Патент на изобретение №2167860
|
||||||||||||||||||||||||||
(54) СПОСОБ ПОЛУЧЕНИЯ ЛАКТАМА
(57) Реферат: Изобретение относится к получению алифатических лактамов из динитрилов. Способ включает две стадии, на первой из которых осуществляют полугидрирование динитрила водородом с гетерогенным катализатором до соответствующего аминонитрила, а на второй стадии проводят гидролиз водой в паровой фазе в присутствии катализатора или без него с одновременной циклизацией до лактама. Промежуточный аминонитрил очищают от воды и/или растворителя и органических примесей перегонкой при пониженном давлении. В качестве катализатора на стадии гидролиза могут быть использованы цеолит, силикат или другие молекулярные сита кислого или амфотерного типа либо фосфат металла общей формулы (PO4)nHh(Imp)p, где (Imp) – импрегнирующее соединение на основе щелочного или щелочноземельного металла. В результате увеличиваются выход и чистота конечного продукта. 14 з.п.ф-лы. Настоящее изобретение относится к способу получения лактамов из динитрилов. Алифатические лактамы, такие как эпсилон-капролактам, являются основным сырьем для получения полиамидов (полиамид-6 из капролактама). В промышленности капролактам получают из циклогексанона, который превращают в оксим. Оксим затем превращают в капролактам в присутствии сильной кислоты, такой как серная кислота, а избыток ее затем нейтрализуют. Такой способ обладает большим недостатком, заключающимся в том, что образуются очень большие количества сульфата аммония, доходящие до нескольких тонн на тонну полученного капролактама. Другой способ получения этих лактамов состоит в осуществлении частичного гидрирования динитрилов до соответствующих аминонитрилов, преимущественно адипонитрила до аминокапронитрила, затем тщательно очищают аминонитрил от всех примесей, таких как различные имины или амины, образующиеся во время гидрирования, а также от диамина, и полимеризуют до полиамида очищенный аминокапронитрил. Вышеописанный способ, привлекательный на первый взгляд, требует, однако, тщательной очистки аминонитрила. В самом деле, полимеризация возможна только в том случае, если аминонитрил не содержит диамин, и особенно различные имины или амины в качестве побочных продуктов, которые ограничивают степень полимеризации и вызывают появление окрасок и образование разветвленного полимера. Однако близость свойств аминонтрила к удаляемым иминам или аминам и, более того, существование равновесия между различными формами побочных продуктов делает такое разделение трудным для осуществления, поэтому необходимо добавлять одно или несколько соединений, предназначенных для превращения иминов, перед их удалением. Так, в патенте ВОИС-А- 9316984 предлагают добавлять к обрабатываемой смеси до 10 мас.% карбонильного соединения, такого как альдегид или кетон. В патенте ВОИС-А-9314064 рекомендуют вводить в реакционную смесь перед ее перегонкой метиленовое соединение, такое как малонитрил, дициклопентадиен, циклопентадиен, нитрометан, нитроэтан, инден. Введение соединений к уже сложной смеси является недостатком указанного способа. Настоящее изобретение относится к способу получения лактама, объединяющему две стадии, из которых одна представляет собой полугидрирование динитрила до аминонитрила, другая – гидролиз с циклизацией аминонитрила, прошедшего только одну операцию очистки. Более конкретно, способ получения лактама заключается в том, что: – гидрируют алифатический динитрил до аминонитрила с помощью водорода и в присутствии катализатора; – перегоняют полученный аминонитрил, чтобы оставить содержание динитрила менее или равное 10 мас.%, а содержание побочных продуктов с имино- или аминофункцией ниже или равным 10 мас.%; – перегнанный аминонитрил в паровой фазе вводят во взаимодействие с водой в присутствии катализатора или без него. Путем перегонки можно было бы получить аминонитрил с массовым содержанием динитрила 0,0050%, но это незначительное содержание достигалось бы в ущерб количеству обрабатываемого аминонитрила, так как пришлось бы отделять промежуточные фракции перегонки, годные для рециркулирования, на новую операцию полугидрирования. Поэтому обычно предпочитают направлять на стадию гидролиза с циклизацией аминонитрил, содержащий 0,005-5 мас.% динитрила. Обычно за счет перегонки трудно снизить в аминонитриле общее содержание других побочных продуктов до менее 0,2%, но поскольку эта величина не является нижним критическим пределом для способа изобретения, то на стадии гидролиза с циклизацией используют аминонитрил, содержащий 0,2- 5 мас.% побочных продуктов с имино- или аминофункцией. Соответствующий используемому динитрилу диамин не мешает стадии гидролиза с циклизацией. В настоящем изобретении его не рассматривают как вышеуказанный побочный продукт с аминофункцией. Алифатические динитрилы, которые могут быть использованы на первой стадии способа изобретения, представляют собой динитрилы общей формулы (I): NC – R – CN (I) в которой R обозначает алкиленовую или алкениленовую, линейную или разветвленную, группу с 1-12 атомами углерода. Предпочтительно используют динитрилы формулы (I), в которой R обозначает алкиленовый, линейный или разветвленный, радикал с 1 – 6 атомами углерода. В качестве примеров таких динитрилов можно назвать адипонитрил, метилглутаронитрил, этилсукцинонитрил, малононитрил, сукцинонитрил и глутаронитрил и их смеси, например смеси адипонитрила и/или метилглутаронитрила и/или этилсукцинонитрила, которые могут образовываться в одном и том же способе синтеза адипонитрила. Практически динитрилы, в которых R=(CH2)4, являются наиболее типичными, так как они соответствуют адипонитрилу (АДН), используемому в настоящем способе. Полугидрирование динитрила до соответствующего аминонитрила с помощью водорода обычно осуществляют в присутствии катализатора на основе никеля Ренея, кобальта Ренея, причем никель Ренея или кобальт Ренея содержат активирующий элемент, выбираемый среди элементов групп IV б, VI б, VII б и VIII периодической системы элементов в том виде, как она опубликована в Handbook of Chemistry and Physics, 51-е изд. (1970- 1971), и сильного неорганического основания, происходящего от щелочного или щелочноземельного металла. Исходная среда для гидрирования включает, по крайней мере, один растворитель, способный, по крайней мере, частично солюбилизировать динитрил, так как гидрирование лучше протекает тогда, когда вышеуказанный динитрил растворен. Эта растворяющая среда может быть образована водой в количестве, по крайней мере, 0,5 мас.