Патент на изобретение №2167875
|
||||||||||||||||||||||||||
(54) ПОЛУЧЕНИЕ ПРОИЗВОДНЫХ 2-ОКСАЗОЛИДИНОНА В ОДНОМ РЕАКТОРЕ
(57) Реферат: В настоящем изобретении предложен усовершенствованный способ получения (S)-4-{ [3-[2-(диметиламино)-этил] -1H-индол -5-ил]метил}-2-оксазолиндинона, который включает следующие стадии: а) образование карбамата формулы (III) из метил-4-нитро-(L)-фенилаланината гидрохлорида; б) восстановление соединения формулы (III) с получением метил-(S)-N-бутоксикарбонил-4-аминофенилаланината формулы (IV); с) восстановление сложноэфирной метильной группы в соединении формулы (IV) с получением (S)-N-бутоксикарбонил-4-аминофенилаланинола формулы (V); d) замыкание кольца в соединении формулы (V) с получением (S)-4(-аминобензил-2-оксазолидинона гидрохлорида) формулы (VII); е) образование соли диазония из соединения формулы (VI) с последующим восстановлением и получением гидразина-(S)-4-(4-гидразинобензил)-2-оксазолидинона гидрохлорида формулы (VII); f) реакцию Фишера соединения формулы (VII) с получением (S)-4-{ [3-[2-(диметиламино)этил] -1Н-индол-5-ил] метил} -2-оксазолидинона. Целевой продукт является агонистом 5-НТ1-подобных рецепторов, которые эффективно используются при лечении мигрени. Технический результат – повышение выхода и чистоты целевого продукта, упрощение технологии процесса. 15 с. и 10 з.п. ф-лы. 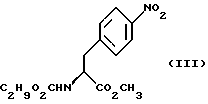 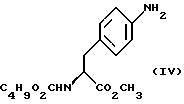 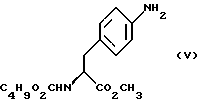 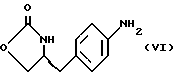 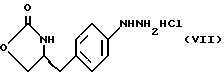
Изобретение относится к усовершенствованному способу получения замещенных производных индола, полезных при лечении и профилактике мигрени. Более точно, настоящее изобретение обеспечивает усовершенствованный способ получения (S)-4-{ [3-[2-(диметиламино) этил]-1Н-индол-5-ил]метил}-2-оксазолидинона, агониста 5-НТ1- подобных рецепторов, которые, как известно, эффективно используются при лечении мигрени. Известно, что селективные агонисты 5-НТ1-подобных рецепторов являются полезными терапевтическими агентами. Такой рецептор медиирует сужение кровеносных сосудов и тем самым изменяет кровоток в каротидном сосудистом ложе. В Европейском патенте 0313397 описан класс специфических агонистов 5-НТ1-подобных рецепторов, полезных для лечения и профилактики заболеваний, которые характеризуются как приводящие к сужению кровеносных сосудов в каротидном сосудистом русле, например, мигрени (состояния, связанного с чрезмерной дилатацией каротидной сосудистой сети). В Международной заявке WO 91/18897 описан еще один класс соединений, обладающих исключительной агонистической активностью по отношению к “5-НТ1-“подобным” рецепторам и превосходным всасыванием после орального приема. Эти свойства делают соединения, описанные в Международной заявке WO 91/18897, весьма полезными для некоторых медицинских показаний, в особенности для лечения и профилактики мигрени, “гистаминовой” головной боли и головной боли, связанной с сосудистыми заболеваниями, которые далее получили в настоящем описании собирательное название “мигрень”. Одно в особенности предпочтительное соединение, описанное в Международной заявке WO 91/18897, представляет собой (S)-N, N-диметил-2-[5- (2-оксо-1,3-оксазолидин-4-ил-метил)-1H-индол-3-ил] этиламин, который также известен как (S)-4-{[3-[2-(диметиламино)этил] –  – индол-5-ил]метил}-2-оксазолидинон и может быть представлен формулой (I): – индол-5-ил]метил}-2-оксазолидинон и может быть представлен формулой (I):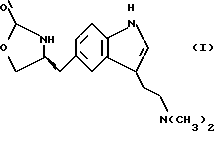 Соединение формулы (I) может существовать в виде его (S)- или (R)-энантиомера и пример его получения описан в Международной заявке WO 91/18897. В этой заявке также предлагается целый ряд возможных реакционных маршрутов для получения соединения формулы (I). Авторами изобретения разработан новый способ получения соединения формулы (I). Предложенный способ обладает преимуществом, по сравнению с процессом, описанным в заявке WO 91/18897, которое состоит в том, что позволяет получить чистый конечный продукт с высоким выходом на промышленной установке благодаря осуществлению процесса в одном реакторе, избежав тем самым необходимости в проведении длительного и дорогостоящего выделения промежуточных продуктов. Кроме того, новый способ позволяет избежать необходимости в использовании таких опасных реагентов, как фосген, или реагентов, представляющих опасность для окружающей среды, например, хлорида олова. Согласно первому аспекту настоящего изобретения предложен способ получения (S)-4-{ [3-[2-(диметиламино)этил] –  индол-5-ил] метил}-2-оксазолидинона, который включает следующие стадии: индол-5-ил] метил}-2-оксазолидинона, который включает следующие стадии:а) образование карбамата из метил-4-нитро-(L)-фенилаланината гидрохлорида формулы (II) 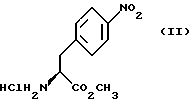 путем добавления карбоната или гидрокарбоната натрия н- бутилхлорформиата и получение в результате реакции метил-(S)  – бутоксикарбонил-4-нитрофенилаланината формулы (III) – бутоксикарбонил-4-нитрофенилаланината формулы (III)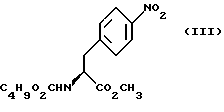 b) восстановление соединения формулы (III) с получением метил-(S)  бутоксикарбонил-4-аминофенилаланината формулы (IV) бутоксикарбонил-4-аминофенилаланината формулы (IV)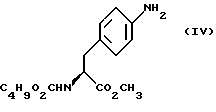 с) восстановление сложноэфирной метильной группы -CO2CH3 в соединении формулы (IV) с получением (S)-N- бутоксикарбонил-4-аминофенилаланинола формулы (V) 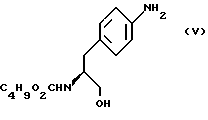 d) замыкание кольца в соединении формулы (V) с получением (S)-4-(4-аминобензил)-2-оксазолидинона формулы (VI) 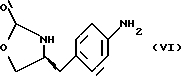 е) получение соли диазония из соединения формулы (VI) с последующим восстановлением и получением гидразина (S)-4-(4- гидразинобензил) -2-оксазолидинона гидрохлорида формулы (VII) 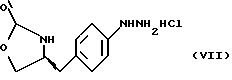 f) реакцию Фишера соединения формулы (VII) с