Патент на изобретение №2167870
|
||||||||||||||||||||||||||
(54) СЕЛЕКТИВНОЕ ХЛОРИРОВАНИЕ 1-(2-ФТОРФЕНИЛ)-4,5-ДИГИДРО-3-МЕТИЛ-5-ОКСО-1Н-1,2,4-ТРИАЗОЛА
(57) Реферат: Описывается новый способ хлорирования 1-(2-фторфенил)-4,5-дигидро-3-метил-5-оксо-1Н-1,2,4-триазола в 4-положении фенильного кольца. Он заключается в том, что а) добавляют 0,8-1,6 молярных эквивалентов хлора к перемешиваемой суспензии 1-(2-фторфенил)-4,5-дигидро-3-метил-5-оксо-1Н-1,2,4-триазола в растворителе, выбранном из ацетонитрила, N,N-диметилформамида, нитрометана и нитробензола, при температуре в интервале температуры от окружающей среды до 50°С при такой скорости, чтобы температура не превышала 50°С, и продолжают перемешивание при 30-50°С 1 – 10 ч; b) удаляют большую часть побочного продукта хлористого водорода в течение 1 – 6 ч путем снижения давления в реакционном сосуде так, чтобы растворитель кипел, при поддержании температуры 30-50°С; с) необязательно реакционный сосуд продувают азотом для снижения концентрации хлористого водорода в реакционной смеси до ниже 1%; d) повторяют стадии а), b) и с) дважды; и е) выделяют 1-(4-хлор-2-фторфенил)-4,5-дигидро-3-метил-5-оксо-1 Н-1,2,4-триазол. Технический результат – увеличение выхода целевого продукта. 3 з.п.ф-лы. Настоящее изобретение относится к хлорированию фенильного кольца. В частности, раскрывается способ, посредством которого атом хлора вводят в 4-положение 1-(2-фторфенильной) группы, связанной с 4,5-дигидро-3-метил-5- оксо-1H-1,2,4-триазолом, промежуточным соединением при получении гербицида этил-  -2-дихлор-5-[4-(дифторметил)-4,5- дигидро-3-метил-5-оксо-1H-1,2,4-триазол-1-ил] -4-фторбензол- пропаноата (“Целевой гербицид”).
Более ранние попытки достичь эффективного получения целевого гербицида сконцентрированы на способе, раскрытом в Примере 1 Патента США N 4818275, в котором 1-(5-амино-2-фтор-4-хлорфенил)-4-дифторметил-4,5-дигидро-3-метил- 5-оксо-1H-1,2,4-триазол получают из 2-фторанилина восьмистадийным путем. В этом способе хлорирование фенильного кольца в 4-положении проводят перед замыканием триазольного кольца посредством реакции 2-фторацетанилида с хлористым сульфурилом в п-диоксане. Однако скоро выяснилось, что этот восьмистадийный путь синтеза не является удовлетворительным из-за избыточного числа стадий и низких выходов целевых продуктов.
Хотя эффективность многостадийного способа для получения сложной молекулы может быть улучшена посредством оптимизации выхода каждой стадии, даже более высокая эффективность может быть достигнута при нахождении пути синтеза с меньшим числом стадий. В настоящее время установлено, что целевой гербицид может быть получен более эффективно новым путем, включающим только шесть стадий. На первой стадии 2-фторгидразин циклизуют до 4,5-дигидро-1-(2-фторфенил)-3- метил-5-оксо-1H-1,2,4-триазола, который затем хлорируют до 1-(4- хлор-2-фторфенил)-4,5-дигидро-3-метил-5-оксо-1H-1,2,4-триазола. На третьей стадии хлорированный продукт дифторметилируют с образованием 1-(4-хлор-2-фторфенил)-4-дифторметил-4, 5-дигидро- 3-метил-5-оксо-1H-1,2,4-триазола. В качестве альтернативы, хотя этот метод менее предпочтителен, 4,5-дигидро-1-(2-фторфенил)-3- метил-5-оксо-1H-1,2,4-триазол сначала может быть дифторметилирован, а затем хлорирован. Промежуточный продукт третьей стадии нитруют с образованием 1-(4-хлор-2-фтор-5- нитрофенил)-4-дифторметил-4,5-дигидро-3-метил-5-оксо-1H-1,2,4- триазола, который восстанавливают до соответствующего 1-(5-амино- 4-хлор-2-фторфенил)-4-дифторметил-4,5-дигидро-3-метил-5-оксо-1H- 1,2,4-триазола. На последней стадии 5-аминозамещенное производное подвергают диазотированию/арилированию с образованием целевого гербицида.
Настоящее изобретение направлено на получение ключевого промежуточного соединения в указанном способе, продукта второй стадии 1-(4-хлор-2-фторфенил)-4,5-дигидро-3-метил-5-оксо- 1H-1,2,4-триазола, который неожиданно был получен с хорошим выходом путем хлорирования 4,5-дигидро-1-(2-фторфенил)-3-метил-5- оксо-1H-1,2,4-триазола по новому способу, раскрытому в описании и заявленному в формуле изобретения. В качестве альтернативы, хотя это менее желательно, можно осуществлять реакцию хлорирования калиевой соли исходного соединения.