% от всех жидких соединений реакционной среды. В дополнение к воде или вместо воды реакционная среда может содержать, по крайней мере, один другой растворитель, такой как спирт и/или амид и/или амин и/или аммиак. Преимущественно являются алканолы, такие как метанол, этанол, пропан-1-ол, пропан-2-ол и бутан-1-ол; диолы, такие как этиленгликоль и пропиленгликоль; полиолы или смеси вышеуказанных спиртов. В случае, если растворителем является амид, то выбирают, в частности, диметилформамид и диметилацетамид. Из аминов, которые могут быть использованы в качестве растворителя, можно применять, например, диамин или аминонитрил, соответствующий динитрилу, который гидрируют. Если используют воду, то растворитель превышает по весу в 2-4 раза вес воды. Согласно предпочтительному варианту осуществления стадии полугидрирования динитрила, исходная реакционная среда содержит диамин и/или аминонитрил, которые могут образовываться из гидрируемого динитрила, а также непревращенный динитрил в количестве 80 – 99,5%. Степень превращения динитрила предпочтительно составляет, по крайней мере, 70%. Сильное неорганическое основание обычно представляет собой гидроксиды, карбонаты и алканоляты щелочного или щелочноземельного металла. Его предпочтительно выбирают среди гидроксидов, карбонатов и алканолятов щелочного металла. Преимущественно используемое сильное неорганическое основание выбирают среди следующих соединений: LiOH, NaOH, КОН, RbOH, CsOH и их смесей. Практически чаще всего используют NaOH и КОН, которые создают хороший компромисс между стоимостью и результатом, хотя RbOH и CsOH могут давать очень хорошие результаты. Реакционная среда имеет состав, изменяющийся в зависимости от типа применяемого способа. Так, если способ осуществляют периодически, например, в условиях лаборатории или на маленьких периодически действующих производствах, то исходная среда постепенно обогащается аминонитрилом и, в меньшей степени, диамином, тогда как концентрация динитрила может либо уменьшаться, если загружают все количество или большую часть вышеуказанного динитрила с момента начала полугидрирования, либо оставаться относительно постоянной, если динитрил вводят постепенно в процесс реакции. Напротив, если способ осуществляют непрерывно, то состав реакционной среды на выходе из реактора зависит от селективности реакции. Вода обычно присутствует в количестве ниже или равном 20%. Предпочтительно содержание воды в реакционной среде составляет 2-15 мас.% от массы всех жидких компонентов вышеуказанной среды. При непрерывном осуществлении способа средний состав определяется соотношением селективности по аминонитрилу к селективности по диамину и скоростью введения динитрила. Количество сильного неорганического основания предпочтительно выше или равно 0,1 моль/кг катализатора. Предпочтительно оно составляет 0,1 – 3 моля на кг катализатора и еще более предпочтительно 0,3-2 моль/кг катализатора. Используемым катализатором может быть никель Ренея и кобальт Ренея, которые содержат кроме никеля или кобальта и остаточного количества металла, который удаляется из исходного сплава при получении катализатора, обычно это алюминий, один или несколько элементов, часто называемых активаторами, таких как, например, хром, титан, молибден, вольфрам, железо, цинк. Наиболее предпочтительными активирующими элементами являются хром и/или железо и/или титан. Эти активаторы обычно присутствуют в количестве 0-15 мас.%, предпочтительно 0-10 мас.% от массы никеля. Количество используемого катализатора может изменяться в широких пределах и зависит от типа осуществления способа или от выбранных реакционных условий. Так, если динитрил вводят постепенно в реакционную среду, то массовое соотношение катализатор/гидрируемый динитрил намного более высокое, чем если бы весь динитрил вводили в начале реакции. Так, можно использовать 0,5 – 50 мас.% катализатора в расчете на всю массу реакционной среды, и чаще всего 1 – 35 %. Для конкретного катализатора и для конкретной степени превращения динитрила выход аминонитрила проходит через максимум, определяемый соотношением основание/Ni или основание/Со. Оптимальность выхода аминонитрила при постоянной степени превращения динитрила зависит от природы и количества активатора, количества воды в реакционной смеси и температуры. Стадию полугидрирования согласно изобретению обычно осуществляют при температуре ниже или равной 150oC, предпочтительно ниже или равной 120oC и, еще более предпочтительно, ниже или равной 100oC. Более конкретно, эта температура составляет величину между комнатной температурой (около 20oC) и 100oC. Без технологических проблем можно работать при температуре ниже 20oC, однако это не представляет интереса в связи с более низкой производительностью реакции. Предварительно одновременно или после нагревания в емкости, где проводят реакцию, устанавливают соответствующее давление водорода, от 1 бар (0,10 МПа) до 100 бар (10 МПа), и предпочтительно 5 бар (0,5 МПа) – 50 бар (5 МПа). Продолжительность реакции изменяется в зависимости от реакционных условий и катализатора. При периодическом способе осуществления продолжительность реакции может изменяться от нескольких минут до нескольких часов. При непрерывном способе работы, который представляет собой предпочтительную форму промышленного осуществления способа, продолжительность, конечно, не представляет собой постоянный параметр. Перед тем, как направить полученный на стадии полугидрирования аминонитрил на стадию гидролиза необходимо удалить большую часть воды и/или растворителя, которые в известных случаях присутствуют, а также непрореагировавший динитрил, образовавшийся диамин и побочные продукты реакции, такие как соединения иминного или аминного типа. Эта очистка может быть осуществлена путем классической операции перегонки, предпочтительно осуществляемой при давлении ниже атмосферного. Сначала отгоняют воду и/или растворитель, затем образовавшийся диамин, например гексаметилендиамин. После этого идет аминонитрил, например, 6-амино-капронитрил (АКН). Непрореагировавший динитрил также может быть отделен путем перегонки. Полученный путем этой перегонки аминонитрил может содержать, как это было сказано выше, до 10 мас.%, и предпочтительно до 5 мас.% динитрила и до 10 мас. %, и предпочтительно до 5 мас.