получением соединения формулы (I). Надлежащим образом осуществляют одну или более стадий a) -f) по методике, которая предусматривает проведение реакции в одном реакционном сосуде. Сначала в одном реакторе предпочтительно осуществляют стадии a) – d) с последующим выделением соединения (формулы (VI), а затем также в одном реакторе осуществляют стадии е) и f). Стадию а) обычно осуществляют в присутствии растворителя, например, водного этилацетата или диоксана, причем водный этилацетат является предпочтительным. По сравнению с гидрокарбонатом натрия, использование карбоната натрия более предпочтительно, причем его добавление осуществляют до введения н-бутилхлорформиата. Реакцию проводят обычно при неэкстремальных температурах, причем подходящим интервалом является температура 5-60oC. Предпочтительный интервал температур для проведения реакции составляет 15-35oC. В наиболее предпочтительном примере осуществления изобретения добавление карбоната натрия осуществляют при температуре около 20oC, а добавление н-бутилхлорформиата – при температуре около 30oC. Стадию восстановления b) осуществляют в присутствии органического растворителя, например, этилацетата или этанола. Предпочтительно стадию b) осуществляют в одном реакторе с использованием этилацетатного раствора соединения формулы (III), полученного на стадии a). Стадия b) представляет собой реакцию гидрогенизации, предпочтительно проводимую в присутствии катализатора, такого как палладированный уголь. Реакцию можно провести в атмосфере азота с использованием водорода при нормальном атмосферном давлении и при комнатной температуре. Предпочтительные условия проведения гидрогенизации включают давление водорода около 1,4 кг/см2 (20 psi) и повышенную температуру, например, в интервале 30-50oC. Полученный раствор соединения формулы (IV) в этилацетате предпочтительно переводят в бутанольный раствор, который можно использовать непосредственно на стадии с) как продукт соответствующей части технологического процесса, проводимого в одном реакторе. Это превращение удобно осуществлять путем частичной перегонки этилацетатного раствора с последующим добавлением бутанола и фракционированием для удаления этилацетата. Стадию с) восстановления сложного метилового эфира обычно проводят в присутствии растворителя, например SVM или н-бутанола. Предпочтительно стадию с) осуществляют как часть технологического процесса, проводимого в одном реакторе, получая из раствора соединения формулы (IV) в этилацетате н-бутанольный раствор, который далее непосредственно восстанавливают. Восстановление предпочтительно проводят с использованием борогидрида натрия и обычно в условиях неэкстремальных температур в интервале 20-40oC. Предпочтительно восстановление осуществляют в две фазы: первую фазу проводят в атмосфере азота при температуре около 25oC, а вторую фазу проводят при температуре около 30oC. Полученный раствор соединения формулы (V) в н-бутаноле после этого сушат в присутствии соляной кислоты и аммиака. Сухой н-бутанольный раствор можно использовать непосредственно на стадии d) как продукт соответствующей части технологического процесса, проводимого в одном реакторе. Стадию d) предпочтительно проводят в сухом растворе, например, в сухом бутанольном растворе соединения формулы (V). Такой сухой бутанольный раствор может быть с успехом приготовлен путем высушивания н-бутанольного раствора, получаемого на стадии с). Сухой н-бутанольный раствор предпочтительно обесцвечивают с помощью угля перед осуществлением реакции замыкания цикла. Замыкание цикла удобно проводить с использованием метоксида натрия в подходящем спиртовом растворителе, например метаноле. Наиболее предпочтительно реакцию замыкания цикла осуществляют, используя 30%-ый раствор метилата натрия в метаноле. Реакцию предпочтительно проводят при повышенной температуре, подходящие значения которой находятся в интервале 50-120oC. Предпочтительная температура реакции составляет около 85oC. Полученное соединение формулы (VI) затем выделяют стандартными методами центрифугирования, фильтрации и сушки. Стадию е) осуществляют с участием выделенного соединения (VI). Выделение может быть достигнуто, например, при использовании хорошо известных методов центрифугирования, фильтрации и сушки. Получение соли диазония проводят с помощью водного раствора нитрита натрия при пониженной температуре, предпочтительно в присутствии концентрированной соляной кислоты. Предпочтительно солеобразование ведут при пониженной температуре, например в интервале 0-5oC. Гидразин получают из соли диазония с использованием сульфита натрия в качестве восстановителя. Используемый сульфит натрия находится в виде водного раствора. Восстановление выгодно проводить в две фазы: первая фаза включает добавление сульфита натрия, вторая фаза – добавление соляной кислоты. Первую фазу предпочтительно проводят при температуре ниже 10oC, а вторую – при повышенной температуре, например в интервале 55-60oC. Раствор соединения формулы (VII) со стадии е) предпочтительно используют непосредственно на стадии f), проводимой в одном реакторе. Стадия f) представляет собой реакцию Фишера. Установлено, что ее выгодно вести при относительно высоком разбавлении для того, чтобы максимально увеличить чистоту конечного продукта. Поэтому раствор, который получают со стадии е), предпочтительно разбавляют водой, а затем проводят реакцию Фишера путем добавления 4,4-ди-этокси 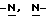 диметилбутиламина в атмосфере азота. Предпочтительно, чтобы при добавлении 4,4- диэтокси диметилбутиламина в атмосфере азота. Предпочтительно, чтобы при добавлении 4,4- диэтокси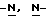 диметилбутиламина разбавленный раствор находился при повышенной температуре. Подходящая температура реакции находится в интервале 75-105oC, а ее предпочтительное значение составляет около 90oC. Предпочтительно реакция протекает при температуре образования флегмы.