Когда впервые рассматривался указанный выше шестистадийный способ, то полагали, что будет трудно осуществить крупномасштабное хлорирование 4,5-дигидро-1-(2- фторфенил)-3-метил-5-оксо-1H-1,2,4-триазола с хорошим выходом. Попытки хлорировать 4,5-дигидро-1-(2-фторфенил)-3-метил-5-оксо-1H- 1,2,4-триазол хлористым сульфурилом, хлорирующим агентом, используемым при аналогичном хлорировании в восьмистадийном способе получения, оказались неудачными, давая очень низкие выходы. Например, в одном из опытов выход, определенный методом газовой хроматографии (ГХ), составлял после 18 часов только примерно 8 процентов площади. (В реакциях хлорирования, описываемых в описании, процент площади не представляет действительного выхода продукта, так как побочные продукты не определяются методом ГХ. Однако он служит указанием на степень протекания реакции, т.е. процент превращения исходного материала). Предшествующие попытки осуществить хлорирование элементарным хлором в лаборатории дали реальные выходы порядка семидесяти процентов даже при использовании значительного молярного избытка хлора. Считалось, что реакция не подходит для осуществления на опытной установке.
Неожиданно в настоящее время установлено, что хлорирование 4,5-дигидро-1-(2-фторфенил)-3-метил-5-оксо-1H- 1,2,4-триазола тремя отдельными загрузками (подачами сырья) с использованием на каждой примерно одного молярного эквивалента газообразного хлора и последующего удаления побочного продукта хлористого водорода между загрузками, последовательно обеспечивает фактические выходы 4,5-дигидро-1-(4- хлор-2-фторфенил)-3-метил-5-оксо-1H-1,2,4-триазола 82-87% в лабораторных условиях, 75-80% или выше на опытной установке (вплоть до 97% конверсии) при чистоте 95% или выше.
В способе по настоящему изобретению общее количество в три молярных эквивалента элементарного хлора подают в реакционную смесь в трех отдельных загрузках, примерно один эквивалент на каждой. Обычно превращения в доминирующий 1-(4-хлор-2-фторфенил)-4,5-дигидро- 3-метил-5-оксо-1H-1,2,4-триазол после каждой из трех загрузок хлора составляют в процентах площади примерно 50%, 75% и более 97%, соответственно.
Ключом к таким высоким выходам является удаление газообразного хлористого водорода, образующегося на стадиях хлорирования, после каждой из загрузок одного молярного эквивалента хлора. Хлористый водород, если он остается в реакционной смеси, будет останавливать реакцию и/или реагировать с растворителем ацетонитрилом, приводя к пониженным выходам продукта. Хлористый водород удаляют из реакционной смеси до возможной степени, используя, во-первых, удаление в вакууме, а затем, необязательно, продувку азотом. Пониженная концентрация хлористого водорода также помогает уменьшить коррозионные свойства реакционной смеси, что особенно важно в процессе реакции, на стадии фильтрации и стадиях обработки. В предпочтительном способе по настоящему изобретению, 4,5-дигидро-1-(2-фторфенил)-3-метил- 5-оксо-1H-1, 2,4-триазол (один эквивалент) помещают в растворитель, такой как ацетонитрил, N,N-диметилформамид, нитрометан или нитробензол, предпочтительно ацетонитрил. Концентрация этого 2-фторфенильного производного в растворителе, в весовых процентах 2-фторфенильного производного к объему растворителя, находится в интервале примерно 5-70%, предпочтительно примерно 10-35% и более предпочтительно 15-25%. Используемый растворитель может быть свежим, рециркулированным из предыдущих циклов реакции хлорирования или комбинацией свежего и рециркулированного растворителей. Полученную суспензию перемешивают во время стадии хлорирования при температуре в пределах от примерно 0oC до примерно 75oC, предпочтительно от температуры окружающей среды (например, 23oC) до 50oC и более предпочтительно в интервале 30oC – 40oC. При осуществлении способа по настоящему изобретению в масштабе опытной установки добавление хлора проводят в закрытой системе под вакуумом примерно 300 – 500 мм Hg, что помогает смягчить рост давления при добавлении газообразного хлора. При осуществлении способа в лабораторных условиях хлорирование обычно проводят при атмосферном давлении без видимых негативных последствий. Для обеспечения надлежащей абсорбции важна скорость подачи газообразного хлора. В масштабе опытной установки давление в реакторе зависит от скорости добавления газообразного хлора в сравнении со скоростью реакции. Предпочтительно поддерживать давление в реакторе ниже 15 psig (103,2 кПа) посредством скорости добавления газообразного хлора, предпочтительно при примерно 0,5 фунта/минуту (0,227 кг/минуту). При том, что реакционная смесь поддерживается в предпочтительном температурном интервале, 0,8-1,6 молярных эквивалентов, предпочтительно 0,9-1,5 молярных эквивалентов, газообразного хлора добавляют ниже поверхности реакционной смеси при скорости, которая будет поддерживать реакционную смесь при температуре ниже 50oC, предпочтительно между 30oC и 40oC. Время, требуемое для завершения первой загрузки газообразного хлора при поддержании вышеуказанных условий, составляет примерно от 10 минут до 2 часов, предпочтительно от 20 минут до одного часа. После завершения первой загрузки газообразного хлора реакционную смесь доводят до температуры примерно 30oC – 50oC, предпочтительно 30oC – 40oC, при которой ее