% других побочных продуктов, образующихся во время гидрирования динитрила. Когда динитрилом является адипонитрил, то этими побочными продуктами могут быть в частности гексаметиленимин (ГМИ), аминометилциклопентиламин (АМЦПА), азациклогептен (АЗЦГе), 1-имино-2-циано-циклопентан (ИЦЦП), диаминоциклогексан (ДЦГ), бис(гексаметилентриамин) (БГГ), продукт конденсации азациклогептена с 6-амино-капронитрилом [6- (6′-амино-гексаметилен-имино)гексанитрил], или, если полугидрирование проводят в спирте, различные соединения, образующиеся за счет реакции этого спирта с промежуточными продуктами реакции. Стадия гидролиза с циклизацией очищенного аминонитрила с целью получения соответствующего лактама заключается во взаимодействии, в паровой фазе, вышеуказанного алифатического аминонитрила общей формулы (I): N  C-R-CH2-NH2(II), C-R-CH2-NH2(II),в которой R обозначает алкиленовый или алкениленовый, линейный или разветвленный, радикал с 1-12 атомами углерода, с водой, предпочтительно в присутствии твердого катализатора. Предпочтительно в формуле (II) аминонитрила R обозначает алкиленовый, линейный или разветвленный, радикал с 1-6 атомами углерода. Твердый катализатор может быть очень разнообразной природы. Можно использовать молекулярное сито (кристаллическое соединение с микропористостью около 3-10 ангстрем), нецеолитное молекулярное сито, фосфат металла или монолитный оксид, кислый или амфотерный. Молекулярные сита представляют собой силикаты и кислые цеолиты. Под цеолитом понимают кристаллический тектосиликат природного или синтетического происхождения, кристаллы которого образованы трехмерным соединением тетраэдрических единиц SiO4 и TO4, где T обозначает трехвалентный элемент, такой как алюминий, галлий, бор и железо, предпочтительно алюминий. Чаще всего используют цеолиты алюмосиликатного типа. В кристаллической решетке цеолиты имеют систему пустот, связанных между собой каналами определенного диаметра, которые называют порами. Цеолиты могут иметь одномерную, двухмерную или трехмерную кристаллическую решетку. Из цеолитов можно использовать природные цеолиты, как, например, оффретит, клиноптилотит, эрионит, шабазит, филипсит. Хорошо зарекомендовали себя также синтетические цеолиты. В качестве примеров синтетических цеолитов с одномерной кристаллической решеткой можно назвать, в частности, цеолит ZSM-4, цеолит L, цеолит ZSM-12, цеолит ZSM-22, цеолит ZSM-23, цеолит ZSM-48. В качестве примеров цеолитов с двухмерной кристаллической решеткой, которые используют предпочтительно, можно назвать бета-цеолит, морденит, ферриерит. Что касается цеолитов с трехмерной кристаллической решеткой, то преимущественно можно указать цеолит Y, цеолит X, цеолит ZSM-5, цеолит ZSM-11, оффретит. Из синтетических цеолитов предпочтение отдают следующим формам: – маззит с молярным соотношением Si/Al = 3,4; – цеолит L с молярным соотношением Si/Al = 1,5-3,5; – морденит с молярным соотношением Si/Al = 5-15; – ферриерит с молярным соотношением Si/Al = 3-10; – оффретит с молярным соотношением Si/Al = 4-8,5; – бета-цеолиты с молярным соотношением Si/Al = 15-25; – цеолиты Y, в особенности цеолиты, получаемые после дезалюминирующей обработки (например, гидрообработка, промывка с помощью соляной кислоты или обработка с помощью SiCl4), преимущественно цеолиты US-Y с молярным соотношением Si/Al выше 3, предпочтительно 6-60; – цеолит X фожазитного типа с молярным соотношением Si/Al = 0,7-1,5: – цеолиты ZSM – 5 или силикалит алюминия с молярным соотношением Si/Al = 10-2000; – цеолит ZSM-11 с молярным соотношением Si/Al = 5-30. Используемые в настоящем способе цеолиты представляют собой известные продукты, описанные в литературе [см. “Atlas of zeolites structure types” W. M. Meier и D.H.Olson, опубликованный the Structure Commission of the International Zeolite Association (1978)]. Можно использовать цеолиты, имеющиеся в продаже, или синтезировать их описанными в литературе способами. Можно ссылаться на вышеуказанный Атлас и преимущественно для получения нижеописанных можно сослаться: – цеолита L – на публикацию Barrer R.M. и др., Z.Kristallogr. 128, с. 352 и последующие (1969); – цеолита ZSM-12 -на патент США 3832449 и статью Lapierre и др., Zeolites, 5, с. 346 и последующие (1985); – цеолита ZSM-22 – на публикацию Kokotailo G.T. и др., Zeolites, 5, с. 349 и последующие (1985); – цеолита ZSM-23 – на патент США 4076842 и статью Rohrman А.С. и др., Zeolites, 5, с. 352 и последующие (1985); – цеолита ZSM – 48 – на работы Schlenker J.L. и др., Zeolites, 5, с. 355 и последующие (1985); – бета-цеолита – на патент США 3308069 и статью Caullet P. и др., Zeolites, 12, с. 240 и последующие (1992); – морденита – на работы Itabashi и др., Zeolites, 6, с. 30 и последующие (1986); – цеолитов X и Y – соответственно на патенты США 4076842 и 3130007; – цеолита ZSM -5 – на патент США 3702886 и статью Shiralkar V.P. и др., Zeolites, 9, с. 363 и последующие (1989); – цеолита ZSM-11 – на работы Harrison I.D. и др., Zeolites, 7, с. 21 и последующие (1987). Цеолиты могут быть использованы в различных формах: порошок, формованные продукты, такие как грануляты (например, цилиндры или шарики), таблетки, монолиты (блоки в форме пчелиных сот), которые получают экструзией, отливкой, прессованием или любым другим известным методом. Практически в промышленности употребляют формы гранул, шариков или монолитов, которые обладают преимуществами как в плане эффективности, так и в плане удобства использования. Изобретение не исключает использования связующих во время формования цеолита, например оксидов алюминия или глин. Каков бы ни был выбранный цеолит, осуществляют обработку, если необходимо, которая делает его кислым. С этой целью прибегают к классическим обработкам. Так, можно обменивать катионы щелочных металлов, подвергая цеолит обработке, которую осуществляют с помощью раствора аммиака и которая приводит таким образом к обмену катиона щелочного металла на ион аммония, затем прокаливанию цеолита с ионами аммония для разложения катиона аммония и замены его на ион H+. Количество используемого раствора аммиака, по крайней мере, равно количеству, необходимому для обмена всех катионов щелочных металлов на ионы аммония. Следовательно, вводят, по крайней мере, 10-5 – 5  10-3 моль раствора аммиака на грамм цеолита.
Из силикатов используют преимущественно силикалит типа I структуры, аналогичной ZSM – II, и бета-силикалит.
Термин “молекулярное нецеолитное сито”, или НЦМС, в настоящем тексте означает молекулярные сита SAPO, описанные в патенте США 4440871; молекулярные сита ELAPSO, описанные в европейском патенте A-O 159624, и некоторые кристаллические алюмофосфаты, металлоалюмофосфаты (MeAPO), ферроалюмофосфаты (FeAPO), титаналюмофосфаты (TAPO), описанные в нижеуказанных патентах. Кристаллические алюмофосфаты описаны в патенте США 4310440; кристаллические металлоалюмофосфаты MeAPO, где Me обозначает, по крайней мере, один металл, выбираемый среди Mg, Mn, Со и Zn, описаны в патенте США 4567029; кристаллические ферроалюмофосфаты FeAPO описаны в патенте США 4554143: титаналюмофосфаты TAPO описаны в патенте США 4500651; другие нецеолитные молекулярные сита ELAPO описаны в европейских патентах A-O 158976 и А-0 158349.