По окончании реакции соединение формулы (I) экстрагируют стандартными методами. Полученный после перегонки продукт реакции охлаждают, доводят его pH до 7 с помощью, например, гидроокиси натрия и затем экстрагируют этилацетатом. Водный слой отделяют и доводят его pH до 10 с помощью гидроокиси натрия. После этого продукт снова экстрагируют приблизительно при температуре 50oC с последующими стандартными операциями обесцвечивания, фильтрации, перегонки, центрифугирования и сушки.
Наиболее предпочтительная схема реакции получения соединения формулы (I) имеет вид, представленный в конце описания.
Согласно другому аспекту настоящего изобретения предлагается способ очистки (S)-4-{ [3-[2-(диметиламино)этил] диметилбутиламина разбавленный раствор находился при повышенной температуре. Подходящая температура реакции находится в интервале 75-105oC, а ее предпочтительное значение составляет около 90oC. Предпочтительно реакция протекает при температуре образования флегмы.
По окончании реакции соединение формулы (I) экстрагируют стандартными методами. Полученный после перегонки продукт реакции охлаждают, доводят его pH до 7 с помощью, например, гидроокиси натрия и затем экстрагируют этилацетатом. Водный слой отделяют и доводят его pH до 10 с помощью гидроокиси натрия. После этого продукт снова экстрагируют приблизительно при температуре 50oC с последующими стандартными операциями обесцвечивания, фильтрации, перегонки, центрифугирования и сушки.
Наиболее предпочтительная схема реакции получения соединения формулы (I) имеет вид, представленный в конце описания.
Согласно другому аспекту настоящего изобретения предлагается способ очистки (S)-4-{ [3-[2-(диметиламино)этил] 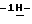 индол-5-ил] -метил}-2-оксазолидинона, который включает следующие стадии: индол-5-ил] -метил}-2-оксазолидинона, который включает следующие стадии:а) растворение сырого (S)-4-{[3-[2-(диметиламино)этил] 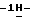 индол-5-ил]-метил} -2-оксазолидинона в смеси этанола в этилацетате, находящейся при температуре образования флегмы, и последующая фильтрация горячего раствора; индол-5-ил]-метил} -2-оксазолидинона в смеси этанола в этилацетате, находящейся при температуре образования флегмы, и последующая фильтрация горячего раствора;b) медленное охлаждение отфильтрованного раствора до температуры около 5oC; с) центрифугирование продукта со стадии b), промывка этилацетатом и последующая сушка; и d) обработка ацетоном для удаления сольватированного этилацетата. Предпочтительно смесь при температуре рефлюкса представляет собой 10%-ый этанол в этилацетате. Горячий раствор должным образом обесцвечивают с помощью обесцвечивающего (активированного) угля перед фильтрацией, проводимой с использованием вспомогательного фильтрующего материала – ускорителя фильтрования. Перед центрифугированием охлажденный отфильтрованный раствор со стадии b) перемешивают в течение продолжительного времени, которое предпочтительно составляет около 18 часов. Стадию сушки с) предпочтительно ведут под вакуумом. Продукт сушат при повышенной неэкстремальной температуре, например, в интервале 40-60oC, предпочтительная величина которой составляет около 50oC. Сухой твердый продукт со стадии с) удобно обработать смесью 20%-ого ацетона в воде в условиях неэкстремальных температур, предпочтительно в интервале 15-30oC, например при комнатной температуре. Полученную суспензию охлаждают до не очень низкой температуры, предпочтительно до 5oC, и перемешивают. После этого продукт центрифугируют, промывают этилацетатом и сушат, предпочтительно под вакуумом, при температуре около 45oC. Полученный продукт представляет собой несольватированное твердое вещество высокой чистоты. Согласно третьему аспекту настоящего изобретения предлагается несольватированный очищенный (S)-4-{[3-[2-(диметиламино)этил] 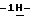 индол-5-ил]-метил}-2-окса-золидинон.