перемешивают в течение времени выдерживания от одного до 10 часов, предпочтительно от трех до шести часов, после которого конверсия 4,5-дигидро-1-(2-фторфенил)-3-метил-5-оксо-1H- 1,2,4-триазола в 1-(4-хлор-2-фторфенил)-4,5-дигидро-3-метил-5- оксо-1H-1,2,4-триазол, как определено методами газовой хроматографии, составляет примерно 50% (% площади). После завершения времени выдерживания реакционную смесь поддерживают при предпочтительной температуре 30oC – 40oC и размещают в условиях пониженного давления. В масштабе опытной установки понижение давления составляет до примерно 100-200 мм Hg, предпочтительно 135-165 мм Hg. В лабораторных условиях понижение давления составляет до примерно 10 – 30 мм Hg, предпочтительно 15 – 25 мм Hg. В масштабе опытной установки кипячение в описанных выше условиях продолжается в течение времени от одного до шести часов, предпочтительно от двух до четырех часов, во время которого следовые количества остаточного хлора и примерно 99% побочного продукта хлористого водорода отгоняются. Если в реакционной смеси остается более 1% хлористого водорода, то реакционную смесь продувают газообразным азотом для того, чтобы довести уровень хлористого водорода до менее 1%. При проведении реакции в лабораторных условиях кипячение в описанных выше условиях продолжается в течение примерно от 20 минут до двух часов, предпочтительно 30 – 50 минут, затем реакционную смесь продувают газообразным азотом в течение периода времени примерно 10 – 30 минут, предпочтительно 15 -25 минут. По завершении первой загрузки газообразного хлора и последующего удаления побочного продукта хлористого водорода, способ повторяют со второй загрузкой газообразного хлора в количествах и при условиях, описанных выше. Конверсия 4,5-дигидро-1-(2-фторфенил)-3-метил-5-оксо-1H-1,2,4- триазола в 1-(4-хлор-2-фторфенил)-4,5-дигидро-3-метил-5-оксо-1H-1, 2,4-триазол после второй загрузки, как определено методами газовой хроматографии, составляет примерно 75% (% площади). Последующее удаление побочного продукта хлористого водорода снова осуществляют в описанных выше условиях. Таким же образом проводят третью загрузку хлора и удаление побочного продукта хлористого водорода. После завершения третьей загрузки хлора и последующего удаления песочного продукта хлористого водорода конверсия 4,5- дигидро-1-(2-фторфенил)-3-метил-5-оксо-1H-1,2,4-триазола в 1-(4- хлор-2-фторфенил)-4,5-дигидро-3-метил-5-оксо-1H-1,2,4-триазол, как определено методами газовой хроматографии, составляет примерно 97% (% площади) или выше. После удаления хлористого водорода реакционную смесь охлаждают и твердый продукт собирают фильтрацией или центрифугированием. При осуществлении способа на опытной установке реакционную смесь охлаждают до примерно 0oC – 15oC, предпочтительно 3oC – 10oC, чтобы максимизировать осаждение любого продукта в растворе и выдерживают при этой температуре в течение периода примерно от 30 минут до двух часов, предпочтительно один час. Твердый продукт собирают центрифугированием и промывают холодным растворителем, предпочтительно ацетонитрилом. При осуществлении способа в масштабе опытной установки выходы 1-(4-хлор-2-фторфенил)-4,5- дигидро-3-метил-5-оксо-1H-1,2,4-триазола составляют примерно 80% с чистотой 98-100%. При соответствующем осуществлении в лабораторных масштабах получают выходы 82 – 89% и чистоту 98 – 100%. Растворитель ацетонитрил, удаляемый из продукта фильтрацией или ценрифугированием, может быть перегнан для использования при последующих хлорированиях по данному способу.
Попытки катализировать описанные выше стадии хлорирования рядом катализаторов не привели к существенному увеличению скорости реакции или не направили ее дополнительно до завершения. Испытанные катализаторы включали п-толуолсульфоновую кислоту, трифторметансульфонат иттербия, уксусную кислоту, гидрокси(4- метилбензолсульфонато-O) фенилиод, триэтилфосфит, воду, серную кислоту, 2,6-ди-трет-бутил-4-метилфенол, элементарный иод, трис [2-(2-метоксиэтокси)этил] амин, хлористый алюминий, хлорид тетрабутиламмония, бромид тетрабутиламмония, диметил- аминопиридин и хлорид железа (3). Также испытывали ряд основных реагентов в стехиометрических количествах для удаления (захвата) побочного продукта хлористого водорода. Они включали ацетат натрия, поли (4-винил) пиридин, триэтиламин и 1, 8- диазабицикло-[5.4.0]ундец-7-ен. Хотя эти реагенты нейтрализовали образующийся хлористый водород, ни один из них не увеличивал значительно скорость реакции или не направлял реакцию дополнительно к завершению.
Хлорирование способами, описанными в целом выше, проводили с калиевой солью 4,5-дигидро-1-(2- фторфенил)-3-метил-5-оксо-1H-1,2,4-триазола или с 4-дифторметил- 4,5-дигидро-1-(2-фторфенил)-3-метил-5-оксо-1H-1,2,4-триазолом. Обычно эти реакции не протекают так быстро, как в случае предпочтительного 4,5-дигидро-1-(2-фторфенил)-3-метил-5-оксо- 1Н-1,2,4-триазола, хотя обе приводили к соответствующему хлорированному продукту. Ряд реакций хлорирования с использованием 4-дифторметил-4,5-дигидро-1-(2-фторфенил)-3-метил-5-оксо-1,2,4- триазола для завершения реакции требовал избыточное количество хлора и длительного времени реакции. Для увеличения скорости реакции использовали перечисленные выше катализаторы, но эти попытки не дали никакого эффекта.