Фосфат металла более конкретно может представлять собой фосфат металла общей формулы (III): 10-3 моль раствора аммиака на грамм цеолита.
Из силикатов используют преимущественно силикалит типа I структуры, аналогичной ZSM – II, и бета-силикалит.
Термин “молекулярное нецеолитное сито”, или НЦМС, в настоящем тексте означает молекулярные сита SAPO, описанные в патенте США 4440871; молекулярные сита ELAPSO, описанные в европейском патенте A-O 159624, и некоторые кристаллические алюмофосфаты, металлоалюмофосфаты (MeAPO), ферроалюмофосфаты (FeAPO), титаналюмофосфаты (TAPO), описанные в нижеуказанных патентах. Кристаллические алюмофосфаты описаны в патенте США 4310440; кристаллические металлоалюмофосфаты MeAPO, где Me обозначает, по крайней мере, один металл, выбираемый среди Mg, Mn, Со и Zn, описаны в патенте США 4567029; кристаллические ферроалюмофосфаты FeAPO описаны в патенте США 4554143: титаналюмофосфаты TAPO описаны в патенте США 4500651; другие нецеолитные молекулярные сита ELAPO описаны в европейских патентах A-O 158976 и А-0 158349.
Фосфат металла более конкретно может представлять собой фосфат металла общей формулы (III):(PO4)nHhM, (Imp)p, (III) в которой: М обозначает двухвалентный, трехвалентный, четырехвалентный или пятивалентный элемент, выбираемый в группах 2а, 3б, 4б, 5б, 6б, 7б, 8, 2б, 3а, 4а и 5а периодической системы элементов, или смесь нескольких из этих элементов или М=0, когда М обозначает некоторые пятивалентные элементы; Imp обозначает импрегнирующее соединение основного характера, образованное щелочным или щелочноземельным металлом, или смесью нескольких из этих металлов, ассоциированное с противоанионом для обеспечения электронейтральности; “n” обозначает 1, 2 или 3; “h” обозначает 0, 1 или 2; “p” обозначает число от 0 до 1/3 и соответствует молярному соотношению между импрегнирующим соединением Imp и импрегнированным соединением (PO4)nHh М. Из аминонитрилов формулы (II) наиболее важным являются такие, которые приводят к лактамам, служащим исходным материалом для получения полиамидов-4, -5, -6 и -10, т.е. такие, в формуле которых символ R обозначает линейный алкиленовый радикал с 2, 3, 4 или 8 атомами углерода. Предпочтительным соединением формулы (II) является 6-амино-капронитрил (или эпсилон-капронитрил), который приводит к капролактаму, полимеризация которого дает полиамид-6. Из металлов групп 2а, 3б, 4б, 5б, 6б, 7б, 8, 2б, 3а, 4а и 5a периодической системы элементов особенно можно назвать бериллий, магний, кальций, стронций, барий, алюминий, бор, галлий, индий, иттрий; лантаниды, такие как лантан, церий, празедиум, неодим, самарий, европий, гадолиний, тербий, диспрозий, гольмий, эрбий, тулий, иттербий и лютеций; цирконий, титан, ванадий, ниобий, железо, германий, олово, висмут. Среди фосфатов лантанидов можно выделить первую группу, которая включает ортофосфаты легких редкоземельных элементов, также называемых редкими цериевыми землями, включающими лантан, церий, празеодим, неодим, самарий и европий. Эти ортофосфаты являются диморфными. Они имеют гексагональную структуру и эволюционируют к моноклинной кристаллической структуре, если их нагревают при температуре 600 – 800oC. Вторую группу фосфатов лантанидов образуют ортофосфаты гадолиния, тербия и диспрозия. Эти ортофосфаты имеют такую же структуру, что и ортофосфаты редких цериевых земель, но, кроме того, содержат третью кристаллическую фазу квадратичной структуры при высокой температуре (около 1700oC). Третью группу фосфатов лантанидов образуют ортофосфаты тяжелых редких земель, называемых также иттриевыми редкими землями, включающими иттрий, гольмий, эрбий, тулий, иттербий и лютеций. Эти соединения кристаллизуются только в квадратичной форме. Из различных семейств вышеуказанных ортофосфатов редких земель предпочтительно используют ортофосфаты цериевых редких земель. Можно использовать фосфаты металлов формулы (II), которые представляют собой смеси фосфатов нескольких вышеуказанных металлов или смешанные фосфаты нескольких вышеуказанных металлов или смешанные фосфаты, содержащие один или несколько вышеуказанных металлов и один или несколько других металлов, таких как щелочные или щелочноземельные металлы. Противоанионы, входящие в формулу импрегнирующего соединения Imp, имеют основной характер. Особенно можно использовать гидроксид-, фосфат-, гидрофосфат-, дигидрофосфат-, хлорид-, фторид-, нитрат-, бензоат-, оксалат- ионы, причем это перечень не является ограничительным. Молярное соотношение “p” предпочтительно составляет 0,02 – 0,2. Если сослаться на общие методы получения фосфатов (такие, как описаны, в частности, в “PASCAL P. Nouveau traite de chimie minerale” т. X (1956), с. 821-823; и в “GMELINS Handbuch der anorganischen Chemie” (8-издание), т.16 (С), с. 202-206 (1965)), то можно выделить два основных пути получения фосфатов: с одной стороны, осаждение растворимой соли металла (хлорид, нитрат) с помощью гидрофосфата аммония или фосфорной кислоты; с другой стороны, растворение оксида или карбоната металла (нерастворимые) с помощью фосфорной кислоты, обычно при нагревании, с последующим переосаждением. Вышеуказанные фосфаты, полученные согласно одному из указанных путей, могут быть высушены, обработаны с помощью органического (такого как гидроксид аммония) или неорганического (такого как гидроксид щелочного металла) основания и могут быть подвергнуты прокаливанию, причем эти три операции могут быть реализованы в указанном порядке или в другом порядке. Фосфаты металлов формулы (III), для которых символ “p” выше 0, могут быть получены путем импрегнирования соединения (PO4)nHh М, полученного согласно одному из вышеуказанных способов, с помощью раствора или суспензии Imp в летучем растворителе, таком как предпочтительно вода. Результаты тем лучше, чем более растворимо соединение Imp и чем более свежеприготовленным является соединение (PO4)nHhM. Таким образом предпочтительный способ получения фосфатов формулы (II) состоит в том, что: а) проводят синтез соединения (PO4)nHhМ; затем предпочтительно без выделения (PO4)nHhМ из реакционной среды; б) вводят импегнирующее соединение Imp в