Патент на изобретение №2167849
|
||||||||||||||||||||||||||
(54) НАФТИЛПРОИЗВОДНЫЕ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ ВЕЩЕСТВА
(57) Реферат: Изобретение относится к новым нафтилпроизводным ф-лы (I), где R1 и R2 – Н, -ОН или -O(С1-С4-алкил); R3 – 1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 4-морфолинил, диалкиламино- или 1-гексаметилен-иминогруппа; n = 2 или 3, или их фармацевтически приемлемым солям. Соединения ф-лы (I) ингибируют проявление симптомов постклимактерического синдрома и могут найти применение в качестве средства для облегчения симптомов постклимактерического синдрома, представляющих собой остеопороз, сердечно-сосудистые заболевания, гиперлипидемию или гормонально зависимое раковое заболевание. Описываются также промежуточные соединения ф-лы (II), где R1a – Н или OR5, R2a – Н или OR6, R5 и R6 – гидроксизащитная группа, R4 – -ОН или – СНО. 4 с. и 9 з.п. ф-лы, 6 табл. 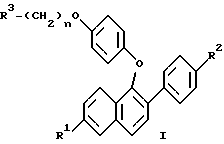 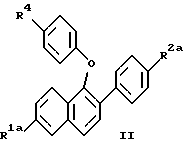
Изобретение относится к области фармацевтической и органической химии и направлено на создание новых нафтилсодержащих соединений, которые пригодны для лечения различных медицинских показаний, связанных с постклимактерическим синдромом, а также фибромы матки, эндометриоза и пролиферации клеток гладких мышц аорты. Настоящее изобретение относится также к промежуточным продуктам, применяемым для получения фармацевтически активных соединений настоящего изобретения, и фармацевтическим композициям. “Постклимактерический синдром” является термином, применяемым для описания различных патологических состояний, часто поражающих женщин, которые вступили в период или завершили период физиологической трансформации, известный как менопауза. Хотя при применении этого термина рассматриваются различные патологии, источником наибольшего долговременного медицинского интереса являются три основных эффекта постклимактерического синдрома: остеопороз, сердечно-сосудистые последствия, например гиперлипидемия, и эстрогензависимый рак, в частности рак молочной железы и матки. Остеопороз описывает группу заболеваний, которые возникают из разных этиологий, но которые характеризуются чистой потерей костной массы на единицу объема. Следствием этой потери костной массы и происходящего за этим костного перелома является недостаточность скелета для обеспечения адекватной структурной опоры для тела. Одним из наиболее общих типов остеопороза является тип, связанный с менопаузой. Большинство женщин теряют от около 20% до около 60% костной массы в трабекулярной части кости в течение 3 – 6 лет после прекращения менструации. Такая быстрая потеря обычно связана с повышением костной резорбции и костеобразования. Однако резорбционный цикл более доминантный и результатом этого является потеря костной массы. Остеопороз является обычным и серьезным заболеванием среди постклимактерических женщин. Подсчитано, что только в Соединенных Штатах имеется 22 миллиона женщин, которые поражены этим заболеванием. Результаты остеопороза персонально опасны и являются также причиной больших экономических потерь, обусловленных его хроническим характером и необходимостью экстенсивной и долговременной поддержки (госпитализации и медицинской помощи на дому). Это особенно справедливо для более пожилых пациентов. Дополнительно, хотя остеопороз обычно не считается угрожающим жизни состоянием, 20 – 30% смертности связано с переломом костей тазобедренного сустава у пожилых женщин. Большой процент этой смертности может быть непосредственно связан с постклимактерическим остеопорозом. Наиболее восприимчивой к воздействию постклимактерического остеопороза тканью в кости является трабекулярная часть кости. Эту ткань часто относят к губчатому слою кости или губчатому веществу кости, она, в частности, концентрируется около окончаний костей (вблизи суставов) и в позвонках позвоночника. Трабекулярная ткань характеризуется небольшими остеоидными структурами, которые соединены друг с другом, а также более твердой и плотной кортикальной тканью, которая составляет наружную поверхность и центральную стержневую часть кости. Эта сеть трабекул образует латеральную опору для наружной кортикальной структуры и критична для биомеханической прочности общей структуры. В постклимактерическом остеопорозе основным является резорбция сетчатой структуры и потеря трабекул, что ведет к повреждению и перелому кости. В свете потери трабекул у постклимактерических женщин не является неожиданным тот факт, что наиболее обычными переломами являются переломы, связанные с костями, которые в большей степени зависят от трабекулярной опоры, например позвонков, шейки выдерживающих нагрузку костей, например бедренной кости и предплечья. Действительно, переломы бедер, переломы нижнего конца лучевой кости и вертебральные переломы с раздроблением являются характерными признаками постклимактерического остеопороза. В настоящее время единственным обычно приемлемым способом лечения постклимактерического остеопороза является эстроген-заместительная терапия. Хотя эта терапия в общем успешна, пациент редко соглашается с этой терапией в основном вследствие того, что лечение эстрогеном часто вызывает нежелательные побочные эффекты. На всем протяжении предклимактерического периода времени большинство женщин имеют меньшую частоту сердечно-сосудистого заболевания, чем сравнимые по возрасту мужчины. После менопаузы, однако, степень заболевания женщин сердечно-сосудистыми болезнями медленно повышается в сравнении со степенью заболевания, наблюдаемой у мужчин. Эту потерю защиты связывают с потерей эстрогена и, в частности, с потерей способности эстрогена регулировать уровень сывороточных липидов. Природа способности эстрогена регулировать сывороточные липиды не вполне понятна, но имеющиеся налицо данные указывают, что эстроген может регулировать (в сторону повышения) рецепторы липидов низкой плотности (LDL) в печени для удаления избытка холестерина. Дополнительно, эстроген, по-видимому, оказывает некоторое влияние на биосинтез холестерина и другие благотворные влияния на состояние сердечно-сосудистой системы. В литературе сообщается, что у постклимактерических женщин, получающих эстроген-заместительную терапию, происходит понижение (возврат) содержания сывороточных липидов до концентраций липидов у женщин предклимактерического состояния. Таким образом, эстроген, по- видимому, должен быть целесообразным лечением для этого состояния. Однако побочные эффекты эстроген-заместительной терапии не приемлемы для многих женщин, что таким образом ограничивает применение этой терапии. Идеальная терапия для этого состояния должна проводиться со средством, которое могло бы регулировать уровень сывороточного липида, как это делает эстроген, но должно быть лишено побочных эффектов и риска, связанного с терапией эстрогеном. Третьей основной патологией, связанной с постклимактерическим синдромом, является эстрогензависимый рак молочной железы и, в меньшей степени, эстрогензависимые раковые заболевания других органов, в частности матки. Хотя пациенты с такими опухолями не ограничиваются исключительно постклимактерическими женщинами, эти опухоли более широко распространены у более старой, постклимактерической части женского населения. Современная химиотерапия этих раковых заболеваний основана в основном на применении антиэстрогенных соединений, например тамоксифена. Хотя такие смешанные агонисты-антагонисты оказывают благотворное воздействие при лечении этих раковых заболеваний и эстрогенные побочные эффекты переносимы при острых, угрожающих жизни ситуациях, они не идеальны. Например, эти средства могут оказывать стимулирующее действие на некоторые популяции раковых клеток в матке из-за их эстрогенных (агонистических) свойств, и они могут, следовательно, в некоторых случаях оказывать противоположное действие. Лучшим средством для лечения этих раковых заболеваний могло быть средство, которое является антиэстрогенным соединением, обладающим незначительными или не обладающим эстроген-агонистическими свойствами на репродуктивных тканях. В ответ на очевидную необходимость новых фармацевтических средств, которые способны облегчить симптомы, inter alia, постклимактерического синдрома, настоящее изобретение направлено на создание новых бензотиофеновых соединений, их фармацевтических композиций и способов применения таких соединений для лечения постклимактерического синдрома и других имеющих отношение к эстрогену патологических состояний, например состояний, указанных ниже. Фиброз матки (фиброзное заболевание матки) является старой и всегда существующей клинической проблемой, который проходит под различными названиями, включая фиброзное заболевание матки, маточную гипертрофию, маточные лиеомиомы, гипертрофию миометрия, фиброзную матку и фиброзный метрит. По существу, фиброз матки является состоянием, когда имеется аномальное депонирование фиброзной ткани на стенке матки. Это состояние является причиной дисменореи и бесплодия у женщин. Точная причина этого состояния недостаточно понятна, но данные дают возможность предположить, что она является неуместной реакцией фиброзной ткани на эстроген. Такое состояние появляется у кроликов при ежедневном введении им эстрогена в течение 3 месяцев. У морских свинок такое состояние появлялось в результате ежедневного введения эстрогена в течение четырех месяцев. Кроме того, аналогичную гипертрофию эстроген вызывает у крыс. Наиболее обычное лечение фиброза матки включает хирургические операции, которые дорого стоят, а также иногда являются причиной осложнений, например спайкообразования и инфекции. У некоторых пациентов первоначальная хирургия является только временным лечением и фибромы возобновляют рост. В этих случаях проводят экстирпацию матки, которая эффективно ликвидирует фиброму, но также обрывает репродуктивную жизнь пациента. Можно также вводить антагонисты гормона, секретирующего гонадотропин, однако их применение регулируется тем фактом, что оно может привести к остеопорозу. Таким образом, существует потребность в новых способах лечения фиброза матки и способы настоящего изобретения удовлетворяют эту потребность. Эндометриоз является состоянием серьезной дисменореи, которая сопровождается сильной болью, кровотечением в эндометриальную массу или брюшинную полость и часто ведет к бесплодию. Причиной симптомов этого