Дополнительные аспекты настоящего изобретения включают соединения формул (III), (IV), (V) и (VI), определенные выше.
Дополнительные аспекты настоящего изобретения включают также и следующие способы получения соединений формул (III), (IV), (V) и (VI): индол-5-ил]-метил}-2-окса-золидинон.
Дополнительные аспекты настоящего изобретения включают соединения формул (III), (IV), (V) и (VI), определенные выше.
Дополнительные аспекты настоящего изобретения включают также и следующие способы получения соединений формул (III), (IV), (V) и (VI):соединение (III): стадия а) способа по первому аспекту настоящего изобретения; соединение (IV): стадия b) способа по первому аспекту настоящего изобретения; соединение (V): стадия с) способа по первому аспекту настоящего изобретения; и соединение (VI): стадия d) способа по первому аспекту настоящего изобретения. Изобретение дополнительно иллюстрируется следующими примерами. Пример 1 Способ получения (S)-4-{[3-[2-(диметиламино)этил] – 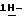 индол-5-ил]-метил}-2-оксазолидинона в массе.
Стадия 1. Получение метил-4-нитро-(L)-фенилаланината гидрохлорида (реакцию см. в конце описания).
Метанольный раствор хлористого водорода получают путем пропускания газообразного хлористого водорода в реактор, содержащий метанол, поддерживая в нем температуру ниже 25oC. Реактор загружают 4-нитро- индол-5-ил]-метил}-2-оксазолидинона в массе.
Стадия 1. Получение метил-4-нитро-(L)-фенилаланината гидрохлорида (реакцию см. в конце описания).
Метанольный раствор хлористого водорода получают путем пропускания газообразного хлористого водорода в реактор, содержащий метанол, поддерживая в нем температуру ниже 25oC. Реактор загружают 4-нитро- 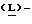 фенилаланином и кипятят с обратным холодильником в течение 1 часа. Смесь охлаждают до 0oC, полученный продукт (метил-4-нитро- фенилаланином и кипятят с обратным холодильником в течение 1 часа. Смесь охлаждают до 0oC, полученный продукт (метил-4-нитро- 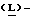 фенилаланината гидрохлорид) центрифугируют, промывают метанолом и сушат в вакууме при температуре 50oC.
Стадия 2. Получение метил-(S)-N-бутоксикарбонил-4- нитрофенил-аланината (реакцию см. в конце описания).
В реактор загружают деминерализованную воду, метил- 4-нитро-(L)-фенилаланината гидрохлорид, карбонат натрия и этилацетат и содержимое реактора охлаждают при перемешивании до температуры около 20oC. К реакционной смеси добавляют н-бутилхлорформиат при поддержании температуры около 30oC и перемешивают около 30 минут. Водный слой отделяют, этилацетатный слой промывают водой, получая этилацетатный раствор метил- фенилаланината гидрохлорид) центрифугируют, промывают метанолом и сушат в вакууме при температуре 50oC.
Стадия 2. Получение метил-(S)-N-бутоксикарбонил-4- нитрофенил-аланината (реакцию см. в конце описания).
В реактор загружают деминерализованную воду, метил- 4-нитро-(L)-фенилаланината гидрохлорид, карбонат натрия и этилацетат и содержимое реактора охлаждают при перемешивании до температуры около 20oC. К реакционной смеси добавляют н-бутилхлорформиат при поддержании температуры около 30oC и перемешивают около 30 минут. Водный слой отделяют, этилацетатный слой промывают водой, получая этилацетатный раствор метил- 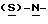 бутокси-карбонил-4-нитрофенил-аланината, который непосредственно используют на следующей стадии.
Стадия 3. Получение метил-(S)-N-бутоксикарбонил-4- аминофенилаланината (реакцию см. в конце описания).
В реактор загружают 5%-ный палладированный уголь в качестве катализатора, этилацетатный раствор метил- бутокси-карбонил-4-нитрофенил-аланината, который непосредственно используют на следующей стадии.
Стадия 3. Получение метил-(S)-N-бутоксикарбонил-4- аминофенилаланината (реакцию см. в конце описания).
В реактор загружают 5%-ный палладированный уголь в качестве катализатора, этилацетатный раствор метил- 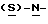 бутоксикарбонил-4-нитрофенилаланината и проводят гидрогенизацию при давлении водорода около 1,4 кг/см2 (20 psi), поддерживая температуру в интервале 30-50oC. По окончании реакции отфильтровывают катализатор через вспомогательный фильтрующий материал и промывают этилацетатом, после чего этилацетатный раствор промывают водным раствором карбоната натрия. Далее раствор метил- бутоксикарбонил-4-нитрофенилаланината и проводят гидрогенизацию при давлении водорода около 1,4 кг/см2 (20 psi), поддерживая температуру в интервале 30-50oC. По окончании реакции отфильтровывают катализатор через вспомогательный фильтрующий материал и промывают этилацетатом, после чего этилацетатный раствор промывают водным раствором карбоната натрия. Далее раствор метил- 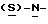 бутокси-карбонил-4-аминофенилаланината в этилацетате частично отгоняют, добавляют бутанол и смесь фракционируют для удаления этилацетата. Бутанольный раствор непосредственно используют на следующей стадии.
Стадия 4. Получение (S)-N-бутоксикарбонил-4-аминофенилаланинола (реакцию см. в конце описания).