реакционную среду; в) отделяют возможную остаточную жидкость от полученного в результате реакции твердого вещества; г) высушивают и в случае необходимости прокаливают. Характеристики катализатора формулы (III) и особенно его устойчивость к дезактивации могут быть улучшены за счет прокаливания. Температура прокаливания предпочтительно составляет 300-1000oC и преимущественно 400-900oC. Продолжительность прокаливания может изменяться в широких пределах. Для сведения, она составляет обычно от 1 часа до 24 часов. Из предпочтительных в способе изобретения катализаторов формулы (III) можно назвать преимущественно фосфат лантана; прокаленный фосфат лантана; фосфат лантана, ассоциированный с производным цезия, рубидия или калия: прокаленный фосфат церия; фосфат церия, ассоциированный с соединением цезия, рубидия или калия; фосфат самария, ассоциированный с соединением цезия, рубидия или калия; фосфат алюминия; фосфат алюминия, ассоциированный с соединением цезия, рубидия или калия, прокаленный фосфат ниобия; фосфат ниобия, ассоциированный с соединением цезия, рубидия или калия; прокаленный гидрофосфат циркония; гидрофосфат циркония, ассоциированный с соединением цезия, рубидия или калия. Кислые монолитные оксиды, которые могут служить в качестве твердых катализаторов на стадии гидролиза с циклизацией, представляют собой, в частности, оксиды металлов, смеси оксидов металлов или оксиды металлов, модифицированные для того, чтобы сделать их кислыми, особенно путем воздействия галогена, галогенида аммония или кислоты, такой как серная кислота или галогенводородные кислоты. Галогеном, в случае необходимости вводимым для подкисления монолитного оксида, предпочтительно является хлор или фтор. Амфотерными монолитными оксидами являются оксиды с амфотерным характером или те, которые приобретают амфотерность в процессе способа их получения или путем последующей обработки. В качестве неограничивающих примеров кислых или амфотерных монолитных оксидов можно назвать смеси SiO2/Al2O3, SiO2/Ga2O3; SiO2/Fe2O3; SiO2/B2O3; галогенированные оксиды алюминия, такие как хлорированные и фторированные глиноземы; сульфатированный диоксид циркония; оксид ниобия; оксид вольфрама; оксид тория; диоксид циркония; диоксид титана; диоксид церия; диоксиды кремния; оксиды алюминия. Среди этих монолитных оксидов оксиды алюминия, которые могут быть использованы в качестве твердого катализатора на стадии гидролиза с циклизацией, имеют очень разнообразную структуру. Однако предпочтительно выбирают среди активных оксидов алюминия такие, которые менее всего дезактивируются. Поэтому предпочтительно выбирают оксиды алюминия, имеющие удельную поверхность, измеряемую по методу БЭТ, выше или равную 5 м2/г и еще более предпочтительно более равную 10 м2/г. Предпочтительно оксид алюминия, используемый в способе согласно изобретению, имеет удельную поверхность, равную или ниже 500 м2/г. Оксиды алюминия, которые могут быть использованы в настоящем способе, представляют собой оксиды алюминия, имеющие удельную поверхность выше или равную 10 м2/г и ниже или равную 280 м2/г, а объем пор с диаметром более 500 ангстрем выше или равный 10 мл/100 г. Удельная поверхность, определенная по методу БЭТ, представляет собой удельную поверхность, определяемую путем адсорбции азота в соответствии с нормой АСТМ Д 3663-78, основанной на методе BRUNAUER – ЕММЕТТ – TELLER, описанном в периодическом издании “The Journal of the American Society”, 60, 309 (1938). Объем пор с диаметром более 500 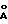 обозначает общий объем, создаваемый всеми порами, имеющими диаметр выше 500 обозначает общий объем, создаваемый всеми порами, имеющими диаметр выше 500 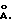 Этот объем измеряют по методу пенетрации ртути, в котором применяют закон Кельвина.
Предпочтительно оксиды алюминия этой первой группы имеют объем пор, диаметр которых выше 500 Этот объем измеряют по методу пенетрации ртути, в котором применяют закон Кельвина.
Предпочтительно оксиды алюминия этой первой группы имеют объем пор, диаметр которых выше 500 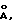 выше или равный 20 мл/100 г или более предпочтительно выше или равный 30 мл/100 г.
Предпочтительно также, чтобы оксиды алюминия этой первой группы имели удельную поверхность выше или равную 50 м2/г.
Оксиды алюминия, которые могут использоваться в настоящем способе, представляют собой также оксиды алюминия, обладающие удельной поверхностью выше или равной 50 м2/г и ниже или равной 280 м2/г, а объем пор, диаметр которых выше 70 ангстрем, выше или равный 30 мл/100 г.
Предпочтительно, чтобы оксиды алюминия этой второй группы имели объем пор, диаметр которых выше 70 выше или равный 20 мл/100 г или более предпочтительно выше или равный 30 мл/100 г.
Предпочтительно также, чтобы оксиды алюминия этой первой группы имели удельную поверхность выше или равную 50 м2/г.
Оксиды алюминия, которые могут использоваться в настоящем способе, представляют собой также оксиды алюминия, обладающие удельной поверхностью выше или равной 50 м2/г и ниже или равной 280 м2/г, а объем пор, диаметр которых выше 70 ангстрем, выше или равный 30 мл/100 г.
Предпочтительно, чтобы оксиды алюминия этой второй группы имели объем пор, диаметр которых выше 70 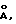 выше или равный 45 мл/100 г.
Также предпочтительно, чтобы оксиды алюминия этой второй группы имели удельную поверхность выше или равную 80 м2/г.
Оксиды алюминия, которые могут быть использованы в настоящем способе, также представляют собой оксиды алюминия, имеющие удельную поверхность выше или равную 280 2/г, а общий объем пор выше или равный 15 мл/100 г.
Предпочтительно, оксиды алюминия этой третьей группы имеют общий объем пор выше или равный 22 мл/100 г и более предпочтительно выше или равный 30 мл/100 г.
Оксиды алюминия также характеризуются своей кислотностью.
Эта кислотность может быть измерена по тесту изомеризации 1-бутена в 2-бутен.
Этот тест основан на реакции изомеризации 1-бутена до смеси цис-2-бутена и транс-2-бутена при температуре Т (Т= 400oC в настоящем случае).
