шприц 23 G; 0,6 мм × 30 мм) приводило к образованию монолитной жидкокристаллической фазы (структура I2). Пример 43: Препараты с клиндамицином против акне Препараты получали, как и в предыдущих примерах, путем смешивания полусинтетического антибиотика клиндамицина (свободное основание или соль) со смесью GDO, PC, этанола и PG в следующих соотношениях (по массе).
Образующиеся препараты-предшественники представляют собой маловязкие жидкости, которые после нанесения являются устойчивыми к воде, поту и пр. Препараты наносят местно на кожу в виде геля или путем распыления, и он является биоадгезивным с хорошими пленкообразующими свойствами. Пример 44: Дополнительные примеры вязкости смесей PC/GDO при добавлении сорастворителя Смеси PC/GDO и сорастворителя получали согласно способам из примера 1 и примера 3 в соотношениях, указанных в приведенной ниже таблице. Пробы оставляли уравновешиваться в течение нескольких суток перед проведением измерения вязкости с использованием реометра Physica USD 200 при 25°C.
Этот пример дополнительно демонстрирует необходимость в растворителе со свойствами понижения вязкости для получения инъецируемых препаратов. Смеси, содержащие глицерин (проба 19) или воду (пробы 20 и 21), являются слишком вязкими, чтобы быть инъецируемыми при концентрациях растворителя, эквивалентных образцам, содержащим EtOH (сравните с образцами 13, 14 и 17). Пример 45: Композиции препарата октреотида Препараты получали, как в примере 1, путем смешивания активного пептида октреотида со смесью GDO (одной из нескольких уровней чистоты) или токоферола, PC, этанола и возможно диолеил-PG в следующих соотношениях (по массе),
где ОСТ представляет собой октреотид, ЕtOН представляет собой этанол, PC представляет собой фосфатидилхолин сои LIPOID S100, GDO представляет собой глицериндиолеат, ТР представляет собой Качество GDO (согласно АС)
Препарат Р (композицию см. выше) вводили крысе путем подкожной инъекции в количестве 1 мл препарата на кг массы тела, что соответствует 30 мг/кг октреотида. Уровни октреотида в плазме отслеживали в течение 5 суток после введения для того, чтобы исследовать любой взрывной профиль. Наивысшая наблюдаемая концентрация в плазме была менее чем в три раза выше средней концентрации в плазме на протяжении первых 5 суток. Результаты данного исследования показаны на Фиг.6. Пример 46: Солнцезащитные препараты,Препараты получали, как и в примере 1, путем смешивания каждого из нескольких УФ-поглощающих/рассеивающих агентов со смесью GDO, PC и этанола в следующих соотношениях (по массе),
где TIOVEIL CM (Uniqema) содержит циклометикон (и) диоксид титана (и) диметикона сополиол (и) стеарат алюминия (и) оксид алюминия, SPECTRAVEIL FIN (Uniqema) содержит оксид цинка (и) С12-С15алкилбензоат (и) полигидроксистеариновую кислоту, SOLAVEIL CT-100 (Uniqema) содержит С12-С15алкилбензоат (и) диоксид титана (и) полигидроксистеариновую кислоту (и) стеарат алюминия (и) оксид алюминия, и TIOVEIL 50 MOTG (Uniqema) содержит диоксид титана (и) каприловый/каприновый триглицерид (и) минеральное масло (и) полигидроксистеариновую кислоту (и) стеарат алюминия (и) оксид алюминия. Образующиеся предшественники препаратов демонстрируют низкую вязкость при приготовлении в виде препаратов и легко наносятся посредством пульверизатора. При контакте с поверхностями тела образуется эластичный слой, защищающий от УФ-света. Пример 47: Периодонтальные депо с хлоргексидином Препараты получали, как в примере 1, путем смешивания противоинфекционного агента хлоргексидина диглюконата со смесью GDO, PC и этанола в следующих соотношениях (по массе)
Препараты-предшественники депо с хлоргексидином имеют низкую вязкость и легко вводятся в периодонтальный карман. Данные композиции обеспечивают лучшее распространение и распределение активного вещества по всему периодонтальному карману по сравнению с продуктами, используемыми в настоящее время, такими как Periochip®. Депо, образующееся после нанесения, обеспечивает защиту против повторного инфицирования кармана. Данное депо также имеет превосходные биоадгезивные свойства и приклеивается к поверхностям слизистых оболочек, зубам и костей. Исследовали высвобождение хлоргексидина диглюконата из 250 мг препарата А (см. выше) в 0,9%-ном водном NaCl (500 мл). Данный препарат держали в цилиндрической металлической чашке, которую помещали в тефлоновый держатель на дне стандартной USP бани для высвобождения. Площадь контакта между препаратом и окружающим физиологическим раствором составляла 2,4 см2, и раствор перемешивали лопастной мешалкой со скоростью 100 об/мин. Кривая высвобождения, показанная на Фиг.7, демонстрирует замедленное и по существу постоянное высвобождение хлоргексидрина из препарата в течение промежутка времени 24 часа. Пример 48: Исследование in vivo высвобождения из депо-препарата, содержащего в качестве смеси со-растворителей смесь спирта-моноола и сульфоксида 1. Материалы и методы 1.1. Препарат Препарат, содержащий активное вещество бупренорфин (BUP от Jansse, Belgium), соевый фосфатидилхолин (SPC от Lipoid, Germany), глицерин диолеат (GDO от Danisco, Denmark), этанол (ЕtOН от Kemetyl, Sweden) и диметилсульфоксид (DMSO от Merck KgaA, Germany), получали согласно Таблице ниже путем отвешивания соответствующих компонентов в стерильный сосуд для инъекций с последующим непрерывным вращением при комнатной температуре до получения полностью прозрачного и гомогенного препарата. Затем препарат подвергали стерилизации фильтрованием под давлением азота 2,5 бар (250 кПа) через PVDF фильтр 0,22 мкм от Millipore.