Конкретные примеры применения способа по данному изобретению приведены ниже.
Пример 1 -2-дихлор-5-[4-(дифторметил)-4,5- дигидро-3-метил-5-оксо-1H-1,2,4-триазол-1-ил] -4-фторбензол- пропаноата (“Целевой гербицид”).
Более ранние попытки достичь эффективного получения целевого гербицида сконцентрированы на способе, раскрытом в Примере 1 Патента США N 4818275, в котором 1-(5-амино-2-фтор-4-хлорфенил)-4-дифторметил-4,5-дигидро-3-метил- 5-оксо-1H-1,2,4-триазол получают из 2-фторанилина восьмистадийным путем. В этом способе хлорирование фенильного кольца в 4-положении проводят перед замыканием триазольного кольца посредством реакции 2-фторацетанилида с хлористым сульфурилом в п-диоксане. Однако скоро выяснилось, что этот восьмистадийный путь синтеза не является удовлетворительным из-за избыточного числа стадий и низких выходов целевых продуктов.
Хотя эффективность многостадийного способа для получения сложной молекулы может быть улучшена посредством оптимизации выхода каждой стадии, даже более высокая эффективность может быть достигнута при нахождении пути синтеза с меньшим числом стадий. В настоящее время установлено, что целевой гербицид может быть получен более эффективно новым путем, включающим только шесть стадий. На первой стадии 2-фторгидразин циклизуют до 4,5-дигидро-1-(2-фторфенил)-3- метил-5-оксо-1H-1,2,4-триазола, который затем хлорируют до 1-(4- хлор-2-фторфенил)-4,5-дигидро-3-метил-5-оксо-1H-1,2,4-триазола. На третьей стадии хлорированный продукт дифторметилируют с образованием 1-(4-хлор-2-фторфенил)-4-дифторметил-4, 5-дигидро- 3-метил-5-оксо-1H-1,2,4-триазола. В качестве альтернативы, хотя этот метод менее предпочтителен, 4,5-дигидро-1-(2-фторфенил)-3- метил-5-оксо-1H-1,2,4-триазол сначала может быть дифторметилирован, а затем хлорирован. Промежуточный продукт третьей стадии нитруют с образованием 1-(4-хлор-2-фтор-5- нитрофенил)-4-дифторметил-4,5-дигидро-3-метил-5-оксо-1H-1,2,4- триазола, который восстанавливают до соответствующего 1-(5-амино- 4-хлор-2-фторфенил)-4-дифторметил-4,5-дигидро-3-метил-5-оксо-1H- 1,2,4-триазола. На последней стадии 5-аминозамещенное производное подвергают диазотированию/арилированию с образованием целевого гербицида.
Настоящее изобретение направлено на получение ключевого промежуточного соединения в указанном способе, продукта второй стадии 1-(4-хлор-2-фторфенил)-4,5-дигидро-3-метил-5-оксо- 1H-1,2,4-триазола, который неожиданно был получен с хорошим выходом путем хлорирования 4,5-дигидро-1-(2-фторфенил)-3-метил-5- оксо-1H-1,2,4-триазола по новому способу, раскрытому в описании и заявленному в формуле изобретения. В качестве альтернативы, хотя это менее желательно, можно осуществлять реакцию хлорирования калиевой соли исходного соединения.
Когда впервые рассматривался указанный выше шестистадийный способ, то полагали, что будет трудно осуществить крупномасштабное хлорирование 4,5-дигидро-1-(2- фторфенил)-3-метил-5-оксо-1H-1,2,4-триазола с хорошим выходом. Попытки хлорировать 4,5-дигидро-1-(2-фторфенил)-3-метил-5-оксо-1H- 1,2,4-триазол хлористым сульфурилом, хлорирующим агентом, используемым при аналогичном хлорировании в восьмистадийном способе получения, оказались неудачными, давая очень низкие выходы. Например, в одном из опытов выход, определенный методом газовой хроматографии (ГХ), составлял после 18 часов только примерно 8 процентов площади. (В реакциях хлорирования, описываемых в описании, процент площади не представляет действительного выхода продукта, так как побочные продукты не определяются методом ГХ. Однако он служит указанием на степень протекания реакции, т.е. процент превращения исходного материала). Предшествующие попытки осуществить хлорирование элементарным хлором в лаборатории дали реальные выходы порядка семидесяти процентов даже при использовании значительного молярного избытка хлора. Считалось, что реакция не подходит для осуществления на опытной установке.
Неожиданно в настоящее время установлено, что хлорирование 4,5-дигидро-1-(2-фторфенил)-3-метил-5-оксо-1H- 1,2,4-триазола тремя отдельными загрузками (подачами сырья) с использованием на каждой примерно одного молярного эквивалента газообразного хлора и последующего удаления побочного продукта хлористого водорода между загрузками, последовательно обеспечивает фактические выходы 4,5-дигидро-1-(4- хлор-2-фторфенил)-3-метил-5-оксо-1H-1,2,4-триазола 82-87% в лабораторных условиях, 75-80% или выше на опытной установке (вплоть до 97% конверсии) при чистоте 95% или выше.
В способе по настоящему изобретению общее количество в три молярных эквивалента элементарного хлора подают в реакционную смесь в трех отдельных загрузках, примерно один эквивалент на каждой. Обычно превращения в доминирующий 1-(4-хлор-2-фторфенил)-4,5-дигидро- 3-метил-5-оксо-1H-1,2,4-триазол после каждой из трех загрузок хлора составляют в процентах площади примерно 50%, 75% и более 97%, соответственно.