В реактор загружают бутанольный раствор метил- бутокси-карбонил-4-аминофенилаланината в этилацетате частично отгоняют, добавляют бутанол и смесь фракционируют для удаления этилацетата. Бутанольный раствор непосредственно используют на следующей стадии.
Стадия 4. Получение (S)-N-бутоксикарбонил-4-аминофенилаланинола (реакцию см. в конце описания).
В реактор загружают бутанольный раствор метил- 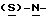 бутоксикарбонил-4-аминофенилаланината со стадии 3, разбавляют н-бутанолом до необходимого объема и охлаждают содержимое реактора примерно до 25oC. После этого в токе азота прибавляют половинное количество борогидрида натрия при поддержании температуры реакции около 25oC и перемешивают в течение 3 часов, после чего добавляют другую половину борогидрида натрия. Смесь перемешивают дополнительно в течение 5 часов, нагревают до температуры 35oC, продолжают перемешивание в течение 12 часов и медленно прибавляют водную соляную кислоту, поддерживая температуру около 30oC с тем, чтобы осуществить разложение любого избытка борогидрида натрия. Далее добавляют воду, нагревают до 35oC и поводят pH примерно до 10 с помощью раствора аммиака. Водный слой отделяют и при поддержании температуры около 35oC промывают органический слой водой. Часть бутанола отгоняют с одновременным обезвоживанием азеотропной перегонкой раствора. Сухой бутанольный раствор непосредственно используют на следующей стадии.
Стадия 5. Получение (S)-4-(4-аминобензил)-2-оксазолидинона (реакцию см. в конце описания).
В реактор загружают сухой н-бутанольный раствор бутоксикарбонил-4-аминофенилаланината со стадии 3, разбавляют н-бутанолом до необходимого объема и охлаждают содержимое реактора примерно до 25oC. После этого в токе азота прибавляют половинное количество борогидрида натрия при поддержании температуры реакции около 25oC и перемешивают в течение 3 часов, после чего добавляют другую половину борогидрида натрия. Смесь перемешивают дополнительно в течение 5 часов, нагревают до температуры 35oC, продолжают перемешивание в течение 12 часов и медленно прибавляют водную соляную кислоту, поддерживая температуру около 30oC с тем, чтобы осуществить разложение любого избытка борогидрида натрия. Далее добавляют воду, нагревают до 35oC и поводят pH примерно до 10 с помощью раствора аммиака. Водный слой отделяют и при поддержании температуры около 35oC промывают органический слой водой. Часть бутанола отгоняют с одновременным обезвоживанием азеотропной перегонкой раствора. Сухой бутанольный раствор непосредственно используют на следующей стадии.
Стадия 5. Получение (S)-4-(4-аминобензил)-2-оксазолидинона (реакцию см. в конце описания).
В реактор загружают сухой н-бутанольный раствор 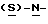 бутоксикарбонил-4-аминофенилаланинола со стадии 4 и добавляют обесцвечивающий активированный уголь. Сухой раствор обрабатывают при температуре около 85oC путем медленного добавления к нему раствора метилата натрия в метаноле. Реакционную смесь нагревают при 85oC при медленном добавлении к ней раствора метилата натрия в метаноле, после чего продолжают нагревание еще 30 минут и фильтруют в горячем виде через вспомогательный фильтрующий материал. После охлаждения раствора по крайней мере в течение 8 часов при 5-10oC смесь центрифугируют, отфильтрованный продукт промывают н-бутанолом и сушат в вакууме при температуре около 50oC.
Стадия 6А. Получение (S)-4-{[3-[2-(диметиламино)этил]-1H-индол-5-ил]- метил}-2-оксазолидинона (реакцию см. в конце описания).
В реактор загружают концентрированную соляную кислоту, деминерализованную воду и бутоксикарбонил-4-аминофенилаланинола со стадии 4 и добавляют обесцвечивающий активированный уголь. Сухой раствор обрабатывают при температуре около 85oC путем медленного добавления к нему раствора метилата натрия в метаноле. Реакционную смесь нагревают при 85oC при медленном добавлении к ней раствора метилата натрия в метаноле, после чего продолжают нагревание еще 30 минут и фильтруют в горячем виде через вспомогательный фильтрующий материал. После охлаждения раствора по крайней мере в течение 8 часов при 5-10oC смесь центрифугируют, отфильтрованный продукт промывают н-бутанолом и сушат в вакууме при температуре около 50oC.
Стадия 6А. Получение (S)-4-{[3-[2-(диметиламино)этил]-1H-индол-5-ил]- метил}-2-оксазолидинона (реакцию см. в конце описания).
В реактор загружают концентрированную соляную кислоту, деминерализованную воду и 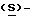 4-(4-аминобензил)-2-оксазолидинон. Содержимое реактора охлаждают до температуры 0-5oC и прибавляют водный раствор нитрита натрия, поддерживая температуру ниже 5oC. После перемешивания смеси около 30 минут к охлажденному водному раствору сульфита натрия добавляют раствор соли диазония, поддерживая температуру ниже 10oC. После перемешивания около 15 минут полученную смесь медленно нагревают до 55-60oC, затем также медленно прибавляют соляную кислоту и выдерживают при температуре около 60oC около 18 часов.