1.2. Исследование in vivo и биоанализ В исследовании использовали 6 самцов крыс SPF Sprague-Dawley (SPRB MOL, M&B Taconic, Lille Skensved, Denmark) с массой более 300 г при получении. Животным обеспечивали 5-дневный восстановительный период перед включением в исследование. Животных содержали по две особи в клетках (Macrolon® III; Scanbur BK A/S, Karlslunde, Denmark) с осиновыми стружками в качестве подстилки (Tapei, Finland) и со свободным доступом к стандартному гранулированному корму для крыс (RM 1 SQC (Expanded), Special Diet Services, England) и водопроводной воде. Под легкой анестезией стандартными методами с помощью иглы для инъекций 5G и одноразовых стерильных шприцов на 1 мл осуществляли подкожные инъекции (1,5 мл/кг, что приблизительно эквивалентно 0,5 мл на животное) препарата, описанного в Таблице. Образцы крови для фармакокинетических анализов забирали перед осуществлением инъекции, через 3 часа, 6 часов, 9 часов, 1 сутки, 2 суток, 7 суток, 14 суток, 21 сутки и 28 суток после инъекции. Образцы крови объемом 0,4 мл собирали посредством сублингвального забора в предварительно обработанные EDTA (этилендиаминотетрауксусная кислота) пробирки. Концентрацию BUP в плазме анализировали в образцах EDTA плазмы с помощью набора для ELISA (твердофазный иммуноферментный анализ) от Neogen, USA. Этот набор для ELISA представляет собой набор для качественного одноступенчатого анализа, предназначенный для обнаружения BUP в образцах от человека. Способ был модифицирован и откалиброван для количественного определения BUP в EDTA плазме крыс. 2. Результаты Полученный профиль концентрации в плазме представлен на Фиг.8. Он демонстрирует пролонгированное и контролируемое высвобождение BUP на протяжении по меньшей мере 28 суток и отношение Сmах/С28 суток BUP в плазме, составляющее приблизительно 5, что свидетельствует о профиле с низким взрывом. В целом эти in vivo данные с очевидностью подтверждают полученные ранее данные о благоприятных свойствах высвобождения in vivo жидких депо-препаратов различных активных агентов. 3. Заключение Данные, представленные на Фиг.8, свидетельствуют о том, что депо-препарат BUP, содержащий как спирт-моноол (EtOH), так и диметилсульфоксид (DMSO), обеспечивает очень удачный фармакокинетический профиль с немедленным началом, низким начальным высвобождением (взрывом) и большой длительностью in vivo.