Ключом к таким высоким выходам является удаление газообразного хлористого водорода, образующегося на стадиях хлорирования, после каждой из загрузок одного молярного эквивалента хлора. Хлористый водород, если он остается в реакционной смеси, будет останавливать реакцию и/или реагировать с растворителем ацетонитрилом, приводя к пониженным выходам продукта. Хлористый водород удаляют из реакционной смеси до возможной степени, используя, во-первых, удаление в вакууме, а затем, необязательно, продувку азотом. Пониженная концентрация хлористого водорода также помогает уменьшить коррозионные свойства реакционной смеси, что особенно важно в процессе реакции, на стадии фильтрации и стадиях обработки. В предпочтительном способе по настоящему изобретению, 4,5-дигидро-1-(2-фторфенил)-3-метил- 5-оксо-1H-1, 2,4-триазол (один эквивалент) помещают в растворитель, такой как ацетонитрил, N,N-диметилформамид, нитрометан или нитробензол, предпочтительно ацетонитрил. Концентрация этого 2-фторфенильного производного в растворителе, в весовых процентах 2-фторфенильного производного к объему растворителя, находится в интервале примерно 5-70%, предпочтительно примерно 10-35% и более предпочтительно 15-25%. Используемый растворитель может быть свежим, рециркулированным из предыдущих циклов реакции хлорирования или комбинацией свежего и рециркулированного растворителей. Полученную суспензию перемешивают во время стадии хлорирования при температуре в пределах от примерно 0oC до примерно 75oC, предпочтительно от температуры окружающей среды (например, 23oC) до 50oC и более предпочтительно в интервале 30oC – 40oC. При осуществлении способа по настоящему изобретению в масштабе опытной установки добавление хлора проводят в закрытой системе под вакуумом примерно 300 – 500 мм Hg, что помогает смягчить рост давления при добавлении газообразного хлора. При осуществлении способа в лабораторных условиях хлорирование обычно проводят при атмосферном давлении без видимых негативных последствий. Для обеспечения надлежащей абсорбции важна скорость подачи газообразного хлора. В масштабе опытной установки давление в реакторе зависит от скорости добавления газообразного хлора в сравнении со скоростью реакции. Предпочтительно поддерживать давление в реакторе ниже 15 psig (103,2 кПа) посредством скорости добавления газообразного хлора, предпочтительно при примерно 0,5 фунта/минуту (0,227 кг/минуту). При том, что реакционная смесь поддерживается в предпочтительном температурном интервале, 0,8-1,6 молярных эквивалентов, предпочтительно 0,9-1,5 молярных эквивалентов, газообразного хлора добавляют ниже поверхности реакционной смеси при скорости, которая будет поддерживать реакционную смесь при температуре ниже 50oC, предпочтительно между 30oC и 40oC. Время, требуемое для завершения первой загрузки газообразного хлора при поддержании вышеуказанных условий, составляет примерно от 10 минут до 2 часов, предпочтительно от 20 минут до одного часа. После завершения первой загрузки газообразного хлора реакционную смесь доводят до температуры примерно 30oC – 50oC, предпочтительно 30oC – 40oC, при которой ее перемешивают в течение времени выдерживания от одного до 10 часов, предпочтительно от трех до шести часов, после которого конверсия 4,5-дигидро-1-(2-фторфенил)-3-метил-5-оксо-1H- 1,2,4-триазола в 1-(4-хлор-2-фторфенил)-4,5-дигидро-3-метил-5- оксо-1H-1,2,4-триазол, как определено методами газовой хроматографии, составляет примерно 50% (% площади). После завершения времени выдерживания реакционную смесь поддерживают при предпочтительной температуре 30oC – 40oC и размещают в условиях пониженного давления. В масштабе опытной установки понижение давления составляет до примерно 100-200 мм Hg, предпочтительно 135-165 мм Hg. В лабораторных условиях понижение давления составляет до примерно 10 – 30 мм Hg, предпочтительно 15 – 25 мм Hg. В масштабе опытной установки кипячение в описанных выше условиях продолжается в течение времени от одного до шести часов, предпочтительно от двух до четырех часов, во время которого следовые количества остаточного хлора и примерно 99% побочного продукта хлористого водорода отгоняются. Если в реакционной смеси остается более 1% хлористого водорода, то реакционную смесь продувают газообразным азотом для того, чтобы довести уровень хлористого водорода до менее 1%. При проведении реакции в лабораторных условиях кипячение в описанных выше условиях продолжается в течение примерно от 20 минут до двух часов, предпочтительно 30 – 50 минут, затем реакционную смесь продувают газообразным азотом в течение периода времени примерно 10 – 30 минут, предпочтительно 15 -25 минут. По завершении первой загрузки газообразного хлора и последующего удаления побочного продукта хлористого водорода, способ повторяют со второй загрузкой газообразного хлора в количествах и при условиях, описанных выше. Конверсия 4,5-дигидро-1-(2-фторфенил)-3-метил-5-оксо-1H-1,2,4- триазола в 1-(4-хлор-2-фторфенил)-4,5-дигидро-3-метил-5-оксо-1H-1, 2,4-триазол после второй загрузки, как определено методами газовой хроматографии, составляет примерно 75% (% площади). Последующее удаление побочного продукта хлористого водорода снова осуществляют в описанных выше условиях. Таким же образом проводят третью загрузку хлора и удаление побочного продукта хлористого водорода. После завершения третьей загрузки хлора и последующего удаления песочного продукта хлористого водорода конверсия 4,5- дигидро-1-(2-фторфенил)-3-метил-5-оксо-1H-1,2,4-триазола в 1-(4- хлор-2-фторфенил)-4,5-дигидро-3-метил-5-оксо-1H-1,2,4-триазол, как определено методами газовой хроматографии, составляет примерно 97% (% площади) или выше. После удаления хлористого водорода реакционную смесь охлаждают и твердый продукт собирают фильтрацией или центрифугированием. При осуществлении способа на опытной установке реакционную смесь охлаждают до примерно 0oC – 15oC, предпочтительно 3oC – 10oC, чтобы максимизировать осаждение любого продукта в растворе и выдерживают при этой температуре в течение периода примерно от 30 минут до двух часов, предпочтительно один час. Твердый продукт собирают центрифугированием и промывают холодным растворителем, предпочтительно ацетонитрилом. При осуществлении способа в масштабе опытной установки выходы 1-(4-хлор-2-фторфенил)-4,5- дигидро-3-метил-5-оксо-1H-1,2,4-триазола составляют примерно 80% с чистотой 98-100%. При соответствующем осуществлении в лабораторных масштабах получают выходы 82 – 89% и чистоту 98 – 100%. Растворитель ацетонитрил, удаляемый из продукта фильтрацией или ценрифугированием, может быть перегнан для использования при последующих хлорированиях по данному способу.