Реакционную смесь разбавляют водой и нагревают примерно до 90oC. Далее в атмосфере азота медленно прибавляют 4,4-диэтокси- 4-(4-аминобензил)-2-оксазолидинон. Содержимое реактора охлаждают до температуры 0-5oC и прибавляют водный раствор нитрита натрия, поддерживая температуру ниже 5oC. После перемешивания смеси около 30 минут к охлажденному водному раствору сульфита натрия добавляют раствор соли диазония, поддерживая температуру ниже 10oC. После перемешивания около 15 минут полученную смесь медленно нагревают до 55-60oC, затем также медленно прибавляют соляную кислоту и выдерживают при температуре около 60oC около 18 часов.
Реакционную смесь разбавляют водой и нагревают примерно до 90oC. Далее в атмосфере азота медленно прибавляют 4,4-диэтокси- 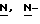 диметилбутиламин и нагревают при температуре образования флегмы около 3 часов. Смесь охлаждают, доводят pH примерно до 7 с помощью раствора гидроокиси натрия и экстрагируют этилацетатом. Водный слой отделяют и доводят pH отделенного слоя до 10 также с помощью раствора гидроокиси натрия. Далее экстрагируют продукт этилацетатом при температуре около 50oC, этилацетатные экстракты (содержащие продукт) объединяют, обрабатывают для обесцвечивания активированным углем и фильтруют через вспомогательный (фильтрующий) материал. Большую часть растворителя отгоняют и полученную суспензию охлаждают примерно до 5oC. Сырой продукт центрифугируют, промывают этилацетатом и сушат в вакууме при температуре 50oC.
Стадия 6В. Очистка (S)-4-{[3-[2-(диметиламино)этил]- 1Н-индол-5-ил]-метил}-2-оксазолидинона диметилбутиламин и нагревают при температуре образования флегмы около 3 часов. Смесь охлаждают, доводят pH примерно до 7 с помощью раствора гидроокиси натрия и экстрагируют этилацетатом. Водный слой отделяют и доводят pH отделенного слоя до 10 также с помощью раствора гидроокиси натрия. Далее экстрагируют продукт этилацетатом при температуре около 50oC, этилацетатные экстракты (содержащие продукт) объединяют, обрабатывают для обесцвечивания активированным углем и фильтруют через вспомогательный (фильтрующий) материал. Большую часть растворителя отгоняют и полученную суспензию охлаждают примерно до 5oC. Сырой продукт центрифугируют, промывают этилацетатом и сушат в вакууме при температуре 50oC.
Стадия 6В. Очистка (S)-4-{[3-[2-(диметиламино)этил]- 1Н-индол-5-ил]-метил}-2-оксазолидинонаМатериалы – Количество Этилацетат – 109,4 л Этанол – 12,3 л Активированный уголь – 2,4 кг Этилацетат (для промывки продукта) – 5,0 л Ацетон – 11,8 л Вода (деминерализованная) – 47,3 кг Вода (деминерализованная для 10,0 кг промывки продукта) – 10,0 кг Вспомогательный фильтрующий материал – 2,0 кг Сырой продукт со стадии 6А растворяют в смеси, находящейся при температуре образования флегмы и состоящей из 10% этанола в этилацетате, обрабатывают обесцвечивающим активированным углем и смесь фильтруют горячей через вспомогательный фильтрующий материал. Раствор медленно охлаждают до температуры выше 5oC и перемешивают в течение 18 часов. Очищенный продукт центрифугируют, промывают этилацетатом и сушат под вакуумом при температуре 50oC. Для удаления сольватированного этилацетата сухой твердый остаток прибавляют к смеси из 20% ацетона в воде при температуре окружающей среды и перемешивают в течение 1 часа. Полученную суспензию охлаждают примерно до 5oC в течение часа перед центрифугированием, затем продукт промывают этилацетатом и сушат в вакууме при температуре около 45oC. Пример 2 Альтернативный способ получения метил-(S)- 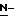 бутоксикарбонил-4-нитрофенилаланината (соединение формулы III) бутоксикарбонил-4-нитрофенилаланината (соединение формулы III)Смесь метил-4-нитро-(L)-фенилаланината гидрохлорида (40,00 г, 0,153 моля) и гидрокарбоната натрия (73 г, 0,870 моля) в 1,4-диоксане (1000 мл) перемешивают при температуре около 10oC в безводной среде. Добавляют раствор бутилхлорформиата (23,12 г, 21,52 мл, 0,169 моля) в 1,4-диоксане (200 мл) в течение 10 минут (температура реакции составляет около 13oC). Полученную суспензию нагревают до комнатной температуры и перемешивают в течение 3 часов. После этого реакцию гасят в воде (1600 мл) и экстрагируют этилацетатом (3 х 650 мл). Объединенные этилацетатные экстракты промывают рассолом (1000 мл), сушат над безводным сульфатом магния, фильтруют и испаряют до получения масла. Остаточный растворитель удаляют с помощью масляного насоса при 50oC с получением сиропа (51,34 г, выход – 103%), который постепенно затвердевает при стоянии. ТСХ (SiO2, EtOAc): гомогенная (Rf = 0,59). 1H ЯМР (60 МГц, CDCl3) – соответствует структуре карбамата. Пример 3 Альтернативный способ получения метил (S) 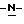 бутоксикарбонил-4-аминофенилаланината (соединение формулы IV) бутоксикарбонил-4-аминофенилаланината (соединение формулы IV)Раствор соединения, полученного по примеру 2 (45,00 г, 0,139 моль) в этаноле (845 мл), добавляют к влажному палладированному углю (Pd/C) (типа 87L, 61,1% H2O) (10%, около 4,5 г) в атмосфере азота. Реакционную смесь оставляют на гидрогенизацию при комнатной температуре и нормальном атмосферном давлении. В течение 9 часов наблюдается равномерное и постоянное поглощение водорода (около 9700 мл). Катализатор фильтруют на гифло (hyflo) и промывают этанолом (100 мл). Фильтрат концентрируют в вакууме (температура водяной бани менее 40oC), последние следы растворителя удаляют с помощью масляного насоса, получая смолу коричневого цвета (41,70 г, 101%). ТСХ (SiO2, EtOAc) подтверждает наличие желаемого продукта (Rf = 0,49) со следами примеси, имеющей более высокую скорость проявления. 