Формула изобретения
1. Состав, образующий in vivo биологически активную депо-композицию, содержащий маловязкую нежидкокристаллическую смесь: 2. Состав, образующий in vivo биологически активную депо-композицию, содержащий маловязкую нежидкокристаллическую смесь: 3. Состав по п.1 или 2, где указанная жидкокристаллическая фазовая структура является биоадгезивной. 4. Состав по п.1 или 2, где компонент (а), по существу, состоит из диацилглицеринов. 5. Состав по п.4, где указанные диацилглицерины содержат глицериндиолеат. 6. Состав по п.1 или 2, где компонент (а), по существу, состоит из по меньшей мере одного токоферола. 7. Состав по п.1 или 2, где компонент (а), по существу, состоит из смеси GDO (глицериндиолеата) и токоферола. 8. Состав по п.1 или 2, имеющий структуру молекулярного раствора фазы L2 и/или L3. 9. Состав по п.1 или 2, имеющий отношение (а) к (б) от 95:5 до 5:95 по массе. 10. Состав по п.1 или 2, содержащий компонент (в) в количестве от 0,5 до 50% по массе от суммы компонентов (а)+(б)+(в). 11. Состав по п.2, где компонент (в) выбран из спиртов, кетонов, сложных эфиров, простых эфиров, амидов, сульфоксидов и их смесей. 12. Состав по п.1 или 2, дополнительно содержащий заряженный амфифил в количестве вплоть до 10% по массе от (а)+(б). 13. Состав по п.1 или 2, где указанный активный агент выбран из лекарственных средств, антигенов, питательных веществ, косметических средств, ароматизаторов, корригентов, диагностических агентов, витаминов, пищевых добавок и их смесей. 14. Состав по п.13, где указанное лекарственное средство выбрано из гидрофильных низкомолекулярных лекарственных средств, липофильных низкомолекулярных лекарственных средств, амфифильных низкомолекулярных лекарственных средств, пептидов, белков, олигонуклеотидов и их смесей. 15. Состав по п.13, где указанное лекарственное средство выбрано из пептидов, родственных соматостатину, интерферонов, глюкагоноподобных пептидов 1 и 2, агонистов GnRH (гонадотропин-высвобождающего гормона), антагонистов GnRH, бисфосфонатов, хлоргексидина и их смесей. 16. Состав по п.1 или 2, который можно вводить посредством инъекции. 17. Состав по п.1 или 2, который можно вводить путем распыления, окунания, промывания, нанесения с помощью прижимного или шарикового ролика, смазывания, закапывания, аэрозольного распыления или распыления нагнетанием. 18. Инъецируемый состав по п.1 или 2, который образует депо, обеспечивающее замедленное высвобождение активного агента в течение по меньшей мере двух недель, где указанный активный агент содержит по меньшей мере один агент, выбранный из: 19. Инъецируемый состав по п.1 или 2, который образует депо, обеспечивающее непрерывное высвобождение активного агента в течение по меньшей мере двух недель, где указанный активный агент содержит по меньшей мере один агент, выбранный из: 20. Местный состав по п.1 или 2 для пероралыюго введения, который образует биоадгезивный продукт с контролируемым высвобождением, где указанный активный агент содержит по меньшей мере один агент, выбранный из: 21. Местный состав по п.1 или 2, пригодный для перорального введения для лечения периодонтальных и местных инфекций, где активный агент представляет собой хлоргексидина глюконат и где данный препарат-предшественник наносят в виде жидкого продукта, который in situ образует поверхностный гель через промежуток времени от 1 с до 5 мин после нанесения. 22. Непарентеральный состав по п.1 или 2 для назального введения в виде спрея, который образует биоадгезивный продукт с контролируемым высвобождением, где указанный активный агент содержит по меньшей мере один агент, выбранный из: 23. Местный состав по п.1 или 2, пригодный для офтальмического введения, где указанный активный агент содержит по меньшей мере один агент, выбранный из диклофенака, пилокарпина, левокабастина гидрохлорида, кетотифена фумарата, тимолола, бетаксолола, картеолола, левобунолола, дорзоламида, бринзоламида, эпинефрина, дипивефрина, клонидина, апраклонидина, бримонидина, пилокарпина, атанопроста, травопроста, биматопроста, унопростона, пилокарпина гидрохлорида, дексаметазона, хлорамфеникола и индометацина. 24. Непарентеральный состав по п.1 или 2 для дерматологического введения, который образует биоадгезивный продукт с контролируемым высвобождением, где активный агент выбран из: 25. Местный состав по п.1 или 2 для дерматологического введения, который образует биоадгезивный продукт с контролируемым высвобождением, где активный агент выбран из косметических средств, ароматизаторов, корригентов, эфирных масел, УФ-поглощающих агентов и их смесей. 26. Способ доставки биоактивного агента в организм человека или животного, не являющегося человеком (предпочтительно млекопитающего), включающий введение состава по любому из пп.1-25, в результате чего после введения, при контакте in vivo с жидкостью тела образуется по меньшей мере одна жидкокристаллическая фазовая структура. 