Попытки катализировать описанные выше стадии хлорирования рядом катализаторов не привели к существенному увеличению скорости реакции или не направили ее дополнительно до завершения. Испытанные катализаторы включали п-толуолсульфоновую кислоту, трифторметансульфонат иттербия, уксусную кислоту, гидрокси(4- метилбензолсульфонато-O) фенилиод, триэтилфосфит, воду, серную кислоту, 2,6-ди-трет-бутил-4-метилфенол, элементарный иод, трис [2-(2-метоксиэтокси)этил] амин, хлористый алюминий, хлорид тетрабутиламмония, бромид тетрабутиламмония, диметил- аминопиридин и хлорид железа (3). Также испытывали ряд основных реагентов в стехиометрических количествах для удаления (захвата) побочного продукта хлористого водорода. Они включали ацетат натрия, поли (4-винил) пиридин, триэтиламин и 1, 8- диазабицикло-[5.4.0]ундец-7-ен. Хотя эти реагенты нейтрализовали образующийся хлористый водород, ни один из них не увеличивал значительно скорость реакции или не направлял реакцию дополнительно к завершению.
Хлорирование способами, описанными в целом выше, проводили с калиевой солью 4,5-дигидро-1-(2- фторфенил)-3-метил-5-оксо-1H-1,2,4-триазола или с 4-дифторметил- 4,5-дигидро-1-(2-фторфенил)-3-метил-5-оксо-1H-1,2,4-триазолом. Обычно эти реакции не протекают так быстро, как в случае предпочтительного 4,5-дигидро-1-(2-фторфенил)-3-метил-5-оксо- 1Н-1,2,4-триазола, хотя обе приводили к соответствующему хлорированному продукту. Ряд реакций хлорирования с использованием 4-дифторметил-4,5-дигидро-1-(2-фторфенил)-3-метил-5-оксо-1,2,4- триазола для завершения реакции требовал избыточное количество хлора и длительного времени реакции. Для увеличения скорости реакции использовали перечисленные выше катализаторы, но эти попытки не дали никакого эффекта.
Конкретные примеры применения способа по данному изобретению приведены ниже.
Пример 1Получение 1-(4-хлор-2-фторфенил)-4,5-дигидро-3-метил-5- оксо-1H-1,2,4-триазола хлорированием 4,5-дигидро-1-(2-фтор- фенил)-3-мeтил-5-oксо-1H-1,2,4-триазола (Лабораторный масштаб) В реакционный сосуд на 1500 мл, оборудованный механической мешалкой, температурным зондом, трубкой ввода газа и конденсатором, загружают 200 г (1.035 моля; 1,0 эквив. ) 4,5-дигидро-1-(2-фторфенил)-3-метил-5-оксо-1H-1,2,4- триазола и 1006 мл ацетонитрила (вес.%/объем триазол/растворитель составляет 19,88%). Перемешиваемую смесь продувают азотом в течение примерно 25 минут, затем температуру доводят до 35oC. Затем в течение одного часа ниже поверхности реакционной смеси барботируют газообразный хлор, примерно 73,7 г (1,035 моля; один эквив.). После завершения добавления реакционную смесь перемешивают при 35oC в течение примерно шести часов, при этом времени газохроматографический анализ реакционной смеси показывает, что реакция прошла до примерно 50% (% площади) конверсии в продукт. При поддержании температуры при 35oC реакционную смесь вакуумируют и выдерживают в вакууме примерно 20 мм Hg в течение 45 минут для удаления побочного продукта хлористого водорода. Затем реакционную смесь продувают в течение 20 минут газообразным азотом для дополнительного удаления побочного продукта хлористого водорода. После удаления хлористого водорода реакционной смеси позволяют охладиться до температуры окружающей среды и перемешивают в течение примерно 16 часов. (Такой период времени был необходим для удобства, а не по необходимости, требуется только примерно три часа). После этого времени второй эквивалент 73,7 г (1,035 моля, один эквив.) газообразного хлора барботируют ниже уровня поверхности реакционной смеси в течение одного часа. После завершения добавления температуру реакционной смеси доводят до 35oC, при которой ее перемешивают в течение 1,5 часов, при этом времени ГХ-анализ указывает, что реакция прошла до примерно 75% (% площади) конверсии в продукт. Сохраняя температуру при 35oC, реакционную смесь снова вакуумируют и выдерживают при примерно 20 мм Hg в течение 1 часа для удаления побочного продукта хлористого водорода. Реакционную смесь снова продувают в течение 20 минут газообразным азотом для дополнительного удаления побочного продукта – хлористого водорода. После удаления хлористого водорода реакционной смеси позволяют остыть до температуры окружающей среды, при которой ее перемешивают в течение примерно 16 часов (снова по соображениям удобства). После этого времени третий эквивалент 73,7 г (1,035 моля, один эквив.) газообразного хлора барботируют ниже уровня поверхности реакционной смеси в течение одного часа. После завершения добавления температуру реакционной смеси доводят до 35oC, при которой