состояния, по-видимому, является эктопические эндометриальные образования, которые неподходящим образом реагируют на нормальное гормональное регулирование и расположены в несоответствующих тканях. По причине неподходящего расположения эндометриальных образований, ткань, по-видимому, инициирует локальные, подобные воспалению ответные реакции, вызывающие инфильтрацию макрофагов и каскад событий, приводящий к инициированию болезненной реакции. Точная этиология этого заболевания недостаточно понятна и его лечение гормональной терапией дает различные результаты, недостаточно изучено и характеризуется различными нежелательными и возможно опасными побочными эффектами. Одно из лечений этого заболевания предусматривает применение низкой дозы эстрогена для подавления эндометриального образования путем отрицательного обратного действия на основную секрецию гонадотропина и последующего овариального продуцирования эстрогена, однако иногда для регулирования этих симптомов необходимо применять эстроген непрерывно, это применение эстрогена может часто приводить к нежелательным побочным эффектам и даже к риску эндометриального рака. Другое лечение заключается в непрерывном введении прогестинов, которые индуцируют аменорею и путем подавления овариального продуцирования эстрогена могут вызвать обратное развитие в эндометриальных образованиях. Применение постоянной терапии прогестином часто сопровождается неприятными побочными эффектами действия прогестинов на центральную нервную систему и часто ведет к бесплодию, обусловленному подавлением функции яичников. Третье лечение заключается во введении слабых андрогенов, которые эффективно регулируют эндометриоз; однако они индуцируют сильные маскулинизирующие эффекты. Несколько из таких лечений эндометриоза вызывают также среднюю степень остеопороза при непрерывной терапии. Следовательно, желательны новые способы лечения эндометриоза. Пролиферация клеток гладких мышц аорты играет важную роль в таких заболеваниях, как атеросклероз и рестеноз. Сосудистый рестеноз после чрескожной пластической операции на полости коронарных сосудов (РТСА), как было показано, является тканевой ответной реакцией, характеризующейся ранней и поздней фазой. Ранняя фаза, проявляющаяся в течение от нескольких часов до нескольких дней после РТСА, обусловлена тромбозом с некоторыми вазоспазмами, тогда как поздняя фаза, как оказалось, является доминирующей из-за избыточной пролиферации и миграции клеток гладких мышц аорты. В этом заболевании повышенная клеточная подвижность и колонизация такими мышечными клетками и макрофагами значительно способствует патогенезу данного заболевания. Избыточная пролиферация и миграция клеток гладких мышц аорты может быть основным механизмом реокклюзии коронарной артерии после РТСА, атеректомии, лазерной пластической операции на сосудах и артериальной хирургии с обходным сосудистым шунтом. Смотри “Intimal Proliferation of Smooth Muscle Cell as an Explanation for Recurrent Coronary Artery Stenosis after Percutaneous Transluminal Coronary Angioplasty”, Austin et al., Journal of the American College of Cardiology, 8:369-375 (Aug. 1985). Сосудистый рестеноз остается основным долговременным осложнением после хирургического вмешательства в закупоренные артерии путем чрескожной пластической операции на полости коронарных сосудов (РТСА), атеректомии, лазерной пластической операции на сосудах и артериальной хирургии с обходным сосудистым шунтом. У 35% пациентов, которым проводили РТСА, реокклюзия имела место в течение от трех до шести месяцев после операции. Современная страгегия для лечения сосудистого рестеноза включает механическое вмешательство при помощи устройств, например стентов, или фармакологические терапии с применением гепарина, низкомолекулярного гепарина, кумарина, аспирина, рыбьего жира, антагониста кальция, стероидов и простациклина. Эти стратегии были не в состоянии преодолеть степень реокклюзии и были неэффективны в лечении и предупреждении сосудистого рестеноза. Смотри “Prevention of Restenosis after Percutaneous Transluminal Coronary Angioplasty: The Search for a “Magic Bullet”, Hermans et al., American Heart Journal, 122: 177-187 (July 1991). В патогенезе рестеноза происходит избыточная клеточная пролиферация и миграция как результат действия факторов роста, продуцированных клеточными компонентами в крови и поврежденными стенками артериальных сосудов, которые медиируют пролиферацию клеток гладких мышц при сосудистом рестенозе. Средства, которые ингибируют пролиферацию и/или миграцию клеток гладких мышц аорты, пригодны для лечения и предупреждения рестеноза. Настоящее изобретение относится к применению соединений в качестве ингибиторов пролиферации клеток гладких мышц аорты и, таким образом, самим ингибиторам рестеноза. Настоящее изобретение относится к соединениям формулы 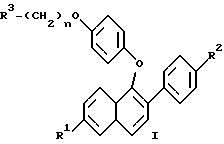 где R1 представляет собой -H, OH, -H(C1-C4-алкил), -OCOC6H5, -OCO(C1-C6-алкил) или -OSO2 (C2-C6-алкил); R2 представляет собой -H, -OH, -O(C1-C4-алкил), -OCOC6H5, -OCO(C1-C6-алкил), -OSO2(C2-C6-алкил) или галоген; R3 представляет собой 1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидино-, 4-морфолино-, диметиламино-, диэтиламино-, диизопропиламино- или 1-гексаметилениминогруппу и n = 2 или 3; или их фармацевтически приемлемым солям. Настоящее изобретение относится также к промежуточным соединениям формулы II, которые пригодны для получения фармацевтически активных соединений настоящего изобретения и показаны ниже 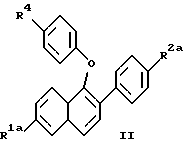 где R1a представляет собой -H или OR5, где R5 представляет собой гидроксизащитную группу, R2a представляет собой -H, галоген или -OR6, где R6 представляет собой гидроксизащитную группу и R4 представляет собой -OH или CHO, или их фармацевтически приемлемым солям. Настоящее изобретение относится также к фармацевтическим композициям, содержащим соединения формулы I и необязательно содержащим эстроген или прогестин, и применению таких соединений, необязательно в комбинации с эстрогеном или прогестином, для облегчения симптомов постклимактерического синдрома, конкретно остеопороза, связанных с сердечно-сосудистой системой патологических состояний и эстрогензависимого рака. В соответствии с приведенным описанием термин “эстроген” включает стероидные соединения, имеющие эстрогенную активность, такие, как например 17  -эстрадиол, эстрон, конъюгированный эстроген (Премарин -эстрадиол, эстрон, конъюгированный эстроген (Премарин ), конский эстроген 17 -этинилэстрадиол и подобные соединения. В соответствии с настоящим описанием термин “прогестин” включает соединения, имеющие гестагенную активность, такие, как например прогестерон, норэтилнодрел, нонгестрел, мегестролацетат, норетиндрон и подобные соединения.
Соединения настоящего изобретения пригодны также для ингибирования фиброзного заболевания матки и эндометриоза у женщин и пролиферации клеток гладких мышц аорты, в частности рестеноза, у людей.
Однако особенность настоящего изобретения включает соединения формулы I ), конский эстроген 17 -этинилэстрадиол и подобные соединения. В соответствии с настоящим описанием термин “прогестин” включает соединения, имеющие гестагенную активность, такие, как например прогестерон, норэтилнодрел, нонгестрел, мегестролацетат, норетиндрон и подобные соединения.
Соединения настоящего изобретения пригодны также для ингибирования фиброзного заболевания матки и эндометриоза у женщин и пролиферации клеток гладких мышц аорты, в частности рестеноза, у людей.
Однако особенность настоящего изобретения включает соединения формулы I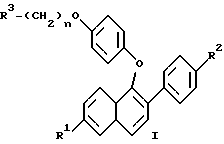 где R1 представляет собой -H, -OH, -O(C1-C4-алкил), -OCOC6H5 -OCO(C1-C6-алкил) или -OSO2(C2-C6-алкил); R2 представляет собой -H, -OH, -O(C1-C4-алкил), -OCOC6H5, -OCO(C1-C6-алкил), -OSO2(C2-C6-алкил) или галоген; R3 представляет собой 1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидино-, 4-морфолино-, диметиламино-, диэтиламино-, диизопропиламино- или 1-гексаметилениминогруппу и n = 2 или 3; или их фармацевтически приемлемые соли. Общие термины, применяемые в описании соединений настоящего изобретения, имеют их обычные значения. Например “C1-C6-алкил” относится к алифатическим цепям нормального или разветвленного строения из 1-6 атомов углерода, включая такие радикалы, как метил, этил, пропил, изопропил, бутил, н-бутил, пентил, изопентил, гексил, изогексил и подобные радикалы. Аналогично термин “C1-C4-алкоксигруппа” обозначает C1-C4-алкильную группу, присоединенную через атом кислорода, и включает такие группы, как например метокси-, этокси- , н-пропокси-, изопропокси- и подобные группы. Из этих алкоксигрупп метоксигруппа наиболее предпочтительна в большинстве случаев. Исходным материалом для получения соединений настоящего изобретения являются соединение формулы III 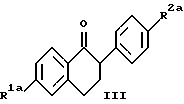 где R1a представляет собой -H или -OR5, где R5 представляет собой гидроксизащитную группу и R2a представляет собой -H, галоген или OR6, где R6 представляет собой гидроксизащитную группу. Соединения формулы III хорошо известны в настоящей области, их получают по методике, описанной Boyle, et al., в патенте США N 4910212, который присутствует здесь в качестве ссылки. Смотри также Collins, D. J. et al. , Aust. J. Chem. 41: 745-756 (1988); и Collins, D.J. et al., Aust. J. Chem. 37: 2279-2294 (1984). При получении соединений настоящего изобретения кетон формулы III обычно ароматизируют, получая фенол формулы IV, который затем реакцией с 4-галогенбензальдегидом превращают в диариловый эфир формулы IIa, который в свою очередь превращают в фенол формулы IIb. Этот синтетический путь приведен в схеме I (см. в конце описания), R1a и R2a имеют указанные выше значения. В первой стадии настоящего способа соединение формулы III превращают в фенол формулы IV через трехстадийное превращение, как описано в работе Wang G. , et al. , M. Syn. Commun., 21:989 (1991). В сущности кетон формулы II енолизируют кипячением с обратным холодильником раствора соединения формулы III в подходящем ацетатном растворителе, в присутствии кислотного катализатора. Полученный ацетат енола непосредственно превращают в ацетат нафтола, который затем гидролизуют в фенол формулы IV. При превращении кетона формулы III в его соответствующий енол можно применять различные известные кислотные