27. Способ по п.26, где указанный состав вводят способом, выбранным из подкожной инъекции, внутримышечной инъекции, внутриполостной инъекции через ткань, внутриполостной инъекции в открытую полость без проникновения сквозь ткань, распыления, нанесения роликом, натирания, намазывания, смазывания, промывания или закапывания. 28. Способ получения жидкокристаллической композиции, при котором на состав по любому из пп.1-25 in vivo воздействует жидкость тела. 29. Способ получения состава, пригодного для введения биоактивного агента (предпочтительно млекопитающему) субъекту, включающий получение нежидкокристаллической маловязкой смеси: 30. Способ получения состава, пригодного для введения биоактивного агента субъекту (предпочтительно млекопитающему), включающий получение нежидкокристаллической маловязкой смеси: 31. Способ по п.29 или 30, где указанный состав представляет собой состав по любому из пп.1-25. 32. Применение состава по любому из пп.1-25 в изготовлении лекарства для использования с замедленным введением активного агента, где активный агент представляет собой лекарственное средство. 33. Применение по п.32 для лечения состояния, выбранного из бактериальной инфекции, грибковой инфекции, болезненности кожи, состояний глаз, болезненности гениталий, инфекции и состояний ногтей пальцев рук и/или ног, укачивания, аддикции, включая аддикцию к никотину, периодонтальной инфекции, конъюнктивита, глаукомы и гормональной недостаточности или дисбаланса. 34. Применение по п.32 для профилактики по меньшей мере одного состояния, выбранного из инфекции во время хирургического вмешательства, инфекций во время имплантации, солнечного ожога, инфекции в месте ожогов, порезов или ссадин, инфекций полости рта, генитальных инфекций и инфекций, являющихся следствием видов деятельности, приводящих к воздействию инфекционных агентов.
РИСУНКИ
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
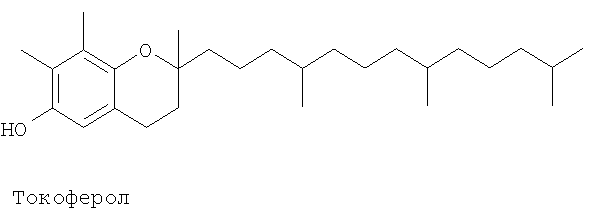
 -лактамы или макроциклические пептидные антибиотики, противогрибковые агенты, такие как полиеновые макролиды (например, амфотерицин В) или азольные противогрибковые средства, противораковые и/или противовирусные лекарственные средства, такие как нуклеозидные аналоги, паклитаксел и его производные, противовоспалительные агенты, такие как нестероидные противовоспалительные лекарственные средства и кортикостероиды, сердечно-сосудистые лекарственные средства, включая агенты, понижающие уровень холестерина и давление крови, анальгетики, антипсихотические средства и антидепрессанты, включая ингибиторы захвата серотонина, простагландины и их производные, вакцины и модуляторы костных процессов. Диагностические агенты включают соединения, меченые радиоизотопами, и контрастирующие агенты, включая агенты, усиливающие контрастность рентгеновских лучей, ультразвука и MRI (магнитно-резонансная томография). Питательные вещества включают витамины, коферменты, биологически активные добавки и пр.
-лактамы или макроциклические пептидные антибиотики, противогрибковые агенты, такие как полиеновые макролиды (например, амфотерицин В) или азольные противогрибковые средства, противораковые и/или противовирусные лекарственные средства, такие как нуклеозидные аналоги, паклитаксел и его производные, противовоспалительные агенты, такие как нестероидные противовоспалительные лекарственные средства и кортикостероиды, сердечно-сосудистые лекарственные средства, включая агенты, понижающие уровень холестерина и давление крови, анальгетики, антипсихотические средства и антидепрессанты, включая ингибиторы захвата серотонина, простагландины и их производные, вакцины и модуляторы костных процессов. Диагностические агенты включают соединения, меченые радиоизотопами, и контрастирующие агенты, включая агенты, усиливающие контрастность рентгеновских лучей, ультразвука и MRI (магнитно-резонансная томография). Питательные вещества включают витамины, коферменты, биологически активные добавки и пр. , I2, L2 и жидкость (раствор). Очень предпочтительно, чтобы композиция по данному изобретению была способна образовать по меньшей мере две и более предпочтительно по меньшей мере три из этих фаз одновременно на переходных стадиях после контакта с водой при физиологических температурах. В частности, весьма предпочтительно, чтобы одна из образовавшихся фаз, по меньшей мере временно, представляла собой фазу I2.
, I2, L2 и жидкость (раствор). Очень предпочтительно, чтобы композиция по данному изобретению была способна образовать по меньшей мере две и более предпочтительно по меньшей мере три из этих фаз одновременно на переходных стадиях после контакта с водой при физиологических температурах. В частности, весьма предпочтительно, чтобы одна из образовавшихся фаз, по меньшей мере временно, представляла собой фазу I2.