ее снова перемешивают в течение шести часов, при этом времени ГХ-анализ показывает, что реакция превращения в целевой продукт прошла примерно на 97% (% площади). Поддерживая температуру при 35oC, реакционную смесь снова выдерживают в вакууме примерно 20 мм Hg в течение одного часа для удаления побочного продукта – хлористого водорода. Затем реакционную смесь продувают в течение 30 минут газообразным азотом для дополнительного удаления хлористого водорода, охлаждают до температуры окружающей среды и фильтруют, получая первую порцию 184,5 г твердого продукта. Перегонка маточного раствора в вакууме примерно 5 мм Hg без нагревания дает 936.5 мл ацетонитрила (93,1% регенерации). Вторую порцию 2,3 г продукта собирают из остатка, остающегося после перегонки. Две порции продукта объединяют, получая 186,8 г (83,5% выход) 1-(4- хлор-2-фторфенил)-4,5-дигидро-3-метил-5-оксо-1H-1,2,4-триазола с чистотой 99,5% (весовые %, определено газохроматографическим методом). Пример 2 Получение 1-(4-хлор-2-фторфенил)-4,5-дигидро-3-метил-5- оксо-1H-1,2,4-триaзoлa хлорированием 4,5-дигидро-1- (2-фторфенил)-3-метил-5-oкco-1H-1,2,4-тpиaзoлa (В масштабе опытной установки на 50 галлонов (189,25 л)) В 50-галлоновый (189,25 л) эмалированный реакционный сосуд, оборудованный конденсатором Hasteloy и стеклянным перемешивающим устройством, загружают 115 фунтов (52,21 кг) ацетонитрила, рециркулированного с предшествующего опыта настоящей реакции), 47,4 фунта (21,5 кг) свежего ацетонитрила (всего 162,4 фунта (73,663 кг) = 94,685 литра) и 40,6 фунта (0,210 фунт-моль (95,302 моля); 1 экв. – 18,416 кг) 4,5-дигидро-1-(2-фторфенил)-3-метил- 5-оксо-1H-1,2,4-триазола (вес. %/об-триазол/растворитель составляет 19,46%). Затем смесь перемешивают и нагревают до 35oC. Во время периода нагревания реакционный сосуд трижды продувают газообразным азотом и герметизируют под вакуумом примерно 300-500 мм Hg. Затем газообразный хлор 21.01 фунта (9,53 кг) (0,296 фунт-моль (134,225 моль); 1,41 эквив.) подают в реакционную смесь ниже поверхности с такой скоростью (примерно 0,5 фунта/мин = 0,227 кг/мин), чтобы поддерживать температуру реакционной смеси при 40oC или меньше и давление в реакционном сосуде ниже 15 psig (103,2 кПа). Время, необходимое для завершения подачи газообразного хлора, составляет примерно 30 минут. После загрузки хлора линии загрузки газообразного хлора продувают азотом, что вызывает некоторое снижение температуры. Температуру реакционной смеси медленно доводят до 40oC при поддержании давления в реакционном сосуде ниже 15 psig (103,2 кПа). Реакционную смесь затем перемешивают в течение времени выдерживания три часа, после этого периода времени анализируют конверсию исходного материала в продукт и содержание побочного продукта хлористого водорода. За трехчасовое время выдерживания конверсия исходного материала в продукт, как определено методами газовой хроматографии, составляла примерно 50 % (% площади). После этого промежутка времени реакционную смесь нагревают с обратным холодильником в течение трех часов при 40oC/150 мм Hg, вызывая удаление побочного продукта хлористого водорода. Приемлемое содержание побочного продукта хлористого водорода менее 1% было достигнуто в конце трехчасового нагревания с обратным холодильником. Затем реакционную смесь охлаждают до 35oC и реакционный сосуд помещают под вакуум примерно 300-500 мм ртутного столба. Вторую загрузку газообразного хлора, 14,01. фунта (6,355 кг) (0,198 фунт-молей (89,507 моль); 0,94 эквив.) затем загружают в реакционную смесь способом, описанным выше для первой загрузки хлора ( 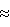 семь часов после первой загрузки). Время, необходимое для завершения второй загрузки газообразного хлора, составляет примерно 30 минут. После окончания загрузки хлора реакционную смесь выдерживают при 40oC/ семь часов после первой загрузки). Время, необходимое для завершения второй загрузки газообразного хлора, составляет примерно 30 минут. После окончания загрузки хлора реакционную смесь выдерживают при 40oC/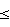 15 psig (103,2 кПа) в течение трех часов, как описано ранее. Затем реакционную смесь анализируют на степень конверсии исходного материала в целевой продукт и на содержание побочного продукта – хлористого водорода. Во время трехчасового выдерживания конверсия исходного материала в продукт, как определено методами газовой хроматографии, составляет примерно 75% (% площади). Реакционную смесь снова нагревают с обратным холодильником в течение трех часов при 40oC/ 150 мм ртутного столба