катализаторы. Предпочтительны неводные кислоты, в частности п-толуолсульфокислота. Приемлемые ацетатные растворители включают например эфиры простых спиртов и уксусной кислоты, в частности изопропенилацетат. Когда настоящую реакцию проводят при кипячении с обратным холодильником, реакция завершается в течение времени от около 6 до около 48 ч. Енольный продукт этой реакции не выделяют, а после завершения реакции полученный раствор обрабатывают подходящим окислителем и нагревают с обратным холодильником, оптимально в течение от около 1 до около 3 ч. Пригодные окислители для этой второй фазы первой стадии реакции, показанной в схеме I, ограничены окислителями, известными в настоящей области, которые могут привести к удалению атома водорода из насыщенной системы для образования ароматизированной системы. Такие окислители включают, например катализаторы гидрирования, например платину, палладий и никель, элементную серу и селен и хиноны. Для настоящей заявки предпочтительны хиноновые окислители, особенно 2,3-дихлор- 5,6-дициано-1,4-бензохинон (DDQ). Для проведения настоящей фазы способа следует применять около от 1 до 2 эквивалентов DDQ на 1 эквивалент субстрата. Полученный продукт настоящей фазы, ацетат нафтола, затем гидролизуют для получения соединения формулы IV, таким образом завершая первую стадию способа получения, показанного на схеме I. Настоящую фазу гидролиза проводят кислотным или основным (щелочным) гидролизом субстрата в полярном протонном растворителе, например воде, или одном или нескольких растворителях, содержащих спирт, например метанол или этанол. В раствор для содействия растворимости можно добавить сорастворитель, например тетрагидрофуран (ТГФ) или диоксан. Пригодные основания для этой фазы включают едкий натрий, едкий кали, гидроксид лития и подобные соединения. Подходящие кислоты включают, например, соляную кислоту, метансульфокислоту, п-толуолсульфокислоту и подобные кислоты. Эту финальную фазу первой стадии, показанной выше на схеме I, можно проводить при комнатной температуре, ее проводят за короткий период времени, обычно от 1 до около 12 ч. Завершение настоящей реакции можно определить путем стандатных хроматографических методик, например тонкослойной хроматографией. Во второй стадии схемы I фенол формулы IV сначала реакцией с основанием, затем добавлением 4-галогенбензальдегида в полярном апротонном растворителе в в инертной атмосфере, например атмосфере азота, превращают в диариловый эфир формулы IIa. Эта реакция хорошо известна в настоящей области, ее проводят по методике, описанной в работе Jeagen, G.W., et al., Synthesis, 63 (1991). Более подробно, сначала проводят реакцию 1 эквивалента соединения формулы IV по меньшей мере с 1 эквивалентом гидрида или карбоната щелочного металла в подходящем растворителе, затем добавляют по каплям 4-галогенбензальдегид в том же растворителе, который применяют для субстрата. Подходящими растворителями для этой реакции являются те растворители или смеси растворителей, которые сохраняют свою инертность в течение реакции. Предпочтителен N,N-диметилформамид (ДМФ), особенно его безводная форма. Предпочтительно гидрид натрия применяют в качестве требуемого основания и 4-фторбензальдегид применяют в качестве предпочтительного 4-галогенбензальдегида. Температура, применяемая в этой стадии настоящего способа должна быть достаточной для завершения этой реакции и не способствует образованию нежелательных побочных продуктов. Предпочтительный температурный предел для этой реакции 30 – 100oC. В предпочтительных условиях реакции соединение формулы IIa можно получить этим способом за время 24 – 48 ч. Последняя реакция, приведенная на схеме I, т.е. превращение альдегидной части соединения формулы IIа в фенольную группу, в результате которой образуется соединение формулы IIa, известна в настоящей области как окисление по Байер-Виллигеру. Смотри, например, Fiesers L., et al., Reagents for Organics Synthesis, 1: 467, Wiley (New Jork, 1967); Hassal, C.H., Organic Reactions, 9:73-106 (Wiley, New Jork 1967). В общем, настоящая реакция включат комбинирование бензальдегида с перкислотой, например перуксусной кислотой или м-хлорпербензойной кислотой, в инертном растворителе, например хлороформе или хлористом метилене. Продукт этой реакции, формиатный эфир, можно затем легко гидролизовать в целевой фенол. Смотри, например Jager et al., supra; Godfrey, I.M., et al., J. Chem. Soc. Perkins. Trans. 1:1353 (1974); и Rue, R. et al., Bull. Soc. Shim. Fr. 3617 (1970). Для настоящей реакции предпочтительный вариант описан Matsumoto, et al., J. Org. Chem. , 49: 4741 (1984). Этот способ включает комбинирование бензальдегида формулы IIa с количеством от по меньшей мере 1 до около 2 эквивалентов 30%-ного пероксида водорода в спиртовом растворителе и в присутствии каталитической кислоты. В этих условиях фенол образуется непосредственно и необходимость дополнительной стадии гидролиза, следовательно, исключается. Предпочтительным растворителем и кислотным катализатором для настоящей реакции является метанол и концентрированная серная кислота соответственно. В этих предпочтительных условиях реакции превращение соединения формулы IIa в соединение формулы IIb завершается после перемешивания в течение от около 12 до около 48 ч при комнатной температуре. Соединения формул IIa и IIb в соответствии с описанием являются новыми промежуточными соединениями формулы II, которые пригодны для получения фармацевтически активных соединений формулы I настоящего изобретения. При получении соединение формулы IIb обрабатывают соединением формулы V R3-(CH2)n-Q, где R3 и n имеют указанные выше значения и Q является бромом или, что предпочтительно, хлором, получая соединение формулы Ia. Соединение формулы Ia затем освобождают от защитных групп, когда присутствуют гидроксизащитные группы, R5 и/или R6, получая соединение формулы Ib. Эти стадии способа показаны на схеме II (см. в конце описания), где R1a, R2a, R3 и n имеют указанные выше значения; R1b представляет собой -H или -OH и R2b представляет собой -H, -OH или галоген; или его фармацевтически приемлемая соль. В первой стадии способа, указанного на схеме II, алкилирование проводят при помощи стандартных методик. Соединения формулы V выпускаются промышленностью или их получают способами, хорошо известными любому специалисту в данной области. Предпочтительно применяют гидрохлоридную соль соединения формулы V, конкретно гидрохлорид 2-хлорэтилпиперидина. Обычно проводят реакцию по меньшей мере около 1 эквивалента субстрата формулы IIb с 2 эквивалентами соединения формулы V в присутствии по меньшей мере около 4 эквивалентов карбоната щелочного металла, предпочтительно карбоната цезия, и подходящего растворителя. Для проведения реакции применяют растворители или смеси растворителей, которые остаются инертными на всем протяжении реакции. Предпочтителен N,N-диметилформамид, особенно его безводная форма. Температура, применяемая в этой стадии, должна быть достаточна для завершения этой реакции алкилирования. Обычно достаточна и предпочтительна комнатная температура. Настоящую реакцию предпочтительно проводят в инертной атмосфере, конкретно в атмосфере азота. В этих предпочтительных реакционных условиях реакция будет завершаться за 16 – 20 ч. Ход реакции можно контролировать при помощи стандартных хроматогарфических методик. В качестве альтернативы для получения соединений формулы Ia проводят реакцию соединения формулы IIb с избытком алкилирующего агента формулы Q-(CH2)n-Q’, где Q и Q’ являются одинаковыми или разными отщепляемыми группами, в щелочном растворе. Подходящие отщепляемые группы включают сульфонаты, например метансульфонат, 4-бромбензолсульфонат, толуолсульфонат, этансульфонат, изопропилсульфонат, 4-метоксибензолсульфонат, 4-нитробензолсульфонат, 2-хлорбензолсульфонат, трифлат и подобные сульфонаты, галогены, например бром, хлор и иод, и другие родственные отщепляемые группы. Предпочтительными отщепляемыми группами являются галогены, особенно предпочтителен бром. Предпочтительный щелочной раствор для этой реакции алкилирования содержит карбонат калия в инертном растворителе, таком, как например метилэтилкетон (МЭК) и ДМФ. В этом растворе 4-гидркосигруппа бензоильной части соединения формулы IIb находится в виде феноксид-иона, который замещает одну из отщепляемых групп алкилирующего агента. Эта реакция проходит лучше, когда щелочной раствор, содержащий реагирующие вещества и реагенты, кипятят с обратным холодильником и оставляют для завершения реакции. Когда в качестве предпочтительного растворителя применяют МЭК, время реакции составляет от около б до около 20 ч. Продукт реакции этой стадии затем подвергают реакции с 1-пиперидином, 1-пирролидином, метил-1-пирролидином, диметил-1-пирролидином, 4-морфолином, диметиламином, диэтиламином или 1-гексаметиленимином по стандартным методикам для образования соединений формулы Ia. Предпочтительно проводят реакцию гидрохлоридной соли пиперидина с алкилирующим соединением формулы IIb в инертном растворителе, например безводном ДМФ, при нагревании до температуры в пределах от около 60oC до около 110oC. Когда смесь нагревают до предпочтительной температуры около 90oC, для завершения реакции требуется только от около 30 мин до около 1 ч. Однако изменения в условиях реакции будут оказывать влияние на время этой реакции, необходимое для ее завершения. Конечно, протекание этой стадии реакции можно контролировать при помощи стандартных хроматографических методик. Соединения формулы Ia, где R5 и/или R6, если они присутствуют, представляют собой C1-C4-алкил, предпочтительно метил, являются новыми соединениями и фармацевтически активны для способов, описанных в изобретении. В соответствии с этим, такие соединения попадают в определение соединений формулы I настоящего изобретения. Предпочтительные соединения формулы I получают отщеплением, когда они присутствуют, гидроксизащитных групп 5 и R6 соединений формулы Ia при помощи хорошо известных методик. Различные реакции для образования и удаления таких отщепляемых групп описываются в ряде стандартных работ, включая, например, “Protective Groups in Organic Chemistry”, Plenum Press (London and New Jork, 1973); Green. T.W., “Protective Groups in Organic Synthesis”, Wiley, (New Jork, 1981); и “The Pepdides”, Vol. 1, Schooder and Lubke, Academic Press (London and New Jork, 1965). Способы удаления предпочтительных гидроксизащитных групп R5 и/или R6, конкретно метила, в основном те, которые описаны в приведенном ниже примере 5. Соединения формулы Ia являются новыми и фармацевтически активны для способов, описанных в настоящем изобретении, они попадают в определение соединений формулы I настоящего изобретения. Другие предпочтительные соединения формулы I получают замещением гидроксигрупп в 6- и/или 4′-положении, когда они присутствуют, группой формулы -O-CO-(C1-C6-алкил) или O-OSO2-(C2-C6-алкил) при помощи хорошо известных методик. Смотри, например, патент США, N 4358593. Например, когда нужно ввести группу -O-CO(C1-C6-алкил), моно- или дигидроксисоединение формулы I подвергают реакции с таким агентом, как ацилхлорид, -бромид, цианид- или -азид, или с соответствующим ангидридом или смешанным ангидридом. Реакции удобно проводят в растворителе основного характера, например пиридине, лутидине, хинолине или изохинолине, или в растворителе третичном амине, например триэтиламине, трибутиламине, метилпиперидине и подобном растворителе. Реакцию можно также проводить в инертном растворителе, например этилацетате, диметилформамиде, диметилсульфоксиде, диоксане, диметоксиэтане, ацетонитриле, ацетоне, метилэтилкетоне и т.