Реакция изомеризации термодинамически равновесна. Она может определяться двумя константами: выше или равный 45 мл/100 г.
Также предпочтительно, чтобы оксиды алюминия этой второй группы имели удельную поверхность выше или равную 80 м2/г.
Оксиды алюминия, которые могут быть использованы в настоящем способе, также представляют собой оксиды алюминия, имеющие удельную поверхность выше или равную 280 2/г, а общий объем пор выше или равный 15 мл/100 г.
Предпочтительно, оксиды алюминия этой третьей группы имеют общий объем пор выше или равный 22 мл/100 г и более предпочтительно выше или равный 30 мл/100 г.
Оксиды алюминия также характеризуются своей кислотностью.
Эта кислотность может быть измерена по тесту изомеризации 1-бутена в 2-бутен.
Этот тест основан на реакции изомеризации 1-бутена до смеси цис-2-бутена и транс-2-бутена при температуре Т (Т= 400oC в настоящем случае).
Реакция изомеризации термодинамически равновесна. Она может определяться двумя константами:– теоретическая константа равновесия KT (Т), определяемая путем расчета:  где [бутен] (равн.) обозначает концентрацию каждого из изомеров в равновесии при температуре Т; – реальная константа равновесия K (T), определяемая по результату измерений: 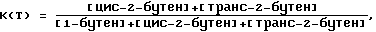 где [бутен] обозначает концентрацию каждого из изомеров при выходе из реактора при температуре Т. Изомеризующую способность А оксидов алюминия определяют по активности относительно равновесия: 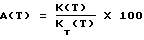 На практике тест осуществляют в парофазном реакторе, работающем в пульсирующем режиме, в который вводят 500 мг размельченного оксида алюминия (частицы размером 400-500 мкм). Оксид алюминия выдерживают в течение 2 часов при 250oC в атмосфере гелия, подаваемого со скоростью 2,5 л/час. Затем оксид алюминия доводят до температуры 400oC и вводят путем инжекции над ним 1 мл 1-бутена в токе гелия. Анализ выходящих газов осуществляют путем хроматографии в газовой фазе и определяют рекуперируемые количества 1-бутена и цис- и транс-2-бутена. Эта изомеризующая способность А корректируется определением изомеризующей способности, получаемой в тех же условиях с пустым реактором. Скорректированная изомеризующая способность Ac обозначает кислотность вышеуказанных оксидов алюминия. Если содержание щелочного или щелочноземельного металла, присутствующего в оксиде алюминия, ниже 60 ммоль на 100 г оксида алюминия, то чем выше величина Ac, тем более кислый оксид алюминия. Обычно оксиды алюминия получают путем дегидратации гиббсита, байерита, нордстрандита или их различных смесей. Можно сослаться, например, на энциклопедию KIRK-OTHMER, т. 2, с. 291-297. Используемые в настоящем способе оксиды алюминия можно получать путем введения в контакт гидратированного оксида алюминия в тонкоизмельченной форме с потоком горячего газа с температурой 400- 1000oC, затем поддержания контакта между гидратом и газами в течение времени от долей секунды до 10 секунд и отделением частично дегидратированного оксида алюминия от горячих газов. В этом отношении особенно можно сослаться на способ, описанный в патенте США 2915365. Можно также осуществлять автоклавирование вышеполученных агломератов оксида алюминия, в водной среде, в случае необходимости в присутствии кислоты, при температуре выше 100oC и предпочтительно при 150-250oC, в течение предпочтительно 1-20 часов, затем их высушивают и их прокаливают. Температуру прокаливания регулируют таким образом, чтобы получались удельные поверхности и объемы пор, находящиеся в указанных интервалах. В соответствии с их основными способами получения оксиды алюминия чаще всего содержат натрий, содержание которого обычно выражается в массовых долях Na2О в расчете на массу оксида алюминия. Катализатор может быть использован в различных формах, таких как порошок, шарики, продукты дробления, экструдаты, таблетки, причем придание формы может быть в случае необходимости реализовано с помощью связующего. Прежде всего речь идет о шариках оксида алюминия, полученных методом коагуляции в капле. Этот тип шариков может быть, например, приготовлен по способу, описанному в европейских патентах NN A-O 015801 и A-O 097539. Контроль за пористостью может быть осуществлен согласно способу, описанному в европейском патенте A-O 097539, путем коагуляции в капле водной суспензии или дисперсии оксида алюминия или раствора основной соли алюминия, находящейся в виде эмульсии, которая образована органической фазой, водной фазой и поверхностно-активным агентом или эмульгатором. Вышеуказанной органической фазой может быть углеводород. Речь также может идти о продуктах дробления оксида алюминия. Эти продукты дробления могут быть получены дроблением любого типа материала на основе оксида алюминия, например, шариков, полученных любыми способами (коагуляция в капле, гранулятор или вращающийся барабан), или экструдатов. Контроль за пористостью этих раздробленных продуктов осуществляется путем выбора материала на основе оксида алюминия, который дробят для получения этих продуктов. Речь также может идти об экструдатах оксида алюминия. Они могут быть получены путем разминания, затем экструзии материала на основе оксида алюминия, причем вышеуказанным материалом может быть продукт быстрой дегидратации гидраргиллита или осаждения геля оксида алюминия. Контроль за пористостью этих экструдатов может быть реализован путем выбора используемого оксида алюминия и за счет условий получения этого оксида алюминия или за счет условий разминания этого оксида алюминия перед экструзией. Например, оксид алюминия может быть смешан во время разминания с порообразователями. В качестве примера экструдаты могут быть получены по способу, описанному в патенте США 3856708. В некоторых случаях бывает целесообразно, чтобы, по крайней мере, часть свободного объема реактора была занята инертным твердым веществом, таким