1H ЯМР (300 МГц, CDCl3) – соответствует структуре продукта и остаточного этанола. Пример 4 Альтернативный способ получения (S) 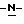 бутоксикарбонил-4-аминофенилаланинола (соединение формулы V) бутоксикарбонил-4-аминофенилаланинола (соединение формулы V)К перемешиваемой суспензии борогидрида натрия (14,80 г, 0,390 моль) в SVM (150 мл) по каплям прибавляют раствор соединения, полученного по примеру 3 (76,40 г, 0,260 моль) в SVM (460 мл) при комнатной температуре. Реакцию оставляют при перемешивании до утра (на время около 18 часов), после чего анализ методом ТСХ указывает на полное потребление исходного продукта. Реакционную смесь подкисляют примерно по pH 4 с помощью 2 М водного раствора соляной кислоты с охлаждением на льду до температуры около 10oC. Полученную смесь концентрируют до получения твердого остатка и медленно добавляют насыщенный водный раствор гидрокарбоната натрия (2000 мл). Водную смесь (с pH около 8) экстрагируют этилацетатом (2 х 750 мл), объединенные органическое экстракты сушат (над сульфатом магния), фильтруют и концентрируют до получения воскообразного твердого остатка бледно-розового цвета (64,56 г, выход – 93%). ТСХ (SiO2, EtOAc) подтверждает наличие желаемого продукта (Rf = 0,33) со следами примесей. 1H ЯМР (60 МГц, CDCl3) – соответствует структуре аланинола. Формула изобретения
 индол-5-ил] метил}-2-оксазолидинона, отличающийся тем, что включает следующие стадии: индол-5-ил] метил}-2-оксазолидинона, отличающийся тем, что включает следующие стадии:а) образование карбамата из метил-4-нитро-(L)-фенилаланината гидрохлорида формулы II 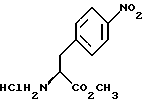 путем добавления карбоната или гидрокарбоната натрия и н-бутилхлор-формиатаиполучениеврезультатереакцииметил-(S) 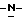 бутоксикарбонил-4-нитрофенилаланината формулы III бутоксикарбонил-4-нитрофенилаланината формулы III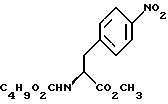 b) восстановление соединения формулы (III) с получением метил-(S) 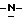 бутоксикарбонил-4-аминофенилаланината формулы IV бутоксикарбонил-4-аминофенилаланината формулы IV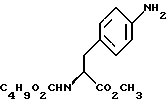 c) восстановление сложноэфирной метильной группы -CO2CH3 в соединении формулы (IV) с получением (S) 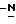 бутоксикарбонил-4-аминофенил аланинола формулы V бутоксикарбонил-4-аминофенил аланинола формулы V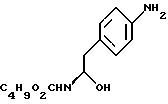 d) замыкание кольца в соединении формулы (V) с получением (s)-4-(4-аминобензил)-2-оксазолидинона формулы VI 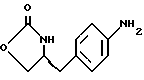 e) получение соли диазония из соединения формулы VI с последующим восстановлением и получением гидразина-(s)-4-(4-гидразинбензил)-2-оксазолидинона гидрохлорида формулы VII 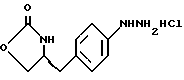 f) реакцию Фишера соединения формулы VII с получением (S)-4-{[3-[2-(диметиламино)-этил]-1Н-индол-5-ил]метил}-2-оксазолидинона. 2. Способ по п. 1, отличающийся тем, что одну или более стадий а)-f) способа осуществляют в одном реакторе. 3. Способ по п.1 или 2, отличающийся тем, что сначала в одном реакторе осуществляют стадии а)-d) с последующим выделением соединения формулы VI, а затем также в одном реакторе осуществляют стадии е)-f). 4. Способ по любому из пп.1 – 3, отличающийся тем, что стадию а) осуществляют в присутствии водного раствора этилацетата с использованием карбоната натрия. 5. Способ по п.4, отличающийся тем, что добавление карбоната натрия на стадии а) осуществляют при температуре около 20oC, а добавление N-бутилхлорформиата осуществляют при температуре около 30oC. 6. Способ по любому из пп. 1 – 5, отличающийся тем, что стадия b) представляет собой реакцию гидрогенизации. 7. Способ по любому из пп.1 – 6, отличающийся тем, что стадию восстановления с) осуществляют с использованием борогидрида натрия. 8. Способ по любому из пп.1 – 7, отличающийся тем, что стадию d) осуществляют на сухом бутанольном растворе соединения формулы V. 9. Способ по любому из пп.1 – 8, отличающийся тем, что реакцию замыкания цикла осуществляют с использованием 30%-ого раствора метилата натрия в метаноле при температуре, которая находится в интервале 50 – 120oC. 10. Способ по любому из пп.1 – 9, отличающийся тем, что стадию е) осуществляют i) взаимодействием соединения формулы VI с нитритом натрия и ii) восстановлением соли диазония, образующейся по реакции i), с помощью сульфита натрия. 11. Способ по любому из пп.1 – 10, отличающийся тем, что реакцию Фишера стадии f) осуществляют при относительно высоком разбавлении. 12. Способ очистки (S)-4-{[3-[2-(диметиламино)этил]- 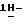 индол-5-ил]метил} -2-оксазолидинона, отличающийся тем, что он включает следующие стадии: индол-5-ил]метил} -2-оксазолидинона, отличающийся тем, что он включает следующие стадии:а) растворение сырого (S)-4-{[3-[2-(диметиламино)этил] 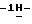 индол-5-ил]-метил} -2-оксазолидинона в смеси этанола в этилацетате при температуре образования флегмы и последующую фильтрацию горячего раствора; b) медленное охлаждение отфильтрованного раствора до температуры около 5oC; с) центрифугирование продукта со стадии b) и промывку этилацетатом с последующей сушкой; d) обработку ацетоном для удаления сольватированного этилацетата.