д., в который добавлен по меньшей мере один эквивалент акцептора кислоты (за исключением указанного ниже), например, третичного амина. При желании можно применять катализаторы ацилирования, например 4-диметиламинопиридин или 4-пирролидинопиридин. Смотри, например, Haslam. et al. , Tetrahedron, 36: 2409-2433 (1980). Эти реакции проводят при умеренных температурах, в пределах от около -20oC до около 100oC, часто в инертной атмосфере, например, атмосфере газообразного азота. Однако для проведения реакции обычно достаточна комнатная температура. Ацилирование гидроксигруппы в 6-положении и/или 4-положении можно проводить катализируемыми кислотами реакциями соответствующих карбоновых кислот в инертных органических растворителях. Применяют кислотные катализаторы, например серную кислоту, полифосфорную кислоту, метансульфокислоту и подобные кислоты. Указанные выше группы R1 и/или R2 соединений формулы I можно также ввести образованием активного эфира соответствующей кислоты, например эфиров, образованных такими известными реагентами, как например, дициклогексилкарбодиимид ацилимидазолы, нитрофенолы, пентахлорфенол, N-гидроксисукцинимид и 1- гидроксибензотриазол. Смотри, например, Bull. Chem. Soc. Japan, 38:1979 (1965) и Chem. Ber., 788 b 2024 (1970). Каждую из указанных выше методик, которые обеспечивают образование групп -O-CO-(C1-C6-алкил), проводят в растворителях, обсужденных выше. Те методики, которые не образуют кислотный продукт в ходе реакции, конечно, не требуют применения акцептора кислоты в реакционной смеси. Когда нужно получить соединение формулы I, у которого гидроксигруппа в 6- и/или 4′-положении соединения формулы I превращена в группу формулы O-OSO2-(C2-C6-алкил), моно- или дигидроксисоединение обрабатывают, например, ангидридом сульфокислоты или производным соответствующей сульфокислоты, например сульфонилхлоридом, -бромидом или сульфониламмониевой солью, как рекомендуется King and Monoir, J. Am. Chem. Soc., 97:2556-2567 (1975). Дигидроксисоединение можно также подвергнуть реакции с ангидридом соответствующей сульфокислоты или смешанными ангидридами сульфокислот. Такие реакции проводят, например, в условиях, указанных выше при обсуждении реакции с галогенангидридами кислот и подобными соединениями. Хотя в способах настоящего изобретения соединения формулы I можно применять в форме свободных оснований, предпочтительно получать и применять их фармацевтически приемлемые соли. Таким образом, соединения, применяемые в способах настоящего изобретения, главным образом образуют фармацевтически приемлемые соли с разнообразными органическими и неорганическими кислотами и включают физиологически приемлемые соли, которые часто применяют в фармацевтической химии. Такие соли также являются частью настоящего изобретения. Типичные неорганические кислоты, применяемые для образования таких солей, включают соляную, бромистоводородную, иодистоводородную, азотную, серную, фосфорную, гипофосфорную и подобные кислоты. Можно также применять соли, полученные из органических кислот, например алифатических моно- и дикарбоновых кислот, фенилзамещенных алкановых кислот, гидроксиалкановых и гидроксиалкандиовых кислот, ароматических кислот, алифатических и ароматических сульфокислот. Такие фармацевтически приемлемые соли, таким образом, включают ацетат, фенилацетат, трифторацетат, акрилат, аскорбат, бензоат, хлорбензоат, динитробензоат, гидроксибензоат, метоксибензоат, метлбензоат, о-ацетоксибензоат, нафталин-2-бензоат, бромид, изобутират, фенилбутират, бета-гидроксибутират, бутин-1,4-диоат, гексин-1,4-диоат, капрат, каприлат, хлорид, циннамат, цитрат, формиат, фумарат, гликоллат, гептаноат, гиппурат, лактат, малат, малеат, гидроксималеат, малонат, манделат, мезилат, никотинат, изоникотинат, нитрат, оксалат, фталат, терефталат, фосфат, вторичный кислый фосфат, первичный кислый фосфат, метафосфат, пирофосфат, пропиолат, пропионат, фенилпропионат, салицилат, себацинат, сукцинат, суберат, сульфат, бисульфат, пиросульфат, сульфит, бисульфит, сульфонат, бензолсульфонат, п-бромфенилсульфонат, хлорбензолсульфонат, этансульфонат, 2-гидроксиэтансульфонат, метансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, п-толуолсульфонат, ксилолсульфонат, тартрат и подобные соли. Предпочтительными солями являются гидрохлориды и оксалаты. Фармацевтически приемлемые соли с кислотами обычно получают реакцией соединения формулы I с эквимолярным или избыточным количеством кислоты. Реагенты обычно смешивают в общем для них растворителе, например диэтиловом эфире или этилацетате. Соль обычно осаждается из раствора в течение от около 1 ч до 10 дней. Ее можно отделить фильтрованием или растворитель можно выпаривать обычным образом. Фармацевтически приемлемые соли обычно имеют повышенную растворимость по сравнению с соединением, из которого их получают, и, таким образом, часто более пригодны для получения готовой лекарственной формы в виде жидкостей или эмульсий. Следующие примеры представлены, чтобы дополнительно иллюстрировать получение соединений настоящего изобретения, Имеется в виду, что любой из следующих примеров не должен ограничивать объем изобретения. Величины ЯМР-спектроскопии следующих примеров получали на ЯМР-приборе GE с 300 МГц. Безводный DMSO-d6 применяли в качестве растворителя, если не оговорено особо. ПРИМЕР 1. Получение [2-(4-метоксифенил)]-6-метоксинафтил-1-ола 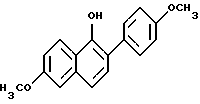 6-Метокси-2-(4-метоксифенил)-3,4-дигидронафталин-1(2H)-он (8,50 г, 30,14 ммоль) растворяли в 50 мл изопропенилацетата вместе с 1 г п-толуолсульфокислоты. Полученную смесь нагревали при кипячении с обратным холодильником в течение 6 ч. После охлаждения до комнатной температуры добавляли 2,3-дихлор- 5,6-дициано-1,4-бензохинон (DDQ) (13,70 г, 60,28 ммоля). Реакционную смесь затем кипятили с обратным холодильником 1,5 ч. После охлаждения до комнатной температуры смесь разбавляли хлористым метиленом (200 мл). Органический слой промывали 0,2 N раствором едкого натра (4 х 200 мл), затем водой (2 х 200 мл). Органический слой сушили (сульфатом натрия) и концентрировали в вакууме до получения темного твердого продукта. Неочищенный ацетат растворяли в 200 мл смеси (1:1) метанола и тетрагидрофурана и обрабатывали избыточным количеством метоксида натрия. Образованный оранжевый осадок отделяли фильтрованием. Фильтрат подкисляли до pH 4 5 N соляной кислотой. Фильтрат далее разбавляли 200 мл воды и затем экстрагировали этилацетатом (3 х 100 мл). Органический слой сушили (сульфатом натрия) и концентрировали в вакууме, получая небелое твердое вещество. Кристаллизацией из смеси гексаны/этилацетат получали 4,24 г (50%) [2-(4-метоксифенил)]-6-метоксинафтил-1-ола в виде белого твердого вещества. Т. пл. 129 – 131oC. 1H ЯМР-спектр (DMSO-d6):  8,19 (д, J= 9,1 Гц, 1H), 7,45 (д,7 = 8,5 Гц, 2H), 7,38 (д, J = 8,0 Гц, 1H), 7,28 (д, J= 8,0 Гц, 1H), 7,18 (дд, J = 9,1, 2,8 Гц, 1H), 7,13 (д, J= 2,8 Гц, 1H), 7,07 (д, J= 8,5 Гц, 2H), 5,76 (ш. с, 1H), 3,94 (с, 3H), 3,88 (с, 3H). FD-масс-спектрометрия: 280. Элементный анализ: Рассчитано для C18H16O3: C 77,12; H 5,75. Найдено C 76,83; H 5,90.
ПРИМЕР 2. (Получение 1-(4-формилфенокси)-2-(4-меткосифенил)-6- метоксинафталина 8,19 (д, J= 9,1 Гц, 1H), 7,45 (д,7 = 8,5 Гц, 2H), 7,38 (д, J = 8,0 Гц, 1H), 7,28 (д, J= 8,0 Гц, 1H), 7,18 (дд, J = 9,1, 2,8 Гц, 1H), 7,13 (д, J= 2,8 Гц, 1H), 7,07 (д, J= 8,5 Гц, 2H), 5,76 (ш. с, 1H), 3,94 (с, 3H), 3,88 (с, 3H). FD-масс-спектрометрия: 280. Элементный анализ: Рассчитано для C18H16O3: C 77,12; H 5,75. Найдено C 76,83; H 5,90.
ПРИМЕР 2. (Получение 1-(4-формилфенокси)-2-(4-меткосифенил)-6- метоксинафталина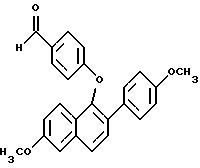 В раствор [2-(4-метоксифенил)] -6-метоксинафтил-1-ола (3,57 г, 12,75 ммоля) в 180 мл безводного N,N-диметилформамида в атмосфере N2 при комнатной температуре добавляли гидрид натрия (535 мг, 13,38 ммоль, 60%-ная дисперсия в минеральном масле) небольшими порциями. После перемешивания в течение 10 мин добавляли 4-фторбензальдегид (3,20 г, 25,50 ммоль). Полученную смесь нагревали до 70oC в течение 36 ч. После охлаждения до комнатной температуры растворитель удаляли в вакууме. Остаток затем распределяли между системой этилацетат/вода. Слои разделяли и органический промывали несколько раз водой. Органический слой в конце сушили (сульфатом натрия) и концентрировали в вакууме до образования масла. Хроматографией его (смесь гексанов и этилацетата, 90:10) получали 2,06 г (48%) 1-(4-формил)фенокси-2- (метоксифенил)-6-метоксинафталин в виде белого твердого вещества, которое кристаллизовали из метанола. Данные для 1-(4-формил) фенокси-2-(4-метоксифенил)-6-метоксинафталина: т. пл. 120 – 121oC. 1H ЯМР-спектр (CDCl3)  9,80 (с, 1H), 7,78 (д, J = 9,2 Гц, 1H), 7,74 (д, J = 8,8 Гц, 1H), 7,67 (д, J= 8,8 Гц, 2H), 7,56 (д, J= 8,8 Гц, 1H), 7,46 (д, J = 8,8 Гц, 2H), 7,21 (д, J =2,6 Гц, 1H), 7,12 (дд, J = 9,2 2,6 Гц, 1H), 6,84 (д, J = 8,8 Гц, 2H). 6,81 (д, J = 8,8 Гц, 2H), 3,95 (с, 3H), 3,78 (с, 3H). FD-масс-спектрометрия: 384. Элементный анализ: Рассчитано для C25H20O4:C 78,11; H 5,24. Найдено C 78,32; H 5,24.