как, например, кварц, с целью благоприятствования испарению и диспергированию реагентов. Как оксиды алюминия, так и вообще твердый катализатор стадии гидролиза с циклизацией используют в форме порошка, таблеток, продуктов дробления, шариков или экструдатов, причем придание вышеуказанной формы в случае необходимости может быть реализовано с помощью связующего. Для реакции гидролиза с циклизацией необходимо наличие воды. Молярное соотношение воды к аминонитрилу, которые вводят, составляет обычно 0,5-50 и предпочтительно 1-20. Значение выше этого соотношения не критическое для изобретения, однако более высокие соотношения почти не представляют интереса по экономическим соображениям. Аминонитрил и воду можно вводить в реактор в виде их смесей в парообразном состоянии или раздельно. Можно осуществить предварительное испарение реагентов, которые затем направляют в смесительную камеру. Можно использовать любой инертный газ в качестве транспортирующего агента, такой как азот, гелий или аргон. Температура, при которой осуществляют стадию гидролиза с циклизацией, должна быть достаточной для того, чтобы реагенты находились в виде паров. Она обычно составляет 200-450oC и предпочтительно 250-400oC. Время контакта между аминонитрилом и катализатором не критическое. Оно может изменяться в зависимости от используемой аппаратуры. Это время контакта предпочтительно составляет 0,5-200 секунд и еще более предпочтительно 1-100 секунд. Давление не является критическим параметром этой стадии способа. Так, можно работать при давлениях от 10-3 бар до 200 бар. Предпочтительно способ осуществляют при давлении 0,1-20 бар. Не исключается использование инертного в реакционных условиях растворителя, такого как, например, алкан, циклоалкан, ароматический углеводород или их галогенированные производные, чтобы обеспечить присутствие жидкой фазы в реакционном потоке. Полученный лактам может быть очищен, обычно путем перегонки, перед полимеризацией. Эта очистка намного более легкая для лактама, чем для аминонитрила. Кроме того, оказалось, что побочные продукты, содержащиеся в аминонитриле, вводимом на стадии гидролиза, большей частью разлагаются в процессе этой стадии и легко удаляются. Перегонку лактама можно осуществлять в присутствии сильного основания, такого как гидроксид щелочного металла, причем присутствие этого основания облегчает отделение лактама от побочных продуктов и аминонитрила, который не провзаимодействовал. Особенно хорошо пригодны гидроксид натрия или гидроксид калия. Нижеследующие примеры иллюстрируют изобретение. Пример 1 В реактор из нержавеющей стали емкостью 3,6 л, снабженный перемешиванием кавитаторного типа (rushtone cavitator), средствами для введения реагентов и водорода и системой для регулирования температуры, загружают: адипонитрил – 1710 г гексаметилендиамин – 574,4 г воду – 253 г КОН – 0,63 г никель Ренея (с 1,7% Cr) – 30,3 г Ni В этом примере имеется 0,4 моля КОН/кг Ni. После продувки реактора азотом, затем водородом реакционную смесь нагревают до 50oC; затем устанавливают давление 2 МПа при этой температуре с помощью непрерывно подаваемого водорода. За ходом реакции следят по поглощению водорода. Когда израсходованы 2,2 эквивалента водорода по отношению к загруженному адипонитрилу, реакцию останавливают путем прекращения перемешивания и охлаждения реакционной смеси. Путем хроматографии в паровой фазе количественно определяют остаточный адипонитрил (АДН) и образовавшийся 6-амино- капронитрил (АКН) и рассчитывают степень превращения (ТТ) АДН и выход АКН по отношению к превращенному АДН (RT). Получают следующие результаты: продолжительность реакции = 118 минут; ТТ АДН = 81%; RT АКН = 60,3%. Эту операцию повторяют 7 раз и объединенные реакционные массы, после отделения катализатора фильтрованием, собирают для перегонки. Аминокапронитрил перегоняют при 120oC при давлении 2660 Па с помощью колонки диаметром 70 мм, с насадкой из нержавеющей стали, имеющей эффективность, которая соответствует 30 теоретическим тарелкам. Таким образом полученный аминокапронитрил содержит: – менее 0,01 мас.% адипонитрила; – 0,1 мас.% гексаметилендиамина; – 98,4 мас.% 6-амино-капронитрила; – 1,5 мас.% различных аминов и иминов, основным из которых является 6-(6′-амино-гексаметилен-имино) гексаннитрил. Стадию гидролиза с циклизацией полученного таким образом 6 – аминокапронитрила (АКН) осуществляют в цилиндрическом реакторе емкостью 20 мл из пирексового стекла, расположенном вертикально и снабженном средствами для нагревания, отверстиями для ввода и вывода газовых потоков и системой введения путем инжекции реагентов. В этот реактор последовательно загружают 10 мл кварца, 1 мл оксида алюминия в виде порошка с гранулометрией 400-500 микрометров и снова 10 мл кварца. Таким образом загруженный реактор нагревают при 400oC в токе воздуха (с дебитом 1,5 л/час) в течение 2 часов. Затем реактор охлаждают до 320oC (выбранная температура реакции) и выдерживают в токе азота (дебит 17,6 мл/мин). После этого с помощью насоса инжектируют смесь АКН с водой (массовое соотношение 50/50, или молярное соотношение вода/АКН = 6,2). Дебит инжекции смеси составляет 1,14 г/час. При выходе из реактора пары конденсируются в стеклянной ловушке при комнатной температуре. Конечную реакционную смесь анализируют с помощью хроматографии в газовой фазе. Определяют степень превращения (ТТ) аминокапронитрила, выход (RT) капролактама (КПЛ) по отношению к превращенному аминокапронитрилу. С помощью различных методов анализа (хроматография в паровой фазе в сочетании с масс-спектрометрией, особенно ядерный магнитный резонанс) констатируют полное исчезновение побочных продуктов в виде аминов и иминов, присутствующих в используемом аминокапронитриле. Получают следующие стабильные характеристики для продолжительности реакции 50 часов: ТТ АКН составляет  99%; 99%;RT КПЛ составляет 98%.