13. Несольватированный очищенный (S)-4-{ [3-[2-(диметиламино)-этил] индол-5-ил]-метил} -2-оксазолидинона в смеси этанола в этилацетате при температуре образования флегмы и последующую фильтрацию горячего раствора; b) медленное охлаждение отфильтрованного раствора до температуры около 5oC; с) центрифугирование продукта со стадии b) и промывку этилацетатом с последующей сушкой; d) обработку ацетоном для удаления сольватированного этилацетата.
13. Несольватированный очищенный (S)-4-{ [3-[2-(диметиламино)-этил] 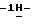 индол-5-ил]-метил}-2-оксазолидинон.
14. Промежуточное соединение метил-(S)-N-бутоксикарбонил-4-нитрофенилаланината формулы III индол-5-ил]-метил}-2-оксазолидинон.
14. Промежуточное соединение метил-(S)-N-бутоксикарбонил-4-нитрофенилаланината формулы III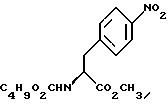 15. Промежуточное соединение метил-(S)-N-бутоксикарбонил-4-аминофенилаланината формулы IV 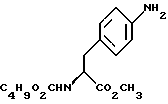 16. Промежуточное соединение (S)-N-бутоксикарбонил-4-аминофенилаланинол формулы V 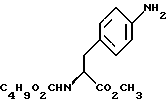 17. Промежуточное соединение (S)-4-(4-аминобензил)-2-оксазолидинон формулы VI 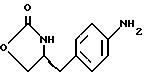 18. Способ получения метил-(S)-N-бутоксикарбонил-4-нитрофенилаланината формулы III 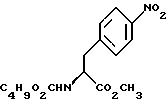 отличающийся тем, что соединение формулы II 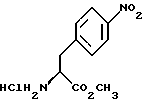 подвергают взаимодействию с карбонатом натрия и н-бутилхлорформиатом. 19. Способ получения метил-(S)-N-бутоксикарбонил-4-аминофенилаланината формулы IV 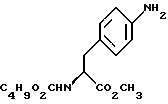 отличающийся тем, что включает восстановление соединения формулы III 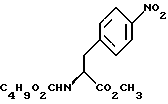 20. Способ получения (S)-N-бутоксикарбонил-4-аминофенилаланинола формулы V 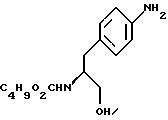 отличающийся тем, что включает восстановление соединения формулы IV 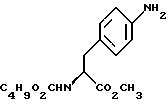 21. Способ получения (S)-4-(4-аминобензил)-2-оксазолидинона формулы VI 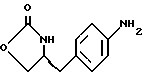 отличающийся тем, что проводят реакцию циклизации соединения формулы V 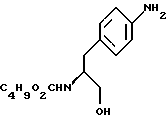 22. Промежуточное соединение по п.14 для получения соединения (S)-4-{ [3-[2-(диметиламино)-этил-1Н-индол-5-ил]метил}-2-оксазолидинона, являющегося полезным терапевтическим агентом для лечения и профилактики мигрени. 23. Промежуточное соединение по п.15 для получения соединения (S)-4-{ [3-[2-(диметиламино)-этил-1Н-индол-5-ил]метил]-2-оксазолидинона, являющегося полезным терапевтическим агентом для лечения и профилактики мигрени. 24. Промежуточное соединение по п.16 для получения соединения (S)-4-{ [3-[2-(диметиламино)-этил-1Н-индол-5-ил]метил}-2-оксазолидинона, являющегося полезным терапевтическим агентом для лечения и профилактики мигрени. 25. Промежуточное соединение по п.17 для получения соединения (S)-4-{ [3-[2-(диметиламино)-этил-1Н-индол-5-ил] , являющегося полезным терапевтическим агентом для лечения и профилактики мигрени. РИСУНКИ
PD4A – Изменение наименования обладателя патента Российской Федерации на изобретение
(73) Новое наименование патентообладателя:
Извещение опубликовано: 10.08.2005 БИ: 22/2005
PC4A – Регистрация договора об уступке патента Российской Федерации на изобретение
Прежний патентообладатель:
(73) Патентообладатель:
Дата и номер государственной регистрации перехода исключительного права: 04.10.2005 № РД0002548
Извещение опубликовано: 20.12.2005 БИ: 35/2005
|
||||||||||||||||||||||||||