ПРИМЕР 3. (Получение 1-(4-гидрокси)фенокси-2-(4-метоксифенил)-6- метокси нафталина 9,80 (с, 1H), 7,78 (д, J = 9,2 Гц, 1H), 7,74 (д, J = 8,8 Гц, 1H), 7,67 (д, J= 8,8 Гц, 2H), 7,56 (д, J= 8,8 Гц, 1H), 7,46 (д, J = 8,8 Гц, 2H), 7,21 (д, J =2,6 Гц, 1H), 7,12 (дд, J = 9,2 2,6 Гц, 1H), 6,84 (д, J = 8,8 Гц, 2H). 6,81 (д, J = 8,8 Гц, 2H), 3,95 (с, 3H), 3,78 (с, 3H). FD-масс-спектрометрия: 384. Элементный анализ: Рассчитано для C25H20O4:C 78,11; H 5,24. Найдено C 78,32; H 5,24.
ПРИМЕР 3. (Получение 1-(4-гидрокси)фенокси-2-(4-метоксифенил)-6- метокси нафталина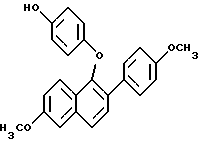 В суспензию 1-(4-формил)фенокси-2-(4-метоксифенил)-6-метоксинафталина (2,70 г, 7,03 ммоль) в 50 мл метанола добавляли пероксид водорода (1,50 мл, 14,1 ммоль, 30%-ный раствор) и затем концентрированную серную кислоту (0,5 мл). Реакционную смесь осторожно нагревали, чтобы помочь растворению, и затем перемешивали при комнатной температуре 36 ч. Для нейтрализации кислоты добавляли твердый бикарбонат натрия и смесь затем распределяли между системой этилацетат/вода (100 мл, равные количества). Слои затем разделяли и органический слой сушили (сульфатом натрия) и концентрировали в вакууме до получения рыжевато-коричневого твердого продукта. Хроматографией (диоксид кремния, 10-30% этилацетат/гексаны) получали 2,08 г (80%) 1-(4-гидроксифенокси)-2-(4-метоксифенил)-6-метоксинафталина в виде белого твердого вещества. Т. пл. 159 – 164oC. 1H ЯМР/спектр (DMSO-d6): 8,91 (с, 1H), 9,76 (д, J = 8,5 Гц, 1H), 7,69 (д, J= 9,2 Гц, 1H), 7,54 (д, J= 8,5 Гц, 1H), 7,48 (д, J= 8,7 Гц, 2H), 7,37 (д, J= 2,4, 1H), 7,09 (дд, J= 9,2, 2,4 Гц, 1H), 6,89 (д, J= 8,7 Гц, 2H), 6,47 (к, JAB= 9,0 Гц, 4H), 3,84 (с, 3H), 3,70 (с, 3H), FD-масс-спектроскопия: 372. Элементный анализ: Рассчитано для C24H20O4 C 77,40; H 5,41. Найдено C 77,19; H 5,70.
ПРИМЕР 4. Получение гидрохлорида 1-[4-[2-(1-пиперидинил)этоксит]фенокси] -2-(4-метоксифенил)-6 -метоксинафталина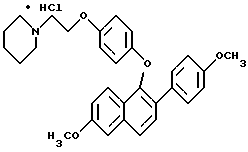 В раствор 1-(4-гидрокси)фенокси-2-(4-метоксифенил)-6- метоксинафталина (865 мг, 2,32 ммоль) в 15 мл безводного N,N-диметилформамида в атмосфере N2 добавляли карбонат цезия (3 г, 9,2 ммоль). После перемешивания в течение 15 мин добавляли гидрохлорид 2-хлорэтилпиперидина (540 мг, 2,90 ммоль). Полученную смесь энергично перемешивали в течение 24 ч, затем распределяли между этилацетатом и водой (200 мл). Слои разделяли и органический слой промывали несколько раз водой. Органический слой затем сушили (сульфатом натрия) и концентрировали в вакууме до получения белого твердого продукта. Неочищенное свободное основание растворяли в 20 мл этилацетата и обрабатывали системой диэтиловый эфир соляная кислота. Образованный осадок отделяли фильтрованием и сушили в вакууме, получая 1,06 г (88%) гидрохлорида 1-[4-[2-(1-пиперидинил) этокси] -фенокси] -2-(4-метоксифенил)-6-метоксинафталина в виде белого твердого вещества. Т. пл. 184 – 188oC. 1H ЯМР-спектр (DMSO-d6): 9,91 (ш.с, 1H), с 7,84 (д, J= 8,5 Гц, 1H), 7,71 (д, J= 9,2 Гц, 1H), 7,61 (д,J = 8,5 Гц, 1H), 7,54 (д, J= 8,8 Гц, 2H), 7,46 (д,J= 2,2 Гц, 1H), 7,15 (дд, J = 9,2, 2,2 Гц, 1H), 6,93 (д,J= 8,8 Гц, 2H), 6,82 (д, J = 9,1 Гц, 2H), 6,61 (a,J = 9,1 Гц, 2H), 4,21 (м, 2H), 3,89 (с, 3H), 3,75 (с, 3H), 3,45-3,33 (м, 4H), 2,92 (м, 2H), 1,80-1,63 (м, 5H), 1,35 (м, 1H). FD-масс-спектроскопия: 483. Элементный анализ: Рассчитано для C31H33NO4 1,0 HCl:C 71,60; H 6,59; N 2,69. Найдено C 71,77; H 6,66; N 2,79. ПРИМЕР 5 Получение гидрохлорида 1-[4-[2-(1-пиперидинил)этокси] фенокси]-2- (4-гидроксифенил)-6-гидроксинафталина 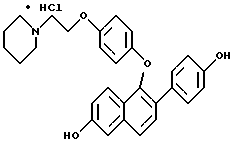 В раствор гидрохлорида 1-[4-[2-(1-пиперидинил)этокси]фенокси]-2-(4-метоксифенил)-6- метоксинафталина (1,00 г, 1,92 ммоль) в 20 мл безводного хлористого метилена в атмосфере N2 при 0oC добавляли трехбромистый бор (0,74 мл, 7,71 ммоль). Полученную смесь выдерживали для нагревания до 8oC и перемешивали 2 ч. Реакционную смесь затем выливали в перемешиваемый холодный насыщенный раствор бикарбоната натрия (200 мл). После прекращения выделения газа водный слой экстрагировали 5% метанол/хлороформ (3 х 100 мл). Органические слои объединяли, сушили (сульфатом натрия) и концентрировали в вакууме до образования масла. Неочищенное свободное основание растворяли в 20 мл этилацетата и обрабатывали системой диэтиловый эфир соляная кислота. Образованный осадок отделяли фильтрованием, сушили в вакууме, получая 798 мг (83%) гидрохлорида 1-[4-[2-(1- пиперидинил)этокси]фенокси]-2-(4-гидроксифенил)-6- гидроксинафталина в виде белого твердого вещества. Т. пл. 130 – 136oC. 1H ЯМР-спектр (DMSO-d6): 9,89 (с, 1H), 9,80 (ш, с, 1H), 9,46 (с, 1H), 7,68 (д,J= 8,5 Гц, 1H), 7,64 (д,J= 9,2 Гц, 1H), 7,50 (д, J= 8,5 Гц, 1H), 7,40 (д, J = 8,8 Гц, 2H), 7,20 (д, J= 2,2 Гц, 1H), 7,04 (дд, J= 9,2 2,2 Гц, 1H), 6,82 (д, J= 9,1 Гц, 2н). 6,76 (д, J= 8,8 Гц, 2H), 6,61 (д, J= 9,1 Гц, 2H), 4,21 (м, 2H), 3,45-3,33 (м, 4H), 2.92 (м, 2H), 1,80-1,63 (м, 5H), 1,35 (м, 1H), FD-масс-спектроскопия: 456. Элементный анализ: Рассчитано для C29H29NO4 1,0 HCl:C 70,80; H 6,15; N 2,85. Найдено: C 70,52; H 6,19; N 2,55.
МЕТОДИКА ИСПЫТАНИЯОбщая препаративная методика В примерах, иллюстрирующих способы, применяли постклимактерическую модель, на которой определяли влияние различных методик лечения на циркулирующие липиды. Самок крыс Sprague Dawley возраста семьдесят пять дней (масса в пределах 200 – 225 г) получают от Charles River Laboratories (Portage, MI), Животных подвергают билатеральной овариэктомии (OVX) или хирургической операции Sham b Charles Rivers Laboratories и затем через неделю отправляют. После прибытия их поселяют в металлических подвешенных клетках группами по 3 или 4 животных на клетку и дают вволю корм (содержание кальция приблизительно 0,5%) и воду в течение одной недели. Комнатную температуру поддерживают 22,2 + 1,7 oC с минимальной относительной влажностью 40%. Фотопериод в комнате составляет 12 час, 12 час в комнате темнота. Отбор тканей при проведении лечебной схемы. После периода акклиматизации в течение одной недели (следовательно, через две недели после OVX) животным начинают давать суточную дозу испытываемого соединения, 17  -этинилэстрадиол или испытываемое соединение вводят, если не оговорено особо, перорально в виде суспензии в 1% карбоксиметилцеллюлозе или раствора в 20% циклодекстрине. Животные получают суточные дозы в течение 4 дней. После проведения схемы лечения животных взвешивают и анестезируют смесью кетамина и ксилазина (2:1, об./об.) и при помощи сердечной пункции отбирают пробу крови. Животных затем умерщвляют асфиксией CO2, матку удаляют разрезом по средней линии и определяют сырой вес матки.
Анализ холестерина.
Пробы крови оставляют для свертывания при комнатной температуре в течение 2 ч, сыворотку получают после центрифугирования в течение 10 мин при 3000 об/мин. Холестерин сыворотки определяют с применением высокоэффективного анализа на холестерин для диагностирования по Boeringer Mannheim. Короче говоря, холестерин окисляют в холест-4-ен-3-он и пероксид водорода. Затем в результате реакции пероксида водорода с фенолом и 4- аминофеназоном в присутствии пероксидазы образуется п-хинониминовый краситель, который определяют спектрофотометрически у 500 нм. Концентрацию холестерина затем подсчитывают по сравнению со стандартной кривой. Весь анализ автоматизируют при помощи Biomek Automated Workstation.
Анализ маточной пероксидазы эозинофилов (ЕРО).
Матку выдерживают при 4oC до времени ферментативного анализа. Матку затем гомогенизируют в 50 объемах 50 мМ трис буфера (pH 8,0), содержащего 0,005% тритона Х-100. При добавлении 0,01% пероксида водорода и 10 мМ о-фенилендиамина (конечные концентрации) в трис- буфер повышение поглощения наблюдают в течение 1 мин у 450 нм. Присутствие эозинофилов в матке указывает на эстрогенную активность соединения. Максимальную скорость с интервалом 15 с определяют в начальной, линейной части реакционной кривой.
Источник соединения: 17 этинилэстрадиол получали от фирмы Sigma Chemical Co., St. Louis, MO.
Методика тестирования остеопороза.