для удаления побочного продукта хлористого водорода до содержания менее 1%. Затем реакционную смесь доводят до ранее описанных условий и проводят третью загрузку 14,01 футов (6,355 кг) (0,198 фунт-моль (89,507 моль); 0,94 эквив.) газообразного хлора (~ 14,5 часа после первой загрузки). По завершении добавления третьей загрузки газообразного хлора реакционную смесь снова выдерживают при 40oC в течение четырех часов. Конверсия исходного материала в продукт 96% (% площади) или более была осуществлена за четырехчасовой промежуток выдержки. Реакционную смесь снова нагревают с обратным холодильником при 40oC/ 15 psig (103,2 кПа) в течение трех часов для удаления побочного продукта хлористого водорода. После завершения нагревания с обратным холодильником (общее время реакции: ~ 22,5 часа) реакционную смесь охлаждают до 5oC в течение 30 минут и перемешивают при этой температуре в течение одного часа. Затем реакционную смесь переносят в подходящую центрифугу, где ее вращают в течение 30 минут для удаления маточного раствора. Маточный раствор помещают в отдельный приемник для регенерации ацетонитрила посредством перегонки. Фильтровальный осадок промывают сначала 35 фунтами (15,89 кг) холодного (0-5oC) ацетонитрила, загруженного непосредственно в центрифугу. Смесь вращают в течение 30 минут для удаления промывного ацетонитрила. Вторую порцию в 35 фунтов (15,89 г кг) холодного ацетонитрила загружают в реакционный сосуд, где его перемешивают в течение пяти минут для удаления каких-либо остатков реакционной смеси. Промывной ацетонитрил затем переносят в центрифугу, где его вращают, как описано ранее. Фильтровальный осадок удаляют из центрифуги и сушат при 60oC/25 мм ртутного столба в течение 24 часов, получая 38,27 фунта (17,37 кг) (выход 80,2%) 1-(4-хлор-2- фторфенил)-4,5-дигидро-3-метил-5-оксо-1H-1,2,4-триазола, который имел 99,9% чистоту (весовые %, как определено методами газовой хроматографии). 15 psig (103,2 кПа) в течение трех часов, как описано ранее. Затем реакционную смесь анализируют на степень конверсии исходного материала в целевой продукт и на содержание побочного продукта – хлористого водорода. Во время трехчасового выдерживания конверсия исходного материала в продукт, как определено методами газовой хроматографии, составляет примерно 75% (% площади). Реакционную смесь снова нагревают с обратным холодильником в течение трех часов при 40oC/ 150 мм ртутного столба для удаления побочного продукта хлористого водорода до содержания менее 1%. Затем реакционную смесь доводят до ранее описанных условий и проводят третью загрузку 14,01 футов (6,355 кг) (0,198 фунт-моль (89,507 моль); 0,94 эквив.) газообразного хлора (~ 14,5 часа после первой загрузки). По завершении добавления третьей загрузки газообразного хлора реакционную смесь снова выдерживают при 40oC в течение четырех часов. Конверсия исходного материала в продукт 96% (% площади) или более была осуществлена за четырехчасовой промежуток выдержки. Реакционную смесь снова нагревают с обратным холодильником при 40oC/ 15 psig (103,2 кПа) в течение трех часов для удаления побочного продукта хлористого водорода. После завершения нагревания с обратным холодильником (общее время реакции: ~ 22,5 часа) реакционную смесь охлаждают до 5oC в течение 30 минут и перемешивают при этой температуре в течение одного часа. Затем реакционную смесь переносят в подходящую центрифугу, где ее вращают в течение 30 минут для удаления маточного раствора. Маточный раствор помещают в отдельный приемник для регенерации ацетонитрила посредством перегонки. Фильтровальный осадок промывают сначала 35 фунтами (15,89 кг) холодного (0-5oC) ацетонитрила, загруженного непосредственно в центрифугу. Смесь вращают в течение 30 минут для удаления промывного ацетонитрила. Вторую порцию в 35 фунтов (15,89 г кг) холодного ацетонитрила загружают в реакционный сосуд, где его перемешивают в течение пяти минут для удаления каких-либо остатков реакционной смеси. Промывной ацетонитрил затем переносят в центрифугу, где его вращают, как описано ранее. Фильтровальный осадок удаляют из центрифуги и сушат при 60oC/25 мм ртутного столба в течение 24 часов, получая 38,27 фунта (17,37 кг) (выход 80,2%) 1-(4-хлор-2- фторфенил)-4,5-дигидро-3-метил-5-оксо-1H-1,2,4-триазола, который имел 99,9% чистоту (весовые %, как определено методами газовой хроматографии).
Формула изобретения
MM4A – Досрочное прекращение действия патента СССР или патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 21.11.2009
Извещение опубликовано: 20.09.2010 БИ: 26/2010
|
||||||||||||||||||||||||||