Пример 2В металлический реактор, снабженный перемешиванием кавитаторного типа, средствами для введения реагентов и водорода и различными системами регулирования, вводят следующие загрузки: адипонитрил – 2856 кг гексаметилендиамин – 1151 кг вода – 588 кг КОН – 0,83 кг никель Ренея (с 1,7% Cr) – 37 кг В этом примере имеется 0,4 моль КОН/кг Ni. Работают в условиях, описанных в примере 1. Получают следующие результаты: продолжительность реакции = 3,5 часа; ТТ АДН = 86%; RT АКН = 64%. После отделения катализатора путем фильтрования, около 6 кг реакционной смеси перегоняют при 120oC при давлении 2660 Па с помощью колонки диаметром 75 мм, с насадкой из нержавеющей стали, которая имеет эффективность, соответствующую 11 теоретическим тарелкам. Для второй стадии способа используют фракцию предыдущей перегонки, которая содержит; 0,02 мас.% адипонитрила; 0,36 мас.% гексаметилендиамина; 97,1 мас.% 6-амино-капронитрила; 2,5 мас.% различных аминов и иминов, основным из которых является 6- (6′-амино-гексаметилен-имино) гексаннитрил. Для того чтобы лучше показать, что способ согласно изобретению позволяет использовать на стадии гидролиза с циклизацией относительно мало очищенный 6-амино-капронитрил, в эту фракцию амино-капронитрила (АКН), которая перегнана, добавляют снова различные дополнительные примеси. Таким образом получают АКН, содержащий; 1 мас.% адипонитрила; 1 мас.% гексаметилендиамина; 2,5 мас. % различных аминов и иминов, из которых основным является 6-(6′-амино-гексаметилен-имино) гексаннитрил; 1 мас.% 1-имино-2-циано-циклопентана; 1 мас. % гексаметиленимина (ГМИ); 1 мас.% бис-(гексаметилентриамина) (БГТ); 92,5 мас.% 6-амино-капронитрила. Стадию гидролиза с циклизацией таким образом полученного АКН осуществляют в цилиндрическом реакторе емкостью 20 мл из пирексового стекла, расположенного вертикально и снабженного средствами для нагревания, отверстиями для ввода и вывода газовых потоков и системой инжекции реагентов. В этот реактор последовательно загружают 3 мл кварца, 2 мл (0,87 г) оксида алюминия (удельная поверхность по БЭТ составляет 130 м2/г) в виде порошка с гранулометрией 300-600 микрометров и снова 5 мл кварца. Таким образом загруженный реактор нагревают при 400oC в токе воздуха (с дебитом 1,5 л/час) в течение 2 часов. Затем реактор охлаждают до 320oC (выбранная температура реакции) и выдерживают в токе азота (дебит 88 мл/мин). После этого с помощью насоса инжектируют смесь АКН с водой (молярное соотношение вода/АКН = 1,1). Дебит инжекции смеси составляет 11 г/час. При выходе из реактора пары конденсируются в стеклянной ловушке при комнатной температуре. Конечную реакционную смесь анализируют путем хроматографии в газовой фазе. Определяют степень превращения (ТТ) амино-капронитрила, выход (RT) капролактама (КПЛ) по отношению к превращенному аминокапронитрилу. С помощью различных методов анализа (хроматография в газовой фазе в сочетании с масс-спектрометрией, особенно ядерный магнитный резонанс) констатируют полное исчезновение побочных продуктов в виде аминов и иминов, присутствующих в используемом аминокапронитриле. Получают следующие стабильные характеристики для продолжительности реакции 7 часов: ТТ АКН = 63%, RT КПЛ = 100%. Формула изобретения
NC-R-CN, в которой R обозначает алкиленовую или алкениленовую, линейную или разветвленную группу с 1 – 12 атомами углерода и предпочтительно с 1 – 6 атомами углерода. 3. Способ по любому из пп.1 и 2, отличающийся тем, что полугидрирование динитрила до соответствующего аминонитрила осуществляют в присутствии катализатора на основе никеля Ренея, кобальта Ренея, причем никель Ренея или кобальт Ренея содержат активирующий элемент, выбираемый среди элементов групп IVб, VIб, VIIб и VIII Периодической системы элементов, и сильного неорганического основания, происходящего от щелочного или щелочноземельного металла. 4. Способ по п.3, отличающийся тем, что используют катализатор, в котором сильное неорганическое основание выбирают среди гидроксидов, карбонатов и алканолятов щелочного или щелочноземельного металла и предпочтительно среди гидроксидов, карбонатов и алканолятов щелочного металла. 5. Способ по п.3, отличающийся тем, что количество используемого катализатора составляет 0,5 – 50 мас.% катализатора в расчете на общую массу реакционной среды, предпочтительно 1 – 35%. 6. Способ по любому из пп.1 – 5, отличающийся тем, что стадию полугидрирования осуществляют при реакционной температуре ниже или равной 150°С, предпочтительно ниже или равной 120°С и более предпочтительно ниже или равной 100°С. 7. Способ по любому из пп.1 – 6, отличающийся тем, что стадию полугидрирования осуществляют при давлении водорода в интервале 1 бара (0,10 МПа) – 100 бар (10 МПа), предпочтительно 5 бар (0,5 МПа) – 50 бар (5 МПа). 8. Способ по любому из пп.1 – 7, отличающийся тем, что перед стадией гидролиза с циклизацией аминонитрил после полугидрирования очищают от большой части воды и/или растворителя, непрореагировавшего динитрила, а также от образовавшегося диамина и побочных продуктов реакции путем классической операции перегонки, предпочтительно осуществляемой при давлении ниже атмосферного, до содержания динитрила до 10 мас.%, предпочтительно до 5 мас.%, а других побочных продуктов до 10 мас.%, предпочтительно до 5 мас.%, в аминонитриле после перегонки. 9. Способ по п.1, отличающийся тем, что гидролиз с циклизацией аминонитрила осуществляют путем взаимодействия в паровой фазе алифатического аминонитрила общей формулы II N C-R-CH2-NH2,в которой R обозначает алкиленовый или алкениленовый, линейный или разветвленный, радикал с 1 – 12 атомами углерода, предпочтительно 1 – 6 атомами углерода, с водой предпочтительно в присутствии твердого катализатора. 10. Способ по п.9, отличающийся тем, что в качестве твердого катализатора используют молекулярные сита, такие, как кислый цеолит или силикалит, нецеолитные молекулярные сита, фосфат металла или монолитные оксиды кислого или амфотерного характера. 11. Способ по п.10, отличающийся тем, что в качестве фосфата металла используют соединение общей формулы III (РO4)nНpМ(lmp)p, в которой М обозначает двухвалентный, трехвалентный, четырехвалентный или пятивалентный элемент, выбираемый в группах 2а, 3б, 4б, 5б, 6б, 7б, 8, 2б, 3а, 4а и 5а Периодической системы элементов, или смесь нескольких из этих элементов; или М = 0, когда М обозначает некоторые пятивалентные элементы; lmp обозначает импрегнирующее соединение основного характера, образованное щелочным или щелочноземельным металлом, или смесями из нескольких из этих металлов, соединенное с противоанионом для обеспечения электронейтральности; n обозначает 1, 2 или 3; h обозначает 0, 1 или 2; p обозначает число от 0 до 1/3 и соответствует молярному соотношению между импрегнирующим соединением lmp и импрегнированным соединением (РO4)nHpМ. 12. Способ по п. 10, отличающийся тем, что монолитный оксид является оксидом алюминия. 13. Способ по п. 12, отличающийся тем, что используют активированный оксид алюминия с удельной поверхностью 5 – 200 м2/г, предпочтительно 10 – 500 м2/г. 14. Способ по любому из пп.1 и 9 – 13, отличающийся тем, что молярное соотношение между подаваемыми водой и аминонитрилом составляет 0,5 – 50, предпочтительно 1 – 20 и более предпочтительно 2 – 20. 15. Способ по любому из пп.1 и 9 – 14, отличающийся тем, что температура, при которой осуществляют стадию гидролиза с циклизацией, составляет 200 – 450°С и предпочтительно 250 – 400°С. |
||||||||||||||||||||||||||