Следуя общей препаративной методике, infra, крыс лечат (обрабатывают) ежедневно в течение 35 дней (6 крыс в подвергаемой лечению группе) и умерщвляют асфилаксией диоксидом углерода на 36-й день. Период времени 35 дней достаточен для максимального восстановления плотности костей, измеренной как описано в изобретении. Во время умерщвления матку удаляют, рассекают для освобождения от посторонней ткани и жидкое содержимое выбрасывают до определения сырого веса, чтобы подтвердить отсутствие эстрогена, связанного с полной овариэктомией. Масса матки, как обычно, снижается на около 75% в ответ на овариэктомию. Матку затем помещают в 10% нейтральный, содержащий буфер формалин, для проведения последующего гистологического анализа.
Вырезают правую бедренную кость и анализируют в рентгеновских лучах, получая оцифрованные результаты анализа. Далее исследование проводят по программе анализа изображения (NIH) для дистального метафиза. Выступающую сторону большеберцовой кости этих животных также сканируют с помощью количественной компьютерной томографии.
В соответствии с указанными выше процедурами, тестируемым животным перорально вводят соединения настоящего изобретения и этинилэстрадиол (ЕЕ2) в 20% гидроксипропил--циклодекстрине.
Определение пролиферации клеток MCF-7 -этинилэстрадиол или испытываемое соединение вводят, если не оговорено особо, перорально в виде суспензии в 1% карбоксиметилцеллюлозе или раствора в 20% циклодекстрине. Животные получают суточные дозы в течение 4 дней. После проведения схемы лечения животных взвешивают и анестезируют смесью кетамина и ксилазина (2:1, об./об.) и при помощи сердечной пункции отбирают пробу крови. Животных затем умерщвляют асфиксией CO2, матку удаляют разрезом по средней линии и определяют сырой вес матки.
Анализ холестерина.
Пробы крови оставляют для свертывания при комнатной температуре в течение 2 ч, сыворотку получают после центрифугирования в течение 10 мин при 3000 об/мин. Холестерин сыворотки определяют с применением высокоэффективного анализа на холестерин для диагностирования по Boeringer Mannheim. Короче говоря, холестерин окисляют в холест-4-ен-3-он и пероксид водорода. Затем в результате реакции пероксида водорода с фенолом и 4- аминофеназоном в присутствии пероксидазы образуется п-хинониминовый краситель, который определяют спектрофотометрически у 500 нм. Концентрацию холестерина затем подсчитывают по сравнению со стандартной кривой. Весь анализ автоматизируют при помощи Biomek Automated Workstation.
Анализ маточной пероксидазы эозинофилов (ЕРО).
Матку выдерживают при 4oC до времени ферментативного анализа. Матку затем гомогенизируют в 50 объемах 50 мМ трис буфера (pH 8,0), содержащего 0,005% тритона Х-100. При добавлении 0,01% пероксида водорода и 10 мМ о-фенилендиамина (конечные концентрации) в трис- буфер повышение поглощения наблюдают в течение 1 мин у 450 нм. Присутствие эозинофилов в матке указывает на эстрогенную активность соединения. Максимальную скорость с интервалом 15 с определяют в начальной, линейной части реакционной кривой.
Источник соединения: 17 этинилэстрадиол получали от фирмы Sigma Chemical Co., St. Louis, MO.
Методика тестирования остеопороза.
Следуя общей препаративной методике, infra, крыс лечат (обрабатывают) ежедневно в течение 35 дней (6 крыс в подвергаемой лечению группе) и умерщвляют асфилаксией диоксидом углерода на 36-й день. Период времени 35 дней достаточен для максимального восстановления плотности костей, измеренной как описано в изобретении. Во время умерщвления матку удаляют, рассекают для освобождения от посторонней ткани и жидкое содержимое выбрасывают до определения сырого веса, чтобы подтвердить отсутствие эстрогена, связанного с полной овариэктомией. Масса матки, как обычно, снижается на около 75% в ответ на овариэктомию. Матку затем помещают в 10% нейтральный, содержащий буфер формалин, для проведения последующего гистологического анализа.
Вырезают правую бедренную кость и анализируют в рентгеновских лучах, получая оцифрованные результаты анализа. Далее исследование проводят по программе анализа изображения (NIH) для дистального метафиза. Выступающую сторону большеберцовой кости этих животных также сканируют с помощью количественной компьютерной томографии.
В соответствии с указанными выше процедурами, тестируемым животным перорально вводят соединения настоящего изобретения и этинилэстрадиол (ЕЕ2) в 20% гидроксипропил--циклодекстрине.
Определение пролиферации клеток MCF-7Клетки адренокарциномы грудной железы MCF-7 (АТСС НТВ 22) сохраняют в MEM (минимальная основная среда, не содержащая фенопрот, Sigma, St. Louis, MO), в которую добавлено 10% фетальная бычья сыворотка (FBS), L-глутамин (2 мМ), пируват натрия (1 мМ), НЕРЕS {(N-[2-гидроксиэтил]пиперазин-N’-[2-этансульфокислота] , 10 мМ} , незаменимые аминокислоты и бычий инсулин (1 мкг/мл) (поддерживающая среда). За десять дней до анализа клетки MCF7 помещают в поддерживающую среду, в которую добавлена 10% фетальная бычья сыворотка, обработанная покрытым декстраном древесным углем (DCC-FBS) (среда для анализа), вместо 10% FBS, для исчерпывания внутренних запасов стероидов. Клетки MCF-7 удаляют из колбы для поддержания при помощи среды для диссоциации клеток (не содержащий Ca++/Mg++HBSS (свободный от фенопрота), в который добавлено 10 мМ HEPES и 2 мМ EDTA). Клетки промывают два раза средой для анализа и концентрацию их устанавливают 80000 клеток/мл. Приблизительно 100 мкл (8000 клеток) добавляют в лунки с плоским дном для микрокультур (Costar 3596) и инкубируют при 37oC в увлажненном 5% CO2, инкубаторе в течение 48 ч, для прикрепления и уравновешивания клеток после переноса. Серийные разведения лекарственных средств или DMSO в качестве контроля на разбавитель готовят в среде для анализа и 50 мкл переносят в тройные микрокультуры и затем добавляют 50 мкл среды для анализа до конечного объема 200 мкл. После дополнительного выдерживания 48 ч при 37oC в увлажненном 5% CO2 инкубаторе микрокультуры пульсируют тритированным тимидином (1 мкКи/лунку) в течение 4 ч. Культивироание завершают замораживанием при -70oC в течение 24 ч с последующим оттаиванием и сбором микрокультур при помощи полуавтоматического скатроновского харвестера клеток. Подсчет импульсов пробы проводят в жидкой фазе с применением -счетчика Wallac Beta Place. Активность соединения формулы I в настоящем анализе показывает, что соединение является потенциальным средством для лечения гормонально зависимого ракового заболевания, конкретно рака молочной железы.
Ингибирование DMBA-индуцированной опухоли молочной железыЭстрогензависимые опухоли молочной железы продуцируют у самок крыс Sprague Dawley, которые приобретают у Harlen Industries, Indianapolis, Indiana. Крысы возраста около 55 дней получают одно пероральное кормление из 20 мг 7,12-диметилбенз/а/антрацена (DMBA). Через 6 недель после введения DMBA молочные железы пальпируют с интервалами в неделю для установления появления опухолей. Всякий раз, когда появляется одна или несколько опухолей, измеряют самый большой и самый малый диаметры каждой опухоли метрическим циркулем, измерения записывают и это животное отбирают для эксперимента. Делается попытка равномерно распределить различные по размерам опухоли по обрабатываемым и контрольный группам так, чтобы опухоли с усредненным размером эквивалентно распределились между тестируемыми группами. Контрольные группы и тестируемые группы для каждого эксперимента содержали 5 – 9 животных. Соединения формулы I вводят внутрибрюшинными инъекциями в 2% акациа или перорально. Перорально вводимые соединения растворяют или суспендируют в 0,2 мл кукурузного масла. Каждое лечение, включая контрольные лечения акациа и кукурузным маслом, проводят однократным ежедневным введением каждому тестируемому животному. После измерения первоначальной опухоли и отбора тестируемых животных опухоли измеряют каждую неделю указанным выше методом. Лечение и измерения животных продолжают 3 – 5 недель, в течение которых определяют конечную площадь опухолей. Определяют изменения в средней опухоли для каждого лечения соединением и контрольного лечения. Методики тестирования фиброза матки Тест 1 От 3 до 20 женщинам, имеющим фиброз матки, вводят соединение настоящего изобретения. Количество введенного соединения составляет 0,1 – 1000,0 мг/день, период введения продолжается 3 месяца. Женщин обследуют в течение периода введения и вплоть до 3 месяцев после окончания введения соединений для определения их действия на фиброз матки. Тест 2 Применяют ту же методику, что в тесте 1, за исключением того, что период введения продолжается 6 месяцев. Тест 3 Применяют ту же методику, что и в тесте 1, за исключением того, что период введения продолжается 1 год. Тест 4 A. Индуцирование фиброзных опухолей у морских свинок. Для индуцирования лейомиомы у половозрелых самок морских свинок применяют пролонгированное стимулирование экстрогеном. Животным вводят эстрадиол 3-5 раз каждую неделю инъекцией в течение 2-4 месяцев или до появления опухолей. Лечения состоят в введении соединения изобретения или наполнителя каждый день в течение 3-16 недель. Затем животных умерщвляют, а матки собирают и анализируют для установления обратного развития опухолей. В. Имплантация маточной фиброзной ткани человека “голым мышам”. Ткань леиомиом человека имплантируют в брюшную полость и/или маточный миометрий половозрелых, кастрированных “самок голых мышей”. Для индуцирования роста трансплантированной ткани вводят экзогенный эстроген. В некоторых случаях собранные опухолевые клетки культивируют in vitro до имплантации. Лечение состоит в том, что соединение настоящего изобретения или наполнитель поставляют путем промывания желудка ежедневно в течение 3 – 16 недель. Имплантаты удаляют и измеряют для определенного роста или обратного развития. Во время умерщвления матки собирают для оценки состояния данного органа. Тест 5 A. Ткани из фиброзных опухолей матки человека собирают и сохраняют in vitro как первичные нетрансформированные культуры. Хирургические образцы протирают через стерильную сетку или сито или альтернативно препарируют при помощи иглы от окружающей ткани для получения суспензии отдельных клеток. Клетки сохраняют в среде, содержащей 10% сыворотку и антибиотик. Определяют скорости роста в присутствии и отсутствие эстрогена. Клетки анализируют на их способность продуцировать компонент С3 комплемента и их ответную реакцию на факторы роста и гормон роста. Культуры in vitro оценивают на их пролиферативную ответную реакцию после обработки прогестином. GnRH (гормон, секретирующий гонадотропин), соединением настоящего изобретения и наполнителем. Содержание рецепторов стероидных гормонов оценивают еженедельно для определения, сохраняются ли in vitro важные свойства клеток. Применяют ткани от 5-25 пациентов. Активность по меньшей мере в одном из указанных выше тестов указывает, что соединения настоящего изобретения являются потенциальными средствами для лечения фиброза матки. Методика тестирования эндометриоза В тестах 1 и 2 можно исследовать влияние 14-дневного и 21-дневного введения соединений настоящего изобретения на рост эксплантированной эндометриальной ткани. Тест 1 От двенадцати до тридцати зрелых самок крыс штамма CD применяют в качестве тестируемых животных. Их разделяют на три группы по равному числу животных в каждой. Проводят мониторинг эстрального цикла всех животных. В день проэструса на каждой самке проводят хирургическую операцию. У самок в каждой группе удаляют левый рог матки, делят его на маленькие квардатики и квадратики свободно нашивают в различных местах по соседству с мезентериальным кровотоком. Кроме того, у самок группы 2 удаляют яичники. На следующий день после хирургической операции животные в группах 1 и 2 получают внутрибрюшенные инъекции воды в течение 14 дней, в то время как животные в группе 3 получают внутрибрюшинные инъекции 1,0 мг соединения настоящего изобретения на 1 кг массы тела в течение такого же времени. После 14 дней лечения самок умерщвляют и удаляют эндометриальные эксплантаты, надпочечники, оставшиеся матки и яичники, если имеются, и подготавливают их для гистологического изучения. Яичники и надпочечники взвешивают. Тест 2 От двенадцати до тридцати зрелых самок крыс штамма CD применяют в качестве тестируемых животных. Их разделяют на две равные группы. Наблюдают эстральный цикл всех животных. В день проэструса на каждой самке проводят хирургическую операцию. У самок в каждой группе удаляют левый рог матки, делят его на маленькие квадратики и квадратики свободно нашивают в различных местах по соседству с мезентеральным кровотоком. Приблизительно через 50 дней после хирургической операции животные группы 1 получают внутрибрюшинные инъекции воды в течение 21 дня, тогда как животные в группе 2 получают внутрибрюшинные инъекции 1 мг соединения настоящего изобретения на 1 кг массы тела в течение того же периода времени. После 21 дней введения каждую самку умерщвляют, удаляют и взвешивают эндометриальные эксплантаты и надпочечники. Эксплантаты измеряют для получения показания по их росту. Проводят мониторинг эстральных циклов. Тест 3 A. Хирургическое индуцирование эндометриоза Для индуцирования эндометриоза у крыс и/или кроликов применяют аутографы эндометриальной ткани. Самок животных в возрасте репродуктивной зрелости подвергают билатеральной овариэктомии и экзогенно снабжают эстрогеном, таким образом обеспечивая конкретный и постоянный уровень гормона. Аутогенную эндометриальную ткань имплантируют в брюшину 5-150 животных и снабжают их эстрогеном для индуцирования роста эксплантированной ткани. Лечение состоит в том, что соединение настоящего изобретения вводят промыванием желудка ежедневно в течение 3 – 16 недель. Имплантаты удаляют и измеряют для определения их роста или обратного развития. Во время умерщвления целый рог матки отделяют для оценки состояния эндометрия. В. Имплантация эндометриальной ткани человека “голым” мышам. Ткань эндометриальных поражений человека имплантируют в брюшину половозрелых, кастрированных самок “голых” мышей. Экзогенный эстроген вводят для индуцирования роста эксплантированной ткани. В некоторых случаях собранные эндометриальные клетки культивируют in vitro до имплантации. Лечение состоит в том, что соединение настоящего изобретения вводят промыванием желудка ежедневно в течение 3 – 16 недель. Имплантаты удаляют и измеряют для определения их роста или обратного развития. Во время умерщвления матку отделяют для оценки состояния интактного эндометрия. Тест 4 A. Ткань эндометриальных повреждений человека отбирают и сохраняют in vitro в качестве первичных нетрасформированных культур. Хирургические образцы протирают через стерильную сетку или сито или альтернативно препарируют при помощи иглы от окружающей ткани для получения суспензий отдельных клеток. Клетки сохраняют в среде, содержащей 10% сыворотку и антибиотик. Определяют скорость роста в присутствии или отсутствии экстрогена. Клетки анализируют на их способность продуцировать компонент С3 комплемента и их ответную реакцию на факторы роста или гормон роста. Культуры in vitro анализируют на их пролиферативную ответную реакцию после обработки прогестинами, GnRH, соединением настоящего изобретения и наполнителем. Уровни рецепторов стероидного гормона оценивают еженедельно для того, чтобы определить, сохраняются ли in vitro. важные свойства клеток. Применяют ткани 5 – 25 пациентов. Активность в любом из этих анализов указывает на то, что соединения настоящего изобретения пригодны для лечения эндометриоза. Методика тестирования ингибирования пролиферации клеток гладких мышц аорты/рестеноза Соединения настоящего изобретения обладают способностью ингибировать пролиферацию клеток гладких мышц аорты. Это можно демонстрировать с применением культивированных клеток гладких мышц аорты кроликов, причем пролиферацию определяют измерением синтеза ДНК. Клетки получают методом эксплантата, как описано в Ross, J. of Cell Biol 50:172 (1971). Клетки помещают в микротитрационные планшеты с 96 лунками на несколько дней. Культуры становятся конфлюэнтными и рост задерживается. Клетки затем переносят в модифицированную по способу Дульбекко среду Игла (DMEM), содержащую 0,5-2% плазму, бедную тромбоцитами, 2 мМ L-глутамина, 100 Е/мл пенициллина, 100 мг/мл стептомицина, 1 мКи/мл 3H-тимидина, 20 нг/мл выделенного из тромбоцитов фактора роста и разные концентрации соединений настоящего изобретения. Основной раствор соединений получают в диметилсульфоксиде и затем разбавляют до подходящей концентрации (0,01-30 мМ) в указанной выше среде для анализа. Клетки затем инкубируют при 37oC в течение 24 ч в атмосфере 5% CO2/95% воздуха. Через 24 ч клетки фиксируют в метаноле. Включение 3H тимидина в ДНК затем определяют подсчетом импульсов, как описано y Bonin, et al., Exp. Cell Res. 181: 475-482 (1989). Ингибирование пролиферации клеток гладких мышц аорты соединениями настоящего изобретения далее демонстрируют путем определения их действия на рост клеток в логарифмической фазе. Клетки гладких мышц аорты кроликов засевают в культуральном планшете с 12 лунками в DMEM, содержащей 10% фетальную бычью сыворотку, 2 мМ L-глутамина, 100 Е/мл пенициллина и 100 мг/мл стрептомицина. Через 24 ч клетки сцепляются и среду заменяют на DMEM, содержащую 10% фетальную бычью сыворотку, 2 мМ L-глутамина, 100 Е/мл пенициллина, 100 мг/мл стрептомицина и желаемые концентрации настоящих соединений. Клетки оставляют расти в течение 4 дней. Клетки обрабатывают трипсином и число клеток в каждой культуре определяют при помощи счетчика ZM-Coulter. Активность в указанных выше тестах указывает, что соединения настоящего изобретения являются потенциальными средствами для лечения рестеноза. Настоящее изобретение предлагает также способ облегчения постклимактерического синдрома у женщин, который содержит указанный выше способ применения соединений формулы I и также предусматривает введение женщине эффективного количества эстрона или прогестина. Такие способы особенно пригодны для лечения остеопороза и снижения содержания сывороточного холестерина, поскольку пациент получает пользу от каждого фармацевтического средства, в то время как соединения настоящего изобретения будут ингибировать нежелательные побочные эффекты эстрогена и прогестина. Активность при лечении такими комбинациями в любых постклимактерических тестах, infra, указывает, что лечения такими комбинациями пригодны для облегчения симптомов постклимактерического синдрома у женщин. Различные формы эстрогена и прогестина коммерчески доступны. Средства на основе эстрогена включают, например, этинилэстроген (0,01-0,03 мг/день), местранол (0,05- 0,15 мг/день) и конъюгированные эстрогенные гормоны, например премарин (Wyerth-Auerst; 0,3-2,5 мг/день). Средства на основе прогестина включают, например метоксипрогестерон, такой как провера (Upjohn; 2,5-10,0 мг/день), норэтилнодрел (1-10 мг/день) и нонэтиндрон (0,5-2,0 мг/день). Предпочтительным соединением на основе эстрогена является премарин, норэтилнодрел и норэтиндрон являются предпочтительными средствами на основе прогестина.
Способ введения такого средства на основе эстрогена или прогестина согласуется со способом введения, который известен в настоящей области. Для большинства способов настоящего изобретения соединения формулы 1 вводят постоянно 1-3 раза ежедневно. Однако можно в частности применять цикличную терапию при лечении эндометриоза или ее можно применять экстренно во время болезненных приступов заболевания. В случае рестеноза терапия может быть ограничена до коротких (1 – 6 месяцев) интервалов после медицинских процедур, например пластической операции на сосудах.
В соответствии с изобретением термин “эффективное количество” обозначает количество соединения настоящего изобретения, которое способно облегчить симптомы различных описанных патологических состояний. Конкретная доза соединения, введенная в соответствии с настоящим изобретением, конечно, будет определяться конкретными обстоятельствами, окружающими данный случай, включая, например, вводимое соединение, путь введения, состояние пациента и патологическое состояние, для которого требуется лечение. Типичная суточная доза будет содержать нетоксичное количество (от около 5 мг до около 600 мг/день) соединения настоящего изобретения. Предпочтительные суточные дозы обычно будут от 15 мг до около 80 мг/день.
Соединения настоящего изобретения можно вводить различными путями, включая пероральный, ректальный, чрескожный, подкожный, внутривенный, внутримышечный и внутриназальный. До введения из соединений предпочтительно приготовляют готовые лекарственные формы, выбор которых будет решаться штатным врачом больницы. Таким образом, другой особенностью настоящего изобретения является фармацевтическая композиция, содержащая эффективное количество соединения формулы I или его фармацевтически приемлемой соли, возможно содержащая эффективное количество эстрогена или прогестина и фармацевтически приемлемый носитель, разбавитель или наполнитель.
Общее содержание активных компонентов в таких готовых лекарственных формах составляет 0,1-99,9% от их массы. Термин “фармацевтически приемлемый” обозначает, что носитель, разбавитель, наполнители и соль должны быть совместимы с другими компонентами готовой формы и не вредны для их реципиента.
Фармацевтические готовые формы настоящего изобретения можно получить различными процедурами, известными в настоящей области науки, с применением хорошо известных и легкодоступных компонентов. Например, из соединений формулы I, с эстрогеном или прогестероном или без них, можно приготовить готовые лекарственные формы с обычными наполнителями, разбавителями или носителями в виде таблеток, капсул, суспензий, порошков и т.д. Примеры наполнителей, разбавителей и носителей, которые пригодны для таких готовых форм, включают наполнители и сухие разбавители, например крахмал, сахар, маннит и кремневые производные; связывающие агенты, например карбоксиметилцеллюлозу и другие производные целлюлозы, альгинаты, желатин и поливинилпирролидон; увлажняющие агенты, например глицерин; дезинтегрирующие агенты, например карбонат кальция и бикарбонат натрия; агенты для замедления растворения, например парафин; ускорители всасывания, например соединения четвертичного аммония; поверхностно-активные вещества, например цетиловый спирт, моностеарат глицерина; адсорбирующие носители, например каолин и бентонит; и смазочные материалы, например тальк, стеаринат кальция и магния и твердые полиэтиленгликоли.
Соединения можно также готовить в составе готовых форм в виде эликсиров или растворов для обычного перорального введения или растворов, пригодных для парентерального введения, например, внутримышечным, подкожным или внутривенным путями. Кроме того, соединения вполне пригодны для приготовления готовых лекарственных дозированных форм с постепенным высвобождением активного компонента и тому подобное. Готовые формы могут быть составлены таким образом, чтобы они могли высвобождать активный компонент только или предпочтительно в конкретном физиологическом месте, возможно в течение некоторого периода времени. Можно изготовлять покрытия, оболочки и защитные матрицы, например из полимерных веществ или восков.
Соединения формулы I, возможно в комбинации с фармацевтическим средством настоящего изобретения, обычно будут вводить в составе стандартной готовой лекарственной формы. Следующие примеры готовых лекарственных форм только иллюстрируют настоящее изобретение и не предназначены для ограничения объема его.
Готовые лекарственные формыВ приведенных ниже готовых лекарственных формах термин “активный компонент” обозначает соединение формулы I или его соль или сольват. Готовая форма 1: Желатиновые капсулы Твердые желатиновые капсулы получают с применением следующих компонентов, мг/капсула%: Активный компонент – 0,1-1000,0 Крахмал, NF – 0-650 Сыпучий порошок крахмала – 0-650 Силиконовая жидкость (350 сСт) – 0-15 Указанную выше готовую форму можно изменить в соответствии с приемлемыми получаемыми разновидностями. Таблетированную готовую форму получают с применением указанных ниже компонентов: Готовая форма 2: Таблетки, мг/таблетка: Активный компонент – 2,5-1000,0 Целлюлоза, микрокристаллическая – 200-650 Диоксид кремния, дымящий – 10-650 Стеариновая кислота – 5-15 Компоненты смешивают и прессуют для образования таблеток. Альтернативно, таблетки, каждая из которых содержит 2,5-1000 мг активного компонента, изготавливают следующим образом: Готовая форма 3: Таблетки, мг/таблетка: Активный компонент – 25-1000 Крахмал – 45 Целлюлоза, микрокристаллическая – 35 Поливинилпирролидон – 4 (в виде 10%-ного раствора в воде) Натриевая соль карбоксиметилцеллюлозы – 4,5 Стеарат магния – 0,5 Тальк – 1 Активный компонент, крахмал и целлюлозу пропускают через сито N 45 меш США и тщательно перемешивают. Раствор поливинилпирролидона смешивают с полученными порошками, которые затем пропускают через сито N 14 меш США. Полученные таким образом гранулы сушат при 50-60oC и пропускают через сито N 18 меш США. Натриевую соль карбоксиметилкрахмала, стеарат магния и тальк, предварительно пропущенные через сито N 60 меш США, затем добавляют в гранулы, которые после перемешивания прессуют на таблеточной машине, получая таблетки. Суспензии, каждая из которых содержит 0,1-1000,0 мг лекарственного средства (активного компонента) в 5 мл дозе, изготавливают следующим образом: Готовая форма 4: Суспензии, мг/5 мл: Активный компонент – 0,1-1000,0 Натриевая соль карбоксиметилцеллюлозы – 50 Сироп – 1,25 Раствор бензойной кислоты, мл – 0,10 Корригент – Сколько нужно Красящее вещество – Сколько нужно Очищенная вода до получения, мл – 5 Лекарственное средство (активный компонент) пропускают через сито N 45 меш и смешивают с натриевой солью карбоксиметилцеллюлозы и сиропом для образования однородной пасты. Раствор бензойной кислоты, корригент и красящее вещество разбавляют некоторым количеством воды и добавляют при перемешивании. Затем добавляют достаточное количество воды, чтобы получить требуемый объем суспензии. Получают аэрозольный раствор, содержащий следующие компоненты: Готовая форма 5: Аэрозоль, мас.%: Активный компонент – 0,25 Этанол – 25,75 Пропеллент 22 (хлордифторметан) – 70,00 Активный компонент смешивают с этанолом и смесь добавляют в часть пропеллента 22, охлаждают до 30oC и переносят в устройство для наполнения. Требуемое количество затем направляют в контейнер из нержавеющей стали и разбавляют оставшимся количеством пропеллента. На контейнер затем прилаживают клапанный блок. Суппозитории получают следующим образом. Готовая форма 6: Суппозитории, мг: Активный компонент – 250 Глицериды насыщенных жирных кислот – 200 Активный компонент пропускают через сито N 60 меш США и суспендируют в глицеридах насыщенных жирных кислот, предварительно расплавленных с применением минимально необходимого тепла. Смесь затем наливают в формы для суппозиторий с номинальной емкостью на 2 г и оставляют для охлаждения. Внутривенную готовую форму получают следующим образом: Готовая форма 7: Внутривенный раствор: Активный компонент, мг – 50 Изотонический солевой раствор, мл – 1000 Раствор указанных выше компонентов вводят пациенту внутривенно со скоростью около 1 мл в мин. Готовая форма 8: Комбинированная капсула I, мг/капсула: Активный компонент – 50 Премарин – 1 Авицел pH 101 – 50 Крахмал 1500 – 117,50 Силиконовое масло – 2 Твин 80 – 0,50 Каб-О-сил – 0,25 Готовая форма 9: Комбинированная капсула II, мг/капсула: Активный компонент – 50 Норэтилнодрел – 5 Авицел pH 101 – 82,50 Крахмал 1500 – 90 Силиконовое масло – 2 Твин 80 – 0,50 Готовая форма 10: Комбинированная таблетка, мг/таблетка: Активный компонент – 50 Премарин – 1 Кукурузный крахмал NF – 50 Повидон К29-32 – 6 Авицел pH 101 – 41,50 Авицел pH 102 – 136,50 Кросповидон XL 10 – 2,50 Стеарат магния – 0,50 Каб-О-Сил – 0,50 Результаты биологической/токсикологической активности для соединения примера 5 представлены в таблицах 1 и 2. Пример 6. Следующие соединения были получены по методике примера 4 или 5 (все полученные соединения сведены заявителем в таблицу, 3 в том числе в таблице указаны уже раскрытые и охарактеризованные в заявке соединения 1 и 2, которые являются соединениями примера 4 и 5 соответственно). Физико-химические данные для полученных соединений представлены в таблице 4, при этом заявитель напоминает, что данные для соединений 1 и 2 ранее представлены в описании. Данные биологической активности для ряда заявленных соединений, испытываемых на моделях после OVX в течение 4 дней, приведены в таблице 5. Результаты испытаний соединений на пролиферацию MCF-7 представлены в таблице 6. Формула изобретения
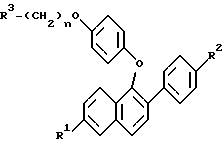 где R1 и R2 – Н, -ОН или -О(С1-С4-алкил); R3 – 1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 4-морфолинил, диметиламино-, диэтиламино-, диизопропиламино- или 1-гексаметилениминогруппа; n = 2 или 3, или их фармацевтически приемлемые соли. 2. Соединение по п.1, где R1 – -ОН, R2 – -ОН, R3 – 1-пиперидинил, n = 2, или его фармацевтически приемлемая соль. 3. Соединение по п.2, где соль представляет собой гидрохлорид. 4. Соединение по п.1, где R1 – Н, -ОН или -ОСН3, R2 – Н, -ОН или ОСН3, n = 2, или его фармацевтически приемлемая соль. 5. Соединение по п.4, где соль представляет собой гидрохлорид. 6. Фармацевтическая композиция, ингибирующая проявление симптомов постклимактерического синдрома, содержащая в качестве активного компонента соединение по любому из пп.1 – 5 и, необязательно, эстроген или прогестин вместе с одним или несколькими фармацевтически приемлемыми носителями, наполнителями или разбавителями. 7. Соединение формулы II 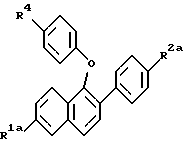 где R1а – Н или ОR5, где R5 – гидроксизащитная группа; R2а – Н или ОR6, где R6 – гидроксизащитная группа; R4 – -ОН или -СНО. 8. Соединение по п.7, где R1а – -ОR5, где R5 – метил, R2а – -ОR6, где R6 – метил. 9. Соединение по п.8, где R4 – -ОН. 10. Способ получения соединений формулы (I) или их фармацевтически приемлемых солей по любому из пп.1 – 5, отличающийся тем, что включает а) взаимодействие соединений формулы IIв 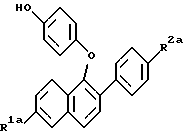 где R1а – Н или -ОR5, где R5 – гидроксизащитная группа; R2а – Н или -ОR6, где R6 – гидроксизащитная группа, с соединением формулы V R3-(СН2)n-Q, где R3 имеет значения, указанные в п.1; Q – уходящая группа; n = 2 или 3, в) необязательно удаление гидроксизащитной группы или групп, с) необязательно образование соли продукта стадии (а) или стадии (в). 11. Соединение формулы (I) по любому из пп.1 – 5 для применения в качестве средств для облегчения симптомов постклимактерического синдрома. 12. Соединение по п.11 для применения в качестве средства для облегчения симптомов постклимактерического синдрома, представляющих собой остеопороз, связанное с ним сердечно-сосудистое заболевание, гиперлипидемию или гормонально зависимое раковое заболевание. 13. Соединение формулы (I) по любому из пп.1 – 5 для применения в качестве средства для ингибирования фиброзного заболевания матки, эндометриоза, пролиферации клеток гладких мышц аорты или рестеноза. РИСУНКИ
MM4A Досрочное прекращение действия патента Российской Федерации на изобретение из-за неуплаты в установленный срок пошлины за поддержание патента в силе
Дата прекращения действия патента: 27.02.2003
Извещение опубликовано: 20.11.2004 БИ: 32/2004
|
||||||||||||||||||||||||||